

SUMMARY
Alveolar epithelial type 2 cell (AEC2) dysfunction is implicated in the pathogenesis of adult and pediatric interstitial lung disease (ILD), including idiopathic pulmonary fibrosis (IPF); however, identification of disease-initiating mechanisms has been impeded by inability to access primary AEC2s early on. Here, we present a human in vitro model permitting investigation of epithelial-intrinsic events culminating in AEC2 dysfunction, using patient-specific induced pluripotent stem cells (iPSCs) carrying an AEC2-exclusive disease-associated variant (SFTPCI73T). Comparing syngeneic mutant versus gene-corrected iPSCs after differentiation into AEC2s (iAEC2s), we find that mutant iAEC2s accumulate large amounts of misprocessed and mistrafficked pro-SFTPC protein, similar to in vivo changes, resulting in diminished AEC2 progenitor capacity, perturbed proteostasis, altered bioenergetic programs, time-dependent metabolic reprogramming, and nuclear factor κB (NF-κB) pathway activation. Treatment of SFTPCI73T-expressing iAEC2s with hydroxychloroquine, a medication used in pediatric ILD, aggravates the observed perturbations. Thus, iAEC2s provide a patient-specific preclinical platform for modeling the epithelial-intrinsic dysfunction at ILD inception.
Graphical Abstract

In brief
Alysandratos et al. differentiate patient-specific iPSCs carrying the ILD-associated SFTPCI73T variant and syngeneic corrected iPSCs into alveolar epithelial type 2 cells (iAEC2s). They find that mutant iAEC2s demonstrate proteostasis perturbations, altered bioenergetic programs, and metabolic reprogramming. They validate these findings in vivo in primary mouse SftpcI73T AEC2s.
INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is the most common and severe form of idiopathic interstitial pneumonia and is characterized by relentless fibrosis leading to disruption of the gas exchange unit and death within an average of 4 years from the time of diagnosis (Lederer and Martinez, 2018; Raghu et al., 2014, 2016; Travis et al., 2013). The poorly understood pathogenesis of IPF, in part due to the lack of human disease models, has been a major hurdle in developing effective therapies. Although a broad, established literature has focused on the role of lung fibroblasts in perpetuating IPF at later disease stages, factors that initiate or drive disease onset at early stages have been particularly hard to identify. With the advent of genome-wide association studies and intensive study of familial forms of pulmonary fibrosis, the lung epithelium has been increasingly implicated as a potential proximal disease driver with variants in gene loci expressed in lung epithelia having been associated with disease risk (Garcia, 2018; Kropski et al., 2015).
Of the many types of epithelia present in the lung, dysfunction of the alveolar epithelial type 2 cell (AEC2), in particular, has been repeatedly implicated in the pathogenesis of interstitial lung disease (ILD), including IPF (Barkauskas and Noble, 2014; Katzenstein, 1985; Winters et al., 2019; Selman and Pardo, 2014). Determining how AEC2 dysfunction leads to disease in humans, however, has been challenging to date. For example, studying AEC2 pathophysiology in primary human cells has been limited by difficulty in accessing patient samples and the tendency of AEC2s to lose their identity in culture (Borok et al., 1998; Foster et al., 2007), particularly in the absence of mesenchymal feeders (Barkauskas et al., 2013).
An ideal model of human AEC2 dysfunction would allow the study of disease by utilizing patient-derived cells to reveal a cascade of mechanistic events associated with the inception as well as progression of lung disease. Here, we present such a model of disease inception employing induced pluripotent stem cells (iPSCs) that we have derived from patients with ILD in order to generate an inexhaustible supply of AEC2-like cells in vitro to study the epithelial-intrinsic events that lead to specific AEC2 perturbations that are then validated in one of the donor patient’s tissue in vivo. To ensure examination of only the cell type responsible for initiating disease without the potential confounding or secondary effects of co-cultured supporting cells, we have developed an epithelial-only model composed solely of purified AEC2-like cells that can be propagated indefinitely without any supporting cells, and we have chosen to focus here on a disease-associated variant in the surfactant protein C (SFTPC) gene because it is known to be expressed postnatally solely in AEC2s and can be gene-edited to provide syngeneic comparator cells, thus providing a reductionist model amenable to discerning the putative epithelial-intrinsic events associated with disease inception.
Heterozygous mutations in SFTPC have been associated with both sporadic and familial pulmonary fibrosis as well as childhood interstitial lung disease (chILD) (Cottin et al., 2011; Crossno et al., 2010; Mulugeta et al., 2015; Nogee et al., 2001; Ono et al., 2011; Thomas et al., 2002; van Moorsel et al., 2010). Among the more than 60 SFTPC variants described to date, the missense mutation g.1286T > C, resulting in substitution of threonine for isoleucine at amino acid 73 in the pro-SFTPC protein (SFTPCI73T), is the focus of our study because it is the predominant disease-associated SFTPC variant. Two recent mouse genetic models, conditionally expressing mutant SFTPC (p.I73T and p.C121G, respectively) from the endogenous Sftpc locus, resulted in polycellular alveolitis and pulmonary fibrosis (Katzen et al., 2019; Nureki et al., 2018). Although previous studies have shown that expression of SFTPCI73T in vitro (Beers et al., 2011; Hawkins et al., 2015) or in mice in vivo (Nureki et al., 2018) leads to SFTPC proprotein misprocessing and lung fibrosis, the cellular mechanisms by which dysfunctional AEC2s initiate the fibrotic cascade in humans remain elusive. Most importantly, elucidating the molecular pathogenesis of human AEC2 dysfunction caused by SFTPCI73T is likely to inform the broader mechanisms by which AEC2 dysfunction leads to IPF.
A critical role for AEC2s in ILD initiation is concordant with their essential role in maintaining distal lung homeostasis. AEC2s function both as facultative progenitors of alveoli as well as secretory cells in the distal lung. In order to meet the high metabolic demands posed by these functions, AEC2s have a larger number of mitochondria compared to other lung cell types (Massaro et al., 1975), and alterations in mitochondrial function, as observed in our model, are likely to lead to a variety of perturbed metabolic programs. Indeed, accumulation of dysmorphic and dysfunctional mitochondria in association with impaired autophagy and mitophagy has been shown in AEC2s from IPF lungs (Bueno et al., 2015). Furthermore, thyroid hormone-mediated restoration of mitochondrial function and mitophagy was recently shown to blunt pulmonary fibrosis in two mouse models of pulmonary fibrosis (Yu et al., 2018).
Because there are significant differences between murine and human pulmonary fibrosis, there is a pressing need for reliable human preclinical disease models that would not only provide further insight into the pathophysiology of human AEC2 dysfunction at ILD inception, but would also serve as a platform to test the safety and efficacy of unproven currently used as well as novel therapies. Here, we show that SFTPCI73T-expressing human iPSC-derived AEC2s (hereafter iAEC2s) provide further insights into disease pathogenesis, allow assessment of currently used unproven treatments, and have the potential to serve as a platform for testing novel therapeutics.
RESULTS
Generation of patient-specific iPSC lines and their differentiation to alveolar epithelium
To examine the mechanisms leading to human AEC2 dysfunction, we generated patient-specific iPSC lines by reprogramming dermal fibroblasts (SPC2) or peripheral blood mononuclear cells (SPC6 and SPC7) from three individuals with lung disease who were heterozygous for the most frequent disease-associated SFTPC variant (SFTPCI73T/WT) (Figure 1A; Table S1). The SPC2 donor in particular suffered from severe ILD (Figure 1B and detailed clinical phenotyping in Table S1) necessitating the need for lung transplantation in early adolescence providing access to histopathological specimens of diseased explanted lung tissue (Figure 1C). The SPC6 and SPC7 donors also suffered from ILD based on chest computed tomography (CT) imaging (Figure S1A; Table S1). To overcome the hurdles imposed by the high variance in differentiation efficiencies or cellular phenotypes associated with different genetic backgrounds of iPSC lines generated from distinct individuals (Bock et al., 2011; Boulting et al., 2011; Kim et al., 2010) and to allow for purification of specific cell types, we engineered gene-corrected syngeneic lines by using activator-like effector nucleases (TALEN)-based gene editing to knock in a tdTomato fluorescent reporter into one allele of the endogenous SFTPC locus of the parental SPC2 iPSC line. This resulted in generation of syngeneic corrected (hereafter SFTPCtdT/WT) and mutant (hereafter SFTPCI73T/tdT) iPSC lines, because the tdTomato cassette is followed by a stop/polyA cassette, preventing expression of the subsequent SFTPC coding sequence from the targeted allele (Figure 1D). Thus, this targeting strategy was designed to result in preparation of a reductionist model where only one SFTPC allele would be expressed in each paired clone, enabling study of only the mutant versus only the normal allele. It also allowed tracking of SFTPCtdTomato+ cells as they emerge through in vitro directed differentiation and purification of corrected and mutant SFTPCtdTomato+ putative iAEC2s for subsequent profiling.
Figure 1. Generation of patient-specific iPSC lines and their differentiation to alveolar epithelium.
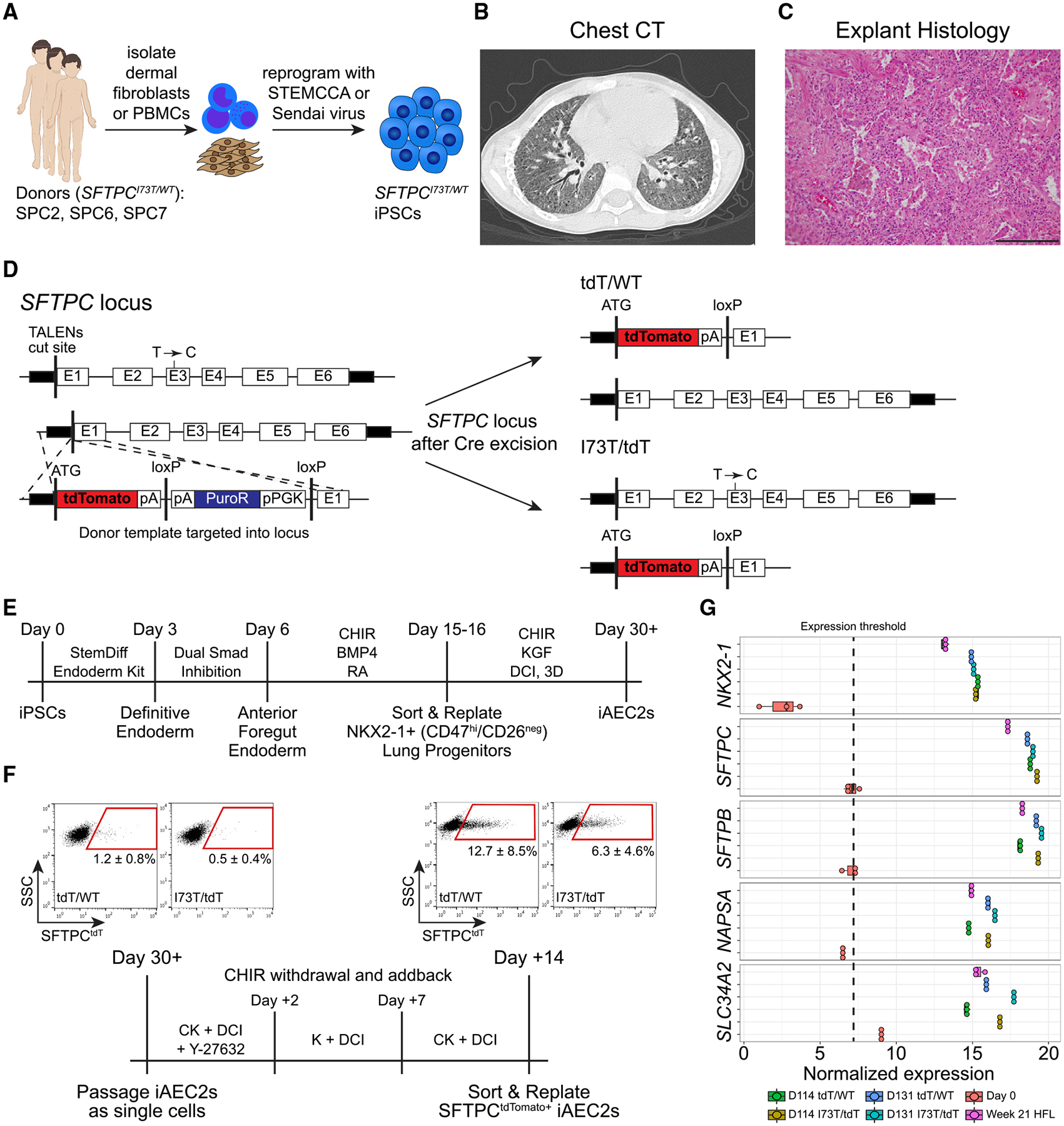
(A) Schematic showing the generation of patient-specific iPSCs from dermal fibroblasts or peripheral blood mononuclear cells (PBMCs) from three individuals carrying the most frequent SFTPC pathogenic variant (SFTPCI73T/WT).
(B) Chest CT of the SPC2 donor reveals diffuse ground glass opacities and traction bronchiectasis.
(C) H&E staining of the SPC2 donor lung explant shows end-stage lung disease with interstitial fibrosis, chronic inflammation, and alveolar remodeling with AEC2 hyperplasia and degenerating macrophages within the residual alveoli. Scale bar, 200 μm.
(D) Transcription activator-like effector nucleases (TALEN) targeting strategy and edited SFTPC loci post Cre-mediated antibiotic cassette excision.
(E) Schematic of directed differentiation protocol from iPSCs to day 30+ monolayered epithelial iAEC2 spheres (alveolospheres).
(F) Schematic of CHIR modulation to achieve iAEC2 maturation and representative flow cytometry dot plots (mean ± SD; n = 6 biological replicates of independent differentiations).
(G) Dot plots depicting the normalized expression level of AEC2-marker genes in day 114 and day 131 SFTPCtdT/WT and SFTPCI73T/tdT iAEC2s compared to day 0 iPSCs and week 21 human fetal distal lung (HFL) controls, by bulk RNA sequencing (boxplots represent mean ± SD; n = 3 experimental replicates of independent wells of a differentiation).
To ensure that any findings from this single allele expression model could be replicated in iAEC2s expressing two active SFTPC alleles, as occurs in vivo, we also engineered an untargeted clone of the SPC2 line (hereafter SFTPCI73T/WT), which then underwent footprint-free correction of the SFTPC variant via CRISPR/Cas9 gene editing (hereafter SFTPCWT/WT) (Figure S1B). Furthermore, to ensure reproducibility across different genetic backgrounds, we utilized the other two donor iPSC lines (SPC6 and SPC7; clinical phenotyping in the Supplemental information) to validate key observations made in the SPC2 iPSC lines.
Derivation of parallel indefinitely self-renewing iAEC2s
Next, we used our previously published lung directed differentiation protocol (Jacob et al., 2017, 2019) to generate corrected and mutant iAEC2s (Figures 1E and 1F), with the goal of producing indefinitely self-renewing patient-specific cells for disease modeling. Corrected and mutant SFTPCtdTomato+ iAEC2s were sorted to purity for further replating, characterization of gene expression (Figure 1G), and culture expansion as self-renewing epithelial-only spheres without the need for subsequent cell sorting (Figures 2A and 2B). The resulting iAEC2s maintained an AEC2-specific transcriptomic profile (Figure 1G); however, mutant iAEC2s demonstrated distinct morphologies and brighter tdTomato reporter expression (Figures 2A and 2B and further discussed below).
Figure 2. Derivation of parallel self-renewing corrected (SFTPCtdT/WT) and mutant (SFTPCI73T/tdT) iAEC2s.
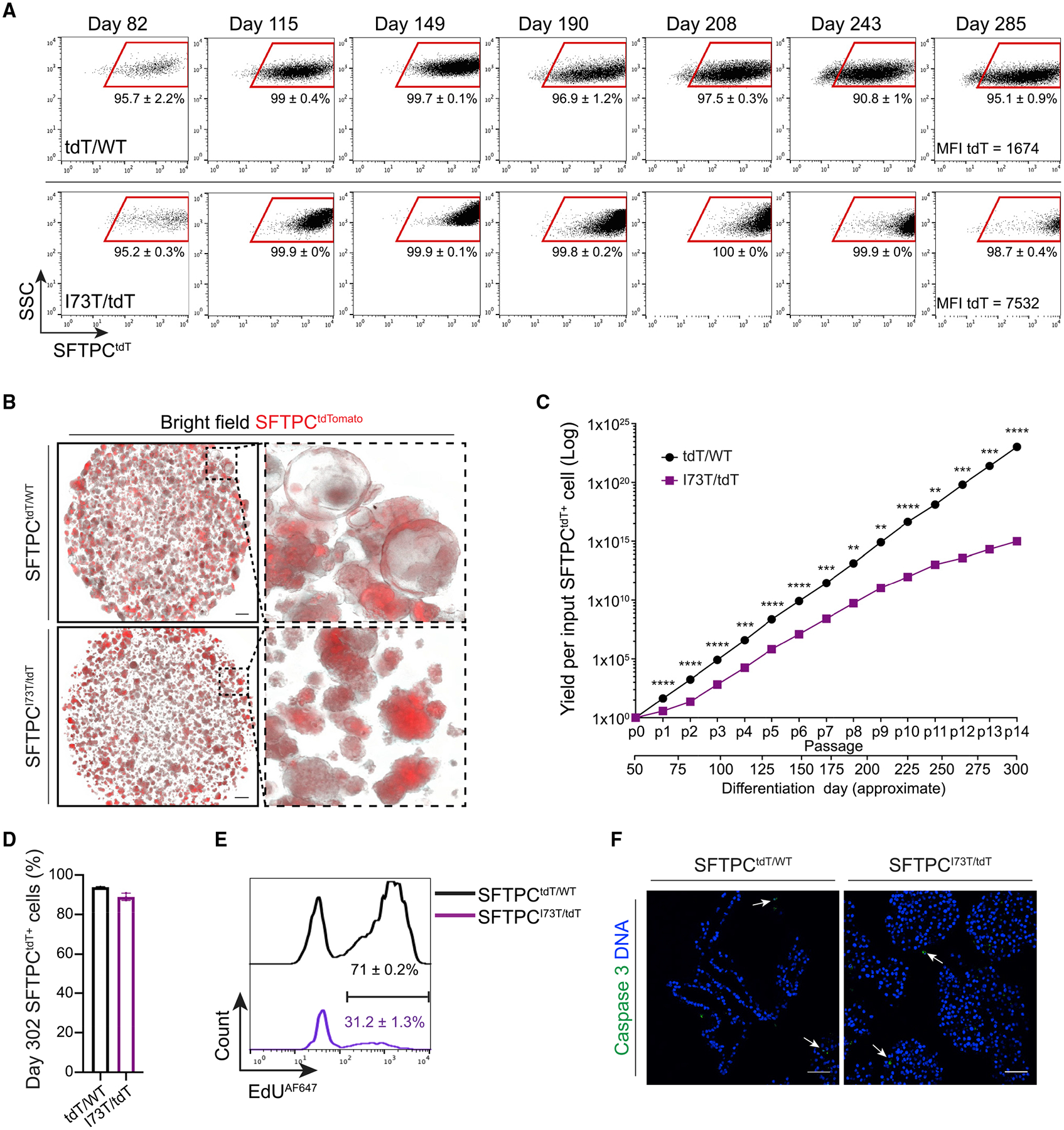
(A) Representative flow cytometry dot plots of day 82 (p2), day 115 (p4), day 149 (p6), day 190 (p8), day 208 (p9), day 243 (p11), and day 285 (p13) SFTPCtdT/WT and SFTPCI73T/tdT iAEC2s. MFI, mean fluorescence intensity.
(B) Representative live-cell imaging of SFTPCtdT/WT and SFTPCI73T/tdT alveolospheres (bright-field/tdTomato overlay; day 149). Scale bars, 500 μm.
(C) Graph showing yield per input tdT/WT or I73T/tdT SFTPCtdTomato+ cell sorted on day 51 and passaged without further sorting. **p < 0.01, ***p < 0.001, ****p < 0.0001 by unpaired, two-tailed Student’s t test.
(D) Bar graph shows retention of the AEC2 cell fate in SFTPCtdT/WT and SFTPCI73T/tdT iAEC2s maintained in culture for 302 days, measured by flow cytometry as the frequency of cells expressing the SFTPCtdTomato reporter.
(E) Histograms show higher proliferation rates in SFTPCtdT/WT compared to SFTPCI73T/tdT iAEC2s, measured by flow cytometry as the frequency of SFTPCtdTomato+ cells that incorporate EdU.
(F) Representative confocal immunofluorescence microscopy of SFTPCtdT/WT and SFTPCI73T/tdT iAEC2s stained for activated caspase 3 (green) and DNA (Hoechst, blue) shows absence of significant apoptosis. Scale bars, 50 μm.
(A and C–E) Mean ± SD is shown; n = 3 experimental replicates of independent wells of a differentiation.
We sought to determine both the stability of SFTPC expression levels as well as the total cell yield arising during serial passaging of mutant versus corrected iAEC2s in epithelial-only 3D cultures. Over a 302-day period, we generated yields of >1023 and >1015 corrected versus mutant iAEC2s per starting sorted tdTomato+ cell, respectively (Figure 2C). Although yields over this time period were significantly lower for mutant iAEC2s, both lines maintained SFTPCtdTomato expression in 94% ± 0.3% and 89.1% ± 1.9% of cells for at least 302 days and maintained a normal karyotype until at least day 214 (Figure 2D). In this self-renewing model, mutant iAEC2s demonstrated significantly lower proliferation rates than corrected iAEC2s assessed by EdU incorporation assays (Figure 2E) and no differences in apoptosis, as measured by immunostaining for activated caspase-3 (Figure 2F). These results suggest SFTPC mutant iAEC2s have diminished self-renewal capacity.
SFTPCI73T iAEC2s display altered apical-basal polarity and aberrant protein trafficking and accumulate large amounts of aberrantly processed SFTPC pro-protein
We further examined the impact of the SFTPCI73T mutation on AEC2 morphology in vitro and in vivo. Although corrected (SFTPCtdT/WT) iAEC2s formed monolayered epithelial spheres (alveolospheres) in culture as expected (Jacob et al., 2017), mutant (SFTPCI73T/tdT) iAEC2s lacked lumens and instead formed ball-like structures when assessed by microscopy of paraffin tissue sections (Figure 3A, panel i), semi-thin plastic sections (Figure 3A, panel ii), or epithelial cell adhesion molecule (EPCAM) immunostained sections (Figure 3B). Similarly, we found morphologic changes in developing AEC2s in vivo in SftpcI73T knockin mice, which do not survive birth (Nureki et al., 2018). For example, we examined E18.5 lungs from these mice and found they displayed arrested lung morphogenesis in late sacculation and complete obliteration of primordial saccules by clumps of HA-tagged pro-SFTPCI73T expressing cells (Figure 3C). Most importantly, analysis of the SPC2 donor lung explant also revealed distal lung morphological changes with areas of hyperplastic or clumped AEC2s that differed from normal lung tissue control sections (Figure 3D; arrowheads), further validating the observations made in vitro in the human iAEC2s. Furthermore, analysis of the same patient lung sections for the AEC1 marker RAGE revealed paucity of AEC1s (Figure S2A), similar to findings described in SftpcI73T knockin mice (Nureki et al., 2018), suggesting disruption of AEC2/AEC1 cell homeostasis as a result of AEC2 dysfunction.
Figure 3. SFTPCI73T/tdT iAEC2s demonstrate distinct cellular morphology and misprocess and mistraffick pro-SFTPC similarly to in vivo SFTPCI73T-expressing AEC2s.
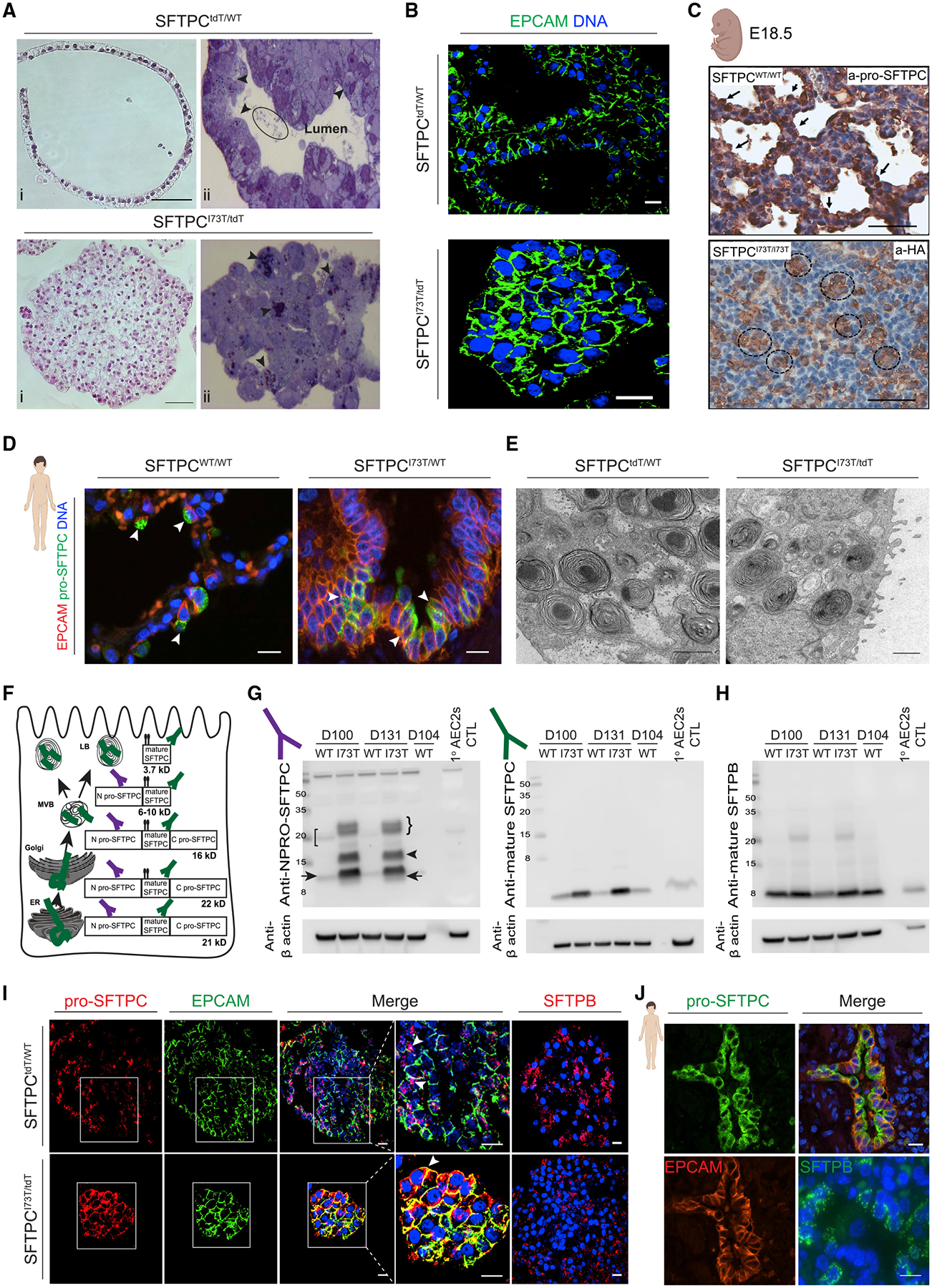
(A) Representative H&E staining of formalin fixed and paraffin embedded sections (i, scale bars, 50 μm) and toluidine blue staining of plastic sections (ii) of SFTPCtdT/WT and SFTPCI73T/tdT iAEC2s. Arrowheads indicate lamellar body-like inclusions; eclipse indicates intraluminal inclusions.
(B) Representative confocal immunofluorescence microscopy of SFTPCtdT/WT and SFTPCI73T/tdT iAEC2s for EPCAM (green) and DNA (Hoechst, blue). Scale bars, 10 μm.
(C) Representative staining of E18.5 CFlp-SFTPCI73T/I73T and CFlp-SFTPCWT/WT embryos for HA-tagged SFTPC. Scale bars, 70 μm.
(D) Representative confocal immunofluorescence microscopy of distal sections of SPC2 donor and healthy donor lung explants stained for EPCAM (red), pro-SFTPC (green), and DNA (Hoechst, blue). Scale bars, 10 μm.
(E) Representative TEM images of SFTPCtdT/WT and SFTPCI73T/tdT iAEC2s depicting lamellar bodies. Scale bars, 1 μm.
(F) Schematic representing the cellular compartments in which pro-SFTPC processing into mature SFTPC occurs. ER, endoplasmic reticulum; MVB, multi-vesicular body; LB, lamellar body.
(G and H) Representative western blots of SFTPCtdT/WT and SFTPCI73T/tdT iAEC2 lysates at the indicated time points were compared to freshly isolated primary human AEC2s lysates for pro-SFTPC (NPRO-SFTPC) (G, left panel), mature SFTPC (G, right panel), and mature SFTPB (H) with β actin as a loading control.
(I) Representative confocal immunofluorescence microscopy of SFTPCtdT/WT and SFTPCI73T/tdT iAEC2s for pro-SFTPC (red; with zoom), EPCAM (green; with zoom), SFTPB (red), and DNA (Hoechst, blue). Scale bars, 10 μm.
(J) Representative confocal immunofluorescence microscopy of distal sections of SPC2 donor and healthy donor lung explants for pro-SFTPC (green), EPCAM (red), SFTPB (green), and DNA (Hoechst, blue). Scale bars, 10 μm.
To screen for potential morphological differences in lamellar bodies between mutant and corrected iAEC2s, we employed toluidine blue-stained plastic sections (Figure 3A, panel ii) as well as transmission electron microscopy (TEM) (Figure 3E). We observed small inclusions reminiscent of lamellar bodies, present intracellularly in both corrected and mutant iAEC2s (Figure 3A panel ii; arrowheads) and extracellularly in the lumens of corrected alveolospheres, possibly reflecting lamellar body exocytosis (Figure 3A panel ii; ellipse), in keeping with our prior publication characterizing surfactant secretion from iAEC2s (Jacob et al., 2017). TEM confirmed these inclusions to be lamellar bodies (Figure 3E), with no appreciable difference in lamellar body ultrastructure observed between mutant and corrected cells, despite the clumped growth pattern of mutant iAEC2s. The altered morphological appearance of mutant iAEC2s included a lack of the characteristic apical-facing “bucket”-like shape of EPCAM immunostaining observed in corrected iAEC2s (Figure 3B), suggesting potential apical-basal polarity defects in mutant cells. To directly assess polarity we immunostained for the apical epithelial markers MUC1, phosphorylated ezrin/radixin/moesin (p-ERM), and CRB3 and the adherens junction protein E-cadherin/CDH1 (Figure S2B). We found that while in corrected iAEC2s MUC1, p-ERM, and CRB3 localized to the luminal apical cell surface and E-cadherin localized on the basal-lateral membrane, mutant iAEC2s exhibited diffuse distribution of these markers across all surfaces (Figure S2B), indicating disrupted apical-basal polarity.
To examine the effects of the SFTPCI73T mutation on human surfactant protein processing, we analyzed protein extracts from normal primary adult AEC2s in comparison to corrected versus mutant iAEC2s. In humans, SFTPC protein is produced exclusively by AEC2s as a 21 kDa pro-protein that undergoes four endoproteolytic cleavages to generate the mature 3.7 kDa form for secretion (Figure 3F) (Beers et al., 1994; Beers and Mulugeta, 2005; Brasch et al., 2002). Complete biosynthesis requires directed anterograde trafficking from Golgi to lamellar bodies, and only cells expressing these organelles are capable of fully processing SFTPC to its mature form. Western blot analysis revealed that mutant iAEC2s accumulated both significant amounts of misprocessed pro-SFTPC forms (Figure 3G, arrowhead) as well as increased amounts of incompletely processed intermediate forms (Figure 3G, right arrow), suggesting aberrant post-Golgi targeting coupled with inefficient posttranslational processing of the SFTPCI73T primary translation product and intermediates (Figure 3G, right bracket and arrow, respectively). In contrast, corrected iAEC2s demonstrated restoration of a normal pro-SFTPC processing pattern (Figure 3G, left bracket and arrow) and production of mature 3.7 kDa SFTPC (Figure 3G, right panel). Despite inefficient turnover of mutant pro-SFTPCI73T, production of mature 3.7 kDa SFTPC was also observed in mutant iAEC2s but accumulated to a greater degree than in corrected iAEC2s, suggesting its turnover is also inefficient (Figure 3G, right panel). By comparison, mutant iAEC2s demonstrated intact SFTPB protein processing generating roughly similar amounts of mature 8 kDa SFTPB (Figure 3H), suggesting that differences in mutant SFTPC posttranslational processing and turnover were likely not a result of a broader AEC2 trafficking dysfunction. In support of this, immunostaining revealed normal pro-SFTPC intracellular localization to lamellar bodies of corrected iAEC2s, observed as distinct cytoplasmic puncta (Figure 3I, arrowheads), whereas in mutant iAEC2s, pro-SFTPC was mislocalized to the plasma membrane (Figure 3I, arrowhead). Mistrafficking of pro-SFTPC to the plasma membrane was also demonstrated in vivo in sections of the SPC2 donor lung explant (Figures 3D and 3J). Similar to the normal SFTPB processing profile, immunostaining revealed normal intracellular localization of SFTPB in both mutant and corrected iAEC2s in vitro (Figure 3I) as well as in the donor patient’s lung tissue in vivo (Figure 3J). Misprocessing and mistrafficking of pro-SFTPC was also validated in the SPC2 line biallelically expressing both the mutant and “footprint-free” corrected SFTPC alleles (SFTPCI73T/WT) (Figures S1C and S1D) as well as the SPC6 and SPC7 iPSC lines (Figure S1E).
Transcriptomic and proteomic profiling of SFTPCI73T iAEC2s and their gene-corrected counterparts identifies differential regulation of the lysosomal/autophagy pathway
We next sought to identify the downstream consequences of pro-SFTPC misprocessing and mistrafficking by profiling mutant and corrected iAEC2s by single-cell RNA sequencing (scRNA-seq), bulk RNA sequencing, and proteomic analyses. Transcriptomes profiled at single-cell resolution were more similar at early time points (day 30) after the initial onset of SFTPC expression but diverged over time at later time points (day 113 or day 114) in two repeated experiments (Figures 4A and S3A; day 114 scRNA-seq experiment includes all live cells irrespective of SFTPCtdTomato expression). For example, only 224 genes were differentially expressed at day 30, whereas 1,107 genes were differentially expressed by day 113 between mutant and corrected iAEC2s (false discovery rate [FDR] <0.05) (Table S2). Cell populations at each time point remained similarly pure in terms of frequencies of expression of NKX2-1, SFTPB (Figures 4B, S3B, and S3C), SFTPC (Figures 4C, 4D, S3B, and S3C), as well as the AEC2 program (Figures 4D, S3B, and S3C), based on quantification of expression of our previously defined 8-gene AEC2 differentiation and 5-gene AEC2 maturation benchmarks established using primary adult AEC2s (Hurley et al., 2020). Importantly, the cells did not detectably assume alternative lung or non-lung fates based on little to no expression of airway (SCGB1A1, FOXJ1, TP63, and KRT5) (Figure 4B) (Hurley et al., 2020), endothelial (CDH5) (Figure 4B), hematopoietic (PTPRC) (Figure 4B), or non-lung endodermal (CDX2, AFP, and ALB) (Figure S3C) transcripts.
Figure 4. Transcriptomic and proteomic/phosphoproteomic analyses identify candidate disease-associated pathways.

(A) Clustering of SFTPCtdT/WT and SFTPCI73T/tdT iAEC2 scRNA-seq transcriptomes at two time points (days (d)30 and 113 of differentiation) using Uniform Manifold Approximation Projection (UMAP).
(B) Average expression levels and frequencies (purple dots) for select genes profiled by scRNA-seq in SFTPCtdT/WT and SFTPCI73T/tdT iAEC2s. Comparison is made to a publicly available adult primary distal lung dataset (Habermann et al., 2020), and genes are selected to indicate AEC2, AEC1, airway, endothelial, epithelial, leukocyte, or proliferation programs.
(C) Heatmap of top 50 upregulated and top 50 downregulated genes comparing d113 SFTPCtdT/WT versus d113 SFTPCI73T/tdT iAEC2s by scRNA-seq (ranked by average log fold-change, FDR <0.05; row-normalized expression Z scores). A subset of differentially expressed genes is highlighted with large font.
(D) Violin plots showing normalized expression for indicated genes or cell cycle phase in d30 and d113 SFTPCtdT/WT versus SFTPCI73T/tdT iAEC2s by scRNA-seq.
(E) Heatmap of the 50 upregulated and top 50 downregulated genes in d114 SFTPCtdT/WT versus d114 SFTPCI73T/tdT iAEC2s by bulk RNA seq (ranked by FDR, FDR <0.05; row-normalized expression Z scores). AEC2-marker genes are highlighted with large font.
(F) Volcano plots indicating differential protein expression in d113 SFTPCtdT/WT versus d113 SFTPCI73T/tdT iAEC2s by mass spectrometry.
(G) Representative confocal immunofluorescence microscopy of distal sections of SPC2 donor and healthy donor lung explants stained for pro-SFTPC (green) and DNA (Hoechst, blue) shows an altered cellular localization pattern and a higher pro-SFTPC fluorescence intensity in the patient’s AEC2s, quantified by relative fluorescence units (RFU). Scale bars, 10 μm (mean ± SD; n = 35 individual SFTPC-positive cells treated as experimental replicates).
(H) Top 10 upregulated pathways in d113 SFTPCI73T/tdT versus d113 SFTPCtdT/WT iAEC2s based on global proteomic analysis.
(I) Integrated gene set enrichment analyses based on scRNA-seq transcriptomic, proteomic, and phosphoproteomic analyses in d113 SFTPCI73T/tdT versus d113 SFTPCtdT/WT iAEC2s with an FDR <0.1 in at least 2 out of 3 datasets. NES, normalized enrichment score; gene ratio, ratio of enriched genes for a given pathway to the total number of genes in the pathway. The last column represents the number of datasets (out of 3) with an FDR <0.05.
**p < 0.01, ***p < 0.001, and ****p < 0.0001 by unpaired, two-tailed Student’s t test for all panels.
Although both mutant and corrected iAEC2s captured for scRNA-seq analysis expressed similar frequencies of the AEC2 program (Figure S3C), mutant cells exhibited significantly higher expression levels of AEC2 marker genes (Figures 4B–4E) and less proliferation (significantly lower MKI67 expression and lower frequencies of cells in G2/S/M cell-cycle phases) (Figures 4B, 4D, and S3B). For example, 11 out of the top 50 differentially upregulated genes in mutant iAEC2s in the day 113 scRNA-seq were composed of transcripts encoding surfactants, lamellar body-related, and other AEC2-marker genes (PGC, SFTPB, SLPI, NAPSA, SLC34A2, LPCAT1, SFTPA1, CEACAM6, MUC1, SFTPC, and TDTOMATO) (Figure 4C). We validated these findings by real-time qPCR quantitation (Figure S3D) as well as in independent experiments analyzed by bulk RNA-seq at 2 time points, day 114 and day 131 of differentiation, again finding AEC2 gene markers comprised 14% of the top 50 most upregulated genes in mutant iAEC2s (Figure 4E).
By global proteomic analysis performed on the same day as scRNA-seq, SFTPC was the most enriched protein overall in mutant versus corrected iAEC2s (ranked by FDR) (Figure 4F), consistent with our SFTPC western blot and immunostaining findings. Additional AEC2 proteins were also over-represented in the top 50 most upregulated proteins in mutant cells, including SFTPB, SFTPA1, SLPI, and PGC, suggesting that both transcripts and proteins encoding multiple surfactants and other AEC2 markers were in the top most upregulated in mutant iAEC2s. Proteomic analysis also validated the less proliferative state of mutant iAEC2s as MKI67 protein was enriched in corrected cells, whereas cell-cycle inhibitors CDKN1C and CDKN2C were upregulated in mutant cells (Figure 4F). Analysis of SPC2 donor’s lung explant sections also revealed a higher in vivo pro-SFTPC immunostaining intensity compared to control distal lung (Figure 4G), suggesting accumulation of mutant pro-SFTPC in the patient’s AEC2s.
Having established that mutant iAEC2s display a less proliferative and more mature AEC2 phenotype than corrected cells with marked accumulation of SFTPC protein, we next sought to determine signaling pathways or biological processes that might differ in mutant cells. The autophagy-related protein ATG2B, essential for lysosomal formation, was in the top 10 most upregulated proteins (ranked by logFC) in mutant cells (Figure 4F) and gene set analysis of GO, KEGG, and HALLMARK terms using both proteomic, scRNA-seq, and bulk RNA-seq analyses all revealed significant upregulation of lysosomal and autophagy-related terms in mutant cells (Figures 4H and S3E). A systems-based approach integrating transcriptomic, proteomic, and phosphoproteomic analyses revealed a high degree of concordant regulation with protein secretion processes being perturbed across all datasets consistent with the build-up of secretory proteins evident on western blots and immunostains. In addition, altered apical junctional processes, glycolysis, STAT3 signaling, MTOR signaling, and inflammatory signaling via nuclear factor κB (NF-κB) implied additional broad perturbations were present across a wide variety of epithelial, metabolic, and inflammatory cellular processes in mutant iAEC2s, whereas proliferation pathways (E2F and MYC targets) were downregulated (Figure 4I). Given that mutant and corrected iAEC2s are (with the exemption of the SFTPCI73T mutation) otherwise genetically identical, the data above suggest that expression of mutant SFTPC drives a transcriptomic program that markedly alters the iAEC2 phenotype and includes alterations in protein expression, quality control, and intracellular signaling.
SFTPCI73T iAEC2s display autophagy perturbations
Given the potential effect of the SFTPCI73T mutation on autophagy suggested by our bioinformatic analyses (Figure 4H, I), we employed a variety of independent static and dynamic approaches to evaluate the autophagy pathway in iAEC2s (Figure 5A). We first assessed the intracellular levels of microtubule-associated protein 1 light chain 3 (LC3), a marker of autophagosome formation (Klionsky et al., 2016). Western blot analysis showed elevated LC3, suggesting an increased autophagosomal mass in mutant compared to corrected iAEC2s (Figure 5B). To distinguish between enhanced autophagosome formation versus reduced degradation, we examined the levels of p62 (SQSTM1). P62 through direct interaction with LC3, facilitates transfer of polyubiquitinated proteins to the completed autophagosome where it is degraded along with its cargo, thus serving as a marker of autophagic degradation. Mutant iAEC2s demonstrated significant accumulation of p62, compared to corrected iAEC2s, consistent with a reduction in autophagosome degradation (Figure 5B). To further delineate if the accumulation of LC3 in mutant iAEC2s was the result of only reduced autophagosome degradation or a concomitant induction in autophagosome formation, we performed autophagic flux studies. We first examined the response to the mTOR inhibitor/autophagy activator torin. When compared to their corrected counterparts, mutant iAEC2s demonstrated significantly lower LC3 turnover following treatment with torin for 18 h (55.2% ± 9.4% versus 26.7% ± 0.3% reduction over baseline, p = 0.05) (Figure 5C). We then examined the effect of bafilomycin A1 (BafA1), a vacuolar-type H+-ATPase (V-ATPase) inhibitor, which inhibits autophagosome degradation via both neutralizing the lysosomal pH and by blocking the fusion of autophagosomes with lysosomes (Klionsky et al., 2016; Yamamoto et al., 1998). Treatment with BafA1 50 nM resulted in earlier accumulation and higher amounts of LC3 in mutant compared to corrected iAEC2s suggesting increased autophagosome formation (Figure 5D). The autophagy perturbations observed in mutant (SFTPCI73T/tdT) iAEC2s were further validated in the SPC2 line biallelically expressing both mutant and corrected SFTPC alleles (SFTPCI73T/WT), as evidenced by the accumulation of both LC3 and p62 in mutant iAEC2s (Figure S1C). Together, these results suggest a concomitant upregulation of autophagosome formation (flux) in mutant iAEC2s as well as a late block in autophagy (autophagosome turnover) consistent with our previous studies in heterologous cell lines stably expressing SFTPCI73T and in vivo in our SftpcI73T mouse model (Hawkins et al., 2015; Nureki et al., 2018).
Figure 5. Autophagy perturbations in SFTPCI73T iAEC2s.

(A) Schematic illustrating the autophagy pathway. Key proteins involved in the pathway (p62/SQSTM1 and LC3) and the mechanisms of action of bafilomycin A1 and torin are depicted.
(B) Representative western blot of SFTPCtdT/WT and SFTPCI73T/tdT iAEC2 lysates for p62 and LC3 with GAPDH and β actin as loading controls with densitometric quantification (mean ± SD; n = 11 for LC3 and n = 7 for p62 independent experiments).
(C) Western blots of cell lysates for LC3 and β actin from autophagic flux studies using SFTPCtdT/WT or SFTPCI73T/tdT iAEC2s treated with either torin (5 μM) for 4, 8, or 18 h or bafilomycin A1 (BafA1) (50 or 100 nM) for 18 h or vehicle (DMSO) for 4 or 18 h (blots shown are representative of n = 2 independent experiments).
(D) A repeat autophagic flux study, as in (C) but using BafA1 (50 nM) for 1, 2, 4, and 18 h or vehicle (DMSO) for 18 h (representative of n = 2 independent experiments).
(E) SFTPCtdT/WT and SFTPCI73T/tdT iAEC2s transduced with a lentiviral LC3:GFP fusion protein and exposed to BafA1 (50 nM) to quantify autophagosomes and autophagolysosomes. Scale bar, 10 μm (representative confocal fluorescence microscopy images of n = 3 experimental replicates of independent wells of a differentiation).
(F) Representative TEM image of SFTPCI73T/tdT iAEC2s shows a double-membrane autophagosome (inset, arrowheads). Scale bar, 1 μm.
(G) Real-time qPCR showing fold change in gene expression compared to WT in primary mouse SftpcI73T AEC2s (mean ± SEM; n = 6 for SftpcI73T and n = 5 for WT mice).
(H) Representative western blots of primary mouse WT and SftpcI73T AEC2 lysates for pro-SFTPC (NPRO), LC3, and p62 with β actin as a loading control (see also Figure 3G) (n = 3 WT and SftpcI73T mice).
(I) Representative live-cell confocal fluorescence microscopy of SFTPCtdT/WT and SFTPCI73T/tdT iAEC2s stained with LysoTracker green. Scale bars, 10 μm.
(J) Quantification of acidic organelles expressed as percentage of covered cell area (mean ± SD; n = 11–14 independent alveolospheres treated as experimental replicates).
(K) Quantification of lysosomal pH based on Lysosensor yellow/blue-dextran ratio (mean ± SD; n = 2 independent experiments with 20–30 cells analyzed per experiment; see also Figure S4).
*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001, by unpaired, two-tailed Student’s t test for all panels.
To further corroborate the observed changes in biochemical markers of autophagy, we next utilized advanced imaging techniques to assess autophagosome dynamics in individual iAEC2s. We first transduced mutant and corrected iAEC2s with a lentiviral construct constitutively expressing GFP fused to LC3 (LC3:GFP) (Twig et al., 2008) to visualize and quantitate autophagosome numbers, which are represented by cytosolic GFP-positive punctae. Consistent with the biochemical profile (Figure 5B), under steady-state conditions, we found more autophagosomes in mutant versus corrected iAEC2s (Figure 5E). The fusion between autophagosomes and lysosomes promotes the degradation of both LC3 and GFP facilitated by low autophagolysosome pH that can be blocked by treatment with BafA1 (Ni et al., 2011). Treatment with BafA1 50 nM for 1 h enhanced both GFP fluorescence and the numbers of GFP-positive punctae in mutant iAEC2s compared to similarly treated corrected iAEC2s consistent with increased autophagic flux (Figure 5E). Similar observations were made in the SPC6 iPSC line where increased numbers of GFP-positive punctae were identified at both steady-state conditions and following treatment with BafA1 (50 nM for 1 h), compared to corrected (SFTPCtdT/WT) iAEC2s (data not shown). Morphologically, the SPC6 iAEC2s lacked lumens and formed ball-like structures (data not shown) similar to SFTPCI73T/tdT iAEC2s (Figures 3A and 3B). The changes in autophagosome dynamics in mutant iAEC2s were further confirmed using ultrastructural analysis by TEM where double membrane intracellular vacuoles characteristic of autophagosomes were easily visualized in mutant iAEC2s, whereas these structures remained sparse in any of the corrected iAEC2s (Figure 5F).
We further compared our iAEC2 in vitro model to in vivo changes that arise soon after induction of the Sftpc mutation in tamoxifen-inducible SftpcI73T knockin mice. Fourteen days after in vivo administration of tamoxifen or vehicle to adult mice, we isolated AEC2s to profile the proliferation state and autophagy pathway. In contrast to iAEC2s after prolonged culture periods, mouse mutant AEC2s were more proliferative compared to wild-type (WT) AEC2s (Figure 5G), possibly attributed to the in vivo setting with alternative lineages present. Nonetheless, mouse mutant AEC2s demonstrated similar pro-SFTPC misprocessing and autophagy perturbations with accumulation of both LC3 and p62 (Figure 5H), suggesting that the differences in proliferation states between mutant and corrected AEC2s did not account for the observed autophagic changes.
Because fusion with acidic lysosomes is required for autophagosome degradation, we considered the possibility that failure of lysosomes to properly acidify might explain the block in autophagy observed in mutant iAEC2s. We therefore stained mutant and corrected iAEC2s with LysoTracker green (a basic dye that stains acidic vesicular organelles) (Figure 5I) and found that mutant iAEC2s contained more acidic organelles compared to corrected cells (Figure 5J). To better assess the iAEC2 lysosomal pH, we used Lysosensor yellow/blue-dextran (Figure S4), a pH-sensitive fluorescent probe targeting the lysosomes with high specificity (Wolfe et al., 2013). Quantitation of probe fluorescence revealed that lysosomes of mutant iAEC2s maintained a lower intraluminal pH when compared to those of corrected iAEC2s (Figure 5K). Together, these data indicate that mutant iAEC2s exhibit an accelerated autophagy flux (autophagosome and autophagolysosome formation) without impairment of vacuolar acidification, suggesting that the enhanced production of mutant SFTPCI73T has saturated the autophagic capacity of mutant iAEC2s without altering lysosomal pH.
Mutant human iAEC2s in vitro and mouse AEC2s in vivo demonstrate metabolic reprogramming
Recognizing that changes in metabolic pathways (i.e., glycolysis) were among the top differentially upregulated pathways in mutant iAEC2s in our integrated analysis (Figure 4I) coupled with our prior work showing that expression of SFTPCI73T could also alter steady-state mitochondrial mass and membrane potential (Hawkins et al., 2015; Nureki et al., 2018), directed us to investigate changes in cellular bioenergetics of mutant iAEC2s. Mitochondrial morphology and mass were assessed using quantitative imaging of MitoTracker dye staining. Distinct mitochondrial morphologies were observed in mutant versus corrected iAEC2s. Although the mitochondria in corrected cells were more elongated forming a network extending from the basal-lateral to the apical cell surface, in mutant cells, mitochondria demonstrated a rounded, fragmented morphology (Figure 6A). Mutant iAEC2s also demonstrated higher mitochondrial mass (Figure 6B) with accumulation of larger (Figure 6C) but more fragmented mitochondria quantified by the aspect ratio (ratio of long over short diameter) (Figure 6D), changes previously described in primary AEC2s from patients with IPF and aged mice (Bueno et al., 2015). The higher mitochondrial mass in mutant iAEC2s was confirmed by the higher levels of mitochondrial protein TOM20, which appeared to be time-dependent with enhanced accumulation as the cells aged in culture (Figure 6E).
Figure 6. SFTPCI73T-expressing AEC2s demonstrate metabolic reprogramming.
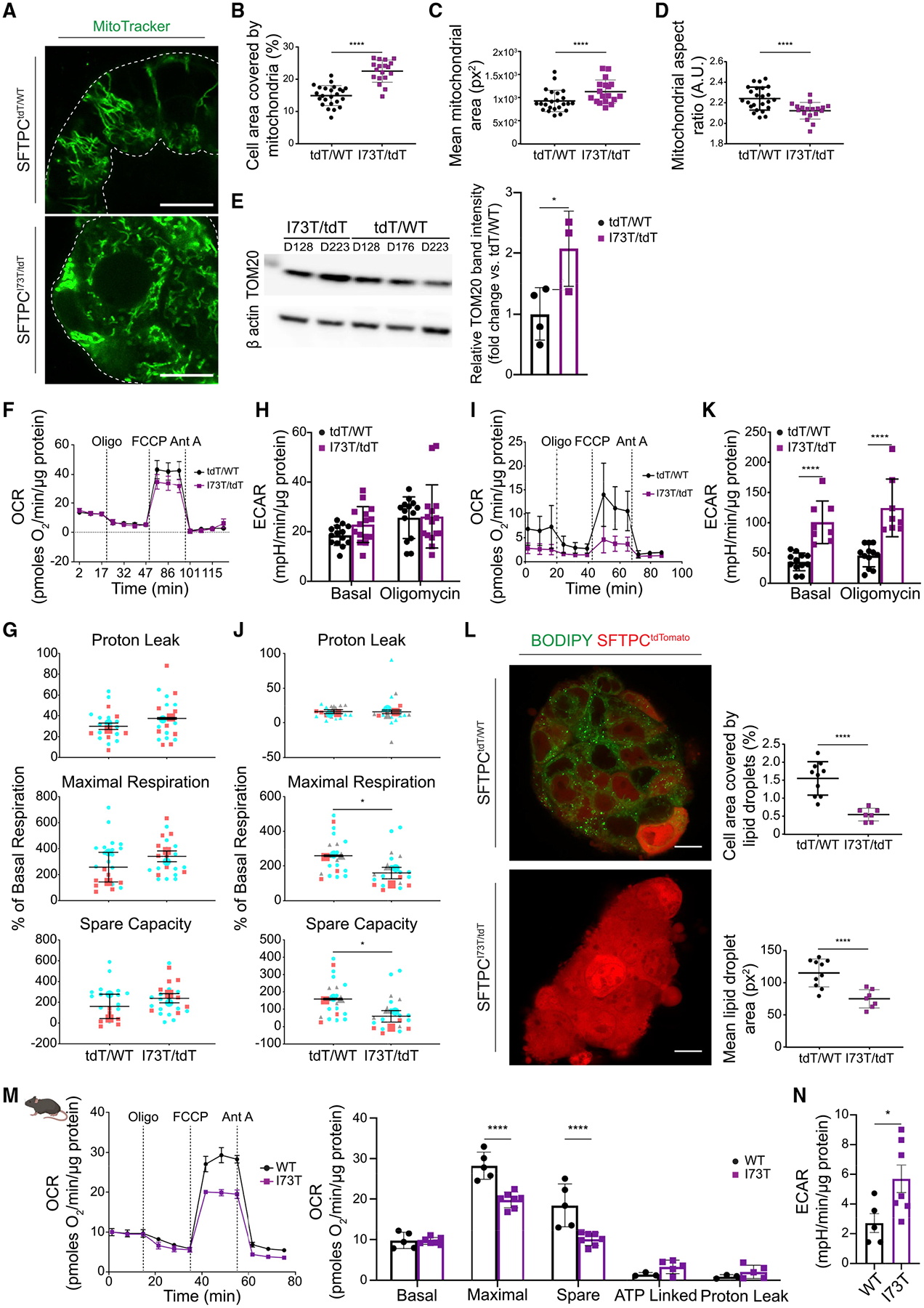
(A) Representative live-cell confocal fluorescence microscopy of SFTPCtdT/WT and SFTPCI73T/tdT iAEC2s stained with MitoTracker green. Scale bars, 10 μm.
(B–D) Quantitative analyses of morphometric data from fluorescence images (mean ± SD; n = 24 SFTPCtdT/WT and n = 18 SFTPCI73T/tdT independent alveolospheres treated as experimental replicates).
(B) Increased mitochondrial mass in SFTPCI73T/tdT iAEC2s, quantified as the percentage of cell area covered by mitochondria.
(C) Increased mitochondrial size in SFTPCI73T/tdT iAEC2s.
(D) SFTPCI73T/tdT iAEC2 mitochondria are more fragmented, assessed by the aspect ratio.
(E) Representative western blot of SFTPCtdT/WT and SFTPCI73T/tdT iAEC2 lysates for TOM20 with β actin as loading control with densitometric quantification (mean ± SD; n = 4 [days 128, 176, 223, 223] SFTPCtdT/WT and n = 3 [days 128, 223, 223] SFTPCI73T/tdT independent experiments).
(F) Early time point OCR was measured under basal conditions followed by addition of oligomycin, FCCP, and antimycin A (Ant A), as indicated.
(G) SFTPCtdT/WT and SFTPCI73T/tdT iAEC2s demonstrate no significant differences in proton leak, maximal respiration, and spare capacity measured as percent change over basal respiration (mean ± SEM; n = 2 independent experiments).
(H) Early time point ECAR under basal conditions and following addition of oligomycin.
(I) Late time point OCR.
(J) SFTPCI73T/tdT iAEC2s demonstrate significantly lower maximal respiration and spare respiratory capacity (mean ± SEM; n = 3 independent experiments) measured as percent change over basal respiration.
(K) Late time point reveals a significantly higher ECAR in SFTPCI73T/tdT iAEC2s (mean ± SD; n = 3 independent experiments).
(L) Representative live-cell confocal fluorescence microscopy of SFTPCtdT/WT and SFTPCI73T/tdT iAEC2s stained with BODIPY 493/503 dye with quantitative analyses. Scale bars, 10 μm (mean ± SD; n = 10 SFTPCtdT/WT and n = 7 SFTPCI73T/tdT independent alveolospheres treated as experimental replicates).
(M) OCR measured under basal conditions and following addition of oligomycin, FCCP, and Ant A, as indicated (mean ± SD; n = 7 SftpcI73T and n = 5 WT mice).
(N) ECAR measured under basal conditions in WT and SftpcI73T mouse AEC2s (mean ± SEM; n = 5 WT mice and n = 7 SftpcI73T). OCR, oxygen consumption rate; ECAR, extracellular acidification rate.
(G and J) Depict super plots: small shapes represent replicate values within each experiment and large shapes represent the average of each independent experiment, tdT/WT and I73T/tdT are color-matched between experiments. *p < 0.05 and ****p < 0.0001 by unpaired, two-tailed Student’s t test for all panels except (M) where a mixed-effects model with Bonferroni’s correction was used.
To investigate whether the changes in mitochondrial morphology between mutant and corrected iAEC2s were associated with changes in mitochondrial function, we performed respirometry experiments at different time points: earlier time points close to our prior transcriptomic/proteomic analyses (differentiation days 128 and 153) and later time points (differentiation days 175, 213, and 291). At the earlier time points, there were no significant differences between mutant and corrected iAEC2s in terms of their basal oxygen consumption rate (OCR), maximal uncoupled mitochondrial respiration (assessed after the administration of FCCP), and spare respiratory capacity (Figures 6F and 6G). Similar to oxidative phosphorylation, no differences were observed in glycolysis between mutant and corrected iAEC2s, as measured by the extracellular acidification rate (ECAR) either at steady-state or following treatment with the ATP synthase inhibitor oligomycin (Figure 6H). However, at later time points, which correspond to further aged iAEC2s, mutant cells demonstrated a significant reduction in maximal uncoupled mitochondrial respiration and spare respiratory capacity (Figures 6I and 6J). Conversely, mutant iAEC2s at later time points demonstrated a higher ECAR (Figure 6K) suggestive of a metabolic switch toward glycolytic metabolism. Because mobilization of lipid stores is required not only for energy utilization but also to sustain the expansion of the surface area of the mitochondrial membranes and autophagosomes, we assessed the lipid content of mutant and corrected iAEC2s. We found significantly lower levels of stored triglycerides in mutant iAEC2s, as assessed by BODIPY493/503 dye (Figure 6L).
To provide in vivo validation of our in vitro findings suggesting metabolic reprogramming of mutant iAEC2s from oxidative phosphorylation to glycolysis and to uncouple these changes from the in vitro differences in proliferation rates between mutant and corrected iAEC2s, we performed additional experiments using the same tamoxifen-inducible SftpcI73T knockin mouse model. AEC2s were isolated 14 days following in vivo tamoxifen or vehicle administration and subjected to respirometry experiments. As shown above (Figure 5G), in this in vivo model mutant AEC2s are more proliferative compared to WT cells. Similar to our in vitro model findings, we found a reduction in maximal uncoupled mitochondrial respiration and spare capacity (Figure 6M) as well as increased ECAR (Figure 6N) in the SftpcI73T mutant AEC2s when compared to WT AEC2s. Together, these data suggest that SFTPC mutant AEC2s demonstrate metabolic reprogramming from oxidative phosphorylation to glycolysis, a process that appears to be time-dependent.
SFTPCI73T iAEC2s elicit an inflammatory response
The proteomic analysis revealed upregulation of the NF-κB pathway in SFTPCI73T mutant iAEC2s compared to their corrected counterparts (Figure 7A). Because autophagy inhibition has been shown to lead to activation of the NF-κB pathway via upregulation of p62 (Hill et al., 2019), we sought to further validate the proteomic findings. We utilized a lentiviral vector (lenti-NF-κB-luc-GFP) (Figure 7B) that we previously engineered to enable independent simultaneous tracking of transduced (GFP+) cells by flow cytometry and real-time assessment of NF-κB activation levels quantified by a luciferase reporter whose expression is regulated by a minimal promoter element preceded by consensus binding sites for the canonical NF-κB p50–p65 heterodimer transcription factor complex (Wilson et al., 2013). Mutant and corrected iAEC2s were infected with the lentiviral vector (MOI 20: Figure 7C), allowed to expand in culture, and transduced (GFP+) SFTPCtdTomato+ mutant and corrected iAEC2s were then sorted and analyzed for luciferase expression (Figure 7D). Mutant iAEC2s demonstrated increased bioluminescence compared to corrected iAEC2s, suggesting increased canonical NF-κB activity in mutant iAEC2s (Figure 7E). Furthermore, mutant iAEC2s secreted higher levels of the NF-κB target proteins GM-CSF, CXCL5, and MMP-1 quantified by Luminex analysis of supernatants, compared to corrected iAEC2s (Figure 7F). Taken together, these findings suggest mutant iAEC2s are an important proinflammatory hub with resultant secretion of inflammatory mediators via activation of the NF-κB pathway.
Figure 7. SFTPCI73T/tdT iAEC2s display activation of the NF-κB pathway and are more susceptible to hydroxychloroquine treatment.
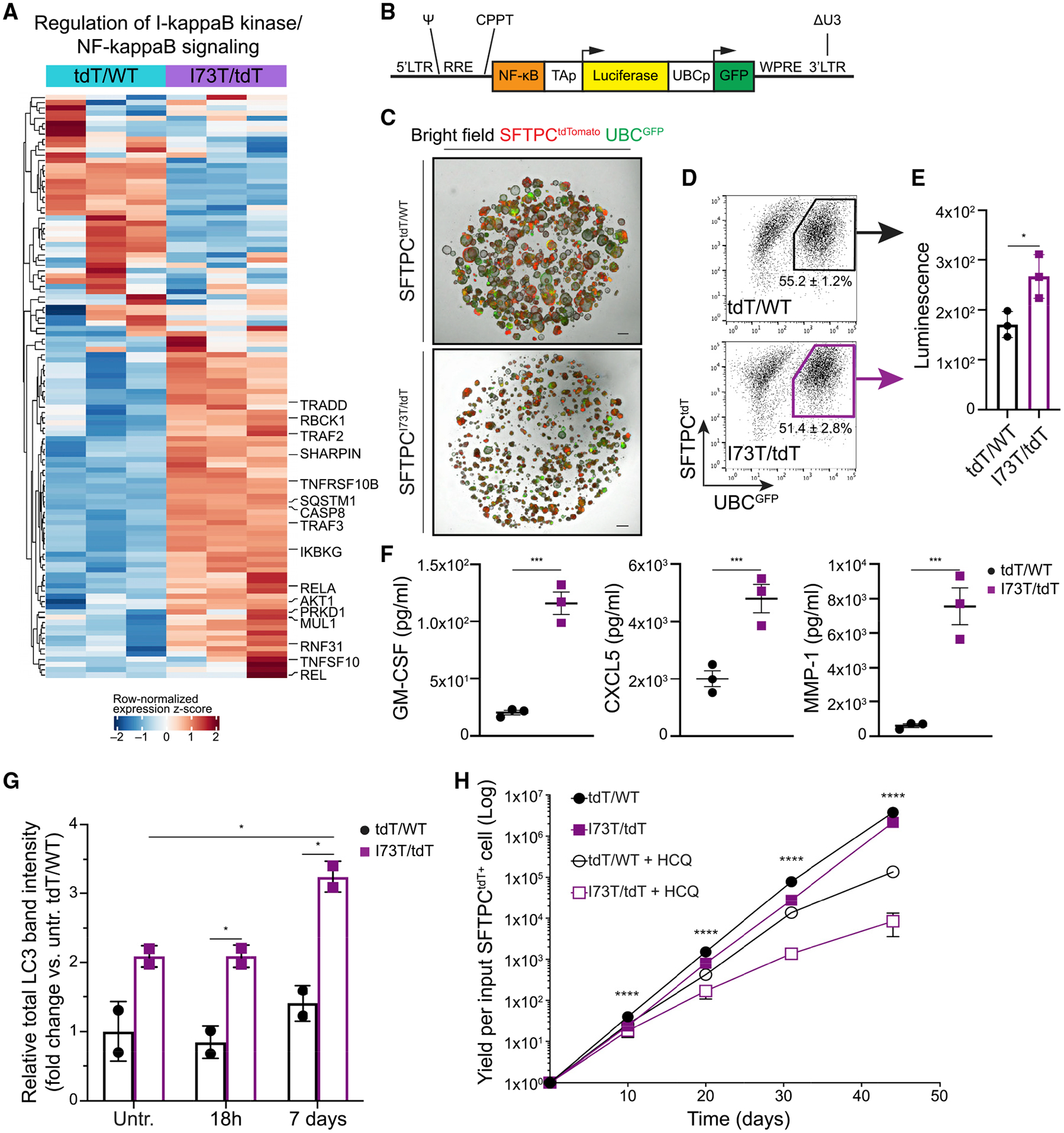
(A) Unsupervised hierarchical clustered heatmap of differentially expressed proteins (FDR <0.05) in the depicted GO biological process gene set (row normalized z-score); a subset of differentially expressed proteins is highlighted with large font.
(B) Schematic of NF-κB-luc-GFP lentiviral construct. LTR, lentiviral long terminal repeats; RRE, rev responsive element; CPPT, central polypurine tract; WPRE, Woodchuck hepatitis virus post-transcriptional regulatory element; ΔU3, deleted U3 region for in vivo inactivation of the viral LTR promoter; Ψ, Psi lentiviral packaging sequence; GFP, green fluorescence protein.
(C) Representative live-cell imaging of lentivirally transduced SFTPCtdT/WT and SFTPCI73T/tdT iAEC2s (bright-field/tdTomato/GFP overlay). Scale bars, 500 μm.
(D) Sort gates used to purify transduced (GFP+) SFTPCtdT/WT and SFTPCI73T/tdT iAEC2s.
(E) Bioluminescence quantification shows increased luciferase activity in SFTPCI73T/tdT versus SFTPCtdT/WT iAEC2s. *p < 0.05, **p < 0.01, ***p < 0.001 by unpaired, two-tailed Student’s t test for all panels.
(F) Luminex analysis of supernatants collected from SFTPCI73T/tdT and SFTPCtdT/WT iAEC2s for the NF-κB target proteins GM-CSF, CXCL5, and MMP-1. *p < 0.05, **p < 0.01, ***p < 0.001 by unpaired, two-tailed Student’s t test for all panels.
(G) SFTPCtdT/WT or SFTPCI73T/tdT iAEC2s were treated with hydroxychloroquine (HCQ; 10 μM) or vehicle (ddH2O) for 18 h or 7 days. Lysates were subjected to western blotting for LC3 with β actin as loading control. Densitometric quantification is shown. See also Figure S5. *p < 0.05, **p < 0.01, ***p < 0.001 by unpaired, two-tailed Student’s t test for all panels.
(H) Graph showing yield in cell number per input SFTPCtdT/WT or SFTPCI73T/tdT iAEC2 with or without treatment with HCQ (10 μM). ****p < 0.0001 across all groups by one-way ANOVA; p < 0.001 for d10, p < 0.01 for d20, p < 0.0001 for d31, and p < 0.05 for d44 for corrected versus mutant iAEC2s by unpaired, two-tailed Student’s t test.
Mean ± SD is shown; n = 3 (E and F) or n = 2 (G) experimental replicates of independent wells of a differentiation or n = 3 (H) biological replicates of independent differentiations.
Treatment of SFTPCI73T iAEC2s with hydroxychloroquine results in aggravation of autophagy perturbations and metabolic reprogramming
To apply our iPSC-derived model system as a preclinical disease model for drug testing, in a proof-of-principle experiment we assessed the effect of hydroxychloroquine (HCQ) on SFTPCI73T-expressing iAEC2s. HCQ is a medication commonly used either alone or in combination with corticosteroids in pediatric patients with genetic disorders of surfactant dysfunction affecting AEC2s (including SFTPCI73T) and resulting in chILD. The use of HCQ in this patient population is based primarily on evidence from case reports or small cohort studies with variable results (Braun et al., 2015; Klay et al., 2018; Kröner et al., 2015). HCQ is a lysosomotropic medication that accumulates in lysosomes inhibiting lysosomal activity and autophagy (Mauthe et al., 2018; Schrezenmeier and Dörner, 2020) raising the possibility it may aggravate the autophagic block already present in SFTPCI73T-expressing iAEC2s. Indeed, treatment of mutant iAEC2s with HCQ (10 μM) for 7 days resulted in increased accumulation of LC3 in HCQ-treated compared to untreated iAEC2s (Figures 7G and S5A). Notably, treatment of corrected iAEC2s with HCQ under the same conditions did not result in significant changes of total LC3 suggesting that mutant iAEC2s are more sensitive to HCQ’s lysosomotropic effect. Long-term treatment of mutant iAEC2s with HCQ (10 μM) resulted in further reduction of the already diminished self-renewal capacity of these cells (Figure 7H), whereas both mutant and corrected iAEC2s retained expression of the SFTPCtdTomato reporter (Figure S5B). Furthermore, mutant iAEC2s treated with HCQ for 7 days demonstrated a significant reduction in OCR/ECAR ratio assessed after the administration of oligomycin and FCCP, compared to untreated iAEC2s (Figure S5C). This suggests a shift toward glycolysis, revealed after pharmacological manipulation of energy supply and demand and reminiscent of the observations made earlier for aged mutant iAEC2s (Figure 6). Similar to LC3 levels, HCQ treatment had no effect on the OCR/ECAR ratio in corrected iAEC2s (Figure S5C).
DISCUSSION
Our results suggest that patient-specific iAEC2s can serve as a human preclinical disease model, revealing key components of the epithelial-intrinsic pathophysiology of human AEC2 dysfunction and providing a platform to test therapeutics. Patient-derived iAEC2s expressing mutant SFTPCI73T in this model exhibit misprocessed and mistrafficked pro-SFTPC protein, recapitulating the mistrafficking occurring in vivo in the donor’s AEC2s. The accumulation of misprocessed pro-SFTPC, as a result of mutant SFTPCI73T expressed from the endogenous human locus in AEC2-like cells, extends similar observations previously made based on forced overexpression of mutant protein in heterologous cell lines (Brasch et al., 2004; Galetskiy et al., 2008; Hawkins et al., 2015; Stewart et al., 2012), profiles of patient bronchoalveolar lavage samples (Brasch et al., 2004), and recently in an in vivo mouse model (Nureki et al., 2018).
The iAEC2 platform also offers the ability to temporally model both the early changes coinciding with accumulation of mutant SFTPC forms and the resulting downstream sequence of events leading to acquisition of a dysfunctional AEC2 phenotype. Multiomic profiling of syngeneically controlled mutant versus corrected iAEC2s identified similarities at an early time point but more distinct phenotypes emerged with culture expansion suggesting a time-dependent phenotype. This time course is in agreement with the SftpcI73T mouse, where an age-dependent lung-diseased phenotype emerged simultaneously with the gradual accumulation of mutant pro-SFTPC forms (Nureki et al., 2018).
Among the many cellular programs disrupted in the iAEC2 model, our findings suggest an early perturbation in autophagy. First, unbiased signaling pathway analysis identified the lysosomal/autophagy pathway as being differentially upregulated in mutant iAEC2s. We validated these observations by a variety of independent static and dynamic approaches (Figure 5) that documented induction of autophagosome formation (flux) and defined a late block in autophagy at the level of autophagosome turnover, evidenced by accumulation of LC3 and p62 and incomplete response to torin in mutant iAEC2s. Importantly, we validated these observations in vivo in primary mouse SftpcI73T AEC2s that are not hypo-proliferative, suggesting that the accumulation of misprocessed and mistrafficked pro-SFTPC was responsible for the observed phenotype rather than the proliferative differences between mutant and corrected iAEC2s. A dysregulated autophagic response has been shown in IPF lungs (Araya et al., 2013; Hill et al., 2019; Patel et al., 2012) and has been postulated to lead to epithelial cell senescence and myofibroblast differentiation (Araya et al., 2013; Hill et al., 2019). Although it remains unclear mechanistically how SFTPC buildup/misprocessing leads to autophagy perturbations, we speculate that it is likely related to failure of lysosomal fusion because lysosomal acidification appears intact in mutant iAEC2s. The resultant proteostasis perturbations and alterations in cellular quality control resulted in diminished progenitor potential as evidenced by reduced self-renewal capacity. Furthermore, the paucity of AEC1s in the donor’s lung explant (Figure S2A), similar to findings described in our SftpcI73T knockin mice (Nureki et al., 2018), point to disruption of normal AEC2/AEC1 homeostasis as a result of AEC2 dysfunction. Ineffectual AEC2-to-AEC1 differentiation and emergence of a transitional cell state have been described in IPF (Jiang et al., 2020; Riemondy et al., 2019). In addition, selective AEC2 ablation (Barkauskas et al., 2013; Garcia et al., 2016; Sisson et al., 2010) or AEC2 senescence induction via conditional deletion of Sin3a (Yao et al., 2020) in mice have resulted in progressive pulmonary fibrosis, either spontaneously or after bleomycin exposure. Therefore, our results agree with the growing body of literature suggesting that dysfunctional AEC2s could initiate the fibrotic cascade.
Because autophagy plays an important role in selective degradation of dysfunctional mitochondria, this may explain our observation that mutant iAEC2s displaying an autophagy phenotype accumulated dysmorphic and dysfunctional mitochondria in a time-dependent manner, changes previously described in primary AEC2s from patients with IPF and aged mice (Bueno et al., 2015). We further validated the mitochondrial dysfunction and metabolic reprogramming observed in mutant iAEC2s in primary mouse SftpcI73T AEC2s. Assessing the bioenergetics of human AEC2s has not been previously possible given the challenges in culturing primary AEC2s.
In addition to detailed alterations in cell quality control and metabolism, our data also provide a plausible explanation of how a dysfunctional AEC2 signals to surrounding cells to initiate inflammatory and profibrotic cascades, because the NF-κB pathway was found to be differentially upregulated in mutant compared to corrected iAEC2s. Furthermore, higher levels of the NF-κB target proteins GM-CSF, CXCL5, and MMP-1 were identified in the supernatants of mutant compared to corrected iAEC2s. Importantly, GM-CSF is a strong chemoattractant for profibrotic Ly6chi monocytes that have been implicated in the pathogenesis of lung fibrosis (Misharin et al., 2017; Venosa et al., 2019).
Finally, in a proof-of-principle experiment, we tested the application of our model system as a preclinical human disease drug testing platform by assessing the effect of HCQ on SFTPCI73T-expressing cells. Treatment of mutant iAEC2s resulted in aggravation of the observed autophagy perturbations, further reduction of the already diminished self-renewal capacity, and metabolic reprogramming toward glycolysis. Taken together, the findings from our epithelial-only model system suggest that treatment with HCQ could be detrimental to patients carrying the SFTPCI73T variant, although a pleiotropic effect resulting from HCQ’s effect on immune cells or beneficial effects on other cell types in vivo cannot be excluded.
Prior reports (Jacob et al., 2017; Korogi et al., 2019; Strikoudis et al., 2019) have provided proof-of-concept that ESC- or iPSC-derived lung epithelia carrying certain mutations could model some aspects of either adult or pediatric ILD, such as inflammatory activation. We believe the current study adds to these prior reports by addressing some of the many mechanisms associated with a cell autonomous, epithelial-intrinsic toxic gain-of-function in purified patient-specific iAEC2s, including detailed profiles of the perturbations in protein trafficking, autophagy, mitochondrial morphology, and function.
In summary, our epithelial-only patient-specific iAEC2 model system not only recapitulates key observations made previously in heterologous cell lines and in vivo mouse models (as well as in vivo in the patient from whom the iPSCs were generated) but also provides new insights into the potential role of AEC2s in the inception of pulmonary fibrosis and serves as a preclinical platform to test therapeutics.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents may be directed to, and will be fulfilled by, the Lead Contact, Darrell Kotton (dkotton@bu.edu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement. Pluripotent stem cell lines generated in this study are available from the CReM iPSC Repository at Boston University and Boston Medical Center and can be found at https://www.bu.edu:443/dbin/stemcells/.
Data and code availability
The scRNA-seq data reported in this publication have been deposited in NCBI’s Gene Expression Omnibus (GEO; accession number GSE160801). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the Proteomics Identifications (PRIDE) partner repository with the dataset identifier PRIDE: PXD022334. All original code has been deposited at GitHub and is publicly available as of the date of publication. DOIs are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal to activated caspase 3 | Sigma Aldrich | Cat# C8487; RRID:AB_476884 |
| Mouse monoclonal to β actin | Sigma Aldrich | Cat# A1978; RRID:AB_476692 |
| Mouse monoclonal to CD26, PE conjugated (clone BA5b) | BioLegend | Cat# 302705; RRID:AB_314289 |
| Mouse monoclonal to CD47, PerCP-Cy5.5 conjugated (clone CC2C6) | BioLegend | Cat# 323110; RRID:AB_940461 |
| Rabbit polyclonal to CRB3 | Szymaniak et al., 2015 | N/A |
| Mouse IgG1 isotype, PE conjugated | BioLegend | Cat# 400113; RRID:AB_326435 |
| Mouse IgG1 isotype, PerCP-Cy5.5 conjugated | BioLegend | Cat# 400149; RRID:AB_893680 |
| Mouse monoclonal to E-Cadherin/CDH1 | BD Biosciences | Cat# 610181; RRID:AB_397580 |
| Mouse monoclonal to EPCAM | Santa Cruz | Cat# Sc-73491; RRID:AB_1122569 |
| Mouse monoclonal to GAPDH | Chemicon | Cat# MAB374; RRID:AB_2107445 |
| Rabbit monoclonal to NKX2-1 (clone EP15847) | Abcam | Cat# ab76013; RRID:AB_1310784 |
| Rabbit monoclonal to Phospho-Ezrin/Radixin/Moesin (p-ERM) | Cell Signaling | Cat# 3726; RRID:AB_10560513 |
| Mouse monoclonal to pro-SFTPC | Santa Cruz | Cat# sc-518029 |
| Rabbit polyclonal to pro-SFTPC | Seven Hills | Cat# WRAB-9337; RRID:AB_2335890 |
| Rabbit polyclonal to mature SFTPC | Seven Hills | Cat# WRAB-76694 |
| Rabbit polyclonal anti-NPRO-SFTPC | Beers et al., 1994 | N/A |
| Rabbit polyclonal anti-mature SFTPB (PT3) | Beers et al., 1992 | N/A |
| Rabbit monoclonal to MUC1 | Abcam | Cat# Ab218998 |
| Rabbit polyclonal to HA tag | Abcam | Cat# Ab20084; RRID:AB_445319 |
| Mouse monoclonal to RAGE | Abcam | Cat# Ab54741; RRID:AB_2242462 |
| Rabbit polyclonal to LC3B | Cell Signaling | Cat# 2775; RRID:AB_915950 |
| Mouse monoclonal to SQSTM1/p62 | Abcam | Cat# Ab56416; RRID:AB_945626 |
| Mouse monoclonal to CPM | Wako | Cat# 014-27501; RRID:AB_2801482 |
| Mouse monoclonal to SLC34A2 | Yin et al., 2008 | N/A |
| AffiniPure Donkey Anti-Rabbit IgG (H+L), AlexaFluor 488 conjugated | Jackson ImmunoResearch | Cat# 711-545-152; RRID:AB_2313584 |
| AffiniPure Donkey Anti-Rabbit IgG (H+L), AlexaFluor 647 conjugated | Jackson ImmunoResearch | Cat# 711-605-152; RRID:AB_2492288 |
| AffiniPure Donkey Anti-Mouse IgG (H+L), AlexaFluor 488 conjugated | Jackson ImmunoResearch | Cat# 715-545-150; RRID:AB_2340846 |
| AffiniPure Donkey Anti-Mouse IgG (H+L), AlexaFluor 647 conjugated | Jackson ImmunoResearch | Cat# 715-605-150; RRID:AB_2340862 |
| AffiniPure Goat Anti-Mouse IgG (H+L), AlexaFluor 488 conjugated | Jackson ImmunoResearch | Cat# 115-545-062; RRID:AB_2338845 |
| AffiniPure Goat Anti-Rabbit IgG (H+L), AlexaFluor 488 conjugated | Jackson ImmunoResearch | Cat# 111-545-144; RRID:AB_2338052 |
| AffiniPure Goat Anti-Mouse IgG (H+L), AlexaFluor 594 conjugated | Jackson ImmunoResearch | Cat# 115-585-146; RRID:AB_2338881 |
| AffiniPure Goat Anti-Rabbit IgG (H+L), AlexaFluor 594 conjugated | Jackson ImmunoResearch | Cat# 111-585-144; RRID:AB_2307325 |
| Goat Anti-Mouse IgG (H+L), HRP conjugated | BioRad | Cat# 0300-0108P; RRID:AB_808614 |
| Goat Anti-Rabbit IgG (H+L), HRP conjugated | BioRad | Cat# 5196-2504; RRID:AB_619908 |
| Chemicals, peptides and recombinant proteins | ||
| Human embryonic stem cell (hESC)- qualified (2D) Matrigel | Corning | Cat# 354277 |
| Growth Factor Reduced (3D) Matrigel | Corning | Cat# 356231 |
| SB431542 | Tocris | Cat# 1614 |
| Dorsomorphin | Stemgent | Cat# 04-0024 |
| CHIR99021 (CHIR) | Tocris | Cat# 4423 |
| Recombinant human FGF10 | R&D Systems | Cat# 345-FG-025 |
| Recombinant Human Keratinocyte Growth Factor (KGF) | R&D Systems | Cat# 251-KG-010 |
| Recombinant Human BMP4 (rhBMP4) | R&D Systems | Cat# 314-BP |
| Retinoic acid | Sigma Aldrich | Cat# R2625 |
| Y-27632 dihydrochloride | Tocris | Cat# 1254 |
| Ascorbic Acid | Sigma Aldrich | Cat# A4544 |
| 1-Thioglycerol (MTG) | Sigma Aldrich | Cat# M6145 |
| BSA 7.5% Stock | Thermo Fisher Scientific | Cat# 15260037 |
| 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (EDTA) | Sigma Aldrich | Cat# E7889 |
| N-(2-Hydroxyethyl)piperazine-N’-(2- ethanesulfonic acid) Solution (HEPES) | Sigma Aldrich | Cat# H0887 |
| Dexamethasone | Sigma Aldrich | Cat# D4902 |
| 8-bromoadenosine 30,50-cyclic monophosphate sodium salt (cAMP) | Sigma Aldrich | Cat# B7880 |
| 3-Isobutyl-1-methylxanthine (IBMX) | Sigma Aldrich | Cat# I5879 |
| Puromycin Dihydrochloride | Thermo Fisher Scientific | Cat# A1113802 |
| Geneticin Sulfate | Life Technologies | Cat# 11811-023 |
| 0.05% trypsin-EDTA | Invitrogen | Cat# 25300-120 |
| Defined Fetal Bovine Serum | Thermo Fisher Scientific | Cat# NC0652331 |
| Dimethyl Sulfoxide (DMSO) | Sigma Aldrich | Cat# D2650 |
| Calcein blue | Life Technologies | Cat# C1429 |
| Hoechst 33342 | Thermo Fisher Scientific | Cat#H3570 |
| DAPI, FluoroPure grade | Invitrogen | D21490 |
| ProLong Diamond Antifade Mountant | Invitrogen | Cat# P36965 |
| LysoTracker Green DND-26 | Thermo Fisher Scientific | Cat# L7526 |
| MitoTracker Green FM | Thermo Fisher Scientific | Cat# M7514 |
| BODIPY-493/503 | Thermo Fisher Scientific | Cat# D3922 |
| LysoSensor Yellow/Blue Dextran | Thermo Fisher Scientific | Cat# L22460 |
| Dispase II | Thermo Fisher Scientific | Cat# 17105-041 |
| Glutaraldehyde | Ladd Research | Cat# 20100 |
| Osmium Tetroxide | Polysciences | Cat# 0223D |
| Uranyl Acetate | Electron Microscopy Sciences | Cat# 22400 |
| EMbed 812 | Electron Microscopy Sciences | Cat# 14120 |
| Paraformaldehyde | Electron Microscopy Sciences | Cat# 19208 |
| Oligomycin | Millipore | Cat# 495455 |
| FCCP | Sigma Aldrich | Cat# C2920 |
| Antimycin A | Sigma Aldrich | Cat# A8674 |
| Bafilomycin A1 | Sigma Aldrich | Cat# B1793 |
| Torin | Tocris | Cat# 4247 |
| Critical commercial assays | ||
| RNeasy Mini Kit | QIAGEN | Cat# 74104 |
| QIAzol Lysis Reagent QIAGEN | QIAGEN | Cat# 79306 |
| TaqMan Fast Universal PCR Master Mix (2X) | Thermo Fisher | Cat# 4364103 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat# 4368814 |
| EZ-TAL TALE Assembly Kit | System Bioscience | Cat# GE120A-1 |
| Click-iT EdU Alexa Fluor 647 Flow Cytometry Assay Kit | Thermo Fisher Scientific | Cat# C10424 |
| Lipofectamine LTX Kit | Thermo Fisher Scientific | Cat# 15338100 |
| P3 Primary Cell 4D-NucleofectorO X Kit S | Lonza | Cat# V4XP-3032 |
| Magnetic Luminex® Performance Assay Human HighSensitivity | R&D Systems | Cat# LXSAHM-31 |
| Deposited data | ||
| Single cell transcriptomic profiling of patient-specific pluripotent stem cell-derived alveolar epithelial type 2 cells expressing the SFTPCI73T variant and their gene-edited corrected counterparts | This paper | GEO (GSE160801) |
| Proteomic and phosphoproteomic profiling of SFTPCI73T expressing patient-specific pluripotent stem cell-derived alveolar epithelial type 2 cells and their gene-edited corrected counterparts. | This paper | PRIDE: PXD022334 |
| Experimental models: Cell lines | ||
| Human: SFTPCI73T/WT donor iPSC line (SPC2) | This paper | http://stemcellbank.bu.edu/Catalog/Item/Home |
| Human: Corrected SFTPC donor iPSC line (SPC2-CorrC1; SFTPCwt/wt) | This paper | http://stemcellbank.bu.edu/Catalog/Item/Home |
| Human: Donor iPSC line targeted with SFTPCtdTomato (SPC2-ST-C11; SFTPCI73T/tdT) | This paper | http://stemcellbank.bu.edu/Catalog/Item/Home |
| Human: Donor iPSC line targeted with SFTPCtdTomato (SPC2-ST-B2; SFTPCtdT/WT) | Kotton Lab; Hurley et al., 2020 | http://stemcellbank.bu.edu/Catalog/Item/Home |
| Oligonucleotides | ||
| See Table S3 | This paper | N/A |
| Recombinant DNA | ||
| EF1a-TALEN_HD; SFTPC recognition sequence of right TALEN 5′-TCA CCG GCG GGC TCT CCA TC-3′. | Kotton Lab; Jacob et al., 2017 | http://www.kottonlab.com |
| EF1a-TALEN_NN; SFTPC recognition sequence of left TALEN: 5’-TAG CAC CTG CAG CAA GAT GG-3′. | Kotton Lab; Jacob et al., 2017 | http://www.kottonlab.com |
| p1303-DV-SFTPC-tdTomato | Kotton Lab; Jacob et al., 2017 | http://www.kottonlab.com |
| pHAGE2 EF1a-Cre-IRES-NeoR-W | Kotton Lab; Jacob et al., 2017 | http://www.kottonlab.com |
| pHAGE2-Cre-IRES-PuroR | Kotton Lab; Somers et al., 2010 | Addgene #30205 |
| Software and algorithms | ||
| FlowJo version 10.5.3 | Becton, Dickinson & Company | https://www.flowjo.com/ |
| ImageJ | National Institutes of Health | https://imagej.nih.gov/ij/ |
| CellProfiler v2.0 | Broad Institute | https://cellprofiler.org |
| Prism | GraphPad | https://www.graphpad.com/ |
| Original code | This paper | https://github.com/CReM-BU/Alysandratos_iAEC2s |
| Other | ||
| StemDiff Definitive Endoderm Kit | StemCell Technologies | Cat# 05110 |
| mTeSR1 | StemCell Technologies | Cat# 05850 |
| Glutamax | Life Technologies | Cat# 35050-061 |
| Gentle Cell Dissociation Reagent | StemCell Technologies | Cat# 07174 |
| Seahorse XF Base Medium Minimal DMEM | Agilent Technologies | Cat# 103193-100 |
| Hank’s Buffered Saline Solution (HBSS; no calcium, no magnesium, no phenol red) | GIBCO | Cat# 14175095 |
| Ham’s F12 Medium | Cellgro | Cat# 10-080-CV |
| Iscove’s Modified Dulbecco’s Medium (IMDM) | Thermo Fisher Scientific | Cat# 12440053 |
| N2 Supplement | Invitrogen | Cat# 17502-048 |
| B27 Supplement | Invitrogen | Cat# 15260-037 |
| Primocin | Invitrogen | Cat# NC9141851 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
iPSC line generation and maintenance
All experiments involving the differentiation of human iPSC lines were performed with the approval of the Institutional Review Board of Boston University (protocol H33122). The Human Research Protection Office of Washington University School of Medicine, St. Louis, MO, approved procurement of dermal fibroblasts for reprogramming with written informed consent. The iPSC line SPC2 was derived by reprogramming these fibroblasts from a patient with childhood interstitial lung disease (chILD) that progressed to end stage lung disease with pulmonary fibrosis requiring lung transplantation (clinical details provided in the supplemental data). Gene sequencing documented a heterozygous SFTPC pathogenic variant in the non-BRICHOS domain, c.218 T > C (p.I73T) (Figure 1). Reprogramming to generate iPSCs was performed using the excisable, floxed lentiviral STEMCCA vector followed by excision of the single integrated lentiviral copy via transient Cre-recombinase exposure and confirmation of successful STEMCCA excision (SPC2-18Cr1 clone; SFTPCI73T/WT) prior to directed differentiation, as previously published (Somers et al., 2010). We have previously published partial characterization of the corrected SPC2 line (SPC2-ST-B2), without disease modeling, including stability of the AEC2 phenotype in the corrected iAEC2s derived from this clone after prolonged cell culture expansion (Hurley et al., 2020). The SPC6 and SPC7 iPSC lines were generated by reprogramming peripheral blood mononuclear cells (PBMCs) from two individuals with the SFTPCI73T variant and clinically phenotyped chILD (clinical details provided in the supplemental data) using the CytoTune-iPS 2.0 Sendai Reprogramming Kit (Invitrogen, Thermofisher, Cat# A16517) according to the manufacturer’s instructions, with detailed methods available for free download from the CReM’s protocols page at https://www.bumc.bu.edu/stemcells. The Institutional Review Board of Boston University approved procurement of these PBMCs and reprogramming into iPSCs with written informed consent.
All iPSC lines (SPC2-ST-B2, SPC2-ST-C11, SPC2-18Cr1, SPC2-CorrC1, SPC6, and SPC7) used in this study demonstrated a normal karyotype (46XY or 46XX) when analyzed by G-banding and/or array Comparative Genomic Hybridization (aCGH) both before and after gene-editing (Cell Line Genetics) (Figure S6A). Pluripotency was confirmed by staining for pluripotency markers (Figure S6B) as previously described (Somers et al., 2010). Culture conditions used for maintenance and editing of undifferentiated iPSCs were as follows: for Transcription activator-like effector nucleases (TALEN) targeting, iPSC lines were maintained on mitomycin C-inactivated mouse embryonic fibroblasts (MEFs) in human iPSC media (WiCell feeder dependent protocol). For CRISPR targeting and prior to directed differentiation, all iPSC lines were maintained in feeder-free conditions, on growth factor reduced Matrigel (Corning) in 6-well tissue culture dishes (Corning), in mTeSR1 media (StemCell Technologies) using gentle cell dissociation reagent (GCDR) for passaging. Further details of iPSC derivation, characterization, and culture are available for free download at https://www.bu.edu:443/dbin/stemcells/protocols.php.
Generation of gene-edited syngeneic iPSC lines
To generate gene-edited corrected syngeneic iPSC lines two approaches were undertaken. In the first approach, we used TALENs to insert a tdTomato fluorescent reporter at the translation initiation (ATG) site of the endogenous SFTPC locus of the parental SPC2 iPSC line, as previously described (Jacob et al., 2017). All TALENs and donor template plasmid maps and sequences are available at www.kottonlab.com. The SFTPC TALEN recognition sequences are: left TALEN 5′-TAG CAC CTG CAG CAA GAT GG-3′ and right TALEN 5′-TCA CCG GCG GGC TCT CCA TC-3′. Between the two binding sites is a 22 bp spacer (ATG TGG GCA GCA AAG AGG TCC T). TALENs were constructed using EZ-TAL TALE Assembly Kit (System Bioscience), according to manufacturer’s instructions, and the resulting SFTPC TALENs encoding plasmids were named EF1a-TALEN_NN (SPC left) and EF1a-TALEN_HD (SPC right), respectively.
To deliver the donor template to the SFTPC locus, we generated a donor vector (p1303 DV-SFTPC-tdTomato) containing the tdTomato coding sequence and a floxed PGK promoter-driven puromycin resistance cassette, flanked by left and right arms of homology to the human endogenous SFTPC locus, as previously described (Jacob et al., 2017). The TALENs and donor vector plasmids were co-transfected into the SPC2 iPSC line using a lipofectamine based transfection protocol. For this purpose, SPC2 iPSCs were plated onto a mitomycin C-inactivated DR4 MEF feeder layer and cultured in human iPSC media (WiCell) in a 6-well plate. After the cells reached 50% confluence, they were transfected with the two TALENs and tdTomato donor vector as follows: 3 μg of donor vector and 1.2 μg of each TALEN were added to 275 μl of IMDM and 4 μl of Plus reagent from the Lipofectamine LTX kit (Thermo Fisher Scientific) and the mixture was incubated at room temperature for 5 minutes. 16 μl of lipofectamine LTX from the same kit was added to another 275 μl of IMDM. 275 μL of the DNA mixture was added to 275 μL of the LTX mixture and incubated at room temperature for 30 minutes. 550 μl of the total mixture was added drop by drop to 1 well of a 6-well plate. Five hours later, the media was changed, and 48 hours later, 0.7 μg/ml puromycin (Thermo Fisher Scientific) was added to the media for 4 days to select antibiotic resistant colonies. After 10 days individual colonies were picked and screened for targeting using the following primer pairs: 5′ GGG TGA GTG AGC TGA TTC GAG 3′, 5′ TGA CCT CCT CGC CCT TGC TCA CCA TG 3′. DNA sequencing using a primer binding in the genome outside any regions included in targeting plasmids confirmed targeting into the endogenous SFTPC locus (5′ GGG TGA GTG AGC TGA TTC GAG 3′). To confirm heterozygous targeting, colonies were screened for a remaining intact SFTPC allele using the following primers: 5′ CTA CGG ACA CAT ATA AGA CCC TGG TC 3′, 5′ CAA ATA GCC ACC ATT CGG ACA CTC 3′. DNA sequencing of the amplified portion of the intact SFTPC allele was performed using the following primer 5′ TAT GGG GAT GGG TAC CAC TGG CTG 3′ to pick colonies of either corrected (SPC2-ST-B2 clone; SFTPCtdT/WT) or mutant (SPC2-ST-C11 clone; SFTPCI73T/tdT) iPSC clones as the tdTomato cassette is followed by a stop/polyA cassette, preventing expression of the subsequent SFTPC coding sequence from the targeted allele (Figure 1D).
Cre-mediated excision of the floxed puromycin resistance cassette was performed using a plasmid containing Cre-recombinase and neomycin resistance (PHAGE2 EF1a-Cre-IRES-NeoR-W; www.kottonlab.com) using the same lipofectamine-based protocol described above, with 4 days of 200 ng/μl geneticin-based (Life Tech) selection for clones that were transfected with Cre-containing plasmid. Excision of the puromycin cassette was confirmed by PCR using the following primers: 5′ ATG ACC GAG TAC AAG CCC ACG 3′, 5′ TCA GGC ACC GGG CTT GC 3′.
In the second approach, we employed the CRISRP/Cas9 gene editing technology to generate a gene-edited corrected iPSC line that expressed two active SFTPC alleles (SPC2-CorrC1 clone; SFTPCWT/WT) as described below. Using the CRISPR/Cas9 technology we targeted the region adjacent to the I73T mutation (c.218 T > C (p.I73T)) in the SFTPC gene locus (Figure S1B) with a guide RNA (specifically recognizing the I73T mutation but not wild-type) that had the following sequence: 5′ CAG GTT CTG GAG ATG AGC AC 3′. gRNA sequences were cloned into the pD1321-AD plasmid backbone (DNA 2.0 ATUM) that contains a M-dasher-GFP sequence fused to Cas9. Delivery of this plasmid to iPSCs enabled co-expression of Cas9, gRNA, and a GFP reporter. To accomplish footprint-free correction of the SFTPC variant, a short single stranded DNA oligo (sequence 5′ GGT GGC TCC ATG CCC TTT CCC CAG GTT CTG GAA ATG TCA ATT GGT GCG CCG GAA GCC CAG CAA CGC CTG GCC CTG AGT GAG 3′) was used as a donor template containing the wild-type SFTPC sequence. SPC2 cells maintained in feeder-free conditions in mTeSR1 media were treated with 10 μM Y-27632 (Tocris) for 3 hours, dissociated in GCDR (StemCell Technologies) for 10 minutes at 37°C and counted using a Luna-II Automated Cell Counter (Logos Biosystems, Annandale, VA). Approximately 5×106 cells were centrifuged at 200 g for 5 minutes, resuspended in a mixture containing P3 solution and supplement (P3 Primary Cell 4D-Nucleofector X Kit L, Lonza) as well as 5 μg CRISPR/Cas9 plasmid and 5 μg oligo donor, and nucleofected using program code CB-150 in the 4D Nucleofector X System (Lonza). Nucleofected cells were replated on 4 wells of a Matrigel-coated 6-well plate in mTeSR1 media, and 10 μM Y-27632 was added for 24 hours. After 48 hours, cells were prepared for sorting. They were treated with 10 μM Y-27632 for 3 hours, dissociated into single cell suspension with GCDR for 10–15 minutes at 37°C, centrifuged at 200 g for 5 minutes, resuspended in mTeSR1 media supplemented with 10 μM Y-27632, and filtered through a 30 μm filter (Falcon). GFP+ cells were sorted into recovery media (1 part mTeSR1 and 1 part conditioned mTeSR1 supplemented with 0.7ng/ml FGF2, plus 10 μM Y-27632) using a high-speed cell sorter (MoFlo Legacy, Beckman Coulter). 1×104 GFP+ sorted cells were plated into a 10 cm tissue culture treated dish pre-coated with growth factor reduced Matrigel (Corning). Recovery media was changed every other day for the first 5 days, with 10 μM Y-27632 added for the first 24 hours. After 5 days, mTeSR1 was used to feed the cells daily, and after 10 days, colonies that emerged were of sufficient size for picking for clonal expansion and screening. Individual colonies were picked and screened for correction using the following primers: 5′ GTC GTG GTG ATT GTG GGA GCC CTG 3′, 5′ CTG GGC ATA CCC ACC TGC TGG TAG 3′. DNA sequencing using the following primer 5′ TAT GGG GAT GGG TAC CAC TGG CTG 3′ confirmed correction of the SFTPC variant, resulting in generation of the SPC2-Corr line.
METHOD DETAILS
iPSC directed differentiation into alveolar epithelial type 2 cells (iAEC2s)
To generate iAEC2s, we performed PSC directed differentiation via definitive endoderm into NKX2-1 lung progenitors using methods we have previously described (Hawkins et al., 2017; Jacob et al., 2017, 2019). In brief, cells maintained in mTeSR1 media were differentiated into definitive endoderm using the STEMdiff Definitive Endoderm Kit (StemCell Technologies) for 3 days (day 0–3) and after the endoderm-induction stage, cells were dissociated with GCDR and passaged into 6-well dishes pre-coated with growth factor reduced Matrigel in “DS/SB” anteriorization media, consisting of complete serum-free differentiation media (cSFDM) base as previously described (Jacob et al., 2017, 2019), supplemented with 2 μM Dorsomorphin (“DS”; Stemgent) and 10 μM SB431542 (“SB”; Tocris). For the first 24 hr after passaging, 10 μM Y-27632 was added to the media. After anteriorization in “DS/SB” media for 3 days (day 3–6), cells were cultured in “CBRa” lung progenitor-induction media until day 15 or 16, as we have previously published (Jacob et al., 2017; Serra et al., 2017). “CBRa” media consists of cSFDM base supplemented with 3 μM CHIR99021 (Tocris), 10 ng/ml recombinant human BMP4 (rhBMP4, R&D Systems), and 100 nM retinoic acid (RA, Sigma). On day 15 or 16 of differentiation, live cells were sorted on a high-speed cell sorter (MoFlo Legacy or MoFlo Astrios EQ) to isolate NKX2-1+ lung progenitors based on CD47hi/CD26neg gating (Hawkins et al., 2017). Sorted lung progenitors were resuspended in undiluted growth factor reduced 3D Matrigel (Corning) at a concentration of approximately 400 cells/μl and distal/alveolar differentiation of cells was performed in “CK+DCI” media, consisting of cSFDM base supplemented with 3 μM CHIR99021, 10 ng/mL rhKGF (CK), and 50 nM dexamethasone (Sigma), 0.1 mM 8-Bromoadenosine 30,50-cyclic monophosphate sodium salt (Sigma) and 0.1 mM 3-Isobutyl-1-methylxanthine (IBMX; Sigma) (DCI). The resulting epithelial spheres were passaged without further sorting on approximate day 30 (day 28–37) of differentiation and a brief period (4–5 days) of CHIR99021 withdrawal followed by one week of CHIR99021 addback was performed (Figure 1F) to achieve iAEC2 maturation, as previously shown (Jacob et al., 2017). After this 14-day period, SFTPCtdTomato+ cells were purified by fluorescence activated cell sorting (FACS) and resuspended in undiluted growth factor reduced 3D Matrigel (Corning) at a concentration of approximately 400 cells/μl in “CK+DCI” media to establish pure long-term iAEC2s cultures. iAEC2s were then maintained through serial passaging as self-renewing monolayered epithelial spheres (“alveolospheres”) by plating in 3D Matrigel (Corning) droplets at a density of 400 cells/μl with refeeding every other day with “CK+DCI” media, according to our previously published protocol (Jacob et al., 2019). iAEC2 culture quality and purity was monitored at each passage by flow cytometry, with > 95% of cells expressing SFTPCtdTomato over time (Figure 2A).
Murine model of SftpcI73T
Tamoxifen inducible SftpcI73T mice were generated and induced following previously reported protocols (Katzen et al., 2019; Nureki et al., 2018). Briefly, the SftpcI73T founder line, expressing a neomycin cassette, was crossed with a mouse line expressing an estrogen receptor (ER)-2 controlled Flp-O recombinase strain knocked into the Rosa26 locus (Jackson Laboratory, Bar Harbor, ME) to generate the inducible-SftpcI73TFlp line. Adult homozygote SftpcI73TFlp mice received tamoxifen (260 or 360 mg/kg in corn oil, oral gavage) at 12–14 weeks of age. AEC2 isolation was performed 14 days after tamoxifen induction.
Mouse AEC2 isolation
Mouse AEC2s were isolated as previously reported (Atochina-Vasserman et al., 2011). Briefly, perfused mouse lungs were digested with dispase (BD Biosciences), mechanically dissociated, and treated with 25 μg/ml DNAase I (Sigma-Aldrich). Negative selection of mesenchymal cells (fibroblasts) was done by differential adherence on plastic culture dishes. CD45-positive cells were removed by negative selection using Dynabeads Untouched Mouse T Cells Kit (11413D) and Dynabeads Mouse DC Enrichment Kit (11429D; Thermo Fisher Scientific).
Flow cytometry and Fluorescence Activated Cell Sorting (FACS)
Preparation of single cell suspensions of 3D Matrigel-embedded iAEC2s for flow cytometry and FACS was achieved by incubation with 2 mg/ml dispase (Thermo Fisher Scientific) for 30–60 minutes at 37°C and subsequent incubation with 0.05% trypsin for 12–15 minutes at 37°C, as previously described (Jacob et al., 2019). Cells were washed with media containing 10% fetal bovine serum (FBS, Thermo Fisher Scientific). Harvested cells were centrifuged at 300 g for 5 minutes at 4°C and resuspended in FACS buffer containing Hank’s Balanced Salt Solution (Thermo Fisher Scientific), 2% FBS and 10 μM Y-27632 (Tocris) and stained with calcein blue AM (Thermo Fisher Scientific) for dead cell exclusion during flow cytometry. Live cells were sorted on a high-speed cell sorter (MoFlo Legacy or MoFlo Astrios EQ, Beckman Coulter, Pasadena, CA) at the Boston University Medical Center Flow Cytometry Core Facility.
Immunostaining of cells in single cell suspension with antibodies against CD47 (Biolegend) and CD26 (Biolegend) and FACS gating of the CD47hi/CD26neg population for purification of lung progenitors was performed as previously described (Hawkins et al., 2017; Jacob et al., 2019). Purification of iAEC2s for the establishment of pure long-term iAEC2 cultures following iAEC2 maturation (via CHIR99021 withdrawal and addback) in the iPSC lines that had not been targeted with any fluorochrome reporters was achieved by staining for carboxypeptidase M (CPM) (SPC2–18Cr1 and SPC2-CorrC1) or SLC34A2 (SPC6 and SPC7) as detailed in Jacob et al. (2019): single cell suspicions were stained on ice in FACS buffer using a primary mouse monoclonal antibody against human CPM (1:200, Fujifilm Wako) or SLC34A2 (MX35 1:200, a generous gift of Dr. Gerd Ritter, Memorial Sloan Kettering) for 30 minutes and subsequently stained with secondary Alexa Fluor 647 conjugated antibody (1:500, Thermo Fisher Scientific) for 20 minutes.
Gating was based on isotype-stained controls or in the case of SFTPCtdTomato on non-lung endoderm outgrowths (day 15–16 sorted CD47lo cells). Flow cytometry staining was quantified using the Stratedigm S1000EXI and analyzed with FlowJo v10.6.2 (FlowJo, Tree Star Inc). Flow cytometry plots shown represent single cells after forward-scatter/side-scatter gating to remove debris as we have previously detailed in Jacob et al. (2019).
Immunofluorescence microscopy
Images of cultured 3D Matrigel-embedded iPSC-derived epithelial spheres were taken on a Keyence BZ-X700 fluorescence microscope (Osaka, Japan). Z stack images were processed using full focus image analysis using Keyence software.
For the analysis of lysosomal, mitochondrial, and lipid droplet morphology, alveolospheres were harvested by incubating with 2 mg/ml dispase (Thermo Fisher Scientific) for 30–60 minutes at 37°C and then stained with Hoechst 33432 (1 μg/ml, Thermo Fisher Scientific) and LysoTracker Green DND-26 (1 μM, Thermo Fisher Scientific), MitoTracker Green (200 nM, Thermo Fisher Scientific), or BODIPY-493/503 (1 μg/ml, Thermo Fisher Scientific) respectively for 45 minutes in CK+DCI media at 37°C and 5% CO2. After 2 washes with fresh media, alveolospheres were plated onto Greiner CELLview glass-bottom 4-compartment dishes (Greiner Bio-One GmbH) in 3D Matrigel (Corning) and CK+DCI media. Alveolospheres were imaged using a Zeiss LSM 880 confocal microscope (Carl Zeiss AG, Oberkochen) with Airyscan super resolution detector using a 40x or 63x oil objective. For all samples Hoechst fluorescence, green fluorescence (LysoTracker, MitoTracker, or BODIPY-493/503), as well as tdTomato fluorescence were recorded. For the duration of the imaging session alveolospheres were kept on a temperature and CO2 controlled humidified stage at 37°C and 5% CO2.
For the analysis of lysosomal pH, alveolospheres were stained with LysoSensor Yellow/Blue Dextran (5 mg/ml, Thermo Fisher Scientific) for 1 hour in CK+DCI media at 37°C and 5% CO2. Subsequently, after 2 washes with fresh media alveolospheres were plated onto Greiner CELLview glass-bottom 4-compartment dishes in 3D Matrigel and CK+DCI media. After a 24-hour chase period alveolospheres were imaged using a Zeiss LSM 880 using 2-photon excitation at 720 nm and a 40x oil objective. LysoSensor fluorescence was recorded as blue emission (401–461 nm) and yellow emission (511–550 nm). Additionally, tdTomato fluorescence was recorded using 561 nm laser excitation and 571–620 nm emission. For the duration of the imaging session alveolospheres were kept on a temperature and CO2 controlled humidified stage at 37°C and 5% CO2. For the determination of pH standard curves, alveolospheres stained with LysoSensor Yellow/Blue Dextran were fixed in 4% paraformaldehyde for 20 minutes. After 3 washes with PBS (pH 7.4), alveolospheres were equilibrated in MES buffers (pH range between 4.5 and 6.0). LysoSensor fluorescence was recorded as above.
For formalin fixation and paraffin embedding, alveolospheres were harvested by incubating with 2 mg/ml dispase (Thermo Fisher Scientific) for 30–60 minutes at 37°C, fixed with fresh 4% paraformaldehyde overnight at 4°C, combined in low melting agarose (SeaPrep) to form a pellet and then paraffin embedded. For immunocytochemistry, sections were rehydrated and antigen retrieval was performed at 95°C for 20 minutes in Dako Target Retrieval Solution (DAKO, S-1699). Sections were permeabilized with 0.5% Triton X-100 (Sigma-Aldrich), blocked with 4% normal donkey serum (NDS) for 1 hour, and incubated in primary antibody in 0.5% Triton X-100 and 4% NDS overnight at 4°C. The staining was detected with secondary Alexa Fluor-conjugated anti-IgG antibodies purchased from Jackson Immunoresearch (donkey anti-rabbit, goat anti-mouse, goat anti-rabbit at 1:400 to 1:500 dilution for 1 hour at room temperature) or Bio-Rad (goat anti-mouse, goat anti-rabbit at 1:10,000 dilution for 1 hour at room temperature) (see Key resources table), as previously described (Hawkins et al., 2015, 2017; Nureki et al., 2018). Nuclear counterstaining was performed with Hoechst dye (1:500, Invitrogen) or DAPI (300 nM, Invitrogen). Coverslips with cells were mounted on glass slides using Prolong Diamond Anti-Fade Mounting Reagent (Thermo Fisher Scientific) and imaged on an Olympus I-70 inverted fluorescent microscope (Olympus, Melville, NY), captured with a Hamamatsu 12-bit coupled-charged camera (Hamamatsu, Japan), and processed using Metamorph, Version 7.8.4.0 (Universal Imaging, West Chester, PA) or imaged using a Zeiss LSM 880 confocal microscope (Carl Zeiss AG, Oberkochen) and processed using ImageJ. Primary antibodies (see Key resources table) include: a polyclonal anti-pro-SFTPC antibody (“NPRO-SFTPC,” 1:200) raised against the Met [10]–Glu [23] domain of rat pro-SFTPC and polyclonal anti-SFTPB (“PT3”) raised against bovine SFTPB both produced in rabbits in house and validated as previously published (Beers et al., 1992, 1994; Beers and Lomax, 1995), as well as a rabbit polyclonal anti-CRB3 antibody (1:1000; Szymaniak et al., 2015). Other commercially obtained antibodies include: anti-activated caspase 3 (rabbit polyclonal, 1:200, Sigma-Aldrich), anti-E-Cadherin (mouse monoclonal, 1:500, BD Biosciences), anti-EPCAM (mouse monoclonal, 1:100, Santa Cruz), anti-HA (rabbit polyclonal, 1:40,000, Abcam), anti-p-ERM (rabbit monoclonal, 1:1000, Cell Signaling), anti-pro-SFTPC (rabbit polyclonal, 1:500, Seven Hills and mouse monoclonal, 1:500, Santa Cruz), anti-MUC1 (rabbit monoclonal, 1:100, Abcam), anti-RAGE (mouse monoclonal, 1:250, Abcam).
Cell proliferation analysis
To assess DNA replication as a method of proliferation analysis, 5-ethynyl-2′-deoxyuridine (EdU) incorporation was evaluated using the Click-iT EdU Alexa Fluor 647 Flow Cytometry Assay Kit (Invitrogen). Corrected and mutant SFTPCtdTomato+ iAEC2s were plated at a concentration of 400 cells/μl in 3D Matrigel in CK+DCI media as described above. Seven days after plating, EdU was added to the cultures (n = 3 independent droplets) for 24 hours at a final concentration of 10 μM. The cells were then dissociated into single cell suspension as described above, fixed, permeabilized, and stained with the Click-iT reaction mixture according to the manufacturer’s instructions. The percentage of SFTPCtdTomato+ cells that incorporated EdU was determined by flow cytometry (Stratedigm).
Transmission electron microscopy
Intact alveolospheres were harvested by incubating with 2 mg/ml dispase (Thermo Fisher Scientific) for 30–60 minutes at 37°C and fixed for 3 hours total in 2.5% glutaraldehyde (Ladd Research) in 0.1% cacodylate buffer (pH 7.4) at room temperature as follows: an equal volume of 5% glutaraldehyde/ 0.1 M cacodylate was added to Eppendorf tubes containing the alveolospheres in known volume of CK+DCI media, fixed for 1.5 hours, and centrifuged gently (300 g for 1 minute) followed by the addition of fresh 2.5% glutaraldehyde/ 0.1 M cacodylate for an additional 1.5 hours at room temperature. The samples were then washed 3 times in 0.1 M cacodylate, post-fixed in 1% Tannic Acid in cacodylate buffer for 5 minutes at room temperature, washed again 3 times in cacodylate buffer, and post-fixed overnight in 1.5% osmium tetroxide (Polysciences) in 0.1 M cacodylate buffer in dark at 4°C. The samples were then washed 3–4 times in 0.05 M Na Maleate buffer (pH 5.2) and block stained in 1.5% Uranyl acetate (Electron Microscopy Sciences, EMS) in 0.025 M Na Maleate buffer (pH 6.0). Next, the samples were dehydrated quickly through acetone on ice, from 70% to 80% to 90%. The samples were then incubated 2 times in 100% acetone at room temperature for 10 minutes each, and in propylene oxide at room temperature for 15 minutes each. Finally, the samples were changed into EMbed 812 (EMS), left for 2 hours at room temperature, changed into fresh Embed 812 and left overnight at room temperature, after which they were embedded in fresh EMbed 812 and polymerized overnight at 60°C. Plastic embedded samples were thin sectioned at 70 nm and grids were stained in 4% aqueous Uranyl Acetate for 5 minutes at 60°C followed by Lead Citrate for 10 minutes at room temperature. Electron microscopy was performed on a Philips CM12 EM operated at 100kV, and images were recorded on a TVIPS F216 CMOS camera with a pixel size of 0.85–3.80 nm per pixel.
Other staining
Toluidine blue staining was performed as follows: 0.5 μm sections (from EMbed plastic blocks) were collected, dried for 30 minutes on a hot plate, and stained 30–60 s in 0.5% Toluidine blue + 0.5% Borax in dH20, rinsed in dH20, dried and coverslipped. For Hematoxylin and Eosin (H&E) staining standard methods were used (AML Labs, Inc, Baltimore, MD).
Quantification of autophagic flux
To quantify autophagosomes, corrected (SFTPCtdT/WT) or mutant (SFTPCI73T/tdT) iAEC2s were transduced with an LC3:GFP encoding lentivirus (Twig et al., 2008) at an MOI of 20 in CK+DCI media containing polybrene (5 mg/ml). iAEC2s were then replated in growth factor reduced 3D Matrigel (Corning) at a concentration of approximately 400 cells/μl in CK+DCI media for expansion. Single cell suspension of transduced iAEC2s was achieved by incubation with 2 mg/ml dispase (Thermo Fisher Scientific) for 30–60 minutes at 37°C and subsequent incubation with 0.05% trypsin for 12–15 minutes at 37°C, as previously described (Jacob et al., 2019). Transduced iAEC2s were plated onto Greiner CELLview glass-bottom 4-compartment dishes (Greiner Bio-One GmbH) pre-coated with growth factor reduced Matrigel (Corning) in CK+DCI media. After 3 days, iAEC2s were imaged at 37°C and 5% CO2, using a Zeiss LSM 710-Live Duo Scan confocal microscope (Carl Zeiss AG, Oberkochen) (488 nm excitation), and GFP+ punctate autophagosomes were quantified by visual inspection in 40–50 cells per group before and after 1 hour of incubation with 50 nM bafilomycin A1 (LC Laboratories). LC3 and p62 were further quantified by western blot as described below.
Western Blot Analysis
Alveolospheres were harvested by incubating with 2 mg/ml dispase (Thermo Fisher Scientific) for 30–60 minutes at 37°C, treated with lysis buffer (RIPA buffer and 1x Roche Complete Protease Inhibitor cocktail), and incubated on ice for 30 minutes. Cellular debris was cleared by centrifugation at 15,000 g for 20 minutes and supernatants were harvested. Protein concentration was measured using Bio-Rad DC Protein Assay. 5–30 μg of lysates were resolved on pre-cast 12% Bis-Tris NuPAGE gels (Invitrogen), transferred to PVDF membranes (Bio-Rad), and blotted with primary antibodies (see below and Key resources table) followed by species-specific secondary antibodies conjugated to IR dyes of either 680 or 800 nm wavelengths (Rockland) at a dilution of 1:10000, as previously described (Hawkins et al., 2015; Maguire et al., 2012). Primary antibodies (see Key resources table) include: surfactant protein B (PT3, a rabbit polyclonal antibody against bovine mature SFTPB; [Beers et al., 1992]; 1:5000), pro-SFTPC (NPRO-SFTPC, a rabbit polyclonal antibody against the Met [10]–Glu [23] domain of rat proSFTPC; [Beers et al., 1994]; 1:3000), mature SFTPC (1:2500, Seven Hills), LC3B (1:750–1:1000, Cell Signaling), SQSTM1/p62 (1:1000, Abcam), β-actin (1:10000, Sigma), and GAPDH (1:5000, Chemicon). Visualization was accomplished using the Odyssey Imaging System (LiCOR Biosciences).
Respirometry assays
Alveolospheres were harvested by incubating with 2 mg/ml dispase (Thermo Fisher Scientific) for 30–60 minutes at 37°C, washed, and resuspended in 150–300 μl of Seahorse XF Base Media Minimal DMEM (Agilent Technologies) containing 2.8 mM glucose and 0.1% FBS (pH 7.4). Organoids were seeded in an XF96e Seahorse plate as described (Assali et al., 2019; Taddeo et al., 2020). Briefly, growth factor reduced Matrigel (1.5 μl/well; Corning) was first pipetted in the center measurement zone of each well. The organoid suspension (5–10 μl) was deposited in the Matrigel-coated measurement zone using a pipette. The plate was then incubated in a non--CO2 incubator for 3.5 minutes to let the Matrigel solidify. Next, 150 μl of pre-warmed Seahorse media was slowly added to each well to avoid dislodging the organoids from the Matrigel. The plate containing the organoids was centrifuged at 500 g for 5 minutes with no brake and subsequently incubated in a non-CO2 incubator for 30–45 minutes prior to running the assay. Oxygen consumption was determined as described (Assali et al., 2019; Taddeo et al., 2020). Port injections were as follows: port A, oligomycin (final concentration 4.5 μM/l); port B, FCCP diluted in a mixture of 80% sodium pyruvate and 20% of 1:1 l-leucine/l-glutamine (final concentration 1 μM/l FCCP in 11.4 mM/l sodium pyruvate and 2.9 mM/l each of leucine/glutamine); and port C, antimycin A (final concentration 2.5 μM/l). Once the Seahorse assay was complete, the plate was kept for mitochondrial content determination. Protein concentration was determined by BCA assay (Thermo Fisher Scientific).
Mitochondrial content determination
Mitochondrial content was assessed using MitoTracker Deep Red (MTDR) as described (Acin-Perez et al., 2020). Briefly, samples (5 μg) were incubated in the presence of 500 nM MTDR for 10 minutes in PBS at 37°C. The plate was centrifuged at 2000 g for 10 minutes and media containing MTDR was discarded using a pipette. Then, 100 μl of PBS was added per well. MTDR fluorescence was measured in a fluorescence plate reader (Tecan, Männedorf, Switzerland) with excitation of 625 nm and emission of 670 nm. Mitochondrial content was referred as MTDR RFU/μg protein.
Quantification of NF-κB signaling
To track the kinetics of the NF-κB pathway in iAEC2s, we utilized a lentiviral vector (lenti-NF-κB-luc-GFP) we have previously published (Wilson et al., 2013) that allows independent simultaneous tracking of transduced (GFP+) cells by flow cytometry and assessment of NF-κB activation levels by luciferase expression. This dual transgenesis vector contains four tandem copies of the canonical NF-κB p50/p65 heterodimer consensus binding sequence located upstream of the minimal TA promoter (TAp), the TATA box of the herpes simplex virus thymidine kinase (HSV-TK) promoter. Located downstream of TAp is the firefly luciferase reporter coding sequence, followed by a constitutively active mammalian ubiquitin C (UBC) promoter driving GFP expression (Figure 7B). Corrected (SFTPCtdT/WT) or mutant (SFTPCI73T/tdT) iAEC2s were transduced with the lentiviral vector at an MOI of 20 in CK+DCI media containing polybrene (5 mg/ml). iAEC2s were then replated in growth factor reduced 3D Matrigel (Corning) at a concentration of approximately 400 cells/μl in CK+DCI media. Thirteen days later, corrected and mutant iAEC2s (n = 3 independent droplets) were dissociated into single cells and sorted for transduced (GFP+) SFTPCtdTomato+ cells which were subsequently resuspended in 75 μl FACS buffer and plated in a 96-well flat bottom black polystyrene microplate (Corning). Cells were then processed according to the Dual-Glo Luciferase Assay System Protocol (Promega). Firefly luminescence was measured using the Tecan Infinite M200 Pro Micro Plate Reader (Tecan, Männedorf, Switzerland). NF-κB target protein levels were further quantified by Luminex analysis as described below.
Luminex analysis
Supernatants of corrected and mutant iAEC2s (n = 3 independent wells) were filtered through 0.22 μm filters (Millipore SCGP00525) and analyzed using a customized Magnetic Luminex® Performance Assay Human HighSensitivity Cytokine Base Kit (R&D Systems, Inc). Mean fluorescence intensity was measured to calculate final concentration in pg/ml using Bioplex200 and Bioplex Manager 5 software (Bio-Rad) and adjusted to cell number.
Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
RNA was extracted by first lysing cells in Qiazol (QIAGEN) and subsequently using RNAeasy mini kit (QIAGEN) according to the manufacturer’s protocol. Complementary DNA (cDNA) was generated by reverse transcription of 150 ng RNA from each sample using MultiScribe Reverse Transcriptase (Applied Biosystems). RT-qPCR was performed using TaqMan Fast Universal PCR Master Mix (Thermo Fisher Scientific) and TaqMan (Applied Biosystems) reagents. The cDNA was diluted 1:4 and 2 μL of cDNA were added to each 20 μL (for Applied Biosystems StepOne 96-well System) or 12 μL (for Applied Biosystems QuantStudio7 384-well system) RT-qPCR reaction. Each sample was run in technical triplicates for 40 cycles of PCR and cycle threshold (Ct) values were averaged between triplicates for analysis. Relative gene expression, normalized to 18S control, was calculated as fold change in 18S-normalized gene expression, compared to baseline, using the 2(−ΔΔCt) method. Baseline expression, defined asfold change= 1, was set to undifferentiated iPSC levels, or if undetected, a cycle number of 40 was assigned to allow fold change calculations. Adult human lung control RNA was extracted from a healthy donor’s distal lung explant. Specifics of primers used are detailed in Key resources table and Table S3.
Isolation of Human Fetal Lung Epithelium
Week 21 human lung tissues were obtained in the Guttentag laboratory under protocols originally reviewed by the Institutional Review Board at the Children’s Hospital of Philadelphia and subsequently reviewed by Vanderbilt University. The cell stocks used in the present studies were donated to the Kotton laboratory for the purpose of providing reference data for this project and their characterization was previously published (Hawkins et al., 2017). “Uncultured naïve lung epithelial cells” were isolated by the overnight culture of lung explants in Waymouth media; a technique that generally yields 86 ± 2% epithelial cells with the remaining cells consisting of fibroblasts with < 1% endothelial cells (Wade et al., 2006).
RNA sequencing and bioinformatic analyses
For bulk RNA sequencing (RNA-seq) comparing corrected (SFTPCtdT/WT) and mutant (SFTPCI73T/tdT) iAEC2s at two different time points (day 114 and day 131), experimental triplicate (n = 3) samples of purified RNA extracts were harvested from each group of samples as follows. Alveolospheres were harvested from 3D Matrigel by incubating with 2 mg/ml dispase (Thermo Fisher Scientific) for 30–60 minutes at 37°C, washed twice with PBS, and RNA was extracted using the Qiazol and miRNeasy mini kit (QIAGEN) according to manufacturer’s instructions. Flow cytometry analysis confirmed that these samples consisted of ≥ 95% SFTPCtdTomato+ cells. mRNA was isolated from each sample using magnetic bead-based poly(A) selection, followed by synthesis of cDNA fragments. The products were end-paired and PCR-amplified to create each final cDNA library. Sequencing of pooled libraries was done using a NextSeq 500 (Illumina). The quality of the raw sequencing data was assessed using FastQC v.0.11.7. Sequence reads were aligned to a combination of the human genome reference (GRCh38) and the tdTomato reporter sequence, using STAR v.2.5.2b (Dobin et al., 2013). Counts per gene were summarized using the featureCounts function from the subread package v.1.6.2. The edgeR package v.3.25.10 was used to import, organize, filter and normalize the data. Genes that were not expressed in at least one of the experimental groups were filtered out (keeping only genes that had at least 10 reads in at least 3 libraries, that is, a worthwhile number of samples as determined by the replicate number in the design matrix. The TMM method was used for normalization. Principal Component Analysis (PCA) and Multidimensional Scaling (MDS) were used for exploratory analysis, to assess sample similarities and potential batch effects. Subsequently, the limma package v3.39.19 (Law et al., 2016) with its voom method, namely, linear modeling and empirical Bayes moderation was used to test differential expression (moderate t tests). P values were adjusted for multiple testing using Benjamini-Hochberg correction (false discovery rate-adjusted p value; FDR). Differentially expressed genes between the groups in each experiment were visualized using Glimma v 1.11.1, and FDR < 0.05 was set as the threshold for determining significant differential gene expression. Gene set analysis was performed with Hallmark, KEGG, and GO Biological Process gene sets using the Camera package (Wu and Smyth, 2012).
For single-cell RNA sequencing (scRNA-seq) comparing corrected (SFTPCtdT/WT) and mutant (SFTPCI73T/tdT) iAEC2s at two different time points (day 30 and day 113), cells were dissociated for sorting as described previously. Cells were sorted as described previously for viable (calcein blue+) SFTPCtdTomato+ cells and resuspended in FACS buffer at a concentration of 800–1000 cells/μl. For the scRNA-seq experiment comparing corrected and mutant iAEC2s on day 114 (Figure S3), all live cells (calcein blue+) irrespective of SFTPCtdTomato expression were included. Single cells were captured for sequencing library preparation using a 10X Chromium (10X Genomics, Pleasanton, CA) instrument. scRNA-seq libraries were prepared according to the Single Cell 3′ v3 Reagent Kits User Guide (10X Genomics). Cellular suspensions were loaded on a Chromium Controller instrument (10X Genomics) to generate single-cell Gel Bead-In-Emulsions (GEMs). Reverse transcription (GEM-RT) was performed in a Veriti 96-well thermal cycler (Thermo Fisher Scientific). After RT, GEMs were harvested and the cDNAs were amplified and cleaned with SPRIselect Reagent Kit (Beckman Coulter). Indexed sequencing libraries were constructed using the Chromium Single-Cell 3′ Library Kit (10X Genomics) for enzymatic fragmentation, end-repair, A-tailing, adaptor ligation, ligation cleanup, sample index PCR, and PCR cleanup. The barcoded sequencing libraries were quantified by quantitative PCR using the KAPA Library Quantification Kit (KAPA Biosystems, Wilmington, MA). Sequencing libraries were loaded on a NextSeq500 (Illumina) with a custom sequencing setting (26bp for Read 1 and 98bp for Read 2), to obtain a sequencing depth of ~50K reads/cell. Fastq files were generated using bcl2fastq v.2.2 and Cellranger v.3.0.2. The sequence files were mapped to the human genome reference (GRCh37) including the TdTomato reporter. We used Seurat v.3 to further process the data. We estimated the doublet rate according to the 10X Chromium guidelines, in proportion to the density of cells loaded. These rates were used to flag potential doublets based on their gene and UMI counts. Cells with fewer than 800 genes detected were also filtered out, as well as cells with high percentage of counts mapping to mitochondrial genes (thresholds set by manual inspection for each dataset, between 25% to 35%). For each experiment, the mutated and corrected samples were merged and then normalized using SCTransform, with cell degradation effect regressed out. After an initial linear dimensionality reduction (PCA), we used UMAP projections to represent the data and the Louvain algorithm was used for clustering. Differential expression tests were done with MAST, applying independent prior to gene testing (minimum detection percentage of 10% in at least one of the populations, minimum average log-fold change of 0.15 between the two populations). Gene signature enrichment scores for iAT2 markers and cell cycle stage were computed using Seurat. The significance of the differences in these scores between corrected and mutant samples was tested using Welch Two Sample t tests. The scRNA-seq data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (GEO; accession number GSE160801).
Proteomic and phosphoproteomic analysis
Cell lysis and trypsin digestion
For proteomic/phosphoproteomic analyses experimental triplicate (n = 3) samples were dissociated for sorting as described previously. Cells were sorted as described previously for viable (calcein blue+) SFTPCtdTomato+ cells, washed with PBS, pelleted, and stored at −80°C. Cell pellets were thawed and individually placed in 300 μl of lysis buffer composed of 8 M guanidinium chloride (GuHCl), 50 mM ammonium bicarbonate, 5 mM dithiothreitol (DTT), phosphatase (PhosSTOP, Roche) and protease (Complete, Mini, EDTA-free Protease Inhibitor Cocktail, Roche) inhibitors and then sonicated on ice using a Branson probe (40 s, in 2 s pulses). Lysates were alkylated in 12 mM iodoacetamide. Protein concentration was quantified by the Bradford protein assay (Bio-Rad) and samples were diluted with 7 volumes of 50 mM ammonium bicarbonate to bring the urea concentration to 1 M. Each sample was then digested overnight at 37°C with 2 μg sequencing-grade trypsin (Pierce Trypsin Protease, MS Grade, Thermo Fisher Scientific).
Tandem Mass Tag (TMT) labeling and reserve phase high performance liquid chromatography (LC)
After digestion, the reactions were terminated by adding trifluoroacetic acid (TFA) to 0.1%, de-salted (Waters Sep-Pak), and resuspended in 100 mM HEPES buffer. Total peptide concentration was quantified using the Pierce Quantitative Colorimetric Peptide Assay (Pierce Quantitative Colorimetric Peptide Assay, Thermo Fisher Scientific). Then 100 μg peptide from each sample (volume adjusted to 100 μl with 100 mM HEPES buffer) was mixed with a unique amine-reactive heavy isotope-coded isobaric TMT (TMT-11plex) reagent, allowing for multiplex of samples prior to LC/MS. After pooling, the samples were desalted and dried, and a total of about 0.6 mg of labeled peptide was suspended in 300 μl buffer containing 0.1% ammonium hydroxide and 2% acetonitrile. This mixture was injected onto a reverse-phase Waters Xbridge C18 HPLC column (Waters) connected to an Agilent 1100 series HPLC system. Peptides were eluted from the column using a gradient of mobile phase A (2% ACN, 0.1% NH4OH) to mobile phase B (98% ACN, 0.1% NH4OH) over 48 minutes. Fractions were collected every minute, and every 12th fraction was pooled, yielding 12 final fractions, each containing approximately 100 μg peptide. Most of each fraction (95%) was set aside for phospho-peptide enrichment using Fe-NTA magnetic beads (Cube Biotech) while the remaining 5% was analyzed separately by mass spectrometry as bulk proteome measurements, totaling 24 injections.
Mass Spectrometry (MS)
Fractionated peptide samples were analyzed using a Q Exactive HFX mass spectrometer connected to Easy nLC 1200 ultra high-pressure chromatography system (Thermo Scientific). Lyophilized peptides were resuspended in mobile phase A solvent (0.1% formic acid and 2% acetonitrile). The samples were loaded onto a reverse-phase nano-trap column with mobile phase A, (75 μm i.d. 3 2 cm, Acclaim PepMap100 C18 3 μm, 100Å, Thermo Scientific) and were separated over an EASY-Spray column, (ES803A, Thermo Scientific) using a gradient (2% to 35% over 120 min) of mobile phase B (0.1% formic acid, 80% acetonitrile) at a flow rate of 250 nl/min. The mass spectrometer was operated in positive ion mode with a capillary temperature of 275°C, and with a potential of 2100V applied to the emitter. All data were acquired with the mass spectrometer operating in automatic data dependent switching mode. A high resolution (60,000) MS precursor ion scan (350–1500 m/z range) was performed to select the 12 most intense ions for subsequent fragmentation and MS/MS analysis using HCD (NCE 33 @ 45,000 resolution).
Peptide identification
Resulting RAW files were converted and searched using the MaxQuant platform (version 1.6.7.0; https://maxquant.org/) under standard settings against the Uniprot human reference protein sequence database (downloaded: April 16, 2019) allowing for two missed trypsin cleavage sites, variable modifications: phosphorylation at S, T, and Y residues, N-terminal acetylation, and methionine oxidation. Carbamidomethylation of cysteine residues was set as a fixed modification. Peptide precursor ions were searched with a maximum mass deviation of 4.5 ppm and fragment ions with a maximum mass deviation of 20 ppm. Peptide and protein identifications were filtered at 1% FDR using the target-decoy database search strategy. The MaxQuant output files designated “Phospho(STY)sites” and “ProteinGroups” were used for data normalization and other statistical analysis.
Data analysis and pathway enrichment
Bioinformatic analysis was performed using R: A language and environment for Statistical Computing (R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org), version 3.6.1. The protein groups and phosphosite tables from MaxQuant were filtered to eliminate entries labeled as reverse hits, potential contaminants, and “only identified by site.” Protein quantitation required at values in at least 70% of TMT channels. The TMT-reporter intensity values were log2 transformed and Loess-normalized. For differential analysis, the LIMMA (Ritchie et al., 2015) R package was used to fit a linear model accounting for the cell lines. Moderated t tests were corrected with the Benjamini-Hochberg method for FDR. Gene set enrichment analysis was performed using the fgsea R package (Korotkevich et al., 2019) using the Hallmark (H) and Curated (C2) gene set libraries from MSigDB (Subramanian et al., 2005) with ranked lists where the gene rank is defined as −log(p value)*sign(log2 fold change) (Reimand et al., 2019). In the case of duplicate proteins mapping to a common gene symbol, the one with the highest absolute value rank was retained. Heatmaps were generated with the ComplexHeatmap R package (Gu et al., 2016). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the Proteomics Identifications (PRIDE) partner repository (Deutsch et al., 2020; Perez-Riverol et al., 2019) with the dataset identifier PRIDE: PXD022334.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical methods relevant to each figure are outlined in the figure legend. In brief, unpaired, two-tailed Student’s t tests were used to compare quantitative analyses comprising two groups of n = 3 or more samples, or one-way ANOVAs with multiple comparisons were used to compare three or more groups. Further specifics about the replicates used in each experiment are available in the figure legends. In these cases, a Gaussian distribution and equal variance between samples was assumed as the experiments represent random samples of the measured variable. The p value threshold to determine significance was set at p = 0.05. p value annotations on graphs are as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Data for quantitative experiments are typically represented as the mean with error bars representing the standard deviation or standard error of the mean, as specified in the figure legends.
Statistical analysis on imaged alveolospheres was performed using Prism 7 (GraphPad Software, San Diego, CA). Each organoid was treated as an experimental replicate and unpaired Student’s t test was used to determine statistically significant differences.
Supplementary Material
Highlights.
Patient-specific iAEC2s serve as a human preclinical platform for modeling ILD
Patient-derived iAEC2s expressing mutant SFTPCI73T have diminished progenitor capacity
SFTPCI73T mutant iAEC2s display proteostasis perturbations and metabolic reprogramming
Mutant iAEC2s are an important proinflammatory hub via activation of the NF-κB pathway
ACKNOWLEDGMENTS
The authors wish to thank all members of the Kotton, Beers, Emili, and Shirihai Labs for insightful discussions. We thank Anne Hinds of the Boston University Pulmonary Center for histology technical support. We thank Yuriy Alekseyev of the Boston University School of Medicine (BUSM) Single Cell Sequencing Core, supported by NIH (1UL1TR001430), and Brian R. Tilton of the BUSM Flow Cytometry Core. We are grateful to Greg Miller and Marianne James of the Boston University Center for Regenerative Medicine (CReM) for maintenance and characterization of patient-specific iPSCs, supported by NIH (NO1 75N92020C00005 and U01TR001810). We thank Michael Morley and Edward E. Morrisey of the University of Pennsylvania for access to their bioinformatics portal for analyses of bulk RNA-seq datasets. The graphical abstract, the autophagy pathway schematic, and the mouse and human cartoons were created with BioRender.com. This work was supported by the I.M. Rosenzweig Junior Investigator Award from The Pulmonary Fibrosis Foundation (to K.-D.A.), an IDEAL Consortium grant from Celgene/Bristol Myers Squibb (D.N.K.), the Albert M. Rose Established Investigator Award from the Pulmonary Fibrosis Foundation (to M.F.B.), the Department of Veterans Affairs Merit Review (1I01BX001176 to M.F.B.), and NIH (U01HL148692, U01HL134745, U01HL134766, U01HL152976, and R01HL095993 to D.N.K., U01HL152970 and RO1HL119436 to M.F.B., and K24HL143281 to L.R.Y.). A.E. acknowledges generous startup funding to the operations of the CNSB from Boston University.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109636.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Acin-Perez R, Benador IY, Petcherski A, Veliova M, Benavides GA, Lagarrigue S, Caudal A, Vergnes L, Murphy AN, Karamanlidis G, et al. (2020). A novel approach to measure mitochondrial respiration in frozen biological samples. EMBO J 39, e104073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya J, Kojima J, Takasaka N, Ito S, Fujii S, Hara H, Yanagisawa H, Kobayashi K, Tsurushige C, Kawaishi M, et al. (2013). Insufficient autophagy in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol 304, L56–L69. [DOI] [PubMed] [Google Scholar]
- Assali EA, Shlomo D, Zeng J, Taddeo EP, Trudeau KM, Erion KA, Colby AH, Grinstaff MW, Liesa M, Las G, and Shirihai OS (2019). Nanoparticle-mediated lysosomal reacidification restores mitochondrial turnover and function in β cells under lipotoxicity. FASEB J 33, 4154–4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atochina-Vasserman EN, Bates SR, Zhang P, Abramova H, Zhang Z, Gonzales L, Tao J-Q, Gochuico BR, Gahl W, Guo C-J, et al. (2011). Early alveolar epithelial dysfunction promotes lung inflammation in a mouse model of Hermansky-Pudlak syndrome. Am. J. Respir. Crit. Care Med 184, 449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkauskas CE, and Noble PW (2014). Cellular mechanisms of tissue fibrosis. 7. New insights into the cellular mechanisms of pulmonary fibrosis. Am. J. Physiol. Cell Physiol 306, C987–C996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, Randell SH, Noble PW, and Hogan BLM (2013). Type 2 alveolar cells are stem cells in adult lung. J. Clin. Invest 123, 3025–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers MF, and Lomax C (1995). Synthesis and processing of hydrophobic surfactant protein C by isolated rat type II cells. Am. J. Physiol 269, L744–L753. [DOI] [PubMed] [Google Scholar]
- Beers MF, and Mulugeta S (2005). Surfactant protein C biosynthesis and its emerging role in conformational lung disease. Annu. Rev. Physiol 67, 663–696. [DOI] [PubMed] [Google Scholar]
- Beers MF, Bates SR, and Fisher AB (1992). Differential extraction for the rapid purification of bovine surfactant protein B. Am. J. Physiol 262, L773–L778. [DOI] [PubMed] [Google Scholar]
- Beers MF, Kim CY, Dodia C, and Fisher AB (1994). Localization, synthesis, and processing of surfactant protein SP-C in rat lung analyzed by epitope-specific antipeptide antibodies. J. Biol. Chem 269, 20318–20328. [PubMed] [Google Scholar]
- Beers MF, Hawkins A, Maguire JA, Kotorashvili A, Zhao M, Newitt JL, Ding W, Russo S, Guttentag S, Gonzales L, and Mulugeta S (2011). A nonaggregating surfactant protein C mutant is misdirected to early endosomes and disrupts phospholipid recycling. Traffic 12, 1196–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock C, Kiskinis E, Verstappen G, Gu H, Boulting G, Smith ZD, Ziller M, Croft GF, Amoroso MW, Oakley DH, et al. (2011). Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell 144, 439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borok Z, Danto SI, Lubman RL, Cao Y, Williams MC, and Crandall ED (1998). Modulation of t1alpha expression with alveolar epithelial cell phenotype in vitro. Am. J. Physiol 275, L155–L164. [DOI] [PubMed] [Google Scholar]
- Boulting GL, Kiskinis E, Croft GF, Amoroso MW, Oakley DH, Wainger BJ, Williams DJ, Kahler DJ, Yamaki M, Davidow L, et al. (2011). A functionally characterized test set of human induced pluripotent stem cells. Nat. Biotechnol 29, 279–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasch F, Ten Brinke A, Johnen G, Ochs M, Kapp N, Müller KM, Beers MF, Fehrenbach H, Richter J, Batenburg JJ, and Bühling F (2002). Involvement of cathepsin H in the processing of the hydrophobic surfactant-associated protein C in type II pneumocytes. Am. J. Respir. Cell Mol. Biol 26, 659–670. [DOI] [PubMed] [Google Scholar]
- Brasch F, Griese M, Tredano M, Johnen G, Ochs M, Rieger C, Mulugeta S, Müller KM, Bahuau M, and Beers MF (2004). Interstitial lung disease in a baby with a de novo mutation in the SFTPC gene. Eur. Respir. J 24, 30–39. [DOI] [PubMed] [Google Scholar]
- Braun S, Ferner M, Kronfeld K, and Griese M (2015). Hydroxychloroquine in children with interstitial (diffuse parenchymal) lung diseases. Pediatr. Pulmonol 50, 410–419. [DOI] [PubMed] [Google Scholar]
- Bueno M, Lai Y-C, Romero Y, Brands J, St Croix CM, Kamga C, Corey C, Herazo-Maya JD, Sembrat J, Lee JS, et al. (2015). PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Invest 125, 521–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottin V, Reix P, Khouatra C, Thivolet-Béjui F, Feldmann D, and Cordier J-F (2011). Combined pulmonary fibrosis and emphysema syndrome associated with familial SFTPC mutation. Thorax 66, 918–919. [DOI] [PubMed] [Google Scholar]
- Crossno PF, Polosukhin VV, Blackwell TS, Johnson JE, Markin C, Moore PE, Worrell JA, Stahlman MT, Phillips JA 3rd, Loyd JE, et al. (2010). Identification of early interstitial lung disease in an individual with genetic variations in ABCA3 and SFTPC. Chest 137, 969–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch EW, Bandeira N, Sharma V, Perez-Riverol Y, Carver JJ, Kundu DJ, García-Seisdedos D, Jarnuczak AF, Hewapathirana S, Pullman BS, et al. (2020). The ProteomeXchange consortium in 2020: enabling ‘big data’ approaches in proteomics. Nucleic Acids Res 48 (D1), D1145–D1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster CD, Varghese LS, Skalina RB, Gonzales LW, and Guttentag SH (2007). In vitro transdifferentiation of human fetal type II cells toward a type I-like cell. Pediatr. Res 61, 404–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galetskiy D, Woischnik M, Ripper J, Griese M, and Przybylski M (2008). Aberrant processing forms of lung surfactant proteins SP-B and SP-C revealed by high-resolution mass spectrometry. Eur. J. Mass Spectrom (Chi-chester) 14, 379–390. [DOI] [PubMed] [Google Scholar]
- Garcia CK (2018). Insights from human genetic studies of lung and organ fibrosis. J. Clin. Invest 128, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia O, Hiatt MJ, Lundin A, Lee J, Reddy R, Navarro S, Kikuchi A, and Driscoll B (2016). Targeted Type 2 Alveolar Cell Depletion. A Dynamic Functional Model for Lung Injury Repair. Am. J. Respir. Cell Mol. Biol 54, 319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- Habermann AC, Gutierrez AJ, Bui LT, Yahn SL, Winters NI, Calvi CL, Peter L, Chung M-I, Taylor CJ, Jetter C, et al. (2020). Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv 6, eaba1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins A, Guttentag SH, Deterding R, Funkhouser WK, Goralski JL, Chatterjee S, Mulugeta S, and Beers MF (2015). A non-BRICHOS SFTPC mutant (SP-CI73T) linked to interstitial lung disease promotes a late block in macroautophagy disrupting cellular proteostasis and mitophagy. Am. J. Physiol. Lung Cell. Mol. Physiol 308, L33–L47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins F, Kramer P, Jacob A, Driver I, Thomas DC, McCauley KB, Skvir N, Crane AM, Kurmann AA, Hollenberg AN, et al. (2017). Prospective isolation of NKX2-1-expressing human lung progenitors derived from pluripotent stem cells. J. Clin. Invest 127, 2277–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill C, Li J, Liu D, Conforti F, Brereton CJ, Yao L, Zhou Y, Alzetani A, Chee SJ, Marshall BG, et al. (2019). Autophagy inhibition-mediated epithelial-mesenchymal transition augments local myofibroblast differentiation in pulmonary fibrosis. Cell Death Dis 10, 591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley K, Ding J, Villacorta-Martin C, Herriges MJ, Jacob A, Vedaie M, Alysandratos K-D, Sun YL, Lin C, Werder RB, et al. (2020). Reconstructed Single-Cell Fate Trajectories Define Lineage Plasticity Windows during Differentiation of Human PSC-Derived Distal Lung Progenitors. Cell Stem Cell 26, 593–608.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob A, Morley M, Hawkins F, McCauley KB, Jean JC, Heins H, Na C-L, Weaver TE, Vedaie M, Hurley K, et al. (2017). Differentiation of Human Pluripotent Stem Cells into Functional Lung Alveolar Epithelial Cells. Cell Stem Cell 21, 472–488.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob A, Vedaie M, Roberts DA, Thomas DC, Villacorta-Martin C, Alysandratos K-D, Hawkins F, and Kotton DN (2019). Derivation of self-renewing lung alveolar epithelial type II cells from human pluripotent stem cells. Nat. Protoc 14, 3303–3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Gil de Rubio R, Hrycaj SM, Gurczynski SJ, Riemondy KA, Moore BB, Omary MB, Ridge KM, and Zemans RL (2020). Ineffectual Type 2-to-Type 1 Alveolar Epithelial Cell Differentiation in Idiopathic Pulmonary Fibrosis: Persistence of the KRT8hi Transitional State. Am. J. Respir. Crit. Care Med 201, 1443–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzen J, Wagner BD, Venosa A, Kopp M, Tomer Y, Russo SJ, Headen AC, Basil MC, Stark JM, Mulugeta S, et al. (2019). An SFTPC BRICHOS mutant links epithelial ER stress and spontaneous lung fibrosis. JCI Insight 4, e126125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzenstein AL (1985). Pathogenesis of “fibrosis” in interstitial pneumonia: an electron microscopic study. Hum. Pathol 16, 1015–1024. [DOI] [PubMed] [Google Scholar]
- Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, Kim J, Aryee MJ, Ji H, Ehrlich LIR, et al. (2010). Epigenetic memory in induced pluripotent stem cells. Nature 467, 285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klay D, Hoffman TW, Harmsze AM, Grutters JC, and van Moorsel CHM (2018). Systematic review of drug effects in humans and models with surfactant-processing disease. Eur. Respir. Rev 27, 170135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korogi Y, Gotoh S, Ikeo S, Yamamoto Y, Sone N, Tamai K, Konishi S, Nagasaki T, Matsumoto H, Ito I, et al. (2019). In Vitro Disease Modeling of Hermansky-Pudlak Syndrome Type 2 Using Human Induced Pluripotent Stem Cell-Derived Alveolar Organoids. Stem Cell Reports 12, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkevich G, Sukhov V, and Sergushichev A (2019). Fast gene set enrichment analysis. bioRxiv 10.1101/060012. [DOI] [Google Scholar]
- Kröner C, Reu S, Teusch V, Schams A, Grimmelt A-C, Barker M, Brand J, Gappa M, Kitz R, Kramer BW, et al. (2015). Genotype alone does not predict the clinical course of SFTPC deficiency in paediatric patients. Eur. Respir. J 46, 197–206. [DOI] [PubMed] [Google Scholar]
- Kropski JA, Blackwell TS, and Loyd JE (2015). The genetic basis of idiopathic pulmonary fibrosis. Eur. Respir. J 45, 1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Alhamdoosh M, Su S, Dong X, Tian L, Smyth GK, and Ritchie ME (2016). RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. F1000Res 5, 1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederer DJ, and Martinez FJ (2018). Idiopathic Pulmonary Fibrosis. N. Engl. J. Med 378, 1811–1823. [DOI] [PubMed] [Google Scholar]
- Maguire JA, Mulugeta S, and Beers MF (2012). Multiple ways to die: delineation of the unfolded protein response and apoptosis induced by Surfactant Protein C BRICHOS mutants. Int. J. Biochem. Cell Biol 44, 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massaro GD, Gail DB, and Massaro D (1975). Lung oxygen consumption and mitochondria of alveolar epithelial and endothelial cells. J. Appl. Physiol 38, 588–592. [DOI] [PubMed] [Google Scholar]
- Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema K-J, Coppes RP, Engedal N, Mari M, and Reggiori F (2018). Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 14, 1435–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, Chen C-I, Anekalla KR, Joshi N, Williams KJN, et al. (2017). Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med 214, 2387–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulugeta S, Nureki S, and Beers MF (2015). Lost after translation: insights from pulmonary surfactant for understanding the role of alveolar epithelial dysfunction and cellular quality control in fibrotic lung disease. Am. J. Physiol. Lung Cell. Mol. Physiol 309, L507–L525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni H-M, Bockus A, Wozniak AL, Jones K, Weinman S, Yin X-M, and Ding W-X (2011). Dissecting the dynamic turnover of GFP-LC3 in the autolysosome. Autophagy 7, 188–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogee LM, Dunbar AE 3rd, Wert SE, Askin F, Hamvas A, and Whitsett JA (2001). A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N. Engl. J. Med 344, 573–579. [DOI] [PubMed] [Google Scholar]
- Nureki S-I, Tomer Y, Venosa A, Katzen J, Russo SJ, Jamil S, Barrett M, Nguyen V, Kopp M, Mulugeta S, and Beers MF (2018). Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J. Clin. Invest 128, 4008–4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono S, Tanaka T, Ishida M, Kinoshita A, Fukuoka J, Takaki M, Sakamoto N, Ishimatsu Y, Kohno S, Hayashi T, et al. (2011). Surfactant protein C G100S mutation causes familial pulmonary fibrosis in Japanese kindred. Eur. Respir. J 38, 861–869. [DOI] [PubMed] [Google Scholar]
- Patel AS, Lin L, Geyer A, Haspel JA, An CH, Cao J, Rosas IO, and Morse D (2012). Autophagy in idiopathic pulmonary fibrosis. PLoS ONE 7, e41394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47 (D1), D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghu G, Chen S-Y, Yeh W-S, Maroni B, Li Q, Lee Y-C, and Collard HR (2014). Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001–11. Lancet Respir. Med 2, 566–572. [DOI] [PubMed] [Google Scholar]
- Raghu G, Chen S-Y, Hou Q, Yeh W-S, and Collard HR (2016). Incidence and prevalence of idiopathic pulmonary fibrosis in US adults 18–64 years old. Eur. Respir. J 48, 179–186. [DOI] [PubMed] [Google Scholar]
- Reimand J, Isserlin R, Voisin V, Kucera M, Tannus-Lopes C, Rostamianfar A, Wadi L, Meyer M, Wong J, Xu C, et al. (2019). Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc 14, 482–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemondy KA, Jansing NL, Jiang P, Redente EF, Gillen AE, Fu R, Miller AJ, Spence JR, Gerber AN, Hesselberth JR, and Zemans RL (2019). Single cell RNA sequencing identifies TGFβ as a key regenerative cue following LPS-induced lung injury. JCI Insight 5, e123637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrezenmeier E, and Dörner T (2020). Mechanisms of action of hydroxychloroquine and chloroquine: implications for rheumatology. Nat. Rev. Rheumatol 16, 155–166. [DOI] [PubMed] [Google Scholar]
- Selman M, and Pardo A (2014). Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. an integral model. Am. J. Respir. Crit. Care Med 189, 1161–1172. [DOI] [PubMed] [Google Scholar]
- Serra M, Alysandratos K-D, Hawkins F, McCauley KB, Jacob A, Choi J, Caballero IS, Vedaie M, Kurmann AA, Ikonomou L, et al. (2017). Pluripotent stem cell differentiation reveals distinct developmental pathways regulating lung- versus thyroid-lineage specification. Development 144, 3879–3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisson TH, Mendez M, Choi K, Subbotina N, Courey A, Cunningham A, Dave A, Engelhardt JF, Liu X, White ES, et al. (2010). Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am. J. Respir. Crit. Care Med 181, 254–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somers A, Jean J-C, Sommer CA, Omari A, Ford CC, Mills JA, Ying L, Sommer AG, Jean JM, Smith BW, et al. (2010). Generation of transgene-free lung disease-specific human induced pluripotent stem cells using a single excisable lentiviral stem cell cassette. Stem Cells 28, 1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart GA, Ridsdale R, Martin EP, Na C-L, Xu Y, Mandapaka K, and Weaver TE (2012). 4-Phenylbutyric acid treatment rescues trafficking and processing of a mutant surfactant protein-C. Am. J. Respir. Cell Mol. Biol 47, 324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strikoudis A, Cieślak A, Loffredo L, Chen Y-W, Patel N, Saqi A, Lederer DJ, and Snoeck H-W (2019). Modeling of Fibrotic Lung Disease Using 3D Organoids Derived from Human Pluripotent Stem Cells. Cell Rep 27, 3709–3723.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymaniak AD, Mahoney JE, Cardoso WV, and Varelas X (2015). Crumbs3-Mediated Polarity Directs Airway Epithelial Cell Fate through the Hippo Pathway Effector Yap. Dev. Cell 34, 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddeo EP, Alsabeeh N, Baghdasarian S, Wikstrom JD, Ritou E, Sereda S, Erion K, Li J, Stiles L, Abdulla M, et al. (2020). Mitochondrial Proton Leak Regulated by Cyclophilin D Elevates Insulin Secretion in Islets at Nonstimulatory Glucose Levels. Diabetes 69, 131–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas AQ, Lane K, Phillips J 3rd, Prince M, Markin C, Speer M, Schwartz DA, Gaddipati R, Marney A, Johnson J, et al. (2002). Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am. J. Respir. Crit. Care Med 165, 1322–1328. [DOI] [PubMed] [Google Scholar]
- Travis WD, Costabel U, Hansell DM, King TE Jr., Lynch DA, Nicholson AG, Ryerson CJ, Ryu JH, Selman M, Wells AU, et al. ; ATS/ERS Committee on Idiopathic Interstitial Pneumonias (2013). An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med 188, 733–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJA, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27, 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Moorsel CHM, van Oosterhout MFM, Barlo NP, de Jong PA, van der Vis JJ, Ruven HJT, van Es HW, van den Bosch JMM, and Grutters JC (2010). Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am. J. Respir. Crit. Care Med 182, 1419–1425. [DOI] [PubMed] [Google Scholar]
- Venosa A, Katzen J, Tomer Y, Kopp M, Jamil S, Russo SJ, Mulugeta S, and Beers MF (2019). Epithelial Expression of an Interstitial Lung Disease-Associated Mutation in Surfactant Protein-C Modulates Recruitment and Activation of Key Myeloid Cell Populations in Mice. J. Immunol 202, 2760–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade KC, Guttentag SH, Gonzales LW, Maschhoff KL, Gonzales J, Kolla V, Singhal S, and Ballard PL (2006). Gene induction during differentiation of human pulmonary type II cells in vitro. Am. J. Respir. Cell Mol. Biol 34, 727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AA, Kwok LW, Porter EL, Payne JG, McElroy GS, Ohle SJ, Greenhill SR, Blahna MT, Yamamoto K, Jean JC, et al. (2013). Lentiviral delivery of RNAi for in vivo lineage-specific modulation of gene expression in mouse lung macrophages. Mol. Ther 21, 825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters NI, Burman A, Kropski JA, and Blackwell TS (2019). Epithelial Injury and Dysfunction in the Pathogenesis of Idiopathic PulmonaryFibrosis. Am. J. Med. Sci 357, 374–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe DM, Lee J-H, Kumar A, Lee S, Orenstein SJ, and Nixon RA (2013). Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur. J. Neurosci 37, 1949–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, and Smyth GK (2012). Camera: a competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res 40, e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, and Tashiro Y (1998). Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct 23, 33–42. [DOI] [PubMed] [Google Scholar]
- Yao C, Guan X, Carraro G, Parimon T, Liu X, Huang G, Mulay A, Soukiasian HJ, David G, Weigt SS, et al. (2020). Senescence of Alveolar Type 2 Cells Drives Progressive Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med 203, 707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin BWT, Kiyamova R, Chua R, Caballero OL, Gout I, Gryshkova V, Bhaskaran N, Souchelnytskyi S, Hellman U, Filonenko V, et al. (2008). Monoclonal antibody MX35 detects the membrane transporter NaPi2b (SLC34A2) in human carcinomas. Cancer Immun 8, 3. [PMC free article] [PubMed] [Google Scholar]
- Yu G, Tzouvelekis A, Wang R, Herazo-Maya JD, Ibarra GH, Srivastava A, de Castro JPW, DeIuliis G, Ahangari F, Woolard T, et al. (2018). Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat. Med 24, 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The scRNA-seq data reported in this publication have been deposited in NCBI’s Gene Expression Omnibus (GEO; accession number GSE160801). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the Proteomics Identifications (PRIDE) partner repository with the dataset identifier PRIDE: PXD022334. All original code has been deposited at GitHub and is publicly available as of the date of publication. DOIs are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal to activated caspase 3 | Sigma Aldrich | Cat# C8487; RRID:AB_476884 |
| Mouse monoclonal to β actin | Sigma Aldrich | Cat# A1978; RRID:AB_476692 |
| Mouse monoclonal to CD26, PE conjugated (clone BA5b) | BioLegend | Cat# 302705; RRID:AB_314289 |
| Mouse monoclonal to CD47, PerCP-Cy5.5 conjugated (clone CC2C6) | BioLegend | Cat# 323110; RRID:AB_940461 |
| Rabbit polyclonal to CRB3 | Szymaniak et al., 2015 | N/A |
| Mouse IgG1 isotype, PE conjugated | BioLegend | Cat# 400113; RRID:AB_326435 |
| Mouse IgG1 isotype, PerCP-Cy5.5 conjugated | BioLegend | Cat# 400149; RRID:AB_893680 |
| Mouse monoclonal to E-Cadherin/CDH1 | BD Biosciences | Cat# 610181; RRID:AB_397580 |
| Mouse monoclonal to EPCAM | Santa Cruz | Cat# Sc-73491; RRID:AB_1122569 |
| Mouse monoclonal to GAPDH | Chemicon | Cat# MAB374; RRID:AB_2107445 |
| Rabbit monoclonal to NKX2-1 (clone EP15847) | Abcam | Cat# ab76013; RRID:AB_1310784 |
| Rabbit monoclonal to Phospho-Ezrin/Radixin/Moesin (p-ERM) | Cell Signaling | Cat# 3726; RRID:AB_10560513 |
| Mouse monoclonal to pro-SFTPC | Santa Cruz | Cat# sc-518029 |
| Rabbit polyclonal to pro-SFTPC | Seven Hills | Cat# WRAB-9337; RRID:AB_2335890 |
| Rabbit polyclonal to mature SFTPC | Seven Hills | Cat# WRAB-76694 |
| Rabbit polyclonal anti-NPRO-SFTPC | Beers et al., 1994 | N/A |
| Rabbit polyclonal anti-mature SFTPB (PT3) | Beers et al., 1992 | N/A |
| Rabbit monoclonal to MUC1 | Abcam | Cat# Ab218998 |
| Rabbit polyclonal to HA tag | Abcam | Cat# Ab20084; RRID:AB_445319 |
| Mouse monoclonal to RAGE | Abcam | Cat# Ab54741; RRID:AB_2242462 |
| Rabbit polyclonal to LC3B | Cell Signaling | Cat# 2775; RRID:AB_915950 |
| Mouse monoclonal to SQSTM1/p62 | Abcam | Cat# Ab56416; RRID:AB_945626 |
| Mouse monoclonal to CPM | Wako | Cat# 014-27501; RRID:AB_2801482 |
| Mouse monoclonal to SLC34A2 | Yin et al., 2008 | N/A |
| AffiniPure Donkey Anti-Rabbit IgG (H+L), AlexaFluor 488 conjugated | Jackson ImmunoResearch | Cat# 711-545-152; RRID:AB_2313584 |
| AffiniPure Donkey Anti-Rabbit IgG (H+L), AlexaFluor 647 conjugated | Jackson ImmunoResearch | Cat# 711-605-152; RRID:AB_2492288 |
| AffiniPure Donkey Anti-Mouse IgG (H+L), AlexaFluor 488 conjugated | Jackson ImmunoResearch | Cat# 715-545-150; RRID:AB_2340846 |
| AffiniPure Donkey Anti-Mouse IgG (H+L), AlexaFluor 647 conjugated | Jackson ImmunoResearch | Cat# 715-605-150; RRID:AB_2340862 |
| AffiniPure Goat Anti-Mouse IgG (H+L), AlexaFluor 488 conjugated | Jackson ImmunoResearch | Cat# 115-545-062; RRID:AB_2338845 |
| AffiniPure Goat Anti-Rabbit IgG (H+L), AlexaFluor 488 conjugated | Jackson ImmunoResearch | Cat# 111-545-144; RRID:AB_2338052 |
| AffiniPure Goat Anti-Mouse IgG (H+L), AlexaFluor 594 conjugated | Jackson ImmunoResearch | Cat# 115-585-146; RRID:AB_2338881 |
| AffiniPure Goat Anti-Rabbit IgG (H+L), AlexaFluor 594 conjugated | Jackson ImmunoResearch | Cat# 111-585-144; RRID:AB_2307325 |
| Goat Anti-Mouse IgG (H+L), HRP conjugated | BioRad | Cat# 0300-0108P; RRID:AB_808614 |
| Goat Anti-Rabbit IgG (H+L), HRP conjugated | BioRad | Cat# 5196-2504; RRID:AB_619908 |
| Chemicals, peptides and recombinant proteins | ||
| Human embryonic stem cell (hESC)- qualified (2D) Matrigel | Corning | Cat# 354277 |
| Growth Factor Reduced (3D) Matrigel | Corning | Cat# 356231 |
| SB431542 | Tocris | Cat# 1614 |
| Dorsomorphin | Stemgent | Cat# 04-0024 |
| CHIR99021 (CHIR) | Tocris | Cat# 4423 |
| Recombinant human FGF10 | R&D Systems | Cat# 345-FG-025 |
| Recombinant Human Keratinocyte Growth Factor (KGF) | R&D Systems | Cat# 251-KG-010 |
| Recombinant Human BMP4 (rhBMP4) | R&D Systems | Cat# 314-BP |
| Retinoic acid | Sigma Aldrich | Cat# R2625 |
| Y-27632 dihydrochloride | Tocris | Cat# 1254 |
| Ascorbic Acid | Sigma Aldrich | Cat# A4544 |
| 1-Thioglycerol (MTG) | Sigma Aldrich | Cat# M6145 |
| BSA 7.5% Stock | Thermo Fisher Scientific | Cat# 15260037 |
| 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (EDTA) | Sigma Aldrich | Cat# E7889 |
| N-(2-Hydroxyethyl)piperazine-N’-(2- ethanesulfonic acid) Solution (HEPES) | Sigma Aldrich | Cat# H0887 |
| Dexamethasone | Sigma Aldrich | Cat# D4902 |
| 8-bromoadenosine 30,50-cyclic monophosphate sodium salt (cAMP) | Sigma Aldrich | Cat# B7880 |
| 3-Isobutyl-1-methylxanthine (IBMX) | Sigma Aldrich | Cat# I5879 |
| Puromycin Dihydrochloride | Thermo Fisher Scientific | Cat# A1113802 |
| Geneticin Sulfate | Life Technologies | Cat# 11811-023 |
| 0.05% trypsin-EDTA | Invitrogen | Cat# 25300-120 |
| Defined Fetal Bovine Serum | Thermo Fisher Scientific | Cat# NC0652331 |
| Dimethyl Sulfoxide (DMSO) | Sigma Aldrich | Cat# D2650 |
| Calcein blue | Life Technologies | Cat# C1429 |
| Hoechst 33342 | Thermo Fisher Scientific | Cat#H3570 |
| DAPI, FluoroPure grade | Invitrogen | D21490 |
| ProLong Diamond Antifade Mountant | Invitrogen | Cat# P36965 |
| LysoTracker Green DND-26 | Thermo Fisher Scientific | Cat# L7526 |
| MitoTracker Green FM | Thermo Fisher Scientific | Cat# M7514 |
| BODIPY-493/503 | Thermo Fisher Scientific | Cat# D3922 |
| LysoSensor Yellow/Blue Dextran | Thermo Fisher Scientific | Cat# L22460 |
| Dispase II | Thermo Fisher Scientific | Cat# 17105-041 |
| Glutaraldehyde | Ladd Research | Cat# 20100 |
| Osmium Tetroxide | Polysciences | Cat# 0223D |
| Uranyl Acetate | Electron Microscopy Sciences | Cat# 22400 |
| EMbed 812 | Electron Microscopy Sciences | Cat# 14120 |
| Paraformaldehyde | Electron Microscopy Sciences | Cat# 19208 |
| Oligomycin | Millipore | Cat# 495455 |
| FCCP | Sigma Aldrich | Cat# C2920 |
| Antimycin A | Sigma Aldrich | Cat# A8674 |
| Bafilomycin A1 | Sigma Aldrich | Cat# B1793 |
| Torin | Tocris | Cat# 4247 |
| Critical commercial assays | ||
| RNeasy Mini Kit | QIAGEN | Cat# 74104 |
| QIAzol Lysis Reagent QIAGEN | QIAGEN | Cat# 79306 |
| TaqMan Fast Universal PCR Master Mix (2X) | Thermo Fisher | Cat# 4364103 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat# 4368814 |
| EZ-TAL TALE Assembly Kit | System Bioscience | Cat# GE120A-1 |
| Click-iT EdU Alexa Fluor 647 Flow Cytometry Assay Kit | Thermo Fisher Scientific | Cat# C10424 |
| Lipofectamine LTX Kit | Thermo Fisher Scientific | Cat# 15338100 |
| P3 Primary Cell 4D-NucleofectorO X Kit S | Lonza | Cat# V4XP-3032 |
| Magnetic Luminex® Performance Assay Human HighSensitivity | R&D Systems | Cat# LXSAHM-31 |
| Deposited data | ||
| Single cell transcriptomic profiling of patient-specific pluripotent stem cell-derived alveolar epithelial type 2 cells expressing the SFTPCI73T variant and their gene-edited corrected counterparts | This paper | GEO (GSE160801) |
| Proteomic and phosphoproteomic profiling of SFTPCI73T expressing patient-specific pluripotent stem cell-derived alveolar epithelial type 2 cells and their gene-edited corrected counterparts. | This paper | PRIDE: PXD022334 |
| Experimental models: Cell lines | ||
| Human: SFTPCI73T/WT donor iPSC line (SPC2) | This paper | http://stemcellbank.bu.edu/Catalog/Item/Home |
| Human: Corrected SFTPC donor iPSC line (SPC2-CorrC1; SFTPCwt/wt) | This paper | http://stemcellbank.bu.edu/Catalog/Item/Home |
| Human: Donor iPSC line targeted with SFTPCtdTomato (SPC2-ST-C11; SFTPCI73T/tdT) | This paper | http://stemcellbank.bu.edu/Catalog/Item/Home |
| Human: Donor iPSC line targeted with SFTPCtdTomato (SPC2-ST-B2; SFTPCtdT/WT) | Kotton Lab; Hurley et al., 2020 | http://stemcellbank.bu.edu/Catalog/Item/Home |
| Oligonucleotides | ||
| See Table S3 | This paper | N/A |
| Recombinant DNA | ||
| EF1a-TALEN_HD; SFTPC recognition sequence of right TALEN 5′-TCA CCG GCG GGC TCT CCA TC-3′. | Kotton Lab; Jacob et al., 2017 | http://www.kottonlab.com |
| EF1a-TALEN_NN; SFTPC recognition sequence of left TALEN: 5’-TAG CAC CTG CAG CAA GAT GG-3′. | Kotton Lab; Jacob et al., 2017 | http://www.kottonlab.com |
| p1303-DV-SFTPC-tdTomato | Kotton Lab; Jacob et al., 2017 | http://www.kottonlab.com |
| pHAGE2 EF1a-Cre-IRES-NeoR-W | Kotton Lab; Jacob et al., 2017 | http://www.kottonlab.com |
| pHAGE2-Cre-IRES-PuroR | Kotton Lab; Somers et al., 2010 | Addgene #30205 |
| Software and algorithms | ||
| FlowJo version 10.5.3 | Becton, Dickinson & Company | https://www.flowjo.com/ |
| ImageJ | National Institutes of Health | https://imagej.nih.gov/ij/ |
| CellProfiler v2.0 | Broad Institute | https://cellprofiler.org |
| Prism | GraphPad | https://www.graphpad.com/ |
| Original code | This paper | https://github.com/CReM-BU/Alysandratos_iAEC2s |
| Other | ||
| StemDiff Definitive Endoderm Kit | StemCell Technologies | Cat# 05110 |
| mTeSR1 | StemCell Technologies | Cat# 05850 |
| Glutamax | Life Technologies | Cat# 35050-061 |
| Gentle Cell Dissociation Reagent | StemCell Technologies | Cat# 07174 |
| Seahorse XF Base Medium Minimal DMEM | Agilent Technologies | Cat# 103193-100 |
| Hank’s Buffered Saline Solution (HBSS; no calcium, no magnesium, no phenol red) | GIBCO | Cat# 14175095 |
| Ham’s F12 Medium | Cellgro | Cat# 10-080-CV |
| Iscove’s Modified Dulbecco’s Medium (IMDM) | Thermo Fisher Scientific | Cat# 12440053 |
| N2 Supplement | Invitrogen | Cat# 17502-048 |
| B27 Supplement | Invitrogen | Cat# 15260-037 |
| Primocin | Invitrogen | Cat# NC9141851 |