

Abstract
L-Dopa-induced dyskinesia (LID) is associated with upregulation of striatal ΔFosB in animal models and patients with Parkinson’s disease (PD). A mechanistic role of ΔFosB is suspected because its transgenic overexpression leads to early appearance of LID in rodents and primates. The present study in rodents is aimed at exploring the therapeutic potential of striatal ΔFosB gene suppression to control LID in patients with PD. To determine the effect of reducing striatal ΔFosB expression, we used RNAi gene knockdown in a rat model of PD and assessed abnormal involuntary movements (AIMs) in response to L-Dopa. Rats with dopamine depletion received striatal injections of rAAV-ΔFosB shRNA or a control virus before exposure to chronic L-Dopa treatment. Development of AIMs during the entire L-Dopa treatment period was markedly inhibited by ΔFosB gene knockdown and its associated molecular changes. The antiparkinsonian action of L-Dopa was unchanged by ΔFosB gene knockdown. These results suggest a major role for ΔFosB in the development of LID, and support exploring strategies to reduce striatal ΔFosB levels in patients with PD.
Introduction
The development of L-Dopa-induced dyskinesia (LID) is associated with significant striatal dysfunction resulting from long-term dopamine (DA) depletion and non-physiologic replacement therapy in PD (1–3). Although the striatal mechanisms underlying these abnormal movements are yet unclear, several molecular markers have been identified (4–8). Particularly, increased levels of striatal ΔFosB are consistently associated with LID across animal models and human studies (7, 9–11). ΔFosB, a truncated splice variant of FosB, is a highly stable transcription factor usually regulated in response to chronic stimuli such as prolonged exposure to drugs (12, 13). In animal models of PD, striatal ΔFosB levels rise after dopamine cell loss, but are further increased following L-Dopa treatment and correlate with LID severity (7, 9). Experiments with transgenic ΔFosB overexpression in parkinsonian rodents and non-human primates provide evidence for an important role of this transcription factor. In both models, high striatal levels of ΔFosB induce LID in animals that have not been chronically exposed to L-Dopa (10, 14). Furthermore, striatal recordings show that ΔFosB overexpression induces neuronal responses to DA that reproduce the changes typically found following chronic L-Dopa treatment. These functional changes occurred in both direct and indirect striatal pathway neurons, suggesting a role of ΔFosB in both pathways (14) in agreement with the participation of both pathways in LID mechanisms (15, 16). Altogether, data suggest that ΔFosB plays a primary role in the pathogenesis of LID (17). Nevertheless, further understanding the ΔFosB role in LID mechanisms is key to determine its potential as a therapeutic target.
Chronic dopaminergic stimulation triggers various molecular changes that may participate cooperatively or independently in LID mechanisms (18, 19). Studies of transgenic induction of ΔFosB overexpression support an independent ΔFosB mechanism for LID occurrence (10, 14). It is possible that other pathways (e.g. PKA-p-DARPP-32 or ERK1/2-MAPK) could induce LID in a similar manner. In fact, L-Dopa priming, a mechanism that establishes persistent dyskinetic responses, is thought to derive from multiple long-term plastic changes in the setting of dopamine depletion (20). Therefore, plastic changes that may be unrelated to ΔFosB could be responsible for LID maintenance after priming as well as LID regeneration after ΔFosB levels are reduced. Both the appearance and maintenance of dyskinesias are important targets to develop effective antidyskinetic therapies. Here, we silenced the ΔFosB gene in the striatum of hemiparkinsonian rats and subsequently exposed the animals to chronic L-Dopa treatment to determine the impact of reduced levels of striatal ΔFosB on LID mechanisms. ΔFosB gene suppression specifically controlled the expression of abnormal involuntary movements (AIMs) during chronic dopamine replacement, thereby supporting further studies of this gene therapy for LID in PD.
Materials and methods
siRNA design and production of viral vectors
ΔFosB lacks the C-terminal 101 amino acids of FosB as a result of alternative splicing leading to a frame shift and premature termination codon (21). Thus, only the junction sequence after splicing can be used to design siRNA specific to ΔFosB (indicated in Fig 1A). Three siRNAs were designed based on this junction sequence and evaluated by transient transfection of PC12 cells followed by Western blot analysis (Fig 1B). PC12 cells were cultured in 12-well plates in Dulbecco’s modified eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) and 5% horse serum in a CO2 incubator at 37°C. To test the efficiency of ΔFosB siRNAs (siRNA#1, siRNA#2 and siRNA#3), cells were transfected with siRNAs using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions for 24 hours. The medium was replaced with fresh medium containing 0.5% FBS for another 24 hours, following by induction of ΔFosB expression with 20% FBS for 2 hours. Cell lysates were then collected for Western blot analysis.
Figure. 1. Generation of viral vector with effective shRNA to ΔFosB.
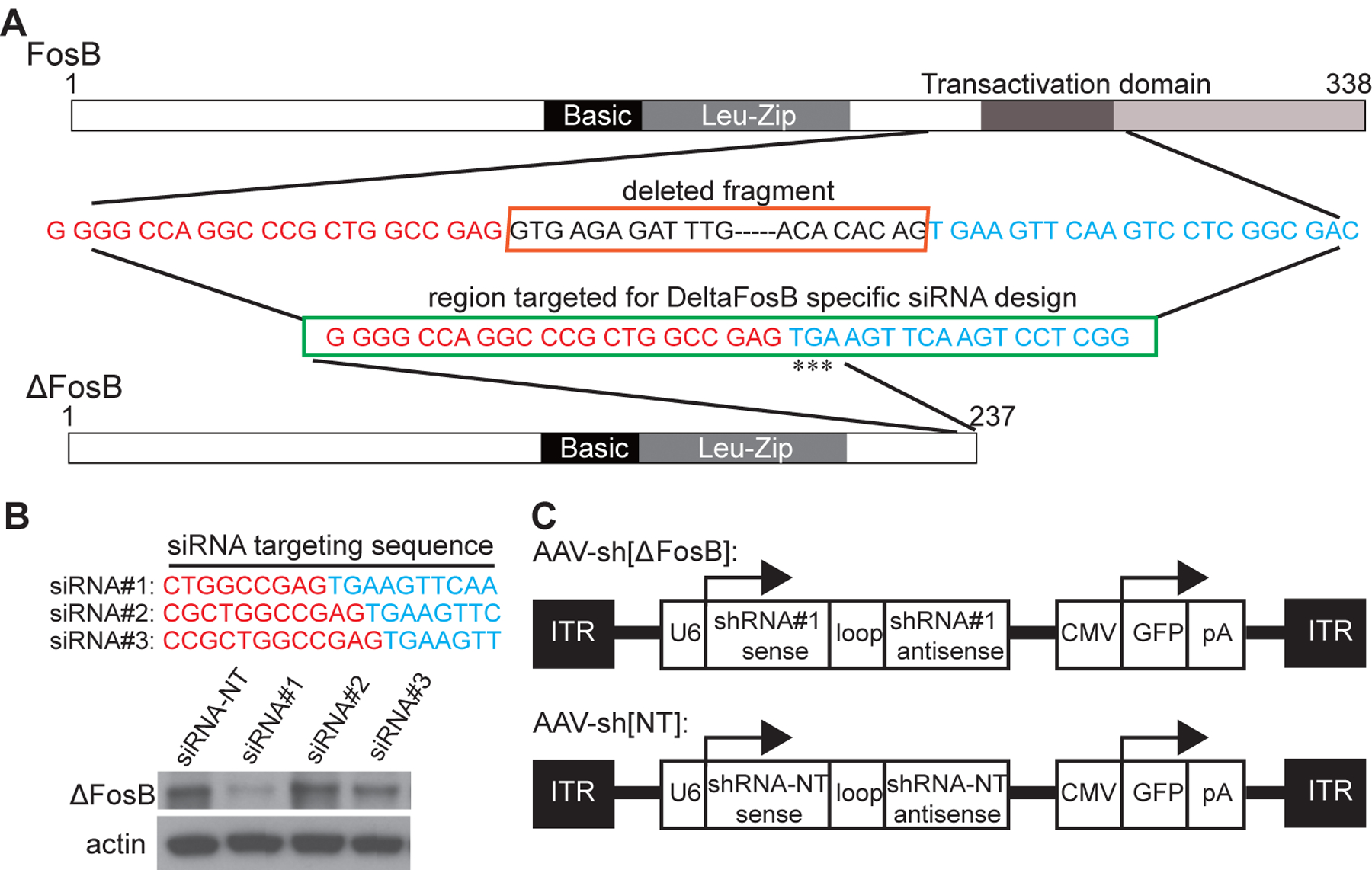
(A) ΔFosB lacks the C-terminal 101 amino acids of FosB as a result of alternative splicing causing frame shift that introduces the termination codon ‘TGA’ (indicated as ‘***’). The deleted sequence is shown in the upper box, and the ΔFosB specific region targeted for siRNA design is shown in the lower box. (B) Double stranded siRNAs corresponding to the target sequences and a control non-targeting (NT) siRNA were synthesized and evaluated by transient transfection of PC12 cells followed by Western blot analysis. siRNA#1 has the best knockdown effect. (C) ΔFosB shRNA (shRNA#1) was designed based on siRNA#1 sequence. An oligonucleotide containing sense and antisense siRNA#1, with a loop (TTCAAGAGA) in between, and flanked by restriction sites was synthesized. This oligonucleotide was then cloned in a rAAV2/5 cis vector under the control of U6 promoter with GFP co-expression driven by the CMV promoter. The control vector AAV-sh[NT] is isogenic to AAV-sh[ΔFosB], except that it expresses a nontargeting shRNA instead of shRNA#1. ITR = inverted terminal repeat; pA = poly(A) tail.
AAV2/5 shRNA viral vectors were prepared at SignaGen Laboratories. Briefly, ΔFosB shRNA (shRNA#1) was designed based on siRNA#1 sequence. An oligonucleotide containing sense and antisense siRNA#1, with a loop (TTCAAGAGA) in between, and flanked by restriction sites was synthesized (Fig 1C). This oligonucleotide was then cloned in a rAAV2/5 cis vector under the control of U6 promoter, with GFP co-expression under the control of CMV promoter. The cis and trans plasmids were then co-transfected into HEK293 cells to produce rAAV-ΔFosB shRNA. The number of genome copies was determined by quantitative PCR. The control vector rAAV-NT shRNA was isogenic to rAAV-ΔFosB shRNA, except that it expressed a nontargeting (NT) shRNA (sense: AGTACTGCTTACGATACGG; antisense: CCGTATCGTAAGCAGTACTT) instead of shRNA#1. The titer of rAAV-ΔFosB shRNA was 1.16 × 1013 vg/ml, and the titer of rAAV-NT shRNA 1.23 × 1013 vg/ml.
Animal model and virus injections
All procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and protocols were approved by the Institutional Animal Care and Use Committee. Adult male Sprague–Dawley rats were housed with free access to food and water, 12 h light/dark cycle, and constant temperature and humidity. To produce a unilateral dopamine lesion, rats were deeply anesthetized with Ketamine/Xylazine for injection of 6-hydroxydopamine (6-OHDA, Sigma-Aldrich, St. Louis, MO, 8 μg dissolved in 4 μl of 0.02% ascorbic acid solution in saline) into the medial forebrain bundle under stereotaxic surgery as described previously (10). Five days after 6-OHDA lesion (see the study timeline in Fig 2), all rats (n=19) were randomly separated in two groups for virus injections into the striatum. rAAV-ΔFosB shRNA or the control virus (rAAV-NT shRNA) were injected at equal particle doses (1 × 1012 vg/mL). Virus injections were guided stereotaxically into the post-commissural area of the striatum (the dorsoposterolateral area that corresponds to the motor territory of the striatum) on the side ipsilateral to the 6-OHDA lesion (stereotaxic coordinates: A: 8.4 and L: 4.4 mm from the middle of the interaural line). To cover the targeted striatal area, the volume was divided in two injections of 3 and 2 μl at 6.2 and 5.2 mm depth from surface of the skull, respectively, using a microinfusion pump at a rate of 0.5 μl/min (10). The viral vectors were thus injected in the middle portion of the posterior striatum to allow for diffusion within the limits of the nucleus.
Figure 2. Timeline of in-vivo experiments.
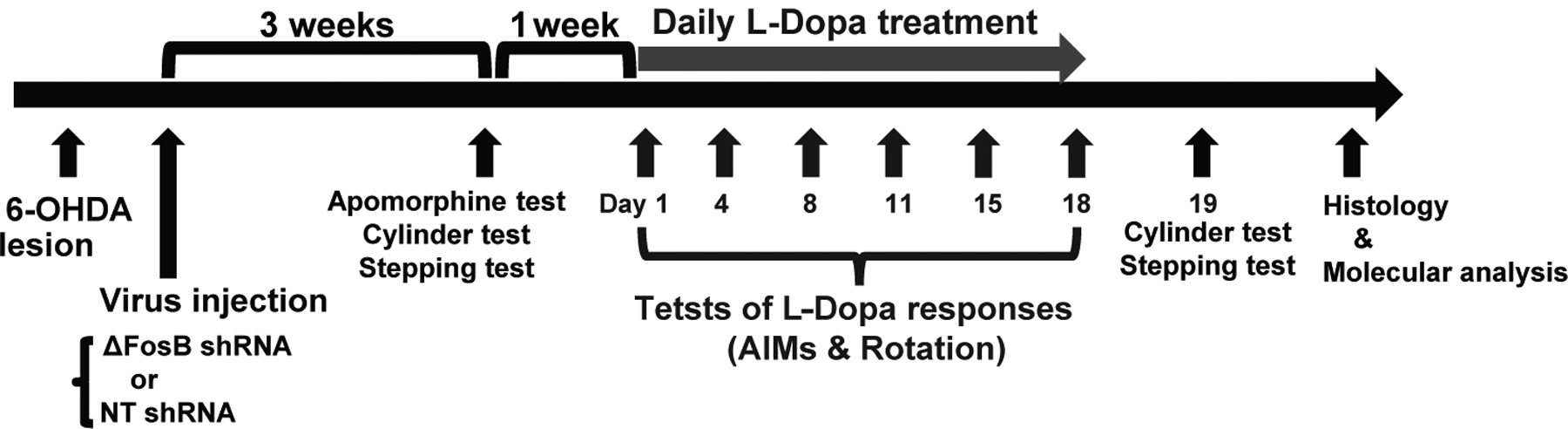
Studies began for all rats with unilateral 6-OHDA lesions of the nigrostriatal pathway, and random assignment to either rAAV injection, rAAV-shRNA ΔFosB or rAAV-NT shRNA. Viruses were injected 5 days after the 6-OHDA lesion. More than 3 weeks after the 6-OHDA lesion and 3 weeks after the virus injection, the apomorphine test was used to confirm complete lesions; animals selected to continue in the study were assessed on the following day with the cylinder and stepping tests to determine baseline motor deficits. One week after this assessment, daily treatment with L-Dopa methyl ester plus benserazide (12/12 mg/kg, i.p.) started for all rats (grey arrow). On days 1, 4, 8, 11, 15 and 18, the L-Dopa motor response was evaluated by rating contralateral rotation and AIMs. On day 19, the cylinder and stepping tests were performed again to determine changes in comparison to baseline. After these tests, animals were euthanized for histological and molecular analyses.
To estimate the extent of the lesion of animals included in each virus group, rats were tested for rotational responses contralateral to the lesion in response to a subthreshold dose of apomorphine (0.05 mg/kg, s.c.) that only induces strong rotation in rats with full lesion. This test was conducted more than 3 weeks after 6-OHDA lesion and 3 weeks after AAV injection (see Fig 2). Contralateral rotations were counted for 60 min post injection, and animals whose rotational behavior reached ≥ 7 turns/min were selected for the study (total n = 13, 7 assigned to the targeting shRNA group and 6 to the NT shRNA group). The total contralateral rotations in response to apomorphine averaged 153 in the rAAV-ΔFosB shRNA group and 144 in the control group (not significant).
Evaluation of motor function
The cylinder (22) and the stepping tests (23) were performed to assess the baseline hypokinesia in both virus groups the day after animals were selected for the study based on the apomorphine test. The cylinder test is designed to evaluate motor asymmetry in this model. Animals are individually placed in a 20 cm-diameter glass cylinder, where they typically exhibit exploratory behavior, and the wall contacts of the forepaws are counted until reaching a total of 20 contacts. Data are presented as percentage of the impaired paw use from the total contacts of both paws. The stepping test consists of measuring the “initiation time” and counting the number of “adjusting steps”. Briefly, the animal is held by the experimenter with one hand restraining the hindlimbs and the other hand restraining one of the forelimbs leaving only one forelimb free to move across a ramp (0.9 m). Initiation time is measured from initial positioning until the first movement of the free paw using 180 sec as break-off point. Adjusting steps are counted for each free paw in the backhand and forehand directions of movement while the rat is moved slowly (5 sec) sideways in the ramp by the experimenter. On the same day, both the cylinder and stepping tests were repeated 30 min after administration of L-Dopa methyl ester (12 mg/kg) plus benserazide (12 mg/kg) given i.p. to determine the baseline response to L-Dopa. Following the 18-day course of chronic L-Dopa treatment, the cylinder and the stepping tests were repeated before and 30 min after L-Dopa injection for comparison with the baseline data (initial versus chronic ΔFosB gene KD). The examiner was blinded to the virus assignment.
Response to chronic L-Dopa treatment
Four weeks after virus injection, all rats entered a course of chronic L-Dopa treatment. L-Dopa methyl ester plus benserazide (12/12 mg/kg) was administered i.p. daily for 18 days. AIMs were assessed using the standardized scale (24) on days 1, 4, 8, 11, 15, and 18 of chronic L-Dopa treatment. Animals were observed and scored directly every 15 min for 120 min after L-Dopa injection by an examiner blinded to the virus assignment. To increase the specificity of behavioral data, contraversive rotation was not included in the analysis of total AIMs. AIMs scores in each interval were added to obtain a total value of the AIMs category (limb, axial and oral AIMs) for each animal in each test. Rotational behavior was assessed using an automated rotometer for the whole duration of the L-Dopa response. The system counted rotations in either direction every 5 min intervals. Full circle rotations contralateral to the lesion were computed for analysis of L-Dopa responses. Peak rotation was taken from the maximum number of contralateral turns in any 5 min interval.
Tissue preparation
All rats were deeply anesthetized and sacrificed by decapitation 24 h after the final L-Dopa treatment for behavioral assessments. Brains were rapidly removed and dissected on dry ice. Dissected posterior striata were quickly frozen on dry ice and stored at −80°C for Western blot analyses. In three rats from each group, the dissection took less of the posterior region of the striatum to leave virus injected areas for immunohistochemistry. The rest of those brains were immersed in 4% paraformaldehyde in PBS for post-fixation, and after 24 hr the brains were immersed in 30% sucrose in PBS until sinking. For immunohistochemistry, coronal sections of the brains were cut serially at 20-μm thickness using a cryostat.
Immunoblotting
Lysates of cultured cells and striatal tissue samples were extracted in 1% SDS in PBS containing phosphatase inhibitor cocktail set II (Calbiochem, La Jolla, CA) and protease inhibitor cocktail set V (Calbiochem). Three mL of buffer was used per 150 mg of striatal tissue. Samples were sonicated and cleared at 140,000 × g for 10 min. Protein concentrations were determined using the BCA assay (Pierce). Lysates (20 μg) were mixed with lithium dodecyl sulfate-sample loading buffer (Life Technologies, Grand Island, NY), separated on a NuPage 4–20% (GenScript, Piscataway, NJ), and transferred onto a polyvinylidine fluoride membrane (Bio-Rad, Hercules, CA). Blots were blocked in 5% BSA (Sigma-Aldrich, St. Louis, MO) in Tris-buffered saline and 0.1% Tween-20 before probing with antibodies. ECL Plus (Perkin-Elmer, Waltham, MA) was used to develop immunoblots, and band intensity was quantified by ImageJ. Primary antibodies used were: FosB (Santa Cruz Biotechnology, Dallas, TX); p-ERK and GFP (Cell Signaling Technology, Danvers, MA); DARPP-32 and Cdk5 (Santa Cruz Biotechnology, Dallas, TX); p-T34-DARPP-32 (Novus Biologicals, Centennial, CO); and β-actin (Sigma-Aldrich, St. Louis, MO).
Immunohistochemistry
Free-floating sections were washed in PBS containing 0.05% Triton X-100 (PBS-T) and then incubated for 30 min with 0.3% H2O2 to quench endogenous peroxidase activity. The sections were soaked with blocking agents and then incubated with primary antibodies dissolved in dilution reagent at 4°C for 24 h. Normal Goat Serum (Vector Laboratories, Burlingame, CA) was used for blocking. The primary antibodies used were mouse monoclonal antibodies against FosB (1:100, sc-48, Santa Cruz Biotechnology), GFP (1:100, ab1218, Abcam), NeuN (1:100, MAB377, Millipore), GFAP (1:100, sc-33673, Santa Cruz Biotechnology), and Iba-1 (1:100, sc-32725, Santa Cruz Biotechnology). Goat biotinylated anti-mouse immunoglobulin (Vector Laboratories) was used as the secondary antibody. After incubating sections for 60 min with VECTASTAIN® ABC reagent, reaction products were visualized using DAB substrate kit (both reagents from Vector Laboratories). The sections were mounted on glass slides and hematoxylin was used to counterstain cell nuclei. For NeuN-, GFAP- or Iba-1-positive cell counting, 10 images were obtained using a digital camera connected to a microscope (40× objective) from each of the 3 animals (25). For immunofluorescence, Alexa Fluor® 488 goat anti-mouse IgG (H + L) (ThermoFisher Scientific, Waltham, MA) was used as the secondary antibody. Images were obtained using ECLIPSE E800 (Nikon, Tokyo, Japan). For GFP-positive cell counting, 10 images were obtained using a digital camera connected to a microscope (40× objective) from each animal (25).
Statistical analysis
Measurements of motor behavior were graded using none integer, and thus composed parametric data that were analyzed using 1- or 2-way analysis of variance (ANOVA) for repeated measures followed by post-hoc Fisher’s Protected Least Significant Difference (PLSD) test when the ANOVA F indicated significance (10). In all analyses, appropriate corrections according to data distribution were made if necessary, such as the Greenhouse-Geisser correction depending on the sample variances in analysis of repeated measures. The relation between each tested molecular marker and AIMs scores was analyzed by linear regression. All data are presented as mean ± S.E.M, and statistical significance was determined at p < 0.05.
Results
Striatal ΔFosB gene suppression inhibited the development of AIMs in rats
The striatal injection of rAAV-ΔFosB shRNA significantly reduced the severity of AIMs, including all categories (limb, masticatory and axial dyskinesias; Fig 3A–D). The pattern of dyskinesia development followed the typical curve, which has a rising slope in the first week and stabilization thereafter. On days 1 and 4 of daily L-Dopa administration, AIMs were mild in all animals including controls (rats injected with rAAV-NT shRNA) due to the sensitization commonly developed during the first week of treatment. Comparison with the control group showed significant differences in AIMs scores starting on day 8 and continuing until the end of the assessment period on day 18 of daily L-Dopa treatment (main effects: total AIMs, F(2.6, 31.8) = 9.1, p < 0.05; limb, F(2.2, 19.8) = 6.5, p < 0.05; axial, F(3.1, 29) = 8.8, p < 0.05; masticatory, F(2, 27.1) = 5.7, p < 0.05). The effect reached more than 65% reduction in the majority of assessed time points. Therefore, ΔFosB gene knockdown (KD) effectively inhibited the development of AIMs in rats, and its effect was maintained chronically (18 days in hemiparkinsonian rats).
Figure 3. Specific reduction of AIMs by striatal ΔFosB gene knockdown.
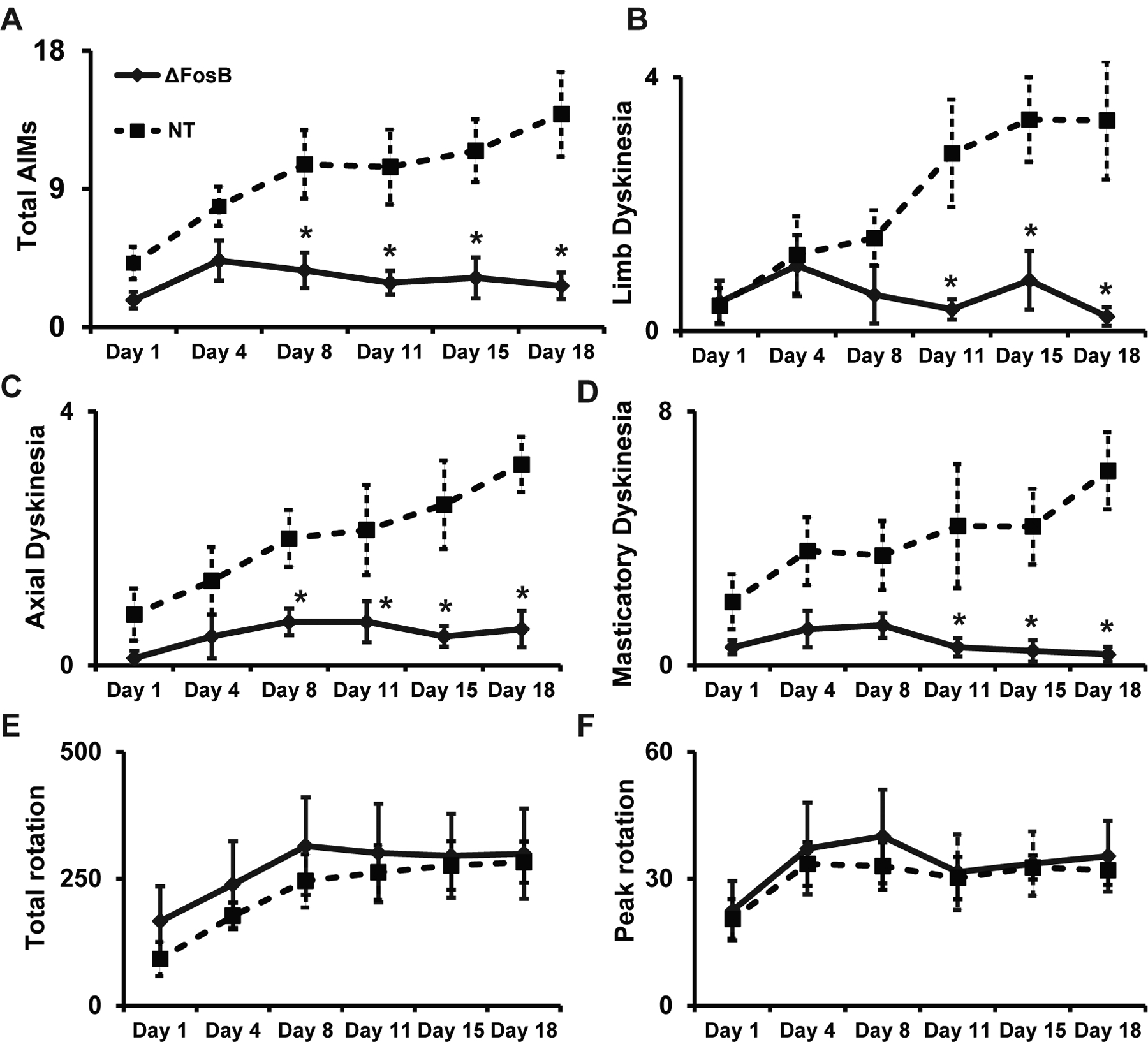
Total AIMs (A), limb dyskinesias (B), axial dyskinesia (C), masticatory dyskinesias (D), and rotations (E-total and F-peak) during chronic L-Dopa treatment. All rats received daily i.p. injections of L-Dopa methyl ester plus benserazide (12/12 mg/kg) and motor responses were assessed on days 1, 4, 8, 11, 15, and 18. (A-D) Significant differences between the two groups in total AIMs scores and each AIM category developed after the first week of L-Dopa treatment. (E, F) No significant difference in total or peak of rotations in response to L-Dopa administration were observed between the two groups. *: p < 0.05 versus control rats (rAAV-NT shRNA group). ANOVAs for repeated measures and Fisher’s PLSD tests (n = 7 and 6 per group). Data are shown as mean ± SEM.
The striatal injection of rAAV-ΔFosB shRNA did not change the rotational response to L-Dopa. At every time point of assessment, the total and peak of contralateral rotations (Fig 3E and F) were similar between rats injected with the rAAV-ΔFosB shRNA and the control group. Therefore, ΔFosB gene KD reduced AIMs without interfering with the antiparkinsonian response to L-Dopa, which manifests as a rotational response in this asymmetric model.
To determine if striatal ΔFosB gene KD modifies basal motor deficits, we assessed the initial contralateral hypokinesia in all rats at 3 weeks post-virus injection (see Fig 2). No differences in either the use of the contralateral limb in the cylinder test (Fig 4A) or initiation time (Fig 4B) and contralateral steps counts in the stepping test (Fig 4C and D) were found between the two virus injected groups. The reversal of contralateral hypokinesia by L-Dopa at baseline was also similar between groups (Fig 4A–D). These results confirmed a similar contralateral parkinsonism and responsiveness to L-Dopa in both animal groups before initiation of chronic L-Dopa administration, as expected after complete dopamine lesion (interaction for L-Dopa effect: cylinder test, F(2.2, 24.5) = 113, p < 0.01; initiation time, F(1.7, 18.4) = 145.4, p < 0.01; step counts, F(2.4, 25.9) = 19.8, p < 0.01). Data also excluded a possible effect of the ΔFosB gene KD on the development of parkinsonism.
Figure 4. Unchanged forelimb hypokinesia after striatal ΔFosB gene knockdown.
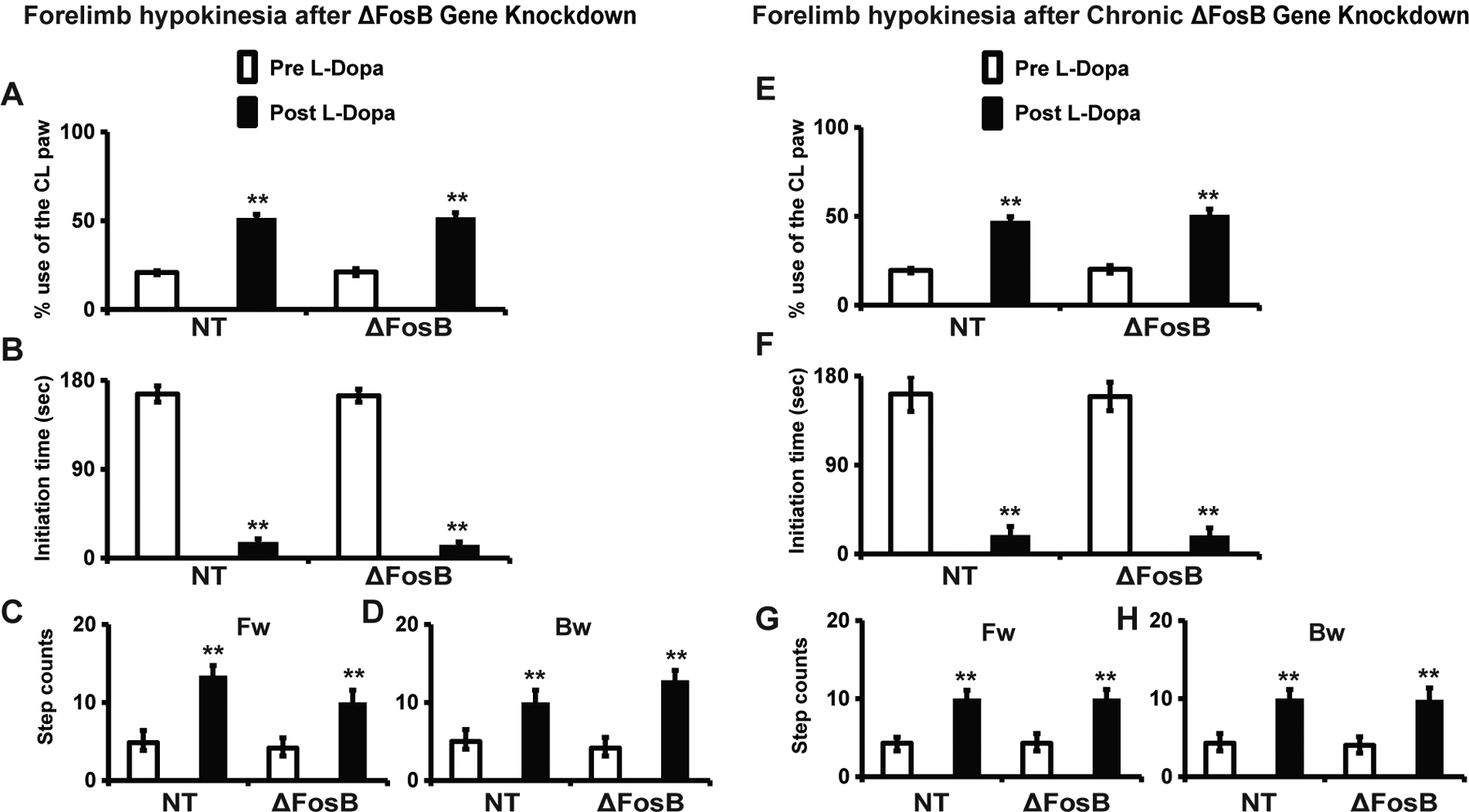
(A-D) Cylinder (A) and stepping tests were used to assess all rats following the apomorphine screening test (i.e. three weeks post virus injection) to assess the initial effects of gene knockdown. Hand touches in the cylinder test (A), initiation time (B), and adjusting steps (C and D) in the stepping test were analyzed pre and post i.p. injection of L-Dopa methyl ester plus benserazide (12/12 mg/kg). (A) The use of the paw contralateral to the lesion (CL, impaired side) expressed as percentage of total touches shows the baseline impairment and its improvement to almost 50 % in response to L-Dopa in both groups (rAAV-ΔFosB shRNA and rAAV-NT shRNA). (B) Initiation time with a cutoff of 180 sec shows that rats could not move their CL forelimb, but markedly improved after L-Dopa injection with movement in less than 20 sec in both groups. (C and D) The step counts of the CL forelimb in the forward (Fw) and backward (Bw) directions were significantly reduced, but also markedly improved by L-Dopa injection in both groups. There was no significant difference in the impairment of the CL limb between the two groups of rats either pre or post L-Dopa injection in any of the measurements (A-D).
(E-H) The same analysis of motor behavior using the cylinder and stepping tests as performed at baseline (initial effects of gene knockdown) was repeated at the end of the assessment period of gene knockdown (day 19, see figure 2). Results showed unchanged impairment of the CL limb in all measurements in both groups of rats (rAAV-ΔFosB shRNA and rAAV-NT shRNA). Cylinder test (E), initiation time (F) and adjusting steps (G-H) of the stepping test performed pre and post L-Dopa methyl ester plus benserazide i.p. injection. Data show no effects of chronic gene knockdown on parkinsonian motor deficits and their responsiveness to L-Dopa after chronic administration.
**: p < 0.01 versus pre L-Dopa in the same group. Two-way ANOVAS for repeated measures (n = 7 and 6 per group). Data are mean ± SEM.
To determine whether chronic KD of the ΔFosB gene could modify hypokinesia, we assessed the animals with the cylinder and stepping tests again at the end of the study (day 19). No differences in these measurements were found between the gene KD and control groups (Fig 4). In addition, comparison of baseline and final scores in both the cylinder and stepping tests showed no difference in animals with or without ΔFosB gene suppression (Fig 4; interaction for L-Dopa effect: cylinder test, F(1, 22) = 1.9, p > 0.05; initiation time, F(1, 22) = 1.2, p > 0.05; step counts, F(1, 22) = 3.5, p > 0.05). Therefore, ΔFosB gene KD had no beneficial or deleterious effects on the hypokinesia developed in rats following a full dopaminergic lesion.
rAAV-ΔFosB shRNA or rAAV-NT shRNA injections resulted in similar viral transduction in striatal cells, which did not extend beyond the striatal boundaries (see Fig 5A). The ΔFosB shRNA viral vector markedly reduced ΔFosB expression in rats, as shown by Western blot analysis of striatal tissue (t-tests, p < 0.001, Fig 5B and C). The analysis of striatal ΔFosB expression level in all rats showed a significant positive correlation with AIMs scores (R2 = 0.6, p < 0.005, Fig 5D).
Figure 5. Regulation of LID molecular markers induced by ΔFosB gene knockdown.
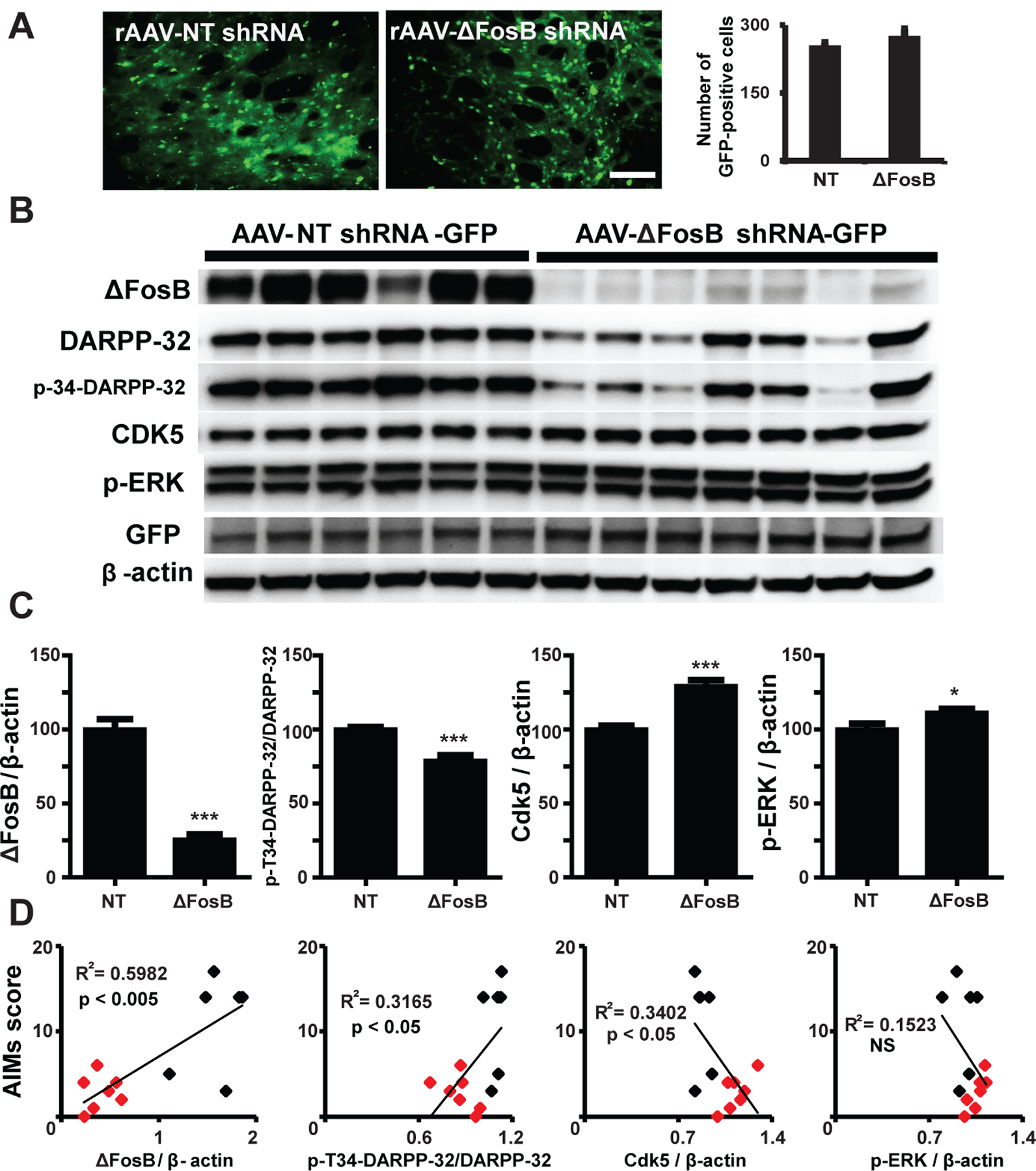
(A) rAAV transduction in the rat striatum. The images show the GFP reporter widely distributed indicating efficient transduction of the striatum with both viruses, ΔFosB targeting or control nontargeting shRNA (scale bar = 100 μm). Cell counting showed nonsignificant difference in expression levels between viruses. (B) Western blot analyses of expression of LID markers and GFP from striatal tissue lysates of each animal. Each lane represents a separate animal. (C) Quantification of each band intensity relative to β-actin comparing marker expression between viral vectors. p-Thr34-DARPP-32 was analyzed as the ratio to total DARPP-32 expression. * p < 0.05 and *** p < 0.001 (unpaired t-tests, n = 7 and 6 per group). Data represent means ± SEM. (D) Correlations between marker expression and AIMs development including all animals (both virus groups; black = NT shRNA group; red = ΔFosB shRNA group). Total AIMs scores taken at the end of the assessment period (18th day), when the highest difference between the ΔFosB targeting and control NT virus group was attained, were correlated with the protein level in each animal. R2 and p values are included in the graphs (linear regression analyses, n=13). Data confirmed a major reduction of ΔFosB expression by shRNA mediated gene knockdown that strongly correlated with AIMs severity, and its impact on the expression of p-T34-DARPP-32.
ΔFosB gene suppression impacted other molecular markers of LID
Striatal ΔFosB gene KD reduced the expression of both dopamine and cyclic AMP-regulated phosphoprotein 32 kDa (DARPP-32) and p-Thr34-DARPP-32 compared to their levels in rats injected with the control virus (Fig 5B). In addition, the ratio of p-Th34-DARPP-32/DARPP-32 decreased by ΔFosB gene KD (t-tests, p < 0.001, Fig 5C). Similar to ΔFosB, the levels of these markers also correlated with the severity of AIMs (R2 = 0.3, p < 0.05, Fig 5D). In contrast, the phosphorylated form of extracellular signal-regulated kinase (p-ERK) was slightly increased by ΔFosB gene KD (t-test, p < 0.05), but its changes had no correlation with AIMs score (p > 0.05, Fig 5B–D). Cyclin-dependent kinase 5 (Cdk5), which was also increased by ΔFosB gene KD (t-test, p < 0.001), correlated inversely with AIMs score (R2 = 0.3, p < 0.05, Fig 5B–D). These results suggest interacting mechanisms between ΔFosB expression and the regulation of other molecules associated with LID development or maintenance.
rAAV-ΔFosB shRNA safely suppressed striatal ΔFosB gene expression
To evaluate the potential toxicity of the method used for gene KD in the striatum (26–28), we analyzed the possibility of neuronal loss and inflammatory reactions in the striatum. Immunostaining for NeuN (neuronal marker), GFAP (astroglial marker), and Iba-1 (activated microglial marker) were analyzed in the striatum of rats randomly selected from each group of virus injections. There was no significant difference in the number of NeuN-, GFAP- or Iba-1-positive cells between the two groups (see the similar cell counts in Fig 6), indicating that no striatal neuronal loss, reactive astrogliosis, or microglial infiltration were observed as a result of rAAV-ΔFosB shRNA injection compared to the control vector injection. These results support that the shRNA-based method of gene KD applied in the rat did not cause structural/cellular damage in the striatum. Daily observation of the animals did not evidence any changes in their normal behavior or overall health.
Figure 6. The striatal tissue remained intact following rAAV injection and ΔFosB gene knockdown.
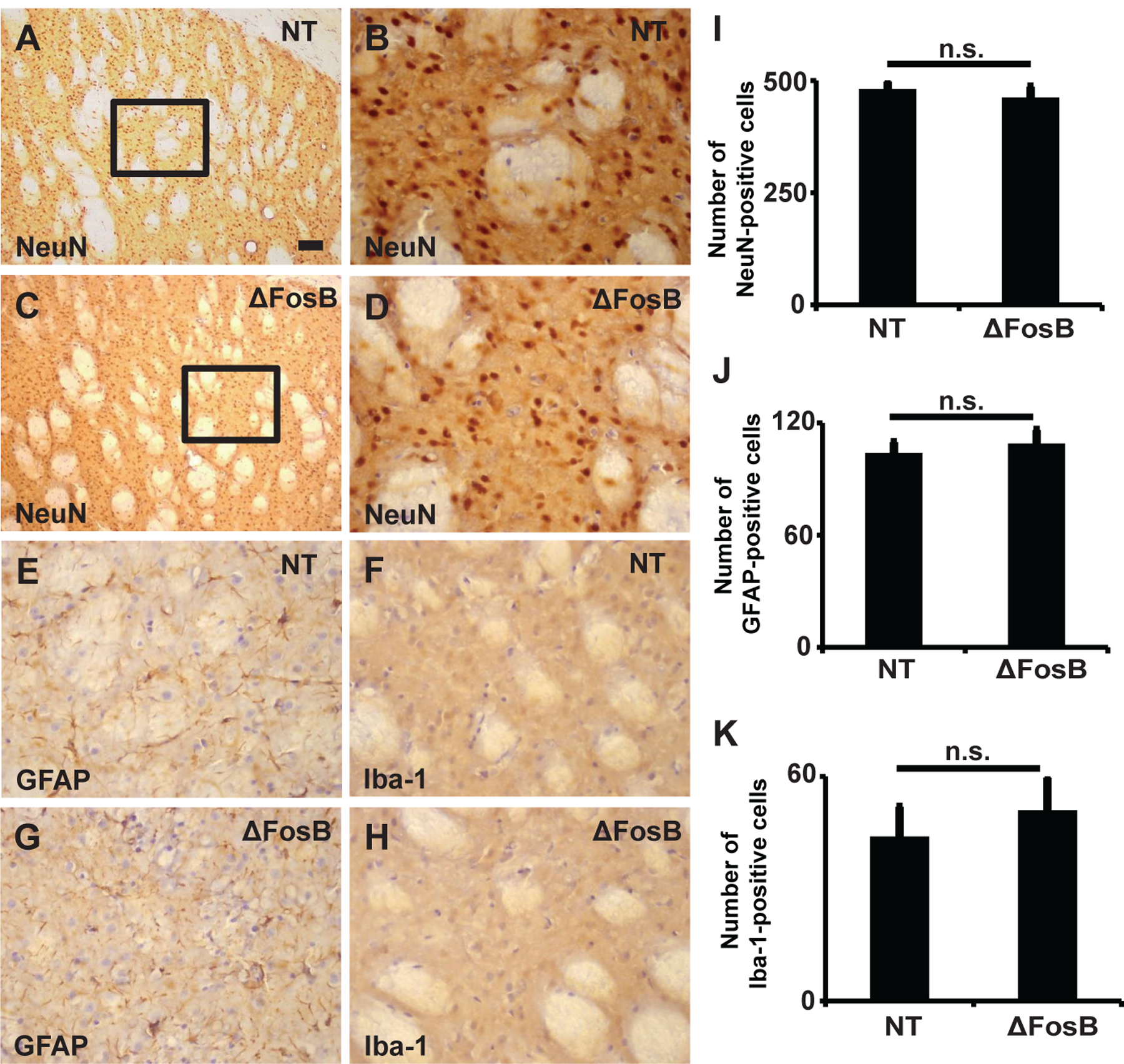
(A-D) NeuN (neuronal marker) staining in the striatum injected with rAAV-NT shRNA (A, B) and rAAV-ΔFosB shRNA (C, D). The inlets in A and C are magnified in B and D, respectively. NeuN stained cells as typical in the striatum and showed no loss of striatal neurons in either group. (E-H) Immunostaining for GFAP (a marker of astrocyte, E and G) or Iba-1 (a marker of microglia, F and H), showed neither active astrocytes nor microglial infiltration in the striatum after injection of targeting (ΔFosB) or control NT virus indicating absence of inflammatory reactions. Scale bar in (A) represents 100 μm (A and C) or 40 μm (B and D-H). (I-K) Cell counts of NeuN- (I), GFAP- (J), and Iba-1-positive cells (K) in the striatum of animals injected with rAAV-NT shRNA (n = 3) and rAAV- ΔFosB shRNA (n = 3). n.s. = not significant..
Discussion
Striatal ΔFosB gene KD in hemiparkinsonian rats effectively inhibited the development of AIMs following chronic L-Dopa treatment. The effect of gene KD was limited to AIMs since the rotational response to L-Dopa was not affected. In addition, ΔFosB gene KD had no impact on motor deficits caused by dopamine cell loss, or on responsiveness to L-Dopa. Behavioral data were obtained before (baseline) and at the end of the evaluation period (day 19 of chronic L-Dopa treatment), which corresponds to the peak of virus transduction, i.e. between 4 and 7 weeks after injection. Therefore, these data indicate that ΔFosB gene KD does not impact parkinsonian motor deficits or their reversal by dopamine replacement, thereby underscoring its specificity to reduce LID. The viral vector carrying ΔFosB shRNA did not cause unwanted behavioral changes or other health changes in the animals. There were no striatal histological changes between animal groups suggesting that rAAV-ΔFosB shRNA did not cause cellular damage, although further studies including cell counts in the absence of viral vector injection are needed to completely rule out such effect. The effective and safe viral transduction and shRNA-mediated gene suppression in the striatum resulted in antidyskinetic effect in rodents that was not only robust (74% reduction on day 18) but also sustainable (maintained beyond two weeks of tests). Although further studies including primate tests are necessary to validate ΔFosB gene therapy for LID, a critical milestone has been reached in this study. The present data show efficacy to block LID development with high specificity by a method of ΔFosB gene KD that has clinical applicability. Furthermore, the impact of ΔFosB gene KD on LID remained stable until the end of the L-Dopa treatment period, and that rules out the potential development of tolerance, a common concern with such strategies to control complex behaviors. ΔFosB gene KD impeded L-Dopa induction of severe LID at any time point after the initial days of treatment (after day 4) suggesting that it could also reduce the severity of established LID. Nevertheless, tests in animals that have already developed severe LID are necessary to determine such effect. These results thus provide the first evidence that targeting ΔFosB expression with a viral vector harboring ΔFosB shRNA could be a useful therapeutic approach for LID in patients with PD.
A primary role of striatal ΔFosB in LID mechanisms has been supported by multiple studies (29), including the latest transgenic overexpression experiment in primates naïve of L-Dopa treatment showing behavioral changes with physiological correlates (14). The present gene KD data showing reduction of LID appearance from the start to the end of L-Dopa treatment have now confirmed that ΔFosB plays such a mechanistic role in LID development in rats. Previous studies have shown antidyskinetic effects using continuous intrastriatal infusion of FosB/ΔFosB antisense (7) or striatal ΔJunD overexpression, which acts as a dominant negative inhibitor of ΔFosB (17). In those models, LID was also reduced without changes in the antiparkinsonian action of L-Dopa. Collectively, these data provide evidence for a ΔFosB role that is specific to LID mechanisms in the rodent. The fact that mild LID was still observed after ΔFosB gene KD could be related to insufficient striatal coverage by the planned virus injections, which were limited to the middle of the posterior striatal region. It is possible that a more extensive gene silencing be necessary to totally eliminate LID in animals with high sensitivity after complete dopamine denervation. Another plausible explanation is that other molecular pathways contributing to LID mechanisms could be responsible for the observed residual AIMs. Further studies of gene KD with multiple targets would be necessary to address the mechanistic role of other molecular pathways. In addition, we suppressed striatal ΔFosB gene expression without neuronal subtype specificity, and thus, we cannot make assumptions on the role of the direct and indirect pathways in the outcome of this gene therapy. A significant amount of data including our previous work support that both striatal pathways participate in LID mechanisms (15, 30, 31). In line with this idea, the present results of gene therapy may be interpreted as the effects of ΔFosB gene KD in both direct and indirect striatal projection neurons.
Gene silencing of striatal ΔFosB also impacted the expression levels of other known LID markers. In particular, there was a significant decrease of the key regulator of striatal neuronal signaling DARPP-32, and even more markedly of its activated form by phosphorylation at Thr34, which was strongly correlated with AIMs scores. Consistent with these results, our studies of striatal ΔFosB overexpression in primates showed significant increases of p-Th34-DARPP-32 (14). Thus, ΔFosB could be a major regulator of the expression level and activation of DARPP-32 (32). Another key molecule is ERK, which is activated primarily following acute L-Dopa administration during priming (4). Striatal p-ERK was slightly increased by ΔFosB gene KD, which could be interpreted as a counteracting (compensatory) mechanism. However, its relationship to LID mechanisms is uncertain because p-ERK changes after ΔFosB gene KD did not correlate with AIMs scores. Also important, our measurements of protein levels at the end of chronic treatment, precisely 24 hs after the last L-Dopa administration, could not reflect all molecular changes. This may be the case of p-ERK that is more tightly dependent on the stimulus, and its upregulation develops during priming but may not be sustained during prolonged L-Dopa administration (33). Congruent with these data, p-ERK did not change after chronic treatment in our previous study of ΔFosB overexpression with marked LID development (14). Therefore, data from ΔFosB gene manipulation do not support a clear link between p-ERK and ΔFosB-mediated mechanisms of LID. Surprisingly, striatal Cdk5, which is a direct target of the transcriptional activity of ΔFosB (34) and has been linked to LID development (35–37), was increased by the ΔFosB gene KD with LID reduction. ΔFosB can act as a positive or negative transcriptional activator for multiple genes (13), and thus we can hypothesize that ΔFosB gene KD impacts the gene expression of other Cdk5 regulators leading to its augmented expression. Yet, Cdk5 levels inversely correlated with AIMs scores, suggesting that the Cdk5 upregulation induced by chronic exposure to L-Dopa may not be a major contributor to LID mechanisms. Altogether, the molecular changes following ΔFosB gene transfer and silencing support the association between ΔFosB and DARPP-32 mechanisms in LID development.
In summary, this study of ΔFosB gene KD in the striatum demonstrated the causative role of this transcription factor in the generation of LID in the rodent. These results are in agreement with previous experimental evidence suggesting such a role in rodent and primate models. ΔFosB is thus posited as a therapeutic target to ameliorate LID in patients with PD. Dyskinesias remain poorly treated with the available pharmacological approaches targeting a variety of mechanisms of unknown specificity. Currently, the drug approved for LID treatment, amantadine, has a diverse binding profile, provides only partial LID improvement in some patients, and its use is largely limited by unwanted effects in many patients (38–40). Furthermore, LID is still a major indication for DBS surgery, which carries its risks and long-term maintenance issues. Here, a strategy for striatal ΔFosB gene suppression based on viral vector delivery of shRNA has shown significant inhibition of LID development with long-lasting effects and free of toxicity. There was no decline of effect over a period of eighteen days of L-Dopa treatment, indicating no development of tolerance to limit the efficacy of ΔFosB gene KD. Brain histology showed no neuronal damage or signs of inflammation induced by ΔFosB shRNA, an important issue with increased mature antisense RNA levels (26, 27) and some types of shRNAs injected into the striatum (26, 28). Therefore, the RNAi used in the present study to reduce striatal ΔFosB levels has proven effective and safe in rodents. These initial rodent data support further development of this gene KD strategy as a therapeutic approach to control dyskinesias in patients with PD.
Acknowledgment
This work was supported by NIH grants NS073994, NS045962, NS110416, RR000165 and OD011132 (S.M.P.); NS073994 (M.M.M.); and Grant-in-Aid for Scientific Research (C) (20K06910) from Japan Society for the Promotion of Science (G.B.). Additionally, M.M.M. is the William Dow Lovett Professor of Neurology and is supported by the Michael J. Fox Foundation for Parkinson’s Research, the American Parkinson Disease Association, the New Jersey Health Foundation, and by NIH grants NS116921, NS101134, and NS096032.
Footnotes
Potential Conflicts of Interest
M.M.M. is a founder of MentiNova, Inc.
References
- 1.Calabresi P, Di Filippo M, Ghiglieri V, Tambasco N, Picconi B. Levodopa-induced dyskinesias in patients with Parkinson’s disease: filling the bench-to-bedside gap. Lancet Neurol 2010;9:1106–17. [DOI] [PubMed] [Google Scholar]
- 2.Lewitt PA, Mouradian MM. Predicting the development of levodopa-induced dyskinesias: a presynaptic mechanism? Neurology 2014;82:1574–5. [DOI] [PubMed] [Google Scholar]
- 3.Beck G, Singh A, Papa SM. Dysregulation of striatal projection neurons in Parkinson’s disease. J Neural Transm 2018;125:449–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Santini E, Valjent E, Usiello A, Carta M, Borgkvist A, Girault JA, et al. Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia. J Neurosci 2007;27:6995–7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Santini E, Alcacer C, Cacciatore S, Heiman M, Herve D, Greengard P, et al. L-DOPA activates ERK signaling and phosphorylates histone H3 in the striatonigral medium spiny neurons of hemiparkinsonian mice. J Neurochem 2009;108:621–33. [DOI] [PubMed] [Google Scholar]
- 6.Nicholas AP, Lubin FD, Hallett PJ, Vattem P, Ravenscroft P, Bezard E, et al. Striatal histone modifications in models of levodopa-induced dyskinesia. J Neurochem 2008;106:486–94. [DOI] [PubMed] [Google Scholar]
- 7.Andersson M, Hilbertson A, Cenci MA. Striatal fosB expression is causally linked with l-DOPA-induced abnormal involuntary movements and the associated upregulation of striatal prodynorphin mRNA in a rat model of Parkinson’s disease. Neurobiol Dis 1999;6:461–74. [DOI] [PubMed] [Google Scholar]
- 8.Picconi B, Centonze D, Hakansson K, Bernardi G, Greengard P, Fisone G, et al. Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat Neurosci 2003;6:501–6. [DOI] [PubMed] [Google Scholar]
- 9.Valastro B, Andersson M, Lindgren HS, Cenci MA. Expression pattern of JunD after acute or chronic L-DOPA treatment: comparison with deltaFosB. Neuroscience 2007;144:198–207. [DOI] [PubMed] [Google Scholar]
- 10.Cao X, Yasuda T, Uthayathas S, Watts RL, Mouradian MM, Mochizuki H, et al. Striatal overexpression of DeltaFosB reproduces chronic levodopa-induced involuntary movements. J Neurosci 2010;30:7335–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tekumalla PK, Calon F, Rahman Z, Birdi S, Rajput AH, Hornykiewicz O, et al. Elevated levels of DeltaFosB and RGS9 in striatum in Parkinson’s disease. Biol Psychiatry 2001;50:813–6. [DOI] [PubMed] [Google Scholar]
- 12.Renthal W, Carle TL, Maze I, Covington HE 3rd, Truong HT, Alibhai I, et al. Delta FosB mediates epigenetic desensitization of the c-fos gene after chronic amphetamine exposure. J Neurosci 2008;28:7344–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nestler EJ. FosB: a transcriptional regulator of stress and antidepressant responses. Eur J Pharmacol 2015;753:66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beck G, Singh A, Zhang J, Potts LF, Woo JM, Park ES, et al. Role of striatal DeltaFosB in l-Dopa-induced dyskinesias of parkinsonian nonhuman primates. Proc Natl Acad Sci U S A 2019;116:18664–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cenci MA, Jörntell H, Petersson P. On the neuronal circuitry mediating L-DOPA-induced dyskinesia. J Neural Transm 2018;125:1157–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jenner P Molecular mechanisms of L-DOPA-induced dyskinesia. Nat Rev Neurosci 2008;9:665–77. [DOI] [PubMed] [Google Scholar]
- 17.Berton O, Guigoni C, Li Q, Bioulac BH, Aubert I, Gross CE, et al. Striatal overexpression of DeltaJunD resets L-DOPA-induced dyskinesia in a primate model of Parkinson disease. Biol Psychiatry 2009;66:554–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sellnow RC, Steece-Collier K, Altwal F, Sandoval IM, Kordower JH, Collier TJ, et al. Striatal Nurr1 Facilitates the Dyskinetic State and Exacerbates Levodopa-Induced Dyskinesia in a Rat Model of Parkinson’s Disease. J Neurosci 2020;40:3675–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steece-Collier K, Stancati JA, Collier NJ, Sandoval IM, Mercado NM, Sortwell CE, et al. Genetic silencing of striatal CaV1.3 prevents and ameliorates levodopa dyskinesia. Mov Disord 2019;34:697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nadjar A, Gerfen CR, Bezard E. Priming for l-dopa-induced dyskinesia in Parkinson’s disease: a feature inherent to the treatment or the disease? Prog Neurobiol 2009;87:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yen J, Wisdom RM, Tratner I, Verma IM. An alternative spliced form of FosB is a negative regulator of transcriptional activation and transformation by Fos proteins. Proc Natl Acad Sci U S A 1991;88:5077–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carlsson T, Carta M, Winkler C, Bjorklund A, Kirik D. Serotonin neuron transplants exacerbate L-DOPA-induced dyskinesias in a rat model of Parkinson’s disease. J Neurosci 2007;27:8011–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olsson M, Nikkhah G, Bentlage C, Bjorklund A. Forelimb akinesia in the rat Parkinson model: differential effects of dopamine agonists and nigral transplants as assessed by a new stepping test. J Neurosci 1995;15:3863–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee CS, Cenci MA, Schulzer M, Bjorklund A. Embryonic ventral mesencephalic grafts improve levodopa-induced dyskinesia in a rat model of Parkinson’s disease. Brain 2000;123:1365–79. [DOI] [PubMed] [Google Scholar]
- 25.Beck G, Shinzawa K, Hayakawa H, Baba K, Sumi-Akamaru H, Tsujimoto Y, et al. Progressive Axonal Degeneration of Nigrostriatal Dopaminergic Neurons in Calcium-Independent Phospholipase A2beta Knockout Mice. PloS One. 2016;11:e0153789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McBride JL, Boudreau RL, Harper SQ, Staber PD, Monteys AM, Martins I, et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc Natl Acad Sci U S A 2008;105:5868–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin JN, Wolken N, Brown T, Dauer WT, Ehrlich ME, Gonzalez-Alegre P. Lethal toxicity caused by expression of shRNA in the mouse striatum: implications for therapeutic design. Gene Ther 2011;18:666–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keiser MS, Monteys AM, Corbau R, Gonzalez-Alegre P, Davidson BL. RNAi prevents and reverses phenotypes induced by mutant human ataxin-1. Ann Neurol 2016;80:754–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engeln M, Bastide MF, Toulme E, Dehay B, Bourdenx M, Doudnikoff E, et al. Selective Inactivation of Striatal FosB/DeltaFosB-Expressing Neurons Alleviates L-DOPA-Induced Dyskinesia. Biol Psychiatry 2016;79:354–61. [DOI] [PubMed] [Google Scholar]
- 30.Liang L, DeLong MR, Papa SM. Inversion of dopamine responses in striatal medium spiny neurons and involuntary movements. J Neurosci 2008;28:7537–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh A, Jenkins MA, Burke KJ Jr., Beck G, Jenkins A, Scimemi A, et al. Glutamatergic Tuning of Hyperactive Striatal Projection Neurons Controls the Motor Response to Dopamine Replacement in Parkinsonian Primates. Cell Rep 2018;22:941–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Teegarden SL, Nestler EJ, Bale TL. Delta FosB-mediated alterations in dopamine signaling are normalized by a palatable high-fat diet. Biol Psychiatry 2008;64:941–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Santini E, Sgambato-Faure V, Li Q, Savasta M, Dovero S, Fisone G, et al. Distinct changes in cAMP and extracellular signal-regulated protein kinase signalling in L-DOPA-induced dyskinesia. PloS One. 2010;5:e12322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen J, Zhang Y, Kelz MB, Steffen C, Ang ES, Zeng L, et al. Induction of cyclin-dependent kinase 5 in the hippocampus by chronic electroconvulsive seizures: role of [Delta]FosB. J Neurosci 2000;20:8965–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aubert I, Guigoni C, Hakansson K, Li Q, Dovero S, Barthe N, et al. Increased D1 dopamine receptor signaling in levodopa-induced dyskinesia. Ann Neurol 2005;57:17–26. [DOI] [PubMed] [Google Scholar]
- 36.Kuan WL, Lin R, Tyers P, Barker RA. The importance of A9 dopaminergic neurons in mediating the functional benefits of fetal ventral mesencephalon transplants and levodopa-induced dyskinesias. Neurobiol Dis 2007;25:594–608. [DOI] [PubMed] [Google Scholar]
- 37.Potts LF, Park ES, Woo JM, Dyavar Shetty BL, Singh A, Braithwaite SP, et al. Dual kappa-agonist/mu-antagonist opioid receptor modulation reduces levodopa-induced dyskinesia and corrects dysregulated striatal changes in the nonhuman primate model of Parkinson disease. Ann Neurol 2015;77:930–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Verhagen Metman L, Del Dotto P, van den Munckhof P, Fang J, Mouradian MM, Chase TN. Amantadine as treatment for dyskinesias and motor fluctuations in Parkinson’s disease. Neurology 1998;50:1323–6. [DOI] [PubMed] [Google Scholar]
- 39.Snow BJ, Macdonald L, McAuley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000;23:82–5. [DOI] [PubMed] [Google Scholar]
- 40.Oertel W, Eggert K, Pahwa R, Tanner CM, Hauser RA, Trenkwalder C, et al. Randomized, placebo-controlled trial of ADS-5102 (amantadine) extended-release capsules for levodopa-induced dyskinesia in Parkinson’s disease (EASE LID 3). Mov Disord 2017;32:1701–9. [DOI] [PMC free article] [PubMed] [Google Scholar]