

Abstract
Aims:
S-glutathionylation is a reversible oxidative modification of protein cysteines that plays a critical role in redox signaling. Glutaredoxin-1 (Glrx), a glutathione-specific thioltransferase, removes protein S-glutathionylation. Glrx, though a cytosolic protein, can activate a nuclear protein Sirtuin-1 (SirT1) by removing its S-glutathionylation. Glrx ablation causes metabolic abnormalities and promotes controlled cell death and fibrosis in mice. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH), a key enzyme of glycolysis, is sensitive to oxidative modifications and involved in apoptotic signaling via the SirT1/p53 pathway in the nucleus. We aimed to elucidate the extent to which S-glutathionylation of GAPDH and glutaredoxin-1 contribute to GAPDH/SirT1/p53 apoptosis pathway.
Results:
Exposure of HEK 293T cells to hydrogen peroxide (H2O2) caused rapid S-glutathionylation and nuclear translocation of GAPDH. Nuclear GAPDH peaked 10–15 minutes after the addition of H2O2. Overexpression of Glrx or redox dead mutant GAPDH inhibited S-glutathionylation and nuclear translocation. Nuclear GAPDH formed a protein complex with SirT1 and exchanged S-glutathionylation to SirT1 and inhibited its deacetylase activity. Inactivated SirT1 remained stably bound to acetylated-p53 and initiated apoptotic signaling resulting in cleavage of caspase-3. We observed similar effects in human primary aortic endothelial cells suggesting the GAPDH/SirT1/p53 pathway as a common apoptotic mechanism.
Conclusions:
Abundant GAPDH with its highly reactive-cysteine thiolate may function as a cytoplasmic rheostat to sense oxidative stress. S-glutathionylation of GAPDH may relay the signal to the nucleus where GAPDH trans-glutathionylates nuclear proteins such as SirT1 to initiate apoptosis. Glrx reverses GAPDH S-glutathionylation and prevents its nuclear translocation and cytoplasmic-nuclear redox signaling leading to apoptosis. Our data suggest that trans-glutathionylation is a critical step in apoptotic signaling and a potential mechanism that cytosolic Glrx controls nuclear transcription factors.
Keywords: Glutaredoxin, S-glutathionylation, trans-glutathionylation, GAPDH, SirT1
Graphical Abstract
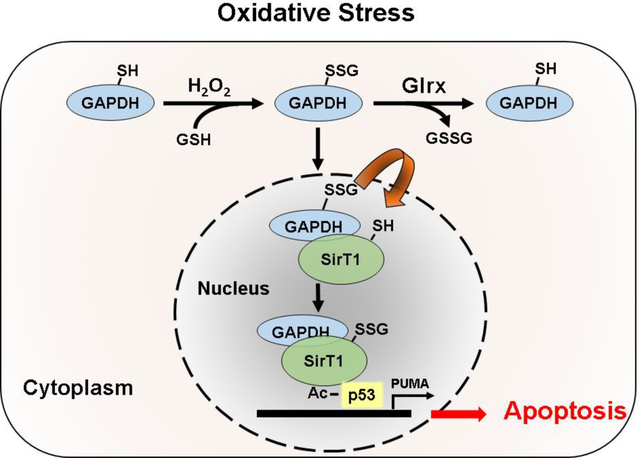
Introduction
Redox regulation plays an essential role in numerous aspects of cellular function. Reversible oxidative post-translational modifications (OPTM), including S-nitrosylation and S-glutathionylation of protein cysteines [1–6], confer biological responses to oxidants in physiology and pathological conditions [7]. Recent studies indicate that S-glutathionylation acts as a regulatory modification resembling transit phosphorylation in cell signaling [8–11]. S-glutathionylation refers to the reversible adduction of the tripeptide glutathione (GSH), an abundant and vital low molecular weight thiol, to protein cysteines. Protein cysteines that exist in the deprotonated thiolate form are strong nucleophiles with high chemical reactivity towards reactive oxygen and nitrogen species (RONS). In a protein, positively charged or coordinating amino acid side chains surrounding the cysteine can stabilize the thiolate making it more reactive. Thus, physiological amounts of RONS only target specific reactive cysteines in a protein leading to reversible OPTMs for redox signaling. The reaction of RONS with protein thiolates results in intermediates that can react further with GSH to form an S-glutathionylated protein [12–16]. Intracellular GSH or thiol transferases, including glutaredoxins, then remove S-glutathionylation [17–21]. Some groups refer to these protein thiolates as peroxidatic cysteines because the net reaction resembles the catalysis of a thiol peroxidase if a peroxide such as hydrogen peroxide (H2O2) controls this signaling mechanism [22].
A reductive cellular environment or glutaredoxins can reduce S-glutathionylation [23,24]. Mammalian cells have two principal isoforms of glutaredoxins. Glutaredoxin-1 (Glrx) mainly localizes in cytoplasm, while glutaredoxin-2 resides in mitochondria and has the specific activity less than 10% of Glrx. Compared to a slow spontaneous chemical reduction, Glrx specifically binds to S-glutathionylated proteins, removes the modification through an exchange reaction, and becomes S-glutathionylated itself. Then, Glrx releases GSH by forming oxidized-GSH (GSSG), back to the reduced active form.
Thus, Glrx does exchange reactions of glutathione, or trans-glutathionylation, which is not restricted to glutaredoxins and can occur between other interacting proteins [25]. Thus, proteins carrying reactive thiolates have the potential to transfer S-glutathionylation, GSH adducts, to the interacting protein and participate in redox signaling cascades. This “redox relay” mechanism would permit oxidative signals to cross cellular compartments and might be involved in the regulation of nuclear transcription factors.
Glyceraldehyde 3-phosphate dehydrogenase (GAPDH), a key enzyme of glycolysis, is involved in many other crucial cellular processes including DNA repair [26,27], tRNA export [28], membrane fusion in ER to Golgi shuttling [29], cytoskeletal dynamics [30–34], and apoptosis [35–41]. Post-translational modifications [36,41] such as S-glutathionylation [42] and S-nitrosylation [16], oligomerization status [43], and subcellular localization partially control the diverse functions of GAPDH [33,39]. S-nitrosylation is a well characterized oxidative post-translational modification of GAPDH [44–47]. Hara and colleagues showed that S-nitrosylation of GAPDH triggered binding to ubiquitin ligase Siah1, promoting nuclear translocation and apoptotic signaling [36]. Furthermore, the cysteine at the active site of GAPDH is required for its redox sensitivity, nuclear translocation, and induction of apoptosis [36,48]. Kornberg and colleagues reported that S-nitrosylated GAPDH can trans-nitrosylated nuclear proteins and suggested that protein trans-nitrosylation might be a general molecular mechanism [49].
Compared to S-nitrosylation, S-glutathionylation, in general, is a more stable oxidative modification [16]. It is not known if S-glutathionylated GAPDH can transfer redox signals to nuclear proteins. S-glutathionylation occurs at the active site cysteine thiolate and inactivates GAPDH activity, leading to conformational changes [42]. This structural transformation leads to opening of the GAPDH protein structure and permits interaction with other proteins and may allow GAPDH to trans-glutathionylate the interacting proteins.
In this study, we show that cytosolic Glrx regulates S-glutathionylation of GAPDH and participates in cytoplasmic-to-nuclear signaling. S-glutathionylation of GAPDH initiated translocation to the nucleus where it interacted, S-glutathionylated, and inactivated Sirtuin-1 (SirT1). S-glutathionylation inactivated SirT1 led to p53 acetylation and apoptosis. Overexpression of Glrx reversed GAPDH S-glutathionylation, prevented nuclear translocation of GAPDH, and attenuated SirT1 inactivation. Our data suggest that GAPDH-glutathionylation is a mechanism to transfer redox signals to the nucleus leading to apoptosis, and thus cytosolic Glrx can control nuclear transcription.
Materials and Methods
Reagents
All biochemical reagents were purchased from Millipore Sigma (St. Louis, MO) and all cell culture reagents from Gibco (Thermo Fisher Scientific, Waltham, MA) and Lonza (Walkersville, MD) unless specified otherwise. FLAG-HA-tagged SirT1 was a kind gift from P. Puigserver (Harvard University, MA, USA). Protein A/G beads (Santa Cruz, Dallas, TX) and antibodies were obtained from the following companies: anti-GAPDH monoclonal antibody (Cell Signaling, Danvers, MA; 1:1000), anti-β-Tubulin monoclonal antibody (Cell Signaling; 1:1000), anti-HA antibody (Cell Signaling; 1:1000), anti-biotin antibody (Millipore Sigma, St. Louis, MO; 1:2000), anti-HDAC1 (Cell Signaling, Danvers, MA; 1:1000), anti-acetylated p53 (Cell Signaling, Danvers, MA 1:1000), anti-p53 (Cell Signaling, Danvers, MA; 1:1000), anti-Glrx1 (Cayman Chemicals, Ann Arbor, MI; 1:1000), anti-SirT1 (Abcam, Cambridge, MA, 1 μg/ml) HA-magnetic beads (Cell Signaling, Danvers, MA). HRP-conjugated secondary antibodies were from GE Healthcare (Pittsburgh, PA) or eBioscience (San Diego, CA) TrueBlot system. Antibody sources and dilutions are presented in Supplemental Table 1.
Cloning, adenovirus generation, and infection
The human GAPDH clone was a kind gift of Dr. S. Snyder, Johns Hopkins University School of Medicine (Baltimore, MD) [49] and cloned into pcDNA3.1 FLAG HA plasmid (Plasmid #52535, Addgene, Cambridge, MA) using EcoRI and XhoI restriction sites. Cysteines were mutated to serines using the QuikChange II site-directed mutagenesis kit (Agilent, Santa Clara, CA) according to the manufacturer’s instructions. Primers for mutagenesis were designed using the QuikChange primer design web application. All clones were verified by sequencing using GENEWIZ (Cambridge, MA). Primer sequences are presented in Supplemental Table 2. Construction of SirT1 mutants and generation of adenoviruses were performed as previously described [10]. In this study the GAPDH triple mutant (C152S, C156S, C247S) as TRI and SirT1 triple mutant (C61S, C318S, C613S) as 3MUT.
Cell culture, transfection, and treatment
HEK 293T (passage 4 to 6) cells were cultured in Dulbecco modified essential medium (DMEM) supplemented with 5% v/v fetal bovine serum and 1% antibiotic and antimycotic solution, maintained in 5% CO2 and 95% humidity. Primary human aortic endothelial cells (HAEC) (Passage 4 to 6) were maintained in EBM-2 Basal Medium (Lonza, Walkersville, MD) supplemented with ascorbic acid, VEGF, rhFGF-B, recombinant IGF-1, heparin, hydrocortisone, rhEGF, gentamicin sulfate, amphotericin-B, and 2% fetal bovine serum as per the manufacturer protocol. Cells were grown in 10 cm or 6-well cell culture dishes to 60% confluency and were transfected with 4 μg of control (empty vector) and expression plasmids using polyethylenimine (PEI; Polyscience, Warrington, PA) at a ratio of 3:1 (PEI:DNA) [47]. Adenoviral transduction of primary cells was performed as per the protocol of Watanabe et al. [10]. Cells were serum starved overnight before the experiments and treated with 500 μM H2O2 to induce oxidative stress.
Immunofluorescent staining
Cells were grown on chamber slides at 37 °C in a cell culture incubator. At the time of fixation, cells were ~50% confluent. Cells were fixed with 4% paraformaldehyde in PBS for 15 minutes at room temperature and permeabilized with 0.5% Triton X-100 in PBS. Samples were blocked with 5% FBS in PBS at room temperature for 1 hour. The primary antibody was diluted to the recommended concentration in 5% FBS in PBS, and 200 μl per well was added to the chamber slides and incubated 2–3 hours at room temperature or overnight at 4 °C. Fluorochrome-conjugated secondary antibodies were prepared in 5% FBS in PBS according to the manufacturer’s recommendations, and 200 μl per well was added to the chamber slides. The samples were incubated for 1 hour at room temperature in the dark. The cells were mounted with mounting medium.
Cysteine labeling assay
The assay was performed as per the protocol established by Boivin et al. with slight modifications [50]. For the GAPDH cysteine labeling assay, the lysis buffer has been prepared in degassed deionized water, containing 50 mM sodium acetate, 150 mM NaCl, and 10 ml glycerol, mixed under vacuum overnight at 4 °C. 1% NP40 was added and the buffer was mixed again for 1 hour under vacuum at room temperature. The degassed buffer was transferred to an InvivO2 300 anaerobic workstation (Baker Ruskinn, Sanford, ME) filled with nitrogen to minimize protein oxidation during cell lysis. Freshly prepared 10 mM iodoacetic acid (IAA), 125 U/ml superoxide dismutase, 250 U/ml catalase, 5 μg/ml leupeptin, and 5 μg/ml aprotinin, were added and the buffer was protected from light. Cells were treated with 500 μM H2O2 for 15 minutes to induce oxidative stress under standard cell culture conditions. After the incubation time was completed, cells were immediately lysed in the hypoxic chamber and lysates were transferred into sterile amber 1.5 ml microcentrifuge tubes, and kept on a shaker for 1 hour at room temperature in the dark. Lysates were spun at 10,000 x g for 10 minutes through Zeba desalting columns (Thermo Fisher Scientific, Waltham, MA) to exchange to the lysis buffer without additions. Lysates were treated with 1 mM tris (2-carboxyethyl) phosphine for 30 minutes at room temperature to reduce the reversibly oxidized protein thiols and then labeled by incubating with 5 mM iodoacetyl-PEG2-biotin at room temperature for 1 hour. Biotinylated proteins were enriched using streptavidin magnetic beads overnight on a rotator at 4 °C. The beads were washed twice with ice-cold buffer, resuspended in Laemmli buffer, heated, and proteins were separated on NuPAGE 4–12% gradient polyacrylamide gels using NuPAGE MES SDS running buffer (Thermo Fisher Scientific, Waltham, MA). Proteins were then transferred onto a PVDF membrane and probed with the GAPDH primary antibody. For SirT1 cysteine labeling, we followed the protocol per Shao et al. [10].
Mass spectroscopy
Wild type (WT)-GAPDH-HA overexpressed HEK 293T cells were treated with or without H2O2. Cells were lysed in a hypoxia chamber using a buffer containing NaCl 150 mM, NP40 1%, HEPES 50 mM (pH 8.0), glycerol 10% and iodoacetamide (IAM) 5 mM. Lysates were alkylated for 30 minutes at room temperature in dark, centrifuge at 10,000 x g for 10 minutes, desalted through Zeba columns to removed excess IAM. WT HA-tag GAPDH (2 mg of total protein) was immunoprecipitated with anti-HA magnetic (10 μl; NEB) beads overnight. We washed beads 3 times with PBS, added 30 μl of the elution buffer containing (6 M Urea, Tris-HCl 20 mM, NaCl 100 mM), and rotated tubes for 30 minutes at room temperature. Proteins were collected using a magnetic stand, diluted to decrease urea concentration from 6 M to 1 M with 50 mM TEAB buffer, added 1 μl trypsin Lys-C protease mix and 3 μl of Rapigest surfactant (Waters). We incubated it overnight on a rotator at 37 °C for digestion and terminated digestion (and cleave acid-labile Rapigest) by adding formic acid to a final concentration of 1%. We desalted peptides using Oasis HLB 1cc (30 mg) reversed-phase cartridges (Waters, Milford, MA) with 0.1% formic acid (buffer A) and 0.1% formic acid, 80% acetonitrile (buffer B), respectively, using a vacuum manifold. Condition cartridges with 3 × 500 μl buffer B followed by equilibration with 4 × 500 μl buffer A. After loading the digests at a reduced flow rate, wash with 3 × 750 μl buffer A. Eluted with three times 500 μl 25% acetonitrile and 0.1% formic acid, followed by a second elution with 30% acetonitrile and 0.1% formic acid and third elution in 35% acetonitrile and 0.1% formic acid and dried in speed vacuum.
Proteomics and mass spectrometry data analysis
Glutathionylation of GAPDH was analyzed using PMI-Byonic version (Protein Metrics) modifications. Data was analyzed with Search GUI-3.3.17 and Peptide Shaker-1.16.44 using the search algorithms X!Tandem, MyriMatch, MS-GF+, and the de novo peptide sequencing Novor and DirecTag. The search parameters for PMI-Byonic were the following: peptide termini, semi-specific; maximum number of missed cleavages, 5; precursor tolerance, 10 ppm; fragment tolerance, 10 ppm; charges applied to charge-unassigned spectra, 2–5; fragmentation type, HCD (Higher-energy C-trap dissociation) or HCD and ETD (Electron transfer dissociation); The coverage of the GAPDH sequence was greater than 85%.
Nuclear and cytoplasmic protein extraction
Cells were transfected or infected and treated with respective conditions and lysed on ice for 10 minutes in a lysis buffer containing 10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, and 0.05% NP40. The lysate was centrifuged at 1000 x g for 10 minutes at 4 °C, and the resulting supernatant was recovered as the cytosolic fraction. Pellets were resuspended in a nuclear extraction buffer containing 5 mM HEPES, 1.5 mM MgCl2, 300 mM NaCl, 0.2 mM EDTA, and 25 % glycerol. Pellets were lysed by sonication and centrifuged at 15,700 x g for 10 minutes at 4 °C. The supernatant contained the nuclear fraction [49].
Immunoblotting and co-immunoprecipitation
Proteins were separated on NuPAGE 4–12% Bis-Tris protein gels using NuPAGE MES SDS running buffer (Thermo Fisher, Waltham, MA) and transferred on PVDF membranes using Trans-Blot Turbo transfer system (Bio-Rad Laboratories, Hercules, CA). Membranes were blocked with 5% nonfat milk, incubated with primary antibodies overnight, washed three times with TBST, and probed with respective secondary antibodies for 1 hour at room temperature.
For immunoprecipitation, cell lysates were rotated at 4 °C overnight with 2 μg of SirT1 antibody that had been pre-coupled with 20 μl of protein A/G beads for 1 hour. Beads were rinsed and proteins separated by SDS-polyacrylamide gel electrophoresis. Overexpressed FLAG-SirT1 was directly immunoprecipitated with anti-FLAG antibody coated magnetic beads. Protein-protein interactions were identified by blotting for the respective interacting protein using GAPDH, SirT1, and Ac-p53 antibodies. Membranes were developed using the KwikQuant Western blot detection kit and images were recorded using the KwikQuant imager (Kindle Biosciences, Greenwich, CT).
GAPDH activity
GAPDH activity was measured with a GAPDH activity kit (Abcam, Cambridge, MA) as per the manufacturer’s protocol. Briefly, HEK 293T or HAEC were treated with or without 500 μM H2O2 for 5–15 minutes, lysed in assay buffer on ice for 10 minutes, centrifuged at 10,000 x g for 5 minutes, and the supernatants were used for GAPDH activity. Samples were diluted in an assay buffer to which GAPDH substrate and developer were added, and the absorbance was read at 450 nm. For experiments involving transfected cells, immunoprecipitated WT and cysteine mutant GAPDH were used.
Statistical analysis
All experiments were performed for a minimum of three times, and the results are presented as mean ± SD. Statistical analysis was performed using Prism 8.1.0 (GraphPad Software, San Diego, CA, USA). Statistical significance was determined using one-way ANOVA with Tukey’s multiple comparison test. Differences were considered statistically significant with a p-value of <0.05. For enzyme kinetics, we used the Michaelis-Menten equation for substrate vs. velocity to determine Km and Vmax.
Results
Oxidative stress-induced S-glutathionylation of GAPDH led to its nuclear translocation
Oxidative and nitrosative stress are known to induce oxidative cysteine modifications of GAPDH, leading to its nuclear translocation. To detect the S-glutathionylation of GAPDH, we performed a cysteine labeling assay and mass spectrometry in HEK 293T cells exposed to H2O2. Hydrogen peroxide increased reversible cysteine oxidation of endogenous GAPDH that was reversed by overexpressed Glrx (Fig. 1A & B). After H2O2 exposure, a significant fraction of endogenous GAPDH maximally translocated to the nucleus in 5–10 minutes (Fig. 1C & D). The cytosolic and nuclear fractions appeared to be minimal cross-contaminated based on the absence of β–tubulin in the nuclear fraction and HDAC1 in cytosolic fraction. Furthermore, we examined nuclear translocation of the triple cysteine-to-serine mutant GAPDH (TRI; C152S, C156S, C247S) (Fig. 1E), which does not have redox-sensitive cysteine residues. The triple mutant GAPDH translocated significantly less to the nucleus as compared to WT GAPDH following H2O2 exposure (Fig. 1E & F). Nuclear translocation appeared not to require GAPDH enzyme activity because H2O2 inhibits GAPDH enzyme activity (Fig. 1G & H).
Figure 1. Oxidative stress induced S-glutathionylation and nuclear translocation of GAPDH in HEK 293T cells.


A) Hydrogen peroxide increased GAPDH S-glutathionylation in HEK 293T cells as measured by cysteine labeling. Glutaredoxin-1 overexpression attenuated GAPDH S-glutathionylation. B) The graph shows the density of biotinylated-(pulled down) GAPDH, control as 1. C) Hydrogen peroxide caused maximal GAPDH translocation to the nucleus in 5–15 minutes based on subcellular fractionation studies. The absence of β–tubulin in the nuclear fraction demonstrated minimal cross contamination. D) Bar graph showing nuclear translocation of GAPDH in a time dependent manner (n=3). For panels A-D, HEK 293T cells were transfected with Glrx or an empty vector (pcDNA 3.1) as a control, exposed with and without 500 μM H2O2 for 15 minutes. E) A cysteine-to-serine mutant GAPDH (TRI) minimally translocated to the nucleus in response to H2O2 compared to WT GAPDH. F) Bar graph showing fold change nuclear translocation WT vs mutant GAPDH compared to control (n=3). Density of nuclear HA-GAPDH after H2O2 exposure is considered as 1. **p<0.01. G) Hydrogen peroxide inhibited endogenous GAPDH activity in HEK 293T cells after 10 minutes. Kinetics were fitted using the Michaelis-Menten plugin in Prism 8.1.0. Data is presented as means ± SD of N = 3. H) Hydrogen peroxide markedly inhibited the GAPDH activity of overexpressed WT GAPDH in HEK 293T cells. Independent on H2O2 exposure, overexpressed mutant GAPDH was catalytically inactive. For panel E-F, HEK 293T cells were transfected with HA-labelled WT or 3 Cys-mutant (TRI) GAPDH or an empty vector (EV) as a control. Cells were treated with 500 μM H2O2 for 15 minutes. n=3. GAPDH activity is presented as a fold change of control. Data are presented as means ± SD of n=3 and analyzed with Mann-Whitney U test (*p<0.05). Statistical significance was determined using one-way ANOVA with Tukey’s multiple comparison test. Differences were considered statistically significant with a p-value of <0.05.
Moreover, our MS data show S-glutathionylation at cysteine 247 position of GAPDH (Fig. 2B), while cysteine 152 and 156 was involved in disulfide bonding following H2O2 treatment (Fig. 2A & C). HCD-ETD did not yield good spectra for sulfinylation and sulfonylation but preserved-well S-glutathionylation of C247 and disulfide formation of C152-C156. The disulfide crosslinked and cyclic section of peptide was more resistant to fragmentation.
Figure 2. Glutathionylation of GAPDH occurs on cysteines in HEK 293T cells.


A) Controls MS spectra is showing carbamidomethylation which represents alkylation of protein cysteines. B) Hydrogen peroxide treatment causes S-glutathionylation at cysteine 247. C) while, cysteine 152 and 156 formed reversible disulfide bonding following hydrogen peroxide exposure.
Glrx prevented GAPDH nuclear translocation and interaction with SirT1
SirT1 is a regulator of cellular stress responses. In many cell types, including HEK 293T and endothelial cells, SirT1, NAD+-dependent histone deacetylase, predominantly resides in the nucleus and regulates acetylation and activity of transcription factors [51]. As shown in Fig. 3A, GAPDH and SirT1 were co-immunoprecipitated after H2O2 treatment. The interaction was absent in untreated cells. Overexpression of Glrx prevented SirT1-GAPDH interaction. These data suggest that S-glutathionylated GAPDH induced GAPDH-SirT1 interaction. It is known that S-glutathionylation of SirT1 inactivates its activity (10).
Figure 3. Glrx prevented the association of GAPDH with SirT1.

A) Hydrogen peroxide induced GAPDH and SirT1 association in HEK 293T cells as demonstrated by co-immunoprecipitation. Glutaredoxin-1 (Glrx) overexpression markedly attenuated this association. B) The cysteine-to-serine mutant SirT1 (3MUT) prevented the H2O2-induced GAPDH-Ac-p53-SirT1 association demonstrated by co-immunoprecipitation. C) Different concentrations of H2O2 increased cleaved caspase-3. Glutaredoxin-1 overexpression abolished caspase-3 cleavage. GAPDH was used as a loading control. D) Quantitative assessment of cleaved-caspase 3 bands is shown as fold changes of caspase 3 activation in different groups (n=3). **p<0.01 compared to control as 1 (white bar). ##p<0.01 compared to between 500 μM H2O2 vs 500 μM H2O2 + Glrx. For panels A, B, and C, HEK 293T cells were transfected with empty vector (pcDNA 3.1), FLAG-SirT1, or Glrx with and without exposure to 500 μM H2O2 for 15 minutes. For caspase activity cells were treated with H2O2 for 8 hrs. Statistical significance was determined using one-way ANOVA with Tukey’s multiple comparison test. Differences were considered statistically significant with a p-value of <0.05.
SirT1 required redox-sensitive cysteines for interacting with GAPDH and acetylated P53 (Ac-p53) under oxidative stress
To determine whether the interaction required redox-sensitive cysteines of SirT1, we overexpressed FLAG-tagged WT or our triple cysteine-to-serine mutant SirT1 (C61S, C318S, C613S; 3MUT) in HEK 293T cells. We previously verified that 3MUT SirT1 is non-oxidizable and maintains its deacetylase activity under oxidative and metabolic stress [10,11]. Normally, SirT1 interacts acetylated p53 (Ac-p53), de-acetylates, and inactivates p53, leading anti-apoptotic signaling. Under control conditions, neither WT nor 3MUT SirT1 interacted with GAPDH or Ac-p53 (Fig. 3B). HEK 293T cells exposed to H2O2 showed a marked increase in the SirT1-GAPDH-Ac-p53 protein association. In contrast, 3MUT SirT1 attenuated protein-protein interaction in cells exposed to H2O2 (Fig. 3B).
Glutaredoxin-1 overexpression diminished the activation of cleaved caspase-3 (Fig. 3C & D), indicating less inactivation of SirT1. In summary, the data indicated the cysteines of SirT1 modulated the deacetylase activity as well as interaction with GAPDH and Ac-p53.
GAPDH exchanged S-glutathionylation with SirT1
To determine if GAPDH transfers S-glutathionylation to SirT1, we performed a series of cysteine labeling experiments overexpressing the triple cysteine-to-serine mutants for GAPDH (TRI) and SirT1 (3MUT) in HEK 293T cells exposed to H2O2.
Hydrogen peroxide induced S-glutathionylation in WT SirT1 but not 3MUT SirT1 in HEK 293T cells (Fig. 4A). The TRI GAPDH decreased endogenous SirT1 thiol modifications compared to WT GAPDH in control and H2O2 exposed cells (Fig. 4B). However, overexpression of WT or 3MUT SirT1 minimally prevented cysteine oxidation of endogenous GAPDH (Fig. 4C). These effects of TRI GAPDH occurred independently of its activity because the TRI GAPDH has negligible catalytic activity (Fig. 1H).
Figure 4. GAPDH trans-glutathionylated SirT1.

A) Hydrogen peroxide reversibly oxidized overexpressed WT SirT1 as measured by cysteine labeling. The overexpressed cysteine-to-serine mutant SirT1 (3MUT) showed only minor oxidation. B) Overexpressed mutant GAPDH (TRI) attenuated H2O2-induced oxidation of endogenous SirT1 in HEK 293T. C) Neither overexpressed WT nor mutant SirT1 (3MUT) affected H2O2-mediated oxidation of endogenous GAPDH. For all panels, HEK 293T cells were transfected with WT or mutant FLAG-SirT1 (3MUT) and WT or mutant HA-GAPDH (TRI).
These experiments demonstrated that S-glutathionylation of GAPDH was up-stream of S-glutathionylation of SirT1, indicating nuclear GAPDH trans-glutathionylated SirT1 during oxidative stress.
Oxidative stress-induced GAPDH S-glutathionylation led to its nuclear translocation in human endothelial cells
Next, we examined whether H2O2 induces S-glutathionylation and nuclear translocation of GAPDH in HAEC to provide additional evidence that this mechanism also occurs in primary human cells. First, we performed a cysteine labeling assay and confirmed an H2O2-dependent increase in S-glutathionylated GAPDH (Fig. 5A). Overexpression of Glrx decreased S-glutathionylation of GAPDH in both control and H2O2 exposed HAEC. Overexpression of Glrx in HAEC inhibited H2O2-induced nuclear translocation of GAPDH, as shown by Western blot (Fig. 5B & C) and immunocytochemistry (Fig 5D & E). H2O2 exposure also decreased endogenous GAPDH activity (Fig. 5F).
Figure 5. Oxidative stress induced S-glutathionylation and nuclear translocation of GAPDH in HAEC.


A) Hydrogen peroxide increased S-glutathionylation of GAPDH as measured by cysteine labeling. Glrx overexpression reversed GAPDH S-glutathionylation. B) Overexpression of Glrx inhibited H2O2-induced nuclear translocation of GAPDH in HAEC as detected by western blot. C) Bar graph showing fold change of nuclear translocation in western blot (n=3). Significance signs (*) compared to control and (#) compared between H2O2 vs Glrx+H2O2. D) immunofluorescence (green; Glrx, red; GAPDH; blue; nucleus, scale bar, 50 μm), representative photos showing H2O2-induced nuclear translocation, which is inhibited by Glrx overexpression (arrow heads). E) Bar graph showing fold change of nuclear translocation in immunofluorescence (n=3). Cell number with nuclear GAPDH in control is shown as 100%. F) Hydrogen peroxide inhibited endogenous GAPDH activity in HAEC. GAPDH activity is presented as a fold change of control. For all panels, HAECs were treated with 500 μM H2O2 for 15 minutes. Data are presented as means ± SD and were analyzed with Mann-Whitney U test. p-value of <0.05 (*), 0.01 (**), 0.001 (***). All the experiments repeated a minimum of three times.
Glrx prevented pro-apoptotic signaling in human endothelial cells exposed to H2O2
Overexpression of Glrx in HAEC decreased GAPDH-SirT1 interaction (Fig. 6A & B) as demonstrated by co-immunoprecipitation. Next, we investigated whether the decreased GAPDH-SirT1 interaction consequently may control SirT1 activity and apoptotic signaling in HAEC. H2O2 markedly increased Ac-p53 indicating SirT1 inactivation, while Glrx overexpression suppressed it (Fig. 6C & D). These results in HAEC further corroborate our findings in HEK 293T cells. To assess proapoptotic signaling initiated by S-glutathionylated GAPDH -SirT1inhibition -Ac-p53, we treated cells with H2O2 for 8 hours with and without Glrx overexpression. Hydrogen peroxide treatment markedly increased cleaved caspase-3 levels that were significantly abolished by Glrx overexpression (Fig. 6E & F).
Figure 6. Glrx attenuated S-glutathionylation of GAPDH and apoptotic signaling in HAEC exposed to H2O2.


A) Hydrogen peroxide induced GAPDH and SirT1 interaction measured by co-immunoprecipitation that was completely abolished by adenoviral Glrx (GFP-Glrx) overexpression (n=3). B) Bar graph showing fold change of GAPDH band after IP with anti-SirT1, indicating GAPDH-SirT1 interaction (P< 0.001). C) Hydrogen peroxide induced Ac-p53 in HAEC, indicating inactive SirT1. Adenoviral Glrx overexpression inhibited the acetylation of p53. Total p53 expression levels remained unchanged as measured by Western blot. D) Bar graph showing ratio of Acy-p53 and total p53 of western blot (n=3) (P< 0.001). E) Hydrogen peroxide caused cleavage of caspase-3 that was attenuated by Glrx overexpression. F) Bar graph showing fold change activation of caspase-3 by densitometric analysis of 19/17 kDa bands (n=3). *p<0.05 compared to other groups. For all panels, HAEC were treated with 500 μM H2O2 for 15 min, except caspase 3 activation in which cells were treated with H2O2 for 8 hrs. GAPDH served as a loading control. Statistical significance was determined using one-way ANOVA with Tukey’s multiple comparison test. Differences were considered statistically significant with a p-value of <0.05 (*), 0.01 (**), 0.001 (***). Significance signs (*) compared to control and (#) compared between H2O2 vs Glrx+H2O2..
Discussion
S-glutathionylation is the reversible addition of glutathione to reactive protein cysteine thiolates that change the structure and function of the modified protein [7]. Under severe oxidative stress, S-glutathionylation may protect essential protein cysteines from irreversible oxidation and degradation [52,53]. S-glutathionylation has emerged as a critical regulator of cell signaling, glucose and lipid metabolism, cell proliferation, and apoptosis.
GAPDH is a glycolytic protein but also has several non-glycolytic functions. We tested the role of GAPDH as a glutathione shuttle to nuclear SirT1 during oxidant-induced SirT1-p53-mediated apoptosis. We showed that oxidants-induced S-glutathionylated GAPDH translocated to the nucleus, interacted with SirT1, and caused S-glutathionylation-mediated SirT1 inactivation and apoptosis. Overexpression of Glrx prevented S-glutathionylation and nuclear translocation of GAPDH, and inhibited GAPDH-SirT1 interaction and apoptotic signaling. Our data indicate that a redox relay involving trans-glutathionylation is a potential signaling mechanism in the apoptosis pathway.
Previous studies suggested S-nitrosylation as a mechanism to regulate nuclear translocation of GAPDH and inactivation of redox-sensitive nuclear proteins [49]. S-nitrosylation or GSNO can also lead to S-glutathionylation of proteins. Brune and coworkers have shown that S-nitrosylated GAPDH with an authentic NO+ donor is unstable, and in the absence of GSH, quickly regains enzyme activity [16]. In contrast, GSNO causes stable S-glutathionylation of GAPDH, inhibiting enzyme activity, that could only be restored by a chemical reductant.
It is possible that strong nucleophilic protein cysteine thiolates (GAPDH has a low pka cysteine thiolate stabilized by an adjacent histidine) attack the S—NO bond of GSNO, catalyzing protein S-glutathionylation. Conversely, weak nucleophilic cysteines cannot break this bond, and GSNO is more likely to S-nitrosylate these cysteines [16]. Thus, GSNO under nitrosative stress also causes GAPDH S-glutathionylation [17]. HEK 293T cells are deficient in NO synthases, and thus, significant S-nitrosylation unlikely occur [56]. Under our experimental conditions using H2O2, the thiolate of GAPDH may form a sulfenic acid that quickly reacts with GSH and becomes S-glutathionylated protein as we showed by mass spectrometry (Fig 2). A recent study by Barinova and colleagues [43] demonstrated that S-glutathionylation introduces a conformational change in GAPDH, opening the structure, which likely facilitates protein interactions and trans-glutathionylation. The precise mechanism of trans-glutathionylation may need a further study. Kornberg et al. showed Thr152Ala (our 154) mutation inhibited GAPDH binding to SirT1 [49]. Therefore, it is unlikely their interaction occurs by disulfide. We found intra-molecule disulfide formation between Cys152 and 156 in GAPDH under oxidative conditions. This intra-molecule disulfide may protect GAPDH from inter-protein disulfide formation with other proteins or further oxidation. We speculate conformational changes, reactivity of other thiols, proximity, and cellular redox status may determine trans-glutathionylation.
Redox relay is an understudied concept that thiol modification can be transferred between proteins via peroxidases and oxidoreductases [25,55,56]. In our best knowledge, this is a first report showing Glrx-controlled S-glutathionylation transfers the signal from cytosol to the nucleus by exchanging GSH adducts between redox-active proteins. In our case, GAPDH exchanges S-glutathionylation with SirT1, a central regulator of the cellular stress response and metabolism [51]. Also, the redox-dead mutant GAPDH had minimal enzyme activity and did not translocate to the nucleus in cells exposed to H2O2. However, inactivated WT GAPDH by S-glutathionylation can translocate into the nucleus, suggesting that S-glutathionylation, not its activity, is required for the nuclear translocation and GAPDH acts upstream to SirT1. We speculate it is a mechanism by which a cytosolic enzyme Glrx controls S-glutathionylation and activity of SirT1 in the nucleus and other redox-sensitive transcription factors. As limitations, S-glutathionylation occurs relatively small portion of a protein. We found 15–20% of total GAPDH translocated to nucleus. Also, a small amount of Cys-mutant GAPDH (TRI) likely translocated in the nucleus. Other modifications may also support GAPDH nuclear translocation [41, 59]. On another note, we cannot exclude that Glrx or Glrx2 resides in the nucleus. Glrx can be intermembrane space of mitochondria [60], but not reported in the nucleus. Glrx2 is a mitochondrial isoform and a weak de-glutathionylation activity compared to Glrx. Glrx in the nucleus may also directly interact with modified SirT1, representing a second check-point for unintended initiation of apoptosis.
In summary, we conclude that trans-glutathionylation is a signaling mechanism to relay redox signals from the cytoplasm into the nucleus. Because GAPDH is an abundant enzyme containing a highly reactive cysteine thiolate, it may function as a redox rheostat to sense oxidative stress. Upon S-glutathionylation, GAPDH can translocate to the nucleus, interact with SirT1 and exchange the modification. S-glutathionylation inactivates SirT1, which interacts with Ac-p53 to form a transcriptionally active complex, initiating proapoptotic signaling and caspase-3 activation. Glrx as glutathione-specific thioltransferase prevents S-glutathionylation of GAPDH and SirT1, inhibiting apoptosis. Thus, Glrx is a promising therapeutic to inhibit excessive apoptosis in pathological conditions such as idiopathic lung fibrosis [57], preeclampsia [58], and non-alcoholic steatohepatitis [10], as well as Glrx may also protect vascular functions by protecting endothelial cell apoptosis in stress conditions.
Supplementary Material
Highlights.
Cytosolic glutaredoxin-1 reverses S-glutathionylation of SirT1 in the nucleus.
S-glutathionylation at Cys247 is required for GAPDH nuclear translocation.
Nuclear GAPDH interacts SirT1 and causes apoptosis via trans-glutathionylation.
Cys-mutant GAPDH attenuates its nuclear translocation and binding to SirT1.
Glutaredoxin-1 inhibits GAPDH nuclear translocation and SirT1-mediated apoptosis.
Acknowledgments
This work was supported by NIH grants R01 DK103750, R01 HL133013, R03 AG 051857 and NIH CTSI award 1UL1TR001430, American Heart Association “Grant in Aid” 16GRNT27660006, the Metabolic Clinical Research Collaborative, pilot funding by the Whitaker Cardiovascular Institute. The article contents are solely the responsibility of the authors and do not necessarily represent the official views of the awarding offices. M.M.B. was supported by the Evans Junior Faculty Research Award by the Department of Medicine of Boston University. We thank Drs. M. Kirber, F. Seta, and L. Deng and the Boston University School of Medicine “Analytical Instrumentation”, “Cellular Imaging”, and “Metabolic Phenotyping” Cores for their technical support.
Abbreviations
- Ac
acetylation
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- Glrx
glutaredoxin-1
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- HAEC
human aortic endothelial cells
- HDAC
histone deacetylase
- H2O2
hydrogen peroxide
- IB
immunoblot
- IP
immunoprecipitation
- SirT1
sirtuin-1 class III NAD+-dependent histone deacetylase
- WB
Western blot
- WT
wild type
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Short JD, Downs K, Tavakoli S, Asmis R, Protein Thiol Redox Signaling in Monocytes and Macrophages, Antioxid. Redox Signal. 25 (2016) 816–835. 10.1089/ars.2016.6697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ullevig S, Kim HS, Asmis R, S-glutathionylation in monocyte and macrophage (dys)function, Int. J. Mol. Sci. 14 (2013) 15212–15232. 10.3390/ijms140815212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gorelenkova Miller O, Mieyal JJ, Sulfhydryl-mediated redox signaling in inflammation: role in neurodegenerative diseases, Arch. Toxicol. 89 (2015) 1439–1467. 10.1007/s00204-015-1496-7. [DOI] [PubMed] [Google Scholar]
- [4].Allen EMG, Mieyal JJ, Protein-thiol oxidation and cell death: regulatory role of glutaredoxins, Antioxid. Redox Signal. 17 (2012) 1748–1763. 10.1089/ars.2012.4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].van der Vliet A, Janssen-Heininger YMW, Anathy V, Oxidative stress in chronic lung disease: From mitochondrial dysfunction to dysregulated redox signaling, Mol. Aspects Med. 63 (2018) 59–69. 10.1016/j.mam.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Janssen-Heininger YMW, Nolin JD, Hoffman SM, van der Velden JL, Tully JE, Lahue KG, Abdalla ST, Chapman DG, Reynaert NL, van der Vliet A, Anathy V, Emerging mechanisms of glutathione-dependent chemistry in biology and disease, J. Cell. Biochem. 114 (2013) 1962–1968. 10.1002/jcb.24551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pimentel D, Haeussler DJ, Matsui R, Burgoyne JR, Cohen RA, Bachschmid MM, Regulation of cell physiology and pathology by protein S-glutathionylation: lessons learned from the cardiovascular system, Antioxid. Redox Signal. 16 (2012) 524–542. 10.1089/ars.2011.4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Murdoch CE, Shuler M, Haeussler DJF, Kikuchi R, Bearelly P, Han J, Watanabe Y, Fuster JJ, Walsh K, Ho Y-S, Bachschmid MM, Cohen RA, Matsui R, Glutaredoxin-1 up-regulation induces soluble vascular endothelial growth factor receptor 1, attenuating post-ischemia limb revascularization, J. Biol. Chem. 289 (2014) 8633–8644. 10.1074/jbc.M113.517219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Watanabe Y, Murdoch CE, Sano S, Ido Y, Bachschmid MM, Cohen RA, Matsui R, Glutathione adducts induced by ischemia and deletion of glutaredoxin-1 stabilize HIF-1α and improve limb revascularization, Proc. Natl. Acad. Sci. U. S. A. 113 (2016) 6011–6016. 10.1073/pnas.1524198113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Shao D, Fry JL, Han J, Hou X, Pimentel DR, Matsui R, Cohen RA, Bachschmid MM, A redox-resistant sirtuin-1 mutant protects against hepatic metabolic and oxidant stress, J. Biol. Chem. 289 (2014) 7293–7306. 10.1074/jbc.M113.520403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Shao D, Han J, Hou X, Fry J, Behring JB, Seta F, Long MT, Roy HK, Cohen RA, Matsui R, Bachschmid MM, Glutaredoxin-1 Deficiency Causes Fatty Liver and Dyslipidemia by Inhibiting Sirtuin-1, Antioxid. Redox Signal. 27 (2017) 313–327. 10.1089/ars.2016.6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Schuppe-Koistinen I, Moldéus P, Bergman T, Cotgreave IA, S-thiolation of human endothelial cell glyceraldehyde-3-phosphate dehydrogenase after hydrogen peroxide treatment, Eur. J. Biochem. 221 (1994) 1033–1037. [DOI] [PubMed] [Google Scholar]
- [13].Niture SK, Velu CS, Bailey NI, Srivenugopal KS, S-thiolation mimicry: quantitative and kinetic analysis of redox status of protein cysteines by glutathione-affinity chromatography, Arch. Biochem. Biophys. 444 (2005) 174–184. 10.1016/j.abb.2005.10.013. [DOI] [PubMed] [Google Scholar]
- [14].Coles SJ, Easton P, Sharrod H, Hutson SM, Hancock J, Patel VB, Conway ME, S-Nitrosoglutathione inactivation of the mitochondrial and cytosolic BCAT proteins: S-nitrosation and S-thiolation, Biochemistry. 48 (2009) 645–656. 10.1021/bi801805h. [DOI] [PubMed] [Google Scholar]
- [15].Giustarini D, Milzani A, Aldini G, Carini M, Rossi R, Dalle-Donne I, S-nitrosation versus S-glutathionylation of protein sulfhydryl groups by S-nitrosoglutathione, Antioxid. Redox Signal. 7 (2005) 930–939. 10.1089/ars.2005.7.930. [DOI] [PubMed] [Google Scholar]
- [16].Mohr S, Hallak H, de Boitte A, Lapetina EG, Brüne B, Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase, J. Biol. Chem. 274 (1999) 9427–9430. [DOI] [PubMed] [Google Scholar]
- [17].Axelsson K, Eriksson S, Mannervik B, Purification and characterization of cytoplasmic thioltransferase (glutathione:disulfide oxidoreductase) from rat liver, Biochemistry. 17 (1978) 2978–2984. [DOI] [PubMed] [Google Scholar]
- [18].Gan ZR, Wells WW, Purification and properties of thioltransferase, J. Biol. Chem. 261 (1986) 996–1001. [PubMed] [Google Scholar]
- [19].Hatakeyama M, Tanimoto Y, Mizoguchi T, Purification and some properties of bovine liver cytosol thioltransferase, J. Biochem. (Tokyo). 95 (1984) 1811–1818. [DOI] [PubMed] [Google Scholar]
- [20].Holmgren A, Hydrogen donor system for Escherichia coli ribonucleoside-diphosphate reductase dependent upon glutathione, Proc. Natl. Acad. Sci. U. S. A. 73 (1976) 2275–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Fernandes AP, Holmgren A, Glutaredoxins: glutathione-dependent redox enzymes with functions far beyond a simple thioredoxin backup system, Antioxid. Redox Signal. 6 (2004) 63–74. 10.1089/152308604771978354. [DOI] [PubMed] [Google Scholar]
- [22].Klomsiri C, Karplus PA, Poole LB, Cysteine-based redox switches in enzymes, Antioxid. Redox Signal. 14 (2011) 1065–1077. 10.1089/ars.2010.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Grek CL, Zhang J, Manevich Y, Townsend DM, Tew KD, Causes and consequences of cysteine S-glutathionylation, J. Biol. Chem. 288 (2013) 26497–26504. 10.1074/jbc.R113.461368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yang Y, c Jao S, Nanduri S, Starke DW, Mieyal JJ, Qin J, Reactivity of the human thioltransferase (glutaredoxin) C7S, C25S, C78S, C82S mutant and NMR solution structure of its glutathionyl mixed disulfide intermediate reflect catalytic specificity, Biochemistry. 37 (1998) 17145–17156. 10.1021/bi9806504. [DOI] [PubMed] [Google Scholar]
- [25].Stöcker S, Van Laer K, Mijuskovic A, Dick TP, The Conundrum of Hydrogen Peroxide Signaling and the Emerging Role of Peroxiredoxins as Redox Relay Hubs, Antioxid. Redox Signal. 28 (2018) 558–573. 10.1089/ars.2017.7162. [DOI] [PubMed] [Google Scholar]
- [26].Meyer-Siegler K, Mauro DJ, Seal G, Wurzer J, deRiel JK, Sirover MA, A human nuclear uracil DNA glycosylase is the 37-kDa subunit of glyceraldehyde-3-phosphate dehydrogenase, Proc. Natl. Acad. Sci. U. S. A. 88 (1991) 8460–8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Perucho M, Salas J, Salas ML, Identification of the mammalian DNA-binding protein P8 as glyceraldehyde-3-phosphate dehydrogenase, Eur. J. Biochem. 81 (1977) 557–562. [DOI] [PubMed] [Google Scholar]
- [28].Singh R, Green MR, Sequence-specific binding of transfer RNA by glyceraldehyde-3-phosphate dehydrogenase, Science. 259 (1993) 365–368. [DOI] [PubMed] [Google Scholar]
- [29].Tisdale EJ, Glyceraldehyde-3-phosphate dehydrogenase is required for vesicular transport in the early secretory pathway, J. Biol. Chem. 276 (2001) 2480–2486. 10.1074/jbc.M007567200. [DOI] [PubMed] [Google Scholar]
- [30].Zala D, Hinckelmann M-V, Yu H, Lyra da Cunha MM, Liot G, Cordelières FP, Marco S, Saudou F, Vesicular glycolysis provides on-board energy for fast axonal transport, Cell. 152 (2013) 479–491. 10.1016/j.cell.2012.12.029. [DOI] [PubMed] [Google Scholar]
- [31].Kumagai H, Sakai H, A porcine brain protein (35 K protein) which bundles microtubules and its identification as glyceraldehyde 3-phosphate dehydrogenase, J. Biochem. (Tokyo). 93 (1983) 1259–1269. [DOI] [PubMed] [Google Scholar]
- [32].Glaser PE, Gross RW, Rapid plasmenylethanolamine-selective fusion of membrane bilayers catalyzed by an isoform of glyceraldehyde-3-phosphate dehydrogenase: discrimination between glycolytic and fusogenic roles of individual isoforms, Biochemistry. 34 (1995) 12193–12203. [DOI] [PubMed] [Google Scholar]
- [33].Reiss N, Oplatka A, Hermon J, Naor Z, Phosphatidylserine directs differential phosphorylation of actin and glyceraldehyde-3-phosphate dehydrogenase by protein kinase C: possible implications for regulation of actin polymerization, Biochem. Mol. Biol. Int. 40 (1996) 1191–1200. [DOI] [PubMed] [Google Scholar]
- [34].Schmitz H-D, Bereiter-Hahn J, Glyceraldehyde-3-phosphate dehydrogenase associates with actin filaments in serum deprived NIH 3T3 cells only, Cell Biol. Int. 26 (2002) 155–164. 10.1006/cbir.2001.0819. [DOI] [PubMed] [Google Scholar]
- [35].Ishitani R, Chuang DM, Glyceraldehyde-3-phosphate dehydrogenase antisense oligodeoxynucleotides protect against cytosine arabinonucleoside-induced apoptosis in cultured cerebellar neurons, Proc. Natl. Acad. Sci. U. S. A. 93 (1996) 9937–9941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, Ferris CD, Hayward SD, Snyder SH, Sawa A, S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding, Nat. Cell Biol. 7 (2005) 665–674. 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- [37].Ishitani R, Kimura M, Sunaga K, Katsube N, Tanaka M, Chuang DM, An antisense oligodeoxynucleotide to glyceraldehyde-3-phosphate dehydrogenase blocks age-induced apoptosis of mature cerebrocortical neurons in culture, J. Pharmacol. Exp. Ther. 278 (1996) 447–454. [PubMed] [Google Scholar]
- [38].Ishitani R, Sunaga K, Hirano A, Saunders P, Katsube N, Chuang DM, Evidence that glyceraldehyde-3-phosphate dehydrogenase is involved in age-induced apoptosis in mature cerebellar neurons in culture, J. Neurochem. 66 (1996) 928–935. [DOI] [PubMed] [Google Scholar]
- [39].Sawa A, Khan AA, Hester LD, Snyder SH, Glyceraldehyde-3-phosphate dehydrogenase: nuclear translocation participates in neuronal and nonneuronal cell death, Proc. Natl. Acad. Sci. U. S. A. 94 (1997) 11669–11674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Saunders PA, Chalecka-Franaszek E, Chuang DM, Subcellular distribution of glyceraldehyde-3-phosphate dehydrogenase in cerebellar granule cells undergoing cytosine arabinoside-induced apoptosis, J. Neurochem. 69 (1997) 1820–1828. [DOI] [PubMed] [Google Scholar]
- [41].Ventura M, Mateo F, Serratosa J, Salaet I, Carujo S, Bachs O, Pujol MJ, Nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase is regulated by acetylation, Int. J. Biochem. Cell Biol. 42 (2010) 1672–1680. 10.1016/j.biocel.2010.06.014. [DOI] [PubMed] [Google Scholar]
- [42].Barinova KV, Serebryakova MV, Muronetz VI, Schmalhausen EV, S-glutathionylation of glyceraldehyde-3-phosphate dehydrogenase induces formation of C150-C154 intrasubunit disulfide bond in the active site of the enzyme, Biochim. Biophys. Acta Gen. Subj. 1861 (2017) 3167–3177. 10.1016/j.bbagen.2017.09.008. [DOI] [PubMed] [Google Scholar]
- [43].Qvit N, Joshi AU, Cunningham AD, Ferreira JCB, Mochly-Rosen D, Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) Protein-Protein Interaction Inhibitor Reveals a Non-catalytic Role for GAPDH Oligomerization in Cell Death, J. Biol. Chem. 291 (2016) 13608–13621. 10.1074/jbc.M115.711630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Foster MW, Hess DT, Stamler JS, Protein S-nitrosylation in health and disease: a current perspective, Trends Mol. Med. 15 (2009) 391–404. 10.1016/j.molmed.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hess DT, Stamler JS, Regulation by S-nitrosylation of protein post-translational modification, J. Biol. Chem. 287 (2012) 4411–4418. 10.1074/jbc.R111.285742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mohr S, Stamler JS, Brüne B, Posttranslational modification of glyceraldehyde-3-phosphate dehydrogenase by S-nitrosylation and subsequent NADH attachment, J. Biol. Chem. 271 (1996) 4209–4214. [DOI] [PubMed] [Google Scholar]
- [47].Haldar SM, Stamler JS, S-nitrosylation: integrator of cardiovascular performance and oxygen delivery, J. Clin. Invest. 123 (2013) 101–110. 10.1172/JCI62854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Nakajima H, Amano W, Fujita A, Fukuhara A, Azuma Y-T, Hata F, Inui T, Takeuchi T, The active site cysteine of the proapoptotic protein glyceraldehyde-3-phosphate dehydrogenase is essential in oxidative stress-induced aggregation and cell death, J. Biol. Chem. 282 (2007) 26562–26574. 10.1074/jbc.M704199200. [DOI] [PubMed] [Google Scholar]
- [49].Kornberg MD, Sen N, Hara MR, Juluri KR, Nguyen JVK, Snowman AM, Law L, Hester LD, Snyder SH, GAPDH mediates nitrosylation of nuclear proteins, Nat. Cell Biol. 12 (2010) 1094–1100. 10.1038/ncb2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Boivin B, Tonks NK, Analysis of the redox regulation of protein tyrosine phosphatase superfamily members utilizing a cysteinyl-labeling assay, Methods Enzymol. 474 (2010) 35–50. 10.1016/S0076-6879(10)74003-9. [DOI] [PubMed] [Google Scholar]
- [51].Feige JN, Auwerx J, Transcriptional targets of sirtuins in the coordination of mammalian physiology, Curr. Opin. Cell Biol. 20 (2008) 303–309. 10.1016/j.ceb.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sabens Liedhegner EA, Gao X-H, Mieyal JJ, Mechanisms of altered redox regulation in neurodegenerative diseases--focus on S--glutathionylation, Antioxid. Redox Signal. 16 (2012) 543–566. 10.1089/ars.2011.4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Zaffagnini M, Michelet L, Marchand C, Sparla F, Decottignies P, Le Maréchal P, Miginiac-Maslow M, Noctor G, Trost P, Lemaire SD, The thioredoxin-independent isoform of chloroplastic glyceraldehyde-3-phosphate dehydrogenase is selectively regulated by glutathionylation, FEBS J. 274 (2007) 212–226. 10.1111/j.1742-4658.2006.05577.x. [DOI] [PubMed] [Google Scholar]
- [54].Hasegawa K, Yoshikawa K, Necdin regulates p53 acetylation via Sirtuin1 to modulate DNA damage response in cortical neurons, J. Neurosci. Off. J. Soc. Neurosci. 28 (2008) 8772–8784. 10.1523/JNEUROSCI.3052-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Peralta D, Bronowska AK, Morgan B, Dóka É, Van Laer K, Nagy P, Gräter F, Dick TP, A proton relay enhances H2O2 sensitivity of GAPDH to facilitate metabolic adaptation, Nat. Chem. Biol. 11 (2015) 156–163. 10.1038/nchembio.1720. [DOI] [PubMed] [Google Scholar]
- [56].Eroglu E, Hallström S, Bischof H, Opelt M, Schmidt K, Mayer B, Waldeck-Weiermair M, Graier WF, Malli R. Real-time visualization of distinct nitric oxide generation of nitric oxide synthase isoforms in single cells. Nitric Oxide. 2017November1;70:59–67. doi: 10.1016/j.niox.2017.09.001. Epub 2017 Sep 4. PMID: 28882669; PMCID: PMC6002809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Anathy V, Lahue KG, Chapman DG, Chia SB, Casey DT, Aboushousha R, van der Velden JLJ, Elko E, Hoffman SM, McMillan DH, Jones JT, Nolin JD, Abdalla S, Schneider R, Seward DJ, Roberson EC, Liptak MD, Cousins ME, Butnor KJ, Taatjes DJ, Budd RC, Irvin CG, Ho Y-S, Hakem R, Brown KK, Matsui R, Bachschmid MM, Gomez JL, Kaminski N, van der Vliet A, Janssen-Heininger YMW, Reducing protein oxidation reverses lung fibrosis, Nat. Med. 24 (2018) 1128–1135. 10.1038/s41591-018-0090-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Guerby P, Swiader A, Augé N, Parant O, Vayssière C, Uchida K, Salvayre R, Negre-Salvayre A, High glutathionylation of placental endothelial nitric oxide synthase in preeclampsia, Redox Biol. 22 (2019) 101126. 10.1016/j.redox.2019.101126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Park J, Han D, Kim K, Kang Y, Kim Y, O-GlcNAcylation disrupts glyceraldehyde-3-phosphate dehydrogenase homo-tetramer formation and mediates its nuclear translocation. Biochim Biophys Acta. 1794 (2009) 254–262. 10.1016/j.bbapap.2008.10.003 [DOI] [PubMed] [Google Scholar]
- [60].Pai HV, Starke DW, Lesnefsky EJ, Hoppel CL, Mieyal JJ. What is the functional significance of the unique location of glutaredoxin 1 (GRx1) in the intermembrane space of mitochondria? Antioxid Redox Signal. 9 (2007) 2027–2033 10.1089/ars.2007.1642. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.