

Abstract
The goal of this review is to look at the role of endothelial damage and dysfunction in the initiation and development of early complications that appear after hematopoietic cell transplantation (HCT). These early complications share overlapping clinical manifestations and the suspicion of underlying endothelial damage. Several studies using different approaches, such as animal and in vitro models, the analysis of soluble biomarkers and clinical findings have provided evidence of this endothelial dysfunction. Historically, the first complication in which the role of endothelial damage was elucidated was the veno-oclusive disease/sinusoidal obstructive syndrome. In the last two decades, increasing evidence of the implication of the endothelium in the pathophysiology of other syndromes such as capillary leak syndrome, transplant-associated microangiopathy, or even graft versus host disease has accumulated. This knowledge opens up potential pharmacologic interventions to prevent/and/or treat endothelial damage and, therefore, to improve the outcome of patients receiving HCT.
Keywords: Endohtelium, Hematopoietic stem cell transplantation, Endothelial dysfunction, Inflammation, GvHD, TA-TMA
1. INTRODUCTION
For over 50 years, hematopoietic cell transplantation (HCT) has been the major curative therapy for several hematologic, metabolic, and neoplastic disorders [1]. However, the efficacy of this procedure is limited by life-threatening complications, the most important being graft versus host disease (GvHD), with high mortality rates. Other common complications are engraftment failure and opportunistic infections. In the last decade, several studies have been published pointing out to the relevant role of the endothelium in the initiation or the development of different HCT complications observed early after cell infusion (Table 1). These studies used different approaches to suggest that the endothelium is an organ to be considered in the prevention and treatment of these complications and, therefore, to improve HCT success. The aim of this review is to integrate experimental and clinical studies on endothelial damage and dysfunction in association with HCT, including in vitro and animal models, and the evaluation of circulating soluble markers.
Table 1.
Early complications of hematopoietic cell transplantation (HCT).
| Vascular Endothelial Syndromes after HCT |
|---|
| Sinusoidal obstruction syndrome (VOD/SOS) |
| Transplant-associated thrombotic microangiopathy (TA-TMA) |
| Diffuse alveolar hemorrhage |
| Engraftment and pre-engraftment syndrome |
| Capillary leak syndrome (CLS) |
| Posterior reversible encephalopathy syndrome |
| Graft versus host disease (GvHD) |
2. ENDOTHELIAL DYSFUNCTION IN HCT: A CHICKEN-AND-EGG SITUATION WHERE THE EGG CAME FIRST
Endothelial cells (ECs) are organized in a monocellular layer that separates all tissues from the circulating blood. The blood vessel endothelium crosses each and every tissue, and in each vascular bed presents unique structural and functional properties. This cellular heterogeneity is reflected by the structural and functional heterogeneity of the whole endothelium as a complex system [2,3]. The EC surface in an adult human is composed of approximately 1 to 6 × 1013 cells, weighs approximately 1 kg, and covers a surface area of more than 3000 square meters [4,5]. Due to its location, ECs are exposed to all kinds of physiological and pathological stimuli, and constitute the first barrier to many drug interventions. This organ is highly active, and has the ability to constantly adapt to environmental changes modulating vasomotor tone, hemostatic balance, and inflammatory reactions, among other responses. Endothelial activation takes place in a graded manner and can reverse to a basal state or progress to an irreversible state known as endothelial dysfunction which [2,3].
So far, endothelial dysfunction has been associated with several diverse pathologies such as obesity [6], chronic kidney disease [7–10], and sepsis [11], among others. It has also been recognized as an important contributor in the development of atherosclerotic disease, underlying plaque formation and growth, and leading to vascular complications and atherosclerotic progression [12]. Therefore, endothelial dysfunction has multiple origins and varies according to the time after HCT and anatomical location. In most cases, endothelial dysfunction implies an increase in leucocyte adhesion and transmigration, molecule extravasation, platelet activation, and cytokine liberation. These phenomena play a synergistic role and generate a positive feedback that worsens the endothelial condition. This complex and systemic reaction makes it difficult to differentiate if endothelial damage is the cause or the consequence of many complications. HCT brings to the researchers a good scenario in which to study the impact of individual inputs on the endothelium. Although a few studies report a slight increase in the levels of some plasma markers of endothelial damage in patients undergoing HCT before the transplant [13], our proposed hypothesis is that before HCT the endothelium does not present a dysfunctional state. Then, during HCT, ECs are activated and damaged by factors such as the drugs used in the conditioning regimen [14–17], radiotherapy [18], cytokines produced by the injured tissues, endogenous microbial products translocated through damaged mucosal barriers, immunosuppressive drugs used during allo-HCT, the engraftment process, and alloreactivity.
3. PRECLINICAL MODELS FOR THE STUDY OF ENDOTHELIAL DYSFUNCTION IN HCT
Several approaches have been applied to investigate endothelial dysfunction associated with HCT. Studies by our group and others have been able to reproduce, in an in vitro model, the endothelial damage, and activation by exposing different types of ECs to sera from patients who received either autologous or allogeneic HCT, with and without associated complications. This in vitro system allowed the evaluation of the specific responses of the endothelium to controlled and well-known stimuli, such as patients sera, but also to molecules reported to be associated with HCT, such as lipopolysaccharide (LPS) or tumor necrosis factor-alpha (TNF-α) [18,19]. Although it has accepted limitations, in vitro models also present several advantages, such as the ability to analyze the response of cells from different locations or to have an accurate control of the inputs under study.
Considerable effort has been put in the search for biomarkers of endothelial damage and dysfunction. A biomarker is defined by Hulka et al. as a cellular, biochemical, or molecular alteration that is measurable in biological media such as human tissues, cells, or fluids [20]. In the context of HCT, the goal has been to find markers with biological or clinical relevance reflecting the initiation and the progression of HCT-related complications, and the potential responses to therapeutic interventions. A major challenge when exploring biomarkers is to limit the sources of noise that interfere with the ability to identify valid molecules [21]. Veno-oclusive disease (VOD), also known as sinusoidal obstructive syndrome (SOS) and GvHD are the two best studied HCT-related complications in the search for biomarkers as a noninvasive method for an early and accurate diagnosis [22,23].
Animal models have been criticized for their lack of resemblance to human illnesses, as there are important species, anatomy, physiology, and microbiota differences, among others, that need to be considered when extrapolating the results to humans [24,25]. Nonetheless, these models are absolutely crucial to investigate complex diseases, as they allow dissecting the contribution of several independent components in the development of the pathology in a complex living system. Allogeneic HCT and its main complication, GVHD, represent a paradigm for the translation of preclinical concepts into the clinical practice, as most of the knowledge on the current therapeutic strategies applied in HCT derives from animal models [26]. For instance, the first understanding of the GVHD nature was obtained using chick embryos and mice [27,28], and the research on canine models was critical for the study of genes involved in histocompatibility [29,30]. These models have allowed the acquisition of the necessary empirical evidence to establish improvements in the clinical practice, such as the reduction in the conditioning intensity and the use of posttransplant cyclophosphamide. Regarding endothelial dysfunction, preclinical models have contributed to reveal its implication in GVHD initiation and development [24,31–33]. In the near future, strategies used in these models will likely also be translated into the human clinical practice such as, for instance, naïve T-cell depletion and the inhibition of certain cytokine and chemokine [34].
4. HCT IS ASSOCIATED WITH ENDOTHELIAL DYSFUNCTION, EVEN IN THE ABSENCE OF ANY RELATED COMPLICATION
Our group has demonstrated the existence of endothelial damage and activation in association with both auto-HCT and allo-HCT, even in the absence of associated complications. Such dysfunction exhibits specific profiles and characteristics in the first weeks after HCT. Macrovascular ECs, obtained from human umbilical veins, were incubated with serum samples from patients undergoing auto and allo-HCT without complications, and several endothelial damage markers were evaluated (Fig. 1). Our findings indicated that an inflammatory reaction, characterized by the activation of p38 MAPK and the increased expression of adhesion receptors on the EC surface, occurred in both auto and allo groups. This inflammatory response occurred together with an increased leukocyte adhesion when ECs were exposed to blood under flow conditions. In addition, a prothrombotic effect was also observed, but only in the allo-HCT group. This result was associated with the production of extracellular matrices enriched with von Willebrand factor (vWF), and with an increased deposition of platelet aggregates when exposed to circulating blood. Interestingly, the activation of the SAPK/JNK pathway, related to apoptosis, was only observed in the allo-HCT group. These results suggest that alloreactivity could be a relevant factor in the induction of endothelial damage.
Figure 1.

Endothelial damage markers follow a different kinetics when comparing the in vitro effect of auto and allo-HCT sera over endothelial cells. Bar diagrams summarize changes in adhesion receptor expression (VCAM-1, ICAM-1, and E-selectin) on the surface of endothelial cells exposed to sera from patients receiving auto (left) or allo (right) HCT. These experiments were performed exposing endothelial cells in culture to the sera from patients receiving autologous or allogeneic HCT for 48 hours. The cells were then fixed, and adhesion receptor expression was assessed by immunofluorescence.
Endothelial damage was especially intense when ECs were exposed to patient's sera collected at day 0, after conditioning treatment and just before the infusion of hematopoietic precursors. These results reflect the deleterious effect of the conditioning regimen and total body irradiation (TBI) on the endothelium [18,35,36]. From day 0, the endothelial damage profile followed different kinetics when comparing the auto-HCT and allo-HCT groups. In the auto-HCT, both engraftment [37] and the use of G-CSF could act as the triggers of the damage seen when ECs were grown in the presence of patients sera obtained at day +14. In the allo-HCT, the alloreactivity could explain the increase in the levels of damage biomarkers in response to sera collected at day +21 [38,39]. These results may be the consequence of the effect on the endothelium of the conditioning treatment, the administration of pro-inflammatory (G-CSF) or immunomodulatory agents, and the onset of donor leukocyte engraftment. The analysis of soluble endothelial damage biomarkers revealed that VWF and soluble tumor necrosis receptor I (sTNFRI) increased progressively before the conditioning to day +14 in auto-HCT, and to day +21 in allo-HCT patients [40,41]. Moreover, the degree of changes in the biomarker levels was associated with the intensity of the conditioning treatment [38], and was more notable in myeloablative compared with reduced-intensity conditioning.
The analysis of circulating markers during has been performed by several groups with the aim of finding a useful diagnostic and/or prognostic tool for HCT-associated complications. VWF, soluble thrombomodulin (TM), TNF-α, plasminogen activator inhibitor type 1, soluble adhesion molecules (sE-selectin, sICAM-1, sVCAM-1), suppression of tumorigenicity-2 (ST2), angiopoietin-2 (ANG2), hyaluronic acid (HA), L-Ficolin, and circulating endothelial cells (CECs) [42,43] are the most commonly evaluated circulating biomarkers. In general, the majority of the studies show an increase in the levels of the different markers with respect to the baseline, especially in patients with HCT complications such as VOD/SOS [22,40,44], thrombotic microangiopathy (TA-TMA), capillary leak syndrome (CLS) [45,46], engraftment syndrome (ES), and GVHD [23,42,43,47–49]. Such increase suggests a potential involvement of the endothelium in the origin and the development of these syndromes.
5. EVIDENCE ON THE ENDOTHELIAL ORIGIN OF VASCULAR ENDOTHELIAL SYNDROMES AFTER HCT
All the syndromes that appear in association with HCT share clinical common features, such as an early onset after progenitor infusion, overlapping clinical manifestations, absence ofwell-defined clinical criteria for diagnosis and well-established treatments, and the tendency to evolve to an irreversible multi-organ dysfunction syndrome. As explained above, the hypothesis that endothelial damage represents a common condition underlying many transplant-related complications is becoming increasingly accepted. Some evidence of this association is summarized below.
VOD/SOSis a potentially fatal complication of HCT that usually occurs within 30 days after cell transfusion. Its diagnosis is based on clinical criteria [50]. The pathogenesis of this complication involves a damage of the sinusoidal ECs and hepatocytes by toxic metabolites generated during the conditioning regimen. The injury on ECs impairs their capacity to regulate the hemostatic balance, reduces nitric oxide production, increases the levels of matrix metalloproteinase [51], and is accompanied by the release of inflammatory cytokines from the injured tissue. The dysfunction of the vascular lining consists of ballooning of ECs, extravasation of cellular and extracellular debris, platelet aggregation in the space of Disse, detachment of ECs, and, eventually, complete sinusoidal occlusion. These phenomena are followed by capillarization, lack of liver sinusoidal EC fenestration, and formation of an organized basement membrane that allows fibrosis [52,53]. The preclinical and clinical evidence of the role of endothelial dysfunction in VOD/SOS development provided a rationale for defibrotide (DF) treatment, a recognized endothelial protective drug, in this setting [50,54]. DF has been used to treat hepatic VOD/SOS with a reported 30–60% complete response rate.
CLS represents a potentially life-threatening complication after HCT. Also called Clarkson syndrome [55], this condition is characterized by transient but severe hypotension that results in vascular collapse and shock, hemoconcentration, and, ultimately, anasarca because of accumulation of fluids and macromolecules in tissues. Allogeneic HCT from an unrelated donor, G-CSF administration, and intensive chemo/radiotherapy have been suggested as possible risk factors for CLS development [56,57]. A recent study in pediatric patients undergoing HCT [58] confirmed historical results suggesting that severe infection is associated with CLS, independently from HCT. This association is supposed to rely upon the endothelial damage induced by the cytokine storm that occurs in severe infection, which is believed to be crucial in the development of CLS. This complication has been postulated to be triggered by a combination of inflammation and reversible microvascular barrier dysfunction [59]. However, as no more than 100 cases of CLS were reported in the literature from 1960 to 2006, and there is a high mortality rate during episodes, the pathophysiology of this entity has been difficult to clarify. The main hypothesis has been that transient episodes were caused, at least in part, by a massive reversible endothelial barrier breach. This hypothesis was confirmed by Xie et al. [46], who demonstrated in an in vitro study that episodic CLS sera directly induce endothelial hyper-permeability and barrier breakdown, and who identified VEGF and Ang2 as the mediators of permeability, whose induction in the circulation is associated with CLS episodes.
HCT-associated thrombotic microangiopathy is a potentially severe complication with a mortality rate of 60–90% despite treatment [60]. In the surviving patients, it is associated with long-termmorbidity including hypertension, chronic kidney disease, gastrointestinal or central nervous system disease, and pulmonary hypertension [61,62]. The TA-TMA incidence is about 10–35% [63], and is more common after allo-HCT than auto-HCT. TA-TMA can manifest as a multi-system disease occurring after an endothelial injury which, in HCT, is known to be multifactorial. The kidney is the organ most commonly affected in TA-TMA, although other organs like the lung, bowel, heart, and brain are also involved. In addition to the endothelial damage component, this complication can include a complement system dysregulation. The latter is well known in the pathophysiology of other thrombotic microangiopathies, such as atypical hemolytic uremic syndrome. The complement system is a major component of the innate immune system, and consists of plasma proteins and membrane regulators and receptors. The plasma proteins interact via three major cascades: the classical, the lectin, and the alternative pathways, which culminate in the formation of the C5b-9 membrane attack complex, which has a lytic activity on target cells, by forming a pore in the cell membrane [64–66]. Eculizumab, a monoclonal antibody directed towards C5, which prevents formation of the C5b-9 membrane attack complex, has been successful in the treatment of several TA-TMA [67]. Furthermore, since TA-TMA is a disorder resulting from endothelial injury, DF has also been used, demonstrating a 55% response rate, as monotherapy or in combination with other therapies [68,69] Novel TA-TMA biomarkers, reflecting predisposition for injury to specific organs, need to be identified in order to aid earlier TA-TMA diagnosis and guide targeted therapies [70].
Graft versus host disease (GvHD) is the major cause of morbidity and mortality following allo-HCT [71,72]. The diagnosis of acute GvHD after allo-HCT remains clinical, taking into consideration patient symptoms, laboratory values, and affected tissue histology. The pathophysiology of aGVHD is complex, and is known to involve aspects of both the adaptive and innate immune responses in subsequent phases. As ECs are the primary barrier separating donor-derived leukocytes and allogeneic target tissue, the endothelium has been increasingly studied and identified as an initial target of an allogeneic acute GVHD [73–75]. It is well known that, in the early phases of this complication (Figs. 2 and 3), ECs are injured by the conditioning regimen [48,76]. Inflammatory angiogenesis [31] and neovascularization [33] are central events in this induced damage. Importantly, experiments of angiogenesis in animal models have demonstrated that metabolic changes trigger alterations in cell mechanics, leading to enhanced migratory and proliferative potential of ECs during the initiation of inflammation. However, this process occurs in the absence of classical endothelial damage markers, such as increase in adhesion receptor expression. Recently, effort has been directed towards identifying potential noninvasive peripheral blood biomarkers with diagnostic and clinical value of acute GvHD, and some promising results have been achieved with markers related to endothelial damage [23,48,77]. This suspected relationship between endothelial damage and acute GVHD has also been corroborated by clinical findings. Corbacioglu et al. performed a phase-3 VOD/SOS using DF for the prevention of VOD/SOS in pediatric patients receiving HCT, and found that the incidence and severity of GVHD, analyzed as an exploratory end point, was significantly lower at days +30 and +100 in DF-treated patients versus controls [78–80]. Similar GVHD findings have been reported in adults, and a phase-2 trial of DF or prevention of GVHD is now recruiting (NCT03339297) [81].
Figure 2.
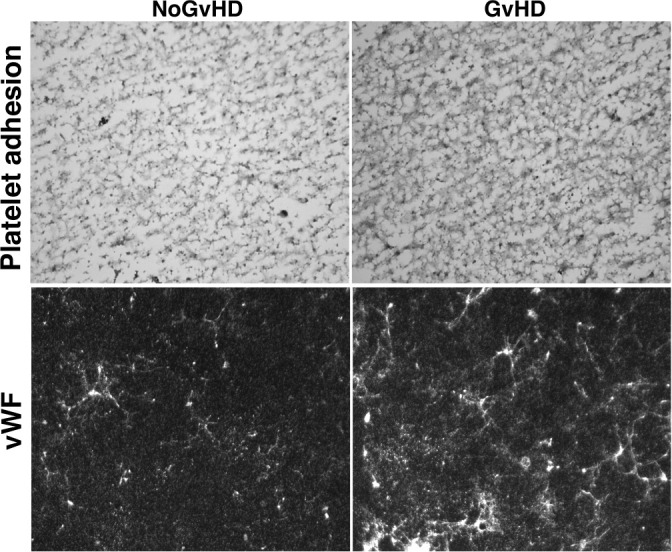
Prothrombotic phenotype of the extracellular matrices (ECM) generated by endothelial cells (ECs) in response to sera from patients with acute graft versus host disease (aGvHD). Representative images of platelet adhesion after exposing ECM to flowing blood (800/s, 5 minutes) (upper images) and presence of von Willebrand Factor (vWF, green staining) on the ECM produced by ECs exposed to sera from both GvHD and No-GvHD patients at day 21 after allo-HCT. Original magnification of × 400 for all micrographs obtained with a fluorescent microscope (LEICA DM4000B).
Figure 3.
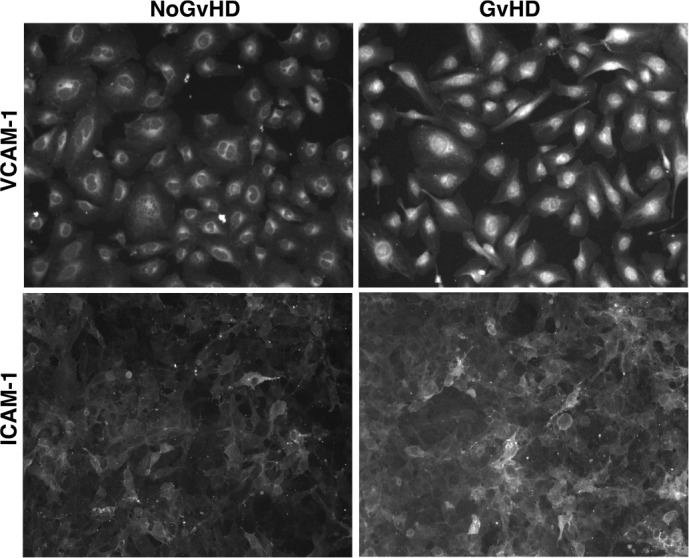
Enhanced expression of the adhesion receptors VCAM-1 and ICAM-1 on endothelial cells exposed to sera from patients developing acute graft versus host disease (GvHD). Micrographs of VCAM-1 and ICAM-1 staining (with red and green colors, respectively) on cell surfaces of microvascular endothelial cells exposed to sera from both GvHD and No-GvHD patients at day 21 after allo-HCT. Original magnification of × 400 for all micrographs obtained with a fluorescent microscope (LEICA DM4000B).
6. SUMMARY AND FUTURE DIRECTIONS
Over the last 60 years, cellular and animal models have played a critical role in constructing our understanding of HCT and the associated complications. The data summarized in this review support the concept that heterogeneous syndromes such as VOD/SOS, TA-TMA, accelerated atherosclerosis, and GvHD share a common denominator in patients treated with allogeneic HCT: a damaged endothelium. Interestingly, some of the evidence comes from clinical findings, such as interventions tested in clinical trials that have been found to be particularly effective for these specific endothelial injury syndromes. To date, no single biomarker has been proved to be valid among independent cohorts for identifying preclinical signs of these HCT-complications, improving the accuracy of the diagnostic, providing opportunities for prevention, and predicting long-term outcomes. Instead, and due to the complexity of the multifactorial nature of the endothelial damage, a panel of biomarkers is more likely to address these important goals in the future. In summary, although the increasing knowledge on the role of the endothelium in HCT-associated complications, achieved through the efforts of hundreds of researchers in the last years, our understanding remains very limited. This limitation constitutes an exciting biomedical research challenge, the results of which could be eventually translated to the improvement of the outcomes of patients receiving HCT.
ACKNOWLEDGMENTS
We would like to thank the Primary Hemostasis laboratory group for their technical support and to German José Carreras Leukaemia Foundation (grant 11R/2016), the Spanish Government (Integrated Project in Health Institutes, PIE15/00027) and Generalitat de Catalunya (2017-SGR671 and CERCA Programme).
Footnotes
Peer review is under the responsibility of IACH
CONFLICT OF INTEREST
M.P., M.D.R. and E.C. declare conflict of interest with Jazz Pharmaceuticals plc/Gentium Inc in the form of speaker's fee for symposia.
REFERENCES
- [1].Ljungman P, Bregni M, Brune M, et al. Allogeneic and autologous transplantation for haematological diseases, solid tumours and immune disorders: current practice in Europe 2009. Bone Marrow Transplant. 2010;45(2):219–34. doi: 10.1038/bmt.2009.141. [DOI] [PubMed] [Google Scholar]
- [2].Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. 2007;100(2):158–73. doi: 10.1161/01.RES.0000255691.76142.4a. [DOI] [PubMed] [Google Scholar]
- [3].Aird WC. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ Res. 2007;100(2):174–90. doi: 10.1161/01.RES.0000255690.03436.ae. [DOI] [PubMed] [Google Scholar]
- [4].Augustin HG, Kozian DH, Johnson RC. Differentiation of endothelial cells: analysis of the constitutive and activated endothelial cell phenotypes. BioEssays. 1994;16(12):901–6. doi: 10.1002/bies.950161208. [DOI] [PubMed] [Google Scholar]
- [5].Cines DB, Pollak ES, Buck CA, et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. 1998;91(10):3527–61. [PubMed] [Google Scholar]
- [6].Hanzu FA, Palomo M, Kalko SG, et al. Translational evidence of endothelial damage in obese individuals: inflammatory and prothrombotic responses. J Thromb Haemost. 2011;9(6):1236–45. doi: 10.1111/j.1538-7836.2011.04285.x. [DOI] [PubMed] [Google Scholar]
- [7].Serradell M, Dı M, Cases A, et al. Uraemic medium accelerates proliferation but does not induce apoptosis of endothelial cells in culture. Haematologica. 2003;18(6):1079–85. doi: 10.1093/ndt/gfg161. [DOI] [PubMed] [Google Scholar]
- [8].Serradell M, Díaz-Ricart M, Cases A, et al. Uremic medium causes expression, redistribution and shedding of adhesion molecules in cultured endothelial cells. Haematologica. 2002;87(10):1053–61. [PubMed] [Google Scholar]
- [9].Carbó C, Arderiu G, Escolar G, et al. Differential expression of proteins from cultured endothelial cells exposed to uremic versus normal serum. Am J Kidney Dis. 2008;51(4):603–12. doi: 10.1053/j.ajkd.2007.11.029. [DOI] [PubMed] [Google Scholar]
- [10].Caballo C, Palomo M, Cases A, et al. NFκB in the development of endothelial activation and damage in uremia: an in vitro approach. PLoS One. 2012;7(8):e43374. doi: 10.1371/journal.pone.0043374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Aird WC. Review article The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Sepsis. 2003;101(10):3765–77. doi: 10.1182/blood-2002-06-1887. [DOI] [PubMed] [Google Scholar]
- [12].Favero G, Paganelli C, Buffoli B, Fabrizio Rodella L, Rezzani R. Endothelium and its alterations in cardiovascular diseases: life style intervention. Biomed Res Int. 2014;2014:801896. doi: 10.1155/2014/801896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Richard S, Seigneur M, Blann A, et al. Vascular endothelial lesion in patients undergoing bone marrow transplantation. Bone Marrow Transplant. 1996;18(5):955–9. [PubMed] [Google Scholar]
- [14].Fusté B, Mazzara R, Escolar G, Merino A, Ordinas A, Díaz-Ricart M. GSF increases expression of adhesion receptors on endothelial cells through activation of p38MAPK. Haematologica. 2004;89(5):578–85. [PubMed] [Google Scholar]
- [15].Mercanoglu F, Turkmen A, Kocaman O, et al. Endothelial dysfunction in renal transplant patients is closely related to serum cyclosporine levels. Transplant Proc. 2004;36(5):1357–60. doi: 10.1016/j.transproceed.2004.05.073. [DOI] [PubMed] [Google Scholar]
- [16].Bouvier N, Flinois JP, Gilleron J, et al. Cyclosporine triggers endoplasmic reticulum stress in endothelial cells: a role for endothelial phenotypic changes and death. Am J Physiol Ren Physiol. 2009;296(1):F160–F9. doi: 10.1152/ajprenal.90567.2008. [DOI] [PubMed] [Google Scholar]
- [17].Carmona A, Díaz-Ricart M, Palomo M, et al. Distinct deleterious effects of cyclosporine and tacrolimus and combined tacrolimus-sirolimus on endothelial cells: protective effect of defibrotide. Biol Blood Marrow Transplant. 2013;19(10):1439–45. doi: 10.1016/j.bbmt.2013.07.001. [DOI] [PubMed] [Google Scholar]
- [18].Eissner G, Kohlhuber F, Grell M, et al. Critical involvement of transmembrane tumor necrosis factor-alpha in endothelial programmed cell death mediated by ionizing radiation and bacterial endotoxin. Blood. 1995;86(11):4184–93. [PubMed] [Google Scholar]
- [19].Eissner G. Fludarabine induces apoptosis, activation, and allogenicity in human endothelial and epithelial cells: protective effect of defibrotide. Blood. 2002;100(1):334–40. doi: 10.1182/blood.V100.1.334. [DOI] [PubMed] [Google Scholar]
- [20].Hulka BS, Wilcosky T. Biological markers in epidemiologic research. Arch Environ Heal An Int J. 1988;43(2):83–9. doi: 10.1080/00039896.1988.9935831. [DOI] [PubMed] [Google Scholar]
- [21].Cassol E, Misra V, Morgello S, Gabuzda D. Applications and limitations of inflammatory biomarkers for studies on neurocognitive impairment in HIV infection. J Neuroimmune Pharmacol. 2013;8(5):1087–97. doi: 10.1007/s11481-013-9512-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Akil A, Zhang Q, Mumaw CL, et al. Biomarkers for diagnosis and prognosis of sinusoidal obstruction syndrome after hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2015;21(10):1739–45. doi: 10.1016/j.bbmt.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Luft T, Benner A, Jodele S, et al. EASIX in patients with acute graft-versus-host disease: a retrospective cohort analysis. Lancet Haematol. 2017;4(9):e414–e23. doi: 10.1016/S2352-3026(17)30108-4. [DOI] [PubMed] [Google Scholar]
- [24].Schroeder MA, DiPersio JF. Mouse models of graft-versus-host disease: advances and limitations. Dis Model Mech. 2011;4(3):318–33. doi: 10.1242/dmm.006668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mestas J, Hughes CCW. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172(5):2731–8. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- [26].Srivastava A, Ahmed Toor A, Moll G, et al. The role of animal models in the study of hematopoietic stem cell transplantation and GvHD: a historical overview. Front Immunol. 2016;7:333. doi: 10.3389/fimmu.2016.00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Simonsen M. The impact on the developing embryo and newborn animal of adult homologous cells. Acta Pathol Microbiol Scand. 1957;40(6):480–500. [PubMed] [Google Scholar]
- [28].Billingham RE. Studies on the reaction of injected homologous lymphoid tissue cells against the host. Ann N Y Acad Sci. 1958;73(3):782–8. doi: 10.1111/j.1749-6632.1959.tb40857.x. [DOI] [PubMed] [Google Scholar]
- [29].Epstein RB, Storb R, Ragde H, Thomas Cytotoxic typing antisera for marrow grafting in littermate dogs. Transplantation. 1968;6(1):45–58. doi: 10.1097/00007890-196801000-00005. [DOI] [PubMed] [Google Scholar]
- [30].Storb R, Rudolph RH, Kolb HJ, et al. Marrow grafts between DL-A-matched canine littermates. Transplantation. 1973;15(1):92–100. doi: 10.1097/00007890-197301000-00014. [DOI] [PubMed] [Google Scholar]
- [31].Riesner K, Shi Y, Jacobi A, et al. Initiation of acute graft-versus-host disease by angiogenesis. Blood. 2017;129(14):2021–32. doi: 10.1182/blood-2016-08-736314. [DOI] [PubMed] [Google Scholar]
- [32].Penack O, Socié G, van den Brink MRM. The importance of neovascularization and its inhibition for allogeneic hematopoietic stem cell transplantation. Blood. 2011;117(16):4181–9. doi: 10.1182/blood-2010-10-312934. [DOI] [PubMed] [Google Scholar]
- [33].Leonhardt F, Grundmann S, Behe M, et al. Inflammatory neovascularization during graft-versus-host disease is regulated by v integrin and miR-100. Blood. 2013;121(17):3307–18. doi: 10.1182/blood-2012-07-442665. [DOI] [PubMed] [Google Scholar]
- [34].Markey KA, MacDonald KPA, Hill GR. The biology of graft-versus-host disease: experimental systems instructing clinical practice. Blood. 2014;124(3):354–62. doi: 10.1182/blood-2014-02-514745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hatoum OA, Otterson MF, Kopelman D, et al. Radiation induces endothelial dysfunction in murine intestinal arterioles via enhanced production of reactive oxygen species. Arterioscler Thromb Vasc Biol. 2006;26(2):287–94. doi: 10.1161/01.ATV.0000198399.40584.8c. [DOI] [PubMed] [Google Scholar]
- [36].Venkatesulu BP, Mahadevan LS, Aliru ML, et al. Radiation-induced endothelial vascular injury: a review of possible mechanisms. JACC Basic Transl Sci. 2018;3(4):563–72. doi: 10.1016/j.jacbts.2018.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Spitzer TR. Engraftment syndrome: double-edged sword of hematopoietic cell transplants. Bone Marrow Transplant. 2015;50(4):469–75. doi: 10.1038/bmt.2014.296. [DOI] [PubMed] [Google Scholar]
- [38].Salat C, Holler E, Kolb HJ, et al. Endothelial cell markers in bone marrow transplant recipients with and without acute graft-versus-host disease. Bone Marrow Transplant. 1997;19(9):909–14. doi: 10.1038/sj.bmt.1700767. [DOI] [PubMed] [Google Scholar]
- [39].Ganster A, Brucker I, Holler E, et al. In vitro monitoring of endothelial complications following hematopoietic allogeneic stem cell transplantation. Bone Marrow Transplant. 2004;33(3):355–7. doi: 10.1038/sj.bmt.1704354. [DOI] [PubMed] [Google Scholar]
- [40].Palomo M, Diaz-Ricart M, Carbo C, et al. Endothelial dysfunction after hematopoietic stem cell transplantation: role of the conditioning regimen and the type of transplantation. Biol Blood Marrow Transplant. 2010;16(7):985–93. doi: 10.1016/j.bbmt.2010.02.008. [DOI] [PubMed] [Google Scholar]
- [41].Matsuda Y, Hara J, Osugi Y, et al. Post-transplant complications serum levels of soluble adhesion molecules in stem cell transplantation-related complications. Bone Marrow Transplant. 2001;27:977–82. doi: 10.1038/sj.bmt.1703026. [DOI] [PubMed] [Google Scholar]
- [42].Erdbruegger U, Dhaygude A, Haubitz M, Woywodt A. Circulating endothelial cells: markers and mediators of vascular damage. Curr Stem Cell Res Ther. 2010;5(4):294–302. doi: 10.2174/157488810793351721. [DOI] [PubMed] [Google Scholar]
- [43].Dignat-George F, Sampol J. Circulating endothelial cells in vascular disorders: new insights into an old concept. Eur J Haematol. 2000;65(4):215–20. doi: 10.1034/j.1600-0609.2000.065004215.x. [DOI] [PubMed] [Google Scholar]
- [44].Catani L, Gugliotta L, Vianelli N, et al. Endothelium and bone marrow transplantation. Bone Marrow Transplant. 1996;17(2):277–80. [PubMed] [Google Scholar]
- [45].Nürnberger W, Michelmann I, Burdach S, Göbel U. Endothelial dysfunction after bone marrow transplantation: increase of soluble thrombomodulin and PAI-1 in patients with multiple transplant-related complications. Ann Hematol. 1998;76(2):61–5. doi: 10.1007/s002770050364. [DOI] [PubMed] [Google Scholar]
- [46].Xie Z, Ghosh CC, Patel R, et al. Vascular endothelial hyperpermeability induces the clinical symptoms of Clarkson disease (the systemic capillary leak syndrome) Blood. 2012;119(18):4321–32. doi: 10.1182/blood-2011-08-375816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Mir E, Palomo M, Rovira M, et al. Endothelial damage is aggravated in acute GvHD and could predict its development. Bone Marrow Transplant. 2017;52(9):1317–25. doi: 10.1038/bmt.2017.121. [DOI] [PubMed] [Google Scholar]
- [48].Major-Monfried H, Renteria AS, Pawarode A, et al. MAGIC biomarkers predict long-term outcomes for steroid-resistant acute GVHD. Blood. 2018;131(25):2846–55. doi: 10.1182/blood-2018-01-822957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Almici C, Skert C, Bruno B, et al. Circulating endothelial cell count: a reliable marker of endothelial damage in patients undergoing hematopoietic stem cell transplantation. Bone Marrow Transplant. 2017;52(12):1637–42. doi: 10.1038/bmt.2017.194. [DOI] [PubMed] [Google Scholar]
- [50].Mohty M, Malard F, Abecassis M, et al. Sinusoidal obstruction syndrome/veno-occlusive disease: current situation and perspectives—a position statement from the European Society for Blood and Marrow Transplantation (EBMT) Bone Marrow Transplant. 2015;50(6):781–9. doi: 10.1038/bmt.2015.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].DeLeve L, Wang X, Kanel GC, et al. Decreased hepatic nitric oxide production contributes to the development of rat sinusoidal obstruction syndrome. Hepatology. 2003;38(4):900–8. doi: 10.1002/hep.1840380416. [DOI] [PubMed] [Google Scholar]
- [52].DeLeve LD, Wang X, Guo Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology. 2008;48(3):920–30. doi: 10.1002/hep.22351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].DeLeve LD. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology. 2015;61(5):1740–6. doi: 10.1002/hep.27376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Corbacioglu S, Greil J, Peters C, et al. Defibrotide in the treatment of children with Veno-Occlusive Disease (VOD): a retrospective multicentre study demonstrates therapeutic efficacy upon early intervention. Bone Marrow Transplant. 2004;33(2):189–95. doi: 10.1038/sj.bmt.1704329. [DOI] [PubMed] [Google Scholar]
- [55].Clarkson B, Thompson D, Horwith M, Luckey EH. Cyclical edema and shock due to increased capillary permeability. Am J Med. 1960;29:193–216. doi: 10.1016/0002-9343(60)90018-8. [DOI] [PubMed] [Google Scholar]
- [56].Deeren DH, Zachee P, Malbrain MLNG. Granulocyte colony-stimulating factor-induced capillary leak syndrome confirmed by extravascular lung water measurements. Ann Hematol. 2005;84(2):89–94. doi: 10.1007/s00277-004-0946-8. [DOI] [PubMed] [Google Scholar]
- [57].Takatsuka H, Wakae T, Mori A, et al. Effects of total body irradiation on the vascular endothelium. Clin Transplant. 2002;16(5):374–7. doi: 10.1034/j.1399-0012.2002.02035.x. [DOI] [PubMed] [Google Scholar]
- [58].Lucchini G, Willasch AM, Daniel J, et al. Epidemiology, risk factors, and prognosis of capillary leak syndrome in pediatric recipients of stem cell transplants: a retrospective single-center cohort study. Pediatr Transplant. 2016;20(8):1132–6. doi: 10.1111/petr.12831. [DOI] [PubMed] [Google Scholar]
- [59].Dhir V, Arya V, Malav IC, et al. Idiopathic Systemic Capillary Leak Syndrome (SCLS): case report and systematic review of cases reported in the last 16 years. Intern Med. 2007;46(12):899–904. doi: 10.2169/internalmedicine.46.6129. [DOI] [PubMed] [Google Scholar]
- [60].Choi CM, Schmaier AH, Snell MR, Lazarus HM. Thrombotic microangiopathy in haematopoietic stem cell transplantation. Drugs. 2009;69(2):183–98. doi: 10.2165/00003495-200969020-00004. [DOI] [PubMed] [Google Scholar]
- [61].Kersting S, Koomans HA, Hené RJ, Verdonck LF. Acute renal failure after allogeneic myeloablative stem cell transplantation: retrospective analysis of incidence, risk factors and survival. Bone Marrow Transplant. 2007;39(6):359–65. doi: 10.1038/sj.bmt.1705599. [DOI] [PubMed] [Google Scholar]
- [62].George JN, Selby GB. Thrombotic microangiopathy after allogeneic bone marrow transplantation: a pathologic abnormality associated with diverse clinical syndromes. Bone Marrow Transplant. 2004;33(11):1073–4. doi: 10.1038/sj.bmt.1704513. [DOI] [PubMed] [Google Scholar]
- [63].Laskin BL, Goebel J, Davies SM, Jodele S. Small vessels, big trouble in the kidneys and beyond: hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Blood. 2011;118(6):1452–62. doi: 10.1182/blood-2011-02-321315. [DOI] [PubMed] [Google Scholar]
- [64].Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344(14):1058–66. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- [65].Walport MJ. Complement. Second of two parts. N Engl J Med. 2001;344(15):1140–4. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- [66].Varela JC, Tomlinson S. Complement: an overview for the clinician. Hematol Oncol Clin North Am. 2015;29(3):409–27. doi: 10.1016/j.hoc.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Jodele S, Fukuda T, Vinks A, et al. Eculizumab therapy in children with severe hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Biol Blood Marrow Transplant. 2014;20(4):518–25. doi: 10.1016/j.bbmt.2013.12.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Uderzo C, Fumagalli M, De Lorenzo P, et al. Impact of thrombotic thrombocytopenic purpura on leukemic children undergoing bone marrow transplantation. Bone Marrow Transplant. 2000;26(9):1005–9. doi: 10.1038/sj.bmt.1702648. [DOI] [PubMed] [Google Scholar]
- [69].Corti P, Uderzo C, Tagliabue A, et al. Defibrotide as a promising treatment for thrombotic thrombocytopenic purpura in patients undergoing bone marrow transplantation. Bone Marrow Transplant. 2002;29(6):542–3. doi: 10.1038/sj.bmt.1703414. [DOI] [PubMed] [Google Scholar]
- [70].Jodele S, Laskin BL, Dandoy CE, et al. A new paradigm: diagnosis and management of HSCT-associated thrombotic microangiopathy as multi-system endothelial injury. Blood Rev. 2015;29(3):191–204. doi: 10.1016/j.blre.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Pidala J. Graft-vs-host disease following allogeneic hematopoietic cell transplantation. Cancer Control. 2011;18(4):268–76. doi: 10.1177/107327481101800407. [DOI] [PubMed] [Google Scholar]
- [72].Barton-Burke M, Dwinell DM, Kafkas L, et al. Graft-versus-host disease: a complex long-term side effect of hematopoietic stem cell transplant. Oncology (Williston Park) 2008;22(11 Suppl Nurse ed):31–45. [PubMed] [Google Scholar]
- [73].Biedermann BC. Vascular endothelium and graft-versus-host disease. Best Pract Res Clin Haematol. 2008;21(2):129–38. doi: 10.1016/j.beha.2008.02.003. [DOI] [PubMed] [Google Scholar]
- [74].Tichelli A, Gratwohl A. Vascular endothelium as ‘novel’ target of graft-versus-host disease. Best Pract Res Clin Haematol. 2008;21(2):139–48. doi: 10.1016/j.beha.2008.02.002. [DOI] [PubMed] [Google Scholar]
- [75].Carreras E, Diaz-Ricart M. The role of the endothelium in the short-term complications of hematopoietic SCT. Bone Marrow Transplant. 2011;46(12):1495–502. doi: 10.1038/bmt.2011.65. [DOI] [PubMed] [Google Scholar]
- [76].Riesner K, Kalupa M, Shi Y, Elezkurtaj S, Penack O. A preclinical acute GVHD mouse model based on chemotherapy conditioning and MHC-matched transplantation. Bone Marrow Transplant. 2016;51(3):410–7. doi: 10.1038/bmt.2015.279. [DOI] [PubMed] [Google Scholar]
- [77].Almici C, Skert C, Verardi R, et al. Changes in circulating endothelial cells count could become a valuable tool in the diagnostic definition of acute graft-versus-host disease. Transplantation. 2014;98(7):706–12. doi: 10.1097/TP.0000000000000385. [DOI] [PubMed] [Google Scholar]
- [78].Corbacioglu S, Cesaro S, Faraci M, et al. Defibrotide for prophylaxis of hepatic veno-occlusive disease in paediatric haemopoietic stem-cell transplantation: an open-label, phase 3, randomised controlled trial. Lancet. 2012;379(9823):1301–9. doi: 10.1016/S0140-6736(11)61938-7. [DOI] [PubMed] [Google Scholar]
- [79].Richardson PG, Soiffer RJ, Antin JH, et al. Defibrotide for the treatment of severe hepatic veno-occlusive disease and multiorgan failure after stem cell transplantation: a multicenter, randomized, dose-finding trial. Biol Blood Marrow Transplant. 2010;16(7):1005–17. doi: 10.1016/j.bbmt.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Richardson PG, Carreras E, Iacobelli M, et al. The use of defibrotide in blood and marrow transplantation. Blood Adv. 2018;2(15):1495–509. doi: 10.1182/bloodadvances.2017008375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Tekgündüz E, Kaya AH, Bozdağ SC, et al. Does defibrotide prophylaxis decrease the risk of acute graft host disease following allogeneic hematopoietic cell transplantation? Transfus Apher Sci. 2016;54(1):30–4. doi: 10.1016/j.transci.2016.01.009. [DOI] [PubMed] [Google Scholar]