

Summary
Exercise is a powerful driver of physiological angiogenesis during adulthood, but the mechanisms of exercise-induced vascular expansion are poorly understood. We explored endothelial heterogeneity in skeletal muscle and identified two capillary muscle endothelial cell (mEC) populations that are characterized by differential expression of ATF3/4. Spatial mapping showed that ATF3/4+ mECs are enriched in red oxidative muscle areas while ATF3/4low ECs lie adjacent to white glycolytic fibers. In vitro and in vivo experiments revealed that red ATF3/4+ mECs are more angiogenic when compared with white ATF3/4low mECs. Mechanistically, ATF3/4 in mECs control genes involved in amino acid uptake and metabolism and metabolically prime red (ATF3/4+) mECs for angiogenesis. As a consequence, supplementation of non-essential amino acids and overexpression of ATF4 increased proliferation of white mECs. Finally, deleting Atf4 in ECs impaired exercise-induced angiogenesis. Our findings illustrate that spatial metabolic angiodiversity determines the angiogenic potential of muscle ECs.
Keywords: single-cell RNA-seq, exercise, endothelial metabolism, endothelial heterogeneity, amino acid metabolism, muscle angiogenesis
Graphical abstract
Highlights
-
•
Capillary mEC populations have differential ATF3/4 and anabolic gene expression
-
•
ATF3/4+ ECs in oxidative muscle areas are metabolically primed for angiogenesis
-
•
Non-essential amino acids and ATF4 activation promote ATF3/4low WmEC proliferation
-
•
EC-specific loss of Atf4 impairs exercise-induced angiogenesis
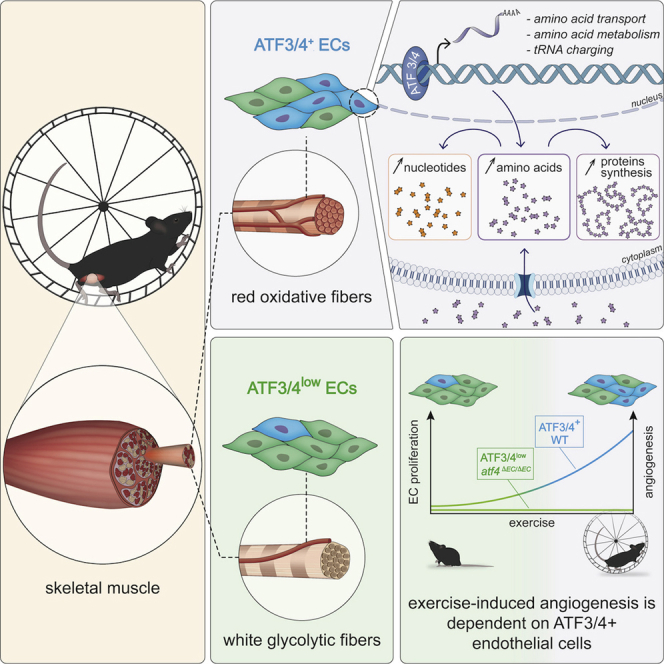
Fan et al. discover two populations of muscle endothelial cells (mECs) that have differential ATF3/4 expression. Notably, ATF3/4+ mECs lie next to oxidative myofibers and have higher expression of amino acid metabolic genes, which metabolically prepare mECs for exercise-induced vascular expansion. Consequently, EC-specific loss of Atf4 impairs exercise-induced angiogenesis.
Introduction
Skeletal muscle is a highly plastic organ that ensures movements ranging from blinking an eye to running and weight lifting, but it is also critical for the maintenance of body metabolic homeostasis (DeFronzo et al., 1981). Muscle is composed of different fiber types, which classically have been defined by their myosin heavy-chain content that determines their contractile properties, but they also substantially differ in their metabolic properties and the nutrients they use for energy generation (Egan and Zierath, 2013; Rowe et al., 2014). Glycolytic (type II) fibers are capable of producing high force at high intensity but fatigue rapidly due to a less efficient means of ATP generation using the glycolytic conversion of glucose to lactate. In contrast, oxidative (type I) fibers produce lower force but are relatively resistant to fatigue. They efficiently generate energy via the oxidative phosphorylation of glucose and lipids within their extensive mitochondrial networks. High concentrations of myoglobin and cytochromes redden these fibers, hence, red oxidative fibers versus white glycolytic fibers. The relative amounts of these fiber types differ between muscles and between individuals, and are even linked to some disease states. For instance, type 2 diabetes is associated with a lower proportion of type I fibers (Stuart et al., 2013).
It is well described that red oxidative fibers are more vascularized when compared with white glycolytic fibers and that myofiber vascularization correlates with mitochondrial density and oxidative capacity (Haas and Nwadozi, 2015). Indeed, muscle metabolic regulators PGC1α/β and ERRα/γ, which activate a broad program of mitochondrial biogenesis and a metabolic switch toward a more oxidative phenotype, integrate muscle metabolism and vascular density by enhancing VEGF levels (Arany et al., 2008; Matsakas et al., 2012; Narkar et al., 2011; Rowe et al., 2011). However, what is less well described is whether in addition to blood vessel density, the functional properties of muscle endothelial cells (mECs) are dependent on the specific fiber that they interact with. Furthermore, whether muscle angiodiversity is controlled by the metabolic microenvironment of the fiber remains to be elucidated.
An increase in muscle capillarization following exercise is a hallmark adaptation to exercise and one of the few non-pathological settings of vascular expansion during adulthood. To meet the metabolic demand of the contracting muscle, the delivery and uptake of oxygen and nutrients increase dramatically during exercise (Ahlborg et al., 1974; Egan and Zierath, 2013). For this reason, blood flow through the vessels surrounding the active muscle fibers is enhanced within seconds (Murrant and Sarelius, 2000) and soon the formation of new vessels is promoted (Bloor, 2005). Increased muscle capillary density should improve blood-tissue exchange properties by increasing the surface area for oxygen diffusion or nutrient uptake as well as faster elimination of toxic waste products. Trained muscle indeed exhibits higher oxygen exchange capacity and improved glucose uptake (Gorski and De Bock, 2019; Prior et al., 2004). The increase in muscle vascularization is an early adaptive event in response to exercise (Andersen and Henriksson, 1977; Waters et al., 2004), but the exact mechanisms through which exercise promotes angiogenesis are poorly understood.
Recent single-cell RNA sequencing (scRNA-seq) approaches revealed that ECs are remarkably heterogeneous (Goveia et al., 2020; Jakab and Augustin, 2020; Kalucka et al., 2020; Vanlandewijck et al., 2018), suggesting that they adapt their transcriptomic signature to meet the specific (metabolic) requirements of their microenvironment. They also revealed common transcriptomic signatures between EC populations during pathological angiogenesis (Rohlenova et al., 2020). Whether physiological angiogenesis is driven by similar mechanisms is not known, but unraveling those would potentially offer targets for regenerative therapy. Here, we show the existence of two capillary subpopulations within the muscle, which are localized in different microenvironments and are characterized by differential expression of ATF3/4. ATF3/4+ mECs lying adjacent to oxidative fibers are metabolically and functionally distinct from ATF3/4low mECs, which are adjacent to glycolytic fibers. Using exercising muscle as a model for physiological angiogenesis, we found that ATF3/4+ mECs proliferate to ensure vascular expansion. Mechanistically, baseline ATF3/4 expression metabolically primes mECs for angiogenesis. Finally, deletion of Atf4 from ECs considerably reduced the transcriptional differences between RmECs and WmECs and prevented the exercise-induced increase in vascular density.
Results
scRNA-seq reveals endothelial cell subpopulations in skeletal muscle
Skeletal muscle is composed of different fiber types with distinct metabolic characteristics. Muscles that consist of mainly oxidative fibers (expressing myosin heavy-chain I or IIa isoforms; MHCI/IIa, from now on oxidative muscle) such as the red parts of m. gastrocnemius (GAS), m. quadriceps femoris (QUAD), and m. soleus (SOL) have high abundance of ECs and capillaries when compared with muscles that contain more glycolytic fibers (expressing MHC IIx or IIb isoforms; MHCIIb/x, glycolytic muscle), such as the white parts of GAS and QUAD, and m. extensor digitorum longus (EDL) (Figures S1A–S1G). To explore muscle EC heterogeneity, we collected CD31+CD45− ECs from GAS of a wild-type (WT) C57BL/6N mouse (8 weeks) and performed scRNA-seq. We used GAS because this muscle contains oxidative as well as glycolytic fibers (Figures S1A and S1E). A single EC suspension was loaded into the 10x Genomics Chromium single-cell controller, and the rest of the cells were reanalyzed by flow cytometry to confirm presence of CD31 and absence of CD45 (Figure S1H). scRNA-seq profiles were obtained from 4,121 cells with a median of 959 genes per cell. All RNA-seq data generated in this paper can be interactively explored at https://shiny.debocklab.hest.ethz.ch/Fan-et-al/. Unsupervised clustering followed by t-distributed stochastic neighbor embedding (t-SNE) revealed six clusters of vascular ECs (Figures 1A and 1B), which all expressed the pan-vascular markers Cdh5 and Pecam1 (Figure 1C) and did not express described marker sets of muscle stem cells and other muscle cell types (Figure S1I) (De Micheli et al., 2020). We detected very few lymphatic ECs characterized by Lyve1, Prox1, and Pdpn, but due to their low number, they were not further analyzed. Next, we used genes that were enriched in specific populations to annotate cell clusters to known vascular EC populations. In agreement with a previously published scRNA-seq dataset that includes ECs isolated from EDL and SOL (Kalucka et al., 2020), P-selectin (Selp) was highly specific for venous ECs. Vwf and Vcam1 were also specific to venous ECs, since only negligible expression was observed in arterial ECs (Figure 1C). Co-staining for SELP and VWF confirmed their co-expression (Figure 1D) in veins. Venous mECs also expressed previously identified markers such as Nr2f2 and Ephb4 (Gerety et al., 1999; Vanlandewijck et al., 2018; You et al., 2005). Arterial and arteriolar mECs expressed known arterial marker genes such as Efnb2 (Gerety et al., 1999; Wang et al., 1998), Gja5 (Lu and Wang, 2017), Sema3g (Kutschera et al., 2011), Fbln5, Hey1, Bmx, and Clu (Ekman et al., 1997; Kalucka et al., 2020). While expressing arterial genes, arteriolar mECs simultaneously expressed genes such as Lpl and its binding partner Gpihbp1, which are specific to capillary ECs (Davies et al., 2010). The expression of arterial markers was strikingly similar between arteriolar and arterial mECs (Figure 1C). Arteriolar mECs, on the other hand, also expressed several capillary marker genes. We found some interesting differences between different populations: for instance, there was high and medium expression of Aqp1 in capillary and arteriolar mECs, respectively, whereas Aqp1 was barely detected in arterial ECs. Indeed, EFNB2+ mECs in larger arteries were devoid of AQP1 protein (Figure 1E). Moreover, capillary mECs selectively expressed carbonic anhydrase 4 (Car4) (Figure 1F) and 8 (Car8), and these genes were absent in arterioles. There was one small population that expressed several EC marker genes but that we could not assign to any well-described EC subtype (xECs). This population was characterized by Itm2a, Tnfaip2, and Esm1 expression, genes that were previously linked to a small capillary population in SOL (Kalucka et al., 2020). Unbiased mapping of our data to the skeletal muscle data of that paper confirmed the overlap (Figure S1J). Subsequently, we focused on the two larger capillary EC populations (CapEC 1 and CapEC 2).
Figure 1.

scRNA-seq reveals EC subpopulations in skeletal muscle
(A) Visualization of mEC populations on a t-SNE plot, color-coded for the identified subpopulations, including venous ECs (vEC), arterial ECs (aEC), arteriolar ECs (arlEC), capillary ECs (CapEC 1 and CapEC 2), and an unknown EC population (xEC).
(B) Heatmap with Z scores showing top 10 enriched marker genes per cell type relative to other subpopulations.
(C) Violin plots of normalized scRNA expression profiles of known EC subtype markers in each mEC cluster.
(D–F) Representative images of GAS cross-sections, stained for (D) an EC marker (CD31, red) combined with α smooth muscle actin (αSMA) (orange), SELP (green), and VWF (gray) (enriched in venous ECs); (E) EFNB2 (green) (enriched in arteries and arterioles) and AQP1 (gray) (low expression in arteries and arterioles; and (F) CAR4 (green) and AQP1 (gray) (enriched in capillaries). Arrows indicate SELP+VWF+ veins in (D) and EFNB2+AQP1− arteries in (E). Scale bars, 50 μm.
See also Figure S1.
Muscle contains two capillary EC populations characterized by distinct Atf3/4 expression
To explore differences between the two capillary subpopulations, we looked at the most differentially expressed genes (DEGs), which included members of the basic leucine zipper transcription factor (bZIP) family as well as genes related to the integrative stress response (Figure 2A). In particular, the expression levels of activating transcription factor 4 (Atf4) and its downstream target Atf3, two master regulators of the integrative response to stress (Hai et al., 2010; Harding et al., 2003; Pakos-Zebrucka et al., 2016), were among the most DEGs (Figure 2A). Even more, while most EC subtypes (CapEC 1, but also non-capillary ECs) expressed Atf3/4, these genes were far less expressed in CapEC 2. Differential Atf3/4 expression levels were not secondary to standard cell dissociation procedures (van den Brink et al., 2017; Wu et al., 2017), since mEC isolation in the presence of actinomycin D (ActD), a transcriptional inhibitor, which prevents the activation of the stress response during cell isolation (Wu et al., 2017), showed that our optimized (10 min) digestion protocol did not affect Atf3/4 or stress response gene expression (Figure S2A). Moreover, staining for ATF3 on GAS cross-sections showed that some, but not all, CD31+ cells were ATF3+ (Figures 2B and 2C). ATF3 immunostainings combined with fiber-type analysis further showed that ATF3(/4)+ ECs are enriched in muscle areas predominantly containing oxidative MHCI/IIa fibers whereas glycolytic MHCIIb/x muscle contained fewer ATF3(/4)+ mECs and most mECs lacked ATF3(/4) (Figures 2B–2D). To further confirm preferential localization of ATF3/4 in capillary ECs from red oxidative muscle areas (from now on red muscle ECs or RmECs) versus ATF3/4low ECs in white glycolytic muscle areas (white muscle ECs or WmECs), we isolated RmECs from SOL and WmECs from EDL (Figures S1C, S1D, and S1G). Gene profiling showed lower Atf3/4 in WmECs (Figure S2B), which was maintained upon ActD treatment (Figure S2C). We also isolated and cultured RmECs and WmECs for 7 days and found that WmECs retained lower Atf3/4 mRNA content (Figure S2D) and fewer WmECs stained positive for ATF3 (Figures 2E and 2F). Thus, ATF3/4low ECs are highly enriched in WmECs whereas ATF3/4+ ECs are enriched in RmECs.
Figure 2.

Muscle contains two capillary EC populations characterized by distinct Atf3/4
(A) Left: heatmap with log fold changes of 50 top marker genes of CapEC 1 relative to CapEC 2. Right: violin plot showing the distribution of Atf3 and Atf4 expression within different mEC populations.
(B) Representative fluorescence images showing ATF3 (gray) and CD31 (red) combined with type I (MHCI, green), type IIa (MHCIIa, blue), and type IIb (MHCIIb, yellow) fiber-type staining on cross-sections in oxidative and glycolytic areas of GAS. Arrows indicate ATF3+CD31+ ECs. Scale bar, 50 μm.
(C) Quantification of the percentage of ATF3+ vessels in oxidative and glycolytic areas of GAS (n = 6).
(D) Representative fluorescence images showing ATF3 (gray) and CD31 (red) combined with type I (MHCI, green), type IIa (MHCIIa, blue), and type IIb (MHCIIb, yellow) fiber-type staining on thick longitudinal sections of GAS. Arrows indicate ATF3+CD31+ ECs. Scale bar, 20 μm.
(E) Representative images of CD31 (red), ATF3 (green), and Hoechst (blue) staining of RmECs and WmECs that were cultured for 7 days. Scale bar, 50 μm.
(F) Quantification of the percentage of ATF3high ECs in RmECs versus WmECs (n = 3).
(G) Heatmap of the top 75 most highly variable genes in RmECs versus WmECs.
(H) TF motif enrichment analysis of upregulated genes in RmECs over WmECs.
(I) ATF4-dependent biological process analysis of upregulated genes in RmECs over WmECs.
Two-tailed unpaired Student’s t test in (C) and (F) (∗p < 0.05). Bar graphs represent mean ± SEM. See also Figure S2 and Data S1.
We next took advantage of the selective enrichment of ATF3/4+ and ATF3/4low ECs in RmECs and WmECs, respectively, to perform a more detailed analysis of the transcriptional differences between RmECs (isolated from SOL) and WmECs (isolated from EDL) using bulk RNA-seq. 782 genes were differentially expressed, and Atf3/4 were present among the most DEGs (Figure 2G; Data S1; https://shiny.debocklab.hest.ethz.ch/Fan-et-al/). Furthermore, unbiased transcription factor (TF) motif enrichment analysis of DEGs showed that the ATF4-binding motif RNMTGATGCAAY was the most enriched one, and an alternative ATF4-binding motif was identified within the ten most enriched TF-binding motifs (Figure 2H). Comparison of our DEG list with the list of verified ATF4 target genes derived from ChIP-seq data (Han et al., 2013) confirmed the presence of 60 ATF4 target genes (Data S1). ATF4 is activated in response to several stressors (Harding et al., 2003; Hayner et al., 2018; Köditz et al., 2007; Quirós et al., 2017; Ron and Walter, 2007; Yang et al., 2018). While ATF4 has an adaptive role under stressful conditions, it can also be activated by pro-growth signals and under such conditions acts as a downstream anabolic effector of the mechanistic target of rapamycin complex 1 (mTORC1), the master regulator of cell growth (Adams, 2007; Ben-Sahra et al., 2016; Torrence et al., 2021). Interestingly, the majority of differentially expressed ATF4-dependent genes encoded enzymes required for non-essential amino acid (NEAA) synthesis, amino acid (AA) uptake, and tRNA charging, and overlapped (49.18% of reported anabolic genes were higher in RmECs) with the set of genes coordinating the ATF4-dependent anabolic response (Torrence et al., 2021) (Figure S2E), whereas only very few (9.23%) ATF4-dependent adaptive genes were activated (Figure S2F). Furthermore, analysis of biological processes executed by known ATF4 genes confirmed activation of “cellular AA synthesis,” “translation,” and “AA transporter” while “endoplasmatic reticulum stress” was not affected (Figure 2I). Since we observed the differential regulation of ATF4-dependent anabolic genes, we subsequently wondered whether ATF3/4 determines the angiogenic potential of mECs.
Atf3/4low WmECs have a lower angiogenic potential in vitro
To explore if ATF3/4low WmECs and ATF3/4+ RmECs have different angiogenic characteristics, we isolated and cultured RmECs and WmECs. EdU assays showed that more RmECs were proliferating when compared with WmECs under standard culture conditions, and RmECs also proliferated faster (Figures 3A–3C). RmECs sprouted better in spheroid assays as they had more and longer sprouts (Figures 3D and 3E). We also embedded muscle explants in collagen matrix, and as soon as 5 days after embedding, we observed a more pronounced outgrowth of isolectin B4-positive (IB4+) capillaries from oxidative muscles containing RmECs (Figures 3F and 3G).
Figure 3.

ATF3/4low WmECs have a lower angiogenic potential
(A) Representative images of EdU incorporation (gray) and EdU intensity in RmECs and WmECs combined with CD31 (red) and ERG (green) staining. Scale bar, 100 μm.
(B) Quantification of the percentage of EdU+ ECs (CD31+) in RmECs versus WmECs (n = 6).
(C) Quantification of (per mEC) EdU median intensity of RmECs versus WmECs (n = 5).
(D and E) Representative bright field pictures (D) and quantification (E) of number of sprouts and average and total sprout length in spheroids composed of RmECs and WmECs (n = 30). Scale bar, 50 μm.
(F and G) Representative images (F) and quantification of the length (G) of vascular sprouts from collagen embedded muscle explants from red oxidative (top row) and white glycolytic (bottom row) muscles or muscle areas. Explants were stained for isolectin B4 (IB4) (red), phalloidin (green), and Hoechst (blue). Scale bar, 100 μm.
(H and I) Representative fluorescence images (H) and quantification (I) of EdU incorporation (red) and Hoechst (blue) in scr versus Atf3/4KD HUVECs (n = 4). Scale bar, 100 μm.
(J and K) Representative pictures (J) and quantifications (K) of scratch wound width in mitomycin C (mitoC)-treated WT (scr) and Atf3/4KD HUVECs (n = 3).
(L and M) Representative pictures (L) and quantifications (M) of total and average sprout length and number of sprouts in WT (scr) and Atf3/4KD HUVEC spheroids (n = 30), with or without mitomycin C (mitoC) treatment. Scale bar, 50 μm.
(N and O) Representative images (N) and quantification (O) of EdU incorporation (gray) in GFP or Atf4 (Atf4OE) overexpressing RmECs and WmECs combined with CD31 (red) and ERG (green). Scale bar, 100 μm.
Two-tailed unpaired Student’s t test in (B), (C), (E), (G), and (I) (∗p < 0.05). One-way ANOVA with Tukey’s multiple comparisons test in (M) and (O) (∗p < 0.05; n.s., not significant). Two-way ANOVA with Sidak’s multiple comparisons test in (K). Bar graphs represent mean ± SEM. See also Figure S3.
To study the functional relevance of ATF3/4 in maintaining the angiogenic potential of RmECs, we silenced Atf3/4 using short hairpin RNAs (shRNAs) against ATF3 as well as ATF4 in human umbilical vein endothelial cells (HUVECs) (Atf3/4KD) (Figure S3A). Lowering ATF3/4 in HUVECs reduced their proliferative capacity (Figures 3H and 3I). We also assessed cell migration using scratch wound assays, but this was not affected by Atf3/4KD (Figures 3J and 3K). Atf3/4KD cells sprouted less (Figures 3L and 3M), and this was caused by lower proliferation since treatment with the proliferation blocker mitomycin C abrogated the sprouting difference (Figures 3L and 3M).
Finally, to prove the functional relevance of ATF4, we used a doxycycline-inducible ATF4 overexpression construct (Torrence et al., 2021), which allowed us to titrate ATF4 expression leading to a 2-fold increase in ATF4 (Figure S3B). Increasing ATF4 in WmECs (but not RmECs) sufficed to increase proliferation (Figures 3N and 3O), despite the lower activation of ATF4 downstream genes when compared with RmECs (Figure S3B). Since metabolism is a crucial mediator of EC function, and since our unbiased RNA-seq analysis already showed that many ATF4-dependent genes involved in AA metabolism were differentially expressed between RmECs and WmECs, we speculated that ATF3/4 maintains angiogenic capacity in RmECs by controlling endothelial AA metabolism.
Atf3/4 rewires AA metabolism to metabolically prime RmECs for angiogenesis
Sufficient AA availability is crucial for protein synthesis during proliferation. While AA metabolism has been extensively studied in cancer cells, several studies have meanwhile highlighted that EC AA metabolism extensively rewires during angiogenesis (Hitzel et al., 2018; Huang et al., 2017; Kim et al., 2017; Vandekeere et al., 2018). Based on our unbiased RNA-seq analysis, we first verified by RT-PCR that neutral AA transporters (SLC1A4, SLC1A5, SLC7A1, and SLC7A5), which were previously shown to be ATF3/4 dependent and which we confirmed as differentially expressed in RmECs versus WmECs (Figure 4A), were downregulated in Atf3/4KD HUVECs (Figure 4B). This coincided with reduced uptake of AA such as leucine and glutamine (Figure 4C). Also, the expression of several enzymes involved in the generation of NEAAs such as asparagine (ASNS) and de novo serine and glycine biosynthesis (PSAT1, PSPH, PHGDH, and SHMT2) was lower in WmECs (Figures 4A and S4A) and Atf3/4KD cells (Figures 4A and S4A). To test whether de novo serine/glycine biosynthesis was reduced in WmECs as well as in Atf3/4KD cells, we supplemented U-13C-glucose, a main substrate for serine/glycine biosynthesis in ECs (Vandekeere et al., 2018), to the medium and traced its incorporation in serine/glycine. We first confirmed that ECs use glucose for de novo synthesis of serine and glycine (Figures 4D and 4E). Additionally, WmECs and Atf3/4KD incorporated less U-13C-glucose into serine and glycine (Figures 4D and 4E). This broad rewiring of AA uptake and synthesis suggested that ATF3/4 are required to maintain intracellular AA balance. Indeed, WmECs and Atf3/4KD HUVECs had reduced levels of most, but not all, AAs (Figures 4F and 4G).
Figure 4.

ATF3/4 rewires AA metabolism and biomass synthesis to metabolically prime RmECs for angiogenesis
(A and B) Gene expression analysis of neutral AA transporters and metabolic genes in RmECs and WmECs (A), as well as WT (scr) and Atf3/4KD HUVECs (B) (n = 3–6).
(C) Leucine and glutamine uptake assay in WT (scr) and Atf3/4KD HUVECs (n = 6).
(D and E) Incorporation of [U-13C]-glucose carbon into serine and glycine in RmECs versus WmECs (D), as well as WT (scr) versus Atf3/4KD HUVECs (E) (n = 4).
(F and G) Intracellular free AA abundance in RmECs versus WmECs (F) (n = 10), and WT (scr) versus Atf3/4KD HUVECs (G) (n = 5).
(H and I) Protein synthesis rate in RmECs versus WmECs (H), as well as WT (scr) versus Atf3/4KD HUVECs (I) (n = 8–12).
(J and K) Intracellular purine levels in RmECs versus WmECs (J), and WT (scr) versus Atf3/4KD HUVECs (K) (n = 5).
(L and M) Total levels and labeling of ADP from [U-13C] glucose in RmECs versus WmECs (L) as well as WT (scr) versus Atf3/4KD HUVECs (M) (n = 4–5).
(N and O) Representative fluorescence images (N) and quantification (O) of EdU incorporation (red) in WT (scr) and Atf3/4KD HUVECs cultured in control conditions (ctrl) and upon supplementation with NEAAs or EAAs (n = 6). Scale bar, 100 μm.
(P and Q) Representative pictures (P) and quantifications (Q) of total sprout length and number of sprouts in WT and Atf3/4KD HUVEC spheroids cultured under control conditions (ctrl) and upon supplementation with NEAAs or EAAs (n = 25). Scale bar, 50 μm.
(R and S) Representative fluorescence images (R) and quantification (S) of EdU incorporation (gray) in RmECs and WmECs (CD31, red; ERG, green) cultured in control (ctrl) and upon supplementation with NEAA (n = 12). Scale bar, 100 μm.
Two-tailed unpaired Student’s t test in (A)–(M) (∗p < 0.05; n.s., not significant). One-way ANOVA with Tukey’s multiple comparisons test in (O), (Q), and (S) (∗p < 0.05). Bar graphs represent mean ± SEM. See also Figure S4.
To understand how reduced AA uptake and synthesis impair proliferation in WmECs and upon Atf3/4KD, we assessed the activity of several anabolic pathways. First, protein synthesis was lower in WmECs and upon Atf3/4KD (Figures 4H, 4I, S4C, and S4D), which was not unexpected because of the lower intracellular AA levels. Moreover, since glycine and one-carbon (1C) units generated during serine-to-glycine conversion are used for de novo purine dNTP synthesis (Labuschagne et al., 2014), we also assessed whether purine synthesis was affected by ATF3/4. Indeed, WmECs and Atf3/4KD had lower purine levels (Figures 4J and 4K). We also found lower contribution of glycine and serine-derived 1C units in purines shown by a lower fraction of purines with mass value of m+6 or higher upon U-13C-glucose feeding (Diehl et al., 2019) (Figures 4L and 4M).
Importantly, metabolic differences between WT and Atf3/4KD ECs as well as RmECs and WmECs were independent of proliferation. Contact-inhibited (CI) ECs had lower levels of Atf3/4 and downstream genes (Figure S4E), suggesting that proliferation coincides with increased ATF3/4 activity. On the other hand, Atf3/4KD CI ECs retained lower expression of AA genes when compared with WT HUVECs despite efficient activation of genes associated with endothelial quiescence pathways (Figure S4E). Under CI conditions, Atf3/4KD ECs retained lower glutamine and leucine uptake (Figure S4F) and intracellular AA levels (Figure S4G), even though differences in AAs under normal culture conditions were small, altogether leading to lower protein synthesis (Figure S4H). Finally, to confirm that intracellular AA levels were different between quiescent non-proliferating RmECs and WmECs in vivo, we isolated and immediately lysed RmECs and WmECs for AA analysis and found lower levels of most AAs in WmECs compared with RmECs (Figure S4I).
To further study whether the metabolic differences between ATF3/4+ RmECs and ATF3/4low WmECs were causing the differences in proliferation/sprouting, we increased AA availability by adding EAAs as well as NEAAs to the normal culture medium. Indeed, it is known that high AA concentrations in culturing media can “rescue” deficits in cellular AA levels (Harding et al., 2003; Torrence et al., 2021). Addition of NEAAs, but not EAAs, completely rescued proliferation (Figures 4N and 4O) as well as sprouting of Atf3/4KD ECs (Figures 4P and 4Q). We also observed increased proliferation of WmECs when cultured in medium containing additional NEAAs, while RmEC proliferation was not affected by NEAA supplementation (Figures 4R and 4S). Of note, adding NEAAs to the culture medium did not affect Atf3/4 expression (Figure S4J). Finally, we also reduced average AA availability in the culture medium to that previously measured in muscle (Felig and Wahren, 1971) by lowering it to 1/3 of full medium AA concentration. Culturing WmECs and Atf3/4KD HUVECs under 1/3 AA levels further reduced intracellular AA levels when compared with full medium conditions (Figures S4K and S4L). In support of this, while WT (Scr) HUVECs were not affected by reduced AA availability, 1/3 AA availability increased the sprouting and proliferation deficit in Atf3/4KD HUVECs (Figures S4M–S4P). Altogether, these data underscore the primary role for AA metabolism in driving the functional differences between RmECs and WmECs.
Ultimately, we aimed at getting further insight into whether ATF3/4 control angiogenesis through interacting with known transcriptional regulators of angiogenesis. Atf3/4KD did not, however, reduce angiogenic activity by increasing the activity of NOTCH or FOXO1 as it did not affect the expression of known FOXO1 or NOTCH targets (Figures S4Q and S4R). Atf3/4KD also did not change the activation of NOTCH target genes upon stimulation with the NOTCH ligand DLL4 (Figure S4R). Lowering ATF3/4 has also been associated with increased P53 stability, but we could not detect P53 stabilization in Atf3/4KD cells (Figure S4S), nor was it identified in our TF analysis of upregulated genes in RmECs over WmECs (Figure 2H). ATF4 is known to form heterodimers with other bZIP TFs, such as C/EBP, in order to control gene expression (Kilberg et al., 2009), but only CHOP-10 showed modest enrichment in our TF motif analysis (Figure 2H). Since the bZIP TF cMYC is a powerful driver of EC metabolism and growth (Wilhelm et al., 2016), we decided to explore whether ATF3/4 control EC proliferation via affecting cMYC. We did not observe altered expression of known cMYC target genes CDK4, CCND2, and CCNB2 upon Atf3/4KD, or altered expression of cMYC itself, suggesting that the proliferative defect under normal conditions was not due to a primary defect in cMYC activity (Figures S4T and S4U). Interestingly, cMYC controls the expression of ATF3/4 (Tameire et al., 2019; Tamura et al., 2005). ATF4 and cMYC also share common target genes in cancer cells, most of which are involved in AA transport and metabolism (Tameire et al., 2019). cMYC overexpression increased ATF3/4 mRNA content and expression of several (ATF3/4-dependent, see above) AA metabolic genes (Figures S4T and S4U). Moreover, while cMYC overexpression in Atf3/4KD cells was still able to activate genes involved in cell-cycle regulation, it failed to activate the AA-related transcriptional signature, showing that ATF3/4 are required for cMYC-mediated induction of genes involved in AA metabolism (Figure S4U). Finally, Atf3/4KD blunted the ability of cMYC to increase sprouting (Figures S4V and S4W) and proliferation capacity (Figures S4X and S4Y) under low mitogenic, 1/3 AA conditions, whereas NEAA supplementation fully rescued proliferation (Figures S4X and S4Y). Thus, basal ATF3/4 metabolically prime ECs for angiogenesis and cMYC-induced proliferation.
Exercise leads to selective expansion of RmECs
To study whether RmECs are also primed for angiogenesis in vivo, we used voluntary wheel running as a model for exercise-induced angiogenesis (Chinsomboon et al., 2009). Single mice were put in cages with free access to running wheels (Figure S5A). After 14 days of exercise, we found an increase in IB4+ vessel area and more ERG+ ECs in the oxidative area of the QUAD, whereas we did not observe increased vessel density in the glycolytic area (Figures 5A–5D). We also detected higher vessel density in oxidative areas of the GAS (Figures S5B and S5C). Subsequently, we performed scRNA-seq on mECs isolated from the GAS of sedentary and exercised mice. Cell clustering showed that there was a shift between the two capillary subpopulations following exercise, as the Atf3/4+ mEC population increased, while Atf3/4low capillary mECs proportionally decreased (Figure 5E). Other mEC populations such as venous, arterial, and arteriolar ECs kept approximately the same cell abundance. Subsequently, we investigated whether Atf3/4+ capillary ECs proliferate themselves or Atf3/4low capillary ECs transformed into Atf3/4+ capillary ECs. To address this question, we applied RNA velocity analysis on our scRNA-seq dataset from sedentary and exercised samples to predict the future state of individual cells (La Manno et al., 2018). No clear extrapolated future states (arrows) were found between Atf3/4+ and Atf3/4low capillary ECs, suggesting no general movement of capillary ECs from Atf3/4low toward Atf3/4+ population (Figure 5F). These data showed that different mEC populations were in a steady-state relationship and suggested that during exercise, the ATF3/4+ RmECs proliferate whereas the Atf3/4low WmECs remain quiescent. To test this, we repeated the running experiment and injected mice with EdU (single injection of 50 μg/g body weight). As expected, proliferation was observed in very few cells in the sedentary group, but it increased substantially in exercised mice (Figures 5G–5I). First, the majority of proliferating cells (EdU+) were ECs (ERG1+). Second, the increase in proliferating mECs (EdU+ERG1+) was largely limited to oxidative areas, whereas mECs within the glycolytic area did not proliferate (Figures 5G–5J). Flow cytometry analysis of RmECs and WmECs confirmed a selective increase in the number of EdU+ RmECs upon exercise (Figures S5D and S5E) and they proliferated faster (Figures S5F and S5G). We also found more ATF3+ ECs in the oxidative area after training, while the number of ATF3+ ECs in the glycolytic area did not change (Figures S5H and S5I). Analysis of spatial distribution of proliferative mECs revealed that EdU+ mECs were present inside the oxidative area. We also found some proliferating ECs in the adjacent peri-oxidative area, which contains a mixture of oxidative and glycolytic fibers, but their number gradually and rapidly decreased with increasing distance from the oxidative area (Figure 5K). Subsequent additional analysis of IB4+ area and number of ERG1+ ECs and EdU+ERG1+ ECs within this peri-oxidative area confirmed that there was an increase in vessel density (Figures S5B, S5C, and S5J–S5L). Thus, upon exercise, ATF3/4+ RmECs, but not ATF3/4low WmECs, become angiogenic.
Figure 5.

Exercise leads to selective expansion of RmECs
(A) Representative fluorescence image showing IB4 (red) combined with type I (MHCI, green), type IIa (MHCIIa, blue), and type IIb (MHCIIb, yellow) fiber-type staining on QUAD cross-sections. Oxidative, peri-oxidative, and glycolytic areas are marked. Scale bar, 400 μm.
(B) Representative fluorescence images of IB4 (red) and ERG (green) staining in oxidative and glycolytic areas of QUAD from sedentary versus exercised (14 days voluntary running) mice. Scale bar, 50 μm.
(C and D) Quantification of IB4+ area (% of total area, C) and number of ERG+ ECs (D) within oxidative and glycolytic areas of sedentary versus exercised mice (n = 3–4).
(E) t-SNE plot derived from scRNA-seq of mECs from sedentary versus exercised mice (14 days voluntary running). Pie charts show the fraction of each population in sedentary (top) and exercised (bottom) mice.
(F) RNA velocity analysis of mECs from sedentary (top) and exercised (bottom) mice (14 days voluntary running). Arrows indicate extrapolated future states of ECs.
(G and H) Representative images of staining of serial sections of QUAD for type I (MHCI, green), type IIa (MHCIIa, blue), and type IIb (MHCIIb, yellow) fibers (first section) and EdU incorporation (gray) co-stained with IB4 (red) and ERG (green) (second section) after 14 days of exercise (G). Scale bar, 200 μm. Magnification of white box (90° rotated) is shown in (H); arrows indicate EdU+ERG+ ECs. Scale bar, 50 μm.
(I) Quantification of % EdU+ERG+ ECs in oxidative and glycolytic areas of QUAD from sedentary versus exercised mice (n = 3).
(J) Representative fluorescence images showing EdU incorporation (gray) and IB4 (red) on thick longitudinal QUAD sections. Arrows indicate EdU+IB4+ ECs. Scale bar, 20 μm.
(K) Quantification of spatial distribution of EdU+ERG+ proliferating ECs in QUAD of sedentary versus exercised mice (n = 6).
Two-tailed unpaired Student’s t test in (C), (D), and (I) (∗p < 0.05; n.s., not significant). Two-way ANOVA with Sidak’s multiple comparisons test in (K) (∗p < 0.05; n.s., not significant). Bar graphs represent mean ± SEM. See also Figure S5.
Deletion of Atf4 in ECs impairs exercise-induced endothelial proliferation and vascular expansion
To confirm the functional role of Atf3/4+ ECs in vivo, we intercrossed Atf4LoxP/LoxP mice (Ebert et al., 2012) with EC-specific inducible Pdgfb-CreERT2 mice (Claxton et al., 2008), to generate Atf4ΔEC/ΔEC mice (Figure 6A). Tamoxifen injection (1 mg/day for 5 days followed by washout of at least 7 days) led to an efficient decrease in Atf4 mRNA (Figure S6A). We subsequently evaluated the transcriptional changes (bulk RNA-seq) induced by loss of ATF4 in RmECs and WmECs isolated from Atf4ΔEC/ΔEC muscles. From the 782 and 140 genes that were differentially expressed between WT RmECs and WmECs, respectively (log fold change > 1 and adjusted p value < 0.05), only 211 and 126 remained differentially expressed between RmECs and WmECs after ATF4 knockout (Figure 6B). The effect of ATF4 knockout was much more pronounced in RmECs than in WmECs, as 73% of DEGs in RmECs were not different any longer after Atf4 deletion, while only 10% of WmECs DEGs were affected by the knockout. Moreover, loss of Atf4 completely wiped out the differential regulation of the anabolic program in RmECs (Figure 6C), and principal component analysis revealed that Atf4-deficient RmECs were almost indistinguishable from WmECs (Figure 6D). These data show that ATF4 is a main transcriptional regulator of RmECs.
Figure 6.

Deletion of Atf4 in ECs impairs exercise-induced EC proliferation and vascular expansion
(A) Scheme showing the generation of Pdgfb-Cre × Atf4Loxp/Loxp (Atf4ΔEC) mice and experimental plan.
(B) Left: heatmap of the top 75 most highly variable genes in WT RmECs versus WT WmECs. Right: relative expression in Atf4ΔEC/ΔEC RmECs versus Atf4ΔEC/ΔEC WmECs.
(C) Left: heatmap showing relative expression of ATF4-dependent anabolic gene set in WT RmECs versus WT WmECs. Right: relative expression in Atf4ΔEC/ΔEC RmECs versus Atf4ΔEC/ΔEC WmECs. Bold gene names refer to DEGs between WT RmECs and WT WmECs (log fold change > 1 and adjusted p value < 0.05).
(D) PCA showing sample distances between RmECs and WmECs (WT and Atf4ΔEC/ΔEC).
(E and F) Representative fluorescence images of serial sections of QUAD from WT and Atf4ΔEC/ΔEC mice stained for type I (MHCI, green), type IIa (MHCIIa, blue), and type IIb (MHCIIb,yellow) fibers (first section) and EdU incorporation (gray) co-stained with IB4 (red) and ERG (green) (second section) after 14 days of exercise (E). Scale bar, 100 μm. Magnification of white box is shown in (F); arrows indicate EdU+ERG+ ECs. Scale bar, 50 μm.
(G) Representative fluorescence images of IB4 vascular staining in glycolytic and oxidative areas of QUAD from WT and Atf4ΔEC/ΔEC mice in sedentary and exercised conditions. Scale bar, 50 μm.
(H and I) Quantification of IB4+ area (% of total area) (H) and the percentage of EdU+ERG+ ECs (I) in oxidative versus glycolytic areas in QUAD of WT versus Atf4ΔEC/ΔEC mice in sedentary and after exercise (n = 6). One-way ANOVA with Tukey’s multiple comparisons test (∗p < 0.05; n.s., not significant). Each dot represents a single mouse.
Bar graphs represent mean ± SEM. See also Figure S6.
To verify that endothelial ATF4 defines the angiogenic potential of RmECs in vivo, we performed exercise experiments. Running distance was not different between genotypes (Figure S6B). While we noticed an increase in IB4+ vascular density upon exercise in WT mice, this was not the case in Atf4ΔEC/ΔEC mice (Figures 6E–6H). This was not due to reduced baseline vascularization, as vascular density of Atf4ΔEC/ΔEC muscle was similar to that of WT littermates. We did not observe more dead mECs under sedentary or exercised conditions (Figure S6D). Rather, exercise-induced proliferation of mECs in the oxidative areas of Atf4ΔEC/ΔEC muscle was reduced (Figures 6E–6I). Lower exercise-induced vascular expansion did not affect short-term (2-week) training adaptations: we found similar increases in exercise performance (Figure S6C) and muscle fiber-type composition (Figure S6E), and the expression of oxidative enzymes was not different between genotypes (Figures S6F–S6H).
Finally, to exclude that reduced exercise-induced proliferation of Atf4ΔEC/ΔEC mECs was secondary to systemic effects caused by widespread deletion of Atf4 in ECs, we performed low-dose tamoxifen injections in Atf4ΔEC/ΔEC mice, which we intercrossed with Rosa26mTmG fate-tracing mice. Low-dose tamoxifen injection (1/100 dilution—0.01 mg in a single injection 5 days before starting the exercise intervention) resulted in mosaic recombination with approximately 20% of ECs being mGFP labeled (Figures S6I and S6J). Subsequently, we verified whether in response to exercise, mGFPWT ECs proliferate and expand whereas mGFPATF4 KO ECs do not. In order to label all proliferating cells, we performed repetitive EdU injections (three times in the last 24 h) (Figures S6I and S6K). Upon exercise, mGFPWT ECs efficiently increased proliferation, as we found 4% of ECs to be EdU+. As expected, mGFPATF4 KO ECs did not increase proliferation: the fraction of mGFPATF4 KO/EdU+ ECs was lower when compared with mTomatoWT non-recombined ECs in the same muscle from the same animal as well as the recombined mGFPWT ECs from Pdgfb-CreERT2RosamTmG mice (which do not carry the Atf4LoxP/LoxP alleles) (Figures S6I and S6K). Consequently, the contribution of mGFPATF4 KO ECs to the total mEC population decreased (Figures S6I and S6J). In conclusion, these data provide further evidence that ATF4 is required for exercise-induced mEC proliferation and vascular expansion.
Discussion
Using scRNA-seq, we identified a specific population of mECs that is characterized by altered levels of Atf3/4 and downstream genes. While all other populations efficiently expressed Atf3/4, levels were very low in one capillary population, which was enriched in the glycolytic muscle areas. It was recently reported in several cell types, including muscle stem cells (van den Brink et al., 2017) and neurons (Wu et al., 2017), that the activation of stress-responsive genes such as Atf3/4 can be artificially evoked during standard cell dissociation procedures. In scRNA-seq datasets (Barry et al., 2019; Goveia et al., 2020), the appearance of subpopulations with high expression of bZIP TFs and stress-responsive genes was attributed to EC activation, either due to isolation (Barry et al., 2019) or tumor-derived factors (Goveia et al., 2020). On the other hand, Atf3/4 have previously been linked to EC biology in vivo: ECs driving regeneration of the EC lining of large arteries express bZIP TFs and ATF3 is required for endothelial regeneration in large arteries (McDonald et al., 2018). High ATF3 has also been linked to diabetic angiopathy (Okamoto et al., 2006). Moreover, increased muscle capillary density upon sulfur AA starvation is associated with increased ATF4 (Longchamp et al., 2018). Isolation of ECs in the presence of actinomycin D, a transcriptional inhibitor, which prevents the activation of stress-responsive genes during cell isolation (Wu et al., 2017), showed that our isolation protocol did not cause differential expression of Atf3/4. Moreover, we found that differences in ATF3+ mECs were present on tissue sections and that culturing RmECs versus WmECs did not wash out the difference in ATF3 protein levels or the expression of Atf3/4. Subsequent bulk RNA-seq profiling of ATF3/4+ RmECs versus ATF3/4low WmECs followed by TF-binding motif enrichment analysis revealed that ATF4-binding motifs were most abundantly present in the promotor sequences of those genes, which were more highly expressed in RmECs, suggesting that ATF4 defines the transcriptional differences between RmECs and WmECs. Genetically removing Atf4 from ECs considerably reduced the transcriptional differences between RmECs and WmECs. Moreover, Atf4ΔEC/ΔEC mice showed blunted exercise-induced angiogenesis, altogether underscoring the functional role of ATF4 as a main transcriptional regulator of RmECs in vivo.
Previous studies already highlighted the importance of AA metabolism for EC biology. ECs take up large amounts of glutamine (Huang et al., 2017; Kim et al., 2017) and use it for the synthesis of biomass. Removing glutamine from the medium increased ATF4 while reducing mTORC1 activation and proliferation/migration (Huang et al., 2017). In contrast, removing methionine and cysteine promoted angiogenesis in vitro in an ATF4-dependent fashion (Longchamp et al., 2018). In addition to those observations, ATF4 can be activated as part of a widespread anabolic program (Adams, 2007; Ben-Sahra et al., 2016; Torrence et al., 2021). Among the most variably expressed genes in RmECs were genes in AA uptake and metabolism that are linked to the anabolic ATF4-dependent program (Torrence et al., 2021). We found that ATF3/4low cells within the glycolytic area did not proliferate in response to exercise and, when brought in culture under angiogenic conditions, showed a significantly lower proliferation rate when compared with ECs isolated from oxidative areas (which have higher ATF3/4). Lowering ATF3/4, either with knockdown approaches in HUVECs or by using WmECs, coincided with a reduced expression of several genes involved in AA uptake and metabolism. Consistently, ATF3/4low cells took up fewer AAs and had lower intracellular levels of both EAAs and NEAAs. We also assessed whether AA synthesis was affected and found that serine/glycine biosynthesis from glucose was lower in primary ATF3/4low WmECs and Atf3/4KD HUVECs. Knockdown of Phgdh, which arrests de novo serine biosynthesis from glucose, reduced EC proliferation and viability even in the presence of serine (Vandekeere et al., 2018). In contrast to the latter observations, we did not observe acute loss of viability under normal culturing conditions upon knockdown of Atf3/4 in HUVECs. We also did not observe lower vascular density in sedentary adult muscle upon deletion of Atf4, or an increased number of dead ECs in Atf4ΔEC/ΔEC mice, which argues against significant loss of EC viability. The reduction in Phgdh and serine/glycine biosynthesis was lower in our study, and the remaining flux through serine biosynthesis likely sufficed to maintain viability while restricting proliferative capacity. Importantly, increasing the availability of NEAAs, but not EAAs, in the culture medium completely restored proliferation and sprouting in Atf3/4KD HUVECs as well as WmECs. This shows that ATF3/4 controls EC proliferation through metabolism. Finally, the metabolic and transcriptional differences were present in quiescent mECs as well as in CI HUVECs, and we found higher levels of AAs in primary isolated (non-cultured) RmECs compared with WmECs. Thus, ATF3/4 metabolically prime mECs for proliferation and angiogenesis by controlling AA availability and metabolism.
ATF4 functions in heterodimers with other bZIP TFs and also co-regulates many of its target genes together with other TFs (Kilberg et al., 2009). Many of those bZIP TFs, such as cMYC (Wilhelm et al., 2016), C/EBP (Kakogiannos et al., 2020; Loinard et al., 2012; Min et al., 2017), JunD (Gerald et al., 2004), and others (Hamik et al., 2006), have been linked to endothelial biology before. Also, ATF3/4 have been previously shown to stabilize P53 (Yan et al., 2005), a known regulator of angiogenesis (Gogiraju et al., 2015). We currently do not know which bZIP TFs co-regulate metabolic gene expression together with ATF4. We only observed that CHOP-10 TF motifs were enriched in RmECs, albeit with low significance. Since ATF4 and CHOP are known to bind the same promotors, the importance of this finding warrants future investigation. Within this study, we focused on the interplay with cMYC because of its known role in promoting angiogenesis through coordinating the expression of genes involved in metabolism, growth, and proliferation (Potente and Carmeliet, 2017). However, it is worthwhile to note that cMYC was not identified in our unbiased TF motif analysis. This is not surprising since cMYC signaling is repressed in quiescent ECs (Wilhelm et al., 2016). Rather, the transcriptional activation of ATF4 by cMYC seems to be a necessary step during angiogenic activation. In this respect, reducing ATF3/4 did not impair the ability of cMYC to activate genes involved in cell-cycle control but clearly blunted the cMYC-induced activation of genes involved in AA metabolism. This selective targeting of metabolic genes resulted in the inability of cMYC to promote proliferation of ECs in culture. These observations underscore once more the powerful role of metabolism as a critical engine of angiogenesis.
Our study largely focused on revealing the molecular mechanisms through which ATF3/4+ mECs maintain angiogenic capacity (or why ATF3/4low ECs lose their angiogenic capacity), and we did not evaluate whether ATF4 is required for maintenance of quiescence or endothelial function in vivo. In this respect, an oxidizing microenvironment as well as AA deprivation activate ATF4 through the integrative stress response leading to the activation of genes involved in adaptation to stress. We observed that a small subset of this adaptive gene set was differentially expressed between RmECs and WmECs in an ATF4-dependent fashion. Future research should evaluate whether ATF3/4 contribute to EC homeostasis and function during quiescence.
It is tempting to speculate about the upstream mechanism leading to lower ATF3/4 in WmECs. Since ATF3/4+ mECs were largely enriched within the red oxidative area, one possibility could be that the oxidative microenvironment of type I/IIa fibers is responsible for maintaining basal ATF3/4 levels over time, while ATF3/4 is gradually lost in glycolytic muscle areas. Indeed, oxidative stress is a known regulator of Atf4 expression (Harding et al., 2003). On the other hand, scRNA-seq of mouse aortic ECs, which presumably have a more uniform metabolic microenvironment when compared with skeletal muscle, also confirmed the presence of ATF3+ cells that expanded upon aortic injury (McDonald et al., 2018). Moreover, it is worthwhile noting that the transcriptional and functional differences between the capillary populations were maintained upon short-term (7 days) culture. RNA velocity analysis also showed that Atf3/4low mECs were not on a trajectory toward acquiring genetic properties of Atf3/4+ cells upon exercise. These observations, however, require confirmation using lineage tracing studies. We only observed expansion of the Atf3/4+ population, suggesting that differences in Atf3/4 are (epi)genetically imprinted in these populations. McDonald et al. further showed that the Atf3+ population is gradually lost during aging and coincides with reduced aortic regeneration, indicating that ECs can permanently lose Atf3/4 under specific conditions such as aging. Future studies should reveal whether long-term exercise is able to maintain ATF3/4 expression into aging and whether this is associated with improved aortic regeneration.
Limitations of study
Angiogenesis is an early adaptive response to training (Andersen and Henriksson, 1977; Waters et al., 2004), and various exercise adaptations are dependent on endothelial function. For instance, the exercise-induced increase in glucose uptake is dependent on an increase in blood perfusion (McConell et al., 2020). Moreover, inhibition of vasodilation and angiogenesis in response to exercise impaired exercise-induced increases in performance (Van der Stede et al., 2021). We did not observe a difference in performance, muscle oxidative parameters, or fiber-type changes. We consider it a limitation of our study that the training period did not go beyond 2 weeks, which is necessary to make sound statements about the interaction between exercise-induced vascular expansion and training adaptations.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat anti-Mouse/Rat CD31/PECAM-1 antibody | R&D Systems | Cat# 3628; RRID: AB_2161028 |
| Rat anti-CD31 antibody | Abcam | Cat# ab7388; RRID: AB_305905 |
| Rabbit anti-CD62P (SELP) antibody | Abcam | Cat# ab59738; RRID: AB_940914 |
| Sheep anti-Von Willebrand Factor (VWF) antibody | Abcam | Cat# ab11713; RRID: AB_298501 |
| Goat anti-Ephrin-B2 Antibody | R&D Systems | Cat# AF496; RRID: AB_2095679 |
| Goat anti-Carbonic Anhydrase IV (CAR4) Polyclonal Antibody | Thermo Fisher Scientific | Cat# PA5-47312; RRID: AB_2607329 |
| Rabbit anti-Aquaporin 1 antibody | Abcam | Cat# ab15080; RRID: AB_2056839 |
| Monoclonal Anti-Actin, alpha-Smooth Muscle - Cy3(TM) antibody produced in mouse | Sigma-Aldrich | Cat# C6198; RRID: AB_476856 |
| PE Rat Anti-Mouse CD31 | BD Biosciences | Cat# 553373; RRID: AB_394815 |
| APC/Fire 750 anti-mouse CD31 Antibody | Biolegend | Cat# 102434; RRID: AB_2629683 |
| PerCP Rat Anti-Mouse CD45 | BD Biosciences | Cat# 557235; RRID: AB_10642171 |
| Anti-mouse CD45, APC/Fire 750, clone 30_F11 | Biolegend | Cat# 103154; RRID: AB_2572115 |
| Anti-mouse CD31, Alexa Fluor 488, clone 390 | Biolegend | Cat# 102414; RRID: AB_493408 |
| APC/Fire 750 anti-mouse CD31 antibody | Biolegend | Cat# 102434; RRID: AB_2629683 |
| Mouse (IgG2b) Myosin Heavy Chain Type I antibody (BA-F8) | DSHB | Cat# BA-F8; RRID: AB_10572253 |
| Mouse (IgG1) Myosin Heavy Chain Type IIA antibody (SC-71) | DSHB | Cat# SC-71; RRID: AB_2147165 |
| Mouse (IgM) Myosin Heavy Chain Type IIB antibody (BF-F3) | DSHB | Cat# BF-F3; RRID: AB_2266724 |
| Rabbit anti-ERG (A7L1G) antibody | Cell Signaling Technology | Cat# 97249; RRID: AB_2721841 |
| Rabbit anti-ATF3 antibody | Sigma-Aldrich | Cat# HPA001562; RRID: AB_1078233 |
| Rabbit anti-PSAT1 antibody | Abcam | Cat# ab96136; RRID: AB_10697763 |
| Rabbit ant-PHGDH antibody | Sigma-Aldrich | Cat# HPA021241; RRID: AB_1855299 |
| Mouse anti-PSPH antibody | Santa Cruz Biotechnology | Cat# sc-271421; RRID: AB_10610605 |
| Rabbit anti-ATF3 antibody [EPR19488] | Abcam | Cat# ab207434; RRID: AB_2734728 |
| Rabbit anti-ATF4 (D4B8) antibody | Cell Signaling Technology | Cat# 11815; RRID: AB_2616025 |
| P21 antibody (EPR362) | Abcam | Cat# ab109520; RRID: AB_10860537 |
| Mouse anti-P53 (1C12) monoclonal antibody | Cell Signaling Technology | Cat# 2524; RRID: AB_331743 |
| c-Myc monoclonal antibody (9E10) | Thermo Fisher Scientific | Cat# MA1-980; RRID: AB_558470 |
| Rabbit Anti-Hexokinase II Monoclonal Antibody, Unconjugated, Clone C64G5 | Cell Signaling Technology | Cat# 2627S; RRID: AB_2232946 |
| Total OXPHOS Rodent WB Antibody Cocktail | Abcam | Cat# ab110413; RRID: AB_2629281 |
| Mouse anti-β-actin antibody | Cell Signaling Technology | Cat# ab181861; RRID: AB_2566811 |
| Donkey anti-Goat IgG (H+L) AF 594 | Thermo Fisher Scientific | Cat# A-11058; RRID: AB_142540 |
| Donkey anti-Rat IgG (H+L) AF 568 | Abcam | Cat# AB175475; RRID: AB_2636887 |
| Donkey anti-Sheep IgG (H+L) AF 350 | Thermo Fisher Scientific | Cat# A-21097; RRID: AB_2535751 |
| Donkey anti-rabbit IgG (H+L) AF 488 | Thermo Fisher Scientific | Cat# A32790; RRID: AB_2762833 |
| Donkey anti-Goat IgG (H+L) AF 488 | Thermo Fisher Scientific | Cat# A32814; RRID: AB_2762838 |
| Donkey anti-rabbit IgG (H+L) AF 568 | Thermo Fisher Scientific | Cat#A10042; RRID: AB_2534017 |
| Donkey anti-rabbit IgG (H+L) AF 350 | Thermo Fisher Scientific | Cat# A10039; RRID: AB_2534015 |
| Donkey anti-rat IgG (H+L) AF 488 | Thermo Fisher Scientific | Cat#A-21208; RRID: AB_2535794 |
| Goat anti-mouse IgG2b AF 488 | Thermo Fisher Scientific | Cat# A-21141; RRID: AB_141626 |
| Goat anti-mouse IgG1 AF 350 | Thermo Fisher Scientific | Cat# A21120; RRID: AB_1500805 |
| Goat anti-mouse IgM AF 568 | Thermo Fisher Scientific | Cat# A-21043; RRID: AB_2535712 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7074S; RRID: AB_2099233 |
| Anti-mouse IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7076; RRID: AB_330924 |
| Anti-streptavidin, HRP-conjugated | Abcam | Cat# ab7403 |
| Chemicals, peptides, and recombinant proteins | ||
| Collagenase IV | Thermo Fisher Scientific | Cat# 17104019 |
| Dispase Ⅱ | Sigma-Aldrich | Cat# D4693 |
| Puromycin | Sigma-Aldrich | Cat# P8833 |
| Endothelial cell growth supplement | Sigma-Aldrich | Cat# E2759 |
| Hoechst | Thermo Fisher Scientific | Cat# 62249 |
| 5-Ethynyl-2'-deoxyuridine (EdU) | Thermo Fisher Scientific | Cat# C10632/4 |
| SYTOX Blue | Thermo Fisher Scientific | Cat# S34857 |
| Alexa-647 Fluor conjugated isolectin B4 | Thermo Fisher Scientific | Cat# I32450 |
| Alexa-568 Fluor conjugated isolectin B4 | Thermo Fisher Scientific | Cat# I21412 |
| [6-3H]-thymidine | PerkinElmer | Cat# NET355 |
| [U-13C]-glucose | Cambridge Isotope Laboratories | Cat# CLM-1396 |
| L-[3,4-3H(N)]-Glutamine | PerkinElmer | Cat# NET551250UC |
| L-[14C(U)]-Leucine | PerkinElmer | Cat# NEC279E050UC |
| EasyTag EXPRESS35S Protein Labeling Mix | PerkinElmer | Cat# NEG772002MC |
| recombinant human Delta-like ligand 4 | R&D Systems | Cat# 1506-D4 |
| EdU (5-ethynyl-20-deoxyuridine) | Thermo Fisher Scientific | Cat# A10044 |
| Tamoxifen | Sigma-Aldrich | Cat# T5648 |
| MEM Non-Essential Amino Acids Solution (100X) | Gibco | Cat# 11140050 |
| MEM Amino Acids Solution (50X) | Gibco | Cat# 11130051 |
| Critical commercial assays | ||
| Click-iT Cell Reaction Buffer Kit | Thermo Fisher Scientific | Cat# C10269 |
| RNeasy Plus Micro Kit | QIAGEN | Cat# 74034 |
| Tyramide SuperBoost Kit | Thermo Fischer Scientific | Cat# B40936 |
| In Situ Cell Death Detection Kit, POD | Sigma-Aldrich | Cat# 11684817910 |
| Click-iT HPG Alexa Fluor 488 Protein Synthesis Assay Kit | Thermo Fisher Scientific | Cat# C10428 |
| Click-iT HPG Alexa Fluor 594 Protein Synthesis Assay Kit | Thermo Fisher Scientific | Cat# C10429 |
| CD31 S-Pluribead anti-ms | PluriSelect | Cat# 29-03100-10 |
| Deposited data | ||
| Single cell RNA-seq raw data for muscle ECs | This paper | GEO: GSE158984 |
| RNA-seq raw data for RmECs versus WmECs, WT and Atf4ΔEC/ΔEC | This paper | GEO: GSE174785 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL6/N wild type | Charles River Laboratories | N/A |
| Mouse: atf4fl/fl | Ebert et al., 2012 | N/A |
| Mouse: pdgfb-CreERT2 | Claxton et al., 2008 | N/A |
| Experimental models: Cell lines | ||
| Primary muscle endothelial cells (mECs) | This paper | N/A |
| Primary human umbilical vein endothelial cells (HUVEC) | PromoCell | Cat# C-12203 |
| 293T | DSMZ | Cat# ACC 635 |
| Oligonucleotides | ||
| pMD2.G plasmid | AddGene | Cat# 12259 |
| psPAX2 plasmid | AddGene | Cat# 12260 |
| GIPZ Lentiviral shRNAs anti-human atf3 | Horizon Discovery | Cat# V3LHS352238 |
| GIPZ Lentiviral shRNAs anti-human atf3 | Horizon Discovery | Cat# V3LHS352240 |
| GIPZ Lentiviral shRNAs anti-human atf3 | Horizon Discovery | Cat# V3LHS405369 |
| GIPZ Lentiviral shRNAs anti-human atf4 | Horizon Discovery | Cat# V2LHS272113 |
| GIPZ Lentiviral shRNAs anti-human atf4 | Horizon Discovery | Cat# V3LHS302002 |
| GIPZ Lentiviral shRNAs anti-human atf4 | Horizon Discovery | Cat# V3LHS302003 |
| pLenti-C-mGFP-P2A-Puro Lentiviral Gene Expression Vector | Origene | CAT# PS100093 |
| Lenti ORF clone of human cMyc | Origene | Cat# RC201611L4 |
| ATF4 doxycycline-inducible overexpression vector | Torrence et al., 2021 | N/A |
| Software and algorithms | ||
| FlowJo Software (version 10.4.2) | Three Star | https://www.flowjo.com/ |
| ImageJ (for image analysis) | NIH | https://imagej.nih.gov/ij/ |
| Prism 8 (version 8.0.0) | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Adobe Illustrator CS6 (version 16.0.4) | Adobe | https://www.adobe.com/ |
| R 4.0.3 | cran.r-project | https://cran.r-project.org/bin/windows/base/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Katrien De Bock (katrien-debock@ethz.ch).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Mice
Wild type C57BL/6N mice were purchased from Charles River (Freiburg im Breisgau, Germany). Atf4LoxP/LoxP mice were generated using homologous recombination in embryonic stem (ES) cells as described in Ebert et al. (2012). To label proliferating cells, an intraperitoneal injection of 5-ethynyl-2’-deoxyuridine (EdU) (E10187, Thermo Fischer Scientific) solution (5 mg/ml in saline) was performed 7 hours before sacrificing the mice. To obtain inducible EC-specific Atf4 knockout (Atf4ΔEC/ΔEC) mice, Atf4LoxP/LoxP mice were intercrossed with Pdgfb-CreERT2 mice, an EC-selective inducible Cre-driver line (Claxton et al., 2008). Rosa26mTmG mice (Muzumdar et al., 2007) intercrossed with Pdgfb-CreERT2 mice were used for in vivo labeling of ECs. Recombination was induced in 8-10 weeks old mice by daily intraperitoneal administration of 1mg tamoxifen (T5648, Sigma-Aldrich) dissolved in 1:10 ethanol: corn oil solution for 5 consecutive days. A wash out period of at least 7 days was allowed before starting the experiments. Tamoxifen-treated Cre-negative littermates were used as control for all experiments. For low dose tamoxifen experiments, a single dose containing a 1/100 diluted tamoxifen solution (final amount: 0.01mg/ mouse in a single injection) was injected five days before starting the exercise intervention. All mouse lines were maintained on a C57BL/6N background.
Mice were randomly allocated to different treatment groups, and the investigator was blinded to the group allocation during the experiment as well as during the analysis. All mice were housed at standard housing conditions (22 °C, 12 h light/dark cycle), with ad libitum access to chow diet (18 % proteins, 4.5 % fibers, 4.5 % fat, 6.3 % ashes, Provimi Kliba SA) and water. Health status of all mouse lines was regularly monitored according to FELASA guidelines. All animal experiments were approved by the local animal ethics committee (Kantonales Veterinärsamt Zürich, licenses ZH211/19, and ZH014/16), and performed according to local guidelines (TschV, Zurich) and the Swiss animal protection law (TschG). All mice used in this study were male, 8 to 12 weeks old.
Isolation of primary muscle endothelial cells (ECs)
mEC isolation for scRNA-seq or mRNA analysis
Mice were euthanized and gastrocnemius muscles from both hindlimbs were immediately dissected and placed in an Petri dish on ice. Muscles were minced with a scalpel until a homogeneous paste-like mash was formed. Thereafter, the mashed muscle was enzymatically digested in digestion buffer containing 2 mg/ml Dispase II (D4693, Sigma-Aldrich, Steinheim, Germany), 2 mg/ml Collagenase IV (17104019, Thermo Fisher Scientific, Zurich, Switzerland), 2 mM CaCl2 and 2% BSA in PBS at 37°C for 10 min, with gentle shaking every 3 min. The reaction was stopped by immediately adding an equal volume of ice cold HBSS containing 20% FBS and the suspension was passed through a 70-μm cell strainer (#431751, Corning, New York, USA) then 40-μm cell strainer (#431750, Corning, New York, USA) to remove tissue debris. Cell suspension was centrifuged at 500 g for 5 min at 4°C, then the pellet was washed with ice cold HBSS (+20% FBS) followed by a centrifugation at 400 g for 5 min in 4°C. Next, the cell pellet was re-suspended in antibody medium (EGM2 CC-3162, Lonza, Basel, Switzerland) with anti-mouse CD31 PE antibody (1:400) (553373, BD Biosciences, Basel, Switzerland) and anti-mouse CD45 PerCP antibody (1:400) (557235 BD Biosciences, Basel, Switzerland) and placed on ice for 20 min in the dark. Before sorting, the cell suspension was washed in FACS buffer (1xPBS+1%BSA) and centrifuged at 400 g for 5 min, 4°C, then the washed cell pellet was re-suspended in FACS buffer containing cell viability dye, SYTOX blue (1:1000) (S34857, Thermo Fischer Scientific, Zurich, Switzerland). Viable endothelial cells (CD31+, CD45-, SYTOX blue-) were sorted by a FACS Aria III (BD Bioscience) sorter. For single cell RNA-seq, ECs were directly sorted (130 μm nozzle) into ice cold collecting medium (EGM2). The collected ECs were placed on ice (no more than 30 min) until loading into10x genomics chromium. For RNA extraction, 200,000 ECs were directly sorted (70 μm nozzle) into 700 μl RNA lysis buffer, and RNA extraction was performed by RNeasy Plus Micro Kit (74034 QIAGEN).
Capillary mEC isolation for culturing
For primary capillary mEC isolation, skeletal muscles from hind-limb were dissected and superficial big vessels were carefully removed. In muscle gastrocnemius and quadriceps femoris, oxidative and glycolytic muscle areas were mechanically separated using a scalpel. Muscles were minced and enzymatically digested as mentioned above at 37°C for 30 min, with gentle shaking every 10 min. The reaction was stopped by adding an equal volume of ice cold HBSS containing 20% FBS and the suspension was passed through a series of 100 and 70 μm cell strainers (#352360, 352350, Corning, New York, USA) to remove tissue debris. After a series of centrifugation and washing steps, the heterogeneous cell population was resuspended in EC culture medium (M+E: 50% full M199 medium+ 50% Endopan 3, details see Cell culture section below) and seeded in collagen type I (125-50, Sigma)-coated plates. Due to the higher expression of P-glycoprotein in ECs compared to other skeletal muscle cells, mECs were selected by adding 4 μg/ml puromycin (P8833, Sigma-Aldrich, St. Louis, USA) to the medium overnight. After 7 days in culture, the purity of mECs was determined by CD31 fluorescence staining and only cultures containing at least 85% of the cells positive for CD31 were used for further experiments.
mEC isolation for metabolomics
Muscles were dissected, minced and enzymatically digested for 10 min in digestion buffer at 37 °C to prepare a single cell suspension (see above). Thereafter, mECs were purified by CD31 pluribeads (29-03100-10, PluriSelect, Leipzig, Germany) according to the manufacturer’s instructions. The purified mEC pellets were immediately lysed in ice cold metabolite extraction buffer (80% methanol, containing 2 μM d27 myristic acid).
Comparison of different mEC dissociation procedures
To avoid the dissociation induced activation of immediate early genes (IEGs) in mECs, we optimized our mEC isolation protocol by comparing different dissociation procedures. Briefly, skeletal muscle were dissected and finely minced on ice cold petri dishes as described above. Minced tissues were enzymatically digested for 40 or 10 min in digestion buffer (mentioned above) at 37 °C. As a control, we compared the dissociation procedure with or without the addition of actinomycin D (ActD) (A1410, Sigma-Aldrich), a transcriptional inhibitor which prevents the activation of IEGs during cell isolation (Wu et al., 2017). Vehicle or ActD (45 μM) was added in the whole dissociation procedure. 40 min digestion protocol without ActD robustly activated atf3 compared with ActD treatment, while 10 min digestion with or without ActD showed no difference in the expression of such IEGs which indicated short digestion time (10 min) did not affect the expression of Atf4 and Atf3 in muscle ECs (Figure S2A). Of note, despite the positive effect concerning transcriptional inhibition during cell dissociation, digestions with ActD had a significant increase of dead cells and a reduction of total mECs yield (data not shown). Therefore, we decided to use the 10 min digestion protocol without ActD for single cell RNA-seq and RNA extraction.
For reasons of consistency, we each time compared RmECs to WmECs from the same mouse.
Muscle explant ex vivo culture
Mouse skeletal muscles were dissected as described above, for muscle gastrocnemius and quadriceps femoris, oxidative and glycolytic muscle areas were mechanically separated using a scalpel. Then muscle tissue was cut into small cubes (1.5 x 1.5 x 1.5 mm), and embedded in collagen gel (1ml gel = 375μl type I collagen (4mg/ml)+ 475μl methyl cellulose stock solution containing 40% FBS+ 150μl NaHCO3 (15.6mg/ml)+10μl NaOH (1M)). EC growth medium (M+E) were pipetted on top of the gel to induce sprouting. 5 days later, muscle explants were fixed with 4% PFA at room temperature and proceeded with staining: Collagen gel with embedded explants were gently taken out with a forceps and placed on a petri dish. Excessive collagen was removed using a scalpel and cut into smaller pieces and transferred to a well of a 24-well plate with PBS containing 3% BSA, 0.2% Triton X-100 for 2h at RT. Gel pieces were then incubated at 4°C overnight with staining cocktail: Isolectin GS-IB4 Alexa Fluor 568 (IB4, 1:250, I21412, Thermo Fisher Scientific) + Phalloidin-iFluor 488 Reagent (1:1000, ab176753, abcam) diluted in PBS containing 3% BSA, 0.05% Triton X-100. After staining, gel pieces were incubated with hoechst (62249, Thermo Fisher Scientific, 1:2000) for 1 hour and washed with PBS + 0.05 %, Triton X-100 12 hours at 4°C. Vascular outgrowth was photographed using an Olympus confocal microscope (FV1200).
Method details
Exercise experiments
For exercise training experiments, mice were individually housed in open cages equipped with an upright running wheel device (TSE Systems, Bad Homburg vor der Höhe, Germany) for the duration of the intervention. Non-running control (sedentary) mice were also single caged in cages of equal dimensions but without running wheels. Continuous recording of wheel movements was performed out of which total distance (km), speed (m/s), and number of running bouts was calculated.
For exercise capacity testing, mice were acclimatized to a treadmill system for 4 sessions (5-lane treadmill, Harvard Apparatus, Panlab) before exercise capacity testing. Each animal rested in the treadmill lane for 5 min, followed by 5min at 7m/min, 10 min at 10m/min and 15min at 12m/min at 5° angle. Following acclimatization, mice underwent an aerobic exercise capacity test to exhaustion on a treadmill with 5° angle. Mice were motivated to run with a shock grid set at 0.4 mA. Starting speed was 5 m/min and was increased by 1 m/min until exhaustion. Speed was capped at a maximum of 25 m/min until exhaustion. Treadmill session was terminated if the mice failed to return to the treadmill after 3 consecutive attempts within the last minute of running. Aerobic capacity is expressed as total time during the test. Access to running wheels was restricted for 24 h before testing.
Single cell RNA-seq and data analysis
FACS sorted muscle-derived ECs were resuspended in 250 μl EGM2 medium (CC-3162, Lonza, Basel, Switzerland), targeting the required 1,000 cells/μl concentration, accounting for a 10–20% loss. We pipetted 9.7 μl cell suspension (concentration of 913 cells/μl, ~8,800 cells), targeting the recovery of ~5,000 cells. Single-cell RNA-seq libraries were obtained following the 10x Genomics recommended protocol, using the reagents included in the Chromium Single Cell 3′v2 Reagent Kit. Libraries were sequenced on the NextSeq 500 (Illumina) instrument, aiming at 50k mean reads per cell.
The 10x Genomics scRNA-seq data was processed using cellranger-2.0.2 with the mouse GRCm38.p5 genome Ensembl release 91. Based on filtered gene-cell count matrix by CellRanger’s default cell calling algorithm, we performed the Seurat workflow. In order to exclude low quality cells and doublets, cells with less than 500 or more than 3000 detected genes were filtered out. Fourteen principal components were chosen in the dimensionality reduction step. A resolution of 0.8 in the unsupervised clustering resulted in the final populations. Lymphatic endothelial cells (Lyve1, Maf, Prox1, Slc45a3, Sema3d, Sema3a, Pdpn) and smooth muscle cells/pericytes (Rgs5, Acta2, Myl9, Myh11) were manually removed. Identification of CapEC 1 top marker genes relative to CapEC 2 were performed using scran and scater packages (Lun et al., 2016; McCarthy et al., 2017).
To compare mECs from sedentary versus exercise mice, we harvested mECs from three mice in each condition. Subsequently, cells with less than 300 or more than 2500 genes, while in exercised mice, we removed cells that have less than 400 or more than 3000 expressed genes. Then two datasets were integrated with by combining the count matrix and processed with standard Seurat workflow. 14 principal components were chosen and a resolution of 0.6 was used in the cell clustering. RNA velocity was estimated by velocyto python module (La Manno et al., 2018) with exonic reads and intronic reads only. Estimated RNA velocity was then overlaid on the t-SNE embedding of the integrated sample.
Bulk RNA-seq and data analysis
mECs were directly sorted (70 μm nozzle) into 700 μl RNA lysis buffer, and RNA extraction was performed using RNeasy Plus Micro Kit (74034 QIAGEN). RNA quality test was performed by Agilent High Sensitivity RNA ScreenTape System (G2964AA). Samples with RNA Integrity Number (RIN) ≥ 8.0 were further analyzed. Bulk RNA-seq libraries were obtained following the Smartseq II recommended protocol. Libraries were sequenced on the Novaseq 6000 (Ilummina) instrument and sequenced data were processed using Kallisto to generate a count file matrix for each individual sample. Samples were pooled together on a single dataset for downstream analysis and genes with one count or less across all samples were filtered out. Variance stabilizing transformation (VST) for negative binomial data with a dispersion-mean trend was used for exploratory analysis such as PCA and identification of highly variable genes. Following DESeq2 analysis workflow (Love et al., 2014), differential expression analysis were performed on the raw counts after estimation of size factors (controlling for differences in the sequencing depth of the samples), the estimation of dispersion values for each gene and fitting a generalized linear model.
Transcription factor motif enrichment analysis were performed using gProfiler online tool (https://biit.cs.ut.ee/gprofiler/gost) were an adjusted p-value ordered list of differentially expressed genes (log fold change > 1 and adjusted p-value < 0.05) were given as input.
Custom enrichment analysis (ATF4 dependent biological process) were performed using a kstest from hypeR package version 1.6.0 (Federico and Monti, 2020) on the adjusted p-value ordered list of differentially expressed genes. ATF4 dependent categories were derived from Han et al. (2013).
Cell culture
Isolated primary mouse skeletal muscle endothelial cells (mECs) and commercially purchased human umbilical vein endothelial cells (HUVECs) from pooled donors (C-12203, PromoCell, Heidelberg, Germany) were routinely cultured in a 1:1 ratio of M199 ((11150059, Thermo Fisher Scientific) supplemented with 20% fetal bovine serum (FBS) (10270-106, Thermo Fisher Scientific), 30 mg/L endothelial cell growth factor supplements (EGCS) (E2759, Sigma-Aldrich), 10 U/ml heparin (H3149 Sigma-Aldrich) and 1% Penicillin-Streptomycin (10,000 U/ml) (15140122, Thermo Fisher Scientific) and Endopan 3 (P04-0010K, PAN BIOTECH, Aidenbach, Germany) (denoted as M+E). 1/3 amino acid (AA) medium was made by mixing full AA DMEM (11966025, Thermo Fischer Scientific) and AA free medium (D9800-27, Biomol, Hamburg, Germany) in a 1:2 ratio. This medium was supplemented with equal amount of endothelial growth factor SingleQuots (CC-4133, Lonza, Basel, Switzerland) and other missing components (i.e., 5.5 mM glucose, 2% FBS, 1% Pen/Strep, 1mM sodium pyruvate) as used for routinely culture. Low mitogenic/AA medium was made by mixing full AA DMEM (11966025, Thermo Fischer Scientific) (supplemented with endothelial growth factor SingleQuots (CC-4133, Lonza, Basel, Switzerland)) and AA free medium (D9800-27, Biomol, Hamburg, Germany) in a 1:2 ratio, resulting in a 1/3 AA and 1/3 growth factor concentration. For 13C tracing experiments with [U-13C]-glucose, glucose-free full AA and 1/3 AA DMEM (same as above) medium (with equal levels of endothelial cell growth factors) was reconstituted with 5.5 mM [U-13C]-glucose (CLM-1396, Cambridge Isotope Laboratories), and cells were incubated for 48 hours to reach steady state. Primary mECs were only used until passage (p)1, HUVECs were only used between p1 and p5. Cells were routinely maintained in 5% CO2 and 95% air at 37 °C, and regularly tested for mycoplasma.
Knock down and overexpression plasmid constructions and lentiviral particle production
Multiple GIPZ Lentiviral shRNAs target human Atf3 (V3LHS352238; V3LHS352240; V3LHS405369) and Atf4 (V2LHS272113; V3LHS302002; V3LHS302003) were purchased from Dharmacon (Horizon Discovery; Waterbeach, United Kingdom). A nonsense scrambled shRNA sequence was used as control. Lentiviral particles were generated by transfection of HEK 293 cells (Cat.# ACC635; DSMZ, Braunschweig, Germany) with Pmd2 (AddGene, Plasmid #12259), lentiviral envelope plasmid psPAX2 (AddGene, Plasmid #12260) and the plasmid containing scramble or against Atf3/4 gene sequences. The inducible ATF4 overexpression vector was a gift from Brendan Manning (Department of Molecular Metabolism, Harvard T.H. Chan school of Public Health, Boston, MA, USA) (Torrence et al., 2021). ATF4 expression was induced with 1ug/ml doxycycline (Sigma-Aldrich, D3447) for 24 hours. The overexpression human plasmid for cMyc (Cat. #RC201611L4) was purchased from Origene. For cMYC overexpression, WT or Atf3/4KD HUVECs were transduced with lentiviral particles expressing the human c-Myc ORF for 16 hours. Thereafter, the medium was changed, and cells were used for experiments. Lentiviral particles were generated by transfection of HEK 293 cells with the respective plasmid and pLenti-C-mGFP-P2A-Puro Lentiviral Gene Expression Vector (Cat. #PS100093, Origene). Lipofectamine 2000 (Cat.# 11668030; Thermo Fisher Scientific) was used for transfection. Viral particles were collected at least 48 hours after incubation. Cells were transduced 24 hours in the presence of 8 μg/ml polybrene and re-fed with fresh medium the next day. Atf3/4KD HUVECs were used in functional assays at least 5 days post-transduction.
Immunohistochemistry and histology
Muscle samples were harvested and embedded in Tissue-Tek and frozen in liquid N2-cooled isopentane. Frozen muscle cross sections (7-10 μm) or longitudinal sections (20 μm) were made using a cryostat (Leica CM 1950) and collected on Superfrost Ultra Plus slides (Thermo Fischer Scientific). Cross sections were permeabilized in PBS with 0.5% Triton X-100 and blocked for 1 h at room temperature in PBS with 0.05% Tween-20, 1% BSA (BS). Primary antibody incubations were performed at 4 °C overnight. Slides were subsequently washed in PBS and incubated for 1 hour in blocking buffer with the appropriate secondary antibodies. Nuclei were stained with Hoechst. The following primary and secondary antibodies were used: anti-CD31 (AF3628, R&D Systems, 1:250) or anti-CD31 (abcam, ab7388, 1:250), anti-CD62P (Selp) (abcam, ab59738 1:250), anti-Von Willebrand Factor (VWF) (abcam, ab11713, 1:500), anti-α smooth muscle actin Cy3 conjugated (αSMA) (1:500, C6198, Sigma-Aldrich), anti-Ephrin-B2 (AF496, R&D Systems,1:200), anti- Carbonic Anhydrase IV (Car4) (PA5-47312 Thermo Fischer Scientific, 1:250), anti-Aquaporin 1 (ab15080, abcam 1:250), Alexa Fluor 594 donkey anti-goat IgG (H+L) (1:250, A-11058, Thermo Fisher Scientific), Alexa Fluor 568 donkey anti-rat IgG (H+L) (1:250, ab175475, abcam), Alexa Fluor 350 donkey anti-sheep IgG (H+L) (1:250, A-21097, Thermo Fisher Scientific), Alexa Fluor 488 donkey anti-rabbit IgG (H+L) (1:250 dilution, A-32790, Thermo Fisher Scientific), Alexa Fluor 488 donkey anti-goat IgG (H+L) (1:250, A-32814, Thermo Fisher Scientific), Alexa Fluor 350 donkey anti-rabbit IgG (H+L) (1:250 dilution, A- 10039, Thermo Fisher Scientific).
Fiber type staining combined with isolectin B4 staining was performed in two steps: First, fiber type staining was performed as described (Masschelein et al., 2020). In short, sections were dried and washed for 5 min in PBS supplemented with 0.05% Triton-X-100 (PBST) and subsequently blocked for 60 min in PBST + 10% goat serum (16210064, Thermo Fisher Scientific). Afterwards a primary antibody cocktail diluted in PBST + 10% goat serum was applied for 120 min against MyHC-I (1:50 dilution, BA-F8 from hybridoma, Iowa City, IA, USA), MyHC-IIa (1:200 dilution, SC-71 from hybridoma) and MyHC-IIb (1:100 dilution, BF-F3 from hybridoma). After washing 3 times for 5 min, a secondary antibody cocktail, diluted in PBST + 10% goat serum, was applied for 60 min. Secondary antibodies were: Alexa Fluor 488 goat anti-mouse IgG2B (1:250 dilution, Thermo Fisher Scientific), Alexa Fluor 350 goat anti-mouse IgG1 (1:250 dilution, A-21120, Thermo Fisher Scientific), Alexa Fluor 568 goat anti-mouse IgM (1:250 dilution, A-21043, Thermo Fisher Scientific). After a 3 x 5 min wash, muscle sections were briefly fixed in 2% PFA for 5 min, and washed with PBS. For additional IB4 vascular staining, muscle sections were incubated overnight at 4 °C with with Isolectin GS-IB4 Alexa Fluor 647 (IB4, 1:500, I32450, Thermo Fisher Scientific) diluted in blocking buffer 0.1% Triton X-100. For ATF3 stainings on muscle cryosections, 7 μm frozen cross / 20 μm longitudinal muscle sections were fixed with 4%PFA for 7 minutes after which an antigen retrieval step was performed with Target Retrieval Solution, (Citrate pH 6.0) (S236984-2, Agilent Dako). After 3x 3 min wash with PBS, sections were treated with 3% H2O2 in PBS for 7min and wash again with PBS 3x 3 min. Thereafter, samples were permeabilized for 30 min in 0.5% TritonX-100 in PBS at RT and blocked with 1%BSA in PBS for 1 hour. Then sections were incubated with ATF3 antibody (1:100, HPA001562, Sigma-Aldrich) overnight at 4 °C and goat anti-rabbit biotinylated secondary antibody(ab6720) was added (90 min in RT: 1/1000 dilution in 1% BSA/PBS). Thereafter sections were incubated in streptavidin-HRP (1:1000 in PBS, ab7403, abcam) for 1 hour at RT and treated with Alexa flour 674/488 (1:100) in amplification diluents for 17 min (Tyramide SuperBoost Kit, streptavidin, B40936, Thermo Fisher Scientific). After 3x 3min PBS wash, the following antibodies were used for further staining: anti-CD31 (abcam, ab7388, 1:250), antibodies for fiber type staining (same as above). Cell nuclei were stained with Hoechst (62249, Thermo Fisher Scientific, 1:2000).
For ATF3 staining in cultured primary muscle ECs, cells were fixed with 4% PFA for 5 min, then permeabilized with 0.5% Triton X-100 for 5 min, and subsequently incubated for 1 h in blocking buffer (PBS with 1%BSA) at RT. Thereafter cells were incubated with following antibodies at 4 °C overnight: Rabbit anti-ATF3 antibody (1:250, ab207434, Abcam); Goat anti-Mouse/Rat CD31/PECAM-1 antibody (1:250, 3628, R&D Systems). Cell nuclei were stained with Hoechst (62249, Thermo Fisher Scientific, 1:2000). In order to assess the number of ATF3high cells, we used a user-defined threshold based on calculating the mean intensity of all cells.
For EdU detection combined with ERG and/or CD31/IB4, EdU was first visualized using the EdU Click-iT Cell Reaction Buffer Kit (C10269, Thermo Fisher Scientific) according to manufacturer's instructions, and subsequently incubated for 1 h in blocking buffer (PBS with 1% BSA) at room temperature (RT). Thereafter, sections were incubated overnight at 4 °C with Isolectin GS-IB4 Alexa Fluor 568 (1:500, I21412, Thermo Fischer Scientific), goat anti-Mouse/Rat CD31/PECAM-1 antibody (1:250, 3628, R&D Systems) and ERG antibody (#97249, Cell Signaling, 1:200) diluted in blocking buffer with 0.1% Triton X-100. For immunohistochemical detection of apoptosis of ECs on muscle cryosections, In Situ Cell Death Detection Kit, POD (11 684 817 910, Roche) was used according to the manufacturer’s instructions, thereafter, sections were further stained with isolectin B4 (647), ERG and Hoechst as mentioned above.
Images were taken with a Zeiss Axio observer Z.1 or an Olympus confocal microscope (FV1200). All images were captured at the same exposure time for one experiment. Composite images were stitched together using the tiles module in the ZEN 2011 imaging software (Zeiss). Fiber type composition, vascular density (% IB4/CD31+ area) and ERG numbers were quantified within the oxidative and glycolytic areas of the muscle using ImageJ software.
RNA extraction and quantitative RT-PCR
RNA of directly FACS sorted mEC was extracted using an RNeasy Plus Micro Kit according to the manufacturer’s instructions (QIAGEN, 74034). RNA of cultured mECs and HUVECs was extracted using PureLink RNA Mini Kit (12183020, Thermo Fischer Scientific). RNA purity and concentration were assed via a spectrophotometer (Spark 10M, Tecan). RNA was reverse-transcribed to cDNA by High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, 43-688-13). A SYBR Green-based master mix (Thermo Fisher Scientific, A25778) was used for real-time qPCR analysis with primers listed in Table S1. To compensate for variations in RNA input and efficiency of reverse-transcription, 18S was used as a housekeeping gene. The delta-delta CT method was used to normalize the data.
In vitro analysis of EC function
Proliferation
Cultured primary mouse muscle ECs (no longer than 8 days, p1) or HUVECs were incubated in growth medium containing 10 μM 5-ethynyl-2’-deoxyuridine (EdU) for 15 hours. For nonessential and essential amino acids (NEAA and EAA) supplementation experiments in HUVECs, 1xMEM NEAA (11140-050, Gibco) or 1xMEM EAA (11130-036, Gibco) was added for 48 hours. For NEAA and EAA supplementation in W/RmECs, 1xMEM NEAA (11140-050, Gibco) was added from the day of isolation (day 0) till day 5, and medium was changed every day. As a measure of proliferation, incorporation EdU was assessed using the Click-iT Cell Reaction Buffer Kit (C10269, Thermo Fisher Scientific), according to the manufacturer’s instructions. Briefly, after EdU incorporation, cells were fixed with 4% paraformaldehyde for 10 min and permeabilized for 20 min in 0.5% Triton X-100 with 3% BSA in PBS, followed by reaction with the Click-iT reaction cocktail for 45 min in dark at room temperature. Thereafter, cells were washed briefly and counterstained with Hoechst (#62249, Thermo Fisher Scientific,1:2000) and antibodies against CD31 (AF3628, R&D Systems, 1:250) or ERG (#97249, Cell Signaling, 1:250). Cells were imaged using a Zeiss Axio Observer.Z1 fluorescence microscope (Carl Zeiss, Oberkochen, Germany). EdU+ cells and ERG+ ECs were counted in at least 5 random fields and the percentage of EdU+ cells in ECs was calculated.
Scratch wound assay
A scratch wound was applied on confluent EC monolayers (pre-treated with 1 μg/ml mitomycin C for 24 hrs where indicated) using a 200 μl tip. After scratch wounding (T0) and photography using a Leica DMI6000 B inverted microscope (Leica Microsystems, Mannheim, Germany), the cultures were further incubated in growth medium and fixed with 4% PFA 24 hours after first scratch (T24). Cells were photographed again (T24) and gap areas at both time points were measured using the Fiji software package (https://fiji.sc) in order to calculate the percentage of wound closure using the following expression: (1 – (T24gap area/T0gap area)) x 100.
Spheroid capillary sprouting assay
Spheroids were prepared as previously described (De Bock et al., 2013) with minor modifications. Briefly, spheroids containing 1000 HUVECs, or mECs per 25 μl droplet were plated overnight as hanging drops in a 20% methylcellulose (9004-67-5, Sigma-Aldrich) in EGM2 mixture. The next day, spheroids were collected in 10% FBS in PBS, concentrated using several centrifugation steps and embedded in a Fibrinogen gel (5 mg/ml fibrinogen (F8630, Sigma-Aldrich) dissolved in EGM2 plus 1U/ml thrombin (T4648, Sigma-Aldrich). To assess tip cell competition, cells were mixed at the indicated ratio. Growth medium (with or without mitomycin C) were pipetted on top of the gel to induce sprouting. 24 hours later, spheroids were fixed with 4% PFA at room temperature and photographed using a Leica DM IL LED microscope (Leica Microsystems GmbH, Wetzlar, Germany). Dll4 stimulation: culture plates were coated with 1 μg/ml recombinant human Delta-like ligand 4 (rhDll4, cat.1506-D4 R&D Systems) with 0.1% gelatin. The control plates were coated with 0.1% gelatin supplemented with 0.02% BSA. Prior to EC seeding, excessive coating solution was removed by aspiration and ECs were seeded at a density of 30.000 cells/cm2. Cells were harvested 24 hours after seeding. Contact inhibition: ECs were seeded in 50% EGM2 / 50% full M199 medium at a density of 15,000 cells/cm2 and were further cultured for 3 days until contact inhibition was reached. To generate the corresponding proliferative control, contact inhibited cells were trypsinized and cultured for 24 hr to re-initiate proliferation.
Immunoblot analysis
Endothelial cells
Cells were collected and lysed with [50 mM Tris–HCl pH 7.0, 270 mM sucrose, 5 mM EGTA, 1 mM EDTA, 1 mM sodium orthovanadate, 50 mM glycerophosphate, 5 mM sodium pyrophosphate, 50 mM sodium fluoride, 1 mM DTT, 0.1% Triton-X 100 and a complete protease inhibitor tablet (C755C25, Roche Applied Science)]. Lysates were centrifuged at 10,000 g for 10 min at 4°C. Supernatant was collected, and protein concentration was measured using the DC protein assay kit (5000116, Bio-rad). 5-10 μg of total protein was loaded in a 10-well pre-casted gradient gel (456-8086, Bio-Rad). After electrophoresis, a picture of the gel was taken under UV-light to determine protein loading using stain-free technology. Proteins were transferred onto a PVDF membrane (Bio-rad, 170-4156) with a semi-dry system and subsequently blocked for 1 h at room temperature with 5% milk in 0.1% TBS-Tween. Membranes were incubated overnight at 4 °C with primary antibodies listed in key resources table. The appropriate HRP-linked secondary antibodies (see key resources table) were used for chemiluminescent detection of proteins. Membranes were scanned with a Chemidoc imaging system (Bio-rad) and quantified using Image Lab software (Bio-rad).
Muscle samples
samples were homogenized in ice cold lysis buffer (1:10, w/v) (modified RIPA buffer: 50 mM Tris-HCl (pH 7.5), 1 mM EDTA, 10% (v/v) glycerol, 1% (v/v) Triton-X, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM DTT, and protease/phosphatase inhibitor cocktail) using an OMNI-THq Tissue homogenizer (OMNI International, Kennesaw, GA, USA) for 20 s until a consistent homogenate was formed. Samples were centrifuged at 4 °C at 10,000 g for 10 min and the supernatant with proteins collected. Protein concentration was determined using the DC assay protein method to equalize the amount of protein. Samples were prepared 3:4 with 4× laemmli buffer containing 10% 2-mercaptoethanol. An amount of 20–40 μg of total protein was loaded in a 15-well pre-casted gradient gel (Bio-rad, #456–8086). After electrophoresis, a picture of the gel was taken under UV-light to determine protein loading using stain-free technology. Proteins were transferred via semi-dry transfer onto a polyvinylidene fluoride membrane (Bio-rad, #170-4156) and subsequently blocked for 1 h at room temperature with 5% milk in TBS- Tween. Membranes were incubated overnight at 4 °C with primary antibodies: Hexokinase-II (Cell Signaling #2867S) and Oxphos (unboiled samples, Abcam #110413). The appropriate secondary antibodies (1:5000) for anti-rabbit and anti-mouse IgG HRP-linked antibodies were used for chemiluminescent detection of proteins. Membranes were scanned with a chemidoc imaging system (Bio-rad) and quantified using Image lab software (Bio-rad).
Amino acid uptake
HUVECs were starved in amino acid free DMEM (supplemented with endothelial cell growth factors) for 1 hour after which L-[3,4-3H(N)]-Glutamine (NET551250UC, PerkinElmer) or L-[14C(U)]-Leucine (NEC279E050UC, PerkinElmer) was added in the medium to a final concentration of 400 nM (Glutamine) or 300 nm (Leucine). After 15 min of incubation at 37 °C, cells were carefully washed three time with ice cold PBS and were lysed in 0.1 N NaOH. The resulting suspension was measured by liquid scintillation counting to determine the intensity of radioactivity. The cells with labeling medium covered and removed immediately were used as background.
Protein synthesis
Protein synthesis rate was determined by Click-iT HPG Alexa Fluor 488/594 Protein Synthesis Assay Kits (C10428/ C10429, Thermo Fisher Scientific) according to the manufacturer’s instructions (2 hours HPG medium incubation), or by assessing radioactive amino acid incorporation into proteins. Briefly, cells were incubated with EC growth medium containing 2 μCi/ml EasyTag EXPRESS35S Protein Labeling Mix (NEG772002MC, PerkinElmer) for 6 hours. Thereafter, the medium was removed, and the cells were washed with ice cold PBS. After protein precipitation with 10% TCA for at least 1 hr, the suspension was transferred to an eppendorf tube and the protein pellet was collected by a centrifugation at 15,000 rpm for 15 min. Finally, the protein pellet was dissolved in 0.5 M NaOH containing 0.1% Triton X. The amount of [35S]-Protein Labeling Mix incorporated into protein was measured by scintillation counting and corrected for total protein content.
Mass spectrometry analysis
Metabolites were extracted from 300K cells in 300 μl of extraction buffer (80% methanol, containing 2 μM d27 myristic acid). Extraction samples were then centrifuged in 4°C for 15 min at 20,000 g and the supernatant was transferred to LC-MS vials. The pellet was used to measure protein levels for normalization purposes. Targeted measurements of amino acids and nucleotides were performed using a Dionex UltiMate 3000 LC System (Thermo Scientific) coupled to a Q Exactive Orbitrap mass spectrometer (Thermo Scientific) operated in negative ionisation mode. Separation of metabolites prior to MS measurement was performed using a Dionex UltiMate 3000 LC System (Thermo Scientific) coupled to a Q Exactive Orbitrap mass spectrometer (Thermo Scientific) operating in negative ion mode. Practically, 15 μl of the sample was injected on a C18 column (Aquility UPLC HSS T3 1.8μm 2.1x100mm) and the following gradient was performed by solvent A (H2O, 10mM Tributyl-Amine, 15mM acetic acid) and solvent B (100% Methanol). Chromatographic separation was achieved with a flow rate of 0.25 ml/min and the following gradient elution profile: 0min, 0%B; 2min, 0%B; 7min, 37%B; 14min, 41%B; 26min, 100%B; 30min, 100%B; 31min, 0%B; 40min, 0%B. The column was placed at 40°C throughout the analysis. The MS operated in full scan mode using a spray voltage of 3.2 kV, capillary temperature of 320°C, sheath gas at 40.0, auxiliary gas at 10.0. The AGC target was set at 3e6 using a resolution of 70.000, with a maximum IT of 128 ms. For 13C-glucose tracing experiments (incorporation into serine and glycine) we corrected for naturally occurring isotopes (Fernandez et al., 1996). For relative metabolite levels, the total ion count was normalized to the protein content.
Flow Cytometry
For endothelial cell percentage analysis in oxidative and glycolytic muscles, different muscle parts were dissected, separated and enzymatically digested as described above, cells were incubated in dark for 30 min with anti-mouse CD31 PE antibody (553373, BD Biosciences) and anti-mouse CD45 PerCP antibody (557235 BD Biosciences) (1:400 diluted in FACS buffer (1xPBS+1% FBS)). Cells were washed with FACS buffer before loading. For EdU proliferation experiments, cells from EdU-injected mice (7 hours labeling) were labeled with the click-iT plus EdU Alexa Fluor 647 Flow Cytometry Assay Kit (C10634, Thermo Fischer Scientific) according to the manufacturer’s instructions and thereafter stained with antibodies for cell surface markers CD31 (same as above) and APC/Fire 750 anti-mouse CD45 antibody (103154, BioLegend, 1:400). For analysis of mTomato+ EC and mGFP+ EC analysis upon low dose tamoxifen injection, muscles were dissected, separated and enzymatically digested as described above, cells were labeled with the click-iT plus EdU Alexa Fluor 647 Flow Cytometry Assay Kit (C10634, Thermo Fischer Scientific) according to the manufacturer’s instructions and thereafter stained with APC/Fire 750 anti-mouse CD31 Antibody (1:100, 102434, BioLegend) overnight at 4°C. Cells were analyzed using a FACS Aria III (BD Bioscience) sorter. Data were analyzed using FlowJo 10 software (Tree Star). A complete list of all antibodies and staining reagents used can be found in key resources table.
Quantification and statistical analysis
The images presented in the manuscript are representative of the data (quantification of image is approximately the group average) and the image/staining quality. All data represent mean ± SEM. GraphPad Prism software (version 8.0.0) was used for statistical analyses. Investigators were always blinded to group allocation. All datasets passed normal (Gaussian) distribution test by Shapiro-Wilk/ Kolmogorov-Smirnov method with significance level (alpha) 0.05. Unless otherwise indicated, when comparing two group means, Student’s t test was used in an unpaired two-tailed fashion. For more than two groups, one-way ANOVA with Tukey’s multiple comparisons test was used and for experimental set-ups with a second variable, two-way ANOVA with Sidak’s multiple comparisons test was used. The statistical method used for each experiment is indicated in each figure legend. Asterisks in figure legends denote statistical significance. No experiment-wide multiple test correction was applied. p > 0.05 is considered non-significant (n.s.). p < 0.05 is considered significant (∗).
Acknowledgments
This manuscript is dedicated to James R. Mitchell (1971–2020). We thank him for his critical feedback and helpful suggestions and will remember him forever. We thank Veronique Juvin from SciArtWork (https://sciartwork.com/) for help with the graphical abstract, and the Functional Genomics Center Zürich (FGCZ) as well as the ETH Flow Cytometry Core Facility (E-FCCF) for excellent technical support. This project was funded by a European Research Council (ERC) starting grant (716140), the Swiss National Science Foundation (SNF 310030B_182829), and grant #18C167 from the Novartis Foundation for Medical-Biological Research. A.A.-D. is supported by a PhD fellowship from the National Research and Technology Council of Mexico (CONACyT). K.D.B. is endowed by the Schulthess Foundation.
Author contributions
Z.F. designed and performed the experiments. G. Turiel, M.G., and G. Tan performed the bioinformatics analysis. R.A., T.K., G.F., T.G., A.A.-D., J.Z., E.M., and P.G. contributed to data analysis and performing experiments. C.M.A. provided the Atf4 floxed mice and commented on the manuscript. B.G. supervised the metabolite analysis. K.D.B. conceptualized the study, supervised the experiments, acquired funding, and wrote the paper.
Declaration of interests
These authors declare no competing interests.
Published: August 5, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cmet.2021.07.015.
Supplemental information
Data and code availability
The mouse single cell RNA-seq data reported in this study are publicly available at the Gene Expression Omnibus (GEO) repository under the accession number (GSE158984). The bulk RNA-seq data are publicly available at GEO under accession number (GSE174785). Please check https://shiny.debocklab.hest.ethz.ch/Fan-et-al/ forhttps://shiny.debocklab.hest.ethz.ch/Fan-et-al/ for data visualization. Comparisons between RmECs and WmECs (bulk RNA-seq) can be downloaded from the app. All other data are available from the corresponding author on request.
All original codes are available from the lead author upon request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Adams C.M. Role of the transcription factor ATF4 in the anabolic actions of insulin and the anti-anabolic actions of glucocorticoids. J. Biol. Chem. 2007;282:16744–16753. doi: 10.1074/jbc.M610510200. [DOI] [PubMed] [Google Scholar]
- Ahlborg G., Felig P., Hagenfeldt L., Hendler R., Wahren J. Substrate turnover during prolonged exercise in man. Splanchnic and leg metabolism of glucose, free fatty acids, and amino acids. J. Clin. Invest. 1974;53:1080–1090. doi: 10.1172/JCI107645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen P., Henriksson J. Capillary supply of the quadriceps femoris muscle of man: adaptive response to exercise. J. Physiol. 1977;270:677–690. doi: 10.1113/jphysiol.1977.sp011975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany Z., Foo S.-Y., Ma Y., Ruas J.L., Bommi-Reddy A., Girnun G., Cooper M., Laznik D., Chinsomboon J., Rangwala S.M. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. 2008;451:1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- Barry D.M., McMillan E.A., Kunar B., Lis R., Zhang T., Lu T., Daniel E., Yokoyama M., Gomez-Salinero J.M., Sureshbabu A. Molecular determinants of nephron vascular specialization in the kidney. Nat. Commun. 2019;10:5705. doi: 10.1038/s41467-019-12872-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sahra I., Hoxhaj G., Ricoult S.J.H., Asara J.M., Manning B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science. 2016;351:728–733. doi: 10.1126/science.aad0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloor C.M. Angiogenesis during exercise and training. Angiogenesis. 2005;8:263–271. doi: 10.1007/s10456-005-9013-x. [DOI] [PubMed] [Google Scholar]
- Chinsomboon J., Ruas J., Gupta R.K., Thom R., Shoag J., Rowe G.C., Sawada N., Raghuram S., Arany Z. The transcriptional coactivator PGC-1alpha mediates exercise-induced angiogenesis in skeletal muscle. Proc. Natl. Acad. Sci. USA. 2009;106:21401–21406. doi: 10.1073/pnas.0909131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claxton S., Kostourou V., Jadeja S., Chambon P., Hodivala-Dilke K., Fruttiger M. Efficient, inducible Cre-recombinase activation in vascular endothelium. Genesis. 2008;46:74–80. doi: 10.1002/dvg.20367. [DOI] [PubMed] [Google Scholar]
- Davies B.S., Beigneux A.P., Barnes R.H., 2nd, Tu Y., Gin P., Weinstein M.M., Nobumori C., Nyrén R., Goldberg I., Olivecrona G. GPIHBP1 is responsible for the entry of lipoprotein lipase into capillaries. Cell Metab. 2010;12:42–52. doi: 10.1016/j.cmet.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bock K., Georgiadou M., Schoors S., Kuchnio A., Wong B.W., Cantelmo A.R., Quaegebeur A., Ghesquière B., Cauwenberghs S., Eelen G. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154:651–663. doi: 10.1016/j.cell.2013.06.037. [DOI] [PubMed] [Google Scholar]
- De Micheli A.J., Spector J.A., Elemento O., Cosgrove B.D. A reference single-cell transcriptomic atlas of human skeletal muscle tissue reveals bifurcated muscle stem cell populations. Skelet. Muscle. 2020;10:19. doi: 10.1186/s13395-020-00236-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFronzo R.A., Jacot E., Jequier E., Maeder E., Wahren J., Felber J.P. The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes. 1981;30:1000–1007. doi: 10.2337/diab.30.12.1000. [DOI] [PubMed] [Google Scholar]
- Diehl F.F., Lewis C.A., Fiske B.P., Vander Heiden M.G. Cellular redox state constrains serine synthesis and nucleotide production to impact cell proliferation. Nat. Metab. 2019;1:861–867. doi: 10.1038/s42255-019-0108-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert S.M., Dyle M.C., Kunkel S.D., Bullard S.A., Bongers K.S., Fox D.K., Dierdorff J.M., Foster E.D., Adams C.M. Stress-induced skeletal muscle Gadd45a expression reprograms myonuclei and causes muscle atrophy. J. Biol. Chem. 2012;287:27290–27301. doi: 10.1074/jbc.M112.374777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan B., Zierath J.R. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013;17:162–184. doi: 10.1016/j.cmet.2012.12.012. [DOI] [PubMed] [Google Scholar]
- Ekman N., Lymboussaki A., Västrik I., Sarvas K., Kaipainen A., Alitalo K. Bmx tyrosine kinase is specifically expressed in the endocardium and the endothelium of large arteries. Circulation. 1997;96:1729–1732. doi: 10.1161/01.cir.96.6.1729. [DOI] [PubMed] [Google Scholar]
- Federico A., Monti S. hypeR: an R package for geneset enrichment workflows. Bioinformatics. 2020;36:1307–1308. doi: 10.1093/bioinformatics/btz700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felig P., Wahren J. Amino acid metabolism in exercising man. J. Clin. Invest. 1971;50:2703–2714. doi: 10.1172/JCI106771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez C.A., Des Rosiers C., Previs S.F., David F., Brunengraber H. Correction of 13C mass isotopomer distributions for natural stable isotope abundance. J. Mass Spectrom. 1996;31:255–262. doi: 10.1002/(SICI)1096-9888(199603)31:3<255::AID-JMS290>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Gerald D., Berra E., Frapart Y.M., Chan D.A., Giaccia A.J., Mansuy D., Pouysségur J., Yaniv M., Mechta-Grigoriou F. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118:781–794. doi: 10.1016/j.cell.2004.08.025. [DOI] [PubMed] [Google Scholar]
- Gerety S.S., Wang H.U., Chen Z.F., Anderson D.J. Symmetrical mutant phenotypes of the receptor EphB4 and its specific transmembrane ligand ephrin-B2 in cardiovascular development. Mol. Cell. 1999;4:403–414. doi: 10.1016/s1097-2765(00)80342-1. [DOI] [PubMed] [Google Scholar]
- Gogiraju R., Xu X., Bochenek M.L., Steinbrecher J.H., Lehnart S.E., Wenzel P., Kessel M., Zeisberg E.M., Dobbelstein M., Schäfer K. Endothelial p53 deletion improves angiogenesis and prevents cardiac fibrosis and heart failure induced by pressure overload in mice. J. Am. Heart Assoc. 2015;4:e001770. doi: 10.1161/JAHA.115.001770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski T., De Bock K. Metabolic regulation of exercise-induced angiogenesis. Vasc. Biol. 2019;1:H1–H8. doi: 10.1530/VB-19-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goveia J., Rohlenova K., Taverna F., Treps L., Conradi L.C., Pircher A., Geldhof V., de Rooij L.P.M.H., Kalucka J., Sokol L. An integrated gene expression landscape profiling approach to identify lung tumor endothelial cell heterogeneity and angiogenic candidates. Cancer Cell. 2020;37:421. doi: 10.1016/j.ccell.2020.03.002. [DOI] [PubMed] [Google Scholar]
- Haas T.L., Nwadozi E. Regulation of skeletal muscle capillary growth in exercise and disease. Appl. Physiol. Nutr. Metab. 2015;40:1221–1232. doi: 10.1139/apnm-2015-0336. [DOI] [PubMed] [Google Scholar]
- Hai T., Wolford C.C., Chang Y.S. ATF3, a hub of the cellular adaptive-response network, in the pathogenesis of diseases: is modulation of inflammation a unifying component? Gene Expr. 2010;15:1–11. doi: 10.3727/105221610x12819686555015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamik A., Wang B., Jain M.K. Transcriptional regulators of angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2006;26:1936–1947. doi: 10.1161/01.ATV.0000232542.42968.e3. [DOI] [PubMed] [Google Scholar]
- Han J., Back S.H., Hur J., Lin Y.H., Gildersleeve R., Shan J., Yuan C.L., Krokowski D., Wang S., Hatzoglou M. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013;15:481–490. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding H.P., Zhang Y., Zeng H., Novoa I., Lu P.D., Calfon M., Sadri N., Yun C., Popko B., Paules R. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- Hayner J.N., Shan J., Kilberg M.S. Regulation of the ATF3 gene by a single promoter in response to amino acid availability and endoplasmic reticulum stress in human primary hepatocytes and hepatoma cells. Biochim. Biophys. Acta Gene Regul. Mech. 2018;1861:72–79. doi: 10.1016/j.bbagrm.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitzel J., Lee E., Zhang Y., Bibli S.I., Li X., Zukunft S., Pflüger B., Hu J., Schürmann C., Vasconez A.E. Oxidized phospholipids regulate amino acid metabolism through MTHFD2 to facilitate nucleotide release in endothelial cells. Nat. Commun. 2018;9:2292. doi: 10.1038/s41467-018-04602-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H., Vandekeere S., Kalucka J., Bierhansl L., Zecchin A., Brüning U., Visnagri A., Yuldasheva N., Goveia J., Cruys B. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. 2017;36:2334–2352. doi: 10.15252/embj.201695518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakab M., Augustin H.G. Understanding angiodiversity: insights from single cell biology. Development. 2020;147:dev146621. doi: 10.1242/dev.146621. [DOI] [PubMed] [Google Scholar]
- Kakogiannos N., Ferrari L., Giampietro C., Scalise A.A., Maderna C., Ravà M., Taddei A., Lampugnani M.G., Pisati F., Malinverno M. JAM-A acts via C/EBP-α to promote Claudin-5 expression and enhance endothelial barrier function. Circ. Res. 2020;127:1056–1073. doi: 10.1161/CIRCRESAHA.120.316742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalucka J., de Rooij L.P.M.H., Goveia J., Rohlenova K., Dumas S.J., Meta E., Conchinha N.V., Taverna F., Teuwen L.-A., Veys K. Single-cell transcriptome atlas of murine endothelial cells. Cell. 2020;180:764–779.e20. doi: 10.1016/j.cell.2020.01.015. [DOI] [PubMed] [Google Scholar]
- Kilberg M.S., Shan J., Su N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol. Metab. 2009;20:436–443. doi: 10.1016/j.tem.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B., Li J., Jang C., Arany Z. Glutamine fuels proliferation but not migration of endothelial cells. EMBO J. 2017;36:2321–2333. doi: 10.15252/embj.201796436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köditz J., Nesper J., Wottawa M., Stiehl D.P., Camenisch G., Franke C., Myllyharju J., Wenger R.H., Katschinski D.M. Oxygen-dependent ATF-4 stability is mediated by the PHD3 oxygen sensor. Blood. 2007;110:3610–3617. doi: 10.1182/blood-2007-06-094441. [DOI] [PubMed] [Google Scholar]
- Kutschera S., Weber H., Weick A., De Smet F., Genove G., Takemoto M., Prahst C., Riedel M., Mikelis C., Baulande S. Differential endothelial transcriptomics identifies semaphorin 3G as a vascular class 3 semaphorin. Arterioscler. Thromb. Vasc. Biol. 2011;31:151–159. doi: 10.1161/ATVBAHA.110.215871. [DOI] [PubMed] [Google Scholar]
- La Manno G., Soldatov R., Zeisel A., Braun E., Hochgerner H., Petukhov V., Lidschreiber K., Kastriti M.E., Lönnerberg P., Furlan A. RNA velocity of single cells. Nature. 2018;560:494–498. doi: 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labuschagne C.F., van den Broek N.J., Mackay G.M., Vousden K.H., Maddocks O.D. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014;7:1248–1258. doi: 10.1016/j.celrep.2014.04.045. [DOI] [PubMed] [Google Scholar]
- Loinard C., Zouggari Y., Rueda P., Ramkhelawon B., Cochain C., Vilar J., Récalde A., Richart A., Charue D., Duriez M. C/EBP homologous protein-10 (CHOP-10) limits postnatal neovascularization through control of endothelial nitric oxide synthase gene expression. Circulation. 2012;125:1014–1026. doi: 10.1161/CIRCULATIONAHA.111.041830. [DOI] [PubMed] [Google Scholar]
- Longchamp A., Mirabella T., Arduini A., MacArthur M.R., Das A., Treviño-Villarreal J.H., Hine C., Ben-Sahra I., Knudsen N.H., Brace L.E. Amino acid restriction triggers angiogenesis via GCN2/ATF4 regulation of VEGF and H2S production. Cell. 2018;173:117–129.e14. doi: 10.1016/j.cell.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X.J., Wang H.T. Reduced Gja5 expression in arterial endothelial cells impairs arteriogenesis during acute ischemic cardiovascular disease. Exp. Ther. Med. 2017;14:4339–4343. doi: 10.3892/etm.2017.5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun A.T., McCarthy D.J., Marioni J.C. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Res. 2016;5:2122. doi: 10.12688/f1000research.9501.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masschelein E., D'Hulst G., Zvick J., Hinte L., Soro-Arnaiz I., Gorski T., von Meyenn F., Bar-Nur O., De Bock K. Exercise promotes satellite cell contribution to myofibers in a load-dependent manner. Skelet. Muscle. 2020;10:21. doi: 10.1186/s13395-020-00237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsakas A., Yadav V., Lorca S., Evans R.M., Narkar V.A. Revascularization of ischemic skeletal muscle by estrogen-related receptor-γ. Circ. Res. 2012;110:1087–1096. doi: 10.1161/CIRCRESAHA.112.266478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy D.J., Campbell K.R., Lun A.T., Wills Q.F. Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics. 2017;33:1179–1186. doi: 10.1093/bioinformatics/btw777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConell G.K., Sjøberg K.A., Ceutz F., Gliemann L., Nyberg M., Hellsten Y., Frøsig C., Kiens B., Wojtaszewski J.F.P., Richter E.A. Insulin-induced membrane permeability to glucose in human muscles at rest and following exercise. J. Physiol. 2020;598:303–315. doi: 10.1113/JP278600. [DOI] [PubMed] [Google Scholar]
- McDonald A.I., Shirali A.S., Aragón R., Ma F., Hernandez G., Vaughn D.A., Mack J.J., Lim T.Y., Sunshine H., Zhao P. Endothelial regeneration of large vessels is a biphasic process driven by local cells with distinct proliferative capacities. Cell Stem Cell. 2018;23:210–225.e6. doi: 10.1016/j.stem.2018.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min Y., Li J., Qu P., Lin P.C. C/EBP-delta positively regulates MDSC expansion and endothelial VEGFR2 expression in tumor development. Oncotarget. 2017;8:50582–50593. doi: 10.18632/oncotarget.16410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrant C.L., Sarelius I.H. Coupling of muscle metabolism and muscle blood flow in capillary units during contraction. Acta Physiol. Scand. 2000;168:531–541. doi: 10.1046/j.1365-201x.2000.00706.x. [DOI] [PubMed] [Google Scholar]
- Muzumdar M.D., Tasic B., Miyamichi K., Li L., Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- Narkar V.A., Fan W., Downes M., Yu R.T., Jonker J.W., Alaynick W.A., Banayo E., Karunasiri M.S., Lorca S., Evans R.M. Exercise and PGC-1α-independent synchronization of type I muscle metabolism and vasculature by ERRγ. Cell Metab. 2011;13:283–293. doi: 10.1016/j.cmet.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto A., Iwamoto Y., Maru Y. Oxidative stress-responsive transcription factor ATF3 potentially mediates diabetic angiopathy. Mol. Cell. Biol. 2006;26:1087–1097. doi: 10.1128/MCB.26.3.1087-1097.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakos-Zebrucka K., Koryga I., Mnich K., Ljujic M., Samali A., Gorman A.M. The integrated stress response. EMBO Rep. 2016;17:1374–1395. doi: 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potente M., Carmeliet P. The link between angiogenesis and endothelial metabolism. Annu. Rev. Physiol. 2017;79:43–66. doi: 10.1146/annurev-physiol-021115-105134. [DOI] [PubMed] [Google Scholar]
- Prior B.M., Yang H.T., Terjung R.L. What makes vessels grow with exercise training? J. Appl. Physiol. (1985) 2004;97:1119–1128. doi: 10.1152/japplphysiol.00035.2004. [DOI] [PubMed] [Google Scholar]
- Quirós P.M., Prado M.A., Zamboni N., D'Amico D., Williams R.W., Finley D., Gygi S.P., Auwerx J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017;216:2027–2045. doi: 10.1083/jcb.201702058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohlenova K., Goveia J., García-Caballero M., Subramanian A., Kalucka J., Treps L., Falkenberg K.D., de Rooij L.P.M.H., Zheng Y., Lin L. Single-cell RNA sequencing maps endothelial metabolic plasticity in pathological angiogenesis. Cell Metab. 2020;31:862–877.e14. doi: 10.1016/j.cmet.2020.03.009. [DOI] [PubMed] [Google Scholar]
- Ron D., Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Rowe G.C., Jang C., Patten I.S., Arany Z. PGC-1beta regulates angiogenesis in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2011;301:E155–E163. doi: 10.1152/ajpendo.00681.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe G.C., Safdar A., Arany Z. Running forward: new frontiers in endurance exercise biology. Circulation. 2014;129:798–810. doi: 10.1161/CIRCULATIONAHA.113.001590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart C.A., McCurry M.P., Marino A., South M.A., Howell M.E., Layne A.S., Ramsey M.W., Stone M.H. Slow-twitch fiber proportion in skeletal muscle correlates with insulin responsiveness. J. Clin. Endocrinol. Metab. 2013;98:2027–2036. doi: 10.1210/jc.2012-3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tameire F., Verginadis I.I., Leli N.M., Polte C., Conn C.S., Ojha R., Salas Salinas C., Chinga F., Monroy A.M., Fu W. ATF4 couples MYC-dependent translational activity to bioenergetic demands during tumour progression. Nat. Cell Biol. 2019;21:889–899. doi: 10.1038/s41556-019-0347-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Hua B.Y., Adachi S., Guney I., Kawauchi J., Morioka M., Tamamori-Adachi M., Tanaka Y., Nakabeppu Y., Sunamori M. Stress response gene ATF3 is a target of c-myc in serum-induced cell proliferation. EMBO J. 2005;24:2590–2601. doi: 10.1038/sj.emboj.7600742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrence M.E., MacArthur M.R., Hosios A.M., Valvezan A.J., Asara J.M., Mitchell J.R., Manning B.D. The mTORC1-mediated activation of ATF4 promotes protein and glutathione synthesis downstream of growth signals. eLife. 2021;10:e63326. doi: 10.7554/eLife.63326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Brink S.C., Sage F., Vértesy Á., Spanjaard B., Peterson-Maduro J., Baron C.S., Robin C., van Oudenaarden A. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods. 2017;14:935–936. doi: 10.1038/nmeth.4437. [DOI] [PubMed] [Google Scholar]
- Van der Stede T., Blancquaert L., Stassen F., Everaert I., Van Thienen R., Vervaet C., Gliemann L., Hellsten Y., Derave W. Histamine H1 and H2 receptors are essential transducers of the integrative exercise training response in humans. Sci. Adv. 2021;7:eabf2856. doi: 10.1126/sciadv.abf2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandekeere S., Dubois C., Kalucka J., Sullivan M.R., García-Caballero M., Goveia J., Chen R., Diehl F.F., Bar-Lev L., Souffreau J. Serine synthesis via PHGDH is essential for heme production in endothelial cells. Cell Metab. 2018;28:573–587.e13. doi: 10.1016/j.cmet.2018.06.009. [DOI] [PubMed] [Google Scholar]
- Vanlandewijck M., He L., Mäe M.A., Andrae J., Ando K., Del Gaudio F., Nahar K., Lebouvier T., Laviña B., Gouveia L. A molecular atlas of cell types and zonation in the brain vasculature. Nature. 2018;554:475–480. doi: 10.1038/nature25739. [DOI] [PubMed] [Google Scholar]
- Wang H.U., Chen Z.F., Anderson D.J. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell. 1998;93:741–753. doi: 10.1016/s0092-8674(00)81436-1. [DOI] [PubMed] [Google Scholar]
- Waters R.E., Rotevatn S., Li P., Annex B.H., Yan Z. Voluntary running induces fiber type-specific angiogenesis in mouse skeletal muscle. Am. J. Physiol. Cell Physiol. 2004;287:C1342–C1348. doi: 10.1152/ajpcell.00247.2004. [DOI] [PubMed] [Google Scholar]
- Wilhelm K., Happel K., Eelen G., Schoors S., Oellerich M.F., Lim R., Zimmermann B., Aspalter I.M., Franco C.A., Boettger T. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature. 2016;529:216–220. doi: 10.1038/nature16498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.E., Pan L., Zuo Y., Li X., Hong W. Detecting activated cell populations using single-cell RNA-seq. Neuron. 2017;96:313–329.e6. doi: 10.1016/j.neuron.2017.09.026. [DOI] [PubMed] [Google Scholar]
- Yan C., Lu D., Hai T., Boyd D.D. Activating transcription factor 3, a stress sensor, activates p53 by blocking its ubiquitination. EMBO J. 2005;24:2425–2435. doi: 10.1038/sj.emboj.7600712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Xia R., Yue C., Zhai W., Du W., Yang Q., Cao H., Chen X., Obando D., Zhu Y. ATF4 regulates CD4+T cell immune responses through metabolic reprogramming. Cell Rep. 2018;23:1754–1766. doi: 10.1016/j.celrep.2018.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You L.R., Lin F.J., Lee C.T., DeMayo F.J., Tsai M.J., Tsai S.Y. Suppression of Notch signalling by the COUP-TFII transcription factor regulates vein identity. Nature. 2005;435:98–104. doi: 10.1038/nature03511. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mouse single cell RNA-seq data reported in this study are publicly available at the Gene Expression Omnibus (GEO) repository under the accession number (GSE158984). The bulk RNA-seq data are publicly available at GEO under accession number (GSE174785). Please check https://shiny.debocklab.hest.ethz.ch/Fan-et-al/ forhttps://shiny.debocklab.hest.ethz.ch/Fan-et-al/ for data visualization. Comparisons between RmECs and WmECs (bulk RNA-seq) can be downloaded from the app. All other data are available from the corresponding author on request.
All original codes are available from the lead author upon request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.