

Abstract
Human papillomaviruses (HPVs) cause persistent infections in stratified cutaneous and mucosal epithelia. In these infections, the viral DNA replicates as low copy number, extrachromosomal double-stranded DNA circular plasmids in the nucleus of the dividing basal cells. When the infected cells begin the process of differentiation, the viral DNA amplifies to a high copy number and virions are assembled in the superficial cells. To study HPV DNA replication, our laboratory generates primary keratinocyte cell lines that contain replicating extrachromosomal HPV genomes. Here, we describe protocols to culture human keratinocytes, to transfect viral DNA into cells using electroporation, to determine the efficiency of genome establishment in cells with a colony forming assay, and to measure the copy number and extrachromosomal status of viral genomes using Southern blotting. These methods can be used to study DNA replication of different oncogenic Alphapapillomavirus HPV types.
Introduction:
There are only a few clinically derived cell lines available in the HPV field that contain extrachromosomal HPV DNA. For example, W12 20863 cells contain extrachromosomal HPV16 (Jeon, Allenhoffmann, & Lambert, 1995; Stanley, Browne, Appleby, & Minson, 1989) and CIN612-9E cells contain extrachromosomal HPV31 (Hummel, Hudson, & Laimins, 1992). Similar cell lines can be generated in the laboratory by transfecting viral DNA into keratinocytes. This is advantageous as different sources of keratinocytes can be used and they can be transfected with genomes from different HPV types, and/or genomes containing specific mutations. Here we describe protocols for developing extrachromosomal HPV keratinocyte cell lines, for evaluating the efficiency of establishment of HPV genomes, and analysis of the extrachromosomal status and copy number of HPV genomes.
Basic Protocol 1: Electroporation to transfect keratinocytes with recircularized HPV genomes
For HPV replication studies, keratinocytes must be grown using the traditional Rheinwald-Green co-culture method (Rheinwald & Green, 1977) (as opposed to commercially available serum free medium) or viral genomes will be lost or will integrate into the cellular genome. In the Rheinwald-Green method, human keratinocytes are grown in co-culture with irradiated J2 3T3 murine fibroblasts (see Supporting Protocol 1 for co-culture methods). Feeder cells secrete growth factors and enhance keratinocyte growth by generating extracellular matrix.
The efficiency of generating HPV-containing keratinocyte lines depends on several parameters. Lipid based transfection can be used to introduce HPV genomes into keratinocytes (Lace, Turek, Anson, & Haugen, 2014), but we find that electroporation is more efficient. Also, different HPV types can yield varying numbers of keratinocyte colonies (Lace et al., 2014). The source of keratinocytes used for transfection can also give determine the efficiency of generating cell lines. The efficiency may also vary among keratinocytes from different donors, the age of the donor of the keratinocytes (neonatal versus adult), or the tissue source (e.g. foreskin versus cervical). The G418 selection conditions used in this protocol (200μg/ml) may need to be optimized for different keratinocyte strains (Lace et al., 2014). Here we describe generation of cell lines containing oncogenic HPV genomes in primary human foreskin keratinocytes, but cells derived from oral, vaginal or cervical epithelium can also be used. Transient selection with G418 selects for transfected cells. Expression of the viral E6 and E7 oncogenes adds another level of selection for HPV containing cells once the transient G418 selection has been removed. Cell lines can also be generated in immortalized cell lines such as NIKS (Allen-Hoffmann et al., 2000; Flores, Allen-Hoffmann, Lee, Sattler, & Lambert, 1999).
Equipment
Nucleofector 2b device (Lonza AAB-1001)
Refrigerated benchtop centrifuge with swing bucket rotor (e.g. Thermoscientific Sorvall Legend XTR, 75-217-420)
Cell counter or hemocytometer
5% and 10% CO2 Incubators
Tissue culture facility (hood, etc)
15 ml polypropylene conical tubes (Sarstedt 62.554.100)
50 ml polypropylene conical tubes (Corning 352070)
Reagents (see relevant recipes in Reagents preparation)
Versene (Invitrogen 15040-066)
Trypsin (Invitrogen 25200-114)
Recovery medium
Human keratinocyte nucleofector kit (Lonza VPD-1002)
Recircularized HPV genomes (see Supporting Protocol 2 and Figure 1)
pRSVneo plasmid (ATCC 37198 and Figure 1)
Keratinocyte F-medium
50 mg/ml G418 (Gibco 10131-027)
J2 3T3 murine fibroblast feeder cells (Kerafast EF3003)
Neomycin resistant J2 3T3 fibroblast feeders (can be generated by transfecting J2 3T3 cells with the pRSVneo plasmid and isolating resistant cells with G418 selection) (Stepp, Meyers, & McBride, 2013).
Early passage primary human keratinocytes such as adult epidermal keratinocytes (Lonza 00192627 or ThermoFisher C0055C) or normal, neonatal foreskin keratinocytes (ATCC PCS-200-010 or ThermoFisher C0015C [single source] and A13401 [pooled]).
Figure 1.
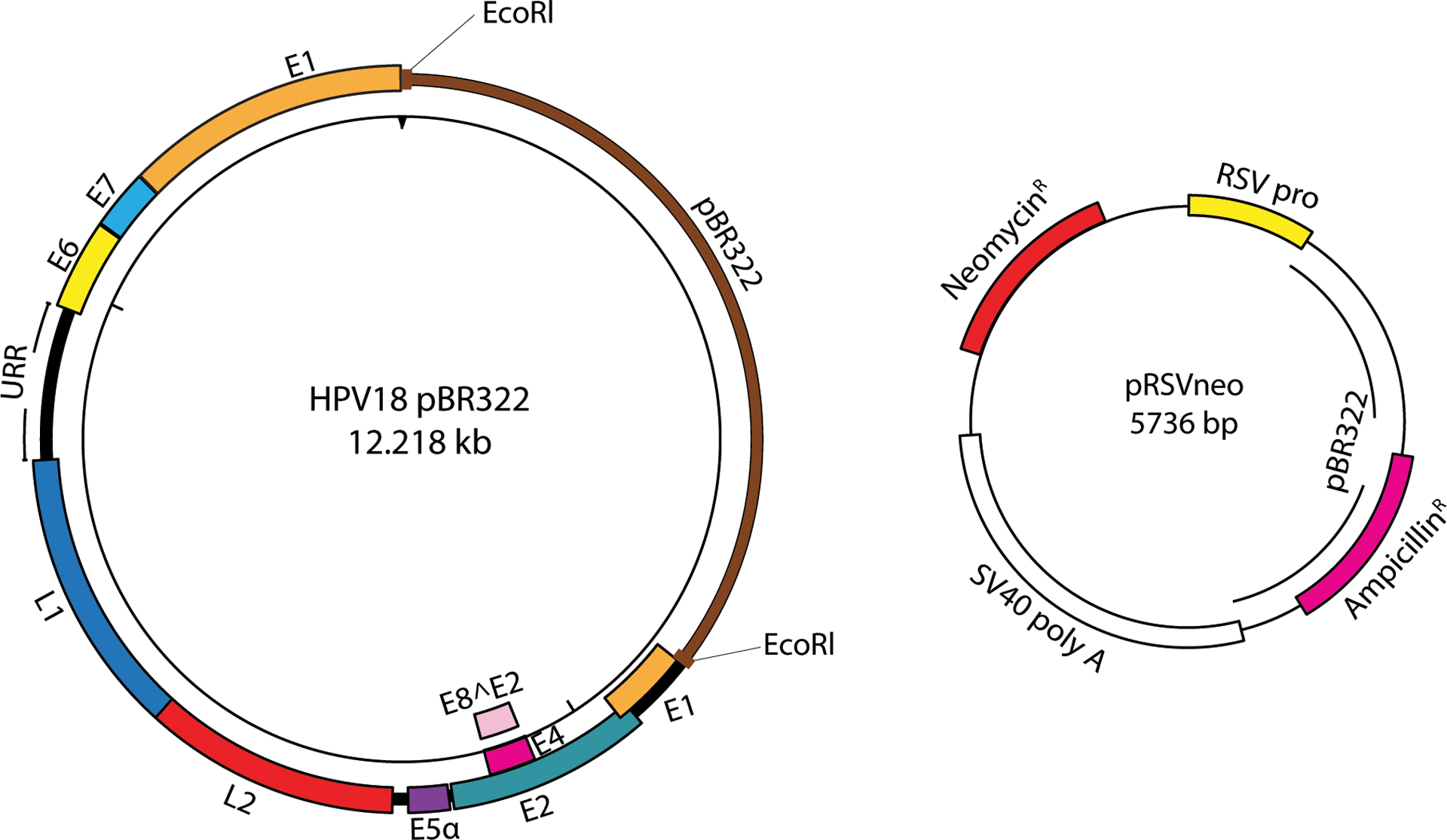
Plasmid maps of HPV18 (cloned in pBR322) and selectable marker plasmid (pRSVneo). The HPV genome is cloned in pBR322, a bacterial plasmid vector, for propagation in bacteria. A restriction digest (the EcoRl site is highlighted in red to release the HPV18 genome from pBR322 vector) releases the HPV genome from this vector for HPV genome recircularization (Support Protocol 2) prior to transfection (Basic Protocol 1). The pRSVneo plasmid is co-transfected with the recircularized HPV genome and enables transient G418 selection of transfected cells (Basic Protocol 1).
- At least two hours prior to transfection, replace medium on 10cm plates containing irradiated feeders with 10 ml of Recovery medium. These plates will be used to culture the keratinocytes after electroporation.
- For keratinocyte and feeder co-culture protocol see Supporting Protocol 1.
To isolate keratinocytes to be used for transfection, aspirate medium from 15cm plates containing feeders and keratinocytes that were prepared in advance for transfection. Add 5 ml Versene to each plate and incubate for 2 minutes at room temperature. Gently pipette Versene across the surface of the monolayer to dislodge the feeders. Check that feeders are dislodged, and keratinocyte colonies remain attached, in a microscope. Aspirate Versene containing detached feeders.
Rinse keratinocyte plates with 10 ml of 1X PBS.
Add 5 ml of trypsin to 15cm plate of keratinocytes and incubate for a few minutes at 37°C. Examine the cells every few minutes in the microscope until they are rounded. Sharply tap the sides of plate to dislodge the keratinocytes. Do not over trypsinize.
Neutralize the trypsin by adding an equivalent volume of keratinocyte F-medium and transfer the suspended cells to a 50 ml centrifuge tube. Pellet cells in bench top centrifuge at 235 × g for 5 minutes.
-
Resuspend cells in 10 ml of Recovery medium and count using a hemocytometer or automated cell counter. Pellet 1×106 keratinocytes, per transfection, individually in 15 ml centrifuge tubes at 235 × g for 5 minutes. Remove supernatant and leave cell pellets on ice until ready to transfect.
Each electroporation requires 1×106 cells. When planning experiments consider whether cells will be used to generate a cell line, to assay colony number, or for short term replication or other assays and scale up accordingly. Examples:- Development of HPV containing cell lines: Plate 0.5×105 cells for passing and 0.5×105 cells for staining colonies.
- If the cells will be used for assays at earlier passes, then best to scale up with duplicate transfections.
- Quantitative Colony forming assay: Plate 0.5×105, 0.3×105 and 0.2×105 cells for staining colonies (see Basic Protocol 2).
- Transient replication assays: Plate 1.6×106 cells.
Prepare a master mix of the human keratinocyte nucleofector reagent A (18 μl per reaction) and B (82 μl per reaction).
Prepare a 15 ml conical tube containing 1 ml of Recovery medium (at room temperature) for every electroporation. If duplicate electroporations are to be performed (2×106 keratinocytes total), prepare a 15 ml conical containing 2 ml of Recovery medium.
Prepare a master mix of the DNA used for each transfection. A total of 2 μg of recircularized HPV genomes should be mixed with 1 μg of pRSVneo plasmid to transfect 1×106 keratinocytes. A control DNA mix should include 2 μg of pUC19 plasmid mixed with 1 μg of pRSVneo. An additional control would be 3 μg of pUC19 plasmid.
When ready to transfect, resuspend cell pellets in 100 μl of nucleofector transfection master mix. Add DNA prepared for transfection from step 9 to resuspended cells and mix by pipetting up and down.
Transfer resuspended cells and DNA mix into the cuvette supplied in the kit. Ensure that the cell suspension is at the bottom of the cuvette by tapping on a flat surface. Insert cuvette into the Nucleofector 2b and electroporate cells using the pre-installed program T-007 (high viability, human keratinocytes).
Using the supplied plastic Pasteur pipette, transfer the transfected cell suspension drop by drop into 15 ml tubes containing Recovery medium, as prepared in step 8. For duplicate transfections, repeat the electroporation step with another sample and combine in Recovery medium.
Use a 1 ml pipette to resuspend transfected cells and drop onto 10cm plates.
Approximately 24 hours post-transfection, remove Recovery medium from plates and replace with 10 ml of F-medium containing 200 μg/ml of G418.
After two days replace medium on plates with fresh F-medium containing 200 μg/ml of G418.
Two days later replace medium with F-medium without G418. Continue to expand cells on plates for an additional 2 weeks or until keratinocyte colonies are visible.
Trypsinize colonies before they become to large (2–3mm) and pass onto a plate of feeders to establish a cell line. A portion of cells should be frozen at every pass for future use. High-risk HPV genomes (e.g. HPV16, 18, and 31)will immortalize the keratinocyte cells; these cells maintain the viral genome as an extrachromosomal element for multiple passages. Cells transfected with unstable (e.g. HPV16 E8^E2 mutant) or low-risk HPV genomes (e.g. HPV 11) show a reduction in viral genome copy number per passage. These latter transfected cell lines should only be analyzed at a low passage post-transfection.
Support Protocol 1: Rheinwald-Green method of co-culture of irradiated J2 3T3 feeders and human keratinocytes
Keratinocytes can be isolated from donor tissues such as neonatal foreskin and adult skin biopsies. The age and tissue origin of the donor tissue can influence the rate of proliferation and senescence of keratinocytes grown in culture. Co-culturing keratinocytes with a monolayer of growth arrested J2 fibroblast feeder cells greatly increases the proliferation and growth of keratinocytes (Fu, Quintero, & Baker, 2003). Feeder cells secrete growth factors and enhance keratinocyte adhesion by generating extracellular matrix. Hence, the confluency and age of feeder cells used in co-culture with keratinocytes can influence keratinocyte growth and proliferation (https://www.kerafast.com/item/1100/3t3-j2-cell-line). Like keratinocytes, low passage feeder cells improve the proliferation of keratinocytes but can also increase the efficiency of recovery of transfected keratinocytes.
Keratinocyte proliferation is greatly boosted by treatment with the Rho-kinase inhibitor, Y27632 (Chapman, Liu, Meyers, Schlegel, & McBride, 2010). This treatment allows keratinocytes to achieve stem-cell like levels of proliferation and growth. Y27632 treatment allows investigators to maintain keratinocytes in an early pass like state and inhibits keratinocyte differentiation. While cells can be grown in Y27632 before electroporation, we do not recommend adding it to the medium afterwards as it may interfere with establishment of HPV genomes.
Equipment:
Cell irradiator (alternatively may use Mitomycin C treatment for growth arrest (Fehrmann & Laimins, 2005)).
Tissue culture incubators set at 37°C. Feeders and keratinocytes grow under 10% and 5% CO2 conditions, respectively.
Refrigerated benchtop centrifuge with swing bucket rotor (e.g. Thermoscientific Sorvall Legend XTR, 75-217-420)
Cell counter or hemocytometer
Reagents (see relevant recipes in Reagent preparation)
Feeder growth medium (see recipe in Reagents and Solutions)
Keratinocyte F-medium (see recipe in Reagents and Solutions)
0.25% Trypsin-EDTA (Invitrogen 25200-114)
Versene (Invitrogen 15040-066)
1X PBS without calcium and magnesium (Lonza 17-516F)
Tissue culture flasks (T-75 [VWR 658175] and T-175 [VWR 660175])
Tissue culture dishes, 100×20mm (Corning 353003) and 150×25mm (Corning 353025)
As an alternative to using an irradiator: Mitomycin C (Sigma 10107409001)
Cell Lines
J2 3T3 murine fibroblast feeder cells (Kerafast EF3003)
Neomycin resistant J2 3T3 fibroblast feeder cells (can be generated by transfecting non-resistant feeders with pRSVneo plasmid and culturing recovering cells with G418 selection) (Stepp et al., 2013)
Keratinocyte cells (e.g. foreskin keratinocytes) isolated from a donor tissue or purchased (see Basic Protocol 1)
Add ~1×106 (freshly thawed or passed) J2 3T3 fibroblast feeder cells to a T-75 flask containing 10 ml of feeder growth medium.
Incubate cells at 37°C in 10% CO2 for several days.
Once cells reach ~80% confluence (~3 days), remove feeder medium and rinse cells with 3 ml of 1X PBS.
Aspirate PBS and add 4 ml of trypsin to each flask. Incubate flasks at 37°C for 2 minutes.
Tap the sides of the flask to dislodge feeder cells from the flask. After confirming that feeders are in suspension, neutralize the trypsin by adding 4 ml of feeder medium into each flask.
Transfer the neutralized suspension of cells into a 15 ml tube and pellet cells in a centrifuge for 5 minutes at 235 × g at room temperature.
Remove supernatant and resuspend cells in 1 ml of fresh feeder medium. Use a cell counter or hemocytometer to quantify the number of feeder cells.
Add 1×106 feeder cells to a T-175 flask containing 25 ml of feeder medium. Gently rock flasks up and down, left and right, to disperse cells evenly throughout the flask and incubate cells at 37°C in 10% CO2.
- Scale up the number of flasks needed depending on the size of the experiment.
- Growth arrested feeders are always needed when co-culturing with keratinocytes. For transfection, at least two 10cm plates of neomycin resistant feeders will be needed for each DNA sample transfected. Each plate will need 1×106 growth arrested feeder cells for co-culture with keratinocytes.
- Feeders must be growth arrested by irradiation or Mitomycin C treatment before co-culture with keratinocytes. Plate 1×106 of growth arrested feeders on a 10cm plate in a total of 10 ml of feeder growth medium and disperse cells evenly on plate. Plate growth arrested feeders on plates at least 24 hours before plating keratinocytes. Growth arrested feeders are good for 5–6 days.
- Irradiation (6000 RADs of gamma irradiation) is required to induce growth arrest in feeder cells (McBride, Chapman, Stepp, & Terunuma, 2014).
- Alternatively, adherent feeders can be treated for 2 hours with 4 μg/ml of Mitomycin C, at 37 °C, to induce growth arrest when an irradiator is not available. Following mitomycin treatment wash feeder cells on plates several times with 4 ml of 1X PBS, making sure to wash the walls of the culture dish (Lambert et al., 2005).
After allowing the growth arrested cells to attach overnight, replace feeder medium with 10 ml of keratinocyte F-medium and drop 0.5–1×106 cells of low passage keratinocytes, thawed from a vial stored in liquid nitrogen, onto plates. Incubate cells at 37°C in 5% CO2
Replace the keratinocyte F-medium every two days and incubate until cells are about 70–80% confluence. See Figure 2 for examples of keratinocytes grown in co-culture with feeders.
If keratinocytes have not reached 70–80% confluence by five days, the irradiated feeders should be replaced to support keratinocyte growth. The feeders are easily removed by gentle pipetting of the medium across the surface of the cell monolayer. Feeders should be removed by aspiration and 1×106 freshly irradiated feeder cells added in 10 ml keratinocyte F-medium. See Figure 2 for an image of keratinocytes on the plate after removing feeders.
- Prior to transfection, the number of keratinocytes can be scaled up by plating 0.5–0.6×106 keratinocytes on 15cm plates containing 3×106 growth arrested feeders and 30 ml of keratinocyte F-medium. Keratinocytes will typically reach 70–80% confluence in 4–5 days.
- A 15cm plate will produce 8–12×106 keratinocytes at 70–80% confluence. This number may vary with the keratinocyte cell type used. For example, NIKS cells are larger in size and thus may result in fewer overall cells harvested from a 15cm plate used for transfection.
Figure 2.
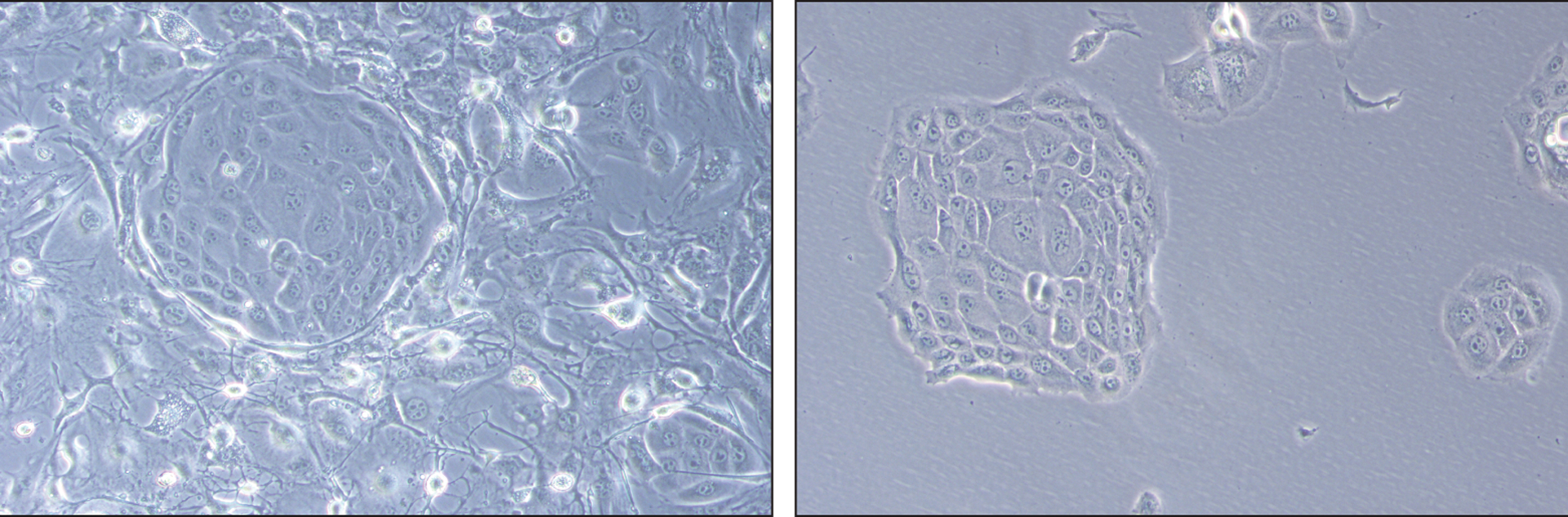
Keratinocyte colonies co-cultured with fibroblast feeders. An image was taken of a colony of proliferative keratinocytes pushing surrounding feeders away as the colony continues to grow. Feeders and keratinocyte cells are indicated. The same colony is visualized on the right after fibroblast feeders are removed with Versene.
Support Protocol 2: Recircularization of HPV genomes
This protocol, adapted from (Schowalter, Pastrana, & Buck, 2011) Schowalter et al. (2011), can be used to release the ~8 kb HPV genomes from the bacterial plasmid vector and recircularize the viral DNA by intramolecular ligation. The principle behind this protocol is to digest the HPV genome from the bacterial plasmid by restriction enzyme digestion and perform a dilute ligation to promote intra‐molecular ligation and generate covalently closed circular genomes. The protocol will produce a mixture of DNA products: covalently closed bacterial plasmids and monomeric, covalently closed HPV genomes. Additional products include religated HPV-bacterial plasmid products (>10kb), and concatemeric HPV genome or plasmid products (>12kb). If the majority of ligated products are monomeric, covalently closed HPV genomes this will be sufficient for transfection.
See Porter and McBride (2020) and Toots et al. (2017) for alternative protocols for obtaining religated genomes as well as recircularized HPV genomes using minicircle technology.
Equipment
Heat block capable of cooling to 16°C, such as ThermoMixer C (Eppendorf #5382000023), or a water bath heated to 16°C in a cold room.
UV light box
Floor model centrifuge (e.g. Sorvall RC6) and a swing bucket rotor (e.g. HS-4)
Additional reagents and equipment for agarose gel electrophoresis
50 ml conical tubes (Labcon 3181-345-008-9)
Reagents
Cloned HPV viral DNA genome prepared using transfection grade plasmid kit (see Table 1 for reference HPV genomes and Figure 1 for sample plasmid maps). HPV genomes can be acquired from The HPV Reference Center.
10X New England Biolabs buffer compatible with selected restriction enzyme (see Table 1)
Restriction enzyme to release HPV genome (see Table 1)
10X Ligation Buffer (500mM Tris-HCl pH7.5, 100mM MgCl2, 100mM DTT)
10mM ATP, pH7.5
T4 DNA ligase (2,000,000 U/ml) (NEB #M0202S)
7.5 M Ammonium acetate
95% Ethanol
70% Ethanol
TE buffer, pH 8.0 (Quality Biological #351‐011‐131)
6X Gel Loading Dye, Purple (NEB #B7024S)
SeaKem GTG Agarose (Lonza #50074)
Ethidium bromide (10 mg/ml) (Invitrogen #15585011)
50X TAE buffer (Tris‐acetate‐EDTA; Thermo Fisher #B49)
Size markers (use both a 1kb plus DNA ladder [NEB N3200S] and HPV plasmid, released from the bacterial vector, as appropriate size markers).
Table 1.
Commonly used HPV plasmid genome DNAs and enzymes used for recircularization.
| HPV | Plasmid name | Bacterial Plasmid Vector | Restriction enzyme and buffer used for recircularization | Reference |
|---|---|---|---|---|
| 16 | pEFHPV-16W12E | pUC | BamHl (NEB R0136L); 10X CutSmart Buffer (NEB B7204S) | (Flores et al., 1999) |
| 18 | pBR322-HPV18 | pBR322 | EcoRl (NEB R0101L); 10X Buffer 2.1 (NEB B7202S) | (Boshart et al., 1984) |
| 31 | pBR322-HPV31 | pBR322 | EcoRl (NEB R0101L); 10X Buffer 2.1 (NEB B7202S) | (Frattini, Lim, & Laimins, 1996) |
Prepare a 200 μl restriction digestion reaction in a 1.7 ml microfuge tube containing 50 μg HPV plasmid DNA. Add 20 μl of the appropriate 10X NEB Buffer, nuclease free water to reach a final volume of 200ul, and 50U of the appropriate restriction enzyme (see Table 1).
Incubate reaction at 37°C for 60 minutes.
- Inactivate enzyme by incubating at the manufacturer’s recommended temperature for 20 minutes.
- If the enzyme cannot be heat‐inactivated, use a DNA cleanup kit to remove the enzyme. Elute digested DNA in a final volume of 199 μl.
Take a 1 μl aliquot of the digest (0.25 μg) and transfer it to a separate tube. Combine with 7.3 μl H2O and 1.7 μl 6X Purple Gel Loading Dye and separate by electrophoresis on a 0.8% agarose TAE gel containing 0.5 μg/ml of EtBr. Visualize on a UV lightbox and confirm digestion is 100% complete before proceeding to step 5.
Prepare a 9 ml ligation reaction with 199 μl of digested DNA, 900 μl of 10X ligation buffer, 900 μl of 10mM ATP, 6,994 μl H2O, and 6 μl of T4 DNA ligase.
Incubate overnight at 16°C.
Precipitate DNA by adding 4.5 ml of 7.5 M ammonium acetate and 35 ml of 95% ethanol. Mix well after each addition.
Incubate overnight at 4°C.
- Allow sample to warm to room temperature.
- This will make the solution less viscous and facilitate pelleting the DNA by centrifugation.
Centrifuge for 1 hour at 4811 × g, room temperature.
Using a serological pipette remove most of the supernatant. Use a 1 ml pipette to carefully remove the remainder of the ethanol, being carefully not to disturb the pellet.
Wash pellet with 10 ml of 70% ethanol.
Centrifuge at 4811 × g for 10 minutes at room temperature.
Repeat step 11–13.
Remove most of the supernatant with a serological pipette. Remove the remainder of the supernatant with a 1 ml pipette tip. Centrifuge the sample briefly to pellet any remaining 70% ethanol and remove the supernatant with a 1 ml pipette tip.
Allow the DNA pellet in the 50 ml conical to dry briefly for 5 minutes.
- Resuspend the pelleted DNA in the 50 ml conical in 50 μl of 1X TE. Allow pellet to dissolve in TE at room temperature for 10 minutes.
- A larger volume of TE may be used to resuspend the pellet, but it is best to use a smaller volume to maintain a higher DNA concentration.
Separate ~ 100 ng of the ligation products by electrophoresis on a 0.8% agarose TAE gel containing 0.5 μg/ml EtBr (Voytas, 2001) and visualize on a UV lightbox.
Alternate Support Protocol 1: Use of HPV replicon containing selection marker
The pRSVneo plasmid supports transient selection of established HPV genomes in keratinocyte colonies in Basic Protocol 1. Alternatively, an HPV18-derived replicon that expresses the neomycin resistance gene can be used. This replicon contains the HPV18 origin and replicates alongside the HPV18 genome in an E1 and E2 dependent manner. This method allows constant selection of transfected cells as HPV replicons are replicated and partitioned into dividing keratinocytes in tandem with co-transfected HPV genomes (Coursey, Van Doorslaer, & McBride, 2021; Van Doorslaer, Chen, Chapman, Khan, & McBride, 2017).
Reagents
CpGneo vector containing a HPV18central URR and origin DNA element (available from the McBride lab)
Empty CpGneo control vector
Recircularized, HPV18 genome (see Support Protocol 2)
Transfection and cell culture reagents mentioned in Basic Protocol 1
Repeat steps 1–8 as outlined in Basic Protocol 1.
Prepare DNA master mixes for transfection consisting of 1 μg of recircularized HPV genome and 1 μg of HPV replicon (i.e. HPV18 genome and HPV18 replicon) for every 1×106 keratinocytes to be transfected. As a control sample, prepare a DNA master mix of 1 μg of recircularized HPV genome and 1 μg of empty CpGneo vector for every 1×106 keratinocytes to be transfected.
Plate 0.1 × 105 cells onto 10cm plates of irradiated feeders for colony staining and and divide the remainder between two additional 10cm plates of irradiated feeders.
Cells are selected in 400 μg/ml G418 for 4 days, and then in 200 μg/ml G418 until colonies appear.
Trypsinize colonies before they become too large (2–3mm) and pass onto a plate of feeders to establish a cell line. A portion of cells should be frozen at every pass for future use.
Basic Protocol 2: Quantitative Colony formation assay to measure the efficiency of HPV genome establishment
Equipment
White light box imager (e.g. Syngene G-box)
Reagents
Formaldehyde (Macron 5016-02)
Formalin fixation solution (3.7% formaldehyde in 1X PBS)
10X Methylene blue stain stock (1.4g of methylene blue dissolved in 100 ml 95% ethanol)
Methylene blue stain working solution (dilute stock 1:10 with deionized water)
10X PBS
Aldex® Aldehyde Management System (Aldex AMS3010)
After electroporation (Basic Protocol 1), plate keratinocytes onto 10cm plates of irradiated feeders. 1 × 106 electroporated keratinocytes can be divided across three plates with 0.2, 0.3 and 0.5 × 105 on each to give a good range of colony numbers.
Culture cells until colonies appear, changing the medium every two to three days and adding fresh irradiated feeders when necessary as described in Support Protocol 1.
Remove feeders from plates to be stained using Versene (or F-medium currently on the plates) by repeatedly pipetting the Versene across the monolayer until the feeders are dislodged.
Aspirate detached feeders and any remaining media from plates and wash cells with 1X PBS.
Prepare formalin fixation solution, 5 ml per 10cm plate.
Remove PBS from plates and add 5 ml of formalin fixation solution, rocking plates gently on a shaker for 5 minutes at room temperature.
Remove formalin fixation solution and decontaminate in container of crystalline Aldex.
Add 5 ml of the methylene blue stain to each 10cm plate.
-
Rock plates gently on rocker at room temperature for 10 minutes.
Alternatively, plates may be left to stain overnight, rocking with 8 ml of methylene blue stain.
Pour methylene blue stain from plates and rinse plates, several times, under a gentle stream of distilled water until only the colonies on the plates remain blue.
Tap open plates, bottom side up onto a paper towel to dislodge any remaining droplets of dye.
-
Dry plates overnight at room temperature, bottom side up.
An imager with a white light converter (e.g. Syngene G-box) can be used to take images of established colonies. See a final example in Figure 3.
Figure 3.
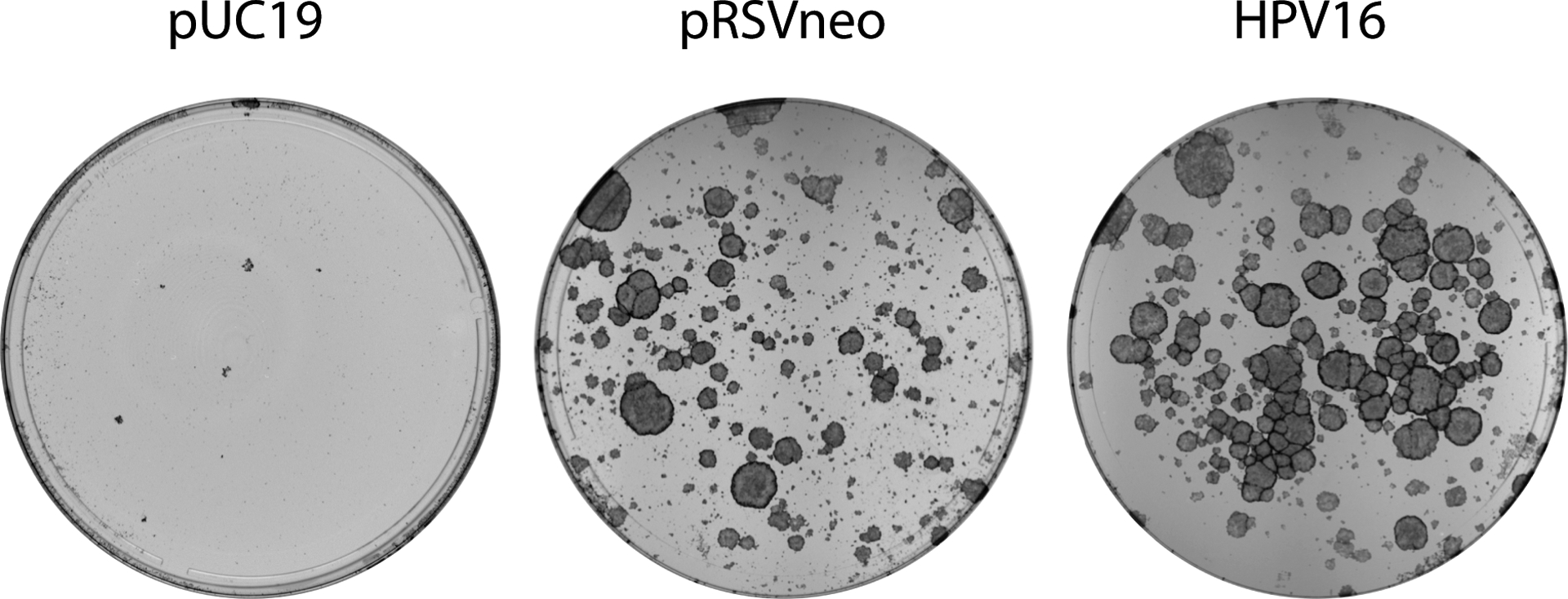
HPV keratinocyte colonies stained with methylene blue 18 days after electroporation. HFK colonies resulting from the method described in Basic Protocol 1 were stained with methylene blue as described in Basic Protocol 2 and imaged on a Syngene white box imager. Comparing the number of colonies formed by transfecting with HPV16 and pRSVneo versus pRSVneo and pUC19, can help determine whether the concentration of G418 used in selection should be increased or decreased. A plate of control plasmid(1.g.pUC19) transfected keratinocytes can help determine the effectiveness of the G418 drug used.
Basic Protocol 3: Southern blot analysis of extrachromosomal viral DNA
Southern blot is a useful tool to study the viral DNA topology present in cells and ensure that they are extrachromosomal. Described here are the steps necessary for southern blot analysis, starting from DNA digestion and ending at visualizing the detected viral DNA using a phosphorimager. Three important distinctions to note in this protocol are the use of top-to-bottom capillary action, the use of a radioactively labeled DNA probe (see Supporting Protocol 3), and the use of a phosphorimager screen and detection device. Although digoxigenin (DIG) probes could theoretically be used in this protocol, we have no experience with this and find that radioactively labeled probes consistently perform well. It is also important to note that while this protocol allows users to visualize viral genome topology and quantify the viral DNA copy number, 2D gel analysis can be used to more rigorously visualize DNA replication intermediates (Henno et al., 2017).
Equipment
Agarose gel electrophoresis equipment (large [20 × 25cm] and small gel [11 × 14cm] sizes are recommended)
Heat block
UV light box and imager
Plastic Tupperware containers (with a fitted lid) to hold agarose gels during depurination and denaturing washes (containers should be at least 27 × 21½ cm in size with a 5cm depth)
Hybridization Oven (e.g. analytykjena UVP Hybridization Oven 95-0030-01)
Hybridization Bottles (VWR 805027 and 805021)
Stratagene UV Stratalinker 1800 or similar
Phosphorimager screen and cassette (e.g. Cytiva BAS Storage Phosphor Screens 28956480)
Phosphorimager (e.g. Cytiva Amersham Typhoon phosphorimager 29238583)
Shaker or rocker
Tabletop centrifuge
Spectrophotometer, such as Nanodrop
DNA sample digestion
NEB restriction enzyme digestion buffer (1.1, 2.1, 3.1, or CutSmart)
Restriction enzyme (various)
SeaKem GTG Agarose (Lonza #50074)
Ethidium bromide
50X TAE buffer (Tris‐acetate‐EDTA; Thermo Fisher #B49)
6X Gel Loading Dye, Purple (NEB #B7024S)
Size markers (use both a 1kb plus DNA ladder [NEB N3200S] and HPV plasmid, released from the bacterial vector and intact, as appropriate size markers).
Transfer to nylon membrane reagents (see relevant recipes in Reagents preparation)
Depurination buffer (0.25M HCl)
Denaturing buffer (3M NaCl, 0.4M NaOH)
Transfer buffer (3M NaCl, 8mM NaOH)
Neutralization buffer (1M phosphate buffer, pH 6.8)
Whatman® Nytran™ SuPerCharge (SPC) TurboBlotter (11×14cm and 15×20cm [MilliporeSigma WHA28415383 and WHA10416316]).
Turboblot refill packs (11×14 [Cytiva 10416306] or 20×25 [Cytiva 10416326]. These pre-cut packs can be substituted with 20 sheets of Whatman GB003/GB005/3MM blotting paper (Cytiva 10547922/10547922/3030–917), 8 sheets of Whatman 3MM Chr blotting paper, a 0.45μm nylon membrane (11×14 [Cytiva 10416230] and 20×25 [Cytiva 10416326]), and a 3MM Chr blotting paper wick, all cut to match the size of the agarose gel.
Hybridization reagents
Hybridization buffer (3X SSC, 2% SDS, 5X Denhardt’s Solution [50X Denhardt’s Solution: 1% Ficoll 400, 1% polyvinylpyrrolidone, 1% bovine serum albumin], 0.2 mg/ml sheared salmon sperm [Sigma 11467140001])
Nylon blot wash (0.1% SDS, 0.1X SSC)
Plastic cling wrap (e.g. Saran wrap)
Day 1: Digestion and gel electrophoresis of DNA samples
There are multiple methods and kits available to isolate cellular DNA (see Support protocols 3 and 4). Care must be taken to ensure that the method or kit chosen purifies both high molecular weight cellular DNA and low molecular weight viral DNA. Analyzing total DNA is important for determining viral genome copy number per cell and the proportion of integrated versus extrachromosomal viral DNA present in cell lines. Alternatively, a modified Hirt extraction method can be used to specifically isolate small, circular, extrachromosomal DNA from cells (Ustav & Stenlund, 1991) The latter method yields a more concentrated viral DNA sample, but it will not isolate viral DNA that is integrated into host chromosomes in the cell line.
Equivalent amounts of DNA must be analyzed, and so cellular DNA must be quantitated (e.g. using a spectrophotometer). Hirt DNA contains viral DNA, mitochondrial DNA, and traces of contaminating cellular nucleic acids and thus, quantitation is not useful. In this case, Hirt DNA is extracted from a specific number of cells.
DNA preparations should be digested with restriction enzymes that have (1) a single restriction site within the HPV genome, and (2) with one that does not cleave viral DNA. The original cloned HPV DNA can be digested from the plasmid vector and used as both a size marker and as a copy number control to quantitate the copy number of extrachromosomal HPV genomes present in the cell line. Restriction enzymes that don’t cut the HPV genome will cleave the cellular DNA and ensure that it separates uniformly in the agarose gel. Undigested cellular DNA can give high molecular weight bands that can trap viral DNA. See Support Protocol 3 and 4, for DNA extraction protocols for the Hirt method and the Qiagen DNeasy Blood and Tissue Kit, respectively.
-
1If total DNA is extracted, digest 1–5 μg of DNA with enzymes that have a single (EcoRl in HPV18) or no restriction sites (Bglll) in the HPV genome.
- Alternatively, if extrachromosomal Hirt DNA is collected, digest equivalent volumes of DNA (e.g. Of a total of 50 ul of Hirt DNA, digest 25 μl with an enzyme that cuts the HPV viral genome once and 25 μl with an enzyme that does not cut the viral genome).
-
2
Mix digested DNA with 6X loading dye to reach a final concentration of 1X.
-
3Prepare a 0.8% agarose gel with 1X TAE. Ensure comb thickness is no greater than 2mm.
- If running extrachromosomal HPV DNA samples, do not add ethidium bromide to the gel as this will cause supercoiled DNA to migrate at a different size.
- If 6–8 spare wells are not available for loading an HPV DNA copy number standard to quantify DNA, an additional comb can be added at the bottom of the gel for loading the standard the next day.
-
4After loading digested DNA samples, load a 1:10 dilution of the 1kb plus DNA ladder along with two dilutions (200 pg and 50 pg) of the linear, HPV DNA size marker.
- Ensure that a gap is present between the DNA ladder and the HPV size markers as radioactive probes will bind to some of the size markers in the ladder.
-
5
Electrophorese the gel overnight (~12–14 hours) using the appropriate voltage. Voltage will vary depending on the size of the gel and whether 1X TAE or TBE buffer is used. Refer to the manufacturer’s instructions of the electrophoresis apparatus used.
Day 2: Depurination, denaturing, and capillary transfer
-
6On the next day run the gel at 100V for 10–15 minutes. This will increase the sharpness of the bands.
- If loading the DNA standard along the bottom of the gel, load samples prior to electrophoresis at 100V for 10–15 minutes.
- Submerge agarose gels containing extrachromosomal HPV in 1X TAE and 0.005mg/ml of ethidium bromide. Shake gels slowly for 15–20 minutes. De-stain agarose gel by exchanging the buffer with 1X TAE without ethidium bromide for 5–10 minutes.
- For all buffers used with the agarose gels, add enough buffer to cover the top of the agarose gels while shaking. Dispose of ethidium bromide according to Institutional safety guidelines.
-
7Visualize the separated DNA using a UV box imager.
- The ethidium bromide image of cellular DNA can serve as a loading control and will reveal if the quantity of DNA samples loaded on the gel are equivalent.
-
8
Rinse gels briefly in distilled water.
-
9If gels contain supercoiled, extrachromosomal HPV genomes, depurinate DNA by soaking the gel for 30 minutes, at room temperature, in 0.25M HCl on a rocker at low speed. Rinse gels with distilled water afterwards.
- DNA depurination is only necessary for supercoiled genomes, otherwise proceed to step 10.
- For all buffers used with agarose gels, add enough buffer to cover the top of the agarose gels while shaking and ensure that gel does not float.
-
10
Gently shake gels in denaturing buffer for 30 minutes at room temperature. Repeat this step once.
-
11
Gently shake the agarose gels in transfer buffer for 15 minutes. While waiting, soak nylon membranes in deionized water to activate membranes.
-
12
Assemble Whatman Nytran SuPerCharge Turboblotter capillary transfer setup as shown in Figure 4 and according to manufacturer’s instructions.
-
13
Place the white “stack tray” of the transfer device on the bench – ensure that it is level.
-
14
Place 20 sheets of dry GB005 blotting paper (thicker pieces) in the stack tray.
-
15
Place 4 sheets of dry 3MM Chr blotting paper (thinner pieces) on top of the stack.
-
16
Prewet one sheet of the 3MM Chr blotting paper with transfer buffer and place it on top.
-
17
Place the transfer membrane on top of the stack and roll out nylon membrane on top of the wet filter paper using a sterile serological pipette and move along the gel as with a rolling pin.
-
18Carefully pick up the agarose gel with gloved hands and place on top of the nylon membrane. Gently roll out any bubbles between the agarose gel and the transfer membrane using a sterile serological pipette and rolling pin motion along the gel.
- Handling 0.8% agarose gels: Consider using a flexible, but sturdy plastic film to move agarose gel to capillary transfer stack.
-
19
Soak the remaining 3 sheets of 3MM Chr blotting paper in transfer buffer and place on top of the gel. Roll out any trapped air bubbles.
-
20
Attach the white transfer buffer reservoir tray of the device to the bottom.
-
21
Soak the 3MM Chr blotting paper wick thoroughly in transfer buffer and place the wick across the stack from left to right and place the two ends of the wick in the buffer tray. Completely fill the transfer buffer reservoir tray with transfer buffer.
-
22
Cover transfer apparatus with plastic cling wrap to stop transfer buffer from evaporating.
-
23
Allow the transfer to proceed overnight or until the bottom of the filter paper stack is soaked through. Allow transfer to proceed for at least 3 hours.
Figure 4.
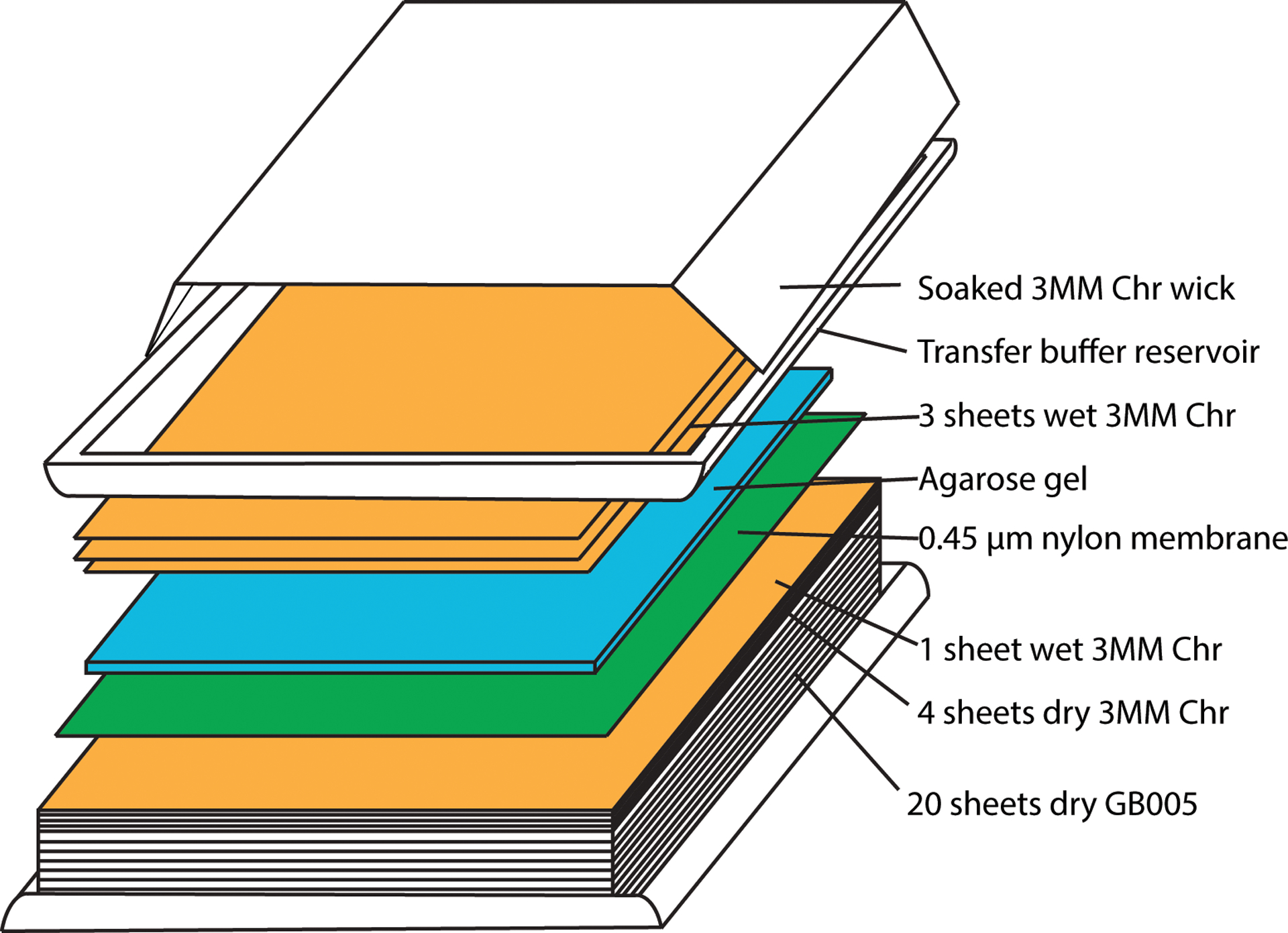
Assembly of capillary transfer unit for southern blot analysis.
Day 3: Membrane probe hybridization
-
24
Following the transfer, remove the nylon membrane and place in a container and gently shake for 5 minutes in enough 1X neutralizing buffer to cover the membrane.
-
25
Place the nylon membrane on a damp Whatman filter paper and crosslink DNA to the membrane using a UV crosslinker. Expose nylon membrane to 1200 microjoules (x100) for 25–50 seconds. Be careful not to let the membrane dry before crosslinking.
-
26
Let the membrane air dry for approximately 30 minutes. The membrane can be stored between two sheets of Whatman filter paper and stored at room temperature or probed immediately.
-
27Meanwhile, prepare prehybridization solution (hybridization buffer without the radioactive probe).
- The amount of prehybridization buffer depends on the size and number of blots. Refer to Table 2 for working amounts and recipe preparation.
-
28Heat denature sheared salmon sperm DNA in a 1.7 ml microfuge tube at 100°C for 10 minutes. Allow samples to briefly cool on ice.
- It is important to use a locking clip to prevent the microfuge cap from popping open during this step and step 34.
-
29
Add the denatured salmon sperm DNA to the prepared hybridization solution.
-
30
Roll up the dry membrane and place it in a hybridization bottle. Add prehybridization buffer to bottles.
-
31Incubate blots in prehybridization buffer in a hybridization oven for at least one hour at 68°C.
- To stop the membrane from rolling up in the bottle, ensure that the outer edge of the membrane is facing the same direction that the tubes are rotated in.
-
32
Combine the radioactive probe (see Supporting Protocol 3) with the necessary amount of sheared salmon sperm DNA (refer to Table 2) as determined by the amount of hybridization buffer needed.
-
33
Incubate the radioactive probe and sheared salmon sperm DNA mix for 10 minutes at 100°C and then chill briefly on ice. Ensure the cap will not pop open at this step by using a locking clip.
-
34
Add the salmon sperm DNA and probe mix to the predetermined amount of hybridization buffer (refer to Table 2).
-
35
Remove the prehybridization solution from the hybridization bottle and replace it with the hybridization solution containing the radioactive probe.
-
36
Incubate in the hybridization oven overnight at 68°C.
Table 2.
Recipe for hybridization buffer (Basic Protocol 3). Small blots (11×14cm) each require 10ml of hybridization buffer while larger blots (15×20cm) require 20mls. It is important to add the buffer constituents in the order shown, otherwise they can precipitate.
| Stock/Total Volume | 10ml | 20ml | 30ml | 50ml | 100ml |
|---|---|---|---|---|---|
| 20X SSC | 1.5ml | 3ml | 4.5ml | 7.5ml | 15ml |
| H2O | 6.3ml | 12.6ml | 18.9ml | 31.5ml | 63ml |
| 50X Denhardts | 1ml | 2ml | 3ml | 5ml | 10ml |
| 20% SDS | 1ml | 2ml | 3ml | 5ml | 10ml |
| 10mg/ml Salmon sperm DNA | 0.2ml | 0.4ml | 0.6ml | 1ml | 2ml |
Day 4: Nylon membrane wash and imaging
-
37
Decant the hybridization solution into a radioactive liquid waste container.
-
38
Wash the membranes by pouring 20 ml of southern wash buffer into hybridization bottle. Incubate hybridization bottles for 20 minutes at 68°C.
-
39
Decant wash solution into a radioactive liquid waste container.
-
40
Move blot into a Tupperware container, add enough 68°C buffer to cover blot, and seal container with a lid. Shake blots with wash solution until container is cooled down to room temperature. Decant wash solution into a radioactive waste container.
-
41Repeat step 40 wash until background radioactivity is no longer detected on the blot.
- Use a Geiger counter to survey blots. Wash blots until there is no detectable signal on regions of the membrane with no DNA sample.
-
42Air dry blots for 30 minutes and wrap blots in plastic cling wrap.
- Note that once blots are dry further washing is not possible.
-
43Place wrapped blot inside a phosphorimager cassette and top with phosphorimager screen.
- Ensure that the phosphorimager screen is blanked by exposing the screen to a white light box for 20 minutes.
-
44Expose the screen to the radioactive blot for 1 hour. Acquire the signal on the phosphorimager screen using a Phosphorimager. This preliminary screen can help determine the optimal time to re-expose the blot to the screen. See an example in Figure 5.
- Alternatively, the signal can be collected on X-ray film by autoradiography
Figure 5.
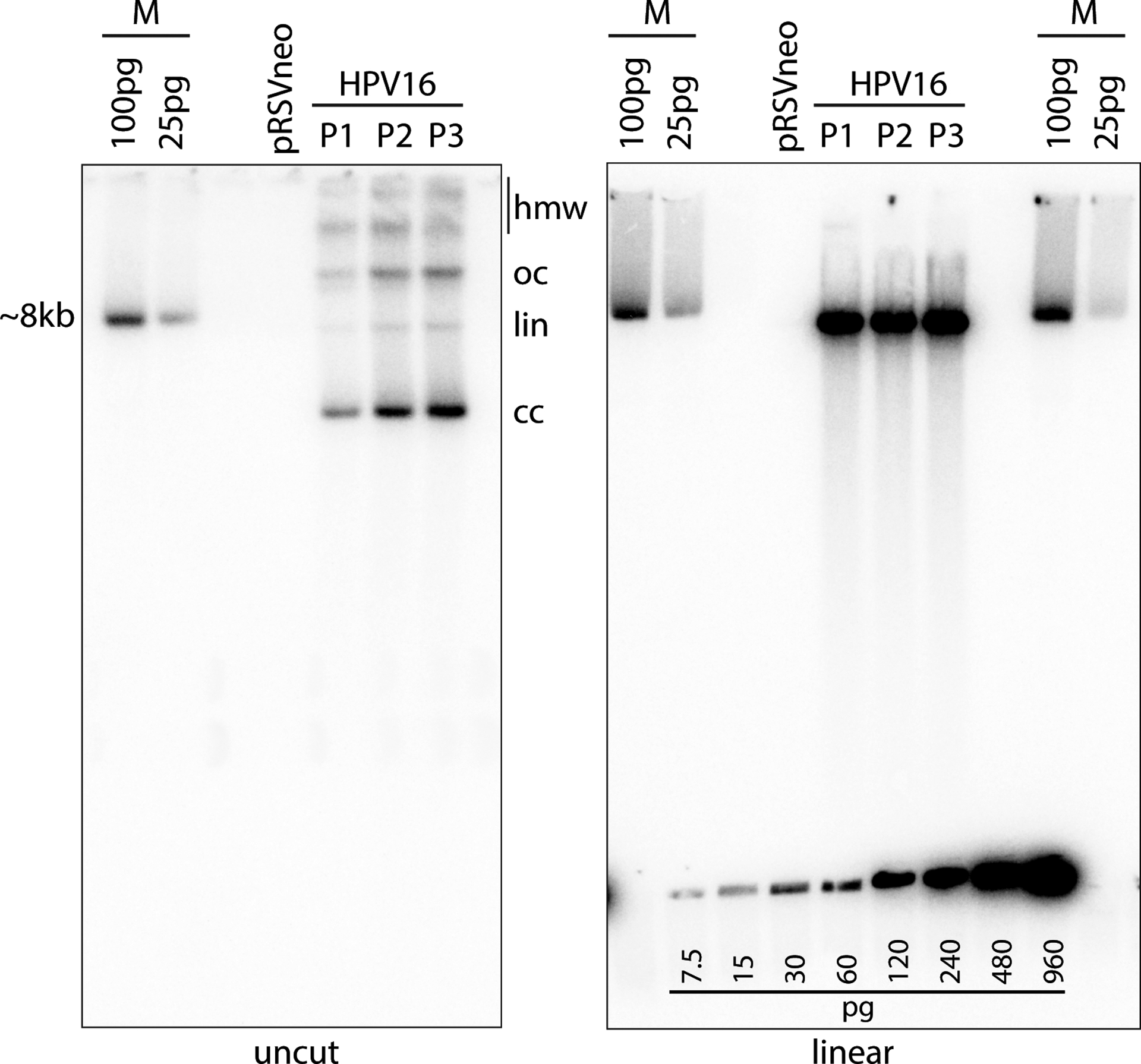
Southern blot analysis. In this example, DNA was extracted from cells passed from the colonies shown in Figure 2 using the Qiagen DNeasy Blood and Tissue Kit (Support Protocol 4). Three micrograms of total cellular DNA was digested with Hindlll (cleaves cellular DNA but does not cut HPV16 viral genome) or BamHl (cuts the viral genome once) and analyzed by Southern blot analysis as detailed in Basic Protocol 3. The left gel shows the HPV DNA isomers detected by the radioactive probe (see Supporting Protocol 3): supercoiled, covalently closed (cc), linear (lin) DNA, open circular/nicked (oc) DNA, and high molecular weight (hmw) forms that may include concatemers. Linearized viral DNA digested with BamHl runs as a single ~8kb band as shown in the gel on the right. The cloned linear HPV16 genome, cleaved from the bacterial vector, is used as a size marker (M). A DNA standard was also created using the HPV16 size marker and was loaded along the bottom of the gel on the right. This DNA standard can be used to estimate the viral DNA copy number. The calculated loaded DNA standard amount should be adjusted to account for the cloned bacterial plasmid and compared to the viral DNA signal in the cell DNA lane. The amount of DNA in a single human cell is ~6pg. Therefore, 3μg cell DNA represents 5 × 105 cells. Each pg of viral DNA (7900bp dsDNA) contains 1.155 ×105 molecules. Thus, the number of viral DNA copies per cell can be easily calculated.
Support Protocol 3: Hirt DNA extraction protocol
Hirt first described an extraction protocol to isolate low molecular weight polyomavirus DNA from mouse cells (Hirt, 1967). The modified method described here is based on the traditional alkaline lysis plasmid preparation protocol used to isolate plasmids from bacterial cells and adapted for eukaryotic cells (Ustav & Stenlund, 1991).
Equipment
Tabletop centrifuge
Heat block
Reagents
Solution 1 (50mM glucose, 25mM Tris-HCl pH8, 10mM EDTA), filter and store at 4°C.
Solution 2 (1% SDS, 0.2M NaOH; make fresh and maintain at room temperature)
Solution 3 (5M Potassium acetate, pH 4.85 [60 ml of 5M potassium acetate and 11.5 ml of glacial acetic acid]); adjust final volume to 100 ml with water. Filter and store at 4°C.
Hirt digestion buffer (20mM Tris [pH8], 100mM NaCl, 10mM EDTA, 2% SDS, 200 μg/ml proteinase K [add fresh]). Maintain at room temperature.
95% ethanol
70% ethanol
Phenol:chloroform:isoamylalcohol (25:24:1) (Invitrogen 15593049)
Chloroform:isoamlalchohol (Sigma-Aldrich C0549-1PT)
5Prime phase log gel-heavy tubes for phenol:chloroform cleanup (Quantbio 10847-802)
Isopropanol
3M sodium acetate, pH5.2 (QBI 351-035-721)
TE buffer, pH 8.0 (Quality Biological #351‐011‐131)
100 mg/ml RNase A (Qiagen 19101)
- Remove feeders, trypsinize, count, and pellet keratinocytes as detailed in Support Protocol 1.
- DNA can be extracted from 1–4×106 cells. When comparing viral DNA levels, DNA should be extracted from an equal number of keratinocytes.
Resuspend pelleted cells in 200 μl of Solution 1 and transfer to a microfuge tube.
Add 400 μl of freshly prepared Solution 2 to lyse resuspended cells. Gently mix tube by inversion until the solution is clear and viscous.
Incubate tubes on ice for 5 minutes exactly.
Add 300 μl of Solution 3 to each tube to precipitate proteins and chromosomal DNA. Mix by inversion until a white precipitate has formed and solution is no longer viscous.
Incubate tubes on ice for 10 minutes. Pellet precipitated chromosomal DNA and proteins out of the lysate by centrifuging samples at top speed (≥ 16,000 × g) at 4°C for 5 minutes.
Transfer the supernatant to a new microfuge tube and precipitate nucleic acids with 0.6 volumes of isopropanol and mix by inversion.
Precipitate low molecular weight nucleic acids in the lysate by centrifuging samples at top speed (≥ 16,000 × g) for 10 minutes at 4°C.
Remove lysate and resuspend pellet in 200 μl of Hirt digestion buffer.
Incubate samples at 37°C for 30 minutes to digest remaining soluble proteins. Following this, incubate samples at 50°C for 30 minutes to self-digest the Proteinase K added in the Hirt digestion buffer.
Prepare column in 5Prime phase log gel-heavy tubes by centrifuging tubes at 12,000–16,000 × g for 20–30 seconds.
After preparing the column in the heavy phase lock tubes, add the 200 μl sample to the tubes.
Next add 200 μl phenol:chloroform:isoamylalcohol to phase lock tube and mix by shaking rapidly for 30 seconds. Do not vortex samples.
Centrifuge samples at ≥ 16,000 × g for 5 minutes.
Repeat step 13 and 14.
Add 200 μl chloroform:isoamylalcohol to the aqueous upper phase in the heavy phase lock tubes and mix by shaking rapidly for 30 seconds. Do not vortex the samples.
Centrifuge at max speed (≥ 16,000 × g) for 5 minutes.
Remove upper aqueous phase and transfer to a new microfuge tube.
Precipitate DNA samples by adding 20 μl of 3M sodium acetate and 3 volumes worth of 95% ethanol. Incubate samples on ice for at least 30 minutes.
Pellet DNA by centrifuging samples ≥ 16,000 × g for 10 minutes. Remove supernatant.
Wash pellet by adding 100 μl of 70% ethanol, flicking the tube to resuspend the pellet, and centrifuging the samples for 5 minutes at max speed (≥ 16,000 × g).
Carefully aspirate remaining ethanol and briefly dry pelleted DNA at room temperature.
Resuspend pellet in 50 μl of 1X TE containing 20μg/ml of RNAse A and incubate samples at 68°C for 20 minutes.
Support Protocol 4: Qiagen DNeasy Blood and Tissue DNA extraction protocol
Equipment:
Tabletop centrifuge
Heat block
Reagents:
Qiagen DNeasy Blood and Tissue Kit (Qiagen 69506)
95% ethanol
70% ethanol
3M sodium acetate (QBI 351-035-721)
TE buffer, pH 8.0 (Quality Biological #351‐011‐131)
RNase A (Qiagen 19101)
- Remove feeders, trypsinize, count, and pellet keratinocytes as detailed in Support Protocol 1.
- Extract DNA from 1–2×106 cells. Any more than 2×106 cells can clog the columns provided with the kit.
Resuspend cell pellet in 200 μl of 1X PBS.
Add 4 μl of 100 mg/ml RNase A and 20 μl of Proteinase K (provided with kit). Invert samples to mix and incubate at room temperature for 2 minutes.
Add 200 μl of DNeasy Blood and Tissue kit lysis buffer AL to samples and mix by vortexing the sample for 2 seconds.
Incubate and shake samples (shaking at ~850rpm) for 10 minutes at 56°C.
Add 200 μl of 95% EtOH and mix by vortexing the sample for 2 seconds.
Place provided spin columns in 2 ml collection tubes and add lysate suspension to columns.
Spin provided columns for 1 minute at 7000 × g and dispose of flow through.
Clean DNA samples bound to column by adding 500 μl of DNeasy Blood and Tissue kit wash buffer AW1 to columns and spinning samples for 1 minute at 7000 × g and dispose of flow through.
Add 500 μl of DNeasy Blood and Tissue kit wash buffer AW2 and spin samples for 3 minutes at max speed (≥ 16,000 × g). Dispose of flow through.
Elute DNA from column by adding 200 μl of DNeasy Blood and Tissue kit buffer AE, incubating samples for 5 minutes at room temperature, and centrifuging samples for 1 minute at 7000 × g.
Repeat step 11 and pool eluted DNA (400 μl total).
Precipitate eluted DNA by adding 40 μl of sodium acetate, mixing briefly by inversion, and adding 3 volumes worth (~1320 μl) of 95% ethanol. Invert samples several times to mix.
Incubate samples for 30 minutes on ice and then pellet DNA for 5 minutes at maximum speed (≥ 16,000 × g).
Wash the DNA pellet by adding 100 μl of 70% EtOH and vortexing the sample briefly. Pellet DNA again for 5 minutes at maximum speed (≥ 16,000 × g).
Carefully aspirate ethanol from tube and allow pellet to air dry briefly.
- Add 50 μl of TE buffer to pellet. Incubate samples overnight at 4°C to dissolve.
- It is critical that samples are not vortexed at this stage. To resuspend DNA in tube flick tube repeatedly.
Support Protocol 5: Generating a radioactive probe
Equipment:
Heat block
Tabletop centrifuge
Liquid scintillation counter
Scintillation vials
Reagents:
α-dCTP 32P 3000Ci/mmol, 10mCi/ml labeled nucleotide (Perkin-Elmer BLU013H250UC)
Linear HPV plasmid DNA specific for DNA samples run for southern blot analysis (e.g. pUC-HPV18) (see Table 1 for reference).
Random Primed DNA labeling kit (Sigma Aldrich 11004760001)
ProbeQuant G-50 Micro Columns Cleanup kit (Sigma Aldrich GE28-9034-08)
Scintillation fluid
Denature 50 ng of the probe/template DNA in total volume of 8 μl at 95°C for 10 minutes. Ensure the cap of the tube will not pop open at this temperature by using a cap lock.
Chill tube briefly on ice and centrifuge microfuge tube briefly to collect DNA at the bottom of the tube.
Mix the following reagents from the Random Primed DNA labeling kit: Reaction buffer (2 μl), 1 μl each of dATP, dGTP, dTTP, 50 μCi of 32P-dCTP, and Klenow enzyme (1 μl). The volume of 32P-dCTP will vary depending on the current radioactive activity. Add water to reach a total volume of 20 μl. Add the Klenow enzyme last.
Incubate mixture in a heat block at 37°C for 30 minutes to 1 hour.
Add 1 μl of 500mM EDTA to inactivate the reaction.
Remove unincorporated radioactive nucleotides using the ProbeQuant G-50 Micro Columns kit as described in the manufacturer’s instructions.
- Measure the specific activity of the radioactive probe in a liquid scintillation counter.
- The specific activity of the probe should be at least 1×108 cpm/μg of DNA. Anything less will require longer exposure to the screen and may result in increased background signal.
Mix the necessary amount of sheared salmon sperm with the radioactive probe (see Table 2) and incubate probe at 100°C for 10 minutes. Ensure the cap of the tube will not pop open at this temperature by using a cap lock.
Incubate briefly on ice and mix probe with predetermined amount of hybridization buffer.
Decant prehybridization solution from roller bottle.
Pipette radioactive probe-hybridization buffer into hybridization bottle and incubate at 68°C overnight, as described in Basic Protocol 3, steps 35–36.
Reagent preparation:
Feeder growth medium (store at 4°C; 2 week shelf life)
500 ml Dulbecco’s modified Eagle’s medium (DMEM, high glucose) (Invitrogen 11960069)
50 ml HyClone Iron-supplemented calf serum (Cytiva SH30072.03)
5 ml L-glutamine (Invitrogen 25030–164)
5 ml of 10,000 U/ml Penicillin-Streptomycin (ThermoFisher 15140163) (optional)
Keratinocyte F-medium (store at 4°C; 2 week shelf life)
355 ml of Ham’s F-12 nutrient mix (Invitrogen 11765062)
118 ml of DMEM, high glucose (Invitrogen 11960069)
25 ml of Fetal bovine serum (ThermoFisher 10439024)
200 μl of 1 mg/ml hydrocortisone (Sigma H4001)
4.2 μl of 1 mg/ml cholera toxin (EMD Millipore 227036)
5 μl of 1 mg/ml epidermal growth factor (Invitrogen PHG0311)
5 ml of 24 μg/ml adenine (Sigma A-2786)
1.25 ml of 2 mg/ml insulin (Gemini 700–112P)
5 ml L-glutamine (Invitrogen 25030–081)
5 ml of 10,000 U/ml Penicillin-Streptomycin (ThermoFisher 15140163) (optional)
NB: If using penicillin-streptomycin in Keratinocyte F-medium be sure to remove replace with no penicillin-streptomycin Keratinocyte F-medium 24 hours prior to transfection.
Transfection Recovery medium (store at 4°C; 2 week shelf life)
338 ml of Ham’s F-12 nutrient mix (Invitrogen 11765062)
110 ml of DMEM, high glucose (Invitrogen 11960069)
50 ml of Fetal bovine serum (ThermoFisher 10439024)
200 μl of 1 mg/ml hydrocortisone (Sigma H4001)
4.2 μl of 1 mg/ml cholera toxin (EMD Millipore 227036)
5 ml of 24 μg/ml adenine (Sigma A-2786)
1.25 ml of 2 mg/ml bovine insulin (Gemini 700-112P)
5 ml L-glutamine (Invitrogen 25030-081)
Formalin (prepare fresh for every use)
3.7% formaldehyde
1X PBS (made from 10X PBS, ThermoFisher 70011044)
H20
10X Methylene blue staining solution (store at room temperature)
1.4% methylene blue stain (made from dissolving 1.4 g methylene blue in 100 ml ethanol)
Dilute in H20 to make 1X working solution
Southern Blot Buffers (store all buffers at room temperature)
Depurination Buffer (0.1M HCl); shelf life >1 year
Denaturing buffer (3M NaCl, 0.4M NaOH); shelf life >1 year
Transfer Buffer (3M NaCl, 8mM NaOH); shelf life >1 year
5X Neutralization Buffer (1M Phosphate Buffer [0.558mM Na2HPO4, 0.437mM NaH2PO4•H2O, pH 6.8]); shelf life >1 year
Hybridization Buffer (3X SSC, 2% SDS, 5X Denhardt’s Solution, 0.2 mg/ml sheared salmon sperm); prepare fresh before use.
Wash Buffer (0.1X SSC, 0.1% SDS); shelf life >1 year.
Commentary
Background Information:
Human papillomaviruses replicate as extrachromosomal plasmids in the nucleus of infected cells. The gneration of keratinocyte cell lines containing extrachromosomal HPV genomes facilitates research on the maintenance and productive phases of the HPV life cycle. Basic protocol 1 presented here uses transient G418 selection of keratinocytes co-transfected with HPV genomes and a selectable pRSVneo plasmid. Alternative protocols used in the HPV field do not use transient drug selection to eliminate untransfected; instead cells transfected with HPV genomes are passaged many times to remove cells from the population whose growth is not supported by HPV oncogene E6 and E7 activity. While both protocols are useful, the protocol presented here vastly reduces the time needed to establish a stable keratinocyte cell line containing extrachromosomal HPV genomes (3 weeks compared to ~3 months). The use of transient G418 selection also allows rearchers to compare the efficiency of viral genome establishment. Viral genome establishment can be visualized and quantified using the quantitative colony formation assay (Basic Protocol 2). As an alternativeto transient selection, an HPV replicon containing a selectable neomycin marker and the HPV upstream regulatory region can be used. Because the replicon replicates alongside the HPV genome, continual selection can be used to select for cells maintaining the HPV genome (Alternative Support Protocol 1). Southern blot analysis is used to determine whether established HPV genomes in transfected keratinocyte cell lines are extrachromosomal or integrated into the host genome. We present here (Basic Protocol 3) an updated southern blot protocol that uses a rapid capillary transfer.
Understand Results:
Rheinwald-Green cell culture method
Proliferative keratinocyte colonies grown in co-coculture with growth arrested feeders should have a cobblestone appearance and push the feeder fibroblasts aside as they expand (see Figure 2, left panel). When passaging cells, feeder cells are completely removed from plates by gently spraying feeders off with Versene. Keratinocyte cells will adhere to plates during feeder removal (see Figure 2, right panel).
Quantitative colony formation assay
Successful establishment of HPV genomes in keratinocyte cell lines should yield multiple colonies of approximately 2–3 mm in size (see Figure 3, right plate). Colonies larger than this will most likely contain cells that have begun to differentiate and/or senesce in the center of the colony and will not grow upon passaging these cells. Cells transfected with the virus should form a greater number colonies than those transfected with the pRSVneo control (compare Figure 3, middle and right plates). Cells transfected with just the pUC19 plasmid should not form any colonies (Figure 3, left plate). If cell transfected with pUC19 alone form colonies this may indicate that the concentration of G418 used for selection may need to be increased.
Southern blot analysis
When running extracted DNA samples on the agarose gel, DNA should be evenly loaded (verified with ethidium bromide staining, Basic Protocol 3, step 7). Uncut, supercoiled viral DNA detected on blots should run at a size between 4–5 kb on a 0.8% agarose gel (Figure 5, left panel). Viral DNA linearized by a single site restriction enzyme should run at ~8kb (Figure 5, right panel). Software (e.g. Cytiva ImageQuant TL 8.1) can be used to measure the amount of the viral DNA standard loaded on the gel (Figure 5, bottom of right panel). A standard curve can be generated by plotting the loaded pg vs detected amount and can be used to calculate the quantity of detected viral DNA (in ng) from each sample.
The DNA standard amount should be adjusted to account for the cloned bacterial plasmid and compared to the viral DNA signal in the cell DNA lane. The amount of DNA in a single human cell is ~6pg. Therefore, 3μg cell DNA represents 5 × 105 cells. Each pg of viral DNA (7900bp dsDNA) contains 1.155 ×105 molecules. Thus, the number of viral DNA copies per cell can be easily calculated. Early passage keratinocyte cell lines containing HPV genomes usually contain between 50–70 viral genome copies per cell.
Troubleshooting:
Few or small colonies; collapsing/aborted colonies or multiple colonies on control plates.
This may occur for a variety of reasons. Using early passage keratinocytes (≤p4 since tissue isolation) greatly enhances the chances of success as older cells are closer to senescence and may not recover as easily from transfection. Different establishment efficiencies may also be due to the original donor cells used for generating keratinocytes. In addition, the quality and concentration of the recircularized HPV genomes and pRSVneo plasmid DNA can also impact the successful establishment of extrachromosomal viral DNA. Lastly, it may be worth considering changing the concentration of G418 used during selection of HPV cell lines as this could differ from one keratinocyte strain to another. It is important to note though that decreasing the G418 concentration may increase the number of cells in the population that do not contain the HPV genome. Comparing the number of colonies transfected with the viral genome to colonies transfected with the pRSVneo plasmid control is critical and can be used to help determine the effects of changing the G418 concentration.
High background or blank spots on southern blots
Non-specific radioactive signal on blots may be an indication that blots were not washed enough. A Geiger counter can be used to survey regions of the blot without DNA to assess the level of background radioactive signal. Blank spots or disrupted signal detected by the phosphorimager screen may be due to incomplete DNA transfer to nylon membranes. Additionally, ensure that the phosphorimager screens are in good condition. Improper maintenance of screens (i.e. exposure to moisture or ungloved hands) may damage screens and impair detection of radioactive signals on blots.
Signal for southern blot is low
Several factors may be responsible for low signal. Extending the phosphorimager exposure time to radioactive blots will increase the signal strength but will result in increased noise (background detection) as well. Increasing the amount of sample loaded into the gel may be necessary. Lastly, ensure that the specific activity of the radioactive probe is at least 1×108 cpm/μg of DNA before probing blots.
Time Considerations:
A general timeline starting from cell culture and HPV genome recircularization to southern blot analysis is presented in Figure 6. Individual time considerations are mentioned in brief below:
Cell culture preparation prior to transfection: approximately 2 weeks
HPV genome recircularization: 3 days
Keratinocyte transfection: 1 day
Colonies arising after transient selection: approximately 14 days
Quantitative colony formation assessment: 1 day
Southern blot analysis: 5 days. Blots containing radioactive signal may be exposed to the phosphorimager screen for longer when necessary.
Figure 6.
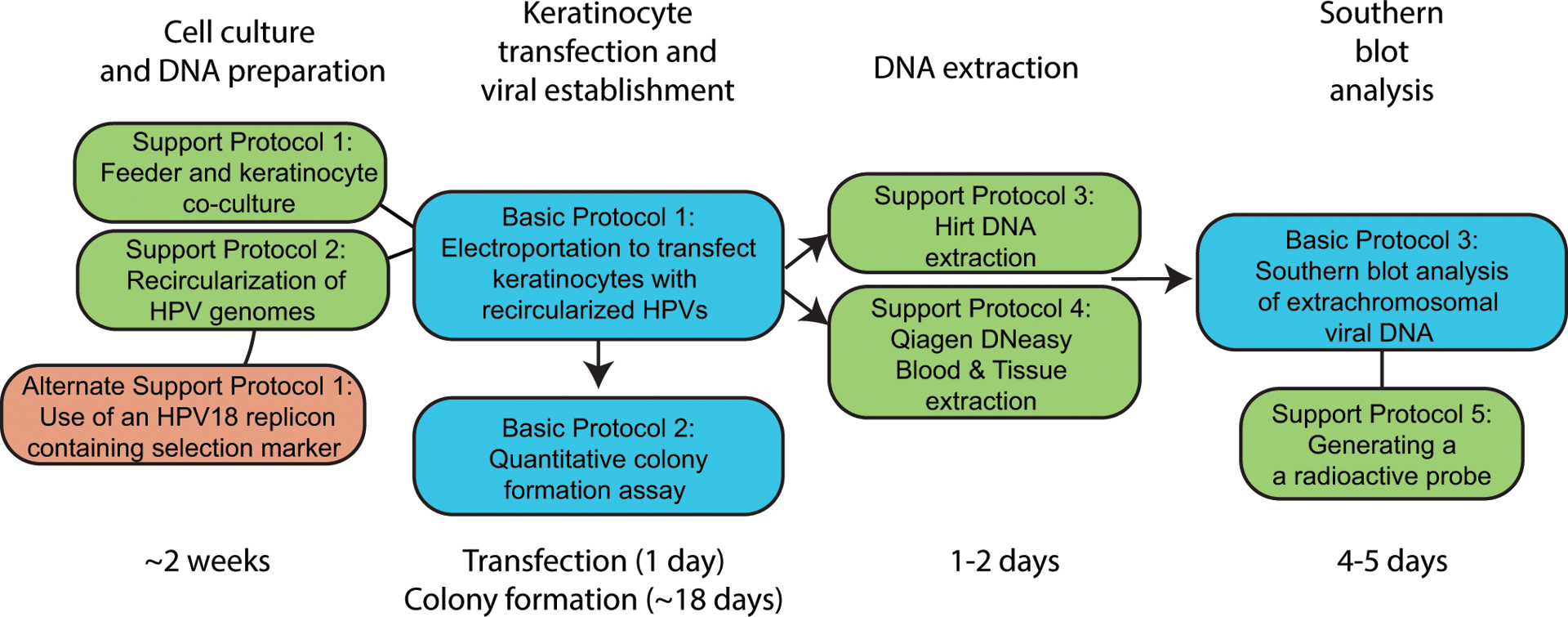
Timeline and overview of protocols. Basic protocols are highlighted in blue, support protocols in green, and alternative protocols in red. General timelines for protocols are listed below.
Acknowledgements
This research was supported by the Intramural Research Division of the National Institutes of Allergy and Infectious Diseases, National Institutes of Health
References:
- Allen-Hoffmann BL, Schlosser SJ, Ivarie CA, Sattler CA, Meisner LF, & O’Connor SL (2000). Normal growth and differentiation in a spontaneously immortalized near-diploid human keratinocyte cell line, NIKS. J Invest Dermatol, 114(3), 444–455. doi: 10.1046/j.1523-1747.2000.00869.x [DOI] [PubMed] [Google Scholar]
- Boshart M, Gissmann L, Ikenberg H, Kleinheinz A, Scheurlen W, & zur Hausen H (1984). A new type of papillomavirus DNA, its presence in genital cancer biopsies and in cell lines derived from cervical cancer. EMBO J, 3(5), 1151–1157. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/6329740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman S, Liu X, Meyers C, Schlegel R, & McBride AA (2010). Human keratinocytes are efficiently immortalized by a Rho kinase inhibitor. J Clin Invest, 120(7), 2619–2626. doi: 10.1172/JCI42297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coursey TL, Van Doorslaer K, & McBride AA (2021). Regulation of HPV18 Genome Replication, Establishment and Persistence by Sequences in the Viral Upstream Regulatory Region. J Virol, JVI0068621. doi: 10.1128/JVI.00686-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehrmann F, & Laimins LA (2005). Human papillomavirus type 31 life cycle: methods for study using tissue culture models. Methods Mol Biol, 292, 317–330. doi: 10.1385/1-59259-848-x:317 [DOI] [PubMed] [Google Scholar]
- Flores ER, Allen-Hoffmann BL, Lee D, Sattler CA, & Lambert PF (1999). Establishment of the human papillomavirus type 16 (HPV-16) life cycle in an immortalized human foreskin keratinocyte cell line. Virology, 262(2), 344–354. doi: 10.1006/viro.1999.9868 [DOI] [PubMed] [Google Scholar]
- Frattini MG, Lim HB, & Laimins LA (1996). In vitro synthesis of oncogenic human papillomaviruses requires episomal genomes for differentiation-dependent late expression. Proc Natl Acad Sci U S A, 93(7), 3062–3067. doi: 10.1073/pnas.93.7.3062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu B, Quintero J, & Baker CC (2003). Keratinocyte growth conditions modulate telomerase expression, senescence, and immortalization by human papillomavirus type 16 E6 and E7 oncogenes. Cancer Res, 63(22), 7815–7824. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/14633708 [PubMed] [Google Scholar]
- Henno L, Tombak EM, Geimanen J, Orav M, Ustav E, & Ustav M (2017). Analysis of Human Papillomavirus Genome Replication Using Two- and Three-Dimensional Agarose Gel Electrophoresis. Curr Protoc Microbiol, 45, 14B 10 11–14B 10 37. doi: 10.1002/cpmc.28 [DOI] [PubMed] [Google Scholar]
- Hirt B (1967). Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol, 26(2), 365–369. doi: 10.1016/0022-2836(67)90307-5 [DOI] [PubMed] [Google Scholar]
- Hummel M, Hudson JB, & Laimins LA (1992). Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J Virol, 66(10), 6070–6080. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/1326657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon S, Allenhoffmann BL, & Lambert PF (1995). Integration of Human Papillomavirus Type-16 into the Human Genome Correlates with a Selective Growth Advantage of Cells. J Virol, 69(5), 2989–2997. doi: 10.1128/Jvi.69.5.2989-2997.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lace MJ, Turek LP, Anson JR, & Haugen TH (2014). Analyzing the Human Papillomavirus (HPV) Life Cycle in Primary Keratinocytes with a Quantitative Colony-Forming Assay. Curr Protoc Microbiol, 33, 14b.12.11–13. doi: 10.1002/9780471729259.mc14b02s33 [DOI] [PubMed] [Google Scholar]
- Lambert PF, Ozbun MA, Collins A, Holmgren S, Lee D, & Nakahara T (2005). Using an immortalized cell line to study the HPV life cycle in organotypic “raft” cultures. Methods Mol Med, 119, 141–155. [DOI] [PubMed] [Google Scholar]
- McBride AA, Chapman S, Stepp WH, & Terunuma A (2014). Conditional Reprogramming of Epithelial Cells with Rho-Kinase Inhibitors. In Hunsberger J& Rao M (Eds.), Intramural Pluripotent Stem Cell Protocol Book (pp. 624–632). Bethesda, MD: NIH Center for Regenerative Medicine,. [Google Scholar]
- Porter SS, & McBride AA (2020). Human Papillomavirus Quasivirus Production and Infection of Primary Human Keratinocytes. Curr Protoc Microbiol, 57(1), e101. doi: 10.1002/cpmc.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rheinwald JG, & Green H (1977). Epidermal growth factor and the multiplication of cultured human epidermal keratinocytes. Nature, 265(5593), 421–424. doi: 10.1038/265421a0 [DOI] [PubMed] [Google Scholar]
- Schowalter RM, Pastrana DV, & Buck CB (2011). Glycosaminoglycans and sialylated glycans sequentially facilitate Merkel cell polyomavirus infectious entry. PLoS Pathog, 7(7), e1002161. doi: 10.1371/journal.ppat.1002161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley MA, Browne HM, Appleby M, & Minson AC (1989). Properties of a non-tumorigenic human cervical keratinocyte cell line. Int.J.Cancer, 43(4), 672–676. Retrieved from http://onlinelibrary.wiley.com/store/10.1002/ijc.2910430422/asset/2910430422_ftp.pdf?v=1&t=ir6unvee&s=f08b645f560d8158e9bd7cd5234e63b9faf64cae [DOI] [PubMed] [Google Scholar]
- Stepp WH, Meyers JM, & McBride AA (2013). Sp100 provides intrinsic immunity against human papillomavirus infection. MBio, 4(6), e00845–00813. doi: 10.1128/mBio.00845-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toots M, Ustav M Jr, Männik A, Mumm K, Tämm K, Tamm T, … Ustav M (2017). Identification of several high-risk HPV inhibitors and drug targets with a novel high-throughput screening assay. PLoS pathogens, 13(2), e1006168. Retrieved from http://journals.plos.org/plospathogens/article/file?id=10.1371/journal.ppat.1006168&type=printable [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ustav M, & Stenlund A (1991). Transient replication of BPV-1 requires two viral polypeptides encoded by the E1 and E2 open reading frames. EMBO J, 10(2), 449–457. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/1846806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Doorslaer K, Chen D, Chapman S, Khan J, & McBride AA (2017). Persistence of an Oncogenic Papillomavirus Genome Requires cis Elements from the Viral Transcriptional Enhancer. MBio, 8(6), e01758–01717. doi: 10.1128/mBio.01758-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voytas D (2001). Agarose gel electrophoresis. Curr Protoc Mol Biol, Chapter 2, Unit2 5A. doi: 10.1002/0471142727.mb0205as51 [DOI] [PubMed] [Google Scholar]