

SUMMARY
During acute malaria, most individuals mount robust inflammatory responses that limit parasite burden. However, long-lived sterilizing anti-malarial memory responses are not efficiently induced, even following repeated Plasmodium exposures. Using multiple Plasmodium species, genetically modified parasites, and combinations of host genetic and pharmacologic approaches, we find that the deposition of the malarial pigment hemozoin directly limits the abundance and capacity of conventional type 1 dendritic cells to prime helper T cell responses. Hemozoin-induced dendritic cell dysfunction results in aberrant Plasmodium-specific CD4 T follicular helper cell differentiation, which constrains memory B cell and long-lived plasma cell formation. Mechanistically, we identify that dendritic cell-intrinsic NLRP3 inflammasome activation reduces conventional type 1 dendritic cell abundance, phagocytosis, and T cell priming functions in vivo. These data identify biological consequences of hemozoin deposition during malaria and highlight the capacity of the malarial pigment to program immune evasion during the earliest events following an initial Plasmodium exposure.
Graphical abstract
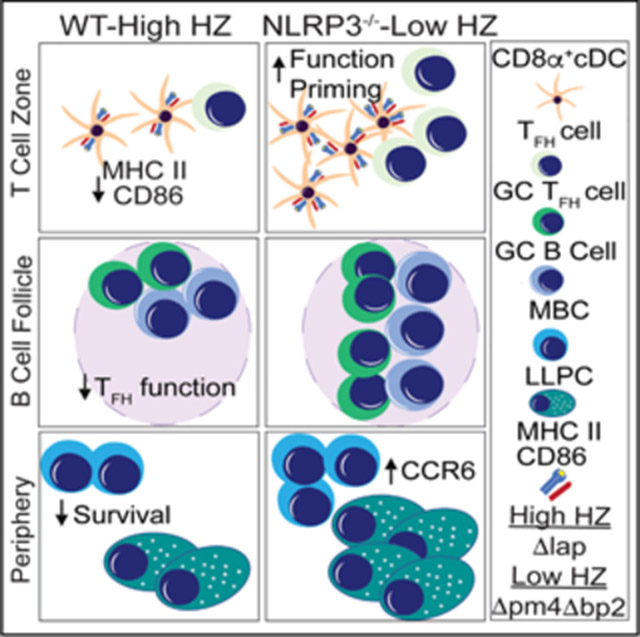
In brief
Using genetic, chimeric, and pharmacologic approaches, Pack et al. demonstrate that the parasite-derived crystal hemozoin erodes conventional CD8α+ type 1 dendritic cell (cDC1) abundance and function in an NLRP3 inflammasome-dependent manner, which results in reduced CD4+ T follicular helper cell differentiation and impaired anti-Plasmodium humoral immunity.
INTRODUCTION
Nearly half of the world’s population lives in areas endemic to the disease syndrome malaria, which is caused by infection with protozoan parasites of the genus Plasmodium. Children under the age of 5 account for two-thirds of all malaria-associated mortality (WHO, 2019). Repeated parasite infections generally fail to elicit sterilizing immunity, even following years of exposure among those living in regions of intense Plasmodium transmission (Tran et al., 2013). The emergence of parasite resistance to front-line antimalarial drugs (Das et al., 2018), coupled with the limited capacity of existing vaccines to elicit protection in endemic populations (Mensah et al., 2016; Rts, 2015), underscores the rationale for continued efforts to close knowledge gaps regarding the mechanisms, whether host or parasite associated, that restrict the development of durable, protective immune memory responses.
Following an initial Plasmodium infection and asymptomatic replication of parasites in the liver, schizont-induced rupture of parasite-infected red blood cells (pRBCs) liberates both host- and parasite-derived factors, including host RBC membranes, parasite antigens, and nucleic acids, that are recognized and often captured by host phagocytes. These stimuli rapidly induce the activation of macrophages and dendritic cells (DCs) that produce reactive oxygen and nitrogen species (Chua et al., 2013; Ranjan et al., 2016; Sobolewski et al., 2005; Sponaas et al., 2009), which are believed to limit infection intensity prior to the induction and orchestration of anti-Plasmodium cellular and humoral immunity. Phagocytes are also responsible, in part, for the production of high levels of proinflammatory cytokines such as interleukin (IL)-1β, IL-12, and interferon (IFN)-γ that both amplify the activation and recruitment of additional effector immune cells (Leisewitz et al., 2004; Perry et al., 2005; Wykes et al., 2007) and contribute to clinical malarial disease syndrome (deWalick et al., 2007). In addition to initiating and amplifying host immunity, DCs also contribute to the initiation of T follicular helper (TFH) cell differentiation via MHC II-restricted antigen presentation and costimulation (Choi et al., 2011; Langenkamp et al., 2000). TFH populations, in turn, are critical for orchestrating protective pathogen-specific humoral responses and deficiencies in anti-malarial humoral immunity have been linked to altered TFH development and function (Hansen et al., 2017; Ryg-Cornejo et al., 2016; Zander et al., 2016; Zander et al., 2015). Whether DCs are distinctly programmed during Plasmodium blood-stage infection such that they inefficiently support the development of anti-Plasmodium TFH responses and humoral immunity is not well defined.
During either in vivo blood-stage infection or following direct in vitro incubation with pRBCs, DCs exhibit an atypical maturation phenotype (Elliott et al., 2007; Götz et al., 2017; Urban et al., 1999), as well as reduced responsiveness to LPS stimulation and impaired capacity to initiate heterologous immune responses during an active Plasmodium infection (Lundie et al., 2010; Millington et al., 2006). Compared to DCs from uninfected subjects, myeloid DCs isolated from the peripheral blood of P. falciparum exposed individuals expressed lower levels of CD80, CD86, and HLA-DR following in vitro stimulation with blood-stage parasites (Turner et al., 2021). In vitro incubation of DCs with intact and dissociated pRBCs, along with RBC ghosts (membranes), identified that hemozoin, a crystalized parasite-derived hemoglobin degradation byproduct and NLRP3 inflammasome and CLEC12A agonist (Kalantari et al., 2014; Raulf et al., 2019; Shio et al., 2009), can alter DC function (Schwarzer et al., 1998; Skorokhod et al., 2004). However, whether modulating hemozoin levels in the context of natural Plasmodium blood-stage infection directly impacts DC-dependent development of anti-Plasmodium humoral responses has not been investigated.
In this report, we combined genetic, chimeric, and pharmacologic approaches to interrogate whether hemozoin exposure during experimental malaria impacts immune programming and the development of protective anti-Plasmodium humoral immune memory responses. Using both genetically modified Plasmodium berghei (Pb) parasites engineered to produce 10-fold lower levels of hemozoin (Lin et al., 2015) and wild-type P. yoelii infection of NLRP3-deficient mice, we show that hemozoin-induced, DC-intrinsic NLRP3-mediated inflammasome activation compromises TFH differentiation, which in turn limits anti-Plasmodium memory B cell and the long-lived plasma cell responses and protective humoral immunity against malaria.
RESULTS
Durable anti-Plasmodium humoral immunity is compromised by hemozoin
To address whether hemozoin formation and deposition influences the generation of durable anti-malarial immunity, we established memory immune responses in wild-type C57BL/6 mice by infecting them with a genetically modified Plasmodium berghei ANKA variant lacking the aspartic proteases plasmepsin 4 and berghepain-2 (Δpm4Δbp2), genes that are necessary for efficient degradation of hemoglobin and crystallization of hemozoin (Lin et al., 2015; Spaccapelo et al., 2010). Mature schizonts of Δpm4Δbp2 parasites produce less hemozoin compared to wild-type (WT) P. berghei ANKA parasites with 50% reductions in hemozoin accumulation within the spleen as early as day 5 post-infection (p.i.) (Figure S1A). Because Δpm4Δbp2 parasites also exhibit reduced growth and replication in vivo compared to WT Pb ANKA parasites (Lin et al., 2015), other groups of mice were infected with a comparison mutant of growth-attenuated P. berghei lacking leucyl aminopeptidase (Δlap), a mutant that is not impaired in hemozoin production (Lin et al., 2015) but exhibits growth and clearance kinetics that are equivalent to Δpm4Δbp2 parasites (Figure S1B). These genetically modified parasites enabled us to directly address the in vivo role of hemozoin and avoid major caveats associated with injection of exogenous hemozoin, including the form (i.e., synthetic or natural), site of accumulation, and dose (Pham et al., 2021). Sixty days after the initial infection with either Δlap or Δpm4Δbp2 parasites, we challenged both groups of mice with WT (virulent) P. berghei ANKA parasites engineered to express the fusion protein GFP-Luciferase (Pb-ANKA-Luc) (Figure 1A). Compared to Δlap-immune mice, Δpm4Δbp2-immune mice exposed to lower levels of hemozoin during primary infection exhibited reduced parasitemia (Figure 1B) and total parasite biomass luminescence (Figure 1C) across all time points examined and whole-body bioluminescent imaging revealed substantial Pb-ANKA-Luc tissue accumulation in Δlap-immune mice that was absent in Δpm4Δbp2-immune mice (Figure 1C). Δpm4Δbp2-immune mice also exhibited 100% survival following WT Pb-ANKA-Luc challenge (Figure 1D).
Figure 1. Hemozoin limits humoral immune memory responses.

(A) C57BL/6 mice were infected with either Δpm4Δbp2 or Δlap parasites. After 60 days, immune mice were challenged with virulent WT Pb-ANKA-luc.
(B) The kinetics of peripheral parasite burden.
(C) Quantification (left) of parasite load as determined by whole-body bioluminescence following virulent Pb-ANKA-luc challenge. Images (right) on day 19 post-challenge.
(D) Survival of Δpm4Δbp2 and Δlap-immune mice following virulent Pb-ANKA-luc challenge.
(E) IgG was purified from the indicated immune mice and either purified IgG or the unbound flow-through was transferred via tail vein to naive WT mice 1 day prior to challenge with virulent Pb-ANKA.
(F) Parasite burden at day 4 post-challenge.
(G and H) Representative flow plots, frequency (G), and (H) number of mature plasma cells (B220negCD19negCD138hiTACIhi) in the bone marrow at day 40 p.i.
(I) Number of MSP11–19-specific plasma cells in the bone marrow at day 40 p.i.
(J and K) Representative flow plots (J) and quantification of (K) splenic IgDnegCD38+CD138hi LLPC precursors expressing CXCR4 at day 14 p.i.
(L and M) Frequency of (L) bone-marrow IgDnegCD38+CD138hiTACIhi plasma cells and (M) MSP11–19-specific IgG secreting cells recovered from the bone marrow of immune mice at day 28 p.i.
Data (mean ± SEM) in (B)–(D) and (mean ± SD) in (I) and (M) are the summary data from two independent experiments. Data (mean ± SD) in (F) and (J)–(L) are representative of two independent experiments with n = 4. Data (mean ± SD) in (G) and (H) are representative of three experiments with n = 4 (see also Figure S1).
Because Δpm4Δbp2-immune mice exhibited enhanced control of WT Pb-ANKA as early as days 3–4 post-challenge, relative to Δlap-immune mice (Figure 1B), we further posited that the observed resistance to virulent Pb-ANKA-Luc challenge could be attributed to pre-existing, protective anti-Plasmodium antibodies. Consistent with this hypothesis, passive transfer of purified immunoglobulin G (IgG) from Δpm4Δbp2-immune donors to WT Pb-ANKA-infected recipient mice (Figure 1E) transiently resulted in a 75% reduction in parasitemia, compared to transfer of an equivalent amount of purified IgG from Δlap-immune donors (Figure 1F), although immune IgG transfer was not sufficient to protect recipients beyond the first week (Figure S1C). Strikingly, antibody titers directed against the 19 kDa fragment of Merozoite Surface Protein 1 (MSP119), an immunodominant and protective blood-stage antigen (John et al., 2004; O’Donnell et al., 2001; Terrientes et al., 1994), were not detectably different between Δlap and Δpm4Δbp2-immune mice (Figure S1D), and we found no significant alterations in the total IgG antibody avidity index or isotype class distributions (Figures S1E and S1F). Nevertheless, our passive transfer data are consistent with an enhanced long-lived antibody response in Δpm4Δbp2-immune mice, compared to Δlap-immune mice, which may be reflected by either quantitative or qualitative (specificity and functional) shifts that register below the sensitivity of these assays.
Secreted anti-Plasmodium antibodies that are present 60 days p.i. likely derive from long-lived plasma cells (LLPCs) that often reside in the bone marrow (Lightman et al., 2019). Given the enhanced protective capacity of Δpm4Δbp2-immune IgG, we quantified parasite-specific LLPC in the bone marrow during the memory phase. By day 40 p.i., we found approximately 2-fold greater numbers of CD138hiTACIhiCD19loB220lo mature resting LLPC (Pracht et al., 2017) (Figures 1G and 1H) and functional MSP119-specific IgG secreting cells (Figure 1I; Figure S1G) in the bone marrow of Δpm4Δbp2-immune mice, compared to Δlap-immune mice. Thus, we next evaluated the kinetics of parasite-specific LLPC formation. As early as day 14 p.i., the frequency and number of splenic CD38+IgDnegCD138hi LLPC precursors expressing CXCR4, a chemokine receptor associated with bone marrow homing (Nie et al., 2004), was 30%–40% greater in Δpm4Δbp2-infected mice (Figures 1J and 1K). By day 28 p.i., the bone marrow of Δpm4Δbp2-immune mice presented with twice as many CD38+IgDnegCD138hiTACIhi LLPC (Figure 1L) and MSP119-specific IgG-secreting cells (Figure 1M), compared to Δlap-immune mice. Taken together, these data support that deposition of hemozoin during a primary blood-stage Plasmodium exposure impairs either the development or function of LLPC and durable anti-Plasmodium humoral responses.
Hemozoin compromises memory B cell responses
Given the deleterious effect of hemozoin on LLPC development and function following primary blood-stage infection, we additionally hypothesized that hemozoin may interfere with the development and function of memory B cell (MBC) populations. Consistent with this hypothesis, splenic CD19+CD38+IgDnegCD73+CD95neg MBC and CD19+CD38+IgDnegCD73+CD80+PD-L2+ MBC, with CD80+PD-L2+ defining those most capable of rapid antibody production (Zuccarino-Catania et al., 2014), were elevated 2-fold by day 30 p.i. in Δpm4Δbp2-immune mice, compared to Δlap-immune mice (Figures 2A-2D). The chemokine receptor CCR6 is reported to enhance cell positioning within secondary lymphoid tissues for superior recall upon pathogen challenge (Elgueta et al., 2015; Suan et al., 2017) and by day 60 p.i., the frequency of splenic MBC cells expressing CCR6 were 2- to 3-fold greater in Δpm4Δbp2-immune mice, compared to Δlap-immune mice (Figures 2E-2G). To interrogate functional MBC responses, we evaluated MSP119-specific serum antibody titers in Δpm4Δbp2 and Δlap-immune mice both prior to and 5 days after challenge with WT Pb-ANKA. In this short interval, increases in MSP119-specific, isotype-switched IgG titers are most likely due to MBC activation and differentiation into plasma cells, rather than de novo humoral immune responses. Although Δpm4Δbp2-immune mice harbor 2-fold higher numbers of MBC (Figures 2A-2D), 5 days after challenge MSP119-specific IgG titers expanded by 10-fold, as compared to 3-fold in Δlap-immune mice (Figure 2H). Together, these data suggest that recall functions of MBC are impaired by the presence and deposition of hemozoin.
Figure 2. Hemozoin constrains the number and function of memory B cells.
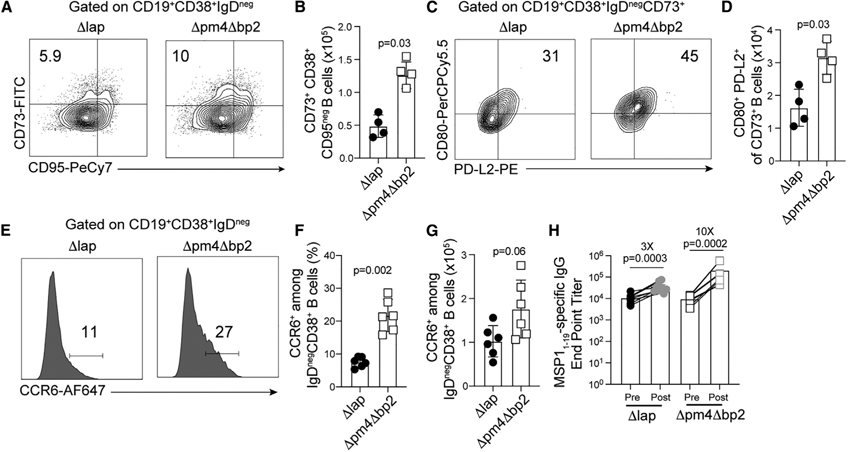
C57BL/6 mice were infected with Δpm4Δbp2 and Δlap parasites.
(A–D) Representative flow plots (A) and quantification (B) of splenic CD19+CD38+IgDnegCD73+CD95neg MBC and the frequency (C) and number (D) of splenic CD19+CD38+IgDnegCD73+CD80+PD-L2+ MBC by day 30 p.i.
(E) Representative histograms describing frequency (F) and number (G) of CCR6+CD19+CD38+IgDneg MBC.
(H) Δpm4Δbp2 and Δlap-immune mice were challenged with virulent Pb-ANKA. Sera were collected before (pre) and 5 days after (post) challenge and MSP11–19-specific IgG endpoint titers were quantified. Data (mean ± SD) in (A)–(D) and are representative of three independent experiments with n = 4–5. Data (mean ± SD) in (E)–(H) are the summary data of two independent experiments.
CD4 T cell help and germinal center B cell responses are impaired by hemozoin
LLPC and MBC populations derive from germinal center (GC) B cell responses (Mesin et al., 2016; Weisel et al., 2016) and hemozoin deposition was associated with reduced LLPC and MBC development as early as days 14 and 30 p.i. in Δpm4Δbp2-infected mice, time points that encompass the peak of GC B cell responses following experimental Plasmodium (Krishnamurty et al., 2016; Vijay et al., 2020). Indeed, by day 14 p.i. the frequency and number of CD95+GL-7+ GC B cells were elevated by ~30% in Δpm4Δbp2-infected mice, compared to Δlap-infected mice (Figures 3A and 3B). Because GC B cell responses depend on the presence and activity of TFH cells (Kräutler et al., 2017), we next postulated that hemozoin impairs either the induction, stepwise differentiation, or function of TFH populations. To test this hypothesis, we transferred Plasmodium-specific CD4 T cell receptor transgenic cells (PbTII [Fernandez-Ruiz et al., 2017]) to WT mice, infected recipients 1 day later with either Δpm4Δbp2 or Δlap parasites and then quantified both polyclonal, endogenous and PbTII CD4 T cells exhibiting characteristics of either mature CXCR5hiPD-1hi GC-TFH cells that localize to follicular GC reactions or their extrafollicular CD150loCXCR5int TFH precursors (Choi et al., 2011; Crotty, 2014). By day 14 p.i., we found 30%–50% increases in the frequencies and numbers of both CD150loCXCR5intTFH precursors (Figures 3C and 3D) and mature CXCR5hiPD-1hi GC-TFH subsets (Figures 3E and 3F) among PbTII cells in Δpm4Δbp2-infected mice, compared to PbTII cells in Δlap-infected mice. We also observed 2-fold greater numbers of CXCR5hiPD-1hi GC-TFH cells among polyclonal endogenous CD4 T cells recovered from Δpm4Δbp2-infected mice (Figures 3G and 3H), supporting the view that this effect extends to the endogenous CD4 T cell receptor repertoire. Bcl-6 is a transcriptional repressor that is essential for TFH differentiation and function (Hatzi et al., 2015) and T-bet+ TH1-like TFH and Foxp3+ T regulatory cells are reported to constrain anti-Plasmodium humoral immunity (Kurup et al., 2017; Obeng-Adjei et al., 2015). However, on 14 p.i., we identified only modestly reduced levels of T-bet and Foxp3, and equivalent expression levels of Bcl-6, in mature GC-TFH cells recovered from Δpm4Δbp2-infected mice, compared to mature GC-TFH cells recovered from Δlap-infected animals (Figure 3I; Figure S2A). Despite these modest cell-intrinsic transcriptional differences, we additionally tested whether mature GC-TFH cells recovered from Δpm4Δbp2-infected mice differed in their capacity to promote GC B cell differentiation. We sort-purified mature CXCR5hiPD-1hiGC-TFH from each group and co-cultured cells with an equivalent number with naive B cells for 5 days (Figure 3J). We observed significant increases in the proportions of B cells exhibiting a CD95+GL-7+ GC phenotype when cells were cultured with mature GC-TFH recovered from Δpm4Δbp2-infected mice, compared to mature GC-TFH cells recovered from Δlap-infected mice (Figures 3K and 3L). Moreover, TFH cells sort-purified on day 7 p.i. from the spleens of Δlap-infected mice were also impaired in their capacity to drive GC responses following transfer to Tcrα−/− recipients, compared to TFH recovered from Δpm4Δbp2-infected mice (Figures 3M-3P; Figures S2B-S2D). Together, these data support that the presence of elevated levels of hemozoin impairs the differentiation and cell-intrinsic function of TFH cells with subsequent reductions in GC B cell responses.
Figure 3. Hemozoin impairs the germinal center response.
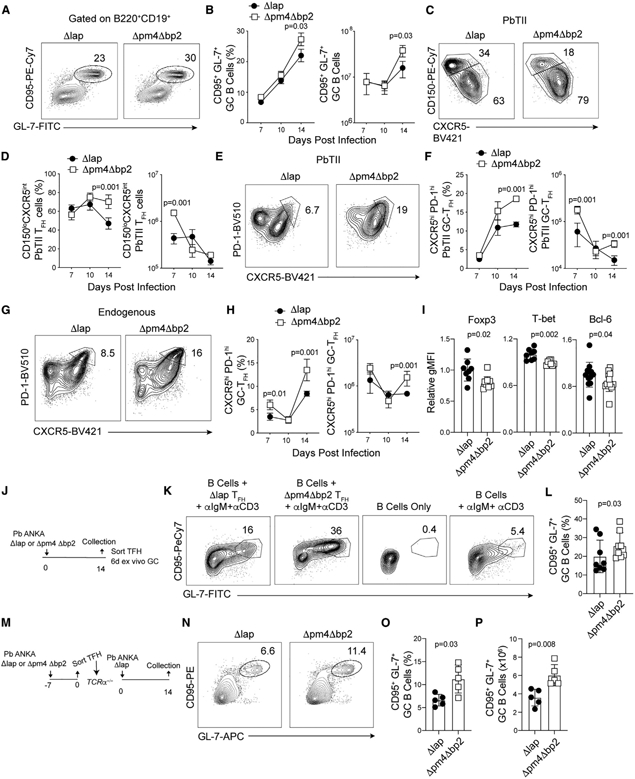
Parasite-specific CD4 T cells (PbTII) were transferred to C57BL/6 mice 1 day prior to infection with either Δpm4Δbp2 or Δlap parasites. Splenic TFH and GC B cell responses were evaluated.
(A–F) Representative plots (A) and frequency and number (B) of GC B cells (B220+CD19+CD95+GL-7+) on d14 p.i. TFH precursors (CD150loCXCR5int) (C and D) and mature GC-TFH (CXCR5hiPD-1hi) expansion (E and F) among donor-derived PbTII cells.
(G and H) Frequency (G) and summary quantification (H) of mature GC-TFH (PD-1hiCXCR5hi) among polyclonal endogenous CD4 T cells.
(I) Relative expression (gMFI) of Foxp3, T-bet, and Bcl-6 in polyclonal GC-TFH cells on day 14 p.i.
(J) Polyclonal GC-TFH cells were sort purified from spleens day 14 p.i. and co-cultured for 6 days with purified naive B cells.
(K and L) Representative flow plots (K) and summary (L) of GC B cell (B220+CD19+CD95+GL-7+) frequency following co-culture.
(M) Polyclonal TFH cells were sort purified from spleens of Δpm4Δbp2 and Δlap-infected mice day 7 p.i. and transferred to Tcrα−/− mice. Tcrα−/− mice were infected with Δlap parasites and GC responses were assessed on 14 p.i.
(N–P) Representative flow plots (N) and summary of GC B cell (B220+CD19+CD95+GL-7+) frequency (O) and number (P).
Data (mean ± SEM) in (A)–(H) are representative of four experiments with n = 4–5. Data (mean ± SD) in (J)–(L) are the summary data from two independent experiments. Data (mean ± SD) in (M)–(P) are representative of two independent experiments with n = 5 (see also Figure S2).
Conventional DC functions are compromised by hemozoin
CD11c+B220neg conventional dendritic cells (cDCs), particularly the CD11bnegCD8α+ conventional type 1 DC (cDC1) subset, are critical for initiating protective T cell responses against blood-stage Plasmodium infection (Fernandez-Ruiz et al., 2017; Voisine et al., 2010). Total CD11c+B220neg cDC numbers, phagocytic functions, and cytokine expression are reported to progressively decline during the course of experimental and clinical malaria, which has been described as immunoparalysis (Loughland et al., 2016; Lundie et al., 2010; Pinzon-Charry et al., 2013; Woodberry et al., 2012). Moreover, purified hemozoin can down-modulate DC reactivity following direct co-incubation ex vivo (Millington et al., 2006; Skorokhod et al., 2004). However, whether the deposition and/or accumulation of hemozoin specifically impairs either total cDC or cDC1 functions during blood-stage Plasmodium infection in vivo has not been directly tested. To investigate this, we examined cDC subset abundance, costimulatory receptor expression, and phagocytic function in Δpm4Δbp2 and Δlap-infected mice (representative gating, Figure S3A). As early as day 4 p.i., we found elevated numbers of CD11c+B220neg cDCs in Δpm4Δbp2-infected mice, compared to their Δlap-infected counterparts (Figures 4A and 4B). These increases were primarily associated with elevated frequencies and numbers of CD11bnegCD8α+ cDC1 in Δpm4Δbp2-infected mice (Figures 4C-4E) expressing elevated levels of MHCII (Figures 4F and 4G). Although expression of the costimulatory molecule CD80 was unaffected by hemozoin abundance (Figures 4H and 4I), CD86 expression was significantly increased on CD11bnegCD8α+ cDC1 recovered from Δpm4Δbp2-infected mice (Figures 4J and 4K). Relative to cells from Δlap-infected mice, similar frequencies of cDC1 from Δpm4Δbp2-infected mice exhibited the capacity to phagocytose fluorescent carboxylate beads and OVA protein, but the quantities of each phagocytosed reagent (gMFI) were significantly elevated (Figures 4L-4O). Enhanced cDC1 function was also observed in antigen processing, as CD11bnegCD8α+ cDC1 DCs recovered from Δpm4Δbp2-infected mice exhibited more efficient ovalbumin (OVA) protein cleavage and endosomal accumulation, compared to cDC1 recovered from Δlap-infected mice (Figures 4P and 4Q). Compromises in cDC antigen accumulation and processing and MHCII and costimulatory receptor expression were further magnified by the second week of primary infection in Δlap-infected mice (Figures S3B-S3I). Of note, CD11b+CD8αneg cDC2 functions were also impacted by the abundance of hemozoin, although these impacts were more modest (Figures S3J-S3L). Finally, and despite the caveats associated with dose, schedule, and formulation, we also found that direct injection of synthetic hemozoin into Δpm4Δbp2-infected mice reversed the enhanced cDC1 abundance (Figures S4A-S4C), MHC II expression (Figures S4D and S4E), and antigen processing (Figures S4F and S4G). These data phenocopy parameters observed in Δlap-infected mice and serve as an additional complementary approach to show that hemozoin impairs cDC1 functions during Plasmodium infection.
Figure 4. Hemozoin limits dendritic cell numbers and functions.
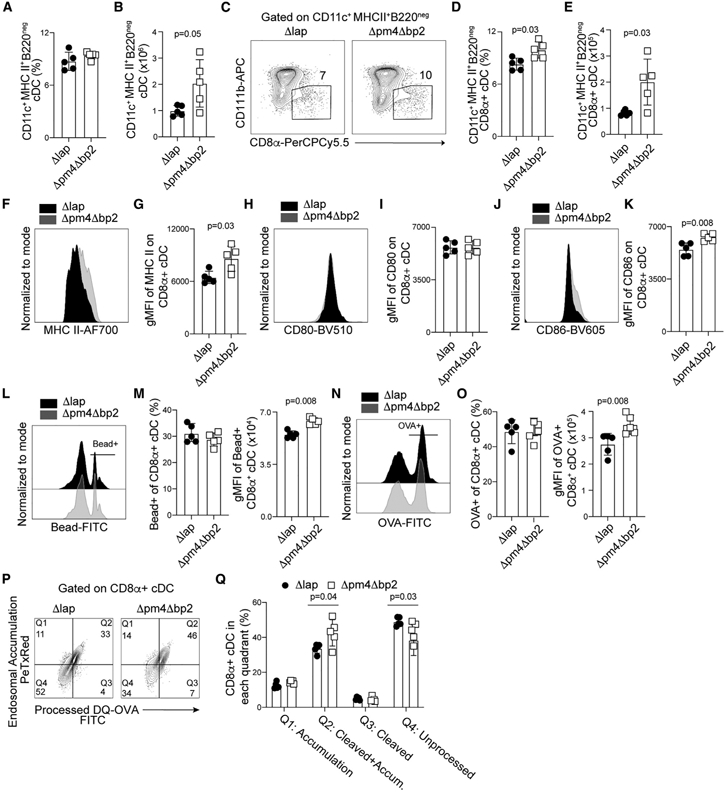
C57BL/6 mice were infected with either Δpm4Δbp2 or Δlap parasites, spleens were harvested on day 4 p.i., and DCs were liberated by digestion as described in STAR Methods.
(A–E) Frequency and number of CD11c+MHCII+B220neg conventional DCs (A and B) and CD11bnegCD8α+ cDC1 (C–E).
(F and G) Representative flow plots (F) and summary of MHC II expression (gMFI) (G) on CD11bnegCD8α+ cDC1.
(H–L) Representative flow plots and summary data of CD80 (H and I) and CD86 (J and K) expression on CD11bnegCD8α+ cDCs.
(L and M) Representative flow plots (L) and quantification of bead+CD11bnegCD8α+ cDCs (left) and fluorescence intensity of phagocytosed microbeads (gMFI) (right) (M).
(N and O) Representative flow plots of OVA-488 uptake (N), frequency of OVA+CD8α+ cDC (O, left), and fluorescence intensity of phagocytosed OVA (gMFI) (O, right).
(P and Q) Flow plots and summary data of DQ-OVA protein processing assay. Data (mean ± SD) in all experiments are representative of two independent experiments with n = 5 (see also Figure S3).
Hemozoin limits early CD4 T cell expansion and differentiation
Given the changes we observed in cDC number, phenotype, and function, we further tested whether hemozoin impaired the capacity of cDC to prime Plasmodium-specific CD4 T cells. PbTII activation and division, as evidenced by CTV dilution, was first evaluated on day 4 p.i., which corresponds to a time point matching our kinetic analyses of impaired cDC function (Figure 4) and the reported development of cDC immunoparalysis (Lundie et al., 2010). Consistent with our cDC phenotypic and functional analyses, reduced levels of hemozoin in Δpm4Δbp2-infected mice increased the capacity of antigen-presenting cells (APCs) to support parasite-specific T cell proliferation (Figure 5A). There were nearly 20% increases in the proportion of PbTII that proliferated and nearly 50% increases in the proportion of divided PbTII cells that differentiated into CD150loCXCR5intTFH precursor cells (Figures 5B and 5C). APC:CD4 T cell interactions are also crucial for helper cell migration into the GC, and this transition is regulated in part by either the strength of TCR stimulation (Benson et al., 2015; Goenka et al., 2011; Moran et al., 2011; Zemmour et al., 2018) or dwell time with APC (González et al., 2005; Govern et al., 2010). To investigate these aspects of cDC function, we transferred Nur77-GFP PbTII reporter cells to both Δpm4Δbp2 and Δlap-infected mice and found that reduced levels of hemozoin in Δpm4Δbp2-infected mice significantly enhanced TCR stimulation strength (Figures 5D and 5E), which likely contributes to the expanded number and proportion of TFH cells in Δpm4Δbp2-infected mice (Figure 3 and Figures 5B and 5C). In contrast to day 14 p.i. CXCR5hiPD-1hi mature GC-TFH (Figure 3I), we found elevated Bcl-6 expression among CD150loCXCR5int TFH precursor cells on days 7 and 10 p.i. (Figure 5F) in mice exposed to reduced levels of hemozoin. Importantly, the skewing of CD4 T cells toward a GC TFH fate in Δpm4Δbp2-infected mice was cDC intrinsic, as ex vivo co-culture of GP66–77 peptide-pulsed cDCs isolated from Δpm4Δbp2-infected mice resulted in both enhanced proliferation (Figures 5G and 5H) and trending upregulation of PD-1 and CXCR5 by GP66–77-specific TCR transgenic SMARTA CD4 T cells (Figures 5I and 5J). Notably, and similar to the cDC datasets, injection of synthetic hemozoin into Δpm4Δbp2-infected mice reversed the enhanced parasite-specific TFH differentiation phenotypes (Figures S4H and S4I). Taken together, these data support that cDC are numerically and functionally compromised by hemozoin and this alters their capacity to optimally induce CD4 T cell responses and TFH differentiation during blood-stage Plasmodium infection.
Figure 5. Optimal TFH expansion is impaired by hemozoin.

CTV-labeled PbTII cells were transferred into mice 1 day prior to infection with either Δpm4Δbp2 or Δlap-infected parasites and harvested 4 days later.
(A) Representative histograms and quantification of CTV dilution among PbTII cells.
(B and C) Flow plots (B) and summary (C) of CD150 and CXCR5 expression by CTV diluted PbTII.
(D) gMFI of Nur77 in CD150loCXCR5intTFH and CD150hiCXCR5loTH1 PbTII CD4+ T cells on day 7 p.i. with light blue corresponding to increased gMFI. Numbers on flow plots are gMFI.
(E) Summary of Nur77 gMFI among CD150loCXCR5intTFH
(F) Normalized Bcl-6 expression by PbTII TFH collected on days 7 and 10 p.i. with Δpm4Δbp2 or Δlap. CTV-labeled SMARTA cells were cocultured for 4 days with GP-66 pulsed cDCs isolated from either Δpm4Δbp2 or Δlap-infected mice.
(G and H) Histogram of CTV dilution (G) and summary (H) data collected from SMARTA cells on d4 of in vitro culture.
(I and J) Flow plots (I) and summary (J) of PD-1 and CXCR5 expression by CTV diluted SMARTA cells.
Data (mean ± SD) are representative of two independent experiments with n = 4–5 (see also Figure S4).
NLRP3 activation prevents the development of optimal TFH and humoral responses
To interrogate the mechanisms by which hemozoin erodes cDC numbers and function, with subsequent impacts in TFH cell development and humoral immunity, we focused on the NLRP3 inflammasome. In vitro data support that hemozoin engages and activates the NLRP3 inflammasome (Kalantari et al., 2014; Shio et al., 2009; Strangward et al., 2018), and we found that serum IL-1β levels were on average 75% lower in mice infected with Δpm4Δbp2 parasites, compared to mice infected with Δlap parasites (Figure S5A). Thus, we next tested whether the enhanced TFH cell differentiation phenotypes we observe in Δpm4Δbp2-infected mice were formally linked to NLRP3 inflammasome activation. To do this, we first transferred WT PbTII CD4 T cells to both C57BL/6 (WT) and NLRP3 deficient (KO) mice and subsequently infected the recipients with either Δlap or Δpm4Δbp2 parasites. Consistent with our previous observations (Figure 3), we found reduced frequencies of both PbTII CD150loCXCR5int TFH precursors and CXCR5hiPD-1hi mature GC-TFH populations in Δlap-infected WT mice, compared to Δpm4Δbp2-infected WT mice (Figure 6A, left panels). Strikingly, PbTII TFH and GC-TFH development in both Δlap-infected NLRP3 KO mice and WT mice treated from days 0 to 6 p.i. with the NLRP3 inflammasome inhibitor MCC950 phenocopied PbTII TFH differentiation in Δpm4Δbp2-infected WT mice (Figure 6A, right panels). NLRP3 activation also impaired the capacity of GC-TFH to promote GC B cell responses, as mature GC-TFH cells sort-purified from Δlap-infected NLRP3 KO mice induced 2-fold greater numbers of GC B cell in ex vivo co-culture assays, compared to GC-TFH cells recovered from Δlap-infected WT mice (Figure 6B). In addition to the deleterious impact of NLPR3 activation on TFH differentiation and function, the frequency and number of splenic GC B cells (Figures S5B and S5C) and CD95negCD73+CD80+PD-L2+ MBC (Figures 6C and 6D) were significantly decreased by either the presence of hemozoin or NLRP3 inflammasome activity.
Figure 6. Hemozoin-mediated impairments in humoral immunity are governed by NLRP3.
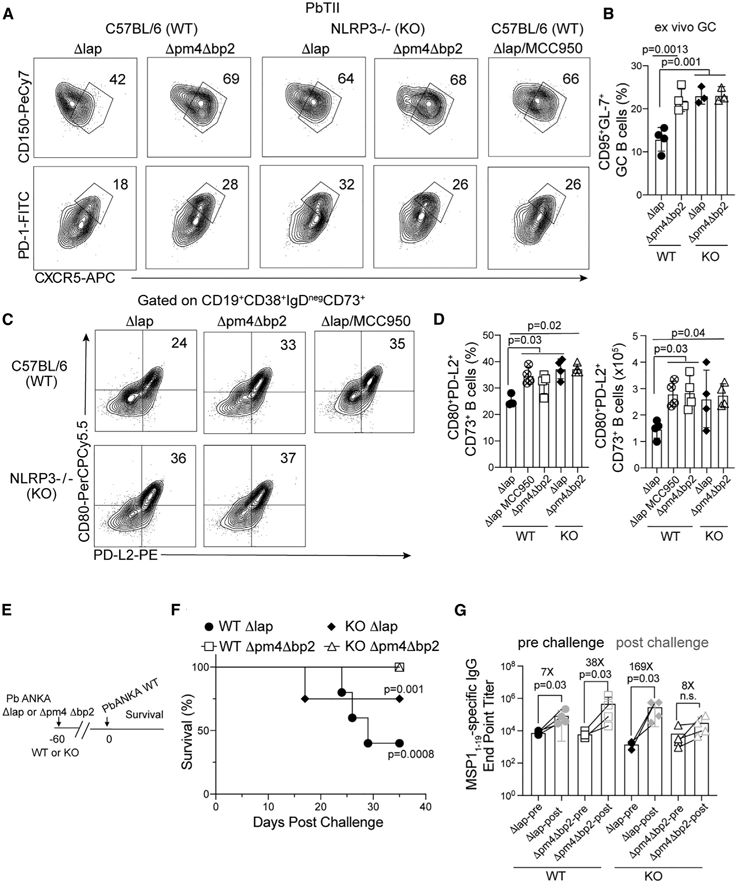
PbTII cells were transferred to C57BL/6 and NLRP3−/− mice 1 day prior to infection with either Δpm4Δbp2 or Δlap parasites. Splenic GC responses were evaluated on day 14 p.i.
(A) Representative flow plots of CD150loCXCR5int TFH and CXCR5hiPD-1hi GC-TFH populations.
(B) CXCR5hiPD-1hi GC-TFH cells were sort purified on day 14 p.i., co-cultured with naive B cells, and the frequency of CD95+GL-7+ GC B cells was quantified after 5 days.
(C and D) Representative flow plots (C) and frequency (D) of CD80+PD-L2+CD73+CD38+CD95neg MBC on day 28 p.i.
(E) C57BL/6 and NLRP3−/− mice were immunized with Δpm4Δbp2 and Δlap parasites 60 days prior to challenge with a virulent Pb-ANKA strain.
(F) Frequency of immune mice that survived virulent Pb-ANKA challenge.
(G) MSP11–19-specific IgG endpoint titers 1 day prior to (pre) and 5 days after (post) virulent Pb-ANKA challenge.
Data (mean ± SD) in (A)–(D) are representative of three experiments with n = 3–4. Data (mean ± SD) in (E)–(G) are representative of two independent experiments with n = 3–4 per experimental group (see also Figure S5).
To directly address whether hemozoin-mediated NLRP3 activation compromises long-term protective immunity in vivo, groups of both WT and NLRP3 KO mice were immunized with either Δpm4Δbp2 or Δlap parasites and then challenged with WT Pb-ANKA after 60 days (Figure 6E). Both WT and NLRP3 KO mice immunized with Δpm4Δbp2 exhibited 100% survival and reduced parasite burden (Figures 6F, S5D, and S5E) through day 35 p.i. However, more than 75% of Δlap-immune NLRP3 KO mice survived, compared to only 30% of Δlap-immune WT mice (Figure 6F). We also found functional MBC recall antibody responses were substantially improved in Δpm4Δbp2-immune WT mice and Δlap-immune NLRP3 KO mice, as evidenced by 38- and 169-fold boosts in parasite-specific IgG titers 5 days after Pb-ANKA challenge, respectively (Figure 6G), in contrast to the modest 7-fold boost in Δlap-immune WT mice. Collectively, these experiments show that the infection of NLRP3-deficient mice with Δlap parasites effectively phenocopies elevated GC-TFH and MBC numbers and functions observed following infection of WT mice with Δpm4Δbp2 parasites, further supporting that hemozoin-mediated NLRP3 activation impairs protective anti-Plasmodium humoral immunity.
Cell-intrinsic NLRP3 activation impairs cDC number and function
Our genetic and pharmacological approaches support that hemozoin-mediated NLRP3 inflammasome activation constrains TFH dependent humoral immunity. To determine whether these phenotypes are mechanistically linked to cDC-intrinsic NLRP3 activity and confirm that our findings extend beyond study of the mutant Pb ANKA parasites, we first evaluated cDC numbers and functions and TFH development in WT and NLRP3 deficient mice infected with Plasmodium yoelii parasites (Figure 7A). We observed that the total numbers of CD11bnegCD8α+ cDC1 (Figures 7B and 7C), and their relative MHC II expression (Figure 7D) were significantly increased in the absence of NLRP3 activation. Moreover, the capacity of cDC1 from P. yoelii-infected mice to both phagocytose multiple carboxylate beads (gMFI) (Figure 7E) and functionally process and accumulate cleaved OVA protein (Figures 7F and 7G) was markedly enhanced in the absence of hemozoin-mediated NLRP3 inflammasome activation, which closely resembles our findings with the mutant Pb ANKA parasites (Figure 4). Both PbTII and polyclonal CD4 T cell responses were also skewed toward a more robust TFH phenotype in P. yoelii-infected NLRP3-deficient mice, compared to P. yoelii-infected WT mice (Figures 7H and 7I). Importantly, P. yoelii-immune NLRP3-deficient mice were substantially more resistant to challenge with WT Pb ANKA (Figures S6A-S6C), phenotypes that additionally associated with enhanced MBC recall function (Figure S6D). Together, these data provide additional support that NLRP3 activation compromises cDC function and results in suboptimal TFH responses and protective humoral immunity.
Figure 7. Hemozoin impairs dendritic functions via cell-intrinsic NLRP3 activity.

(A) Experimental design for samples depicted in (B)–(I) from C57BL/6 (WT) and NLRP3−/− (KO) mice infected with P. yoelii 17XNL parasites.
(B and C) Summary CD11bnegCD8α+ cDC1 frequency (B) and number (C) in WT and NLRP3 KO mice.
(D) Histogram and summary of MHC II expression in CD8α+ cDC1.
(E) Histograms of bead uptake, summary graph of the frequency of bead positive CD8α+ cDC1 (center), and gMFI of phagocytosed beads (right).
(F and G) Flow plots (F) and summary (G) of DQ-OVA antigen processing assay. The frequency of CD11bnegCD8α+ cDC1 in each quadrant is depicted.
(H and I) Flow plots (H) and number (I) of CD150loCXCR5int TFH among parasite-specific (left) and polyclonal (right) CD4 T cells on day 6 p.i.
(J) Experimental schematic for C57BL/6:NLRP3−/− (50:50) bone-marrow chimera generation.
(K) Ratio of CD11b+CD8αneg cDC2 and CD11bnegCD8α+ cDC1 recovered from chimeras on d11 p.i.
(L and M) Histograms and quantification of MHC II expression by total cDC and CD11bnegCD8α+ cDC1.
(N) Flow plots of bead uptake (left), summarized frequency of fluorescent bead+ CD11bnegCD8α+ cDC1 (center), and gMFI of fluorescent beads are quantified (right).
(O) DQ-OVA antigen-processing assay.
(P) The frequency of CD11bnegCD8α+ cDC1 in each quadrant is quantified.
(Q) C57BL/6 (WT) mice were administered either MCC950 or PBS i.p. during the first 6 days of infection, and bone marrow was collected on day 28 for quantification of MSP19 LLPC via ELISPOT.
(R and S) Representative ELISPOT photos (R) and summary data (S).
Data (mean ± SD) in (A)–(I) and (Q)–(S) are representative of two independent experiments with n = 5. Data (mean ± SD) in (K)–(P) are the summary data collected from two independent experiments (see also Figure S6).
Finally, to compare WT and NLRP3 KO cDC1 development and function in the same Plasmodium-infected host, we generated and infected WT:NLRP3 KO (50:50) mixed bone-marrow chimeras with Δlap parasites (Figure 7J). Importantly, this approach controls for potential differences in parasite load, antigen, and inflammation in Plasmodium-infected WT and KO mice. Although only modest shifts in cDC1:cDC2 ratios were observed among NLRP3 KO cells (Figure 7K), we observed that NLRP3 KO cells exhibited significantly elevated MHC II expression among both total cDC and CD11bnegCD8α+ cDC1 populations (Figures 7L and 7M), compared to WT cDC and cDC1 populations. These data provide direct in vivo evidence that hemozoin-mediated cDC-intrinsic NLRP3 activation, rather than extrinsic and environmental cues that include inflammatory cytokines, such as IL-1β, regulate the expression of MHC II. Consistent with these findings, NLRP3 KO CD11bnegCD8α+ cDC1 exhibited significantly enhanced phagocytotic (Figure 7N) and protein processing functions (Figures 7O and 7P), compared to WT cDC1 recovered from the same Plasmodium infection environment. Last, to complement these genetic studies and confirm that early NLRP3 activation leads to impaired humoral immunity, we treated Δlap-infected WT mice with MCC950 during the initial stages of DC activation and CD4 T cell priming (days 0 to 6 p.i.) and measured the development of MSP119-specific plasma cells. Pharmacologic inhibition of NLRP3 inflammasome activity resulted in 2-fold greater numbers of parasite-specific plasma cells by day 28 p.i. (Figures 7Q-7S). Altogether, our data provide direct in vivo evidence that hemozoin-mediated inflammasome activation during an initial blood-stage Plasmodium exposure limits cDC1 numbers and functions, which in turn impairs the development of protective anti-Plasmodium cellular and humoral immunity.
DISCUSSION
The adaptive immune response elicited by Plasmodium infection is robust, but the resilience of humoral memory populations is suboptimal. Waning immune-mediated resistance to malarial disease in individuals who emigrate from endemic areas and the failure of vaccines in endemic populations highlight that new mechanistic understanding of why humoral responses following Plasmodium exposure are suboptimal remains a priority. Our study revealed that these defects may be programmed early after an initial Plasmodium exposure via hemozoin-mediated, NLRP3 inflammasome-dependent impacts on the cDC compartment with significant reductions in phagocytosis, antigen processing, and MHC II expression occurring as early as day 4 p.i. We further linked these cDC re-programming events to impairments in the stepwise differentiation of TFH, which resulted in reduced GC B cell responses and lower numbers of MBC and LLPC capable of producing protective, anti-parasite antibodies. Further investigation supported that humoral memory is suppressed by DC-intrinsic inflammasome activation. Importantly, hemozoin-induced NLRP3 activation limited humoral immunity irrespective of the strain of parasites (P. yoelii or P. berghei ANKA) used to initiate cDC reprogramming, suggesting that sustained hemozoin-induced compromise of CD8α+ cDC numbers and function may be a conserved pathophysiologic feature of blood-stage Plasmodium infection.
Despite the genetic and life-cycle complexity of Plasmodium parasites, hemozoin is currently the only described malaria-associated NLRP3 inflammasome agonist. The capacity for inflammasome activation to alter cDC gene expression and phenotype has been evaluated ex vivo, but studies designed to evaluate the contribution of DC-intrinsic inflammasome activation during vaccination and infection are limited. A recent report showed that inflammasome activation during Salmonella infection is detrimental to T cell priming and that reduced levels of inflammasome machinery limit splenic DCs from undergoing inflammasome-mediated pyroptosis (McDaniel et al., 2020). It was further reported that cDC activation by inflammasomes rarely occurs early during infection because of IRF4 and IRF8-mediated suppression of inflammasome genes, but this observation is not consistent with those from the Plasmodium field where robust expression of inflammasome genes and caspase 1 activation are observed following both rodent and human infection (Ataide et al., 2014). Indeed, the magnitude of inflammasome activation that occurs during the blood stage of Plasmodium infection is supported by high levels of serum IL-1β, which contribute to malarial symptoms that include fever, chills, and malaise. Deleterious functions for hemozoin during malaria are also suggested by strong correlations between hemozoin accumulation and severe clinical manifestations and inflammation in the central nervous system and placenta, where increased accumulation of monocytes and platelets can exacerbate disease (Hochman et al., 2016; Milner et al., 2013; Moore et al., 2004; Nguyen et al., 1995; Sarr et al., 2006). We found that reducing cDC exposure to hemozoin limited splenic IL-1β levels by day 10 p.i. and significantly increased DC maturation kinetics and function. Our in vivo studies showed that hemozoin-induced, NLRP3-dependent inflammasome activation impaired cDC phagocytosis, antigen processing, and expression of MHC II. Reported discrepancies between experimental studies of ex vivo and in vivo contributions of inflammasome activation and resultant programming of DC fate and function may be linked to the severity of malaria or virulence of specific species of Plasmodium parasites, as well as the tissue-specific deposition and abundance of hemozoin. Our complementary genetic, biochemical, and pharmacologic studies support that NLRP3 activation exerts a similarly deleterious impact on humoral immunity following either P. yoelii or P. berghei ANKA primary infections.
The functional defects we observed in cDC following hemozoin-mediated NLRP3 inflammasome activation were further linked to negative impacts on the Plasmodium-specific T cell response. T cell priming, the emergence of CD150loCXCR5intTFH precursors, and the eventual differentiation of mature CXCR5hiPD-1hiGC-TFH cells were all reduced by hemozoin-mediated NLRP3 activation in cDC. Notably, alum is a well-documented NLRP3 agonist and adjuvant reported to promote the initiation of T cell and humoral responses. Alum’s capacity to dissociate from vaccine components upon injection differs significantly from that of hemozoin, which is retained within the DCs. This difference may serve as one explanation for disparities in reported outcomes following examination of NLRP3-mediated DC function. Our high-resolution analyses revealed that, in the absence of NLRP3 activation, parasite-specific T cells exhibited evidence of enhanced TCR stimulation, which is reported to support increased CXCR5 expression and reinforce TFH differentiation. Whether the presence of hemozoin and altered DC programming impairs a limited number of GC-TFH functions or reduces the expression of multiple co-stimulatory molecules and cytokines is not currently known. Elucidation of the precise mechanisms by which hemozoin limits the capacity of cDC to promote the development of optimal GC-TFH function remains of interest and will also be relevant to our understanding of how Plasmodium infection influences responses to either heterologous infection or vaccination.
Our results support the hypothesis that hemozoin and the associated programming of DC number and function contribute to suboptimal humoral responses following Plasmodium infection. During a blood-stage Plasmodium infection with reduced levels of hemozoin, enhancements in humoral immunity begin early in the spleen, where we observed a coordinated shift in CXCR4+CD138+ LLPC precursors and accumulation of mature LLPC in the bone marrow. Our data support a model wherein the abundance of LLPC or reactivation of memory B cells mediate protection from lethal parasite challenge, as opposed to either qualitative shifts in either the isotype or affinity of parasite-specific antibodies in mice with reduced hemozoin exposure. Limitations of our study include the sensitivity of assays used to probe the quantity, quality, and functions of secreted antibody responses, particularly in our studies showing enhanced survival yet equivalent immune memory baseline titers in NLRP3 KO mice and WT mice infected with Δpm4Δbp2 parasites. Future molecular and biophysical studies designed to interrogate critical qualitative attributes and effector functions of the secreted antibody responses are warranted. Another notable aspect of our studies that we did not assess is whether hemozoin accumulation within the bone marrow impacts or erodes the maintenance of either vaccine-induced or pathogen-specific LLPC. The bone marrow serves as an infection reservoir for P. vivax parasites, and the presence of hemozoin in the bone marrow pushes the cytokine environment toward a more proinflammatory state (Lee et al., 2017). Understanding whether either inflammation or cytokine and chemokine perturbations contribute to reductions in the frequency of mature LLPC following infection with Plasmodium may shed light onto additional factors that restrain the quality and/or durability of anti-Plasmodium humoral responses.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Noah Butler (noah-butler@uiowa.edu).
Materials availability
This study did not generate unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This study did not generate/analyze datasets or code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
C57BL/6 wild-type (CD45.2 and CD45.1), C57BL/6-Tg(Nr4a1-EGFP/cre)820Khog/J (Nur77GFP Stock No. 016617) and Nlrp3tm1Bhk (NLRP3 KO stock No. 021302) were either purchased from The Jackson Laboratory or acquired from colonies at the University of Iowa. For the generation of WT: NLRP3−/− chimeras, CD45.1 wild-type recipient mice were irradiated with 475 rads twice, separated by 4 h. Bone marrow cells from CD45.1 (C57BL/6) and CD45.2 (NLRP3 KO) were mixed 50:50 and 1 × 107 were injected i.v. Mice were maintained on a Uniprim diet (Envigo) for 2 weeks. Chimerism was assessed at 8 weeks post-irradiation. Age and sex-matched mice between 6 and 13 weeks were used for all experiments. Ethics oversight for animal use was provided by The University of Iowa IACUC.
Parasite strains and infections
Two wild-type P. berghei ANKA parasite lines were used: the Pb-ANKA line, obtained from MR4 (American Type Culture Collection) and the reporter, reference P. berghei ANKA line 676m1cl1 (Pb-ANKA-Luc), which constitutively express GFP and luciferase that was obtained through BEI Resources (MRA-868) and was contributed by Chris J. Janse and Andrew P. Waters. The Plasmodium berghei ANKA mutants Δpm4Δbp2 and Δlap have been described in Lin et al. (2015) and were obtained from Dr. Chris Janse (Leiden University Medical Center, LUMC, the Netherlands). P. yoelii (clone 17XNL) were obtained from the Malaria Research and Reference Reagent Resource Center (MR4, American Type Culture Collection). All parasite lines were maintained by passage in NIH Swiss Webster mice. Primary infections were initiated by an intravenous (IV) transfer of 1 *× 106 parasitized red blood cells (RBCs) derived from an infected donor mouse. Challenge of mice with virulent Pb-ANKA or Pb-ANKA-luc were initiated by intravenous (IV) injection of 5 × 106 donor-derived parasitized RBCs.
Quantification of parasite biomass
Percentage of infected RBC was monitored in tail blood samples by flow cytometric analysis of the frequency of Hoechst+ Diethydiumbromide+Ter119+CD45neg cells (Malleret et al., 2011). Parasite loads, quantified as total flux (p/s/cm2/sr), following IP injection of 250 μg of D-luciferin (Promega) were determined using an IVIS Lumina S5 and Living Image software (Perkin Elmer).
Treatments
Synthetic hemozoin (sHZ) (InvivoGen; 750 μg in PBS) was injected every second day IV (tail vein). MCC950 (Sigma, 250 μg in PBS) was administered via intraperitoneal (IP) injection every 2 days beginning at the time of infection.
METHOD DETAILS
Immune cell phenotyping
For analysis of splenic cells, single cell suspensions were generated by forcing mouse spleens through a 70 μm mesh. For experiments enumerating cDC subsets, spleens were incubated with collagenase IV (100 U/mL) and DNase I (10 μg/mL) for 30 mins at 37°C prior to preparation of single cell suspensions. RBC were lysed prior to Fc receptor blockade (CD16/32-clone 2.4G2) in FACS buffer (PBS+ 0.09% sodium azide + 2% FCS) for 15 min at 4°C prior to surface staining. For identifying TFH like cells, cells were stained with purified rat anti-mouse CXCR5 (BD Biosciences) in TFH staining buffer (PBS+ 0.09% sodium azide + 2% FCS+ 0.05% BSA+ 2% mouse serum) for 1h at 4 °C. Cells were washed then stained with biotin-conjugated AffiniPure Goat anti-rat IgG (H+L) (Jackson Immunoresearch) in TFH buffer for 30 min. Cells were then washed and surfaced stained in an antibody cocktail containing (Anti-CD4, -PD-1, -CD150, -CD11a, -CD44, streptavidin) for at least 30 min at 4°C. Anti-Bcl-6, -T-bet and -Foxp3 (BioLegend) transcription factor staining was performed using the Foxp3 Staining Kit per manufacturer’s protocol (BD Biosciences). For B cell staining, cells were surface stained in FACS buffer for at least 30 min in the following cocktail solution containing anti-CD19, -B220, -CD267, -CD138, -CD38 (BioLegend), -CD95 (BD PharMingen), -IgD and -CXCR4 (BioLegend) to identify plasma cells, while a cocktail of anti-CD19, -B220, -CD38, -CD80, -CD273, -CD73, -IgD, -CCR6 (BioLegend) was used to identify memory B cells. cDCs were identified using a surface stain mix containing Anti-CD8, -F4/80, -CD80, -CD86, -CD19, -B220, -CD11c (BioLegend) and anti-CD11b and MHC II (Thermofisher) following Fc-block for 10 mins at 4°C.
Standard and antibody affinity ELISA
For standard ELISA, Nunc Immunosorb Plates were coated with 1 μg/mL of recombinant MSP19 overnight at 4°C. Plates were blocked for 2h at RT with PBS+2.5%BSA+ 2%FCS prior to addition of serially diluted sera and incubation overnight at 4°C. Wells were washed with PBS + 0.05% Tween20 prior to incubation with HRP conjugated anti-mouse IgG for 2h at RT. For affinity ELISA, after plates were coated overnight as described above, plates were washed with PBS-Tween prior to blocking with PBS+1% BSA+0.05% Tween for 1h at 37 °C. Diluted serum was incubated for 2h at RT in buffer (2.5% BSA+5% FCS in PBS). Wells were washed extensively with PBS+Tween20 prior to addition of diluted ammonium thiocyanate and incubation for 15 minutes. Wells were washed prior to incubation with HRP conjugated anti-mouse IgG for 1h at 37°C. All plates were reacted with SureBlue TMB Peroxidase and TMB Stop Solution (Sera Care) prior to absorbance measurement at an OD of 450 nm using a plate reader (Biotek). The concentration of serum IL-1β was assessed using an IL-1β LEGENDplex immunoassay (Biolegend) according to the manufacturer’s protocol.
ELISPOT
White plates (Nunc MaxiSorp) were coated with 1 μg/mL of recombinant MSP-19 diluted in 0.1M Na2HPO4 binding solution and incubated overnight at 4°C. Plates were washed with PBS and blocked for 2h at RT with 10% FCS in RPMI 1640. Bone marrow cells were diluted serially in 10% FCS in RPMI 1640 and incubated for 18 h at 37 °C with 5% CO2. Plates were thoroughly washed then incubated with HRP-conjugated anti-mouse IgG in PBS+5% FCS overnight at 4 °C. Spots were then developed with 3-amino-9-ethylcarbazole.
IgG purification
The method for IgG purification from immune serum using Protein G Sepharose4 Fast Flow columns was adapted from manufacturer’s protocol and Akter et al. (Akter et al., 2019). Briefly, following column equilibration with binding buffer (20 mM sodium phosphate), diluted serum (1:4) was ran over column and flow-through was collected prior to complex elution. IgG was eluted from the column using 0.1M glycine+0.01% sodium azide and the pH was neutralized using 1M Tris-HCl. Amicon Ultra-4 Membrane Units were used for buffer exchange to PBS.
CTV labeling of T cells
CTV labeled CD4+ T cells were prepared according to manufacturer’s protocol (Invitrogen) with minor protocol modification. Splenocytes were washed and suspended in serum free media supplemented 1:1000 with Cell Trace Violet. Cells were incubated at 37°C for 20 min prior to dilution with RPMI 1640+10% FCS and incubation for 5 mins. Cells were pelleted and incubated again for 5 mins with RPMI 1640+10% FCS at 37°C prior to surface staining.
Ex vivo cultures
GC-TFH (CXCR5hiPD-1hi) were sort purified from Δpm4Δbp2- and Δlap-infected mice on day 14 p.i. and suspended in IMDM+10% FCS. Naive B cells, enriched from naive mice using a B cell Isolation Kit (Miltenyi Biotec), were mixed with GC-TFH at a 1:1 ratio and co-incubated for 5 days at 37°C with 5% CO2. Cell cultures were supplemented with anti-IgM (5 μg/mL) (Jackson Immunoresearch) and anti-CD3 (5 μg/mL) (Biolegend). For cDC-T cell cultures, magnetically enriched cDCs were pulsed with 1 nM of GP-66 for 1h, washed, and incubated at a 1:10 ratio with CTV-labeled SMARTA cells for 4 days.
Phagocytosis assays
To examine the phagocytic capacity of cDC, splenocyte suspensions were incubated with Fluoresbrite carboxylate microsphere YG (0.5 μm-Polysciences) at a ratio of 1:100 microspheres/cell or in 25 μg/mL solution OVA-488 (Molecular Probes) for 1h at 37°C. To assess antigen processing by cDC, splenocytes were incubated in a 25 μg/mL solution of DQ-OVA (Molecular Probes) for 1h at 37°C. Cells were then stained with Fc-block for 10 mins at 4°C prior to surface staining.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data centers in summary data graphs represent the mean. Data dispersion and tests of statistical significance are detailed in the figure legends. Graphs were generated and statistical analyses were performed using Prism Graphpad 9 software (GraphPad Software). For analyses comparing only two groups, Mann-Whitney non-parametric tests were used to determine p value. For comparison of 3 or more groups, Dunn’s test for nonparametric multiple comparisons were employed and adjusted P values are reported. Details can be found in respective figure legends.
Supplementary Material
KEY RESOURCES TABLE
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Purified anti-mouse CD3 | Biolegend | Cat#100202; RRID: AB_312659 |
| Anti-mouse CD16/32 (Fc-Block) | BioXCell | Cat#BE0307, RRID: AB_2736987 |
| Biotin SP-conjugated Affinipure Goat-anti-IgG (H+L) F(ab) | Jackson Immunoresearch | Cat#112-065-167, RRID: AB_2338179 |
| Purified rat anti-mouse CXCR5 | BD Biosciences | Cat#551961, RRID: AB_394302 |
| Anti-mouse CXCR4-BV605 (clone: L276F12) | Biolegend | Cat#146519, RRID: AB_2687359 |
| Anti-mouse CD150-PeCy7 (clone:TC15-12G12.2) | Biolegend | Cat#115914, RRID: AB_439797 |
| Anti-mouse CD267-PE (clone 8F10) | Biolegend | Cat#133403, RRID: AB_2203542 |
| Anti-mouse IgD-PeDazzle 594 (clone: 11-26c.2a) | Biolegend | Cat#405742, RRID: AB_2571985 |
| Anti-mouse CD86-BV605 (clone:GL1) | Biolegend | Cat#105037, RRID: AB_11204429 |
| Anti-mouse CD80-BV510 (clone:16-10A1) | Biolegend | Cat#104741, RRID: AB_2810337 |
| Anti-mouse CD80-PerCPCy5.5 (clone:16-10A1) | Biolegend | Cat#104722, RRID: AB_2291392 |
| Anti-mouse CD11c-PE Cy7 (clone:N418) | Biolegend | Cat#117318, RRID: AB_493568 |
| Anti-mouse CD38-Pac Blue (clone:90) | Biolegend | Cat#102720, RRID: AB_10613468 |
| Anti-mouse CD95-FITC (clone: Jo2) | BD Biosciences | Cat#554257, RRID: AB_395329 |
| Anti-mouse MHCII-AF700 (clone:M5/114.15.2) | Biolegend | Cat#107622, RRID: AB_493727 |
| Anti-mouse CCR6-AF647 (clone:29-2L17) | Biolegend | Cat#129808, RRID: AB_1227497 |
| Anti-mouse CD138 Pac Blue (clone 281-2) | Biolegend | Cat#142523, RRID: AB_2565621 |
| Anti-mouse B220-PerCP-Cy5.5 (clone:RA3-6B2) | Biolegend | Cat#103236, RRID: AB_893354 |
| Anti-mouse B220-PE (clone:RA3-6B2) | Invitrogen | Cat#12-0452-83, RRID: AB_465672 |
| Anti-mouse CD11b-APC (clone: M17/70) | Biolegend | Cat#101212, RRID: AB_312795 |
| Anti-mouse CD4-PerCP-Cy5.5 (clone:GK1.5) | Biolegend | Cat#100434, RRID: AB_893324 |
| Anti-mouse CD8-PerCP-Cy5.5 (clone: 53-6.7) | Biolegend | Cat#100734, RRID: AB_2075238 |
| Anti-mouse CD19-AF700 (clone: 6D5) | Biolegend | Cat#115528, RRID: AB_493735 |
| Anti-mouse CD45.2-PE (clone: 104) | Biolegend | Cat#109808, RRID: AB_313445 |
| Anti-mouse CD273-PE (clone: TY25) | Biolegend | Cat#107206, RRID: AB_2162011 |
| Anti-mouse F4/80-PE (clone: BMB) | Biolegend | Cat#123110, RRID: AB_893486 |
| Anti-mouse Foxp3-PeCy7 (clone: 3G3) | Tonbo | Cat#60-5773-U100, RRID: AB_2621869 |
| Anti-mouse Bcl-6-AF647 (clone IG191E/A8) | Biolegend | Cat#648305, RRID: AB_2565298 |
| Anti-mouse Tbet-BV605 (clone: 4B10) | Biolegend | Cat#644817, RRID: AB_11219388 |
| Anti-mouse CD95-PeCy7 (clone: Jo2) | BD PharMingen | Cat#557653, RRID: AB_396768 |
| Anti-mouse CD73-FITC (clone: TY/11.8) | Biolegend | Cat#127220, RRID: AB_2716076 |
| Anti-mouse PD-1-BV510 (clone: RPM1-30) | Biolegend | Cat#135241, RRID: AB_2715761 |
| Anti-mouse PD-1-FITC (clone: RPM1-30) | Thermo Fisher Scientific | Cat#11998181, RRID: AB_465466 |
| Anti-mouse CD45.1-PacBlue (clone: A20) | Biolegend | Cat#110722, RRID: AB_492866 |
| Anti-mouse CD90.1-PeDazzle (clone: OX-7) | Biolegend | Cat#202524, RRID: AB_1595635 |
| Anti-mouse CD44-AF700 (clone:IM7) | Biolegend | Cat#103026, RRID: AB_493713 |
| Anti-mouse CD44-FITC (clone:IM7) | Biolegend | Cat#103006, RRID: AB_312957 |
| Anti-mouse CD11a-FITC (clone:M17/4) | Biolegend | Cat#101106, RRID: AB_312779 |
| Anti-mouse GL7-FITC (Clone:GL7) | Biolegend | Cat#144603, RRID: AB_2561696 |
| Chemicals, peptides, and recombinant proteins | ||
| Hemozoin | InvivoGen | Cat#tlrl-hz |
| MCC950 (NLRP3 Inhibitor) | Sigma | Cat#256373-96-3 |
| D-luciferin | Promega | Cat#P1043 |
| Collagenase IV | GIBCO | Cat#17104019 |
| DNase I | Roche | Cat#10104159001 |
| Cell Trace Violet | Invitrogen | Cat#C34557 |
| streptavidin-BV421 | BD Biosciences | Cat#563259 |
| streptavidin-APC | BD Biosciences | Cat#405207 |
| Fluoresbrite carboxylate microsphere YG 0.5 μm | Polysciences | Cat#15700 |
| OVA-488 | Molecular Probes | Cat#O34781 |
| DQ-OVA | Molecular Probes | Cat#D12053 |
| B Cell Isolation Kit | Miltenyi Biotec | Cat#130-090-862 |
| GP66-77 peptide | BioSynthesis Inc. | Cat#Custom |
| Recombinant MSP119 (yPyMSP119XL) | BEI Resources | Cat#MRA-48 |
| Critical commercial assays | ||
| SureBlue TMB Peroxidase | Sera Care | Cat#5120-0077 |
| TMB Stop Solution | Sera Care | Cat#5150-0021 |
| LEGENDplex immunoassay | Biolegend | Cat#740150 |
| Experimental models: Organisms/strains | ||
| C57BL/6J | Jackson Laboratories | Cat#RRID:IMSR_JAX:000664 |
| C57BL/6-Tg(Nr4a1-EGFP/cre)820Khog/J | Jackson Laboratories | Cat#RRID:IMSR_JAX:016617 |
| B6.129S6-Nlrp3tm1Bhk/J | Jackson Laboratories | Cat#RRID:IMSR_JAX:021302 |
| B6.SJL-Ptprca Pepcb/BoyJ | Jackson Laboratories | Cat#RRID:IMSR_JAX:002014 |
| B6.Cg-Ptprca Pepcb Tg(TcrLCMV)1Aox/PpmJ | Jackson Laboratories | Cat#RRID:IMSR_JAX:030450 |
| P. berghei ANKA | BEI-MR4 | Cat#MRA-871 |
| P. berghei ANKA line 676m1cl1 | BEI-MR4 | Cat#MRA-868 |
| P. yoelii (clone 17XNL) | BEI-MR4 | Cat#MRA-593 |
| P. berghei ANKA Δpm4Δbp2 and Δlap | A gift from Dr. Chris Janse (Leiden University) | https://rupress.org/jem/article/212/6/893/41818/Replication-of-Plasmodium-in-reticulocytes-can |
| Software and algorithms | ||
| Flowjo v10.5.0 | Treestar | https://www.flowjo.com/solutions/flowjo/downloads |
| Prism v9 | Graphpad | https://www.graphpad.com |
| Excel v16.16.20 9 | Microsoft | https://www.microsoft.com/en-us/microsoft-365/excel |
| Living Image | Perkin Elmer | https://www.perkinelmer.com/product/spectrum-200-living-image-v4series-1-128113 |
| Other | ||
| Biotek plate reader | Biotek | N/A |
| BD LSR II | BD | N/A |
| BD FACSVerse | BD | N/A |
| IVIS Lumina S5 | Perkin Elmer | N/A |
Highlights.
Hemozoin engages NLRP3 to reduce CD8α+ dendritic cell (DC) number and function
Impaired DC responses compromise the B cell helper functions of CD4+ T cells
The accumulation of hemozoin reduces memory B cell and plasma cell responses
NLRP3 deficiency boosts humoral immune memory-mediated protection
ACKNOWLEDGMENTS
The authors acknowledge the late Shahid M. Khan (University of Leiden) for his contributions to generating and characterizing the mutant parasites used throughout this study. We also thank members of the Butler lab for thoughtful discussions. The research reported in this publication was supported by the NCI (grant number P30CA086862) and the National Center for Research Resources of the NIH (grant number S10OD016199). A.D.P. was supported by the NIH (grant number T32AI007511). P.G. was supported by the NIH (grant number AI148904). Z.R.Z. was supported by the NIH (grant number T32AI007260). K.L.L. was supported by the NIH (grant numbers AI141196 and AI127565). W.R.H. was supported by the Australian Research Council (grant number CE140100011) and the National Health and Medical Research Council (NHMRC; grant numbers 1113293 and 1154457). D.F-R. was supported by the National Health and Medical Research Council (grant number 1139486). N.S.B. was supported by the NIH (grant numbers AI125446 and AI127481).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109586.
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. One or more of the authors of this paper received support from a program designed to increase minority representation in science.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Akter J, Khoury DS, Aogo R, Lansink LIM, SheelaNair A, Thomas BS, Laohamonthonkul P, Pernold CPS, Dixon MWA, Soon MSF, et al. (2019). Plasmodium-specific antibodies block in vivo parasite growth without clearing infected red blood cells. PLoS Pathog. 15, e1007599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ataide MA, Andrade WA, Zamboni DS, Wang D, Souza Mdo.C., Franklin BS, Elian S, Martins FS, Pereira D, Reed G, et al. (2014). Malaria-induced NLRP12/NLRP3-dependent caspase-1 activation mediates inflammation and hypersensitivity to bacterial superinfection. PLoS Pathog. 10, e1003885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson RA, MacLeod MK, Hale BG, Patakas A, Garside P, and Brewer JM (2015). Antigen presentation kinetics control T cell/dendritic cell interactions and follicular helper T cell generation in vivo. eLife 4, e06994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, Lao C, and Crotty S (2011). ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity 34, 932–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua CL, Brown G, Hamilton JA, Rogerson S, and Boeuf P (2013). Monocytes and macrophages in malaria: protection or pathology? Trends Parasitol. 29, 26–34. [DOI] [PubMed] [Google Scholar]
- Crotty S (2014). T follicular helper cell differentiation, function, and roles in disease. Immunity 41, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Saha B, Hati AK, and Roy S (2018). Evidence of Artemisinin-Resistant Plasmodium falciparum Malaria in Eastern India. N. Engl. J. Med 379, 1962–1964. [DOI] [PubMed] [Google Scholar]
- deWalick S, Amante FH, McSweeney KA, Randall LM, Stanley AC, Haque A, Kuns RD, MacDonald KP, Hill GR, and Engwerda CR (2007). Cutting edge: conventional dendritic cells are the critical APC required for the induction of experimental cerebral malaria. J. Immunol 178, 6033–6037. [DOI] [PubMed] [Google Scholar]
- Elgueta R, Marks E, Nowak E, Menezes S, Benson M, Raman VS, Ortiz C, O’Connell S, Hess H, Lord GM, and Noelle R (2015). CCR6-dependent positioning of memory B cells is essential for their ability to mount a recall response to antigen. J. Immunol 194, 505–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott SR, Spurck TP, Dodin JM, Maier AG, Voss TS, Yosaatmadja F, Payne PD, McFadden GI, Cowman AF, Rogerson SJ, et al. (2007). Inhibition of dendritic cell maturation by malaria is dose dependent and does not require Plasmodium falciparum erythrocyte membrane protein 1. Infect. Immun 75, 3621–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Ruiz D, Lau LS, Ghazanfari N, Jones CM, Ng WY, Davey GM, Berthold D, Holz L, Kato Y, Enders MH, et al. (2017). Development of a Novel CD4+ TCR Transgenic Line That Reveals a Dominant Role for CD8+ Dendritic Cells and CD40 Signaling in the Generation of Helper and CTL Responses to Blood-Stage Malaria. J. Immunol 199, 4165–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goenka R, Barnett LG, Silver JS, O’Neill PJ, Hunter CA, Cancro MP, and Laufer TM (2011). Cutting edge: dendritic cell-restricted antigen presentation initiates the follicular helper T cell program but cannot complete ultimate effector differentiation. J. Immunol 187, 1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González PA, Carreño LJ, Coombs D, Mora JE, Palmieri E, Goldstein B, Nathenson SG, and Kalergis AM (2005). T cell receptor binding kinetics required for T cell activation depend on the density of cognate ligand on the antigen-presenting cell. Proc. Natl. Acad. Sci. USA 102, 4824–4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz A, Tang MS, Ty MC, Arama C, Ongoiba A, Doumtabe D, Traore B, Crompton PD, Loke P, and Rodriguez A (2017). Atypical activation of dendritic cells by Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 114, E10568–E10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govern CC, Paczosa MK, Chakraborty AK, and Huseby ES (2010). Fast on-rates allow short dwell time ligands to activate T cells. Proc. Natl. Acad. Sci. USA 107, 8724–8729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen DS, Obeng-Adjei N, Ly A, Ioannidis LJ, and Crompton PD (2017). Emerging concepts in T follicular helper cell responses to malaria. Int. J. Parasitol 47, 105–110. [DOI] [PubMed] [Google Scholar]
- Hatzi K, Nance JP, Kroenke MA, Bothwell M, Haddad EK, Melnick A, and Crotty S (2015). BCL6 orchestrates Tfh cell differentiation via multiple distinct mechanisms. J. Exp. Med 212, 539–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochman SE, Madaline TF, Wassmer SC, Mbale E, Choi N, Seydel KB, Whitten RO, Varughese J, Grau GE, Kamiza S, et al. (2016). Author Correction for Hochman et al. , Fatal Pediatric Cerebral Malaria Is Associated with Intravascular Monocytes and Platelets That Are Increased with HIV Coinfection. mBiol. 7, 02068–02015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John CC, O’Donnell RA, Sumba PO, Moormann AM, de Koning-Ward TF, King CL, Kazura JW, and Crabb BS (2004). Evidence that invasion-inhibitory antibodies specific for the 19-kDa fragment of merozoite surface protein-1 (MSP-1 19) can play a protective role against blood-stage Plasmodium falciparum infection in individuals in a malaria endemic area of Africa. J. Immunol 173, 666–672. [DOI] [PubMed] [Google Scholar]
- Kalantari P, DeOliveira RB, Chan J, Corbett Y, Rathinam V, Stutz A, Latz E, Gazzinelli RT, Golenbock DT, and Fitzgerald KA (2014). Dual engagement of the NLRP3 and AIM2 inflammasomes by plasmodium-derived hemozoin and DNA during malaria. Cell Rep. 6, 196–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kräutler NJ, Suan D, Butt D, Bourne K, Hermes JR, Chan TD, Sundling C, Kaplan W, Schofield P, Jackson J, et al. (2017). Differentiation of germinal center B cells into plasma cells is initiated by high-affinity antigen and completed by Tfh cells. J. Exp. Med 214, 1259–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurty AT, Thouvenel CD, Portugal S, Keitany GJ, Kim KS, Holder A, Crompton PD, Rawlings DJ, and Pepper M (2016). Somatically Hypermutated Plasmodium-Specific IgM(+) Memory B Cells Are Rapid, Plastic, Early Responders upon Malaria Rechallenge. Immunity 45, 402–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurup SP, Obeng-Adjei N, Anthony SM, Traore B, Doumbo OK, Butler NS, Crompton PD, and Harty JT (2017). Regulatory T cells impede acute and long-term immunity to blood-stage malaria through CTLA-4. Nat. Med 23, 1220–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenkamp A, Messi M, Lanzavecchia A, and Sallusto F (2000). Kinetics of dendritic cell activation: impact on priming of TH1, TH2 and nonpolarized T cells. Nat. Immunol 1, 311–316. [DOI] [PubMed] [Google Scholar]
- Lee MSJ, Maruyama K, Fujita Y, Konishi A, Lelliott PM, Itagaki S, Horii T, Lin JW, Khan SM, Kuroda E, et al. (2017). Plasmodium products persist in the bone marrow and promote chronic bone loss. Sci. Immunol 2, 2. [DOI] [PubMed] [Google Scholar]
- Leisewitz AL, Rockett KA, Gumede B, Jones M, Urban B, and Kwiatkowski DP (2004). Response of the splenic dendritic cell population to malaria infection. Infect. Immun 72, 4233–4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightman SM, Utley A, and Lee KP (2019). Survival of Long-Lived Plasma Cells (LLPC): Piecing Together the Puzzle. Front. Immunol 10, 965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JW, Spaccapelo R, Schwarzer E, Sajid M, Annoura T, Deroost K, Ravelli RBG, Aime E, Capuccini B, Mommaas-Kienhuis AM, et al. (2015). Replication of Plasmodium in reticulocytes can occur without hemozoin formation, resulting in chloroquine resistance. J. Exp. Med 212, 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughland JR, Minigo G, Burel J, Tipping PE, Piera KA, Amante FH, Engwerda CR, Good MF, Doolan DL, Anstey NM, et al. (2016). Profoundly Reduced CD1c+ Myeloid Dendritic Cell HLA-DR and CD86 Expression and Increased Tumor Necrosis Factor Production in Experimental Human Blood-Stage Malaria Infection. Infect. Immun 84, 1403–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundie RJ, Young LJ, Davey GM, Villadangos JA, Carbone FR, Heath WR, and Crabb BS (2010). Blood-stage Plasmodium berghei infection leads to short-lived parasite-associated antigen presentation by dendritic cells. Eur. J. Immunol 40, 1674–1681. [DOI] [PubMed] [Google Scholar]
- Malleret B, Claser C, Ong AS, Suwanarusk R, Sriprawat K, Howland SW, Russell B, Nosten F, and Rénia L (2011). A rapid and robust tri-color flow cytometry assay for monitoring malaria parasite development. Sci. Rep 1, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel MM, Kottyan LC, Singh H, and Pasare C (2020). Suppression of Inflammasome Activation by IRF8 and IRF4 in cDCs Is Critical for T Cell Priming. Cell Rep. 31, 107604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mensah VA, Gueye A, Ndiaye M, Edwards NJ, Wright D, Anagnostou NA, Syll M, Ndaw A, Abiola A, Bliss C, et al. ; MVVC group (2016). Safety, Immunogenicity and Efficacy of Prime-Boost Vaccination with ChAd63 and MVA Encoding ME-TRAP against Plasmodium falciparum Infection in Adults in Senegal. PLoS ONE 11, e0167951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesin L, Ersching J, and Victora GD (2016). Germinal Center B Cell Dynamics. Immunity 45, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millington OR, Di Lorenzo C, Phillips RS, Garside P, and Brewer JM (2006). Suppression of adaptive immunity to heterologous antigens during Plasmodium infection through hemozoin-induced failure of dendritic cell function. J. Biol 5, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner D Jr., Factor R, Whitten R, Carr RA, Kamiza S, Pinkus G, Molyneux M, and Taylor T (2013). Pulmonary pathology in pediatric cerebral malaria. Hum. Pathol 44, 2719–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JM, Chaisavaneeyakorn S, Perkins DJ, Othoro C, Otieno J, Nahlen BL, Shi YP, and Udhayakumar V (2004). Hemozoin differentially regulates proinflammatory cytokine production in human immunodeficiency virus-seropositive and -seronegative women with placental malaria. Infect. Immun 72, 7022–7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J, and Hogquist KA (2011). T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J. Exp. Med 208, 1279–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen PH, Day N, Pram TD, Ferguson DJ, and White NJ (1995). Intraleucocytic malaria pigment and prognosis in severe malaria. Trans. R. Soc. Trop. Med. Hyg 89, 200–204. [DOI] [PubMed] [Google Scholar]
- Nie Y, Waite J, Brewer F, Sunshine MJ, Littman DR, and Zou YR (2004). The role of CXCR4 in maintaining peripheral B cell compartments and humoral immunity. J. Exp. Med 200, 1145–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell RA, de Koning-Ward TF, Burt RA, Bockarie M, Reeder JC, Cowman AF, and Crabb BS (2001). Antibodies against merozoite surface protein (MSP)-1(19) are a major component of the invasion-inhibitory response in individuals immune to malaria. J. Exp. Med 193, 1403–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeng-Adjei N, Portugal S, Tran TM, Yazew TB, Skinner J, Li S, Jain A, Felgner PL, Doumbo OK, Kayentao K, et al. (2015). Circulating Th1-Cell-type Tfh Cells that Exhibit Impaired B Cell Help Are Preferentially Activated during Acute Malaria in Children. Cell Rep. 13, 425–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry JA, Olver CS, Burnett RC, and Avery AC (2005). Cutting edge: the acquisition of TLR tolerance during malaria infection impacts T cell activation. J. Immunol 174, 5921–5925. [DOI] [PubMed] [Google Scholar]
- Pham TT, Lamb TJ, Deroost K, Opdenakker G, and Van den Steen PE (2021). Hemozoin in Malarial Complications: More Questions Than Answers. Trends Parasitol. 37, 226–239. [DOI] [PubMed] [Google Scholar]
- Pinzon-Charry A, Woodberry T, Kienzle V, McPhun V, Minigo G, Lampah DA, Kenangalem E, Engwerda C, López JA, Anstey NM, and Good MF (2013). Apoptosis and dysfunction of blood dendritic cells in patients with falciparum and vivax malaria. J. Exp. Med 210, 1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pracht K, Meinzinger J, Daum P, Schulz SR, Reimer D, Hauke M, Roth E, Mielenz D, Berek C, Côrte-Real J, et al. (2017). A new staining protocol for detection of murine antibody-secreting plasma cell subsets by flow cytometry. Eur. J. Immunol 47, 1389–1392. [DOI] [PubMed] [Google Scholar]
- Ranjan R, Karpurapu M, Rani A, Chishti AH, and Christman JW (2016). Hemozoin Regulates iNOS Expression by Modulating the Transcription Factor NF-κB in Macrophages. Biochem. Mol. Biol. J 2, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raulf MK, Johannssen T, Matthiesen S, Neumann K, Hachenberg S, Mayer-Lambertz S, Steinbeis F, Hegermann J, Seeberger PH, Baumgärtner W, et al. (2019). The C-type Lectin Receptor CLEC12A Recognizes Plasmodial Hemozoin and Contributes to Cerebral Malaria Development. Cell Rep. 28, 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RTS,S Clinical Trials Partnership (2015). Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: final results of a phase 3, individually randomised, controlled trial. Lancet 386, 31–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryg-Cornejo V, Ioannidis LJ, Ly A, Chiu CY, Tellier J, Hill DL, Preston SP, Pellegrini M, Yu D, Nutt SL, et al. (2016). Severe Malaria Infections Impair Germinal Center Responses by Inhibiting T Follicular Helper Cell Differentiation. Cell Rep. 14, 68–81. [DOI] [PubMed] [Google Scholar]
- Sarr D, Marrama L, Gaye A, Dangou JM, Niang M, Mercereau-Puijalon O, Lehesran JY, and Jambou R (2006). High prevalence of placental malaria and low birth weight in Sahelian periurban area. Am. J. Trop. Med. Hyg 75, 171–177. [PubMed] [Google Scholar]
- Schwarzer E, Alessio M, Ulliers D, and Arese P (1998). Phagocytosis of the malarial pigment, hemozoin, impairs expression of major histocompatibility complex class II antigen, CD54, and CD11c in human monocytes. Infect. Immun 66, 1601–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shio MT, Eisenbarth SC, Savaria M, Vinet AF, Bellemare MJ, Harder KW, Sutterwala FS, Bohle DS, Descoteaux A, Flavell RA, and Olivier M (2009). Malarial hemozoin activates the NLRP3 inflammasome through Lyn and Syk kinases. PLoS Pathog. 5, e1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skorokhod OA, Alessio M, Mordmüller B, Arese P, and Schwarzer E (2004). Hemozoin (malarial pigment) inhibits differentiation and maturation of human monocyte-derived dendritic cells: a peroxisome proliferator-activated receptor-gamma-mediated effect. J. Immunol 173, 4066–4074. [DOI] [PubMed] [Google Scholar]
- Sobolewski P, Gramaglia I, Frangos JA, Intaglietta M, and van der Heyde H (2005). Plasmodium berghei resists killing by reactive oxygen species. Infect. Immun 73, 6704–6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaccapelo R, Janse CJ, Caterbi S, Franke-Fayard B, Bonilla JA, Syphard LM, Di Cristina M, Dottorini T, Savarino A, Cassone A, et al. (2010). Plasmepsin 4-deficient Plasmodium berghei are virulence attenuated and induce protective immunity against experimental malaria. Am. J. Pathol 176, 205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sponaas AM, Freitas do Rosario AP, Voisine C, Mastelic B, Thompson J, Koernig S, Jarra W, Renia L, Mauduit M, Potocnik AJ, and Langhorne J (2009). Migrating monocytes recruited to the spleen play an important role in control of blood stage malaria. Blood 114, 5522–5531. [DOI] [PubMed] [Google Scholar]
- Strangward P, Haley MJ, Albornoz MG, Barrington J, Shaw T, Dookie R, Zeef L, Baker SM, Winter E, Tzeng TC, et al. (2018). Targeting the IL33-NLRP3 axis improves therapy for experimental cerebral malaria. Proc. Natl. Acad. Sci. USA 115, 7404–7409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suan D, Kräutler NJ, Maag JLV, Butt D, Bourne K, Hermes JR, Avery DT, Young C, Statham A, Elliott M, et al. (2017). CCR6 Defines Memory B Cell Precursors in Mouse and Human Germinal Centers, Revealing Light-Zone Location and Predominant Low Antigen Affinity. Immunity 47, 1142–1153.e4. [DOI] [PubMed] [Google Scholar]
- Terrientes ZI, Kramer K, Herrera MA, and Chang SP (1994). Naturally acquired antibodies against the major merozoite surface coat protein (MSP-1) of Plasmodium falciparum acquired by residents in an endemic area of Colombia. Mem. Inst. Oswaldo Cruz 89 (Suppl 2), 55–61. [DOI] [PubMed] [Google Scholar]
- Tran TM, Li S, Doumbo S, Doumtabe D, Huang CY, Dia S, Bathily A, Sangala J, Kone Y, Traore A, et al. (2013). An intensive longitudinal cohort study of Malian children and adults reveals no evidence of acquired immunity to Plasmodium falciparum infection. Clin. Infect. Dis 57, 40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner TC, Arama C, Ongoiba A, Doumbo S, Doumtabé D, Kayentao K, Skinner J, Li S, Traore B, Crompton PD, and Götz A (2021). Dendritic cell responses to Plasmodium falciparum in a malaria-endemic setting. Malar. J 20, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban BC, Ferguson DJ, Pain A, Willcox N, Plebanski M, Austyn JM, and Roberts DJ (1999). Plasmodium falciparum-infected erythrocytes modulate the maturation of dendritic cells. Nature 400, 73–77. [DOI] [PubMed] [Google Scholar]
- Vijay R, Guthmiller JJ, Sturtz AJ, Surette FA, Rogers KJ, Sompallae RR, Li F, Pope RL, Chan JA, de Labastida Rivera F, et al. (2020). Infection-induced plasmablasts are a nutrient sink that impairs humoral immunity to malaria. Nat. Immunol 21, 790–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voisine C, Mastelic B, Sponaas AM, and Langhorne J (2010). Classical CD11c+ dendritic cells, not plasmacytoid dendritic cells, induce T cell responses to Plasmodium chabaudi malaria. Int. J. Parasitol 40, 711–719. [DOI] [PubMed] [Google Scholar]
- Weisel FJ, Zuccarino-Catania GV, Chikina M, and Shlomchik MJ (2016). A Temporal Switch in the Germinal Center Determines Differential Output of Memory B and Plasma Cells. Immunity 44, 116–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2019). World malaria report 2019 (WHO; ). [Google Scholar]
- Woodberry T, Minigo G, Piera KA, Amante FH, Pinzon-Charry A, Good MF, Lopez JA, Engwerda CR, McCarthy JS, and Anstey NM (2012). Low-level Plasmodium falciparum blood-stage infection causes dendritic cell apoptosis and dysfunction in healthy volunteers. J. Infect. Dis 206, 333–340. [DOI] [PubMed] [Google Scholar]
- Wykes MN, Liu XQ, Beattie L, Stanisic DI, Stacey KJ, Smyth MJ, Thomas R, and Good MF (2007). Plasmodium strain determines dendritic cell function essential for survival from malaria. PLoS Pathog. 3, e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zander RA, Obeng-Adjei N, Guthmiller JJ, Kulu DI, Li J, Ongoiba A, Traore B, Crompton PD, and Butler NS (2015). PD-1 Co-inhibitory and OX40 Co-stimulatory Crosstalk Regulates Helper T Cell Differentiation and Anti-Plasmodium Humoral Immunity. Cell Host Microbe 17, 628–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zander RA, Guthmiller JJ, Graham AC, Pope RL, Burke BE, Carr DJ, and Butler NS (2016). Type I Interferons Induce T Regulatory 1 Responses and Restrict Humoral Immunity during Experimental Malaria. PLoS Pathog. 12, e1005945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemmour D, Zilionis R, Kiner E, Klein AM, Mathis D, and Benoist C (2018). Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat. Immunol 19, 291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, Good-Jacobson KL, and Shlomchik MJ (2014). CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat. Immunol 15, 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This study did not generate/analyze datasets or code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.