

ABSTRACT
The fish pathogen Aliivibrio (Vibrio) salmonicida LFI1238 is thought to be incapable of utilizing chitin as a nutrient source, since approximately half of the genes representing the chitinolytic pathway are disrupted by insertion sequences. In the present study, we combined a broad set of analytical methods to investigate this hypothesis. Cultivation studies revealed that A. salmonicida grew efficiently on N-acetylglucosamine (GlcNAc) and chitobiose [(GlcNAc)2], the primary soluble products resulting from enzymatic chitin hydrolysis. The bacterium was also able to grow on chitin particles, albeit at a lower rate than on the soluble substrates. The genome of the bacterium contains five disrupted chitinase genes (pseudogenes) and three intact genes encoding a glycoside hydrolase family 18 (GH18) chitinase and two auxiliary activity family 10 (AA10) lytic polysaccharide monooxygenases (LPMOs). Biochemical characterization showed that the chitinase and LPMOs were able to depolymerize both α- and β-chitin to (GlcNAc)2 and oxidized chitooligosaccharides, respectively. Notably, the chitinase displayed up to 50-fold lower activity than other well-studied chitinases. Deletion of the genes encoding the intact chitinolytic enzymes showed that the chitinase was important for growth on β-chitin, whereas the LPMO gene deletion variants only showed minor growth defects on this substrate. Finally, proteomic analysis of A. salmonicida LFI1238 growth on β-chitin showed expression of all three chitinolytic enzymes and, intriguingly, also three of the disrupted chitinases. In conclusion, our results show that A. salmonicida LFI1238 can utilize chitin as a nutrient source and that the GH18 chitinase and the two LPMOs are needed for this ability.
IMPORTANCE The ability to utilize chitin as a source of nutrients is important for the survival and spread of marine microbial pathogens in the environment. One such pathogen is Aliivibrio (Vibrio) salmonicida, the causative agent of cold water vibriosis. Due to extensive gene decay, many key enzymes in the chitinolytic pathway have been disrupted, putatively rendering this bacterium incapable of chitin degradation and utilization. In the present study, we demonstrate that A. salmonicida can degrade and metabolize chitin, the most abundant biopolymer in the ocean. Our findings shed new light on the environmental adaption of this fish pathogen.
KEYWORDS: Aliivibrio salmonicida, LPMO, chitinase, lytic polysaccharide monooxygenase, pathogen
INTRODUCTION
Chitin is one of the most abundant biopolymers in nature and is a primary component of rigid structures such as the exoskeleton of insects and crustaceans and the cell walls of fungi and some algae (1–4). Some reports also indicate that chitin is found in the scales and guts of fish (5, 6). This linear polysaccharide consists of N-acetyl-d-glucosamine (GlcNAc) units linked by β-1,4 glycosidic bonds that associate with other chitin chains to form insoluble chitin fibers. Despite the recalcitrance of chitin, the polymer is readily degraded and metabolized by chitinolytic microorganisms in the environment (7, 8).
Most bacteria solubilize and depolymerize chitin by secreting chitinolytic enzymes. Such enzymes include chitinases from family 18 and 19 of the glycoside hydrolases (GH18 and -19, respectively) and lytic polysaccharide monooxygenases (LPMOs) from family 10 of the auxiliary activities (AA10), according to classification by the carbohydrate-active enzyme database (CAZy; http://www.cazy.org/) (9). Whereas chitinases cleave chitin chains by a hydrolytic mechanism (10, 11), LPMOs perform chitin depolymerization by an oxidative reaction (12–14). The latter enzymes usually target the crystalline parts of chitin fibers that are inaccessible for the chitinases. When combined, chitinases and LPMOs act synergistically, providing efficient depolymerization of this recalcitrant carbohydrate (12, 15–17). The products of enzymatic chitin degradation are mainly GlcNAc and (GlcNAc)2 but also native and oxidized chitooligosaccharides, the latter (aldonic acids) arising from LPMO activity.
The chitin degradation pathway is conserved in the Vibrionaceae (18, 19). Here, GlcNAc and (GlcNAc)2 are transported into the periplasm by unspecific porins (20, 21) or by dedicated transport proteins for chitooligosaccharides [(GlcNAc)2–6], named chitoporins (22, 23). Once transported to the periplasm, (GlcNAc)2–6 may be hydrolyzed to GlcNAc by family GH20 N-acetylhexosaminidases or N,N-diacetylchitobiose phosphorylases (24). Transport of GlcNAc or deacetylated GlcN across the inner membrane can occur through phosphotransferase systems, while (GlcNAc)2 may be transported via the action of an ABC transporter (18). Once located in the cytosol, GlcNAc, GlcNAc1P, or GlcN undergo amino-sugar metabolism. It should be noted that the fate of chitooligosaccharide aldonic acid is not known.
Chitin degradation can be achieved by several marine bacteria and can give advantages for survival and proliferation in the marine environment (8, 25). Some pathogens have chitin central in their life cycle, the most prominent example being the human pathogen Vibrio cholerae that uses chitin-containing zooplankton as transfer vectors and nutrition (26, 27). The ability of the Gram-negative marine bacterium Aliivibrio salmonicida (previously Vibrio salmonicida), to utilize chitin or GlcNAc as a nutrient source is controversial. This pathogenic bacterium, which is the causative agent of cold water vibriosis in salmonids, was identified as a new vibrio-like bacterium in 1986 (28). Upon discovery and initial characterization of the pathogen (strain HI 7751), Egidius et al. (28) did not observe degradation of chitin by the bacterium when growing on agar plates containing purified chitin. On the other hand, the monomeric building block of chitin, GlcNAc, was readily consumed by the bacterium. When the genome of the bacterium was sequenced 2 decades later (strain LFI1238), it was shown that insertion sequence (IS) elements caused disruption of almost 10% of the protein-encoding genes (29, 30). Especially effected was the chitin utilization pathway, where seven genes, including those for three chitinases and a chitoporin, were either disrupted or truncated (29). In addition, the gene encoding the periplasmic chitin-binding protein (VSAL_I2576, also called CBP) was disrupted by a frameshift. The CBP ortholog in V. cholerae (VC_0620) has been shown to activate the two-component chitin catabolic sensor/kinase ChiS that regulates chitin utilization (31, 32). The gene encoding the ChiS ortholog in A. salmonicida is intact (29), along with the TfoX-encoding gene, for which the protein product also is involved in the regulation of enzymes related to chitin degradation in the Vibrionaceae (33, 34). Of the putative secreted chitinolytic enzymes, only genes encoding one chitinase and two lytic polysaccharide monooxygenases remained intact in the A. salmonicida genome. It was suggested that such extensive gene disruption could indicate inactivation of this pathway, and indeed, the authors did not observe degradation of insoluble chitin or utilization of GlcNAc as a nutrient source (29).
To obtain a deeper understanding of the roles of the A. salmonicida chitinolytic enzymes, we have analyzed the chitin degradation potential of A. salmonicida LFI1238 by biochemical characterization of the secreted chitinolytic enzymes, gene deletion and cultivation experiments, gene expression analysis, and proteomics.
RESULTS
A. salmonicida can utilize both GlcNAc and (GlcNAc)2 as nutrient sources.
To assess the ability of A. salmonicida LFI1238 (not to be confused with Aeromonas salmonicida) to grow on GlcNAc and (GlcNAc)2, the wild-type strain was cultivated in minimal medium supplemented with 0.2% glucose (11.1 mM; control experiment), 0.2% GlcNAc (9.0 mM), or 0.2% (GlcNAc)2 (4.7 mM) over a period of 92 h. The cultivation experiments showed that A. salmonicida can utilize both GlcNAc and (GlcNAc)2 as sole carbon sources (Fig. 1). Growth rates were compared by calculating the specific rate constants (μ) and generation time across the exponential phase (see Table S1 in the supplemental material), showing little difference between the three carbon sources. To correlate GlcNAc and (GlcNAc)2 consumption with the bacterial growth, the concentrations of these sugars in the culture supernatant were determined at different time points during growth (Fig. 1E and F). The data show decreasing concentrations of GlcNAc during growth and complete depletion within 40 h (Fig. 1E). In comparison, (GlcNAc)2 is utilized at a slower speed, becoming depleted after 80 h (Fig. 1F).
FIG 1.

Utilization of glucose, GlcNAc, and (GlcNAc)2. Growth of A. salmonicida LFI1238 in minimal medium supplemented with 0.2% glucose (A), 0.2% GlcNAc (9.0 mM) (B), or 0.2% (GlcNAc)2 (4.7 mM) (C). (D) Growth in defined medium without supplementation of carbon source (negative control). Growth results are shown as mean values from three biological replicates, and the standard deviation is indicated. Depletion of soluble substrates by A. salmonicida, determined by sampling of the culture supernatant of one replicate at different time points through the growth time period and quantification of GlcNAc (E) or (GlcNAc)2 (F) by ion exclusion chromatography. Results are shown as the mean values from three technical replicates.
Sequence analysis and homology modeling.
Since A. salmonicida was able to utilize both GlcNAc and (GlcNAc)2, the major products of enzymatic chitin degradation, it was of interest to analyze the chitinolytic potential of the bacterial genome, investigating the details of both intact genes and pseudogenes. A previous study had already identified the presence of three putatively secreted chitinolytic enzymes (29). Annotation of putative CAZy domains of these three enzymes using the dbCAN server (35) showed that the chitinase sequence, here named AsChi18A, (that contains 881 amino acids, which is unusually large for a chitinase) contains predicted carbohydrate-binding module 5 (CBM5) and CBM73 chitin-binding domains and a C-terminal GH18 domain, the latter modest in size (only 324 amino acids) (Fig. 2A). The protein sequence also shows long regions that were not annotated. Attempts to functionally annotate these regions with other sequence analysis servers such as InterPro, Pfam, and SMART were inconclusive. The relatively small size of the GH18 catalytic domain indicates an enzyme stripped of most subdomains that often are in place to form a substrate binding cleft. Indeed, homology modeling using Swiss-Model (36) revealed a model structure with a shallow substrate binding cleft, reminiscent of a nonprocessive endochitinase, which is clearly observed compared to the processive exochitinase SmChi18A from Serratia marcescens that has a deep substrate binding cleft and the shallow-clefted nonprocessive chitinase ChiNCTU2 from Bacillus cereus (37) (Fig. 2B). AsChi18A also shows an arrangement of active site residues that is similar to that of the latter enzyme (see Fig. S1).
FIG 2.

Predicted domains and three-dimensional structures of the A. salmonicida chitinase and LPMOs. (A) Prediction of CAZy domains of the chitinolytic enzymes was performed using the dbCAN server. Numbers indicate the position in the sequence. The theoretical molecular weights of the proteins calculated by the ProtParam tool (in the absence of the predicted signal peptide) are 87.4, 52.5, and 41.2 kDa for AsChi18A, AsLPMO10A, and AsLPMO10B, respectively. Signal peptides were determined by the SignalP 4.0 server (http://www.cbs.dtu.dk/services/SignalP/) and represent residues 1 to 29, 1 to 25, and 1 to 26 for AsChi18A, AsLPMO10A, and AsLPMO10B, respectively. The GenBank protein identifiers for the enzymes are CAQ78442.1 (AsChi18A, also called endochitinase A), CAQ80888.1 (AsLPMO10A, also called chitin-binding protein) and CAQ80971.1 (AsLPMO10B, also called chitinase B”). (B) The homology model of AsChi18A (left) and the structures of SmChi18A deep clefted exochitinase from S. marcescens (middle) and the Bacillus cereus GH18 ChiNCTU2 shallow clefted chitinase (37) (right) are shown in light brown surface representation with the catalytic acids colored red (or indicated by a red star for SmChiA, as it is concealed by other amino acids). Ligands are shown in stick representation with gray- (chitooctaose; SmChi18A) and purple-colored (chitobiose; ChiNCTU2) carbon atoms. Subsites are indicated by numbering. Ligands shown in the AsChi18A substrate binding cleft are derived from structural superimpositions of the AsChi18A model with SmChi18A or ChiNCTU2 and are provided for illustrational purposes only. The template used for modeling the AsChi18A catalytic GH18 domain was PDB ID 3N1A (apoenzyme structure of ChiNCTU2 from B. cereus) and gave a Q mean value of −1.99, which represents a good quality model. (C) The crystal structure of CBP21 (PDB ID 2BEM) and the homology models for AsLPMO10A and AsLPMO10B are shown in cartoon representation. For CBP21, the side chains of the amino acids that have been shown to be involved in substrate binding by experimental evidence (42, 43, 103) are shown in stick representation. The corresponding amino acids in AsLPMO10A and AsLPMO10B are also shown in stick representation. One exception is W46 of AsLPMO10B, which is not present in the two other enzymes. The latter residue is positioned on an insertion that potentially extends the putative binding surface (indicated by rectangle with dashed lines). In CBP21, Ser58 is shown with two alternative side chain conformations. Swiss-Model was used with default parameters to generate the homology models of AsLPMO10A and -B, using PDB structures 2XWX (66.5% sequence identity to AsLPMO10A) and 4YN2 (43.6% sequence identity to AsLPMO10B), respectively, as the templates. The Q mean scores obtained were −1.65 for the AsLPMO10A model and −3.34 for AsLPMO10B.
Annotation of the LPMO sequences showed that both proteins contained an N-terminal catalytic AA10 domain and a C-terminal CBM73 or CBM5 chitin-binding domain in AsLPMO10A or –B, respectively (Fig. 2A). Like the chitinase, both LPMOs displayed regions in the sequence that were not possible to annotate using standard bioinformatics tools. Pairwise sequence alignment of the two LPMOs revealed only 20% identity between the catalytic domains. BLAST search and modeling by homology of the individual catalytic domains showed that the catalytic module of AsLPMO10A was similar to CBP21 from S. marcescens (49.5% identity) (Fig. 2C) (38, 39) and to the catalytic AA10 domain of GbpA, a Vibrio cholerae colonization factor (40) (65.6% identity). The similarity of full-length AsLPMO10A to V. cholerae GbpA (61% sequence identity) and their similar multimodular architectures (both have an N-terminal AA10 LPMO domain, followed by a “GbpA2” domain, an unannotated domain, and a C-terminal CBM73 domain) indicate the possibility of functionally similar roles. The catalytic AA10 domain of AsLPMO10B is, as already noted, unlike that of AsLPMO10A. From sequence database searches, orthologs were identified in a large variety of species from the Vibrionaceae family and also in other marine bacteria such as Shewanella and Pseudoalteromonas. None of these related enzymes have hitherto been biochemically characterized. When searching for similar sequences in the PDB database, the most similar structure to the AsLPMO10B catalytic domain belongs to the viral proteins called “spindolins” (43.5% identity, but the alignment contains many insertions/deletions). No activity data exists for spindolins, but it is assumed that they are active toward chitin (41). It is therefore not straightforward to assign an activity to AsLPMO10B based on sequence analysis. To analyze the putative structural difference between the LPMO domains, homology models were made using Swiss-Model homology modeling software (36). Compared to CBP21, one of the best-characterized family AA10 LPMOs, both A. salmonicida LPMOs show several differences that may influence both substrate binding and catalysis (Fig. 2C). AsLPMO10A is relatively similar to CBP21 but displays some differences that may be of functional relevance: amino acids W62, R119, and K195 in AsLPMO10A correspond to amino acids Y54, T111, and N185 in CBP21 that all have been shown to influence substrate binding and the functional stability of the enzyme (42, 43). AsLPMO10B shows an active site environment similar to that of CBP21 but has an extension of the putative binding surface that positions a putatively solvent-exposed Trp (W46) further away from the active site histidines than those for Y54 in CBP21 and W62 in AsLPMO10A. Whether these differences are important for the substrate binding properties of the enzymes is not straightforward to interpret based on the data presented in this study, since both A. salmonicida proteins have CBMs that very likely contribute to chitin binding.
Analysis of pseudogenes related to chitin catabolism.
In addition to the intact genes encoding the chitinase AsChi18A and LPMOs AsLPMO10A, and -B, the genome of A. salmonicida LFI1238 harbors multiple pseudogenes encoding truncated or fragmented enzymes related to chitin catabolism that are assumed to be nonfunctional (open reading frame [ORF] identifiers VSAL_I0763, VSAL_I0902, VSAL_I1108, VSAL_I1414, and VSAL_I1942) (see Fig. S2 for detailed illustration of the genomic organization of the truncated genes and the associated insertion sequence [IS] elements). Interestingly, transcription of A. salmonicida pseudogenes (including chitinase-related pseudogenes) has been observed (44–46). In addition, A. salmonicida is motile despite two flagellar synthesis genes (fliF/VSAL_I2308 and (flaG/VSAL_I12316) being disrupted by premature stop codons (29). Thus, we performed a deeper analysis of the putative protein-encoding parts of the A. salmonicida pseudogenes related to the chitinolytic machinery to investigate their putative functionality (Table 1). All proteins encoded by the truncated genes were essentially identical to sections in orthologous proteins in the closely related bacterium Aliivibrio logei 5S-186, showing >95% sequence identity of the aligned regions. In more detail, VSAL_I0763 encoded a protein containing 202 amino acids that was 99% identical to a region in a putative family GH18 chitinase. However, VSAL_I0763 did not contain the catalytic DXXDXDXE sequence motif characteristic of GH18 chitinases. VSAL_I0902, VSAL_I1108, and VSAL_I1942 also appeared to be truncated family GH18 chitinases, but contrary to VSAL_I0763, they were largely intact and all contained the catalytic sequence motif. It should be noted that VSAL_I0902 and VSAL_I0763 are fragments belonging to the same chitinase, as they are essentially identical (99% sequence identity) to separate parts of the family GH18 chitinase sequence encoded by the Aliivibrio logei 5S-186 OCH20886.1 gene (see Fig. S11 and S12). Finally, VSAL_I1414 proved to be a family 19 chitinase with a minor truncation at the C terminus.
TABLE 1.
Sequence analysis of truncated genes in the A. salmonicida genome related to chitin catabolism
| Gene IDa | Proteinb | Size (no. of aa)c | dbCAN2d | Prosited | Similaritye | Seq IDf (%) | Coverageg (%) |
|---|---|---|---|---|---|---|---|
| VSAL_I0763 | 202 | No hit | No hit | Chitinase (Aliivibrio logei 5S-186), OCH20886.1, 733 aa | 99 | 27.6 | |
| VSAL_I0902 | AsChi18Bp | 533 | GH18 | GH18 catalytic motif | Chitinase (Aliivibrio logei 5S-186), OCH20886.1, 733 aa | 99 | 72.7 |
| VSAL_I1108 | AsChi18Dp | 880 | GH18 | GH18 catalytic motif | WSD1 family O-acyltransferase (Aliivibrio sp. SR45-2)h, WP_065610756.1, 1,051 aa | 98 | 83.7 |
| VSAL_I1414 | AsChi19p | 506 | GH19 | No hit | Carbohydrate-binding protein (Aliivibrio logei), WP_065612067.1, 558 aa | 99 | 90.7 |
| VSAL_I1942 | AsChi18Cp | 720 | GH18 | GH18 catalytic motif | Chitinase (Aliivibrio logei), WP_065612067.1, 844 aa | 99 | 81.4 |
ID, identifier of the truncated gene sequence obtained from the A. salmonicida genome sequence (GenBank no. FM178379.1).
All proteins that were putative active enzymes if translated were named according to CAZy nomenclature, indicating the enzyme family and activity but also with the letter “p” at the end indicating “putative.”
Number of amino acids (aa) encoded by the gene.
dbCAN2 and Prosite columns show the CAZy annotation obtained by analysis of the truncated protein sequence by the dbCAN2 annotation tool (http://bcb.unl.edu/dbCAN2/) and annotation by the Prosite database (https://prosite.expasy.org), respectively.
Sequence of the most similar protein obtained by protein BLAST, its identifier, and the number of amino acids in the complete protein. Genes were translated to protein sequences using the Expasy Translate tool.
Percentage of identical amino acids obtained from pairwise sequence alignment of the truncated A. salmonicida protein and the most similar match obtained by protein BLAST using EMBOSS Needle sequence alignment tool.
Extent of coverage obtained by the truncated protein toward the complete protein in the pairwise sequence alignment. The pairwise sequence alignments used for the analysis shown in this table are available in Fig. S11 to S16 in the supplemental material.
This gene seems to be incorrectly annotated as analysis by dbCAN2; Prosite and Intepro all indicated that the protein encoding sequence is a family GH18 chitinase and not an O-acyltransferase.
Based on the pseudogene analysis, VSAL_I0902, VSAL_I1108, VSAL_I1942, and VSAL_I1414 can result in functional protein if translated and properly folded. To make the interpretation of results that include these pseudoproteins more convenient, they were named according to CAZy nomenclature indicating enzyme family, putative function, and the letter “p” indicating pseudoprotein (VSAL_I0902, AsChi18Bp; VSAL_I1942, AsChi18Cp; VSAL_I1108, AsChi18Dp; and VSAL_I1414, AsChi19p) (Table 1).
AsChi18A and AsLPMO10A and -B bind chitin.
To determine the biochemical properties of putatively chitinolytic enzymes (the pseudogene-encoded chitinases were not expressed and characterized), AsChi18A and AsLPMO10A and -B were cloned, expressed, and purified (see Fig. S3). The presence of putative chitin-binding modules on all three chitinolytic enzymes prompted investigation of the substrate binding properties of the proteins. Using purified protein, α-chitin and β-chitin were used as the substrates in particle sedimentation assays (Fig. 3). All proteins showed binding to the substrate particles, and AsLPMO10B seems to bind slightly weaker to the substrates used than AsLPMO10A.
FIG 3.
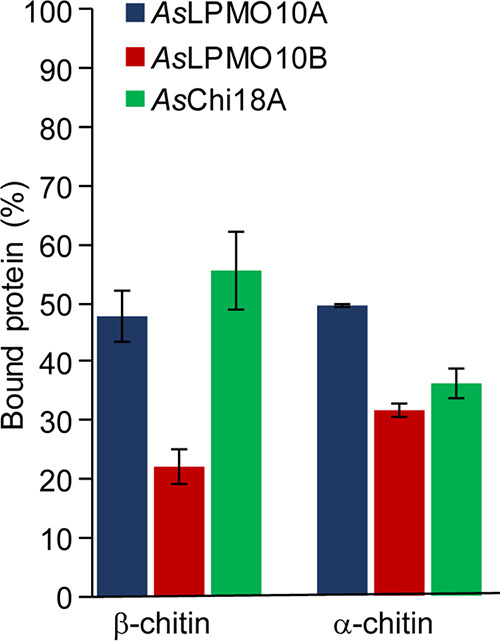
Substrate binding of AsChi18A and AsLPMO10A and -B. Each bar shows the percentage of bound proteins after 2 h of incubation at 30°C. Reaction mixtures contained 10 mg/ml of substrate, 0.75 μM (LPMOs) or 0.50 μM (AsChi18A) of enzymes, and 10 mM Tris-HCl buffer at pH 7.5. All reactions were run in triplicates, and the standard deviations are indicated by error bars.
AsChi18A displays low chitinolytic activity.
Since all three enzymes bound to chitin, the catalytic properties of the purified chitinase and two LPMOs were analyzed. Using β-chitin as the substrate, the activity and operational stability of AsChi18A was followed over several hours at temperatures ranging from 10 to 60°C. The progress curves observed for AsChi18A indicate an optimal operational stability, i.e., the highest temperature for which enzyme activity remains stable over time, at approximately 30°C (Fig. 4A). Similar to that for other GH18 chitinases, the dominant product of chitin hydrolysis by AsChi18A was (GlcNAc)2, with small amounts of GlcNAc (<5%).
FIG 4.
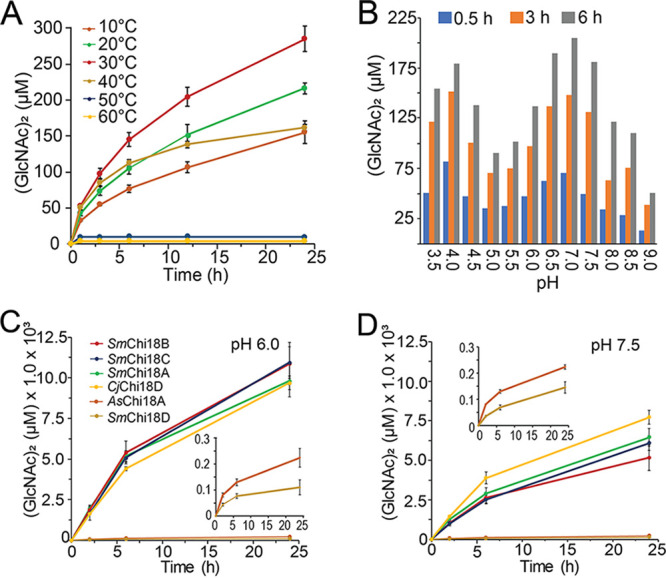
Enzymatic properties of AsChi18A. Production of (GlcNAc)2 by AsChi18A analyzed at various temperatures (A) and pH values (B). The activity of AsChi18A was also compared to that of the chitinases from Serratia marcescens (SmChi18A, -B, -C, and -D) and C. japonicus (CjChi18D) at pH 6.0 (C) and 7.5 (D). All reaction conditions included 10 mg/ml β-chitin and 0.5 μM enzyme. For data displayed in panel A, reactions were carried out at pH 7.5. For the data displayed in panel B, all reaction mixtures were incubated at 30°C. Buffers used were as follows: formic acid, pH 3.5; acetic acid, pH 4.0 and 4.5; ammonium acetate, pH 4.5 and 5.0; morpholineethanesulfonic acid (MES), pH 5.5, 6.0, and 6.5; bis-Tris-HCl, pH 7.0; Tris-HCl, pH 7.5 and 8.0; and bicine, pH 8.5 and 9.0. The amounts of (GlcNAc)2 presented are based on the average from three independent reaction mixtures containing 10 mg/ml β-chitin, 0.5 μM enzyme, and 10 mM buffer. The insets in panels C and D show magnified views of reactions catalyzed by AsChi18A and SmChi18D. Standard deviations are indicated by error bars (n = 3).
To compare AsChi18A activity with that of other well-characterized chitinases, the chitin degradation potential of the enzyme was compared with those of the four GH18 chitinases of S. marcescens (SmChi18A, -B, -C, and -D) (47–49) and CjChi18D, which is the most potent chitinase of Cellvibrio japonicus (50). Activities were monitored at pH 6.0 (Fig. 4C), which is the pH where the S. marcescens and C. japonicus chitinases have their optima (47, 51, 52), and at pH 7.5 (Fig. 4D), which is a typical pH of seawater and the near pH optimum of AsChi18A (Fig. 4B). Strikingly, SmChi18A, -B, and -C and CjChi18D yielded more than 50-fold more (GlcNAc)2 than AsChi18A after 24 h of incubation at pH 6. At pH 7.5, the differences in yields were lower (in the range of 25- to 40-fold larger yields, except for SmChi18D), most likely reflecting the difference in pH optima. It should be noted that the presence of NaCl in concentrations similar to that in seawater (∼0.6 M) only marginally influenced AsChi18A activity (see Fig. S4).
AsLPMO10A and -B are active toward chitin.
Both A. salmonicida LPMOs were able to oxidize α- and β-chitin, yielding aldonic acid chitooligosaccharide products with degree of polymerization ranging from 3 to 8 (see Fig. S5). Such product profiles are commonly observed for family AA10 LPMOs that target chitin (12, 14, 53). The two enzymes displayed slightly different operational stabilities when probed at temperatures ranging from 10 to 60°C (Fig. 5). AsLPMO10A showed an operational stability similar to that of AsChi18A, being approximately 30°C (Fig. 5A and B). In contrast, AsLPMO10B showed an operational stability lower than 30°C (Fig. 5C and D). Comparison of the LPMO activities showed that AsLPMO10A seems generally more active than AsLPMO10B, the former enzyme yielding approximately twice as much soluble oxidized product than the latter (Fig. 5B and D).
FIG 5.
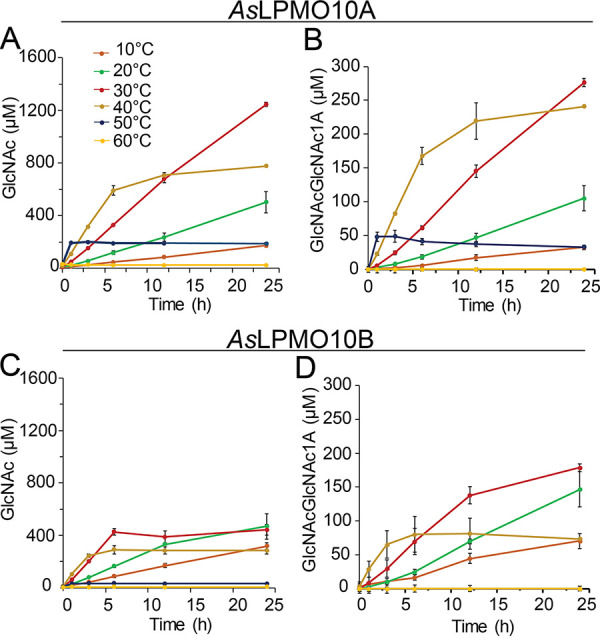
Operational temperature stability of A. salmonicida LPMOs. The activity of AsLPMO10A (A) and AsLPMO10B (B) is indicated by the production of GlcNAc. Since the end product of chitin degradation by the LPMOs is oxidized chitooligosaccharides (see Fig. S5 in the supplemental material) that are inconvenient to quantify, the reaction products obtained from the reactions were depolymerized by chitobiase that completely converts the oligosaccharide mixture to GlcNAc and oxidized (GlcNAc)2 (i.e., GlcNAcGlcNAc1A). (B and D) Quantities of the latter products formed by the LPMOs. The amounts presented are based on the average from three independent reactions, which contained 10 mg/ml of β-chitin, 1 μM enzyme, 1 mM ascorbic acid, and 10 mM Tris-HCl buffer at pH 7.5, incubated at different temperatures between 10 and 60°C (color code provided in panel A). Standard deviations are indicated by error bars.
Combination of the chitinase and LPMOs shows enzyme synergies.
For the putative chitinolytic system of A. salmonicida, the situation was different than that for any other chitinolytic system studied, since the chitin degradation potential of the chitinase was substantially lower than that of the LPMOs (Fig. 4C and D and 5). Usually, the chitinase of a chitinolytic system is substantially more efficient in substrate solubilization than the LPMO. Nevertheless, synergies were observed when combining the AsChi18A with AsLPMO10B, giving an almost double yield than the sum of products calculated by adding the sum of their individual yields, for both β- and α-chitin (Fig. 6). AsLPMO10A, on the other hand, showed a weaker synergy when combined with AsChi18A.
FIG 6.
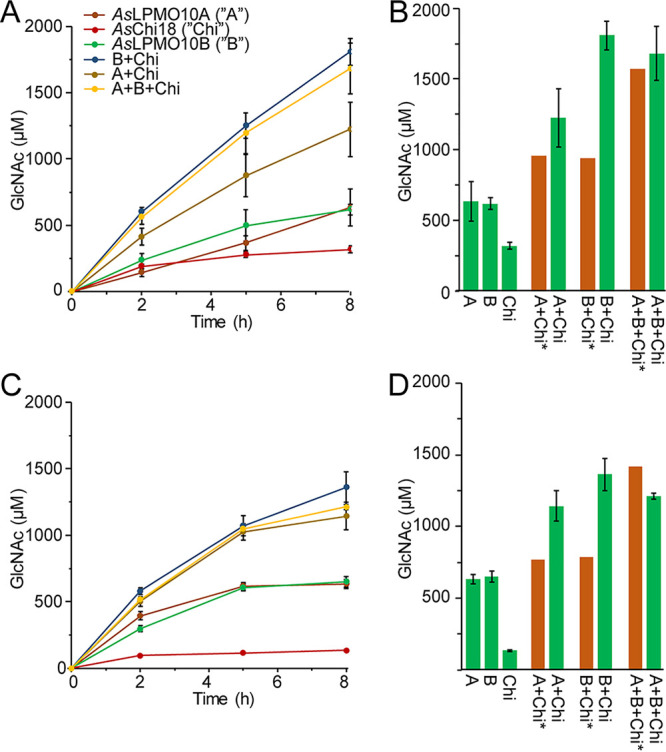
Synergistic activity of AsLPMO10s and AsChi18A on chitin. Production of GlcNAc by the individual and combined enzymes on β-chitin (A) and α-chitin (C). (B and D) Theoretical calculated amounts of GlcNAc based on the sum of its production by the individual enzymes (brown bars) and the detected amounts of GlcNAc by combining the enzymes after 8 h (green bars). The amounts presented are based on the averages from three independent reaction mixtures containing 10 mg/ml of chitin substrate, 1 μM LPMOs and/or 0.5 μM GH18, 1 mM ascorbic acid, and 10 mM Tris-HCl buffer at pH 7.5 incubated at 30°C for 8 h. Standard deviations are indicated by error bars (n = 3).
AsChi18A is important for growth of A. salmonicida on chitin.
Since the A. salmonicida chitinase and LPMOs were able to depolymerize both α- and β-chitin to soluble sugars that are metabolizable for the bacterium [GlcNAc and (GlcNAc)2], the ability of the bacterium to utilize chitin particles as a carbon source was assessed. For this experiment, β-chitin was used for its higher purity and lower recalcitrance than those of α-chitin. To unravel the roles of AsChi18A and AsLPMO10A and -B in chitin degradation, A. salmonicida gene deletion strains were included in the cultivation experiments. The two single-LPMO-deletion strains showed a moderate decrease of the growth rate compared to that of the wild type, displaying a 30% increase in generation time (Fig. 7A and Table 2). In contrast to the biochemical assays that showed stronger synergy between recombinant AsChi18A and AsLPMO10B than between AsChi18A and AsLPMO10A, the cultivation assays showed that deletion of the single LPMOs resulted in the same growth reduction as deletion of both LPMOs. Deletion of the AsChi18A gene decreased growth to a larger extent than observed for the LPMO mutant strains (Fig. 7A), indicating that AsChi18A is more important than the LPMOs for the ability of A. salmonicida to utilize chitin as a carbon source. The triple deletion mutant (ΔAΔBΔChi) was least able to utilize chitin as a source of nutrients, which also was clear from an agar plate chitin solubilization assay where only a marginal disappearance of chitin was observed (see Fig. S6). Growth of the ΔAΔBΔChi mutant and the wild type on LB25 medium was, on the other hand, similar (see Fig. S7), indicating that the gene deletions only influenced chitin utilization and not metabolism in general.
FIG 7.
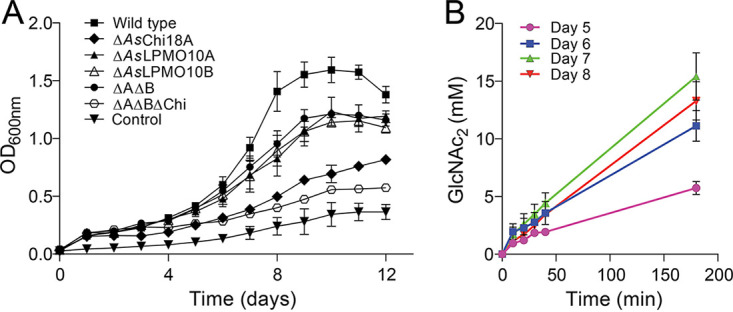
Growth of A. salmonicida LFI1238 and derivate gene deletion strains on β-chitin. (A) Growth of A. salmonicida LFI1238 at 12°C in minimal medium supplemented with 1% β-chitin. (B) Chitinase activity in the culture supernatant of A. salmonicida growing on β-chitin. The chitinase activity was assayed by mixing a sample of the culture supernatant sampled at various time points with 15 mM chitopentaose and quantifying the (GlcNAc)2 resulting from hydrolysis over a period of 180 min. Error bars indicate standard deviations (n = 3).
TABLE 2.
Growth rate and maximum cell density of A. salmonicida and derivative mutant strains
| Strain | Rate constant (μ [h−1]) | Generation time (h) | Max cell density (OD600) |
|---|---|---|---|
| Wild type | 0.43 ± 0.01 | 17.5 ± 0.4 | 1.60 ± 0.08 |
| ΔAsChi18A | NAa | NA | 0.82 ± 0.03 |
| ΔAsLPMO10A | 0.27 ± 0.07 | 29.1 ± 8.2 | 1.25 ± 0.08 |
| ΔAsLPMO10B | 0.28 ± 0.01 | 26.8 ± 1.1 | 1.15 ± 0.04 |
| ΔAΔB | 0.28 ± 0.02 | 26.8 ± 1.7 | 1.24 ± 0.04 |
| ΔAΔBΔChi | NA | NA | 0.58 ± 0.02 |
| Wild-type control medium | NA | NA | 0.37 ± 0.05 |
NA, not available.
It should be noted that the wild-type bacteria incubated in the minimal medium (Asmm) without added chitin obtained growth to an optical density (OD) of 0.37 ± 0.05 after 7 days of incubation (Fig. 7A and Table 2) due to the presence of essential amino acids and traces of the LB25 preculture medium. Furthermore, it was also observed that all bacterial strains incubated in the defined medium supplemented with chitin showed an increase in the OD of ∼0.1 within the first 24 h. This was most likely caused by the presence of chitin monomers, dimers, oligosaccharides, or other nutrients in the chitin substrate that could be utilized by the bacteria without need of the chitinase or LPMOs.
To evaluate whether growth of the bacterium correlated with chitinolytic activity, the culture supernatant of the wild type growing on β-chitin was sampled once a day in the period of highest growth (days 5 to 8) and analyzed for hydrolytic activity toward the soluble chitooligosaccharide chitopentaose. Indeed, the chitin hydrolytic potential of the culture supernatant increased from day 5 to day 8 (Fig. 7B), indicating secretion of one or more chitinases (only dimeric and trimeric products were observed; large concentrations of GlcNAc would indicate the presence of a secreted N-acetylhexosaminidase).
Gene expression analysis by PCR amplification of cDNA.
Encouraged by the biochemically functional chitinolytic machinery of A. salmonicida and the ability of the bacterium to metabolize chitin degradation products and chitin particles, it was of interest to couple these traits to transcription of genes representing the enzymes in the chitinolytic machinery. The pseudogene encoding parts of a family GH18 chitinase (VSAL_I0902; AsChi18Bp) was also included in the analysis. RNA was isolated from A. salmonicida LFI1238 grown on glucose, GlcNAc, (GlcNAc)2, and β-chitin (same cultures as shown in Fig. 1 and 7), from both exponential and stationary phases. Gene expression was assessed qualitatively by agarose gel chromatography (Table 3). The gene expression was assessed as positive if the target gene was amplified in two of three biological replicates and, at the same time, no amplification was observed in PCR samples obtained in the control reactions having no reverse transcriptase during cDNA synthesis (examples shown in Fig. S8). The resulting data indicated that AsChi18A, AsLPMO10B, and, surprisingly, the chitinase pseudogene AsChi18Bp were expressed in the exponential phase during growth on all carbon sources. Similarly, expression of AsChi18A and AsLPMO10A was detected in the stationary phase, however, not under all conditions. Expression of AsLPMO10B was only detected in the exponential phase during growth on GlcNAc.
TABLE 3.
Gene expression resultsa
| Geneb | GlcNAc |
(GlcNAc)2 |
Glucose |
β-Chitin |
||||
|---|---|---|---|---|---|---|---|---|
| Exp | Stat | Exp | Stat | Exp | Stat | Exp | Stat | |
| AsChi18A | + | + | + | − | + | + | + | + |
| AsLPMO10A | + | + | + | + | + | − | + | − |
| AsLPMO10B | + | − | − | − | − | − | − | − |
| AsChi18B p | + | − | + | − | + | − | + | − |
Exp, exponential phase, Stat, stationary phase. Data shown as positive (“+” in bold) or negative (“−”) detection of expression, based on three biological replicates.
AsChi18A (VSAL_I0757), AsLPMO10A (VSAL_II0134), AsLPMO10B (VSAL_II0217), and AsChi18Bp (VSAL_I0902).
Proteomic analysis of expressed carbohydrate-active enzymes.
To obtain a more complete understanding of chitin degradation by A. salmonicida during growth, label-free quantitative proteomics was used to identify and quantify proteins secreted by the bacterium when growing on this insoluble polysaccharide. Guided by the gene expression analysis (Table 3), cultures were grown to exponential phase on 1% β-chitin before harvest and separation into supernatant and cell pellet fractions for analysis of both secreted and intracellular proteins. For analysis of bacteria and proteins binding to chitin, chitin from the growing culture was collected and boiled directly in sample buffer. These samples are referred to as “chitin-bound” samples and are enriched in proteins with high affinity for chitin. In total, 1,179 proteins were identified (see Data Set 1A), from which 20 were annotated as carbohydrate-active enzymes (CAZymes) (Data Set 1B), including glycoside hydrolases, those with transferase activities, and those involved in lipid biosynthesis, glycogen metabolism, peptidoglycan (murein), and carbohydrate metabolic processes (Fig. 8; see also Table S2). In more detail, both LPMOs (AsLPMO10A and AsLPMO10B) and AsChi18A were identified, albeit not in all samples and at variable intensities. AsLPMO10A was present at highest abundance among the CAZymes, especially in the chitin-bound samples. The protein was identified in all three biological replicates under all sampled conditions except in the bacterial pellet obtained from growth on glucose, where the protein was only identified in one biological replicate (Fig. 8).
FIG 8.

CAZymes expressed by A. salmonicida LFI1238. Heat map presentation of identified CAZymes (Data Set 1B) and label-free quantification intensities ranging from low intensity (gray) to medium intensity (red) and high intensity (white). The data are presented as three biological replicates. Conditions are as follows: proteins eluted from chitin obtained from the culturing experiment (Chitin), culture supernatant proteins from the chitin cultivation experiment (ChitinS), proteins extracted from the bacterial cells obtained from the chitin cultivation experiment (ChitinP), culture supernatant proteins obtained from culturing the bacterium on glucose (GlucoseS), and proteins extracted from bacterial cell pellet from the glucose cultivation experiment (GlucoseP).
AsChi18A and AsLPMO10B were only detected in the culture supernatant in one or two of the biological replicates obtained from growth on glucose and in two of three replicates of the chitin-bound samples. AsChi18A was only identified in the chitin-bound sample and the culture supernatant of the glucose grown samples. However, the chitinase was found at a noticeable higher intensity in the chitin-bound samples than in the supernatant samples obtained from cultivation on glucose.
Importantly, VSAL_I2989, a family GH20 β-N-acetylhexosaminidase, was identified among the CAZymes. This enzyme is vital for hydrolyzing (GlcNAc)2 into two GlcNAc units but also has the ability to depolymerize longer chitooligosaccharides (even aldonic acid chitooligosaccharides resulting from LPMO activity) (53). Sequence analysis revealed 58% identity between VSAL_I2989 (∼100% sequence coverage) and the biochemically characterized VhNAG1 (a family GH20 β-N-acetylhexosaminidase) from Vibrio harveyi 650 (54). The amino acids involved in catalysis and substrate binding are conserved (see Fig. S9), indicating a function of VSAL_I2989 in chitin catabolism. It should be noted that N,N-diacetylchitobiose phosphorylases can also perform a role similar to that of β-N-acetylhexosaminidases. Interestingly, a family 3 glycosyl hydrolase (GH3), annotated as NagZ, was also identified. GH3s have a broad range of substrate specificities, which mostly involves peptidoglycan recycling pathways. However, the marine bacteria Pseudoalteromonas piscicida, Vibrio furnissii, and Thermotoga maritima harbor GH3s that are believed to participate in intracellular chitin metabolism (55–57). NagZ was detected at similar levels in both glucose and chitin cultures, indicating that it is not dependent on chitin degradation. Also, the amino acid sequence of NagZ was similar to those of other NagZ orthologs in this GH family (e.g., 67% sequence identity to NagZ of V. cholerae) that remove β-N-acetylglucosamine from ends of peptidoglycan fragments (58). MalQ (a family GH77 4-alpha-glucanotransferase,) and MltD (a family GH23 membrane-bound lytic murein transglycosylase) were only detected when the bacterium was grown on glucose. VSAL_I1407 (putative family 2 glycosyl transferase family 2 GT2) was only detected in the chitin substrate fraction. GTs are generally involved in biosynthesis by transferring sugar moieties from activated donor molecules to specific acceptor molecules, forming glycosidic bonds.
Analysis of the chitin catabolic pathway in A. salmonicida.
To assess the chitin catabolic pathway used by the bacterium, the proteomics data were scrutinized with the aim of identifying expressed proteins with a putative role in uptake, transport, or downstream processing of chitin degradation products. An illustration of relevant findings and the suggested pathway is shown in Fig. 9 (proteomics data and protein identifiers are shown in Data Set 1C). Guided by the biochemical assays and cultivation experiments, secreted AsChi18A, AsLPMO10A, and AsLPMO10B are indicated to hydrolyze and cleave chitin into smaller oligosaccharides. It must be noted that AsChi18Bp, AsChi19Ap, and AsChi18Cp are illustrated in context with AsChi18A based on conserved domains rather than evidence of participating in extracellular hydrolysis of chitin. Interestingly, AsChi18Bp is one of few proteins exclusively identified in chitin samples. VSAL_I2989, the family GH20 β-N-acetylhexosaminidase, which shows an ∼3-fold increase in abundance during growth on chitin compared to that on glucose (P = 0.0082, paired two-tailed t test) (see Fig. S10), is indicated to hydrolyze (GlcNAc)2 into GlcNAc in the periplasmic space.
FIG 9.

Putative chitin utilization pathway by A. salmonicida LFI1238. Illustration of detected proteins by label-free proteomics (Data Set 1C) aligned with their putative roles in the utilization pathway and the MaxLFQ intensities. Enzymes acting on chitin include AsChi18A, AsLPMO10A, AsLPMO10B, and putative pseudogene chitinases (AsChi18Bp, AsChi18Cp, and AsChi19Ap). Transport across membranes involves phosphotransferase system (PTS) components (BglI, Crr, VSAL_I1659, and VSAL_II0823) and NagE (PTS permease for N-acetylglucosamine and glucose). Hydrolysis of (GlcNAc)2 into GlcNAc involves VSAL_I2989 (family GH20 beta-N-acetylhexosaminidase). Amino sugar metabolism involves NagK (N-acetyl-d-glucosamine kinase), NagB (glucosamine-6-phosphate deaminase), GlmU (N-acetylglucosamine-1-phosphate uridyltransferase/UDP-N-acetylglucosamine pyrophosphorylase), and GlmM (phosphoglucosamine mutase). A bar chart comparing the log2 LFQ values of the putative chitinolytic enzymes is shown in Fig. S10.
Utilization of extracellular sugars requires uptake and transportation across both the outer and inner membranes. With the lack of a functional chitoporin, other proteins relevant for outer membrane transport were investigated. Of the proteins related to transport through the outer membrane, 14 proteins were identified, including outer membrane assembly factors and outer membrane proteins of the OmpA family, OmpU, and TolC. These proteins are not generally known for sugar transport but cannot be excluded. For transport of sugars across the inner membrane, the most relevant transporters identified were 9 proteins assigned to the phosphoenolpyruvate-dependent sugar phosphotransferase system and two N-acetylglucosamine and glucose permeases (annotated NagE and NagE* in Fig. 9 and Data Set 1C). These transporters are likely contributing to translocation of GlcNAc across the inner membrane and showed increased abundance in chitin samples compared to that of glucose (Fig. 9). Two phosphotransferase system (PTS) IIA components (Crr and BglI) and two lactose/cellobiose-specific IIB subunits (VSAL_I1659 and VSAL_II0823) were identified, of which the lactose/cellobiose-specific subunits likely contribute to sugar transportation across the inner membrane, were found upregulated during growth on chitin compared to that on glucose. It should be noted that no ABC transporter proteins specific for (GlcNAc)2 or GlcNAc-specific subunits were identified, although these are common in transport of such sugars (59–61).
In terms of downstream processing of GlcNAc and amino sugar metabolism, the monosaccharide is most likely converted into GlcNAc-6-phosphate (GlcNAc-6P) by the permease NagE or the N-acetylglucosamine kinase NagK (Fig. 9). Deacetylation of GlcNAc-6P by the N-acetylglucosamine 6-phosphate deacetylase NagA (not identified in the present experiment) would result in GlcN-6P, a product further processed into fructose-6-phosphate (Fru-6P) by the glucosamine-6-phosphate deaminase NagB, an enzyme which was found at higher abundance than glucose in the chitin pellet samples (Fig. 9). Alternatively, GlcN-6P can be processed (in three steps) by the phosphoglucosamine mutase GlmM or by the bifunctional N-acetylglucosamine-1-phosphate uridyltransferase/UDP-N-acetylglucosamine pyrophosphorylase GlmU into UDP-GlcNAc. The latter sugar nucleotide can be processed to other UDP sugars or utilized in pathways such as lipopolysaccharide biosynthesis or peptidoglycan synthesis. GlmM and GlmU were found under all conditions analyzed (Fig. 9).
DISCUSSION
Knowing whether A. salmonicida is able to utilize chitin as a source of carbon (and nitrogen) is important for understanding the ecology of the bacterium and its implications for pathogenicity. The literature contains conflicting information about this topic, but in the present study, we clearly demonstrate that A. salmonicida is capable of degrading chitin to soluble chitooligosaccharides and utilizing these as a nutrient source. This capability is dependent on the single chitinase in the A. salmonicida genome despite the low in vitro activity of the chitinase and the ability of the LPMOs to degrade chitin. In the absence of AsChi18A, only products from LPMO activity will be available to the bacterium. These products are oxidized chitooligosaccharides with a high degree of polymerization that most likely cannot be taken up by the bacterium due to the absence of a specific outer membrane transporter (chitoporin). The fact that minor growth of the bacterium is still achieved in the absence of the chitinase is most likely due to the presence of a family GH20 N-acetylhexosaminidase in the culture supernatant that can depolymerize LPMO-generated chitooligosaccharides to GlcNAc, which can be taken up and catabolized by the bacterium. Another explanation may be that the chitooligosaccharides are cleaved by secreted pseudochitinases, proteins indeed observed by the proteomics data. In support of the latter hypothesis, minor growth on β-chitin and indications of degradation of colloidal chitin were observed for the A. salmonicida ΔAΔBΔChi variant (Fig. 7 and Fig. S5 in the supplemental material, respectively). Notably, the importance of a single chitinase for growth on chitin is not unique to A. salmonicida LFI1238. In C. japonicus, CjChi18D is essential for the degradation of α-chitin despite the expression of three additional chitinases and two LPMOs (50). Similarly, a systematic genetic dissection of chitin degradation and uptake in Vibrio cholerae found that the chitinase ChiA2 is critical for growth on chitin but not sufficient alone (62).
Both A. salmonicida LPMOs are required for obtaining maximum growth on chitin, an observation that is different than for the efficient chitin degrader C. japonicus, where deletion of the chitin-active LPMO resulted in delayed growth but did not affect the growth rate (50). This may be explained by the 50-fold lower activity of AsChi18A than of CjChi18D of C. japonicus. In C. japonicus, the contribution of the LPMOs to chitin solubilization is most likely minor compared to that in A. salmonicida, for which the rate of depolymerization is almost equal for the LPMOs and the chitinase. AsLPMO10A and -B are distinctly different in domain organization and sequence, and the former enzyme is more active toward β-chitin than the latter. This may be related to the chitin binding properties of the enzymes, as AsLPMO10A binds better than AsLPMO10B to both α- and β-chitins (Fig. 3). Alternatively, the difference in activity can be related to the ability of the components in the reaction mixture to generate reactive oxygen species such as hydrogen peroxide, e.g., by the oxidase activity of LPMOs, as shown in several studies (63–65). In such a scenario, the discovery that LPMOs can use H2O2 as a cosubstrate and that the concentration of H2O2 in solution may be rate limiting for LPMO reactions (13, 66, 67) may account for activity differences between LPMOs when no external H2O2 is added to the enzyme reaction (only reductant).
The contribution of the LPMOs to chitin utilization by A. salmonicida is most likely related to the synergy obtained when combining the LPMOs with the chitinase. Such synergy can be explained by the ability of AsLPMO10s to cleave chitin chains that are inaccessible to AsChi18A (i.e., in the crystalline regions of the substrate). The newly formed chitin chain ends formed by LPMO activity represent new points of attachment for the chitinases, thereby increasing substrate accessibility. Indeed, several studies have demonstrated this phenomenon (16, 68–70), including a study on the virulence-related LPMO from Listeria monocytogenes (71).
A surprising observation was made when combining both LPMOs and the chitinase in a chitin degradation reaction (Fig. 6B and D). Here, no synergy was observed for β-chitin degradation, and a lower-than-theoretical yield was obtained for α-chitin. This was unexpected, since the bacterial cultivation assay indicated a cooperative relationship between the LPMOs, as the reduced growth observed for two single-LPMO-deletion strains was similar to that observed for the double-LPMO mutant strain (AsΔLPMO10A-ΔLPMO10B). The explanation for the lack of synergy is not straightforward, but it may be that a total concentration of 2 μM LPMO is too much for these reactions, giving rise to less bound enzyme to the substrate and thereby production of harmful reactive oxygen species (ROS) by the nonbound LPMO molecules. It is well established that LPMOs not bound to the substrate are more prone to autooxidation (13, 43, 72). Another explanation could be that a nonoptimal enzyme stoichiometry could create competition for substrate binding sites. Indeed, both LPMOs were expressed during growth on β-chitin, although AsLPMO10A was detected in substantially higher abundance. As a matter of fact, AsLPMO10A was the protein showing the highest abundance among the detected CAZymes, also when the bacterium was cultivated on glucose. This could imply that this LPMO has additional functions (this is discussed in more detail below). All three chitinolytic enzymes were observed in highest abundance in the samples obtained from the chitin particles, indicating high affinity of the enzymes toward chitin, a trait corroborated by the substrate binding experiments.
The proteomic analysis identified peptides from three pseudogenes. Interestingly, AsChi18Bp was only identified during growth on chitin, in contrast to the gene expression analysis where it was detected during growth in all carbon sources. This suggests a regulatory mechanism of translation influenced by the presence of chitin particles and that the relevant transcription factor regulating this gene is still functional. It is not uncommon that bacterial pseudogenes are expressed (73, 74), and Kuo and Ochman have hypothesized that this may be related to the regulatory region of the pseudogenes remaining intact (74). It must be noted that translation of a pseudogene does not necessarily equate to a functional protein. Indeed, our data showing a large growth impairment upon AsChi18A deletion suggest that translation of pseudogenes is insufficient for chitin degradation, although, as previously noted, minor growth can also be observed for the triple-knockout strain. Pseudogenes have long been considered to only represent dysfunctional outcomes of genome evolution, and the multitude of pseudogenes in A. salmonicida LFI1238 possibly reflects its adaption to a pathogenic lifestyle. On the other hand, there is increasing evidence indicating that pseudogenes can have functional biological roles, and recent studies have shown that pseudogenes potentially regulate the expression of protein-coding genes (reviewed in references 75 and 76).
An intriguing observation of chitin catabolism by A. salmonicida is the absence of key regulatory proteins such as ChiS and TfoX in the proteomics data. These regulatory proteins are important for chitin catabolism in other bacterial species in the Vibrionaceae family (18, 31, 33, 34). There is no doubt that A. salmonicida is capable of chitin catabolism; thus, the bacterium may have evolved an alternative mechanism for regulating the chitin utilization loci. In support of this hypothesis, the gene encoding the periplasmic chitin-binding protein, which activates ChiS when bound to (GlcNAc)2 (31), is disrupted in the A. salmonicida genome (29).
Although the A. salmonicida chitinolytic system clearly is active and functional, there are some observations that may indicate other or additional functions of the chitinolytic enzymes. First, the activity of the chitinase is substantially lower than what would be expected for an enzyme dedicated to chitin hydrolysis. Second, the dominantly expressed LPMO (AsLPMO10A) is not essential for chitin degradation and is also abundantly expressed when the bacterium is cultivated on glucose. These observations could be associated with the adaption of a pathogenic lifestyle where the need for chitin as a nutrient source has been reduced but could also indicate other or additional functions, for example, roles in virulence. The notion of chitinases having additional functions has been suggested by several studies, for example, those showing cleavage of mucin glycans by the V. cholerae chitinase Chi2A (77) and hydrolysis of LacdiNAc (GalNAcβ1-4GlcNAc) and LacNAc (Galβ1-4GlcNAc) by the L. monocytogenes and Salmonella enterica serovar Typhimurium chitinases (78). Such substrates were not evaluated by activity assays with AsChi18A. Moreover, incubation of AsChi18A with mucus collected from Atlantic salmon skin revealed an unidentifiable product (different from the negative control), but determination of its identity was unsuccessful.
Compared to other virulence-related chitinases, AsChi18A has a similar size but different modular architecture. For example, ChiA2 from V. cholerae, which has been shown to improve survival of the bacterium in the host intestine, also contains around 800 amino acids, but the GH18 domain is close to the N terminus and CBM44 and CBM5 chitin-binding domains are present on the C-terminal side. As already noted, ChiA2 has been shown to cleave intestinal mucin (releasing GlcNAc) but has a deep substrate binding cleft and resembles an exochitinase (85% sequence identity to the structurally resolved exochitinase of Vibrio harveyi [79]). An unusual property of AsChi18A is its double pH optimum, shown by enzyme activity to be approximately equal at pHs 4 and 7 (Fig. 4B). Chitinases usually display a single pH optimum, but double pH optima are not uncommon for hydrolytic enzymes, e.g., such as phytase from Aspergillus niger (80) and β-galactosidase from Lactobacillus acidophilus (81). It is possible that this property is associated with the chitinase being utilized in environments that vary in pH. If the A. salmonicida chitinase has evolved a role in addition to chitin degradation, the same question applies for the LPMOs. Both LPMOs are active toward chitin, but it is not certain that this is the intended substrate of these enzymes. For instance, GbpA, an LPMO from V. cholerae, has activity toward chitin (53), but its main function seems to be related to bacterial colonization of transfer vectors (e.g., zooplankton), the host epithelium (e.g., human intestine), or both (82, 83). The LPMO of L. monocytogenes is also active toward chitin (71), but the gene encoding this enzyme is not expressed when the bacterium grows on chitin (on the other hand, the L. monocytogenes chitinase-encoding genes are expressed when the bacterium is grown on chitin [71, 84]). The LPMO of the human opportunistic pathogen Pseudomonas aeruginosa, CbpD, was recently shown to be a chitin-active virulence factor that attenuates the terminal complement cascade of the host (85). In the present study, both LPMOs were expressed in the presence of chitin but also under the glucose control condition, indicating that regulation is not controlled by chitin or soluble chitooligosaccharides. Thus, chitin may represent a potential substrate for these LPMOs but is possibly not the (only) biologically relevant substrate.
On the other hand, some LPMOs are designed to only disrupt and disentangle chitin fibers rather than to contribute to their degradation in a metabolic context, namely, the viral family AA10 LPMOs (also called spindolins) (41). These LPMOs are harbored by insect-targeting entomopox- and baculoviruses and have been shown to disrupt the chitin-containing peritrophic matrix that lines the midguts of insect larvae (86). The main function proposed for the viral LPMOs is to destroy the midgut lining in order to allow the virus particles to access the epithelial cells that are located underneath. Since the scales and guts of fish are indicated to contain chitin (5, 6), it is tempting to speculate that the role of the fish-pathogenic LPMOs is similar to that of viral LPMOs, namely, to disrupt this putatively protective chitin layer in order to provide an entry point to the bacteria for infection.
In conclusion, the present study shows that A. salmonicida LFI1238 can degrade and catabolize chitin as a sole carbon source, despite possessing a chitinolytic pathway assumed to be incomplete. Our findings imply that the bacterium can utilize chitin to proliferate in the marine environment, although possibly not as efficiently as other characterized chitinolytic marine bacteria. Nevertheless, it is likely that this ability can be of relevance for the spread of this pathogen in the ocean. Finally, our discovery that pseudogenes are actively transcribed and translated indicates that such genes cannot be disregarded as being functionally important.
MATERIALS AND METHODS
Bacterial strains and culturing conditions.
A. salmonicida strain LFI1238 originally isolated from the head kidney of diseased farmed cod (Gadus morhua [29]) and mutant strains (see below) were routinely cultivated at 12°C in liquid Luria Broth (LB) supplemented with 2.5% sodium chloride (LB25; 10 g/liter tryptone, 5 g/liter yeast extract, 25 g/liter NaCl) or solid LB25 supplemented with 15 g/liter agar powder (LA25) and, if applicable, 2% (wt/vol) colloidal chitin made from α-chitin (gift from Silje Lorentzen). Growth analysis was performed at 12°C in A. salmonicida-specific minimal medium [Asmm; 100 mM KH2PO4, 15 mM NH4(SO4)2, 3.9 μM FeSO4·7H2O, 2.5% NaCl, 0.81 mM MgSO4·7H2O, 2 mM valine, 0.5 mM isoleucine, 0.5 mM cysteine, 0.5 mM methionine, and 40 mM glutamate]. Prior to inoculation of Asmm, strains were grown for up to 48 h in 10 to 15 ml LB25 at 200 rpm. One milliliter of bacteria was harvested by centrifugation at 6,000 × g for 1 min, followed by immediate resuspension of the pellet in 1 ml Asmm. The cell suspension was transferred to the final cultures by a 1:50 dilution in medium supplemented with 0.2% glucose, 0.2% N-acetyl-d-glucosamine, 0.2% diacetyl-chitobiose (Megazyme, Bray, Ireland) or 1% β-chitin from squid pen purchased from France Chitine (Orange, France; batch 20140101). Culture volumes ranged from 5 to 50 ml. Final cultures were incubated at 12°C with shaking at 175 rpm. Growth was measured by optical density at 600 nm (OD600) using an Ultrospec 10 cell density meter (Biochrom). The baseline was set by using sterile Asmm with or without 1% β-chitin. OD600 measurements of the β-chitin cultures were obtained by allowing the cultures to settle for 30 s before collecting 1 ml for measurement.
Generation of gene deletion strains.
LFI1238 derivative in-frame deletion mutants ΔAsChi18A, ΔAsLPMO10A, ΔAsLPMO10B, ΔAsLPMO10A-ΔLPMO10B, and ΔLPMO10A-ΔLPMO10B-ΔChi18A (also referred to as the ΔAΔBΔChi mutant) were constructed by allelic exchange as described by others (87, 88). For clarification, Table 4 lists the target genes, their associated protein names, predicted carbohydrate-active enzyme family (CAZyme family), and corresponding CAZyme annotated names applied throughout this study.
TABLE 4.
Description of target genes
| Gene name | Protein name | CAZy family | CAZyme name |
|---|---|---|---|
| VSAL_I0757 chiA | Endochitinase ChiA | GH18 | AsChi18A |
| VSAL_II0134 gbpA | GlcNAc-binding protein A | AA10 | AsLPMO10A |
| VSAL_II0217 | Chitinase B | AA10 | AsLPMO10B |
Primers were ordered from Eurofins Genomics (Ebersberg, Germany) and designed with restriction sites and regions complementary to the pDM4 cloning vector to allow for in-fusion cloning. Table 5 lists primers used for construction of the deletion alleles. For construction of the ΔAsChi18A mutant, the flanking regions upstream and downstream of the AsChi18A gene were amplified using primer pairs GH18_YF/GH18_IR and GH18_IF/GH18_YR, respectively. The two PCR fragments were fused by overlapping extension PCR, where complementarity in the 5′ regions of the primers resulted in linkage of the AsChi18A-flanking regions. ΔAsLPMO10A and ΔAsLPMO10B were constructed in the same manner as described for ΔAsChi18A using the listed primers (Table 5).
TABLE 5.
Primers used for construction of in-frame deletion mutants
| Primer | Sequence 5′→3′ |
|---|---|
| AsGH18_YF | GAAGGGCCCCACTAGTCGCACACTGATTTATCACACT |
| AsGH18_IR | GTTCATTAATGTCAGACTGTTAATGAAAATCCGTTTCAT |
| AsGH18_IF | CATTAACAGTCTGACATTAATGAACGCTCAATAA |
| AsGH18_YR | ACCGTCGACCCTCGAGGTGTTCTAATAGCGGGCATT |
| AsLPMO10A_YF | GAAGGGCCCCACTAGTGGGTACAAGATTGTTGCTTTT |
| AsLPMO10A_IR | ATCCCAAGCCATCGTTGAGCATTTATTCATCATTTATTC |
| AsLPMO10A_IF | AAATGCTCAACGATGGCTTGGGATAAAATCTAACCA |
| AsLPMO10A_YR | ACCGTCGACCCTCGAGGTGTACGGATGTTCTAACATC |
| AsLPMO10B_YF | GAAGGGCCCCACTAGTCCGTCAATCATCAACTAGAGA |
| AsLPMO10B_IR | TCCCCATTCTATTGTATTTGTCATATTTCATCCTTGTCT |
| AsLPMO10B_IF | AATACAATA GAATGGGGAGTATGGCGA |
| AsLPMO10B_YR | ACCGTCGACCCTCGAGTTTCTTGTCACCCATGATCAC |
The final PCR products were inserted into the suicide vector pDM4 by In-Fusion HD cloning (TaKaRa Bio USA, Inc.). In short, pDM4 linearized with SpeI and XhoI was gently mixed with 5× In-Fusion HD enzyme premix, purified PCR fragment (purified using NucleoSpin gel and PCR clean-up; MACHEREY-NAGEL GmbH & Co. KG), and H2O to the final volume. The ratio of the insert to linearized vector was determined using the online tool “In-Fusion molar ratio calculator” (TaKaRa Bio USA, Inc.). The reaction mix was incubated at 50°C for 15 min. Following incubation, the reaction mix was placed on ice for 20 min and transformed into Escherichia coli S17-1 λpir by standard transformation techniques.
Conjugation was performed as described by others (87–90). In brief, pelleted cells from 1 ml E. coli S17-1 donor cells (OD600 of 0.60 to 0.80) and 1 ml A. salmonicida LFI1238 recipient cells (OD600 of 1.00 to 1.40) were washed in LB, mixed, and transferred to LA1 as a spot. The spot plate was incubated 6 h at room temperature and ∼17 h at 12°C. The next day, the cell spot was collected and resuspended in 2 ml LB25, grown for 24 h with shaking, and spread onto LA25 containing chloramphenicol (2 μl/ml) (2CAM). Potential transconjugants were restreaked on LA25 2CAM, incubated for 3 to 5 days, and tested for integration of the pDM4 construct by colony PCR using a combination of primers annealing within and outside the integrated plasmid (Table 6). Next, confirmed transconjugants were grown in LB25 to an OD600 of 0.4 and spread onto LA25 containing 5% sucrose. Colonies appearing within 5 days were tested for excision of the integrated plasmid by sequentially patching single colonies onto LA25 plates containing 2CAM or 5% sucrose. Mutants showing loss of resistance to CAM and presence of gene deletion product (colony PCR using primer pairs AsΔChi18A_For/AsΔChi18A_Rev) were confirmed by GATC Biotech Sanger sequencing (Eurofins Genomics, Germany).
TABLE 6.
Primers designed for construction of flanking regions and fusion product and for sequencing and selection/verification of transconjugants and mutants
| Primer | Sequence 5′→3′ |
|---|---|
| AsGH18_For | GCTGATGGCGTGATCAAC |
| AsGH18_Rev | GGCGCGTGCTAATTTCAA |
| AsLPMO10A_For | GGCTGCTATTGTCACAGAATA |
| AsLPMO10A_Rev | AAGCCTAATAAAGCACACCCA |
| AsLPMO10B_For | GATGAGGTGTACCATCTTGAA |
| AsLPMO10B _Rev | TGTAATAGAATGTCACCAGCA |
| pDM4_Seq_F | CGGGAGAGCTCAGGTTAC |
| pDM4_Seq_R | GGCTTCTGTTTCTATCAGCT |
Mutant strains containing multiple gene deletions were generated in a stepwise manner. Specifically, LFI1238 ΔAsLPMO10A was the recipient for pDM4-ΔAsLPMO10B. Similarly, the resulting ΔAsLPMO10A/ΔLPMO10B strain was the recipient for pDM4-ΔAsChi18A, thus generating the triple mutant strain ΔLPMO10A/ΔLPMO10B/ΔChi18A. All strains and vectors are listed in Table 7.
TABLE 7.
Complete list of strains and vectors
| Strain or plasmid | Comment | Reference or source |
|---|---|---|
| Strains | ||
| LFI1238 | Aliivibrio salmonicida strain LFI1238 | N-9291a |
| S17-1 λpir | Escherichia coli conjugation donor strain S17-1 λpir | 102 |
| AsΔChi18A | LFI1238 containing gene deletion ΔChi18A | This study |
| AsΔLPMO10A | LFI1238 containing gene deletion ΔLPMO10A | This study |
| AsΔLPMO10B | LFI1238 containing gene deletion ΔLPMO10B | This study |
| AsΔLPMO10A-Δ10B | LFI1238 containing gene deletions ΔLPMO10A and ΔLPMO10B | This study |
| AsΔLPMO10A-Δ10B-ΔChi | LFI1238 containing gene deletions ΔLPMO10A, ΔLPMO10B, and ΔChi18A | This study |
| Plasmids | ||
| pDM4 | pDM4 SacB suicide plasmid/cloning vector | 90 |
| pDM4-AsΔChi18A | pDM4 construct designed for allelic exchange and deletion of AsChi18A | This study |
| pDM4-AsΔLPMO10A | pDM4 construct designed for allelic exchange and deletion of AsLPMO10A | This study |
| pDM4-AsΔLPMO10B | pDM4 construct designed for allelic exchange and deletion of AsLPMO10B | This study |
Originally isolated by the Norwegian Institute of Fisheries and Aquaculture Research, N-9291, Tromsø, Norway, but provided by Simen Foyn Nørstebø for this study.
Cloning, expression, and purification.
Codon-optimized genes encoding AsLPMO10A (residues 1 to 491; UniProt identifier [ID] B6EQB6), AsLPMO10B (residues 1 to 395; UniProt ID B6EQJ6), and AsChi18A (residues 1 to 846; UniProt ID B6EH15) from A. salmonicida (LFI1238) were purchased from GenScript (Piscataway, NJ, USA). Gene-specific primers (Table 8) with sequence overhangs corresponding to the prelinearized pNIC-CH expression vector (Addgene, Cambridge, MA, USA) were used to amplify the genes in order to insert them into the vector by a ligation-independent cloning method (91). All the cloned genes contained their native signal peptides. Sequence-verified plasmids were transformed into ArcticExpress (DE3) competent cells (Agilent Technologies, CA, USA) for protein expression. Cells harboring the plasmids were inoculated and grown in Terrific broth (TB) medium supplemented with 50 μg/ml of kanamycin (50 mg/ml stock). Cells producing the full-length AsLPMO10s were cultivated in flask media at 37°C until an OD of 0.700, cooled down for 30 min at 4°C, induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG), and incubated for 44 h at 10°C with shaking at 200 rpm. Cells producing AsChi18A were grown in a Harbinger LEX bioreactor system (Epiphyte Three Inc., Toronto, Canada) using the same procedure described above, although the cells were cultured for a shorter time period (12 h) and air was pumped into the culture by spargers. Successively, cells were harvested by centrifugation, and the periplasmic extracts were generated by osmotic shock (92). The periplasmic fractions, containing the mature proteins (signal peptide free), were sterilized by filtration (0.2 μm) before purification (see below).
TABLE 8.
Cloning primers for AsLPMO10A and -B and AsChi18A
| Cloning primers | Sequence (5′→3′) |
|---|---|
| pNIC-CH/AsLPMOA (forward) | TTAAGAAGGAGATATACTATGATGAATAAATGCAGTACCAA |
| pNIC-CH/AsLPMOA (reverse) | AATGGCTTGGGACAAAATCTAAGCGCACCATCATCACCACCATT |
| pNIC-CH/AsLPMOB (forward) | TTAAGAAGGAGATATACTATGACCAACACGATTAAAATCAATTC |
| pNIC-CH/AsLPMOB (reverse) | AATGGGGTGTGTGGCGCTAAGCGCACCATCATCACCACCATT |
| pNIC-CH/AsGH18A (forward) | TTAAGAAGGAGATATACTATGAAACGTATCTTTATTAACAGT |
| pNIC-CH/AsGH18A (reverse) | TGATGAATGCGCAAGCGCACCATCATCACCACCATT |
AsLPMO10A and AsLPMO10B were purified by anion-exchange chromatography using a 5-ml HiTrap DEAE FF column (GE Healthcare) followed by hydrophobic interaction chromatography (HIC) using a 5-ml HiTrap phenyl FF (HS) column (GE Healthcare). For the ion exchange procedure, proteins in the periplasmic extract were applied to the column using a binding buffer containing 50 mM bis-Tris-HCl (pH 6.0). After all nonbound proteins had passed through the column, bound proteins were eluted by applying a linear gradient (0% to 100% in 20 column volumes with a flow rate of 1 ml/min), using an elution buffer containing bis-Tris-HCl (pH 6.0) and 500 mM NaCl. Fractions were collected and analyzed for the presence of LPMO using SDS-PAGE. Fractions containing LPMO were pooled and adjusted to 1 M (NH4)2SO4 and applied on the HIC column using a binding buffer consisting of 50 mM Tris-HCl (pH 7.5) and 1 M (NH4)2SO4. Following elution of unbound proteins, bound proteins were eluted by applying a linear gradient (0% to 100% over 20 column volumes with a flow rate of 1.5 ml/min) using an elution buffer containing 50 mM Tris-HCl (pH 7.5). In addition, AsLPMO10B was further purified by size exclusion chromatography using a HiLoad 16/60 Superdex 75 column operated at 1 ml/min and with a running buffer containing 1× phosphate-buffered saline (PBS), pH 7.4.
AsChi18A was purified by immobilized metal affinity chromatography using a HisTrap FF 5-ml column (GE Healthcare). The periplasmic extract containing AsChi18A was applied to the column using a binding buffer consisting of 20 mM Tris-HCl (pH 8.0) and 5 mM imidazole, using a flow rate of 3 ml/min. Bound proteins were eluted from the column by applying a linear gradient (0% to 100% over 20 column volumes with a flow rate of 3 ml/min) with an elution buffer containing 20 mM Tris-HCl (pH 8.0) and 500 mM imidazole. Fractions containing the pure protein, identified by SDS-PAGE, were pooled and concentrated using Amicon Ultra centrifugal filters (Millipore, Cork, Ireland).
Protein purity was analyzed by SDS-PAGE. Concentrations of the pure proteins were determined by measuring A280 and using the theoretical molar extinction coefficients of the respective enzyme (calculated using the ExPASy ProtParam tool) to estimate the concentration in milligrams per milliliter. Before use, AsLPMO10A and AsLPMO10B were saturated with Cu(II) by incubation with excess of CuSO4 in a molar ratio of 1:3 for 30 min at room temperature. The excess Cu(II) was eliminated by passing the protein through a PD MidiTrap G-25 desalting column (GE Healthcare) equilibrated with 50 mM Tris-HCl (pH 8.0) and 150 mM NaCl.
Preparation of substrates.
The substrates used in the assays were either squid pen β-chitin (France Chitine, Orange, France), shrimp shell α-chitin purchased from ChitiNor As (Avaldsnes, Norway) and skin mucus of Salmo salar. Skin mucus was collected from freshly killed farmed Atlantic salmon purchased from the Solbergstrand Marine Research Facility (Drøbak, Norway). The mucus was gently scraped off the skin of the fish by using a spatula and stored in plastic sample tubes at −20°C until use.
Enzyme activity assays.
For activity assays, chitin was suspended in 20 mM Tris-HCl (pH 7.5) in 2-ml Eppendorf tubes to yield a final concentration of 10 mg/ml. All reaction mixtures were incubated at 30°C and stirred in an Eppendorf Comfort thermomixer at 700 rpm. For LPMO reactions, the final enzyme concentrations were 1 μM, and reactions were started by the addition of 1 mM ascorbic acid (this activates the LPMOs). Similar reaction conditions were used for AsChi18A, although the final enzyme concentration used was 0.5 μM and ascorbic acid was not added to the reaction mixtures. At regular intervals, samples were taken from the reactions, and the soluble fractions were separated from the insoluble substrate particles using a 96-well filter plate (Millipore) operated with a vacuum manifold. Subsequently, the soluble fraction of AsLPMO10-catalyzed reaction mixtures was incubated with 1.5 μM a chitobiase from S. marcescens (also known as SmCHB or SmGH20A) at 37°C overnight in order to convert LPMO products (oxidized chitooligosaccharides of various degrees of polymerization) to N-acetylglucosamine (GlcNAc) and chitobionic acid (GlcNAcGlcNAc1A) as previously described in references 53 and 93, followed by sample dilution with 50 mM H2SO4 at a ratio of 1:1 prior to quantification by HPLC (see below). The soluble fractions of AsChi18A reactions, were diluted with H2SO4 after the filtration step, which stopped the enzymatic reaction, before quantification of (GlcNAc)2 by high-performance liquid chromatography (HPLC) (see below). Additionally, to collect samples for product profiling by matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) (see below) of the two AsLPMO10-catalyzed reaction mixtures, 5 μl of the soluble fraction was sampled after filtration and kept at −20°C prior to analysis.
Analysis and quantification of native and oxidized chitooligosaccharides, (GlcNAc)2 and GlcNAc.
Qualitative analysis of the native and oxidized products of the AsLPMO10A and -B soluble fractions was performed by MALDI-TOF MS using a method developed by Vaaje-Kolstad et al. (12). For this analysis, 1 μl of sample was mixed with 2 μl 2,5-dihydroxybenzoic acid (9 g liter−1, prepared in 150:350 H2O-acetonitrile), applied to an MTP 384 target plate in ground steel TF (Bruker Daltonics GmbH, Bremen, Germany) and dried under a stream of warm air. The samples were analyzed with an Ultraflex MALDI-TOF/TOF instrument (Bruker Daltonics GmbH) equipped with a nitrogen 337-nm laser beam, using Bruker FlexAnalysis software. Quantitative analysis of all soluble products formed by the chitinolytic enzymes or GlcNAc or (GlcNAc)2 in culture supernatants was performed by ion exclusion chromatography using a Dionex Ultimate 3000 UHPLC system (Dionex Corp., Sunnyvale, CA, USA) equipped with a Rezex RFQ-Fast Acid H+ (8%) 7.8% 100-mm column (Phenomenex, Torrance, CA). The column was preheated to 85°C and was operated by running 5 mM H2SO4 as a mobile phase at a flow rate of 1 ml/min. The products were separated isocratically and detected by UV absorption at 194 nm. The amounts of GlcNAc and (GlcNAc)2 were quantified using standard curves. Pure GlcNAc and (GlcNAc)2 were obtained from Sigma and Megazyme, respectively. To quantify chitobionic acid (GlcNAcGlcNAc1A), a standard was produced in-house by treating chitobiose (Megazyme, Bray, Ireland) with a chitooligosaccharide oxidase (ChitO) from Fusarium graminearum, which yields 100% conversion of chitobiose to chitobionic acid, a method previously described by Loose et al. (53). Standards were regularly analyzed in each run.
Analysis of chitinase activity in culture supernatants.
To analyze the presence of chitinolytic activity in the supernatant of A. salmonicida when growing on β-chitin, a 1-ml sample of wild-type bacterial culture was harvested at various time points during growth on chitin. The sample was centrifuged, and the supernatant was filter sterilized using 0.22-μm sterile Ultrafree centrifugal filters. Five hundred microliters filter-sterilized supernatant was concentrated to 100 μl using Amicon ultra centrifugal filter units with a 3,000-Da cutoff (Merck Millipore, Cork, Ireland) and washed three times in 10 mM Tris (pH 7.5)-0.2 M NaCl (Tris-HCl NaCl). The concentrated supernatants containing secreted enzymes were stored in Tris-HCl at 4°C until use. The presence of chitinolytic activity was assessed by mixing 100 μM chitopentaose with 15 μl enzyme cocktail in 20 mM Tris (pH 7.5)-0.2 M NaCl and incubated at 30°C. The generated products were analyzed and quantified by ion exclusion chromatography as described above.
Protein binding assays.
The binding capacity of AsLPMO10s and AsChi18A on α-chitin and β-chitin was tested by a particle sedimentation assay, suspending 10 mg/ml of substrate in 20 mM Tris-HCl (pH 7.5) to a total volume of 350 μl in 2-ml Eppendorf tubes. Reactions were started by the addition of AsLPMO10A or –B (0.75 μM final concentration) or AsChi18A (0.50 μM), which were incubated in 2-ml Eppendorf tubes at 30°C and stirred in an Eppendorf Comfort thermomixer at 700 rpm. Samples were taken (100 μl) after 2 h and immediately filtrated using a 96-well filter plate (Millipore) operated with a vacuum manifold to obtain the unbound protein fraction. To assess the percentage of bound proteins to the substrate, control samples with only enzyme and buffer were utilized, representing the maximum quantity of protein present in the samples (100%). The protein concentration in each sample was determined using Bradford assays (Bio-Rad, Munich, Germany).
RNA isolation and gene expression analysis.
To analyze the expression of specific genes as previously done by, e.g., Wagner et al. (94), samples were taken at mid-exponential phase (OD of 0.6 to 0.7) and early stationary phase (OD of 1.0 to 1.3), and a 0.1-ml sample of each culture was directly transferred to 2 ml RNAprotect cell reagent (Qiagen, Hilden, Germany). The samples were vortexed 5 s, incubated 5 min at room temperature, and subsequently harvested by centrifugation at 4,000 × g for 10 min at 4°C. The supernatant was carefully decanted, and the cell pellet was stored at −20°C until cell lysis and RNA isolation. RNA isolation was performed using a Qiagen RNeasy minikit (Qiagen) according to the Quick-Start protocol. To disrupt the bacterial cell wall before isolation, the pellet was lysed using 200 μl Tris-EDTA (pH 8.0) supplemented with 1 mg/ml lysozyme, vortexed for 10 s, and subsequently incubated at room temperature for 45 min. Seven hundred microliters buffer RLT (kit buffer; Qiagen) supplemented with 10 μl/ml β-mercaptoethanol was added to the sample and mixed vigorously before proceeding with the protocol. The quantity of isolated RNA was determined using a NanoDrop.
Residual genomic DNA (gDNA) was removed using The Heat&Run gDNA removal kit (ArcticZymes, Tromsø, Norway). Eight microliters of the RNA samples was transferred to an RNase-free Eppendorf tube on ice. For each 10-μl reaction mixture, 1 μl of 10× reaction buffer and 1 μl heat-labile double-strand-specific DNAse (dsDNase) were added. The suspension was gently mixed and incubated at 37°C for 10 min. To inactive the enzyme, samples were immediately transferred to 58°C for 5 min. The RNA concentration was measured by using a NanoDrop before proceeding to cDNA synthesis.
cDNA synthesis was performed using iScript reverse transcription supermix (Bio-Rad, Hercules, CA, USA). For each sample, 100 ng RNA, 4 μl 5× iScript reverse transcription supermix, and RNase-free water to a total volume of 20 μl were assembled in PCR tubes. All samples were additionally prepared with an iScript no-reverse-transcriptase control supermix to account for residual gDNA in downstream analysis. The cDNA synthesis of the samples was performed by using a SimpliAmp thermal cycler (Thermo Fischer Scientific, USA) with the following steps: priming at 25°C for 5 min, reverse transcription at 46°C for 20 min, and inactivation of the reverse transcriptase at 95°C for 1 min. The synthesized cDNA was stored at −20°C until analysis.
The cDNA samples were screened for presence of AsChi18A, AsLPMO10A, AsLPMO10B, and VSAL_I0902/AsChi18Bp by PCR amplification using Red Taq DNA polymerase 2× master mix (VWR, Oslo, Norway) according to the manufacturer’s protocol. The PCR was carried out using 30 cycles with an annealing temperature of 58°C (AsChi18A, AsLPMO10A, or AsLPMO10B) or 56°C (VSAL_I0902/AsChi18Bp) and a 30-s extension. To evaluate gDNA presence, samples prepared with no reverse transcriptase during cDNA synthesis (referred to as −RT control) were applied as the template for primer pairs for AsLPMO10A and VSAL_I0902.
PCR products were visualized by agarose gel electrophoresis of the total 20-μl PCR mix in 1.3% agarose 1× Tris-acetate-EDTA (TAE) electrophoresis buffer (Thermo Scientific, Vilnius, Lithuania). The agarose was supplemented with peqGreen DNA/RNA dye (PEQLAB; VWR, Oslo, Norway) for visualization. After gel visualization, the gene expression was assessed as positive if the target gene was amplified in two of three biological replicates and, at the same time, no amplification was observed in PCR samples prepared with the −RT controls. A complete list of primers used for amplification of target genes is shown in Table 9.
TABLE 9.
Primers applied for amplification of target genes using cDNA
| Primer | Sequence (5→3′) | Product size (bp) |
|---|---|---|
| GH18Expression_F | AGTCAAGCATCAGCCAAGAAAG | 566 |
| GH18Expression_R | TAAGGCAAGGCTCGATCCAG | |
| 10AExpression_F | ATTCGGTCCTGCTGATGG | 565 |
| 10AExpression_R | ATTTGCTTGACCTTGTGTTGC | |
| 10BExpression_F | TCAAGCGTGTCAGTCTGC | 441 |
| 10BExpression_R | TGCCAACGAGTGTAGAGC | |
| I0902Expression_F | ATGCACAAGGTCGATCTG | 297 |
| I0902Expression_R | ATGGGATGTACTTGTCGC |
Sample preparation and proteomic analysis.
Biological triplicates of A. salmonicida LFI1238 were incubated in 50 ml Asmm supplemented with 1% β-chitin. At mid-exponential phase, cultures were harvested and fractioned into supernatant and pellet by centrifugation at 4,000 × g for 10 min at 4°C. β-Chitin aliquots from the culture flasks were transferred to 2-ml Safe-Lock Eppendorf tubes (Eppendorf, Hamburg, Germany) and boiled directly for 5 min in 30 μl NuPAGE LDS sample buffer and NuPAGE sample reducing agent (Invitrogen, CA, USA). Filter-sterilized supernatant was concentrated using Vivaspin 20 centrifugal concentrators (Vivaproducts, Littleton, MA, USA) by centrifugation at 4,000 rpm and 4°C until it reached 1 ml concentrate. The bacterial pellet was lysed in 2 ml 1× BugBuster protein extraction reagent (Novagen) and incubated by slow shaking for 20 min, followed by centrifugation and protein precipitation. Proteins were precipitated by adding trichloroacetic acid (TCA) to 10% and incubation overnight at 4°C. The precipitated proteins were harvested by centrifugation at 16,000 × g and 4°C for 15 min and washed twice in ice-cold 90% acetone-0.01 M HCl. All final samples were boiled in 30 μl NuPAGE LDS sample buffer and sample reducing agent for 5 min and loaded on Mini-PROTEAN TGX Stain-Free gels (Bio-Rad laboratories, Hercules, CA, USA). Electrophoresis was performed at 300 V for 3 min using the Bio-Rad Mini-PROTEAN Tetra System. Gels were stained with Coomassie brilliant blue R250, and 1- by 1-mm cube gel pieces were excised and transferred to 2 ml LoBind tubes containing 200 μl H2O. Sequentially, the gel pieces were washed 15 min in 200 μl H2O and decolored by incubating twice for 15 min in 200 μl 50% acetonitrile and 25 mM ammonium bicarbonate (AmBic). Next, reduction was performed by incubating the gel pieces in dithiothreitol (DTT; 10 mM DTT-100 mM AmBic) for 30 min at 56°C, and alkylation was performed with iodoacetamide (IAA; 55 mM IAA-100 mM AmBic) for 30 min at room temperature. After removal of the IAA solution, the gel pieces were dehydrated using 200 μl 100% acetonitrile and digested using 30 to 45 μl of a 10-ng/μl trypsin solution overnight at 37°C. The next day, digestion was stopped by addition of 40 μl 1% trifluoroacetic acid (TFA). Peptides were extruded from the gel pieces by 15 min of sonication and desalted using C18 ZipTips (Merch Millipore, Darmstadt, Germany), according to the manufacturer’s instructions.
Peptides were analyzed as previously described (95). In brief, peptides were loaded onto a nanoscale HPLC-tandem mass spectrometry (nanoHPLC-MS/MS) system (Dionex Ultimate 3000 UHPLC; Thermo Scientific) coupled to a Q-Exactive hybrid quadrupole Orbitrap mass spectrometer (Thermo Scientific). Peptides were separated using an analytical column (Acclaim PepMap RSLC C18, 2 μm, 100 Å, 75-μm inside diameter [i.d.] by 50 cm, nanoViper) with a 90-min gradient from 3.2% to 44% (vol/vol) acetonitrile in 0.1% (vol/vol) formic acid at flow rate 300 nl/min. The Q-Exactive mass spectrometer was operated in data-dependent mode acquiring one full scan (400 to 1,500 m/z) at a resolution (R) of 70,000 followed by (up to) 10 dependent MS/MS scans at an R of 35,000. The raw data were analyzed using MaxQuant version 1.6.3.3, and proteins were identified and quantified using the MaxLFQ algorithm (96). The data were searched against the UniProt A. salmonicida proteome (UP000001730; 3,513 sequences) supplemented with common contaminants such as human keratin and bovine serum albumin. In addition, reversed sequences of all protein entries were concatenated to the database to allow for estimation of false-discovery rates. The tolerance levels used for matching to the database were 4.5 ppm for MS and 20 ppm for MS/MS. Trypsin/P was used as the digestion enzyme, and 2 missed cleavages were allowed. Carbamidomethylation of cysteine was set as a fixed modification, and protein N-terminal acetylation, oxidation of methionines, and deamidation of asparagines and glutamines were allowed as variable modifications. All identifications were filtered in order to achieve a protein false-discovery rate (FDR) of 1%. Perseus version 1.6.2.3 (97) was used for data analysis, and the quantitative values were log2 transformed and grouped according to carbon source and condition (substrate/supernatant/pellet). Proteins were only considered detected if they were present in at least two replicates under at least one condition. All identified proteins were annotated for putative carbohydrate-active functions as predicted by dbCAN2 (98), biological functions (GO and Pfam) downloaded from UniProt, and subcellular location using SignalP5.0 (99).
Pseudogenes.
Pseudogenes are gene sequences that have been mutated or disrupted into an inactive form over the course of evolution and are commonly thought of as “junk DNA.” The genome of A. salmonicida LFI1238 contains a significant number of IS elements, and several genes are truncated and annotated as such pseudogenes. Since pseudogenes in general are believed to be nonfunctional, putative products of these are commonly not included in proteome databases. Consequently, a proteomic analysis toward the annotated proteome of A. salmonicida LFI1238 will not detect products of these genes. To include these in our analysis, a few required steps were taken. First, pseudogenes of four chitinases and a chitodextrinase were selected as genes of interest based on the publication by Hjerde et al. (29). Next, the truncated nucleotide sequence of a pseudogene was retrieved by searching the complete genome sequence annotation of A. salmonicida LFI1238 chromosome I (FM178379.1) for the specific gene locus. The gene locus of each selected pseudogene is shown in Table 1. The nucleotide sequences were translated to putative protein sequences using the translate tool at ExPASy Bioinformatics Resource Portal (100). The translate tool identifies potential start and stop codons of the query sequence by assessing reading frames 1 to 3 of forward and reverse DNA strands. Manually, putative peptides larger than or equal to 6 amino acids were selected as supplements for the proteomic analysis. Pseudogene products of which unique peptides were identified were assigned a putative CAZy annotation using dbCAN2.
Data availability.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (101) partner repository with the data set identifier PXD021397.
ACKNOWLEDGMENTS
This work was funded by grant 249865 (to G.V.-K., A.S., M.Ø.A., and J.S.M.L.) from the Norwegian Research Council and by a Ph.D. fellowship from the Norwegian University of Life Sciences, Faculty of Chemistry Biotechnology and Food Science, to G. Minniti. B.B. has received the support of the EU in the framework of the Marie-Curie FP7 COFUND People Program, through the award of an AgreenSkills fellowship (under grant agreement no. 267196).
We thank Simen Foyn Nørstebø (Faculty of Veterinary Medicine, Department of Food Safety and Infection Biology, Norwegian University of Life Sciences) for providing bacterial strains for deletion mutagenesis and for valuable advice regarding the protocol and Anne Cathrine Bunæs for assistance in purification of proteins.
A.S. planned experiments, performed experiments, analyzed data, and wrote the paper. J.S.M.L. planned experiments and wrote the paper. G. Minniti planned experiments, performed experiments, analyzed data, and wrote the paper. J.S.M.L. planned experiments and wrote the paper. S.M. performed experiments, analyzed data, and wrote the paper. B.B. performed experiments, analyzed data, and wrote the paper. M.Ø.A. performed experiments, analyzed data, and wrote the paper. G. Mathiesen planned experiments and wrote the paper. G.V.-K. conceptualized the study, planned experiments, analyzed data, wrote the paper, and supervised the study.
We declare no conflict of interest.
Footnotes
Supplemental material is available online only.
Contributor Information
Gustav Vaaje-Kolstad, Email: gustav.vaaje-kolstad@nmbu.no.
Eric V. Stabb, University of Illinois at Chicago
REFERENCES
- 1.Wang Y, Chang Y, Yu L, Zhang C, Xu X, Xue Y, Li Z, Xue C. 2013. Crystalline structure and thermal property characterization of chitin from Antarctic krill (Euphausia superba). Carbohydr Polym 92:90–97. 10.1016/j.carbpol.2012.09.084. [DOI] [PubMed] [Google Scholar]
- 2.Raabe D, Romano P, Sachs C, Fabritius H, Al-Sawalmih A, Yi SB, Servos G, Hartwig HG. 2006. Microstructure and crystallographic texture of the chitin–protein network in the biological composite material of the exoskeleton of the lobster Homarus americanus. Mat Sci Eng A 421:143–153. 10.1016/j.msea.2005.09.115. [DOI] [Google Scholar]
- 3.Acosta N, Jiménez C, Borau V, Heras A. 1993. Extraction and characterization of chitin from crustaceans. Biomass Bioenerg 5:145–153. 10.1016/0961-9534(93)90096-M. [DOI] [Google Scholar]
- 4.Bacon JS, Davidson ED, Jones D, Taylor IF. 1966. The location of chitin in the yeast cell wall. Biochem J 101:36C–38C. 10.1042/bj1010036c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang WJ, Fernandez J, Sohn JJ, Amemiya CT. 2015. Chitin is endogenously produced in vertebrates. Curr Biol 25:897–900. 10.1016/j.cub.2015.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wagner G, Lo J, Laine R, Almeder M. 1993. Chitin in the epidermal cuticle of a vertebrate (Paralipophrys trigloides, Blenniidae, Teleostei). Cell Mol Life Sci 49:317–319. 10.1007/BF01923410. [DOI] [Google Scholar]
- 7.Gooday GW. 1990. The ecology of chitin degradation. Adv Microb Ecol 11:387–430. 10.1007/978-1-4684-7612-5_10. [DOI] [Google Scholar]
- 8.Keyhani NO, Roseman S. 1999. Physiological aspects of chitin catabolism in marine bacteria. Biochim Biophys Acta 1473:108–122. 10.1016/s0304-4165(99)00172-5. [DOI] [PubMed] [Google Scholar]
- 9.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. 2014. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42:D490–D495. 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brameld KA, Shrader WD, Imperiali B, Goddard WA. 1998. Substrate assistance in the mechanism of family 18 chitinases: theoretical studies of potential intermediates and inhibitors. J Mol Biol 280:913–923. 10.1006/jmbi.1998.1890. [DOI] [PubMed] [Google Scholar]
- 11.Terwisscha van Scheltinga AC, Armand S, Kalk KH, Isogai A, Henrissat B, Dijkstra BW. 1995. Stereochemistry of chitin hydrolysis by a plant chitinase/lysozyme and X-ray structure of a complex with allosamidin: evidence for substrate assisted catalysis. Biochemistry 34:15619–15623. 10.1021/bi00048a003. [DOI] [PubMed] [Google Scholar]
- 12.Vaaje-Kolstad G, Westereng B, Horn SJ, Liu Z, Zhai H, Sørlie M, Eijsink VGH. 2010. An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 330:219–222. 10.1126/science.1192231. [DOI] [PubMed] [Google Scholar]
- 13.Bissaro B, Røhr ÅK, Müller G, Chylenski P, Skaugen M, Forsberg Z, Horn SJ, Vaaje-Kolstad G, Eijsink VGH. 2017. Oxidative cleavage of polysaccharides by monocopper enzymes depends on H2O2. Nat Chem Biol 13:1123–1128. 10.1038/nchembio.2470. [DOI] [PubMed] [Google Scholar]
- 14.Gregory RC, Hemsworth GR, Turkenburg JP, Hart SJ, Walton PH, Davies GJ. 2016. Activity, stability and 3-D structure of the Cu(II) form of a chitin-active lytic polysaccharide monooxygenase from Bacillus amyloliquefaciens. Dalton Trans 45:16904–16912. 10.1039/c6dt02793h. [DOI] [PubMed] [Google Scholar]
- 15.Forsberg Z, Mackenzie AK, Sørlie M, Røhr AK, Helland R, Arvai AS, Vaaje-Kolstad G, Eijsink VGH. 2014. Structural and functional characterization of a conserved pair of bacterial cellulose-oxidizing lytic polysaccharide monooxygenases. Proc Natl Acad Sci USA 111:8446–8451. 10.1073/pnas.1402771111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakagawa YS, Kudo M, Loose JS, Ishikawa T, Totani K, Eijsink VGH, Vaaje‐Kolstad G. 2015. A small lytic polysaccharide monooxygenase from Streptomyces griseus targeting α‐and β‐chitin. FEBS J 282:1065–1079. 10.1111/febs.13203. [DOI] [PubMed] [Google Scholar]
- 17.Sabbadin F, Hemsworth GR, Ciano L, Henrissat B, Dupree P, Tryfona T, Marques RDS, Sweeney ST, Besser K, Elias L, Pesante G, Li Y, Dowle AA, Bates R, Gomez LD, Simister R, Davies GJ, Walton PH, Bruce NC, McQueen-Mason SJ. 2018. An ancient family of lytic polysaccharide monooxygenases with roles in arthropod development and biomass digestion. Nat Commun 9:756. 10.1038/s41467-018-03142-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hunt DE, Gevers D, Vahora NM, Polz MF. 2008. Conservation of the chitin utilization pathway in the Vibrionaceae. Appl Environ Microbiol 74:44–51. 10.1128/AEM.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin H, Yu M, Wang X, Zhang XH. 2018. Comparative genomic analysis reveals the evolution and environmental adaptation strategies of vibrios. BMC Genomics 19:135. 10.1186/s12864-018-4531-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uchiyama T, Kaneko R, Yamaguchi J, Inoue A, Yanagida T, Nikaidou N, Regue M, Watanabe T. 2003. Uptake of N,N'-diacetylchitobiose [(GlcNAc)2] via the phosphotransferase system is essential for chitinase production by Serratia marcescens 2170. J Bacteriol 185:1776–1782. 10.1128/JB.185.6.1776-1782.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eisenbeis S, Lohmiller S, Valdebenito M, Leicht S, Braun V. 2008. NagA-dependent uptake of N-acetyl-glucosamine and N-acetyl-chitin oligosaccharides across the outer membrane of Caulobacter crescentus. J Bacteriol 190:5230–5238. 10.1128/JB.00194-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keyhani NO, Li XB, Roseman S. 2000. Chitin catabolism in the marine bacterium Vibrio furnissii - identification and molecular cloning of a chitoporin. J Biol Chem 275:33068–33076. 10.1074/jbc.M001041200. [DOI] [PubMed] [Google Scholar]
- 23.Suginta W, Chumjan W, Mahendran KR, Schulte A, Winterhalter M. 2013. Chitoporin from Vibrio harveyi, a channel with exceptional sugar specificity. J Biol Chem 288:11038–11046. 10.1074/jbc.M113.454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park JK, Keyhani NO, Roseman S. 2000. Chitin catabolism in the marine bacterium Vibrio furnissii - identification, molecular cloning, and characterization of a N-N'-diacetylchitobiose phosphorylase. J Biol Chem 275:33077–33083. 10.1074/jbc.M001042200. [DOI] [PubMed] [Google Scholar]
- 25.Erken M, Lutz C, McDougald D. 2015. Interactions of Vibrio spp. with zooplankton. Microbiol Spectr 3:VE-0003-2014. 10.1128/microbiolspec.VE-0003-2014. [DOI] [PubMed] [Google Scholar]
- 26.Lutz C, Erken M, Noorian P, Sun S, McDougald D. 2013. Environmental reservoirs and mechanisms of persistence of Vibrio cholerae. Front Microbiol 4:375. 10.3389/fmicb.2013.00375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pruzzo C, Vezzulli L, Colwell RR. 2008. Global impact of Vibrio cholerae interactions with chitin. Environ Microbiol 10:1400–1410. 10.1111/j.1462-2920.2007.01559.x. [DOI] [PubMed] [Google Scholar]
- 28.Egidius E, Wiik R, Andersen K, Hoff K, Hjeltnes B. 1986. Vibrio salmonicida sp. nov., a new fish pathogen. Int J Syst Evol Microbiol 36:518–520. [Google Scholar]
- 29.Hjerde E, Lorentzen MS, Holden MT, Seeger K, Paulsen S, Bason N, Churcher C, Harris D, Norbertczak H, Quail MA, Sanders S, Thurston S, Parkhill J, Willassen NP, Thomson NR. 2008. The genome sequence of the fish pathogen Aliivibrio salmonicida strain LFI1238 shows extensive evidence of gene decay. BMC Genomics 9:616. 10.1186/1471-2164-9-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kashulin A, Sørum H, Hjerde E, Willassen NP. 2015. IS elements in Aliivibrio salmonicida LFI1238: occurrence, variability and impact on adaptability. Gene 554:40–49. 10.1016/j.gene.2014.10.019. [DOI] [PubMed] [Google Scholar]
- 31.Klancher CA, Yamamoto S, Dalia TN, Dalia AB. 2020. ChiS is a noncanonical DNA-binding hybrid sensor kinase that directly regulates the chitin utilization program in Vibrio cholerae. Proc Natl Acad Sci USA 117:20180–20189. 10.1073/pnas.2001768117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li X, Roseman S. 2004. The chitinolytic cascade in vibrios is regulated by chitin oligosaccharides and a two-component chitin catabolic sensor/kinase. Proc Natl Acad Sci USA 101:627–631. 10.1073/pnas.0307645100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Debnath A, Mizuno T, Miyoshi SI. 2020. Regulation of chitin-dependent growth and natural competence in Vibrio parahaemolyticus. Microorganisms 8:1303. 10.3390/microorganisms8091303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Metzger LC, Matthey N, Stoudmann C, Collas EJ, Blokesch M. 2019. Ecological implications of gene regulation by TfoX and TfoY among diverse Vibrio species. Environ Microbiol 21:2231–2247. 10.1111/1462-2920.14562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yin Y, Mao X, Yang J, Chen X, Mao F, Xu Y. 2012. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res 40:W445–W451. 10.1093/nar/gks479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Gallo Cassarino T, Bertoni M, Bordoli L, Schwede T. 2014. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42:W252–W258. 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hsieh YC, Wu YJ, Chiang TY, Kuo CY, Shrestha KL, Chao CF, Huang YC, Chuankhayan P, Wu WG, Li YK, Chen CJ. 2010. Crystal structures of Bacillus cereus NCTU2 chitinase complexes with chitooligomers reveal novel substrate binding for catalysis: a chitinase without chitin binding and insertion domains. J Biol Chem 285:31603–31615. 10.1074/jbc.M110.149310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suzuki K, Suzuki M, Taiyoji M, Nikaidou N, Watanabe T. 1998. Chitin binding protein (CBP21) in the culture supernatant of Serratia marcescens 2170. Biosci Biotechnol Biochem 62:128–135. 10.1271/bbb.62.128. [DOI] [PubMed] [Google Scholar]
- 39.Vaaje-Kolstad G, Horn SJ, van Aalten DMF, Synstad B, Eijsink VGH. 2005. The non-catalytic chitin-binding protein CBP21 from Serratia marcescens is essential for chitin degradation. J Biol Chem 280:28492–28497. 10.1074/jbc.M504468200. [DOI] [PubMed] [Google Scholar]
- 40.Wong E, Vaaje-Kolstad G, Ghosh A, Hurtado-Guerrero R, Konarev PV, Ibrahim AFM, Svergun DI, Eijsink VGH, Chatterjee NS, van Aalten DMF. 2012. The Vibrio cholerae colonization factor GbpA possesses a modular structure that governs binding to different host surfaces. PLoS Path 8:e1002373. 10.1371/journal.ppat.1002373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chiu E, Hijnen M, Bunker RD, Boudes M, Rajendran C, Aizel K, Olieric V, Schulze-Briese C, Mitsuhashi W, Young V, Ward VK, Bergoin M, Metcalf P, Coulibaly F. 2015. Structural basis for the enhancement of virulence by viral spindles and their in vivo crystallization. Proc Natl Acad Sci USA 112:3973–3978. 10.1073/pnas.1418798112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aachmann FL, Sørlie M, Skjåk-Bræk G, Eijsink VGH, Vaaje-Kolstad G. 2012. NMR structure of a lytic polysaccharide monooxygenase provides insight into copper binding, protein dynamics, and substrate interactions. Proc Natl Acad Sci USA 109:18779–18784. 10.1073/pnas.1208822109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Loose JSM, Arntzen MO, Bissaro B, Ludwig R, Eijsink VGH, Vaaje-Kolstad G. 2018. Multipoint precision binding of substrate protects lytic polysaccharide monooxygenases from self-destructive off-pathway processes. Biochemistry 57:4114–4124. 10.1021/acs.biochem.8b00484. [DOI] [PubMed] [Google Scholar]
- 44.Khider M, Hansen H, Hjerde E, Johansen JA, Willassen NP. 2019. Exploring the transcriptome of luxI− and ΔainS mutants and the impact of N-3-oxo-hexanoyl-l- and N-3-hydroxy-decanoyl-l-homoserine lactones on biofilm formation in Aliivibrio salmonicida. PeerJ 7:e6845. 10.7717/peerj.6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khider M, Hjerde E, Hansen H, Willassen NP. 2019. Differential expression profiling of ΔlitR and ΔrpoQ mutants reveals insight into QS regulation of motility, adhesion and biofilm formation in Aliivibrio salmonicida. BMC Genomics 20:220. 10.1186/s12864-019-5594-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pedersen HL, Hjerde E, Paulsen SM, Hansen H, Olsen L, Thode SK, Santos MT, Paulssen RH, Willassen NP, Haugen P. 2010. Global responses of Aliivibrio salmonicida to hydrogen peroxide as revealed by microarray analysis. Mar Genomics 3:193–200. 10.1016/j.margen.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 47.Brurberg MB, Nes IF, Eijsink VGH. 1996. Comparative studies of chitinases A and B from Serratia marcescens. Microbiology 142:1581–1589. 10.1099/13500872-142-7-1581. [DOI] [PubMed] [Google Scholar]
- 48.Suzuki K, Sugawara N, Suzuki M, Uchiyama T, Katouno F, Nikaidou N, Watanabe T. 2002. Chitinases A, B, and C1 of Serratia marcescens 2170 produced by recombinant Escherichia coli: enzymatic properties and synergism on chitin degradation. Biosci Biotechnol Biochem 66:1075–1083. 10.1271/bbb.66.1075. [DOI] [PubMed] [Google Scholar]
- 49.Tuveng TR, Hagen LH, Mekasha S, Frank J, Arntzen MØ, Vaaje-Kolstad G, Eijsink VGH. 2017. Genomic, proteomic and biochemical analysis of the chitinolytic machinery of Serratia marcescens BJL200. Biochim Biophys Acta Proteins Proteom 1865:414–421. 10.1016/j.bbapap.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 50.Monge EC, Tuveng TR, Vaaje-Kolstad G, Eijsink VGH, Gardner JG. 2018. Systems analysis of the glycoside hydrolase family 18 enzymes from Cellvibrio japonicus characterizes essential chitin degradation functions. J Biol Chem 293:3849–3859. 10.1074/jbc.RA117.000849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Synstad B, Gåseidnes S, Van Aalten DM, Vriend G, Nielsen JE, Eijsink VGH. 2004. Mutational and computational analysis of the role of conserved residues in the active site of a family 18 chitinase. Eur J Biochem 271:253–262. 10.1046/j.1432-1033.2003.03923.x. [DOI] [PubMed] [Google Scholar]
- 52.Synstad B, Vaaje-Kolstad G, Cederkvist FH, Saua SF, Horn SJ, Eijsink VGH, Sørlie M. 2008. Expression and characterization of endochitinase C from Serratia marcescens BJL200 and its purification by a one-step general chitinase purification method. Biosci Biotechnol Biochem 72:715–723. 10.1271/bbb.70594. [DOI] [PubMed] [Google Scholar]
- 53.Loose JSM, Forsberg Z, Fraaije MW, Eijsink VGH, Vaaje-Kolstad G. 2014. A rapid quantitative activity assay shows that the Vibrio cholerae colonization factor GbpA is an active lytic polysaccharide monooxygenase. FEBS Lett 588:3435–3440. 10.1016/j.febslet.2014.07.036. [DOI] [PubMed] [Google Scholar]
- 54.Suginta W, Chuenark D, Mizuhara M, Fukamizo T. 2010. Novel beta-N-acetylglucosaminidases from Vibrio harveyi 650: cloning, expression, enzymatic properties, and subsite identification. BMC Biochem 11:40. 10.1186/1471-2091-11-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chitlaru E, Roseman S. 1996. Molecular cloning and characterization of a novel β-N-acetyl-d-glucosaminidase from Vibrio furnissii. J Biol Chem 271:33433–33439. 10.1074/jbc.271.52.33433. [DOI] [PubMed] [Google Scholar]
- 56.Choi KH, Seo JY, Park KM, Park CS, Cha J. 2009. Characterization of glycosyl hydrolase family 3 beta-N-acetylglucosaminidases from Thermotoga maritima and Thermotoga neapolitana. J Biosci Bioeng 108:455–459. 10.1016/j.jbiosc.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 57.Tsujibo H, Fujimoto K, Tanno H, Miyamoto K, Imada C, Okami Y, Inamori Y. 1994. Gene sequence, purification and characterization of N-acetyl-beta-glucosaminidase from a marine bacterium, Alteromonas sp. strain O-7. Gene 146:111–115. 10.1016/0378-1119(94)90843-5. [DOI] [PubMed] [Google Scholar]
- 58.Cheng Q, Li H, Merdek K, Park JT. 2000. Molecular characterization of the beta-N-acetylglucosaminidase of Escherichia coli and its role in cell wall recycling. J Bacteriol 182:4836–4840. 10.1128/JB.182.17.4836-4840.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yadava U, Vetting MW, Al Obaidi N, Carter MS, Gerlt JA, Almo SC. 2016. Structure of an ABC transporter solute-binding protein specific for the amino sugars glucosamine and galactosamine. Acta Crystallogr F Struct Biol Commun 72:467–472. 10.1107/S2053230X16007500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Oiki S, Mikami B, Maruyama Y, Murata K, Hashimoto W. 2017. A bacterial ABC transporter enables import of mammalian host glycosaminoglycans. Sci Rep 7:1069. 10.1038/s41598-017-00917-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wei X, Guo Y, Shao C, Sun Z, Zhurina D, Liu D, Liu W, Zou D, Jiang Z, Wang X, Zhao J, Shang W, Li X, Liao X, Huang L, Riedel CU, Yuan J. 2012. Fructose uptake in Bifidobacterium longum NCC2705 is mediated by an ATP-binding cassette transporter. J Biol Chem 287:357–367. 10.1074/jbc.M111.266213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hayes CA, Dalia TN, Dalia AB. 2017. Systematic genetic dissection of chitin degradation and uptake in Vibrio cholerae. Environ Microbiol 19:4154–4163. 10.1111/1462-2920.13866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bennati-Granier C, Garajova S, Champion C, Grisel S, Haon M, Zhou S, Fanuel M, Ropartz D, Rogniaux H, Gimbert I, Record E, Berrin JG. 2015. Substrate specificity and regioselectivity of fungal AA9 lytic polysaccharide monooxygenases secreted by Podospora anserina. Biotechnol Biofuels 8:90. 10.1186/s13068-015-0274-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Isaksen T, Westereng B, Aachmann FL, Agger JW, Kracher D, Kittl R, Ludwig R, Haltrich D, Eijsink VGH, Horn SJ. 2014. A C4-oxidizing lytic polysaccharide monooxygenase cleaving both cellulose and cello-oligosaccharides. J Biol Chem 289:2632–2642. 10.1074/jbc.M113.530196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kittl R, Kracher D, Burgstaller D, Haltrich D, Ludwig R. 2012. Production of four Neurospora crassa lytic polysaccharide monooxygenases in Pichia pastoris monitored by a fluorimetric assay. Biotechnol Biofuels 5:79. 10.1186/1754-6834-5-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hangasky JA, Iavarone AT, Marletta MA. 2018. Reactivity of O2 versus H2O2 with polysaccharide monooxygenases. Proc Natl Acad Sci USA 115:4915–4920. 10.1073/pnas.1801153115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kuusk S, Bissaro B, Kuusk P, Forsberg Z, Eijsink VGH, Sørlie M, Väljamäe P. 2018. Kinetics of H2O2-driven degradation of chitin by a bacterial lytic polysaccharide monooxygenase. J Biol Chem 293:523–531. 10.1074/jbc.M117.817593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vaaje-Kolstad G, Bøhle LA, Gåseidnes S, Dalhus B, Bjørås M, Mathiesen G, Eijsink VGH. 2012. Characterization of the chitinolytic machinery of Enterococcus faecalis V583 and high-resolution structure of its oxidative CBM33 enzyme. J Mol Biol 416:239–254. 10.1016/j.jmb.2011.12.033. [DOI] [PubMed] [Google Scholar]
- 69.Hamre AG, Eide KB, Wold HH, Sørlie M. 2015. Activation of enzymatic chitin degradation by a lytic polysaccharide monooxygenase. Carbohydr Res 407:166–169. 10.1016/j.carres.2015.02.010. [DOI] [PubMed] [Google Scholar]
- 70.Yang Y, Li J, Liu X, Pan X, Hou J, Ran C, Zhou Z. 2017. Improving extracellular production of Serratia marcescens lytic polysaccharide monooxygenase CBP21 and Aeromonas veronii B565 chitinase Chi92 in Escherichia coli and their synergism. AMB Express 7:170. 10.1186/s13568-017-0470-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Paspaliari DK, Loose JS, Larsen MH, Vaaje‐Kolstad G. 2015. Listeria monocytogenes has a functional chitinolytic system and an active lytic polysaccharide monooxygenase. FEBS J 282:921–936. 10.1111/febs.13191. [DOI] [PubMed] [Google Scholar]
- 72.Filandr F, Kavan D, Kracher D, Laurent C, Ludwig R, Man P, Halada P. 2020. Structural dynamics of lytic polysaccharide monooxygenase during catalysis. Biomolecules 10:242. 10.3390/biom10020242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Feng Y, Chien KY, Chen HL, Chiu CH. 2012. Pseudogene recoding revealed from proteomic analysis of salmonella serovars. J Proteome Res 11:1715–1719. 10.1021/pr200904c. [DOI] [PubMed] [Google Scholar]
- 74.Kuo CH, Ochman H. 2010. The extinction dynamics of bacterial pseudogenes. PLoS Genet 6:e1001050. 10.1371/journal.pgen.1001050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pink RC, Wicks K, Caley DP, Punch EK, Jacobs L, Carter DRF. 2011. Pseudogenes: pseudo-functional or key regulators in health and disease? RNA 17:792–798. 10.1261/rna.2658311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Milligan MJ, Lipovich L. 2015. Pseudogene-derived lncRNAs: emerging regulators of gene expression. Front Genet 5:476. 10.3389/fgene.2014.00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mondal M, Nag D, Koley H, Saha DR, Chatterjee NS. 2014. The Vibrio cholerae extracellular chitinase ChiA2 is important for survival and pathogenesis in the host intestine. PLoS One 9:e103119. 10.1371/journal.pone.0103119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Frederiksen RF, Yoshimura Y, Storgaard BG, Paspaliari DK, Petersen BO, Chen K, Larsen T, Duus JØ, Ingmer H, Bovin NV, Westerlind U, Blixt O, Palcic MM, Leisner JJ. 2015. A diverse range of bacterial and eukaryotic chitinases hydrolyzes the LacNAc (Galβ1–4GlcNAc) and LacdiNAc (GalNAcβ1–4GlcNAc) motifs found on vertebrate and insect cells. J Biol Chem 290:5354–5366. 10.1074/jbc.M114.607291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Songsiriritthigul C, Pantoom S, Aguda AH, Robinson RC, Suginta W. 2008. Crystal structures of Vibrio harveyi chitinase A complexed with chitooligosaccharides: implications for the catalytic mechanism. J Struct Biol 162:491–499. 10.1016/j.jsb.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 80.Nagashima T, Tange T, Anazawa H. 1999. Dephosphorylation of phytate by using the Aspergillus niger phytase with a high affinity for phytate. Appl Environ Microbiol 65:4682–4684. 10.1128/AEM.65.10.4682-4684.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Choonia HS, Lele S. 2013. Three phase partitioning of β-galactosidase produced by an indigenous Lactobacillus acidophilus isolate. Sep Purif Technol 110:44–50. 10.1016/j.seppur.2013.02.033. [DOI] [Google Scholar]
- 82.Kirn TJ, Jude BA, Taylor RK. 2005. A colonization factor links Vibrio cholerae environmental survival and human infection. Nature 438:863–866. 10.1038/nature04249. [DOI] [PubMed] [Google Scholar]
- 83.Bhowmick R, Ghosal A, Das B, Koley H, Saha DR, Ganguly S, Nandy RK, Bhadra RK, Chatterjee NS. 2008. Intestinal adherence of Vibrio cholerae involves a coordinated interaction between colonization factor GbpA and mucin. Infect Immun 76:4968–4977. 10.1128/IAI.01615-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Leisner JJ, Larsen MH, Jørgensen RL, Brøndsted L, Thomsen LE, Ingmer H. 2008. Chitin hydrolysis by Listeria spp., including L. monocytogenes. Appl Environ Microbiol 74:3823–3830. 10.1128/AEM.02701-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Askarian F, Uchiyama S, Masson H, Sørensen HV, Golten O, Bunæs AC, Mekasha S, Røhr ÅK, Kommedal E, Ludviksen JA, Arntzen MØ, Schmidt B, Zurich RH, van Sorge NM, Eijsink VGH, Krengel U, Mollnes TE, Lewis NE, Nizet V, Vaaje-Kolstad G. 2021. The lytic polysaccharide monooxygenase CbpD promotes Pseudomonas aeruginosa virulence in systemic infection. Nat Commun 12:1230. 10.1038/s41467-021-21473-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mitsuhashi W, Kawakita H, Murakami R, Takemoto Y, Saiki T, Miyamoto K, Wada S. 2007. Spindles of an entomopoxvirus facilitate its infection of the host insect by disrupting the peritrophic membrane. J Virol 81:4235–4243. 10.1128/JVI.02300-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nørstebo SF, Paulshus E, Bjelland AM, Sørum H. 2017. A unique role of flagellar function in Aliivibrio salmonicida pathogenicity not related to bacterial motility in aquatic environments. Microb Pathog 109:263–273. 10.1016/j.micpath.2017.06.008. [DOI] [PubMed] [Google Scholar]
- 88.Bjelland AM, Sørum H, Tegegne DA, Winther-Larsen HC, Willassen NP, Hansen H. 2012. LitR of Vibrio salmonicida is a salinity-sensitive quorum-sensing regulator of phenotypes involved in host interactions and virulence. Infect Immun 80:1681–1689. 10.1128/IAI.06038-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nørstebo SF, Lotherington L, Landsverk M, Bjelland AM, Sørum H. 2018. Aliivibrio salmonicida requires O-antigen for virulence in Atlantic salmon (Salmo salar L.). Microb Pathog 124:322–331. 10.1016/j.micpath.2018.08.058. [DOI] [PubMed] [Google Scholar]
- 90.Milton DL, O'Toole R, Horstedt P, Wolf-Watz H. 1996. Flagellin A is essential for the virulence of Vibrio anguillarum. J Bacteriol 178:1310–1319. 10.1128/jb.178.5.1310-1319.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Aslanidis C, de Jong PJ. 1990. Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res 18:6069–6074. 10.1093/nar/18.20.6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Manoil C, Beckwith J. 1986. A genetic approach to analyzing membrane protein topology. Science 233:1403–1408. 10.1126/science.3529391. [DOI] [PubMed] [Google Scholar]
- 93.Forsberg Z, Nelson CE, Dalhus B, Mekasha S, Loose JSM, Crouch LI, Røhr ÅK, Gardner JG, Eijsink VGH, Vaaje-Kolstad G. 2016. Structural and functional analysis of a lytic polysaccharide monooxygenase important for efficient utilization of chitin in Cellvibrio japonicus. J Biol Chem 291:7300–7312. 10.1074/jbc.M115.700161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wagner TM, Janice J, Paganelli FL, Willems RJ, Askarian F, Pedersen T, Top J, de Haas C, van Strijp JA, Johannessen M, Hegstad K. 2018. Enterococcus faecium TIR-domain genes are part of a gene cluster which promotes bacterial survival in blood. Int J Microbiol 2018:1435820. 10.1155/2018/1435820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tiukova IA, Brandenburg J, Blomqvist J, Sampels S, Mikkelsen N, Skaugen M, Arntzen MO, Nielsen J, Sandgren M, Kerkhoven EJ. 2019. Proteome analysis of xylose metabolism in Rhodotorula toruloides during lipid production. Biotechnol Biofuels 12:137. 10.1186/s13068-019-1478-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. 2014. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13:2513–2526. 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tyanova S, Temu T, Cox J. 2016. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc 11:2301–2319. 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- 98.Huang L, Zhang H, Wu P, Entwistle S, Li X, Yohe T, Yi H, Yang Z, Yin Y. 2018. dbCAN-seq: a database of carbohydrate-active enzyme (CAZyme) sequence and annotation. Nucleic Acids Res 46:D516–D521. 10.1093/nar/gkx894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Almagro Armenteros JJ, Tsirigos KD, Sonderby CK, Petersen TN, Winther O, Brunak S, von Heijne G, Nielsen H. 2019. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol 37:420–423. 10.1038/s41587-019-0036-z. [DOI] [PubMed] [Google Scholar]
- 100.Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. 2003. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res 31:3784–3788. 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, Perez E, Uszkoreit J, Pfeuffer J, Sachsenberg T, Yilmaz S, Tiwary S, Cox J, Audain E, Walzer M, Jarnuczak AF, Ternent T, Brazma A, Vizcaino JA. 2019. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47:D442–D450. 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Simon R, Priefer U, Puhler A. 1983. A broad host range mobilization system for in vivo genetic-engineering - transposon mutagenesis in Gram-negative bacteria. Nat Biotechnol 1:784–791. 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 103.Vaaje-Kolstad G, Houston DR, Riemen AHK, Eijsink VGH, van Aalten DMF. 2005. Crystal structure and binding properties of the Serratia marcescens chitin-binding protein CBP21. J Biol Chem 280:11313–11319. 10.1074/jbc.M407175200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Set S1. Download AEM.00529-21-s0001.xlsx, XLSX file, 0.3 MB (347.4KB, xlsx)
Figures S1 to S16, Tables S1 and S2. Download AEM.00529-21-s0002.pdf, PDF file, 1.1 MB (1.1MB, pdf)
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (101) partner repository with the data set identifier PXD021397.