

Abstract
BACKGROUND
Helicobacter pylori (H. pylori), a bacterium that infects approximately half of the world’s population, is associated with various gastrointestinal diseases, including peptic ulcers, non-ulcer dyspepsia, gastric adenocarcinoma, and gastric lymphoma. As the burden of antibiotic resistance increases, the need for new adjunct therapies designed to facilitate H. pylori eradication and reduce negative distal outcomes associated with infection has become more pressing. Characterization of the interactions between H. pylori, the fecal microbiome, and fecal fatty acid metabolism, as well as the mechanisms underlying these interactions, may offer new therapeutic approaches.
AIM
To characterize the gut microbiome and metabolome in H. pylori patients in a socioeconomically challenged and underprivileged inner-city community.
METHODS
Stool samples from 19 H. pylori patients and 16 control subjects were analyzed. 16S rRNA gene sequencing was performed on normalized pooled amplicons using the Illumina MiSeq System using a MiSeq reagent kit v2. Alpha and beta diversity analyses were performed in QIIME 2. Non-targeted fatty acid analysis of the samples was carried out using gas chromatography-mass spectrometry, which measures the total content of 30 fatty acids in stool after conversion into their corresponding fatty acid methyl esters. Multi-dimensional scaling (MDS) was performed on Bray-Curtis distance matrices created from both the metabolomics and microbiome datasets and a Procrustes test was performed on the metabolomics and microbiome MDS coordinates.
RESULTS
Fecal microbiome analysis showed that alpha diversity was lowest in H. pylori patients over 40 years of age compared to control subjects of similar age group. Beta diversity analysis of the samples revealed significant differences in microbial community structure between H. pylori patients and control subjects across all ages. Thirty-eight and six taxa had lower and higher relative abundance in H. pylori patients, respectively. Taxa that were enriched in H. pylori patients included Atopobium, Gemellaceae, Micrococcaceae, Gemellales and Rothia (R. mucilaginosa). Notably, relative abundance of the phylum Verrucomicrobia was decreased in H. pylori patients compared to control subjects. Procrustes analysis showed a significant relationship between the microbiome and metabolome datasets. Stool samples from H. pylori patients showed increases in several fatty acids including the polyunsaturated fatty acids (PUFAs) 22:4n6, 22:5n3, 20:3n6 and 22:2n6, while decreases were noted in other fatty acids including the PUFA 18:3n6. The pattern of changes in fatty acid concentration correlated to the Bacteroidetes:Firmicutes ratio determined by 16S rRNA gene analysis.
CONCLUSION
This exploratory study demonstrates H. pylori-associated changes to the fecal microbiome and fecal fatty acid metabolism. Such changes may have implications for improving eradication rates and minimizing associated negative distal outcomes.
Keywords: Gut microbiome, Metabolome, Helicobacter pylori, Antibiotic resistance, Dysbiosis, Eradication
Core Tip: Helicobacter pylori (H. pylori) infects half of the world’s population and is associated with various diseases, including malignancy. Research of microbiome and metabolomic changes associated with H. pylori may hold therapeutic potential. We sought to characterize the fecal microbiome and fatty acid metabolism among H. pylori patients in our community. We observed differences in alpha and beta diversity among H. pylori patients compared to controls, particularly for those over 40 years old. Changes in several fecal fatty acids, including those associated with anti-inflammatory activity, were observed. Our findings may have implications for improving H. pylori eradication and minimizing associated negative distal outcomes.
INTRODUCTION
Helicobacter pylori (H. pylori), a bacterium found in the stomach of roughly half of the world’s population, is associated with various gastrointestinal diseases, including peptic ulcers, non-ulcer dyspepsia, gastric adenocarcinoma, and gastric lymphoma. Although many infections are asymptomatic, eradication of H. pylori has been shown to reduce the incidence of gastric cancer and is thus universally recommended[1]. For decades, clarithromycin-based triple therapy was considered the first-line therapy for H. pylori eradication and associated gastric cancer prevention. Following the global trend in emerging antibiotic resistance, rates of clarithromycin, metronidazole, and levofloxacin resistance among H. pylori strains have increased markedly, causing an alarmingly high rate of eradication failure[2]. In the absence of universally available antibiotic susceptibility testing, current guidelines advocate for the use of empiric quadruple therapy strategies; however, the efficacy of these regimens may be limited by dual resistance, local availability, and patient adherence[3].
As the burden of antibiotic resistance increases, the need for new adjunct therapies designed to facilitate H. pylori eradication and reduce negative distal outcomes associated with infection has become more pressing. The relationship between H. pylori and the microbiome has garnered particular attention of late[4]. With the advent of high throughput 16S rRNA gene sequencing methods and open-source software for subsequent analysis and visualization, researchers have been able to characterize changes in microbial diversity and community structure, as well as specific taxa that are differentially abundant in association with particular phenotypes[5]. Many studies have focused on H. pylori-mediated changes to the gastric microbiome, in which data have demonstrated variable effects on multiple metrics of microbial diversity and community structure[4]. H. pylori-associated gastric dysbiosis is particularly significant in the context of studies which suggest increased relative abundance of particular taxa, including Lactobacillus and Lachnospiraceae, among patients who develop premalignant lesions and gastric cancer[6,7]. H. pylori-associated gastric dysbiosis has also prompted researchers to evaluate the adjunct therapeutic role of probiotics, which may increase the rate of H. pylori eradication and limit negative side effects of antibiotic treatment[8-11].
More recently, researchers have also focused on the impact of H. pylori infection on distal gut, i.e., fecal microbiota. Despite H. pylori’s role as an almost exclusively gastric pathogen, animal and clinical studies have demonstrated alterations in distal gut microbiota associated with H. pylori infection[12-15]. The mechanisms by which gastric H. pylori infection may influence the distal gut microbiome are unclear, although authors have speculated that multiple factors, including alterations in hydrochloric acid secretion, gastrointestinal hormones, and immune regulation, may be involved[12-14]. As stated above, microbiome modulation with probiotics may have a role in improving eradication rates and minimizing adverse effects of antibiotics. Additionally, H. pylori-mediated changes to the distal gut microbiome may be significant in the context of research that implicates gut dysbiosis in the development of colorectal cancer (CRC)[16]. Indeed, authors of a large case-control study found that seropositivity for the H. pylori virulence factor VacA was associated with increased odds of CRC[17]. The well-documented associations between gut dysbiosis, type 2 diabetes, and metabolic syndrome has also prompted speculation about the role of H. pylori-associated gut dysbiosis in diabetes, which has been associated with H. pylori infection[18-21]. Further characterization of the interactions between H. pylori and the gut microbiome, as well as the mechanisms underlying these interactions, may lead to a better understanding of the role of microbiome modulation in H. pylori therapy, as well as the development of novel therapies and screening tools aimed at the negative distal outcomes associated with H. pylori infection, including CRC and metabolic diseases.
Given the association between H. pylori infection and gastrointestinal inflammation, attention has also been paid to compounds with anti-inflammatory activity, including fatty acids. Polyunsaturated fatty acid (PUFA) supplementation in particular may have therapeutic potential for H. pylori treatment via multiple mechanisms, including suppression of inflammatory cytokines and enzymes and direct bacteriocidal effects via disruption of cell membranes[22-24]. 10-hydroxy-cis-12-octadecenoic acid (10-HOE; a linoleic acid derivative) and docosahexaenoic acid (DHA) have both been shown to inhibit in vitro growth of H. pylori and gastric infection in mouse models[25,26]. Nevertheless, the effects of H. pylori infection on fatty acid metabolism in the gastrointestinal tract are not completely understood. Furthermore, the extent to which such metabolomic changes may relate to alterations in the gastrointestinal microbiota are unknown. Notably, short-chain fatty acids (SCFAs) produced by gut microbiota are thought to play a role in intestinal homeostasis, regulation of immune function, and dysbiosis in the setting of inflammatory bowel disease, which is associated with decreased levels of these important molecules[27,28]. Exploration of changes in fatty acid production and metabolism associated with H. pylori-mediated alterations of the fecal microbiome is warranted, as it may reveal strategies to inhibit the cascade of events leading to neoplasia.
Previously, we studied the pattern of H. pylori prevalence and antibiotic resistance in our socioeconomically challenged and underprivileged inner-city community[29]. As a next step, we then sought to characterize the gut microbiome associated with H. pylori infection in our patients. We further explored interactions among H. pylori infection, the gut microbiota, and the gut metabolome by comparing stool fatty acid profiles between subjects with and without H. pylori infection. Our overarching goal is to identify potential targets for intervention that could improve H. pylori eradication rates and limit associated negative distal outcomes.
MATERIALS AND METHODS
Patients and sample collection
Patients presenting to the Cooper Digestive Health Institute in Camden, New Jersey were recruited for our study. H. pylori patients at least 18 years of age and diagnosed via biopsy between June 1, 2017 and December 31, 2019, were eligible for inclusion. Patients less than 18 years old, pregnant women, patients taking antibiotics at the time of recruitment or sampling, and non-English speaking patients were excluded from participation. H. pylori-positive patients are identified in our electronic medical record system (EPIC) by ICD-9 codes of 041.86, 531.90, 535.60, 795.79, 008.47, 531.00, V12.08. Histological examination of the biopsied samples showed positive outcome indicating H. pylori infection. Control participants were recruited from a healthy population, also seen at Cooper Health Institute for well visits. The control subjects were screened for lack of previous history and current documentation for H. pylori infection as well as lack of any GI-related symptoms before recruitment. Their stools were tested by reverse transcription-polymerase chain reaction (PCR) based biprobe assay for presence of H. pylori DNA[30]. None of the stool samples from the control subjects showed presence of H. pylori DNA. Informed consent was obtained by one of several Institutional Review Board (IRB)-approved team members. Surveys about demographic information and relevant medical history were collected. Information on diagnosis, disease features, medication, history of antibiotic use, and treatment regimen were collected from the electronic medical record. Patients were asked to provide a stool sample before initiating eradication therapy, and samples were collected and stored in Stool Nucleic Acid Collection and Preservation Tubes (Cat. No. 45630, 45660, Norgen Biotek Corp., Thorold, Ontario, Canada) prior to freezing at –80 ℃ as described previously[29].
Processing of samples and 16S rRNA gene sequencing
Total genomic bacterial DNA extraction was performed from frozen fecal samples using the Qiagen DNeasy PowerSoil HTP Kit (Cat. No. 12955-4, Qiagen, Valencia, CA, United States). Adequate DNA yield was confirmed using NanoDrop spectrophotometry. For high throughput sequencing, 515F–806R Golay barcodes were used to target the hypervariable V4 region of the 16S rRNA gene, which is highly conserved and ideal for gut microbiome analysis. Marker genes in isolated DNA were PCR-amplified using GoTaq Master Mix (Cat. No. M5133, Promega, Madison, WI, United States) as described in Caporaso et al[31]. PCR products were cleaned and normalized using the SequalPrepTM Normalization Plate Kit (Cat. No. A1051001, ThermoFisher, Waltham, MA, United States) following the manufacturer’s instructions. Purification of PCR product amplicons was performed with the QIAquick PCR Purification Kit (Cat. No. 28104, Qiagen). Quantification of the PCR amplified library was performed with the InvitrogenTM Quant-iTTM PicoGreenTM dsDNA Assay Kit (Cat. No. P7589, ThermoFisher). 16S rRNA gene sequencing was performed on normalized pooled amplicons using the Illumina MiSeq System using a MiSeq reagent kit v2 (300 cycles; Cat. No. MS-102-2002, Illumina Inc.). FASTQ files for reads 1 (forward), 2 (reverse), and the index (barcode) read were generated using the BCL-to-FASTQ file converter bcl2fastq (ver. 2.17.1.14, Illumina, Inc.). Sequencing of the samples was conducted at the University of Colorado Boulder BioFrontiers Next-Gen Sequencing core facility.
Sequencing data analysis
Analysis was performed using Python-based packages (Python 3.6.12), QIIME 2 2019.4 (denoising and taxonomy assignment), Quantitative Insights Into Microbial Ecology (QIIME) 2 2020.11 (statistical testing), and Linear Discriminant Analysis (LDA) using LDA Effect Size (LEfSe)[32,33]. Sequences were de-multiplexed, then filtered and clustered into sub-operational taxonomic units (sOTUs) using QIIME 2 DADA2[34]. A masked phylogenetic tree was created using MAFFT via QIIME2[35,36]. Taxonomy was assigned using a naïve-Bayes classifier based on the latest Greengenes 16S rRNA gene database available as of August 2019 via the QIIME 2 interface (gg_13_8). Additional Python packages (SciPy.stats, Scikit-bio) were used for statistical tests on QIIME 2-generated data. Inter-class comparisons were made based on attributes of our population including: H. pylori status, gender, age group, race, education, and alcohol use. Alpha and beta diversity analyses were performed in QIIME 2, rarefied to an even sampling depth of 22400[37]. Alpha diversity was assessed by Faith’s phylogenetic diversity, chao1 index, and observed OTUs. Differences in alpha diversity between groups were determined by Kruskal-Wallis non-parametric rank test for categorical variables or by Fisher Z transformation on Spearman rho values for differences between alpha diversity correlations with numeric data across groups. Bacterial composition (beta diversity) was analyzed by principal coordinate analyses (PCoA) of both unweighted and weighted unique fraction metric (UniFrac) phylogenetic distance matrices[38-41]. Beta diversity was assessed for statistical significance between groups using Monte Carlo permutations [Adonis and permutational multivariate analysis of variance (PERMANOVA)], and dispersion of group samples was assessed using permutational analysis of multivariate dispersions (PERMDISP)[42,43]. Comparisons of relative abundances of microbial taxa between groups were performed using LEfSe with the Segata et al[44] online interface. Taxa with an LDA value of 2.0 or greater and a two-tailed P value ≤ 0.05 with Kruskal-Wallis and pairwise Wilcoxon analyses were considered significantly enriched. The code is available at https://github.com/sterrettJD/H-pylori-microbiome-analysis.
Non-targeted fatty acid analysis
Collected fecal samples were stored at –80 ℃ until analysis. Sample preparation and analysis were performed using gas chromatography-mass spectrometry (GC-MS) by Metabolon, Inc. (Durham, NC, United States). This method (Metabolon TAM112-002) measures the total content of 30 fatty acids in stool samples after conversion into their corresponding fatty acid methyl esters (FAMEs). The measured concentrations are provided in weight corrected μg/g of sample. NQ (not quantified-below the lower limit of quantification) values were treated as a concentration of 0.001 mg/mL.
Briefly, stool samples were homogenized and the suspension was weighed out in a 100 mg aliquot in a test tube. Liquid/Liquid extraction was performed to extract fatty acids and remove the nucleic acid preservative. A 250 μL aliquot of each extract was transferred to a clean analysis tube. The solvent was removed by evaporation under a stream of nitrogen. Internal standard solution was added to the dried sample extracts, quality controls (QCs), and calibration standards. The solvent was again removed by evaporation under a stream of nitrogen. The dried samples and QCs were subjected to methylation/transmethylation with methanol/sulfuric acid, resulting in the formation of the corresponding methyl esters (FAME) of free fatty acids and conjugated fatty acids. The reaction mixture was neutralized and extracted with hexanes. An aliquot of the hexanes layer was injected onto a 7890A/5975C GC-MS system (Agilent Technologies, Santa Clara, CA, United States). Mass spectrometric analysis was performed in the single ion monitoring positive mode with electron ionization. Quantitation was performed using both linear and quadratic regression analysis generated from fortified calibration standards prepared immediately prior to each run. Raw data were collected and processed using Agilent MassHunter GC-MS Acquisition B.07.04.2260 and Agilent MassHunter Workstation Software Quantitative Analysis for GC-MS B.09.00/ Build 9.0.647.0. Data reduction was performed using Microsoft Office 365 ProPlus Excel. Volcano plot data are included as Supplementary Tables 1-3 (Supplementary Table 1: Volcano plot data with all Helicobacter data set; Supplementary Table 2: Volcano plot data with High Bacteroidetes; Supplementary Table 3: Volcano plot data with High Firmicutes).
Procrustes analysis
Multi-dimensional scaling (MDS, using the Python package Scikit-learn) was performed on Bray-Curtis distance matrices created from both the fecal metabolome and fecal microbiome datasets (using the Python package SciPy.distance), and a Procrustes test was performed on the metabolomics and microbiome MDS coordinates. As described by Peres-Neto and Jackson, a Procrustes randomization test (PROTEST) was performed by randomly sampling the MDS coordinates and performing a Procrustes test on the reordered coordinates 10000 times, and the P value was calculated based on the portion of randomized Procrustes tests with resulting m2 (Gower’s statistic) scores lower than the Procrustes m2 score of the observed datasets[45].
Ethics approval
This study received approval by the Cooper Health System IRB (17-077EX) and all the steps were carried out as per the standards set by the IRB.
RESULTS
Fecal microbiome analysis
Selected demographic attributes of our participants are presented in Table 1. Fecal microbiome analysis of 35 stool samples collected from 19 H. pylori patients and 16 healthy control subjects was carried out. The mean number of reads per sample was 269462 with a minimum of 22483 reads/sample. Rarefaction was set to 22400 for even sampling based on this minimum. We analyzed the association of each of the factors included in Table 1 with alpha and beta diversity (via Kruskal-Wallis and PERMANOVA for nominal variables or via Spearman correlation and Mantel test for numerical variables, respectively). Age was the only factor that showed statically significant differences (P value of ≤ 0.05) in alpha diversity in the H. pylori patients compared to the control participants, with H. pylori patients over 40 years of age having lower richness than control participants over 40 years of age. Significant differences in community composition and group dispersion were seen between H. pylori patients and control participants.
Table 1.
Patient demographics and clinical characteristics
|
|
Total (n = 35)
|
H. pylori
patients (n = 19)
|
Control (n = 16)
|
| Sex, n (%) | |||
| Male | 19 (54) | 13 (68) | 6 (38) |
| Female | 16 (46) | 6 (32) | 10 (62) |
| Age, n (%) | |||
| 18-40 | 15 (43) | 3 (16) | 12 (75) |
| > 40 | 20 (57) | 16 (84) | 4 (25) |
| Highest education, n (%) | |||
| Less than high school | 2 (6) | 2 (11) | 0 (0) |
| High school | 12 (34) | 11 (58) | 1 (6) |
| Undergraduate | 4 (11) | 2 (11) | 2 (13) |
| Graduate | 15 (43) | 3 (16) | 12 (75) |
| Post-graduate | 1 (3) | 0 (0) | 1 (6) |
| Race, n (%) | |||
| African American | 8 (23) | 6 (32) | 2 (12) |
| Asian American | 2 (6) | 0 (0) | 2 (12) |
| Caucasian | 13 (37) | 1 (5) | 12 (75) |
| Non-White Hispanic | 8 (23) | 8 (42) | 0 (0) |
| Other | 3 (9) | 3 (16) | 0 (0) |
| Smoker | 5 (14) | 4 (21) | 1 (6) |
| Past H. pylori treatment | 1 (3) | 1 (5) | 0 (0) |
| Duodenal ulcer | 1 (3) | 1 (5) | 0 (0) |
| Non-ulcer dyspepsia | 14 (40) | 10 (53) | 4 (25) |
H. pylori: Helicobacter pylori.
Information was collected about each participant regarding (1) If they had been treated for H. pylori infection in the past; (2) If so, the date infection was diagnosed; (3) Treatments given; and (4) Outcomes: Eradication testing with dates. Only one patient had H. pylori infection 10 years ago and had successfully completed treatment at that time. History about antibiotic treatments for any infection and proton pump inhibitors (PPIs) use for both H. pylori patients and control subjects in the past five years was also documented and these data values were included in the statistical analyses. Our statistical analyses showed that the differences seen in H. pylori patients were not due to the previous treatments.
Analysis of alpha diversity
Chao1 index alpha diversity between H. pylori patients and control subjects and regression analysis of observed OTUs by age are shown in Figure 1. Alpha diversity, which estimates the diversity of a microbial community within a sample, incorporates the number of different OTUs and their respective abundance in each sample. Alpha diversity was lowest in H. pylori patients over 40 years of age, who had significantly lower Chao1 scores compared to control subjects over 40 years of age (Kruskal-Wallis H = 6.036, P = 0.014). H. pylori patients over 40 years old also had lower Chao1 scores approaching statistical significance when compared to H. pylori patients under 40 (Kruskal-Wallis H = 2.813, P = 0.094). Additionally, the correlation of age with observed OTUs (Figure 1B), Faith’s phylogenetic diversity, and Chao1 was consistently different between H. pylori patients vs control subjects, as evidenced by a Fisher Z transformation on Spearman rank correlation rho values (observed OTUs: H. pylori rho = –0.21 vs control rho = 0.50, Fisher P = 0.041; Faith: H. pylori rho = –0.04 vs control rho = 0.57, Fisher P = 0.067; Chao1: H. pylori rho = –0.15 vs control rho = 0.44, Fisher P = 0.097).
Figure 1.

Chao1 index alpha diversity and observed operational taxonomic units by Helicobacter pylori status stratified by age. A: Chao1 index alpha diversity is compared between Helicobacter pylori (H. pylori) patients (blue) and control subjects (orange), subgrouped by age. H. pylori patients over 40 years old (n = 16) had significantly lower Chao1 scores compared to control subjects over 40 (n = 4) (Kruskal-Wallis H = 6.036, P = 0.014). Boxes represent 1st and 3rd quartiles, and central lines represent median values. Whiskers represent non-outlier high and low values. Points show outliers, which were determined by having a distance from the 1st or 3rd quartile greater than 1.5 times the interquartile range; B: Observed operational taxonomic units (operational taxonomic units) are compared between H. pylori patients (blue, n = 19) and control subjects (orange, n = 16), as a function of age. Shading around the regression lines indicates 95% confidence intervals. OTU: Operational taxonomic units; H. pylori: Helicobacter pylori.
Analysis of beta diversity
Next we analyzed beta diversity of our samples, which estimates how samples differ from each other. An unweighted UniFrac PCoA plot of H. pylori patients vs control subjects is shown in Figure 2. Adonis testing revealed that current H. pylori infection represented 7% of the variation in unweighted UniFrac beta diversity (F = 2.523, R2 = 0.071, P = 0.002). Additionally, unweighted UniFrac PERMANOVAs identified significant differences between H. pylori patients and control subjects (F = 2.523, P = 0.001). Furthermore, unweighted UniFrac PERMDISP did not reveal differences in dispersion among H. pylori patients vs control subjects (F = 0.0506, P = 0.809), but weighted UniFrac PERMDISP did (F = 6.82955, P = 0.017). We also explored the effect of the use of PPIs via beta diversity analysis as PPIs are the medications commonly given to H. pylori patients and we had previously noted their influence on the antibiotic resistance of H. pylori[29]. Unweighted UniFrac PERMANOVA identified differences between PPI users and non-users across all participants (F = 1.939, P = 0.012), though the magnitude of the effect was smaller than that seen for H. pylori infection as described above. It should also be noted that only four H. pylori patients and one control subject were on PPI at the time of study and sample collection, so its contribution to the results described here may not be significant.
Figure 2.

Beta diversity analysis by unweighted UniFrac principal coordinate analysis. Unweighted principal coordinate analysis (PCoA) plot is shown comparing Helicobacter pylori (H. pylori) patients (blue, n = 19) to control subjects (red, n = 16). Significant differences between H. pylori patients and control subjects were identified by unweighted UniFrac PERMANOVA (F = 2.523, P = 0.001). The proportion of variance explained by each principal coordinate axis is denoted in the corresponding axis label; PCo1 explains 16.95% of the variability and PCo2 explains 9.13% of the variability. H. pylori: Helicobacter pylori.
LDA effect size
LEfSe scores for taxa that were differentially distributed across H. pylori patients vs control subjects are shown in Figure 3. Thirty-eight taxa had lower relative abundance in H. pylori patients relative to healthy control subjects, and six taxa had higher relative abundance in H. pylori patients relative to healthy control subjects. Taxa that were enriched in H. pylori patients include Atopobium, Gemellaceae, Micrococcaceae, Gemellales and Rothia (R. mucilaginosa). Figure 4 shows the arcsin square root transformed relative abundances of each phylum, grouped by H. pylori status. Notably, relative abundance of the phylum Verrucomicrobia, and its main constituent, Akkermansia muciniphila, was decreased in H. pylori patients compared to control subjects.
Figure 3.

Linear discriminant analysis effect size scores for taxa enriched in Helicobacter pylori or control participants. Linear discriminant analysis effect size (LEfSe) scores for taxa that were differentially distributed across Helicobacter pylori (H. pylori) patients (n = 19) vs control subjects (n = 16) via Kruskal-Wallis and Wilcoxon tests with two-tailed α = 0.05. Negative values represent taxa that were enriched in H. pylori patients, whereas positive values represent taxa that were enriched in control subjects. Error bars represent a 95% confidence interval and are only present if the listed taxonomic identity was the lowest level classified for multiple operational taxonomic units highlighted by LEfSe. Color represents the phylum. H. pylori: Helicobacter pylori.
Figure 4.

Transformed relative abundances of phyla grouped by Helicobacter pylori infection. Arcsin square root transformed relative abundances of each phylum, grouped by Helicobacter pylori (H. pylori) status. Bacterial relative abundances of H. pylori patients (n = 19) are shown in blue, whereas healthy controls (n = 16) are shown in orange. Boxes represent the 1st and 3rd quartiles, and vertical lines in the middle of the boxes represent median values. Whiskers represent the lowest and highest non-outlier values. Points show outliers, which were determined by having a distance from the 1st or 3rd quartile greater than 1.5 times the interquartile range. Of note, relative abundances of Verrucomicrobia were significantly lower in H. pylori patients compared to healthy controls (Kruskal-Wallis H = 4.455, P = 0.034). H. pylori: Helicobacter pylori.
Procrustes analysis
The Procrustes randomization test was carried out to investigate the relationship between the fecal microbiome and fecal metabolome. Procrustes analysis identified a significant relationship between the two datasets, as the resulting P value of 0.017 indicates that the correspondence of each participant’s fecal microbiome to their fecal metabolome was better than 98% of randomly sampled simulations (Figure 5).
Figure 5.

Procrustes-transformed Bray-Curtis multi-dimensional scaling of microbiome and metabolome. Connections between the fecal microbiome and fecal metabolome Bray-Curtis multi-dimensional scaling plots. The microbiome data shown here have been transformed by the Procrustes analysis to minimize disparity between the two datasets, and lines have been drawn to indicate microbiome and metabolome from the same participants. Orange dots represent the microbiomes, whereas blue triangles represent the metabolomes. The Procrustes randomization test showed a significant relationship between the datasets (m2 = 0.86, P = 0.017). MDS: Multi-dimensional scaling.
Fecal fatty acid analysis
To evaluate the impact of H. pylori infection on fatty acid composition, we performed non-targeted metabolomics analysis of a panel of 30 fatty acids including long chain fatty acids (LCFAs), monounsaturated fatty acids (MUFAs) and PUFAs. We prepared a volcano plot evaluating changes in fatty acid profile for the entire population of H. pylori patient samples relative to healthy control subjects (Figure 6A). In addition, we prepared separate volcano plots for subgroups of the H. pylori patients with high Bacteroidetes and high Firmicutes based on 16S rRNA gene analysis (Figure 6B and C). See Supplementary Tables 1-3 for the full volcano plot data. Volcano plots allow for the plotting of P values vs fatty acid fold change from control samples for all evaluated fatty acids.
Figure 6.

Volcano plots of composition of evaluated fatty acids. A: Control vs entire Helicobacter pylori (H. pylori) patient group; B: Control vs sub-group of H. pylori patients identified as having high ratio of Bacteroidetes to Firmicutes (n = 7 of 19 patients); C: Control vs sub-group of H. pylori patients identified as having high ratio of Firmicutes to Bacteroidetes (n = 4 of 19 patients). Selected long-chain fatty acids (green), monounsaturated fatty acids (blue) and polyunsaturated fatty acids (red) are highlighted. H. pylori: Helicobacter pylori.
Stool samples from H. pylori patients showed increases in several fatty acids including the PUFAs 22:4n6, 22:5n3, 20:3n6 and 22:2n6 while decreases were noted in fatty acids including the PUFA 18:3n6, relative to healthy control subjects. The pattern of changes in fatty acid concentration was correlated to the Bacteroidetes:Firmicutes ratio (determined by 16S rRNA gene analysis). Among the sub-population of H. pylori patients identified to have high Bacteroidetes (Bacteroidetes:Firmicutes ratio ≥ 1 standard deviation above the ratio in control participants, n = 7) we found several fatty acids that were diminished relative to control participants including the LCFAs 24:0, 22:0 and 15:0 along with MUFAs 20:1n9 and 18:1n9. By contrast, in the sub-population of H. pylori patients identified to have high Firmicutes (Firmicutes:Bacteroidetes ratio ≥ 1 standard deviation above ratios in control participants; n = 4) a distinct set of fatty acids were found to be significantly increased and decreased relative to control participants. In this subpopulation of H. pylori patients we observed increases in the PUFA 22:2n6 and decreases in PUFAs 18:3n6 and 20:4n3 relative to control participants.
DISCUSSION
As little is known about the microbiome of H. pylori patients in an underprivileged community, this study is useful due to its exploratory nature. A sample size calculation for microbiome analysis was not thus performed prior to collecting samples. Casals-Pascual et al[46] outline that in human microbiome studies knowledge of the baseline microbiome diversity and composition, along with knowledge of the magnitude of changes that confer clinically relevant outcomes are important for determining sample size. However, this could not easily be predicted in our sample demographic due to lack of previous research. Though the effects of H. pylori and low socioeconomic status (SES) on the gut microbiome have been investigated separately in previous studies, the two states (H. pylori infection and low SES) could potentially have conflicting relationships with diversity and composition, making it difficult to determine the baseline microbiome diversity and composition without large assumptions. Thus, much of this study provides the baseline to characterize the microbiome of H. pylori patients in an underserved population, which was previously understudied to a degree by which sample size calculations would have required a large number of assumptions. Though the power is likely weak in this study, especially for post-hoc analyses such as alpha diversity in H. pylori patients over 40 years of age vs control participants over 40 years of age, multiple lines of evidence, including both the Kruskal-Wallis test with age group and the Spearman correlation with actual age support the relationship in our sample. These results can provide a basis for future study design and power calculations.
Our sample size is modest in part due to the limitations posed by the socioeconomic and cultural attributes of our patient population and logistic challenges associated with sample collection. Camden County has the lowest median income and highest poverty and unemployment rates in southern New Jersey. It also has the lowest educational attainment and the greatest socioeconomic disparities among ethnic minority groups. More than one third of the population in Camden City, where our hospital is located, lives below the national poverty line, and the median household income is USD $26105[47]. Our local community is thus considered one of the poorest and most economically distressed communities in the United States. A significant fraction of adults over 25 years old (almost one quarter) have not completed high school. It is possible that this influences patients’ appreciation of the relevance of the study and willingness to participate. A significant portion of potential participants were excluded due to non-English-speaking status, thus being unable to partake in the informed consent process. Logistic challenges imposed by the process of stool sample collection also limited the number of subjects. A large number of potential participants were unable to return to the clinic for stool sample delivery due to poor transportation access or inability to leave employment or domestic responsibilities during the window for sample collection (after diagnosis but prior to initiation of antibiotics). Nonetheless, the present study yielded meaningful results.
H. pylori infection was associated with significantly decreased fecal microbial diversity relative to over the age of 40 years. Among all participants, correlations between age and multiple metrics of microbial alpha diversity were consistently different between H. pylori patients and control subjects. H. pylori was also shown to account for a significant portion of the variation in beta diversity between infected patients and control subjects. Collectively, these results may suggest an H. pylori-mediated alteration of the fecal microbiome that progresses with age. While other authors have attributed decreased diversity of gastric microbiota to the ability of H. pylori to outcompete other species, secretion of antibacterial peptides, and local pH alterations, the mechanisms by which H. pylori affect distal gut microbiota remain unclear[4,48-50]. Although our study was not designed to uncover these mechanisms, multiple factors hypothesized by other authors, including alterations in hydrochloric acid secretion, gastrointestinal hormones, and immune regulation, may be involved in the mediation of distal gut dysbiosis[12-14]. The impact of H. pylori on fecal microbial diversity observed in our study may be reversible, as a recent study showed that H. pylori eradication could restore gastric microbial diversity to levels seen in uninfected controls[51].
Less likely is the possibility that the data reflect a preceding decrease in gut microbial diversity that facilitates H. pylori infection, potentially mediated by other factors or exposures that accumulate with age. In other words, patients who experience decreased gut microbial diversity as they age, potentially mediated by diet or chronic inflammation, may be more prone to H. pylori infection, particularly by mid-adulthood. In our previous study of 2014 H. pylori patients, we observed that 80% of our patients were above the age of 40 years, consistent with studies showing increased prevalence of H. pylori infection with age in both developing and developed countries[29,52]. Antibiotic exposure is known to alter the gut microbiome, and increased rates of antibiotic-resistant H. pylori among older adults have been attributed to increased antibiotic use compared to younger patients[53,54]. Conceivably, decreased gut microbial diversity induced by cumulative antibiotic exposure could create an intragastric niche for H. pylori, and antibiotic-resistant H. pylori strains in particular, to thrive. However, H. pylori infection is thought to be acquired in infancy and carried through to adulthood, and this observation would not be consistent with preceding microbiome alteration facilitating de novo H. pylori infection in adulthood[55].
LEfSe showed that multiple taxa were differentially enriched or depleted in stool from H. pylori patients compared to controls. The significance of differential expression of these taxa is unknown; however, certain taxa stand out. The enrichment of Atopobium in H. pylori patients is notable in the context of a metagenomic analysis that revealed increased Atopobium parvulum abundance among patients with colorectal intramucosal carcinomas[56]. Although H. pylori is predominantly associated with gastric cancers, a recent meta-analysis suggested a significantly increased risk of CRC as well, and our data suggest that H. pylori-associated increases in Atopobium abundance may have a role in the distal gut carcinogenesis[57].
Over-representation of Rothia mucilaginosa, a commensal oral microbe, has previously been observed in fecal samples from patients with primary sclerosing cholangitis (PSC)[58]. Because of the sensitivity of R. mucilaginosa to gastric fluid, Bajer et al[58] hypothesized that its presence in PSC patients represented contamination of the intestine via previous endoscopic retrograde cholangiopancreatography. The enrichment of Rothia sp. in fecal samples from our H. pylori patients may reflect the potent gastric acid suppression by H. pylori, allowing translocation of swallowed oral flora to the distal gut. The clinical consequences of the colonization of the distal gut by Rothia sp. are unknown, although the bacteria has been implicated as a cause of myriad syndromes, including dental caries, pneumonia, and bacteremia, particularly in immunocompromised patients[59]. The increased abundance of Gemellaceae in H. pylori patients is consistent with the observation of increased organisms from the Gemella genus in patients with current H. pylori infection reported by Gao et al[60]. Increased relative abundance of Gemellaceae in the Crohn’s ileum has been reported by other groups[61,62].
Interestingly, the phylum Verrucomicrobia was significantly depleted in H. pylori patients. Verrucomicrobia includes Akkermansia muciniphila, a mucus-residing commensal bacterium of the large intestine. A. muciniphila is an obligate chemoorganotroph, utilizing mucus as a sole carbon, nitrogen, and energy source that produces SCFAs including acetate, propionate, and, to a smaller extent, 1,2-propanediol[63,64]. Our study was not designed to study this aspect in detail, but the decreased relative abundance of Verrucomicrobia may suggest disruption of the gut mucosal environment by H. pylori. In a mouse stress model, exposure to chronic psychosocial stress resulted in expansion of Helicobacter spp., which has been demonstrated to proliferate in response to glucocorticoid administration as well as psychosocial stress exposure[65]. Specifically, stress-induced increases in Helicobacter spp. were associated with increases in other Proteobacteria spp., including unidentified genera belonging to the Enterobacteraceae and Desulfovibrionaceae families. Concurrently, relative abundance of Mucispirillum, an obligate mucus-residing bacterium, decreased; a decline of Mucispirillum in rodents is associated with early disruption of the gastrointestinal surface mucus layer and a prolonged delay to recovery after the period of pathogen clearance[66]. Potential disruption of the gut mucosa by H. pylori infection is also consistent with decreased relative abundance of Faecalibacterium prausnitzii. F. prausnitzii is an important gram-positive human commensal that produces butyrate and other SCFA through the fermentation of dietary fiber. Reduced relative abundance of F. prausnitzii has been associated with inflammatory conditions including Crohn’s disease, obesity, asthma, and stress-related psychiatric disorders in which inflammation is a risk factor, such as major depressive disorder[67-70].
Strategies to promote increased gastric microbial diversity among patients with H. pylori infection, particularly those over 40 years of age, may hold therapeutic potential in eradicating infection, and some clinical trials with adjunctive probiotic formulas have shown promising results in this regard[71]. Likewise, treatment of gut dysbiosis in patients with H. pylori infection may limit its associated pathologies. Reduced fecal microbiome diversity is associated with multiple disease markers including increased adiposity, insulin resistance, dyslipidemia, and a pro-inflammatory state[72]. H. pylori infection is associated with higher rates of diabetes, specifically, and addressing gut dysbiosis in such patients could conceivably aid primary dietary and pharmacologic strategies to achieve glycemic control and insulin sensitivity[20]. Rehabilitation of the gut microbiota in patients with H. pylori may also have a role in minimizing the risk of malignancy. Mouse studies suggest that the carcinogenic effects of H. pylori partly depend on interactions with the gut microbiome, and persistent infection with H. pylori may create a niche favorable for taxa that are found in increased abundance in gastric cancer, including Lactobacillus and Lachnospiraceae[73,74]. It has been suggested that lactic acid bacteria can influence gastric cancer by a number of mechanisms such as (1) Supply of exogenous lactate, that acts as a fuel source for cancer cells; (2) Production of reactive oxygen species and N-nitroso compounds; and (3) By allowing colonization of carcinogenic non-H. pylori bacteria[75]. Accordingly, reversal of abnormalities in gut microbiota associated with H. pylori may also limit its carcinogenic potential. Finally, consideration must be given to the effects of H. pylori treatment on the gut microbiome, as data suggest that current therapies may be associated with dysbiosis and subsequent adverse effects[76]. Several factors including lifestyle and diet can act as confounders in analysis of the diversity and composition of the gut microbiome. However, given the challenges associated with controlling for such variables, their presence should not prohibit drawing meaningful conclusions from significant observations of gut dysbiosis in association with H. pylori.
While evaluating changes in the LCFAs palmitic (16:0) and stearic (18:0) concentrations, we found that palmitic acid concentrations were increased in our test samples compared to control samples [625.23 ± 598.82 mg/g (n = 18) vs 334.10 ± 284.58 mg/g (n = 12)], whereas concentrations of stearic acid were comparable between populations. Interestingly, when we evaluated the high Firmicutes (> 1.4:1 Firmicutes:Bacteroidetes, n = 4) sub-group compared to the high Bacteroidetes (> 2.7:1 Bacteroidetes:Firmicutes, n = 7) sub-group of our test population, we found the concentrations of both these LCFAs to be diminished in the high Bacteroidetes population while the concentrations of both were enhanced in the high Firmicutes sub-population. This is consistent with previous studies in mice where administration of LCFA-rich diets resulted in an observed increase in Firmicutes to Bacteroidetes ratio[77]. Ktsoyan et al[78] implicated a characteristic profile of serum LCFA concentrations in association with H. pylori infection compared to healthy controls. It is plausible that diet can modulate the impact of H. pylori on the gut microbiome, which in turn may relate to different disease phenotypes, i.e., asymptomatic infection, peptic ulcer disease, and gastric cancer.
The accumulation of the saturated LCFAs is regulated under normal physiological conditions by desaturation of 16:0 to 16:1n7, elongation of 16:0 to 18:0 and/or desaturation of 18:0 to 18:1n9 which can be further elongated to 20:1n9 (Figure 7). While only small changes in concentrations of these MUFAs were noted between our H. pylori patients and control subjects, the changes in MUFA concentrations in our high Firmicutes and high Bacteroidetes sub-populations showed more significant and consistent changes across the series of MUFAs. The concentration of 16:1n7, 18:1n9 and 20:1n9 were decreased in the high Bacteroidetes sub-group, whereas these MUFAs were slightly increased in the high Firmicutes sub-group. Interestingly, this contrasts published data on the benefits of high MUFA diets in animal models and humans. In previous studies, high MUFA diets have been reported to lead to increased ratios of Bacteroidetes to Firmicutes and were correlated with reductions in several disease indicators[77]. Together, these observations may suggest that either the patients in our high Bacteroidetes sub-population have severely limited MUFA production/intake, contrasting previous work on MUFA rich diets or that the endogenous biosynthetic pathways for MUFA production are dysregulated in this patient population.
Figure 7.
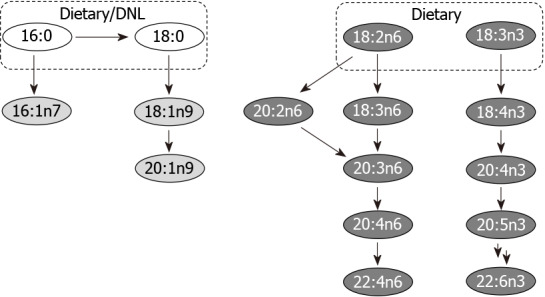
Major metabolic pathways. Selected long-chain fatty acids (no fill), monounsaturated fatty acids (light grey fill) and polyunsaturated fatty acids (dark grey fill) are highlighted. Although many of the fatty acids shown may be supplemented by diet, key parent members of these pathways acquired from de novo lipogenesis and/or diet are indicated. DNL: De novo lipogenesis.
The range of PUFAs observed endogenously have essential physiological roles and are produced from dietary alpha-linolenic acid (18:3n3) and linoleic acid (18:2n6). We observed that both of these dietary PUFAs were increased in our test samples compared to the control samples. In humans, C18:3n3 is converted through multiple steps to eicosapentaenoic acid (20:5n3) and DHA (22:6n3), whereas 18:2n6 proceeds to produce arachidonic acid (AA, 20:4n6) (Figure 7). As these two biosynthetic pathways share enzymes for their respective dehydration and elongation steps, they are antagonistic to each other. In general, a high 18:3n3 (n-3 fatty acid) diet is correlated with positive physiological effects whereas a high 18:2n6 (n-6 fatty acid) diet is correlated with metabolic dysbiosis and negative physiological effects[79,80]. Accordingly a balanced dietary intake of n-6/n-3 fatty acids is recommended to be 1:1-2:1, although the average ratio in a typical Western diet has been reported to be > 15:1[81]. In accordance with published ratios for n-6/n-3 fatty acid intake in Western diets, we find the ratio of 18:2n6/18:3n3 to be 22:1 in our control samples and 15:1 in our H. pylori patient test samples.
Endogenously, the essential dietary fatty acid 18:3n3 undergoes multiple dehydration and elongation steps to produce docosapentaenoic acid (DPA, 22:5n3) and DHA (22:6n3). For all intermediates along this biosynthetic pathway, with exception to 18:3n3 and 22:5n3, we observe concentrations at or below the limit of quantification for the majority of samples making an evaluation of the individual steps of this biosynthesis pathway difficult.
Contrasting the low concentrations observed for 18:3n3 and fatty acids produced along this biosynthetic pathway, higher overall concentrations were observed for fatty acids derived from 18:2n6, perhaps reflecting the higher concentration of dietary n-6 fatty acids as described earlier. In humans, the dietary fatty acid, 18:2n6 undergoes dehydration to produce 18:3n6. This intermediate undergoes a subsequent elongation to produce 20:3n6, which is dehydrated to 20:4n6 (AA). Alternatively, 18:2n6 can undergo elongation first to produce 20:2n6, which is dehydrated to produce 20:3n6 (Figure 7).
We found that the early steps in the metabolism of 18:2n6 were significantly impacted in all of our H. pylori samples by comparison of downstream fatty acid metabolites to controls. Most notably, in all H. pylori test samples the concentration of 18:3n6 was significantly diminished. Concentration of 18:3n6 was 10.7 ± 18.2 mg/g (n = 12) in the control samples and 0.36 ± 1.50 mg/g (n = 18) in the test samples. While we would anticipate that this deficiency in dehydration of 18:2n6 would lead to lower levels of fatty acid metabolites downstream of 18:3n6, this is not the case. In fact, by the following biosynthetic step, i.e., elongation to produce 20:3n6, the fatty acid concentration of 20:3n6 in test samples was found to be higher than in controls. These higher concentrations were also reflected in arachidonic acid (20:4n6) and adrenic acid (22:4n6). It is plausible that the alternative d-8-desaturase mediated route via 20:2n6 may be prominent in these H. pylori patient samples, which are marked by a deficiency in d-6-desaturase mediated pathway via 18:3n6[82]. We observed a striking decrease in the concentration of the PUFA C18:3n6 in H. pylori infected patients. Interestingly, among the multiple liposomal fatty acids evaluated by Thamphiwatana et al[83], only the linolenic acid formulation (C18:3) showed bactericidal activity by impacting the H. pylori membrane structure and stability.
As the burden of antibiotic resistance grows, populations like ours are impacted greatly by the high prevalence of H. pylori colonization. As eradication by a one-size-fits-all approach becomes more and more difficult, it becomes even more important to look towards strategies that can provide insight into community attributes that can help guide targeted therapies, predict likelihood of treatment outcomes, and possibly create strategies towards mitigating the impact of increased antibiotic usage. A greater, more individualized understanding of gut microbiome features among patients experiencing H. pylori colonization, especially in our underserved population, will pave the way for better community impact and reduce healthcare burdens of repeated treatment and mounting antibiotic resistance. As gut dysbiosis is inextricably tied not only to H. pylori infection but also to the use of antibiotics itself, the cycle of treatment and re-treatment may very well only further exacerbate vulnerability to enteric pathogens. Our results add to the growing evidence suggesting that H. pylori is associated with alterations in distal gut microbiota, particularly in those over 40 years of age. Strategies to better understand gut dysbiosis features among affected groups may create opportunities for improving eradication rates and reducing negative associated distal outcomes.
CONCLUSION
This exploratory study demonstrates H. pylori-associated changes to the fecal microbiome and fecal fatty acid metabolism. Such changes may have implications for improving eradication rates and minimizing associated negative distal outcomes.
ARTICLE HIGHLIGHTS
Research background
Helicobacter pylori (H. pylori), a bacterium that infects approximately half of the world’s population, is associated with various gastrointestinal diseases, including peptic ulcers, non-ulcer dyspepsia, gastric adenocarcinoma, and gastric lymphoma. Following the global trend in emerging antibiotic resistance, rates of clarithromycin, metronidazole, and levofloxacin resistance among H. pylori strains have increased markedly, causing high rates of eradication failure.
Research motivation
As the burden of antibiotic resistance increases, the need for new adjunct therapies designed to facilitate H. pylori eradication and reduce negative distal outcomes associated with infection has become more pressing. Characterization of the interactions between H. pylori, the fecal microbiome, and fecal fatty acid metabolism, as well as the mechanisms underlying these interactions, may offer new therapeutic approaches.
Research objectives
The aim of this study is to characterize the fecal microbiome and metabolome in H. pylori patients in a socioeconomically challenged and underprivileged inner-city community.
Research methods
Stool samples from 19 H. pylori patients and 16 control subjects were analyzed. 16S rRNA gene sequencing was performed on normalized pooled amplicons using the Illumina MiSeq System using a MiSeq reagent kit v2. Alpha and beta diversity analyses were performed in QIIME 2. Non-targeted fatty acid analysis of the samples was carried out using gas chromatography-mass spectrometry, which measures the total content of 30 fatty acids in stool after conversion into their corresponding fatty acid methyl esters. Multi-dimensional scaling (MDS) was performed on Bray-Curtis distance matrices created from both the metabolomics and microbiome datasets and a Procrustes test was performed on the metabolomics and microbiome MDS coordinates.
Research results
Fecal microbiome analysis showed that alpha diversity was lowest in H. pylori patients over 40 years of age compared to control subjects of similar age group. Beta diversity analysis of the samples revealed significant differences in microbial community structure between H. pylori patients and control subjects across all ages. Thirty-eight and six taxa were identified by Linear Discriminant Analysis (LDA) using LDA Effect Size to be enriched in H. pylori patients vs control individuals, respectively. Taxa that were enriched in H. pylori patients included Atopobium, Gemellaceae, Micrococcaceae, Gemellales and Rothia (R. mucilaginosa). Notably, relative abundance of the phylum Verrucomicrobia was decreased in H. pylori patients compared to control subjects. Procrustes analysis showed a significant relationship between the microbiome and metabolome datasets. Stool samples from H. pylori patients showed increases in several fatty acids including the polyunsaturated fatty acids (PUFAs) 22:4n6, 22:5n3, 20:3n6 and 22:2n6, while decreases were noted in other fatty acids including the PUFA 18:3n6. The pattern of changes in fatty acid concentration correlated to the Bacteroidetes:Firmicutes ratio determined by 16S rRNA gene analysis.
Research conclusions
The study suggests H. pylori-associated changes to the fecal microbiome and fecal fatty acid metabolism. Such changes may have implications for improving eradication rates and minimizing associated negative distal outcomes.
Research perspectives
Future study with greater power should be directed to confirming the distal gut dysbiosis and changes to fecal fatty acid metabolism seen in our exploratory analysis of patients with H. pylori infection, as well as towards exploring the relationships between such phenomena and eradication success, colorectal cancer, and metabolic disease.
ACKNOWLEDGEMENTS
We thank Drs. Robert Cooper, Anjali Mone and Sanket Patel for their help in the collection of some of the samples for the study; these were also approved in the IRB protocol.
Footnotes
Institutional review board statement: This study received approval by the Cooper Health System Institutional Review Board (IRB) (17-077EX) and all the steps were carried out as per the standards set by the IRB.
Institutional animal care and use committee statement: There are no animals used in the study.
Conflict-of-interest statement: The authors report no conflicts of interest.
Manuscript source: Invited manuscript
Peer-review started: March 31, 2021
First decision: June 23, 2021
Article in press: August 13, 2021
Specialty type: Gastroenterology and hepatology
Country/Territory of origin: United States
Peer-review report’s scientific quality classification
Grade A (Excellent): A
Grade B (Very good): 0
Grade C (Good): C
Grade D (Fair): D, D
Grade E (Poor): 0
P-Reviewer: Krzyżek P, Neri M, Werawatganon D, Zhang J S-Editor: Fan JR L-Editor: A P-Editor: Yuan YY
Contributor Information
Brian White, Department of Biomedical Sciences, Cooper Medical School of Rowan University, Camden, NJ 08103, United States.
John D Sterrett, Department of Integrative Physiology, University of Colorado Boulder, Boulder, CO 80309, United States.
Zoya Grigoryan, Department of Internal Medicine, Lenox Hill Hospital, NYC, NY 10075, United States.
Lauren Lally, Department of Internal Medicine, Thomas Jefferson University Hospital, Philadelphia, PA 19107, United States.
Jared D Heinze, Department of Integrative Physiology, University of Colorado Boulder, Boulder, CO 80309, United States.
Hyder Alikhan, Department of Biological Sciences, Rowan University, Glassboro, NJ 08028, United States.
Christopher A Lowry, Department of Integrative Physiology, University of Colorado Boulder, Boulder, CO 80309, United States.
Lark J Perez, Department of Chemistry and Biochemistry, Rowan University, Glassboro, NJ 08028, United States.
Joshua DeSipio, Department of Gastroenterology, Cooper University Hospital, Camden, NJ 08103, United States.
Sangita Phadtare, Department of Biomedical Sciences, Cooper Medical School of Rowan University, Camden, NJ 08103, United States. phadtare@rowan.edu.
Data sharing statement
The code is available at https://github.com/sterrettJD/H-pylori-microbiome-analysis. Volcano plot data are included as Supplementary Tables 1-3 (Supplementary Table 1: Volcano plot data with all Helicobacter data set; Supplementary Table 2: Volcano plot data with High Bacteroidetes; Supplementary Table 3: Volcano plot data with High Firmicutes).
References
- 1.Lee YC, Chiang TH, Chou CK, Tu YK, Liao WC, Wu MS, Graham DY. Association Between Helicobacter pylori Eradication and Gastric Cancer Incidence: A Systematic Review and Meta-analysis. Gastroenterology. 2016;150:1113–1124.e5. doi: 10.1053/j.gastro.2016.01.028. [DOI] [PubMed] [Google Scholar]
- 2.Savoldi A, Carrara E, Graham DY, Conti M, Tacconelli E. Prevalence of Antibiotic Resistance in Helicobacter pylori: A Systematic Review and Meta-analysis in World Health Organization Regions. Gastroenterology. 2018;155:1372–1382.e17. doi: 10.1053/j.gastro.2018.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fallone CA, Moss SF, Malfertheiner P. Reconciliation of Recent Helicobacter pylori Treatment Guidelines in a Time of Increasing Resistance to Antibiotics. Gastroenterology. 2019;157:44–53. doi: 10.1053/j.gastro.2019.04.011. [DOI] [PubMed] [Google Scholar]
- 4.Sheh A, Fox JG. The role of the gastrointestinal microbiome in Helicobacter pylori pathogenesis. Gut Microbes. 2013;4:505–531. doi: 10.4161/gmic.26205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eun CS, Kim BK, Han DS, Kim SY, Kim KM, Choi BY, Song KS, Kim YS, Kim JF. Differences in gastric mucosal microbiota profiling in patients with chronic gastritis, intestinal metaplasia, and gastric cancer using pyrosequencing methods. Helicobacter. 2014;19:407–416. doi: 10.1111/hel.12145. [DOI] [PubMed] [Google Scholar]
- 7.Aviles-Jimenez F, Vazquez-Jimenez F, Medrano-Guzman R, Mantilla A, Torres J. Stomach microbiota composition varies between patients with non-atrophic gastritis and patients with intestinal type of gastric cancer. Sci Rep. 2014;4:4202. doi: 10.1038/srep04202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang B, Tang L, Huang C, Tian C, Chen L, He Z, Yang G, Zuo L, Zhao G, Liu E, Wang S, Lin H, He J, Yang S. The Effect of Probiotics Supplementation on Gut Microbiota After Helicobacter pylori Eradication: A Multicenter Randomized Controlled Trial. Infect Dis Ther. 2021;10:317–333. doi: 10.1007/s40121-020-00372-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ojetti V, Bruno G, Ainora ME, Gigante G, Rizzo G, Roccarina D, Gasbarrini A. Impact of Lactobacillus reuteri Supplementation on Anti-Helicobacter pylori Levofloxacin-Based Second-Line Therapy. Gastroenterol Res Pract. 2012;2012:740381. doi: 10.1155/2012/740381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Du YQ, Su T, Fan JG, Lu YX, Zheng P, Li XH, Guo CY, Xu P, Gong YF, Li ZS. Adjuvant probiotics improve the eradication effect of triple therapy for Helicobacter pylori infection. World J Gastroenterol. 2012;18:6302–6307. doi: 10.3748/wjg.v18.i43.6302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi X, Zhang J, Mo L, Shi J, Qin M, Huang X. Efficacy and safety of probiotics in eradicating Helicobacter pylori: A network meta-analysis. Medicine (Baltimore) 2019;98:e15180. doi: 10.1097/MD.0000000000015180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heimesaat MM, Fischer A, Plickert R, Wiedemann T, Loddenkemper C, Göbel UB, Bereswill S, Rieder G. Helicobacter pylori induced gastric immunopathology is associated with distinct microbiota changes in the large intestines of long-term infected Mongolian gerbils. PLoS One. 2014;9:e100362. doi: 10.1371/journal.pone.0100362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kienesberger S, Cox LM, Livanos A, Zhang XS, Chung J, Perez-Perez GI, Gorkiewicz G, Zechner EL, Blaser MJ. Gastric Helicobacter pylori Infection Affects Local and Distant Microbial Populations and Host Responses. Cell Rep. 2016;14:1395–1407. doi: 10.1016/j.celrep.2016.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frost F, Kacprowski T, Rühlemann M, Bang C, Franke A, Zimmermann K, Nauck M, Völker U, Völzke H, Biffar R, Schulz C, Mayerle J, Weiss FU, Homuth G, Lerch MM. Helicobacter pylori infection associates with fecal microbiota composition and diversity. Sci Rep. 2019;9:20100. doi: 10.1038/s41598-019-56631-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dash NR, Khoder G, Nada AM, Al Bataineh MT. Exploring the impact of Helicobacter pylori on gut microbiome composition. PLoS One. 2019;14:e0218274. doi: 10.1371/journal.pone.0218274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song M, Chan AT, Sun J. Influence of the Gut Microbiome, Diet, and Environment on Risk of Colorectal Cancer. Gastroenterology. 2020;158:322–340. doi: 10.1053/j.gastro.2019.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Butt J, Varga MG, Blot WJ, Teras L, Visvanathan K, Le Marchand L, Haiman C, Chen Y, Bao Y, Sesso HD, Wassertheil-Smoller S, Ho GYF, Tinker LE, Peek RM, Potter JD, Cover TL, Hendrix LH, Huang LC, Hyslop T, Um C, Grodstein F, Song M, Zeleniuch-Jacquotte A, Berndt S, Hildesheim A, Waterboer T, Pawlita M, Epplein M. Serologic Response to Helicobacter pylori Proteins Associated With Risk of Colorectal Cancer Among Diverse Populations in the United States. Gastroenterology. 2019;156:175–186.e2. doi: 10.1053/j.gastro.2018.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, Peng Y, Zhang D, Jie Z, Wu W, Qin Y, Xue W, Li J, Han L, Lu D, Wu P, Dai Y, Sun X, Li Z, Tang A, Zhong S, Li X, Chen W, Xu R, Wang M, Feng Q, Gong M, Yu J, Zhang Y, Zhang M, Hansen T, Sanchez G, Raes J, Falony G, Okuda S, Almeida M, LeChatelier E, Renault P, Pons N, Batto JM, Zhang Z, Chen H, Yang R, Zheng W, Yang H, Wang J, Ehrlich SD, Nielsen R, Pedersen O, Kristiansen K. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 19.Vrieze A, Van Nood E, Holleman F, Salojärvi J, Kootte RS, Bartelsman JF, Dallinga-Thie GM, Ackermans MT, Serlie MJ, Oozeer R, Derrien M, Druesne A, Van Hylckama Vlieg JE, Bloks VW, Groen AK, Heilig HG, Zoetendal EG, Stroes ES, de Vos WM, Hoekstra JB, Nieuwdorp M. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. 2012;143:913–6.e7. doi: 10.1053/j.gastro.2012.06.031. [DOI] [PubMed] [Google Scholar]
- 20.Jeon CY, Haan MN, Cheng C, Clayton ER, Mayeda ER, Miller JW, Aiello AE. Helicobacter pylori infection is associated with an increased rate of diabetes. Diabetes Care. 2012;35:520–525. doi: 10.2337/dc11-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang YJ, Sheu BS. Metabolic Interaction of Helicobacter pylori Infection and Gut Microbiota. Microorganisms. 2016;4 doi: 10.3390/microorganisms4010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thompson L, Cockayne A, Spiller RC. Inhibitory effect of polyunsaturated fatty acids on the growth of Helicobacter pylori: a possible explanation of the effect of diet on peptic ulceration. Gut. 1994;35:1557–1561. doi: 10.1136/gut.35.11.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Correia M, Michel V, Osório H, El Ghachi M, Bonis M, Boneca IG, De Reuse H, Matos AA, Lenormand P, Seruca R, Figueiredo C, Machado JC, Touati E. Crosstalk between Helicobacter pylori and gastric epithelial cells is impaired by docosahexaenoic acid. PLoS One. 2013;8:e60657. doi: 10.1371/journal.pone.0060657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bergsson G, Steingrímsson O, Thormar H. Bactericidal effects of fatty acids and monoglycerides on Helicobacter pylori. Int J Antimicrob Agents. 2002;20:258–262. doi: 10.1016/s0924-8579(02)00205-4. [DOI] [PubMed] [Google Scholar]
- 25.Correia M, Michel V, Matos AA, Carvalho P, Oliveira MJ, Ferreira RM, Dillies MA, Huerre M, Seruca R, Figueiredo C, Machado JC, Touati E. Docosahexaenoic acid inhibits Helicobacter pylori growth in vitro and mice gastric mucosa colonization. PLoS One. 2012;7:e35072. doi: 10.1371/journal.pone.0035072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsui H, Takahashi T, Murayama SY, Kawaguchi M, Matsuo K, Nakamura M. Protective efficacy of a hydroxy fatty acid against gastric Helicobacter infections. Helicobacter. 2017;22 doi: 10.1111/hel.12430. [DOI] [PubMed] [Google Scholar]
- 27.Parada Venegas D, De la Fuente MK, Landskron G, González MJ, Quera R, Dijkstra G, Harmsen HJM, Faber KN, Hermoso MA. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front Immunol. 2019;10:277. doi: 10.3389/fimmu.2019.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Corrêa-Oliveira R, Fachi JL, Vieira A, Sato FT, Vinolo MA. Regulation of immune cell function by short-chain fatty acids. Clin Transl Immunology. 2016;5:e73. doi: 10.1038/cti.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh H, Chung H, Awan I, Mone A, Cooper R, Patel S, Grigoryan Z, Ishida Y, Papachristou C, Judge T, DeSipio J, Phadtare S. Designing strategies for eradication of Helicobacter pylori based on prevalence patterns of infection and antibiotic resistance in a low-income, medically underserved community in the United States. Helicobacter. 2021;26:e12769. doi: 10.1111/hel.12769. [DOI] [PubMed] [Google Scholar]
- 30.Redondo JJ, Keller PM, Zbinden R, Wagner K. A novel RT-PCR for the detection of Helicobacter pylori and identification of clarithromycin resistance mediated by mutations in the 23S rRNA gene. Diagn Microbiol Infect Dis. 2018;90:1–6. doi: 10.1016/j.diagmicrobio.2017.09.014. [DOI] [PubMed] [Google Scholar]
- 31.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, Koester I, Kosciolek T, Kreps J, Langille MGI, Lee J, Ley R, Liu YX, Loftfield E, Lozupone C, Maher M, Marotz C, Martin BD, McDonald D, McIver LJ, Melnik AV, Metcalf JL, Morgan SC, Morton JT, Naimey AT, Navas-Molina JA, Nothias LF, Orchanian SB, Pearson T, Peoples SL, Petras D, Preuss ML, Pruesse E, Rasmussen LB, Rivers A, Robeson MS 2nd, Rosenthal P, Segata N, Shaffer M, Shiffer A, Sinha R, Song SJ, Spear JR, Swafford AD, Thompson LR, Torres PJ, Trinh P, Tripathi A, Turnbaugh PJ, Ul-Hasan S, van der Hooft JJJ, Vargas F, Vázquez-Baeza Y, Vogtmann E, von Hippel M, Walters W, Wan Y, Wang M, Warren J, Weber KC, Williamson CHD, Willis AD, Xu ZZ, Zaneveld JR, Zhang Y, Zhu Q, Knight R, Caporaso JG. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McDonald D, Clemente JC, Kuczynski J, Rideout JR, Stombaugh J, Wendel D, Wilke A, Huse S, Hufnagle J, Meyer F, Knight R, Caporaso JG. The Biological Observation Matrix (BIOM) format or: how I learned to stop worrying and love the ome-ome. Gigascience. 2012;1:7. doi: 10.1186/2047-217X-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Price MN, Dehal PS, Arkin AP. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weiss S, Xu ZZ, Peddada S, Amir A, Bittinger K, Gonzalez A, Lozupone C, Zaneveld JR, Vázquez-Baeza Y, Birmingham A, Hyde ER, Knight R. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome. 2017;5:27. doi: 10.1186/s40168-017-0237-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. UniFrac: an effective distance metric for microbial community comparison. ISME J. 2011;5:169–172. doi: 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vázquez-Baeza Y, Pirrung M, Gonzalez A, Knight R. EMPeror: a tool for visualizing high-throughput microbial community data. Gigascience. 2013;2:16. doi: 10.1186/2047-217X-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Legendre P, Legendre L. Numerical Ecology. 3rd ed. Elsevier, 2012. [Google Scholar]
- 42.Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecology . 2001;26:32–46. [Google Scholar]
- 43.Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Stevens MHH, Szoecs E, Wagner H. vegan: Community Ecology Package, 2018. [cited 10 March 2021]. Available from: https://CRAN.R-project.org/package=vegan .
- 44.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peres-Neto PR, Jackson DA. How well do multivariate data sets match? Oecologia. 2001;129:169–178. doi: 10.1007/s004420100720. [DOI] [PubMed] [Google Scholar]
- 46.Casals-Pascual C, González A, Vázquez-Baeza Y, Song SJ, Jiang L, Knight R. Microbial Diversity in Clinical Microbiome Studies: Sample Size and Statistical Power Considerations. Gastroenterology. 2020;158:1524–1528. doi: 10.1053/j.gastro.2019.11.305. [DOI] [PubMed] [Google Scholar]
- 47.Data USA: Camden City, New Jersey. QuickFacts: United States Census Bureau, 2019. [cited 10 March 2021]. Available from: https://gistbok.ucgis.org/bok-topics/united-states-census-data .
- 48.Noto JM, Peek RM Jr. The gastric microbiome, its interaction with Helicobacter pylori, and its potential role in the progression to stomach cancer. PLoS Pathog. 2017;13:e1006573. doi: 10.1371/journal.ppat.1006573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao Y, Gao X, Guo J, Yu D, Xiao Y, Wang H, Li Y. Helicobacter pylori infection alters gastric and tongue coating microbial communities. Helicobacter. 2019;24:e12567. doi: 10.1111/hel.12567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Das A, Pereira V, Saxena S, Ghosh TS, Anbumani D, Bag S, Das B, Nair GB, Abraham P, Mande SS. Gastric microbiome of Indian patients with Helicobacter pylori infection, and their interaction networks. Sci Rep. 2017;7:15438. doi: 10.1038/s41598-017-15510-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo Y, Zhang Y, Gerhard M, Gao JJ, Mejias-Luque R, Zhang L, Vieth M, Ma JL, Bajbouj M, Suchanek S, Liu WD, Ulm K, Quante M, Li ZX, Zhou T, Schmid R, Classen M, Li WQ, You WC, Pan KF. Effect of Helicobacter pylori on gastrointestinal microbiota: a population-based study in Linqu, a high-risk area of gastric cancer. Gut. 2020;69:1598–1607. doi: 10.1136/gutjnl-2019-319696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pounder RE, Ng D. The prevalence of Helicobacter pylori infection in different countries. Aliment Pharmacol Ther. 1995;9 Suppl 2:33–39. [PubMed] [Google Scholar]
- 53.Ji Z, Han F, Meng F, Tu M, Yang N, Zhang J. The Association of Age and Antibiotic Resistance of Helicobacter Pylori: A Study in Jiaxing City, Zhejiang Province, China. Medicine (Baltimore) 2016;95:e2831. doi: 10.1097/MD.0000000000002831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zullo A, Perna F, Hassan C, Ricci C, Saracino I, Morini S, Vaira D. Primary antibiotic resistance in Helicobacter pylori strains isolated in northern and central Italy. Aliment Pharmacol Ther. 2007;25:1429–1434. doi: 10.1111/j.1365-2036.2007.03331.x. [DOI] [PubMed] [Google Scholar]
- 55.Jones NL, Sherman PM. Helicobacter pylori infection in children. Curr Opin Pediatr. 1998;10:19–23. doi: 10.1097/00008480-199802000-00005. [DOI] [PubMed] [Google Scholar]
- 56.Yachida S, Mizutani S, Shiroma H, Shiba S, Nakajima T, Sakamoto T, Watanabe H, Masuda K, Nishimoto Y, Kubo M, Hosoda F, Rokutan H, Matsumoto M, Takamaru H, Yamada M, Matsuda T, Iwasaki M, Yamaji T, Yachida T, Soga T, Kurokawa K, Toyoda A, Ogura Y, Hayashi T, Hatakeyama M, Nakagama H, Saito Y, Fukuda S, Shibata T, Yamada T. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat Med. 2019;25:968–976. doi: 10.1038/s41591-019-0458-7. [DOI] [PubMed] [Google Scholar]
- 57.Yang F, Xu YL, Zhu RF. Helicobacter pylori infection and the risk of colorectal carcinoma: a systematic review and meta-analysis. Minerva Med. 2019;110:464–470. doi: 10.23736/S0026-4806.19.05942-1. [DOI] [PubMed] [Google Scholar]
- 58.Bajer L, Kverka M, Kostovcik M, Macinga P, Dvorak J, Stehlikova Z, Brezina J, Wohl P, Spicak J, Drastich P. Distinct gut microbiota profiles in patients with primary sclerosing cholangitis and ulcerative colitis. World J Gastroenterol. 2017;23:4548–4558. doi: 10.3748/wjg.v23.i25.4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramanan P, Barreto JN, Osmon DR, Tosh PK. Rothia bacteremia: a 10-year experience at Mayo Clinic, Rochester, Minnesota. J Clin Microbiol. 2014;52:3184–3189. doi: 10.1128/JCM.01270-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gao JJ, Zhang Y, Gerhard M, Mejias-Luque R, Zhang L, Vieth M, Ma JL, Bajbouj M, Suchanek S, Liu WD, Ulm K, Quante M, Li ZX, Zhou T, Schmid R, Classen M, Li WQ, You WC, Pan KF. Association Between Gut Microbiota and Helicobacter pylori-Related Gastric Lesions in a High-Risk Population of Gastric Cancer. Front Cell Infect Microbiol. 2018;8:202. doi: 10.3389/fcimb.2018.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song SJ, Yassour M, Morgan XC, Kostic AD, Luo C, González A, McDonald D, Haberman Y, Walters T, Baker S, Rosh J, Stephens M, Heyman M, Markowitz J, Baldassano R, Griffiths A, Sylvester F, Mack D, Kim S, Crandall W, Hyams J, Huttenhower C, Knight R, Xavier RJ. The treatment-naive microbiome in new-onset Crohn's disease. Cell Host Microbe. 2014;15:382–392. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haberman Y, Tickle TL, Dexheimer PJ, Kim MO, Tang D, Karns R, Baldassano RN, Noe JD, Rosh J, Markowitz J, Heyman MB, Griffiths AM, Crandall WV, Mack DR, Baker SS, Huttenhower C, Keljo DJ, Hyams JS, Kugathasan S, Walters TD, Aronow B, Xavier RJ, Gevers D, Denson LA. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J Clin Invest. 2014;124:3617–3633. doi: 10.1172/JCI75436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Derrien M, Vaughan EE, Plugge CM, de Vos WM. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int J Syst Evol Microbiol. 2004;54:1469–1476. doi: 10.1099/ijs.0.02873-0. [DOI] [PubMed] [Google Scholar]
- 64.Ottman N, Davids M, Suarez-Diez M, Boeren S, Schaap PJ, Martins Dos Santos VAP, Smidt H, Belzer C, de Vos WM. Genome-Scale Model and Omics Analysis of Metabolic Capacities of Akkermansia muciniphila Reveal a Preferential Mucin-Degrading Lifestyle. Appl Environ Microbiol. 2017;83 doi: 10.1128/AEM.01014-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hemmings SMJ, Malan-Müller S, van den Heuvel LL, Demmitt BA, Stanislawski MA, Smith DG, Bohr AD, Stamper CE, Hyde ER, Morton JT, Marotz CA, Siebler PH, Braspenning M, Van Criekinge W, Hoisington AJ, Brenner LA, Postolache TT, McQueen MB, Krauter KS, Knight R, Seedat S, Lowry CA. The Microbiome in Posttraumatic Stress Disorder and Trauma-Exposed Controls: An Exploratory Study. Psychosom Med. 2017;79:936–946. doi: 10.1097/PSY.0000000000000512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Belzer C, Gerber GK, Roeselers G, Delaney M, DuBois A, Liu Q, Belavusava V, Yeliseyev V, Houseman A, Onderdonk A, Cavanaugh C, Bry L. Dynamics of the microbiota in response to host infection. PLoS One. 2014;9:e95534. doi: 10.1371/journal.pone.0095534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, Grangette C, Vasquez N, Pochart P, Trugnan G, Thomas G, Blottière HM, Doré J, Marteau P, Seksik P, Langella P. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA. 2008;105:16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Newton RJ, McLellan SL, Dila DK, Vineis JH, Morrison HG, Eren AM, Sogin ML. Sewage reflects the microbiomes of human populations. mBio. 2015;6:e02574. doi: 10.1128/mBio.02574-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiang H, Ling Z, Zhang Y, Mao H, Ma Z, Yin Y, Wang W, Tang W, Tan Z, Shi J, Li L, Ruan B. Altered fecal microbiota composition in patients with major depressive disorder. Brain Behav Immun. 2015;48:186–194. doi: 10.1016/j.bbi.2015.03.016. [DOI] [PubMed] [Google Scholar]
- 70.Martín R, Miquel S, Benevides L, Bridonneau C, Robert V, Hudault S, Chain F, Berteau O, Azevedo V, Chatel JM, Sokol H, Bermúdez-Humarán LG, Thomas M, Langella P. Functional Characterization of Novel Faecalibacterium prausnitzii Strains Isolated from Healthy Volunteers: A Step Forward in the Use of F. prausnitzii as a Next-Generation Probiotic. Front Microbiol. 2017;8:1226. doi: 10.3389/fmicb.2017.01226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ji J, Yang H. Using Probiotics as Supplementation for Helicobacter pylori Antibiotic Therapy. Int J Mol Sci. 2020;21 doi: 10.3390/ijms21031136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto JM, Kennedy S, Leonard P, Li J, Burgdorf K, Grarup N, Jørgensen T, Brandslund I, Nielsen HB, Juncker AS, Bertalan M, Levenez F, Pons N, Rasmussen S, Sunagawa S, Tap J, Tims S, Zoetendal EG, Brunak S, Clément K, Doré J, Kleerebezem M, Kristiansen K, Renault P, Sicheritz-Ponten T, de Vos WM, Zucker JD, Raes J, Hansen T MetaHIT consortium, Bork P, Wang J, Ehrlich SD, Pedersen O. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500:541–546. doi: 10.1038/nature12506. [DOI] [PubMed] [Google Scholar]
- 73.Lofgren JL, Whary MT, Ge Z, Muthupalani S, Taylor NS, Mobley M, Potter A, Varro A, Eibach D, Suerbaum S, Wang TC, Fox JG. Lack of commensal flora in Helicobacter pylori-infected INS-GAS mice reduces gastritis and delays intraepithelial neoplasia. Gastroenterology. 2011;140:210–220. doi: 10.1053/j.gastro.2010.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lertpiriyapong K, Whary MT, Muthupalani S, Lofgren JL, Gamazon ER, Feng Y, Ge Z, Wang TC, Fox JG. Gastric colonisation with a restricted commensal microbiota replicates the promotion of neoplastic lesions by diverse intestinal microbiota in the Helicobacter pylori INS-GAS mouse model of gastric carcinogenesis. Gut. 2014;63:54–63. doi: 10.1136/gutjnl-2013-305178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vinasco K, Mitchell HM, Kaakoush NO, Castaño-Rodríguez N. Microbial carcinogenesis: Lactic acid bacteria in gastric cancer. Biochim Biophys Acta Rev Cancer. 2019;1872:188309. doi: 10.1016/j.bbcan.2019.07.004. [DOI] [PubMed] [Google Scholar]
- 76.Hsu PI, Pan CY, Kao JY, Tsay FW, Peng NJ, Kao SS, Wang HM, Tsai TJ, Wu DC, Chen CL, Tsai KW Taiwan Acid-related Disease (TARD) Study Group. Helicobacter pylori eradication with bismuth quadruple therapy leads to dysbiosis of gut microbiota with an increased relative abundance of Proteobacteria and decreased relative abundances of Bacteroidetes and Actinobacteria. Helicobacter. 2018;23:e12498. doi: 10.1111/hel.12498. [DOI] [PubMed] [Google Scholar]
- 77.Machate DJ, Figueiredo PS, Marcelino G, Guimarães RCA, Hiane PA, Bogo D, Pinheiro VAZ, Oliveira LCS, Pott A. Fatty Acid Diets: Regulation of Gut Microbiota Composition and Obesity and Its Related Metabolic Dysbiosis. Int J Mol Sci. 2020;21 doi: 10.3390/ijms21114093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ktsoyan ZA, Beloborodova NV, Sedrakyan AM, Osipov GA, Khachatryan ZA, Kelly D, Manukyan GP, Arakelova KA, Hovhannisyan AI, Olenin AY, Arakelyan AA, Ghazaryan KA, Aminov RI. Profiles of Microbial Fatty Acids in the Human Metabolome are Disease-Specific. Front Microbiol. 2010;1:148. doi: 10.3389/fmicb.2010.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fu Y, Wang Y, Gao H, Li D, Jiang R, Ge L, Tong C, Xu K. Associations among Dietary Omega-3 Polyunsaturated Fatty Acids, the Gut Microbiota, and Intestinal Immunity. Mediators Inflamm. 2021;2021:8879227. doi: 10.1155/2021/8879227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kaliannan K, Wang B, Li XY, Kim KJ, Kang JX. A host-microbiome interaction mediates the opposing effects of omega-6 and omega-3 fatty acids on metabolic endotoxemia. Sci Rep. 2015;5:11276. doi: 10.1038/srep11276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Simopoulos AP. The importance of the ratio of omega-6/omega-3 essential fatty acids. Biomed Pharmacother. 2002;56:365–379. doi: 10.1016/s0753-3322(02)00253-6. [DOI] [PubMed] [Google Scholar]
- 82.Zárate R, El Jaber-Vazdekis N, Tejera N, Pérez JA, Rodríguez C. Significance of long chain polyunsaturated fatty acids in human health. Clin Transl Med. 2017;6:25. doi: 10.1186/s40169-017-0153-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Thamphiwatana S, Gao W, Obonyo M, Zhang L. In vivo treatment of Helicobacter pylori infection with liposomal linolenic acid reduces colonization and ameliorates inflammation. Proc Natl Acad Sci USA. 2014;111:17600–17605. doi: 10.1073/pnas.1418230111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The code is available at https://github.com/sterrettJD/H-pylori-microbiome-analysis. Volcano plot data are included as Supplementary Tables 1-3 (Supplementary Table 1: Volcano plot data with all Helicobacter data set; Supplementary Table 2: Volcano plot data with High Bacteroidetes; Supplementary Table 3: Volcano plot data with High Firmicutes).