

SUMMARY
Chemokines are chemotactic cytokines that regulate the migration of immune cells. Chemokines function as cues for the coordinated recruitment of immune cells into and out of tissue and also guide the spatial organization and cellular interactions of immune cells within tissues. Chemokines are critical in directing immune cell migration necessary to mount and then deliver an effective anti-tumor immune response; however, chemokines also participate in the generation and recruitment of immune cells that contribute to a pro-tumorigenic microenvironment. Here, we review the role of the chemokine system in anti-tumor and pro-tumor immune responses and discuss how malignant cells and the tumor microenvironment regulate the overall chemokine landscape to shape the type and outcome of immune responses to cancer and cancer treatment.
INTRODUCTION
Chemokines are 8- to 12-kDa secreted proteins that regulate directed cell migration (chemotaxis), adhesion, cell positioning, and cell-cell interactions by binding to Gαi-protein-coupled seven-transmembrane-spanning receptors (GPCRs), so-called classical chemokine receptors (Griffith et al., 2014). Chemokines also bind to atypical chemokine receptors (ACKRs) that are also seven-transmembrane-spanning receptors but are not G-protein coupled and do not induce chemotaxis (Bonecchi and Graham, 2016). The chemokine system consists of nearly 50 chemokine ligands, 20 signaling GPCRs, and 4 ACKRs and plays essential roles during development, homeostasis, inflammation, infection, and pathological processes, including tumorigenesis.
The process of tumorigenesis is shaped by cancer cells, tissue-resident cells, and recruited immune cells that express a broad array of chemokine ligands and chemokine receptors (Balkwill, 2004; Chow and Luster, 2014; Nagarsheth et al., 2017). Chemokines act on tumor cells regulating their stem-like cell properties, proliferation, and invasiveness as well as on stromal cells to regulate neoangiogenesis, neurogenesis, and fibrogenesis (Mukaida et al., 2014). Moreover, chemokines regulate the phenotype and function of immune cells by regulating their localization and cellular interactions in lymphoid tissues and the tumor microenvironment (TME). Major breakthroughs in our understanding of the role of the immune system during tumorigenesis has led to the development of novel immunotherapeutic approaches to treat a variety of cancers that has greatly benefited cancer patients. Immunotherapy remains one of the most exciting therapeutic promises of the 21st century.
The role of chemokines in tumor immunity is multifaceted, given the diverse and dynamically regulated expression of chemokine ligands and chemokine receptors by immune cells, stromal cells, and tumor cells. Chemokines regulate critical aspects of immune cell biology during tumorigenesis, including immune cell activation, recruitment, phenotype, and function. The role of chemokines in immune cell recruitment and immune cell activation is well appreciated. However, chemokine-dependent control of immune cell localization and cellular interactions within the TME that programs immune cell phenotype and function has only started to be uncovered.
Adding to the complexity, the very same chemokine systems contribute to anti- and pro-tumorigenic immune responses. The balance between these disparate functions is likely dependent on the stage of tumorigenesis, the state of immune cell activation, the balance of effector and regulatory response, and the relative expression of chemokine receptors on effector and regulatory target cells. Given the existence of chemokine-dependent positive feedback loops, initial small differences in anti- and pro-tumorigenic responses might be enhanced over time, contributing to consolidation of the tumor immune contexture. For example, often the recruitment of pro-tumorigenic immune cell types prompts chemokine-dependent infiltration of other immunosuppressive cells. Certain chemokine systems are predominantly associated with anti-tumorigenic immune responses, including the XCR1 and CXCR3 chemokine axes, while other chemokine systems are predominantly associated with pro-tumorigenic responses, including the CCR4 and CCR8 chemokine axes. Chemokines that mediate the recruitment of anti-tumorigenic immune cells and help maintain their functionality within TME are attractive therapeutic targets to explore in order to improve responses to cancer therapies. Likewise, inhibition of chemokines that recruit and promote the suppressive function of immune cells are also interesting potential therapeutic targets to improve responses to therapies. Further, the induction or transduction of chemokine receptors on adoptively transferred anti-tumor T cells that facilitate their ability to enter deep into the tumor and license their functionality is another therapeutic strategy that is being explored to improve responses to cancer therapies.
Here, we review recent advances in our understanding of chemokine function in the immune response to cancer. We discuss the role of chemokines within TME in the context of the heterogeneity of solid tumors at the spatial and cellular level, including the potential involvement of chemokines in regulation of molecular and cellular cues critical for preservation of immune cell states that correlate with potent anti-tumor immunity or pro-tumorigenic immune cell phenotypes. We also discuss the current understanding of how anti-tumorigenic chemokine systems operate in certain tumors but are silenced in others, including tumor-intrinsic and extrinsic factors that regulate the chemokine landscape during tumorigenesis, the regulation of the tumor immune contexture by oncogenic driver mutations and ongoing immune pressure, and the role of chemokines in the context of currently available immunotherapies. We underscore the complexity of the chemokine system and highlight how assessment of the chemokine landscape in the context of other immunological parameters might guide treatment strategies and predict responsiveness to available immunotherapies.
Chemokines in the generation, delivery, and function of pro- and anti-tumor immune cells
Tumors develop from normal cells through the acquisition of genetic changes that enable their uncontrolled proliferation (Hanahan and Weinberg, 2011). Tumor progression is often supported by tissue-resident immune cells, fibroblasts, endothelial cells, and neurons that are co-opted by cancer for its benefit. Furthermore, tumor escape is promoted by recruited immune cells with suppressive phenotypes, which are similar to the cellular phenotypes seen in the resolution phase of wound healing or tissue remodeling (Coussens et al., 2013; Hua and Bergers, 2019). These cell types include tumor-associated macrophages (TAMs), tumor-associated neutrophils (TANs), myeloid-derived suppressor cells (MDSCs), and regulatory T (Treg) cells (Figure 1). Despite the self-origin of neoplastic cells, tumors can be controlled by responses of natural killer (NK) cells that have strong cytolytic function against physiologically stressed cells. Furthermore, CD4+ T helper type 1 (Th1) cells and CD8+ effector T (Teff) cells that are specific for tumor-associated antigens (TAA) or neoantigens can lyse cancer cells. The initiation of anti-tumor T cell responses depends on antigen-presenting cells (APCs), mainly dendritic cells (DCs), that capture and process TAAs for presentation to T cells (Figure 2). During tumorigenesis, chemokines regulate the activation, recruitment, phenotype, and function of immune cells within the TME. Therefore, chemokines and their receptors have a significant impact on the quality of pro- and anti-tumorigenic immune responses.
Figure 1. Chemokines in pro-tumor immunity.
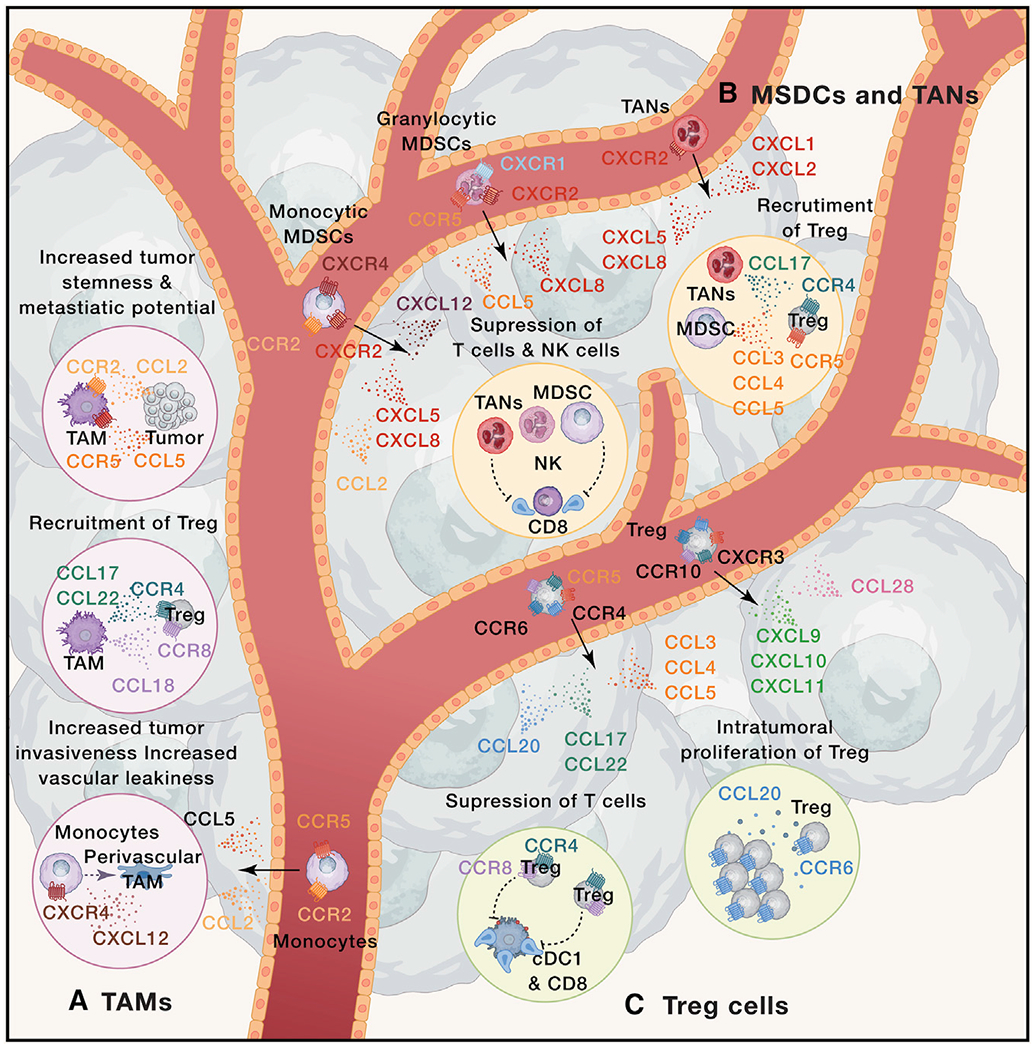
Immune cells, including (A) tumor-associated macrophages (TAMs), (B) myeloid-derived suppressor cells (MDSC), tumor-associated neutrophils (TANs), and (C) regulatory T cells (Treg) contribute to tumor growth. (A) TAMs differentiate from monocytes that are recruited into tumors in response to the CCR5-CCL5 and CCR2-CCL2 chemokine axes. CXCL12 signals through CXCR4 to induce monocyte differentiation towards perivascular macrophages, which contribute to tumor intravasation and vascular leakiness. TAMs contribute to tumor progression through the production of CCL17, CCL22, and CCL18 that attract CCR4+ and CCR8+ Treg cells. CCR2-CCL2 and CCR5-CCL5 chemokine axes regulate cross-talk between TAMs and tumor cells that contributes to tumor stemness and metastatic potential. (B) The CXCR4-CXCL12, CXCR2-CXCL5/CXCL8, and CCR2-CCL2 chemokine axes contribute to the recruitment of monocytic-MDSC, whereas CXCR1-CXCL8, CXCR2-CXCL8, and CCR5-CCL5 axes attract granulocytic-MDSCs. TANs are predominantly recruited through CXCR2 that responds to CXCL1, CXCL2, CXCL5 and CXCL8. Within the TME, MDSCs and TANs suppress T cells and NK cells, as well as contribute to the recruitment of Treg by release of CCL3, CCL4, and CCL5 (MO-MDSCs) or CCL17 (TANs). (C) Treg cells can be recruited by multiple chemokine systems, including CXCR3-CXCL9/CXCL10/CXCL11, CCR10-CCL27, CCR6-CCL20, CCR4-CCL17/CCL22, and CCR5-CCL3/CCL4/CCL5. CCR8+ CCR4+ highly suppressive Treg cells contribute to tumor growth by suppressing T cell responses within TME. Intratumoral Treg cell responses are enhanced by chemokines, such as the CCR6-CCL20 axis, which promotes Treg cell proliferation. Black arrows represent chemokine-mediated cell recruitment. Dotted colored arrows represent cell differentiation and proliferation. Dotted lines represent cell-mediated activation (arrow end) or inhibition (blunt end).
Figure 2. Chemokines in anti-tumor immunity.
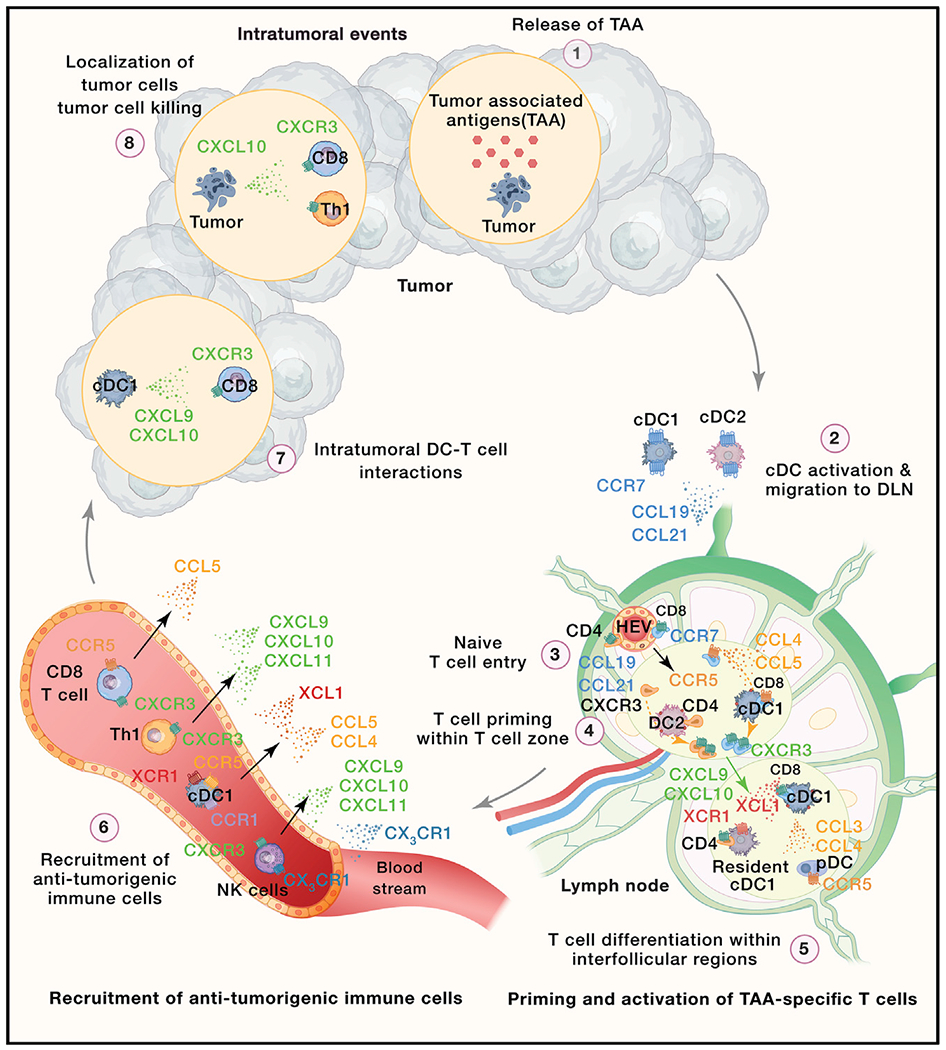
The generation of anti-tumor immunity requires that (1) tumor cells release tumor-associated antigens (TAAs) and neoantigens. (2) TAAs and neoantigens are then taken up and processed by professional antigen-presenting cells (APCs), such as intratumoral conventional type 1 dendritic cells (cDC1s) and cDC2s. In the presence of inflammatory cues, cDCs mature and upregulate CCR7, which promotes their migration into tumor-draining lymph nodes (TDLN) in response to gradients of CCL19 and CCL21. (3) The CCR7-CCL19/CCL21 chemokine axis also controls entry of naive CD8+ and CD4+ T cells into TDLN. (4) Within TDLN, naive TAA-specific CD8+ and CD4+ T cells engage in interactions with cDC1s and cDC2s, respectively. The CCR5-CCL5 chemokine axis facilitates such encounters in the case of naive TAA-specific CD8+ T cells. Activation of CD4+ and CD8+ T cells is accompanied by CXCR3 upregulation, which drives migration of activated T cells into interfollicular regions (IFRs) of the LN. (5) Within IFRs, activated T cells engage in secondary interactions with specific immune cells. For instance, activated CD8+ T cells and migratory cDC1s release chemokines, such as XCL1 and CCL3 and CCL4, to attract LN resident XCR1+ cDC1s and CCR5+ plasmacytoid DCs (pDCs), respectively. TAA-specific CD4+ T cells engage in CXCR3-dependent secondary interactions with DCs that drive their differentiation toward Th1 cells. Moreover, TAA-specific CD4+ T cells and TAA-specific CD8+ T cells interact with the same XCR1+ cDC1s that facilitate the delivery of CD4+ T cell help to primed CD8+ T cells. (6) Various anti-tumorigenic immune ceils, such as NK cells, cDC1 s, Th1 cells, and CD8+ T cells are recruited from the blood into the tumor microenvironment (TME) in response to distinct chemokine gradients. For example, NK cell recruitment is driven by CX3CR1-CX3CL1 and CXCR3-CXCL9/CXCL10/CXCL11. XCR1-XCL1, CCR5-CCL4/CCL5, and CCR1-CCL4 mediate the recruitment of cDC1s. Th1 cells and CD8+ T cells can be recruited through the CXCR3-CXCL9/CXCL10/CXCL11 axis. In addition, CD8+ T cell infiltration can also be triggered by CCR5-CCL5. (7) cDC1s release CXCL9 and CXCL10, contributing to the recruitment of CXCR3+ CD8+ T cells. Moreover, this chemokine system facilitates intratumoral DC-CD8+ T cell interactions, promoting the effector functions of intratumoral Teff cells. (8) Finally, activated CD8+ T cells and Th1 cells localize in the proximity of tumor cells, possibly in response to tumor-derived CXCL10. CD8+ T cells and Th1 cells produce cytokines or directly lyse tumor cells, contributing to tumor eradication. Black arrows represent chemokine-mediated cell recruitment. Gray arrows represent the flow of cells in the tumor immunity cycle. The green arrow represents chemokine-mediated positioning within LNs. Dotted colored arrow represents sequence of cellular events preceding T cell activation and division.
Our knowledge regarding the role of chemokines in immune cell activation during tumorigenesis stems primarily from models of viral and bacterial infection (Table 1). It is assumed that the same chemokine systems operate during tumorigenesis to coordinate the activation of immune cells. However, the activity of the chemokine axes involved in anti-tumorigenic responses is likely reduced or even silenced by tumors, whereas the activity of the chemokine axes driving activation of pro-tumorigenic immune cells is likely enhanced. For instance, conventional type 1 DC (cDC1) maturation induced by the processing of TAAs and sensing of inflammatory cues triggers upregulation of CCR7, which is critical for cDC1 migration from the tumor into tumor-draining lymph nodes (TDLNs) (Figure 2). CCR7 deficiency, specifically in the cDC1 subset, results in impaired anti-tumoral T cell responses and tumor outgrowth (Roberts et al., 2016). Furthermore, tumor-derived ligands for liver X receptor (LXR) can interfere with the capacity of DCs to migrate to TDLNs (Villablanca et al., 2010). Recent data suggest that the ability of cDC2s to migrate from the TME to TDLNs also depends on CCR7 and is critical for priming of anti-tumorigenic CD4+ T c (Figure 2). However, cDC2 recruitment is often blocked due to the suppressive activity of intratumoral Treg cells (Binnewies et al., 2019). Growing tumors trigger changes in chemokine expression not only within the TME but also in surrounding tissues and lymphoid organs, interfering with immune cell activation. TDLNs are a primary site of anti-tumor T cell activation. Efficient priming of TAA-specific CD8+ T c and TAA-specific CD4+ T cells requires a series of chemokine-mediated interactions with migratory and LN-resident DCs in specific regions of the LN (Lian and Luster, 2015) (Figure 2). Importantly, tumor-derived factors can silence expression of CCL21 by stroma cells within TDLN. CCL21 is required for recruitment of T cells and DCs into the LN and reduction of its expression in TDLN correlates with altered positioning of immune cell populations, which likely contributes to poor T cell activation (Riedel et al., 2016). Tumor-derived factors interfere with normal myelopoiesis in the bone marrow, increasing generation of granulocytic MDSC (Giese et al., 2019). The CXCR4-CXCL12 chemokine axis plays a vital role in the retention of developing and mature immune cells within the bone marrow (Griffith et al., 2014). Solid tumors increase CXCL12 expression in bone marrow stromal cells, leading to accumulation of CXCR4-expressing myeloid cells. Thus, distant tumors might interfere with hematopoiesis (Chiodoni et al., 2020). It remains to be defined if tumor-derived factors affect chemokine expression in other lymphoid tissues, such as spleen and thymus, and how it affects immune cell generation and activation.
Table 1.
Chemokines and chemokine receptors
| Receptor | Cell expression | Chemokine ligand(s) | General immune function |
|---|---|---|---|
| Classical chemokine receptors | |||
| CXCR1 | neutrophils >> monocytes, NK cells, mast cells, basophils, CD8+ Teff cells, and endothelial cells | CXCL6 and CXCL8 | neutrophil trafficking |
| CXCR2 | neutrophils >> monocytes, NK cells, mast cells, basophils, CD8+ T cells, and endothelial cells | CXCL1–CXCL3 and CXCL5–CXCL7 | B cell lymphopoiesis, neutrophil egress from bone marrow, and neutrophil trafficking |
| CXCR3 | Th1 cells, CD8+ Tcm and Tem cells, NK cells, NKT cells, pDCs, B cells, Treg cells, and Tfh cells | CXCL9, CXCL10, and CXCL11 | type 1 adaptive immunity |
| CXCR4 | most leukocytes and endothelial cells | CXCL12 | hematopoiesis, organogenesis, and bone marrow homing |
| CXCR5 | B cells, Tfh cells, Tfr cells, and CD8+ Tem cells | CXCL13 | B cell and T cell trafficking in lymphoid tissue to the B cell zone or follicles |
| CXCR6 | Th1 cells, Th17 cells, γδ T cells, ILCs, NKT cells, NK cells, plasma cells, and endothelial cells | CXCL16 | ILC function and adaptive immunity |
| CCR1 | monocytes, macrophages, neutrophils, Th1 cells, basophils, and DCs | CCL3, CCL3L1, CCL5, CCL8 (human), CCL14 (human), CCL15 (human), and CCL16 (human) | innate immunity and adaptive immunity |
| CCR2 | monocytes, macrophages, Th1 cells, immature DCs, basophils, NK cells, and endothelial cells | CCL2, CCL7, CCL8 (human), CCL12 (mouse), CCL13 (human), and CCL16 (human) | monocyte trafficking and type 1 adaptive immunity |
| CCR3 | eosinophils >> basophils and mast cells | CCL5, CCL7, CCL11, CCL13 (human), CCL15, (human) CCL24, and CCL26 | type 2 adaptive immunity, eosinophil distribution, and trafficking |
| CCR4 | Th2 cells, skin-homing and lung-homing T cells, Treg cells >> Th17 cells, CD8+ T cells, monocytes, B cells, and immature DCs | CCL17 and CCL22 | homing of T cells to the skin and lungs and type 2 immune responses |
| CCR5 | monocytes, macrophages, Th1 cells, NK cells, Treg cells, CD8+ T cells, DCs, and neutrophils | CCL3, CCL3L1, CCL4, CCL4L1, CCL5, and CCL16 (human) | type 1 adaptive immunity |
| CCR6 | Th17 cells >> immature DCs, γδ T cells, NKT cells, NK cells, Treg cells, and Tfh cells | CCL20 | immature DC trafficking, GALT development, and type 17 adaptive immune responses |
| CCR7 | naive T cells, Tcm cells, Trcm cells, mature DCs, and B cells | CCL19 and CCL21 | mature DCs, B cell and T cell trafficking in lymphoid tissue to the T cell zone, and egress of DCs and T cells from tissue |
| CCR8 | Th2 cells, Treg cells, skin Trm cells, γδ T cells, monocytes, and macrophages | CCL1, CCL8 (mouse), and CCL18 (human) | immune surveillance in skin, type 2 adaptive immunity, and thymopoiesis |
| CCR9 | gut-homing T cells, thymocytes, B cells, DCs, and pDCs | CCL25 | homing of T cells to the gut, GALT development and function, and thymopoiesis |
| CCR10 | skin-homing T cells and IgA plasmablasts | CCL27 and CCL28 | homing immunity at mucosal sites and immune surveillance in the skin |
| XCR1 | cross-presenting CD8+ DCs and thymic DCs | XCL1 and XCL2 | antigen cross-presentation by CD8+ DCs |
| CX3CR1 | resident monocytes, macrophages, microglia, Th1 cells, CD8+ Tem cells, NK cells, γδ T cells, and DCs | CX3CL1 | patrolling monocytes in innate immunity, microglial cell and NK cell migration, and type 1 adaptive immunity |
| AKCRs | |||
| ACKR1 (DARC) | red blood cells and endothelial cells | CXCL1–CXCL 3, CXCL5–CXCL8, CXCL11, CXCL13, CCL2, CCL7, CCL14, CCL17, and CCL22 | chemokine transcytosis and chemokine scavenging |
| ACKR2 (D6) | DC, B cells, and lymphatic endothelium | CCL2–CCL5, CCL7, CCL8, CCL11, CCL14, CCL17, and CCL22 | chemokine scavenging |
| ACKR3 (CXCR7) | B cells and stromal cells | CXCL11 and CXCL12 | shaping chemokine gradients for CXCR4 |
| ACKR4 (CCRL1;CCX-CKR) | thymic epithelium | CXCL13, CCL19, CCL21, and CCL25 | chemokine scavenging |
DC, dendritic cell; pDC, plasmacytoid dendritic cell; GALT, gut-assosciated lymphoid tissue; IgA, immunoglobulin A; ILC, innate lymphoid cell; NK, natural killer; NKT cell, natural killer T cell; Tcm cell, central memory T cell; Teff cell, effector T cell; Tern cell, effector-memory T cell; Tfh cell, follicular helper T cell; Tfr cell, follicular regulatory T cell; Th cell, T helper cell; Trcm cell, recirculating memory T cell; Treg cell, regulatory T cell; Trm cell, resident-memory T cell.
There is an extensive literature on the role of chemokines in the recruitment of distinct immune cell subsets into the tumor bed, which is summarized in Table 2 and depicted in Figures 1 and 2. Interestingly, the very same chemokine systems are often involved in recruitment of pro- and anti-tumorigenic immune cells. For instance, CXCR3 and its ligands CXCL9 and CXCL10, and CCR5 and its ligand CCL5, recruit both Treg cells and CD8+ T cells. Therefore, the kinetics of expression of chemokines within the TME and changes in chemokine receptor expression on immune cells is a likely determinant of TME immune cell composition. For example, intratumoral Treg cells are heterogeneous, and different Treg subsets dominate at distinct stages of tumorigenesis (Li et al., 2019). During infection, the ability of Treg cells to suppress T cell responses depends on the expression of transcription factors and chemokine receptors characteristic for Th1 (Tbet, CXCR3), Th2 (Gata3, CCR4), Th17 (RORγt, CCR6), or follicular helper T (Tfh) cells (Bcl6, CXCR5). The Th1, Th17, and Tfh-like Treg subsets were shown to suppress responses of corresponding conventional CD4+ Th subsets (Wing et al., 2019). CXCR3+ Treg cells accumulate within the TME earlier compared to CCR4+ CCR8+ Treg cells (Li et al., 2019). It remains to be defined whether the influx of CXCR3+ Treg cells occurs in response to proliferation of CXCR3+ Th1 and CD8+ T cells. Likewise, the functional roles of distinct Treg subsets during tumorigenesis have to be defined. The CD27low and CD27hi subsets of NK cells that have distinct functional features show differential expression of CXCR3 and CX3CR1. Thus, chemokines expressed in the TME and expression of cognate chemokine receptors on NK cells may affect the mobilization efficiency of specific NK cell subsets into the tumor, which in turn could influence the overall clinical outcome. Interestingly, the recruitment of specific immune cell subsets into the TME is strongly dependent on the presence of the immune cell populations. For instance, recruitment of cDC1s into tumors depends on NK-cell-derived chemokines, such as XCL1 and CCL5 (Böttcher et al., 2018) (Figure 2). Importantly, tumor-infiltrating cDC1s are one of the primary producers of CXCL9 and CXCL10, chemokine ligands pivotal for CD8+ Teff cell recruitment into the TME (Mikucki et al., 2015; Spranger et al., 2017). In comparison to other chemokine axes that often play redundant roles, the XCL1-XCR1 axis is uniquely involved in the recruitment of cDC1. Therefore, its therapeutic targeting might represent a specific way to promote cDC1 and T cell accumulation. Moreover, it might promote interactions between cDC1 and CD8+T cells to facilitate T cell expansion and acquisition of effector functions (Dorner et al., 2009). In contrast, the CCL2-CCR2 and CXCL1/CXCL8-CXCR2 chemokine axes are predominantly involved in the recruitment of suppressive cells, such as monocytes and monocytic-MDSCs or TANs and granulocytic-MDSCs, respectively. These pro-tumorigenic immune cell subsets subsequently contribute to release of CCL18, CCL17, and CCL22 to drive infiltration of highly suppressive CCR4+ Treg cells (Figure 1). In addition, abundance of MDSCs correlates with reduced expression of CXCL11 and decreased infiltration of CD8+ T cells (Yang et al., 2020). Thus, chemokine-mediated cellular feedback loops might enhance initial small differences in anti- and pro-tumorigenic responses during tumor progression.
Table 2.
Chemokines in cancer immunity and treatment
| Chemokine | Receptor | Roles in anti-tumor immunity | Roles in pro-tumor immunity | Effects on cancer treatment |
|---|---|---|---|---|
| CXCL1, CXCL2, CXCL3, and CXCL5 | CXCR2 | ND | recruitment of MDSCs | contributes to chemoresistance via recruitment of MDSCs |
| CXCL9 and CXCL10 | CXCR3 | recruitment of NK and Teff cells | ND | recruitment of Teff cells during chemotherapy; facilitates interactions between DCs and T cells during immunotherapy |
| CXCL12 | CXCR4 | ND | recruitment and differentiation of TAMs and recruitment of pDCs | ND |
| CXCL16 | CXCR6 | ND | ND | homing of Teff cells during radiotherapy |
| CCL1 and CCL8 | CCR8 | ND | recruitment of Treg cells | ND |
| CCL2 | CCR2 | recruitment of NK cells | recruitment of monocytes, TAMs, and Th17 cells | recruitment of APCs during chemotherapy |
| CCL3 | CCR1 and CCR5 | recruitment on NK cells | recruitment and retention of TAMs and recruitment of CCR5+ Treg cells | ND |
| CCL4 | CCR5 | recruitment of cDC1 | recruitment of CCR5+ Treg cells | ND |
| CCL5 | CCR5 | recruitment of cDC1, NK cells, Teff cells | recruitment of CCR5+ Treg cells, TAMs, and Th17 cells | recruitment of Teff cells during chemotherapy; contributes to chemoresistance via recruitment of monocytes and MDSCs |
| CCL11 | CCR3 | recruitment of eosinophils | recruitment of TAM | ND |
| CCL17 | CCR4 | ND | recruitment of Treg cells and upregulation of OX40L and ICOSL expression on pDCs | ND |
| CCL20 | CCR6 | recruitment of cDCs | recruitment of Th17 cells, immature DCs, Treg cells, and TAMs | ND |
| CCL22 | CCR4 | ND | recruitment of Treg cells and upregulation of OX40L and ICOSL expression on pDCs | ND |
| CCL26 | CCR3 | ND | recruitment of MDSCs | ND |
| CCL28 | CCR10 | ND | recruitment of Treg cells | ND |
| XCL1 | XCR1 | recruitment of cDC1s | ND | ND |
| CX3CL1 | CX3CR1 | recruitment of NK and Teff cells | ND | ND |
ND, not determined; MDSC, myeloid-derived suppressor cell; TAM, tumor-associated macrophage.
The role of chemokines in the regulation of immune cell phenotypes and function within the TME is only just beginning to be uncovered. The emerging picture suggests that simple characterization of cells as pro- or anti-tumorigenic does not fully reflect the cellular complexity of the TME. For instance, the phenotype of anti-tumor CD8+ T cells can change with tumor progression, and only specific CD8+ T cell signatures (TCF-1+ and CX3CR1+) correlate with effective anti-tumor immunity (Miller et al., 2019; Sade-Feldman et al., 2018; Siddiqui et al., 2019). Likewise, Treg cell signatures can either correlate with tumor progression (CCR8+ Treg cells) (De Simone et al., 2016; Plitas et al., 2016) or responsiveness to PD-1 immunotherapy (interferon γ [IFNγ]+ Treg cells) (Di Pilato et al., 2019). There is also a significant level of heterogeneity and plasticity within TAMs and TANs (Azizi et al., 2018; Gubin et al., 2018; Lavin et al., 2017) with extreme pro- (M1 TAMs and N2 TANs) and anti-tumorigenic phenotypes (M2 TAMs and N1 TANs) and likely many intermediate cell states.
It remains poorly defined how such heterogenous cell states are maintained within the TME and to what extent they are susceptible to therapeutic modulation. The TME is heterogenous in regard to factors such as oxygen availability, the abundance of nutrients, and acidity that are known to control phenotype and function of immune cells. Thus, localization of immune cells within the TME controlled by chemokines might be one of the key determinants of immune cell phenotype. Differentiation and maturation of immune cells is often associated with major reprogramming of chemokine receptor expression. For instance, as stem-like TCF-1+ CD8+ T cells differentiate into effector-like CD8+ T cells and ultimately to exhausted T cells, they downregulate certain chemokine receptors (CXCR5 and CXCR3) and upregulate others (CXCR6 and CX3CR1) (He et al., 2016; Philip et al., 2017). Moreover, stem-like TCF-1+ CD8+T cells locate in niches rich in APCs within human tumors (Jansen et al., 2019). Higher expression of CXCR3 on stem-like TCF-1 + cells might be critical for their recruitment toward CXCL9 and CXCL10 chemokine ligands derived from cDC1s, promoting their reactivation and expansion within TME (Broz et al., 2014; Maurice et al., 2019) and acquisition of effector functions through exposure to interleukin-12 (IL-12) (Ruffell et al., 2014) (Figure 2). Likewise, CXCR3 and its ligands might regulate effector cell localization in the proximity of tumor cells (Hickman et al., 2015)(Figure 2).
The phenotype of TAMs and TANs is likely the result of niche-specific signaling within TME. For instance, within the TME, the CXCR4-CXCL12 axis facilitates the differentiation of newly recruited TAMs into perivascular TAMs. These stationary TAMs promote vascular leakiness and tumor cell intravasation (Arwert et al., 2018) (Figure 1). Furthermore, suppressive functions of TAMs within TME can be regulated by the CCL5-CCR5 axis, as blocking of CCR5 alters TAM phenotype toward anti-tumorigenic and leads to objective clinical responses in colorectal cancer patients (Halama et al., 2016). Moreover, chemokine axes, such as CCR2-CCL2 and CCR5-CCL5, mediate cross-talk of tumor cells with TAMs, promoting tumor stemness, metastasis, and resistance to therapy (Frankenberger et al., 2015; Pastushenko et al., 2018) (Figure 1). Treg cell suppressive functions in the TME are dependent on antigen recognition (Bauer et al., 2014), which might be regulated by CCR8 and/or CCR4 chemokine-dependent recruitment of Treg cells in the proximity of DCs within the TME. Expression of CCR8 on Treg cells contributes to the maintenance of their suppressive phenotype, as CCL1-CCR8 signaling enhances the expression of Foxp3 (Barsheshet et al., 2017), and Foxp3 levels correlate with the stability of the Treg phenotype (Charbonnier et al., 2019; Di Pilato et al., 2019). Furthermore, CCR8+ Treg cells show elevated expression of suppressive markers, such as CD25, CTLA4, CD39, TIGIT, PD1, ICOS, OX40, and Helios, compared with their CCR8− counterparts (Wang et al., 2019b). Alternatively, CCR4, which regulates Treg cell/DC interactions within LNs, might play a similar role within the TME (Rapp et al., 2019). Within the TME, CCR6 signaling can potentiate in situ proliferation of Treg cells (Xu et al., 2011) (Figure 1). Understanding how chemokine cues contribute to the formation of cellular niches within the TME represents an exciting avenue for future research. Even if chemokines and chemokine receptors represent difficult therapeutic targets, they might help to dissect cellular components critical for induction and maintenance of pro- and anti-tumorigenic immune cell states.
Chemokines and the tumor immune contexture
The tumor immune contexture, understood as the density, composition, functional state, location, and type of immune cells within the tumor, is relevant for prognosis and response to treatment. The accumulation of anti-tumorigenic immune cells, such CD8+ Teff and NK cells, at the invasive margin of primary tumor, together with decreased abundance of myeloid cells, often correlates with improved prognosis (Galon and Bruni, 2020; Mlecnik et al., 2016). The immune profile of tumors is highly diverse not only across tumor types but also among patients bearing the same tumor type and even in different tumor sites within the same patient (Mlecnik et al., 2018; Zhang et al., 2019). This diversity results from the combination of various cancer-intrinsic and extrinsic factors, likely including variations in tumor genetics, microbiome, and tissue of origin. Moreover, the tumor immune contexture undergoes dynamic changes during disease progression or recurrence (Bindea et al., 2013). Importantly, developing tumors shape the chemokine milieu within the TME to control the abundance and function of pro- and anti-tumorigenic immune cells. Expression of chemokines and chemokine receptors is often regulated by oncogenic driver mutations and ongoing immune pressure that contributes to the dynamic modulation of the tumor immune contexture. Thus, the chemokine system is an important component of the immune contexture.
Based on the quantification and localization of CD3+ and CD8+ T cells within the tumor core and the invasive margin, four main categories of tumors have been described: (1) cold tumors (lack of CD8+ Teff cell infiltration); (2) altered, immune-excluded tumors (CD8+ Teff cell infiltration at the invasive margin but exclusion from the tumor core); (3) altered, immunosuppressed tumors (low level of CD8+ Teff cell infiltration); and (4) hot, inflamed tumors (high level of CD8+ Teff cell infiltration within tumor core and at the invasive margin) (Galon and Bruni, 2019). Cold tumors might show myeloid cell infiltration, but they uniformly show a limited number or absence of CD8+ Teff cells. This tumor profile is likely reflecting inefficient generation of anti-tumor immunity. Multiple distinct mechanisms might contribute to the establishment of cold tumors, such as low antigenicity of tumors or impaired T cell priming within TDLNs. Cross-presenting tissue-resident cDC1s excel at orchestrating anti-tumor CD8+ T cell responses, yet tumor-intrinsic factors interfere with cDC1 recruitment into the tumor, thereby impacting early T cell priming and infiltration into the TME. For instance, melanomas with little or no T cell infiltration have activated Wnt/β-catenin signaling (Spranger et al., 2015). Using an autochthonous mouse model, it was demonstrated that melanoma-cell-intrinsic activated β-catenin suppressed production of CCL4 via upregulation of the transcriptional repressor ATF3, restricting cDC1 migration into tumors (Spranger et al., 2015). The recruitment of cDC1s into tumors is mediated by tumor-infiltrating NK cells that release CCL5 and XCL1 (Böttcher et al., 2018). Moreover, NK cells promote cDC1 survival by production of FMS-related tyrosine kinase 3 ligand (Barry et al., 2018). Therefore, it is likely that this axis might be disrupted by tumors to evade immune recognition. For example, cyclooxygenase 2 (COX2), which is frequently overexpressed by cancer cells, is involved in the synthesis of prostaglandin E2 (PGE2) (Zelenay et al., 2015). PGE2 reduces NK cell viability and limits their capacity to produce pro-inflammatory chemokines, which in turn limits cDC1 density favoring tumor progression (Böttcher et al., 2018). Interestingly, distinct tissues differ in the steady-state abundance of cross-presenting DCs (Salmon et al., 2019). It remains to be determined how chemokine cross-talk between NK and cDC1 cells is regulated at distinct tissue locations and how tumors originating from distinct organs might interfere with this axis. Moreover, the density of cDC1s within tumors tends to decrease over time (Binnewies et al., 2018), suggesting that progressing tumors might acquire the ability to silence the production of cDC1-recruiting chemokines. In order to trigger productive T cell responses, cDC1s need to be activated, and tumors might interfere with this process. For instance, tumor-derived LXR ligands can impair CCR7 expression on DCs, and this impairs cDC1 migration into TDLNs (Villablanca et al., 2010). It remains unknown if a different threshold of activation must be reached to induce CCR7-dependent cDC1 migration into TDLNs in tumors originating from sterile tissues, such as kidney and liver, versus tissues with environmental interfaces, such as skin, lung, and gut. ACKR4, a scavenger receptor for CCL19, CCL21, CCL25, and CXCL13, also plays a role in tumorigenesis. By controlling CCL19 and CCL21 chemokine bioavailability and maintaining functional chemotactic gradients, ACKR4 has been demonstrated to regulate DC migration into lymphatics as well as into T cell areas of draining LNs (Bryce et al., 2016; Heinzel et al., 2007; Ulvmar et al., 2014). It remains to be determined if tumors interfere with ACKR4 function to disrupt DC migration into TDLNs.
Altered, immune-excluded tumors are characterized by CD8+ T cell infiltration that remains localized at the edge of the tumor. This immune-excluded phenotype reflects the ability of the host to generate anti-tumor immunity but suggests the existence of mechanisms within the tumor to physically hinder T cell infiltration into the core of the tumor, resulting in immune escape. One reason for T cell exclusion might stem from the absence of T-cell-recruiting signals within the core of the tumor, such as potent T-cell-attracting chemokines (i.e., CXCL9, CXCL10, CXCL11, CX3CL1, CCL2, and CCL5). This deficiency in chemokine production can result from tumor-induced modulation of pathways that control their expression. For instance, CXCR3 ligands can be epigenetically silenced in certain tumors. Polycomb repressive complex 2 (PRC2) components, which trimethylate histone 3 lysine 27 (H3K27me3), have been shown to repress the expression of CXCL9 and CXCL10, but not other IFNγ-induced genes in colon cancer and ovarian cancer (Nagarsheth et al., 2016; Peng et al., 2015). In contrast, Jumonji-C-domain-containing protein (JMJD3), an H3K27-specific demethylase, has been shown to activate the expression of CXCL9 and CXCL10. Thus, H3K27me3-specific methyltransferases and demethylases can regulate CXCL9 and CXCL10 expression. Furthermore, using pharmacological drugs to target epigenetic inhibition of chemokines leads to significantly higher chemokine production and increases CD8+ Teff cell recruitment into tumor. This increase in Teff cell migration in vivo results in slower tumor growth (Figure 3). In contrast, in a Wnt1-overexpressing lung adenocarcinoma model, active β-catenin was increased in cDCs, and this repressed the production of T-cell-recruiting chemokines, such as CCL2, CXCL9, and CXCL10 (Kerdidani et al., 2019). Cleavage and inactivation of T-cell-recruiting chemokines by proteases is another potential mechanism that contributes to immune evasion and T cell exclusion. In the HER2-driven breast cancer murine model and the B16F10 melanoma murine model, the activity of proteolytic enzymes cleaves CXCL9 and CXCL10. N-terminal proteolytic cleavage by DPP4 (Barreira da Silva et al., 2015) or MMP-9 (Juric et al., 2018) decreases the activity of CXCR3 ligands, resulting in the impaired infiltration of CD4+ and CD8+ T cells into the TME and tumor progression. Consequently, inhibition of MMP-9 or DDP4 in these tumor models results in reduced tumor burden and enhanced infiltration of T cells into the tumor. Interestingly, another study found that CXCL9 and CXCL10 can induce the upregulation of cathepsin B in breast cancer patients. This cysteine protease was shown to cleave CXCR3 ligands in vitro, resulting in their reduced activity. Thus, breast cancer cells may dampen the activity of the CXCR3 chemokine system by cathepsin-B-mediated degradation of CXCR3 ligands in vivo. CCL2 and CCL5 were reported to be modified by peroxynitrite, and reactive nitrogen species within the TME can induce CCL2 chemokine nitration (Molon et al., 2011; Thompson et al., 2017). Nitration of CCL2 reduces its affinity for CCR2 and impairs the recruitment of CD8+ T cells into the TME, resulting in tumor evasion.
Figure 3. Chemokine systems in cancer therapy.
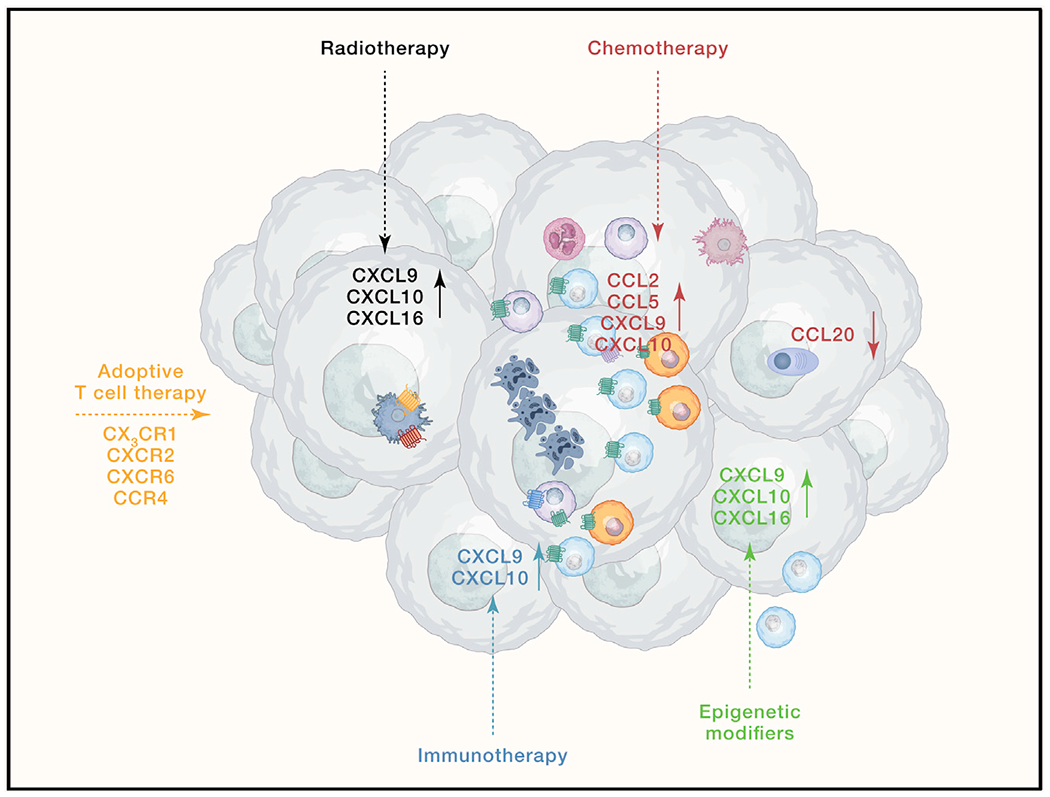
Induction of certain chemokines and downregulation of others (solid arrows) in the TME contribute to the efficacy of cancer treatments (dotted arrows), such as radiotherapy, chemotherapy, epigenetic modifiers, and immunotherapy. Moreover, expression of specific chemokine receptors on adoptively transferred T cells plays a critical role in their infiltration into the TME.
Most solid tumors consist of two distinct compartments that include the tumor parenchyma and the surrounding stroma. Stromal cell organization can impact the ability of T cells to infiltrate into the tumor core, impairing immunosurveillance regardless of inherent tumor immunogenicity. For instance, tumor hypoxia and transforming growth factor β (TGF-β) signaling activate cancer-associated fibroblasts (CAFs). Activated CAFs can secrete high amounts of extracellular matrix molecules, creating a collagen-rich fiber that physically limits immune cell infiltration. These effects can be disrupted by using TGF-β neutralizing antibodies (Mariathasan et al., 2018; Tauriello et al., 2018). CAFs can also release factors that mediate immune cell differentiation and recruitment. For instance, CXCL12 production by CAFs favors T cell exclusion in mouse pancreatic and lung tumor models through a mechanism that has yet to be elucidated (Feig et al., 2013). In addition to stromal cells, there is also a network of TAMs along the tumor margin that can trap tumor-specific T cells and lead to their suppression (Beatty et al., 2015). The role of chemokines in this process remains to be determined.
Some tumors show a low degree of CD8+ Teff cell infiltration but prominent recruitment of other immune cells, often with immunosuppressive properties, such as TAMs, MDSCs, and Treg cells. These tumors show upregulation of inflammatory mediators that promote pro-tumorigenic inflammation and shape TME toward a tumor-permissive state. In some types of cancer, inflammation precedes cancer formation. Conversely, in other types, genetic alterations in cancer cells or tumorigenesis-induced cell death and oxidative stress trigger inflammatory pathways and inflammatory cell recruitment. Many of these pathways are mechanistically linked to the increased production of chemokines (Greten and Grivennikov, 2019; Mantovani et al., 2008). For example, loss of the tumor suppressor p53 results in the increased expression of nuclear factor κB (NF-κB)-dependent inflammatory genes, among them chemokines. Several studies demonstrated that NF-κB-mediated induction of chemokines contributes to tumor progression, tumor recurrence, and resistance to therapy (Taniguchi and Karin, 2018). For example, NF-κB activity can induce the expression of CCL5 that drives the recruitment of CCR5-expressing macrophages, which supplies breast tumor cells with collagen that promotes their proliferation (Walens et al., 2019). In pancreatic adenocarcinoma, NF-κB induces CXCL5 expression that drives CXCR2-dependent recruitment of granulocytic-MDSCs, promoting tumor progression (Chao et al., 2016). NF-κB can also trigger CCL2 production, promoting monocyte recruitment and the establishment of a pre-metastatic niche (Keklikoglou et al., 2019). In TAMs, activation of NF-κB induces CCL22 production that leads to the recruitment of CCR4+ Treg cells (Sun et al., 2016). In addition, TAMs recruit CCR6+ Treg cells through an NF-κB-dependent induction of CCL20 (Wu et al., 2019).
Increased activation of oncogenes is frequent in tumorigenesis and leads to expression of chemokines that favor tumor growth. For instance, activation of protein tyrosine kinase RET due to chromosome rearrangement is an early event in the development of papillary thyroid carcinoma. Uncontrolled activation of RET results in the induction of a transcriptional program that upregulates chemokines that attract monocytes and DCs, such as CCL2 and CCL20, as well as chemokines that promote angiogenesis, such as CXCL8 (Borrello et al., 2005). Another oncogene, K-Ras, regulates expression of CXCL3, a chemokine important for MDSC recruitment (Liao et al., 2019). MYC, a transcription factor that is overexpressed in many human tumors, elicits the production of chemokines, including CCL2 and CCL5, which attract inflammatory cells, such as mast cells, which promote angiogenesis and tumor growth (Soucek et al., 2007). Similarly, the BRAFV600E mutation increases the expression of CCL17 and CCL22 to drive infiltration of CCR4-expressing Treg cells into tumors, suppressing tumor immunity (Shabaneh et al., 2018).
Oxidative stress, amino acid deprivation, and endoplasmic reticulum stress frequently occur during tumorigenesis and lead to upregulation of ATF4, which has an established role in cancer progression and treatment resistance (Singleton and Harris, 2012). ATF4 can contribute to the progression of cancer by induction of CCL2 that facilitates the recruitment of TAMs into the TME (Liu et al., 2017). Under normal oxygen conditions, tumor suppressor von Hippel-Lindau (VHL) protein ubiquitinates and targets HIF-1 for degradation, thereby decreasing CXCR4 expression on cancer cells. In contrast, under hypoxic conditions, common in many tumors, VHL is inactivated and HIF-1 is stabilized, inducing the expression of CXCR4 on tumor cells (Staller et al., 2003). CXCR4 is upregulated in human cancers and is involved in metastasis (Mortezaee, 2020). In addition, hypoxia in ovarian cancer triggers the transcription of CCL28 that recruits CCR10-expressing Treg cells into tumors, promoting tumor tolerance and angiogenesis (Facciabene et al., 2011). In hepatocellular carcinoma, hypoxia induces CCL26 expression that recruits MDSCs into tumors (Chiu et al., 2016). Furthermore, necroptosis-induced release of CXCL1 from pancreatic cancer cells promotes recruitment of MDSCs and TAMs, resulting in myeloid-cell-induced adaptive immune suppression (Seifert et al., 2016).
ACKR1 and ACKR2 act as negative regulators of inflammation. ACKR1 can sequester CXCL1 and CXCL2, whereas ACKR2 is a scavenger for most CC chemokines. Little is known about their regulation during tumorigenesis, but it is likely that their activity is reduced in tumors, promoting the development of an immunosuppressive TME. Indeed, ACKR1 expression in mice correlated with reduced tumor burden (Shen et al., 2006). In contrast, ACKR2 expression on lymphatic endothelial cells limits inflammation in dextran-sulfate-sodium- and azoxymethane-induced inflammation-associated colon cancer by scavenging CCL2, CCL3, and CCL5, which is associated with a decreased incidence of colon cancer (Vetrano et al., 2010).
Tumor-intrinsic genetic alterations and tumorigenesis-induced extrinsic stress signals lead to production of inflammatory cytokines and chemokines that initiate inflammation in many cancers. Induction of tumor-promoting inflammation occurs with distinct kinetics and can appear before or after tumor initiation. In some instances, it might be evident only at later stages of tumorigenesis. Therefore, it might actively promote tumor growth very early or remain silent until late stages of metastasis or therapy resistance (Greten and Grivennikov, 2019). In addition to recruited immunosuppressive cells, tissue-resident macrophages can also promote tumor outgrowth and metastasis. It remains to be determined if certain chemokine cues that orchestrate cross-talk between tissue-resident macrophages and epithelial and mesenchymal cells during normal organogenesis can be co-opted by developing cancer to promote its progression and metastasis.
A key feature of immune-inflamed, hot tumors is the abundance of CD4+ and CD8+ T cells and their localization in the proximity of tumor cells. Therefore, the efficient priming of anti-tumor T cells within TDLNs and sufficient production of T-cell-recruiting chemokines is likely to be an important feature of inflamed tumors. Indeed, expression of CCL2, CCL4, CCL5, CXCL9, and CXCL10 correlates with T cell presence (Harlin et al., 2009). Moreover, induction of T-cell-recruiting chemokines by activation of lymphotoxin-β-receptor signaling through TNFSF14 was sufficient to create a T-cell-inflamed microenvironment (Tang et al., 2016). Data suggest that the initial influx of CD8+ T cells might be critical for further enhancement of T cell recruitment. Indeed, CD8+ Teff cell recruitment into tumors might be induced by chemotactic agents released upon destruction of tumor cells by CD8+ Teff cells already present within the TME. Dying tumor cells stimulate a TLR3-dependent, cancer-cell-autonomous type I IFN response that triggers the production of CXCL10 (Boissonnas et al., 2007; Sistigu et al., 2014).
Hot tumors are usually characterized by high mutational burden, which is thought to increase the occurrence of TAAs, thereby promoting immune recognition of tumor cells. Presence of an anti-tumor T cell response within the TME is often accompanied by infiltration of Treg and myeloid cells. In contrast to non-inflamed tumors, hot tumors show enrichment for transcripts encoding indoleamine-2,3-dioxyenase (IDO), Foxp3, and PD-L1. These suggest that these immunosuppressive pathways might be triggered by active anti-tumor immunity rather than being an intrinsic property of the tumor. Indeed, activated CD8+ Teff cells can produce CCL22 that attract CCR4+ Treg cells (Spranger et al., 2013). As a result of chronic exposure to tumor antigens, unproductive interactions with DCs within the TME, and exposure to an immunosuppressive environment, T cells often progressively lose their effector functions, transitioning into T cell exhaustion. T cell dysfunction and upregulation of immune checkpoint inhibitors seems to be the major obstacle for efficient tumor control in these types of tumors. T cell exhaustion develops over time and might be controlled by yet-unidentified chemokine cues operating within the TME.
In sum, silencing or activation of certain chemokine systems at distinct stages of tumorigenesis contributes to the establishment of the immune contexture within the TME, which influences the natural history of a tumor and its response to therapy. It is unlikely that chemokine expression alone is sufficient to define the immune contexture. The delicate balance and timing of multiple mechanisms, such as tumor mutations, epigenetic changes, host genetics, tissue of origin, microbiome, and cancer immune editing, shape the tumor immune contexture. Nevertheless, the cytokine and chemokine milieu, together with the type of immune and non-immune tumor-associated cell types at any given time, determine the balance between anti-tumor and pro-tumor responses. Therefore, analysis of chemokine expression, together with other critical immune parameters such as numbers, phenotype, and positioning of immune cells, represents a valuable prognostic biomarker, which might allow for the tailored treatment for individual patients.
Role of chemokines in cancer treatment
Immunotherapy harnesses the patient’s immune system to destroy tumors by relieving effector cell dysfunction and inhibiting suppressive immune cell populations. The immune contexture predicts responsiveness to immunotherapy, with hot tumors being the most responsive and cold tumors being the least responsive. There is growing interest in combining different cancer treatment approaches to overcome tumor resistance and sensitize cold and altered tumors for more effective immunotherapy. Modulation of chemokine expression during cancer treatment contributes to the efficacy of as well as the resistance to therapy and will be reviewed in this section (Table 2).
Checkpoint blockade therapy targets immunosuppressive molecules on the surface of T cells to restore their effector function, enabling better tumor control. The efficacy of checkpoint blockade therapy strongly correlates with the preexisting immune response (Tumeh et al., 2014). Patients with T-cell-inflamed tumors that are enriched for T-cell-recruiting chemokines, such as CCL5, CXCL9, CXCL10, and CXCL11, are most likely to benefit from checkpoint blockade therapy (Ayers et al., 2017; Dangaj et al., 2019; Gao et al., 2016; Tumeh et al., 2014). However, responses are not guaranteed in these patients, indicating that immune cell infiltration into the TME is necessary, but not sufficient, for a clinical response. Indeed, recent data correlate responsiveness to checkpoint blockade therapy with the presence of a specific subset of T cells that are characterized by expression of the transcription factor TCF-1 (Miller et al., 2019; Sade-Feldman et al., 2018; Siddiqui et al., 2019). Moreover, our group has recently found that the chemokine receptor CXCR3 and its ligand, CXCL9, were important for the response to anti-PD-1 therapy in mouse tumor models (Chow et al., 2019) (Figure 3). Upregulation of CXCL9 expression by cDC1 following PD-1 blockade enabled cDC1 to specifically activate CXCR3-expressing CD8+ T cells that are not terminally exhausted, thereby facilitating the generation of an effective anti-tumor response to eliminate tumor cells. CXCL9 is not only essential for the clinical response to anti-PD-1 treatment but also has been demonstrated to be pivotal for the efficacy of TIM-3 blockade therapy in mouse models (de Mingo Pulido et al., 2018). Anti-TIM-3 promoted the expression of CXCL9 by cDC1s, which triggered the intratumoral response of CD8+ T cells, enhancing the responsiveness to paclitaxel chemotherapy. Therefore, lack of response in some patients with T-cell-inflamed tumors might be a result of low abundance of TCF-1+ cells and/or impaired cross-talk with cDC1s within the TME.
Adoptive cell transfer therapy using chimeric antigen receptor T cell therapy and in-vitro-expanded autologous T cells has also shown clinical promise. However, one of the significant hurdles that limits the efficacy of adoptive cell transfer therapy in the treatment of solid tumors is the restriction of adoptively transferred T cell infiltration into the tumor bed due to abnormal tumor vessels and an immunosuppressive TME. Thus, adoptive cell transfer therapy alone might not be sufficient to treat patients with cold and altered tumor profiles. To endow T cells with a more exceptional ability to migrate into tumors, different chemokine systems, including CCR4 (Di Stasi et al., 2009; Rapp et al., 2015), CXCR2 (Idorn et al., 2018; Jin et al., 2019; Peng et al., 2010), CX3CR1 (Siddiqui et al., 2016), and CXCR6 (Lesch et al., 2019), have been tested in the context of adoptive cell transfer therapy in mouse tumor models. However, the translation of these preclinical studies is urgently needed to determine if manipulation of the chemokine system can enhance the efficacy of adoptive cell transfer therapy for cancer patients.
Radiotherapy and chemotherapy are designed to induce death in rapidly proliferating malignant cells. Although the efficacy of radiotherapy and chemotherapy was initially attributed to the direct cytotoxic effects on malignant cells, it is now well appreciated that it is mediated, at least in part, through the activation of an anti-tumor immune response. Radiotherapy and chemotherapy can trigger the induction of an immunogenic cell death of cancer cells, promoting the cross-presentation of tumor-derived antigens and subsequent activation of adaptive anti-tumor T cell responses. Therefore, there is growing interest in developing novel optimal radiotherapy and chemotherapy protocols to efficiently trigger T cell responses. Moreover, several trials testing the synergy of checkpoint inhibitors with radiotherapy or chemotherapy are ongoing. The efficacy of radiotherapy and chemotherapy likely rely on the infiltration of cDC1s into the TME. Indeed, recent findings suggest that CCR7-dependent migration of cDCs from the TME into TDLNs is critical for efficacy of chemotherapy (Sharma et al., 2018). Therefore, cold tumors that interfere with cDC1 recruitment might show limited benefit from these treatment regimens unless the treatment itself enhances APC accumulation in the TME, as demonstrated in a murine model of fibrosarcoma. In this cancer type, treatment with anthracycline induces the expression of CCL2 in the TME, which recruits functional APCs and stimulates the generation of functional anti-tumor T cell responses (Ma et al., 2013, 2014). Nevertheless, chemotherapy and radiotherapy might be more beneficial in cold and altered tumors that do not interfere with cDC1 recruitment. In these tumor types, chemotherapy and radiotherapy might unleash the expression of T-cell-recruiting chemokines within the TME. For instance, in a murine model of breast cancer, irradiation-induced CXCL16 expression was critical for the homing of CXCR6-expressing CD8+ Teff cells into the tumor (Matsumura et al., 2008) (Figure 3). Furthermore, another chemokine axis was found to mediate a similar effect in a mouse model of melanoma in which irradiation triggers the production of type I and II IFNs, upregulation of CXCL9 or CXCL10, and subsequent recruitment of CXCR3-expressing Teff cells (Lim et al., 2014; Lugade et al., 2008) (Figure 3). Similar to radiotherapy, treatment of tumor-bearing mice with chemotherapy leads to intratumoral expression of chemokines, such as CXCL9, CXCL10, and CCL5, which drives recruitment of CD4+ and CD8+ T cells into the tumor bed (Hong et al., 2011) (Figure 3). These chemokines are also upregulated in patients with melanoma who responded to chemotherapy, and their expression correlated with CD4+ and CD8+ T cell infiltration, tumor control, and patient survival (Hong et al., 2011). In addition, tumor cells increase expression of CXCL10 in response to anthracycline-based chemotherapy, which is important for anti-tumor T cell responses (Sistigu et al., 2014). Another mechanism of action of chemotherapy might include interference with chemokine-mediated immunosuppressive pathways. For example, treatment of non-small cell lung cancer patients with docetaxel results in significant reduction in CCL20 expression (Figure 3). Thus, chemotherapy might contribute to anti-tumor immune responses by inhibiting the CCL20-CCR6+ Treg axis (Zhang et al., 2015).
The resistance of cancer cells to therapy remains a significant problem in the treatment of local and metastatic disease. Resistance can be driven by the tumor immune contexture at the start of treatment or might be acquired as the adaptation of the tumor to therapy and can be mediated by both tumor- and host-derived factors. Therapy-induced changes in the expression of chemokines contribute to tumor resistance or tumor recurrence. For instance, upregulation of CCL2 and CCL5 post-radiotherapy results in the recruitment of immunosuppressive cells, such as CCR2+CCR5+ monocytes, MDSCs, and CCR2+ Treg cells, leading to cancer outgrowth (Connolly et al., 2016; Kalbasi et al., 2017; Liang et al., 2017; Mondini et al., 2019). In addition, CCR5-dependent recruitment of macrophages post-radiotherapy contributes to the local recurrence of cancer (Rafat et al., 2018). The CCL2-CCR2 axis is likely involved in resistance to BRAF inhibitor therapy in melanoma patients, as high expression of CCL2 in melanoma patients correlates with a lack of response (Vergani et al., 2016). Murine tumor models suggest that the outgrowth of BRAF-inhibitor-resistant melanomas might be associated with the recruitment of CCR2+ MDSCs into the TME (Steinberg et al., 2017). Recent data revealed a novel mechanism of chemoresistance that is also mediated by CCL2. Chemotherapeutic drugs can induce the release of tumor-derived extracellular vesicles (EVs) with enhanced pro-metastatic potential. These tumor-derived EVs trigger CCL2 expression in endothelial cells that facilitates the recruitment of pro-tumorigenic CCR2+ Ly6C+ monocytes (Keklikoglou et al., 2019). Interestingly, the efficacy of immune checkpoint therapy is improved in CCR2-deficient mice. These findings suggest the potential contribution of the CCL2-CCR2 axis and MDSCs to resistance to immune checkpoint therapy (Kim et al., 2019). The CCR6-CCL20 axis contributes to resistance to various treatments, most likely through the recruitment of CCR6+ Treg cells. In colorectal cancer patients, high expression of CCL20 and CCR6+ Treg cells is closely associated with chemoresistance (Wang et al., 2019a). Likewise, high CCL20 expression correlates with chemoresistance in triple-negative breast cancer patients (Chen et al., 2018). In hepatocellular carcinoma, the accumulation of CCR6+ Treg cells in response to CCL20 produced by TAAs or tumor cells contributes to resistance to PD-L1 immunotherapy (Wu et al., 2019). In other tumors, the infiltration of suppressive cells and resistance to therapy is dependent on CXCL1 and CXCL2. For instance, in breast cancer patients, increased activity of NF-κB post-chemotherapy drives enhanced CXCL1 and CXCL2 expression in cancer cells, contributing to the recruitment of MDSCs into the TME and ultimately the survival of cancer cells and chemoresistance (Acharyya et al., 2012). Likewise, in a murine Kras-driven model of lung cancer, resistance to PD-L1 is driven by CXCL2-dependent recruitment of Gr1+ tumor-infiltrating neutrophils (Faget et al., 2017). The resistance to adoptive T cell therapy is common, and mechanisms underlying this phenomenon are still poorly understood. The establishment of TMEs with low expression of T-cell-attracting chemokines might play a critical role in the low efficacy of the treatment. For instance, highly glycolytic tumors are poorly infiltrated by T cells, which correlates with low levels of CXCL10 (Cascone et al., 2018).
In sum, therapeutic strategies that induce expression of T-cell-recruiting chemokines within TME, promote recruitment of cDC1s into the TME and TDLNs, and/or interfere with the chemokine suppressive axis that drives the recruitment of MDSCs and Treg cells should improve the responsiveness of cold and altered tumors to immunotherapy. In contrast, the responsiveness of T-cell-inflamed tumors to immunotherapy should be enhanced by facilitating CXCL9-mediated cross-talk of PD-L1-responsive T cells with cDC1s within the TME.
Chemokines as biomarkers
Cancer biomarkers can be classified into the following three categories: (1) predictive biomarkers that predict response to specific therapeutic intervention; (2) prognostic biomarkers that inform about overall cancer progression and cancer recurrence, regardless of therapy; and (3) diagnostic biomarkers that can be used to identify the disease. Data suggest that chemokines and their receptors have the potential to serve as predictive, prognostic, and diagnostic cancer biomarkers. Changes in the expression of chemokine receptors and their ligands often correlate with overall survival and disease-free survival of patients, suggesting that the chemokine system has the potential to serve as a prognostic biomarker for certain cancers. Upregulation of certain chemokines occurs in specific cancers, making chemokines potential useful diagnostic biomarkers. Lastly, changes in chemokine levels post-treatment or their basal level at the start of therapy might predict responses of patients to therapy, suggesting that chemokines can potentially be used as predictive biomarkers.
Analysis of current data demonstrates that the same chemokine axis can have either an anti-tumorigenic or a pro-tumorigenic role depending on the immune context within which it operates. For instance, in ovarian carcinoma, patients with higher CXCL9 levels in ascites (Lieber et al., 2018) or CXCL9 and CXCL10 levels in solid tumor tissue (Bronger et al., 2016) show significantly longer relapse-free survival. High levels of CXCL9 and CXCL10 were also significantly correlated with longer overall survival in advanced pancreatic ductal adenocarcinoma patients receiving chemotherapy (Qian et al., 2019) and in colorectal cancer(Mlecnik et al., 2010). In contrast, increased CXCL9 levels predict poor prognosis in oral cavity squamous cell carcinoma (Chang et al., 2013), nasopharyngeal carcinoma (Hsin et al., 2013), and clear-cell renal carcinoma (Liu et al., 2016). In addition, patients with higher CXCL10 levels had significantly poorer overall survival and disease-free survival in hepatocellular carcinoma (Li et al., 2017). It is not known why the CXCL9-CXCR3 axis is pro-tumorigenic in these settings, but it has been proposed that CXCR3 signaling in these types of cancer cells and/or the recruitment of CXCR3-expressing Treg cells is pro-tumorigenic. Therefore, high-dimensional deep profiling of immune cells at a single-cell level within human tumors will allow the simultaneous use of multiple parameters to predict a patient’s prognosis and response to treatment. As an example, tumor mutational burden (TMB) and the T-cell-inflamed gene expression profile (GEP) are emerging predictive biomarkers for the response to anti-PD-1 therapy. The T cell-inflamed GEP is composed of 18 inflammatory genes, among which are three chemokines (CXCR6, CXCL9, and CCL5). Patients with a high TMB and GEP show the most robust objective response rate and longer progression-free survival, pointing to the possible importance of these chemokines in the anti-PD-L1 immune response (Cristescu et al., 2018)
Concluding remarks
Given their role in regulating the migration and cell-cell interactions of immune cells, chemokines are intimately involved with both anti-tumor and pro-tumor immune responses as well as the response and resistance to cancer therapies. Chemokines participate in the generation of anti-tumor immune responses in TDLNs and in the delivery of these responses to the TME. Similarly, chemokines regulate the recruitment of pro-tumorigenic immune responses to the TME and their cellular interactions within TME that promote immunosuppression. The fact that chemokines (in some cases the very same chemokines) can contribute to both pro-tumor and anti-tumor responses reflects the complexity of the chemokine system, as a given chemokine can recruit and activate both anti-tumor effector cells and pro-tumor regulatory cells. Thus, the integrated chemokine landscape of a tumor will need to be considered in the context of the qualities of the tumor immune response to understand its role in the overall response. Nonetheless, expression of specific chemokines can be associated with response to therapy and as such can be used as a biomarker for predicting and monitoring the response to therapy. Further, the induction of chemokines that mediate the recruitment of cDCs, Th1 cells, and Teff cells into the TME is an attractive therapeutic strategy to explore to improve responses to cancer therapies. Likewise, the induction or transduction of chemokine receptors on adoptively transferred anti-tumor T cells is another attractive therapeutic strategy that is being explored to improve responses to cancer therapies.
REFERENCES
- Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, Manova-Todorova K, Leversha M, Hogg N, Seshan VE, et al. (2012). A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 150, 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arwert EN, Harney AS, Entenberg D, Wang Y, Sahai E, Pollard JW, and Condeelis JS (2018). A unidirectional transition from migratory to perivascular macrophage is required for tumor cell intravasation. Cell Rep. 23, 1239–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, et al. (2017). IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest 127, 2930–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, Nainys J, Wu K, Kiseliovas V, Setty M, et al. (2018). Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell 174, 1293–1308.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkwill F (2004). Cancer and the chemokine network. Nat. Rev. Cancer 4, 540–550. [DOI] [PubMed] [Google Scholar]
- Barreira da Silva R, Laird ME, Yatim N, Fiette L, Ingersoll MA, and Albert ML (2015). Dipeptidylpeptidase 4 inhibition enhances lymphocyte trafficking, improving both naturally occurring tumor immunity and immunotherapy. Nat. Immunol 16, 850–858. [DOI] [PubMed] [Google Scholar]
- Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, Nelson AE, Loo K, Kumar R, Rosenblum MD, et al. (2018). A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat. Med 24, 1178–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsheshet Y, Wildbaum G, Levy E, Vitenshtein A, Akinseye C, Griggs J, Lira SA, and Karin N (2017). CCR8+FOXp3+ Treg cells as master drivers of immune regulation. Proc. Natl. Acad. Sci. USA 114, 6086–6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer CA, Kim EY, Marangoni F, Carrizosa E, Claudio NM, and Mempel TR (2014). Dynamic Treg interactions with intratumoral APCs promote local CTL dysfunction. J. Clin. Invest 124, 2425–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty GL, Winograd R, Evans RA, Long KB, Luque SL, Lee JW, Clendenin C, Gladney WL, Knoblock DM, Guirnalda PD, and Vonderheide RH (2015). Exclusion of T cells from pancreatic carcinomas in mice is regulated by Ly6C(low) F4/80(+) extratumoral macrophages. Gastroenterology 149, 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, et al. (2013). Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39, 782–795. [DOI] [PubMed] [Google Scholar]
- Binnewies M, Mujal AM, Pollack JL, Combes AJ, Hardison EA, Barry KC, Tsui J, Ruhland MK, Kersten K, Abushawish MA, et al. (2019). Unleashing type-2 dendritic cells to drive protective antitumor CD4(+) T cell immunity. Cell 177, 556–571.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. (2018). Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med 24, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissonnas A, Fetler L, Zeelenberg IS, Hugues S, and Amigorena S (2007). In vivo imaging of cytotoxic T cell infiltration and elimination of a solid tumor. J. Exp. Med 204, 345–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonecchi R, and Graham GJ (2016). Atypical chemokine receptors and their roles in the resolution of the inflammatory response. Front. Immunol 7, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrello MG, Alberti L, Fischer A, Degl’innocenti D, Ferrario C, Gariboldi M, Marchesi F, Allavena P, Greco A, Collini P, et al. (2005). Induction of a proinflammatory program in normal human thyrocytes by the RET/PTC1 oncogene. Proc. Natl. Acad. Sci. USA 102, 14825–14830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, Rogers NC, Sahai E, Zelenay S, and Reis E Sousa C (2018). NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell 172, 1022–1037.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronger H, Singer J, Windmüller C, Reuning U, Zech D, Delbridge C, Dorn J, Kiechle M, Schmalfeldt B, Schmitt M, and Avril S (2016). CXCL9 and CXCL10 predict survival and are regulated by cyclooxygenase inhibition in advanced serous ovarian cancer. Br. J. Cancer 115, 553–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, et al. (2014). Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 26, 638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryce SA, Wilson RAM, Tiplady EM, Asquith DL, Bromley SK, Luster AD, Graham GJ, and Nibbs RJB (2016). ACKR4 on stromal cells scavenges CCL19 to enable CCR7-dependent trafficking of APCs from inflamed skin to lymph nodes. J. Immunol 196, 3341–3353. [DOI] [PubMed] [Google Scholar]
- Cascone T, McKenzie JA, Mbofung RM, Punt S, Wang Z, Xu C, Williams LJ, Wang Z, Bristow CA, Carugo A, et al. (2018). Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab. 27, 977–987.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KP, Wu CC, Fang KH, Tsai CY, Chang YL, Liu SC, and Kao HK (2013). Serum levels of chemokine (C-X-C motif) ligand 9 (CXCL9) are associated with tumor progression and treatment outcome in patients with oral cavity squamous cell carcinoma. Oral Oncol. 49, 802–807. [DOI] [PubMed] [Google Scholar]
- Chao T, Furth EE, and Vonderheide RH (2016). CXCR2-dependent accumulation of tumor-associated neutrophils regulates T-cell immunity in pancreatic ductal adenocarcinoma. Cancer Immunol. Res 4, 968–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbonnier LM, Cui Y, Stephen-Victor E, Harb H, Lopez D, Bleesing JJ, Garcia-Lloret MI, Chen K, Ozen A, Carmeliet P, et al. (2019). Functional reprogramming of regulatory T cells in the absence of Foxp3. Nat. Immunol 20, 1208–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Qin Y, Wang D, Zhou L, Liu Y, Chen S, Yin L, Xiao Y, Yao XH, Yang X, et al. (2018). CCL20 triggered by chemotherapy hinders the therapeutic efficacy of breast cancer. PLoS Biol. 16, e2005869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiodoni C, Cancila V, Renzi TA, Perrone M, Tomirotti AM, Sangaletti S, Botti L, Dugo M, Milani M, Bongiovanni L, et al. (2020).Transcriptional profiles and stromal changes reveal bone marrow adaptation to early breast cancer in association with deregulated circulating microRNAs. Cancer Res. 80, 484–498. [DOI] [PubMed] [Google Scholar]
- Chiu DK-C, Xu IM-J, Lai RK-H, Tse AP-W, Wei LL, Koh H-Y, Li LL, Lee D, Lo RC-L, Wong C-M, et al. (2016). Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology 64, 797–813. [DOI] [PubMed] [Google Scholar]
- Chow MT, and Luster AD (2014). Chemokines in cancer. Cancer Immunol. Res 2, 1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow MT, Ozga AJ, Servis RL, Frederick DT, Lo JA, Fisher DE, Freeman GJ, Boland GM, and Luster AD (2019). Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti-PD-1 therapy. Immunity 50, 1498–1512.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly KA, Belt BA, Figueroa NM, Murthy A, Patel A, Kim M, Lord EM, Linehan DC, and Gerber SA (2016). Increasing the efficacy of radiotherapy by modulating the CCR2/CCR5 chemokine axes. Oncotarget 7, 86522–86535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens LM, Zitvogel L, and Palucka AK (2013). Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science 339, 286–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, Sher X, Liu XQ, Lu H, Nebozhyn M, et al. (2018). Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 362, eaar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dangaj D, Bruand M, Grimm AJ, Ronet C, Barras D, Duttagupta PA, Lanitis E, Duraiswamy J,Tanyi JL, Benencia F, et al. (2019). Cooperation between constitutive and inducible chemokines enables T cell engraftment and immune attack in solid tumors. Cancer Cell 35, 885–900.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mingo Pulido Á, Gardner A, Hiebler S, Soliman H, Rugo HS, Krummel MF, Coussens LM, and Ruffell B (2018). TIM-3 regulates CD103+ dendritic cell function and response to chemotherapy in breast cancer. Cancer Cell 33, 60–74.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, Bonnal RJP, Provasi E, Sarnicola ML, Panzeri I, et al. (2016).Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating T regulatory cells. Immunity 45, 1135–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Pilato M, Kim EY, Cadilha BL, Prüßmann JN, Nasrallah MN, Seruggia D, Usmani SM, Misale S, Zappulli V, Carrizosa E, et al. (2019). Targeting the CBM complex causes Treg cells to prime tumours for immune checkpoint therapy. Nature 570, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, Heslop HE, Brenner MK, Dotti G, and Savoldo B (2009). T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 113,6392–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorner BG, Dorner MB, Zhou X, Opitz C, Mora A, Güttler S, Hutloff A, Mages HW, Ranke K, Schaefer M, et al. (2009). Selective expression of the chemokine receptor XCR1 on cross-presenting dendritic cells determines cooperation with CD8+ T cells. Immunity 31, 823–833. [DOI] [PubMed] [Google Scholar]
- Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, Gimotty PA, Gilks CB, Lal P, Zhang L, and Coukos G (2011). Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 475, 226–230. [DOI] [PubMed] [Google Scholar]
- Faget J, Groeneveld S, Boivin G, Sankar M, Zangger N, Garcia M, Guex N, Zlobec I, Steiner L, Piersigilli A, et al. (2017). Neutrophils and snail orchestrate the establishment of a pro-tumor microenvironment in lung cancer. Cell Rep. 21, 3190–3204. [DOI] [PubMed] [Google Scholar]
- Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, et al. (2013). Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 110, 20212–20217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankenberger C, Rabe D, Bainer R, Sankarasharma D, Chada K, Krausz T, Gilad Y, Becker L, and Rosner MR (2015). Metastasis suppressors regulate the tumor microenvironment by blocking recruitment of prometastatic tumor-associated macrophages. Cancer Res. 75, 4063–4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galon J, and Bruni D (2019). Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov 18, 197–218. [DOI] [PubMed] [Google Scholar]
- Galon J, and Bruni D (2020). Tumor immunology and tumor evolution: intertwined histories. Immunity 52, 55–81. [DOI] [PubMed] [Google Scholar]
- Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. (2016). Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 167, 397–404.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese MA, Hind LE, and Huttenlocher A (2019). Neutrophil plasticity in the tumor microenvironment. Blood 133, 2159–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greten FR, and Grivennikov SI (2019). Inflammation and cancer: triggers, mechanisms, and consequences. Immunity 51, 27–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith JW, Sokol CL, and Luster AD (2014). Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu. Rev. Immunol 32, 659–702. [DOI] [PubMed] [Google Scholar]
- Gubin MM, Esaulova E, Ward JP, Malkova ON, Runci D, Wong P, Noguchi T, Arthur CD, Meng W, Alspach E, et al. (2018). High-dimensional analysis delineates myeloid and lymphoid compartment remodeling during successful immune-checkpoint cancer therapy. Cell 175, 1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halama N, Zoernig I, Berthel A, Kahlert C, Klupp F, Suarez-Carmona M, Suetterlin T, Brand K, Krauss J, Lasitschka F, et al. (2016). Tumoral immune cell exploitation in colorectal cancer metastases can be targeted effectively by anti-CCR5 therapy in cancer patients. Cancer Cell 29, 587–601. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, and Gajewski TF (2009). Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 69, 3077–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He R, Hou S, Liu C, Zhang A, Bai Q, Han M, Yang Y, Wei G, Shen T, Yang X, et al. (2016). Follicular CXCR5-expressing CD8(+) T cells curtail chronic viral infection. Nature 537, 412–428. [DOI] [PubMed] [Google Scholar]
- Heinzel K, Benz C, and Bleul CC (2007). A silent chemokine receptor regulates steady-state leukocyte homing in vivo. Proc. Natl. Acad. Sci. USA 104, 8421–8426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman HD, Reynoso GV, Ngudiankama BF, Cush SS, Gibbs J, Bennink JR, and Yewdell JW (2015). CXCR3 chemokine receptor enables local CD8(+) T cell migration for the destruction of virus-infected cells. Immunity 42,524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M, Puaux A-L, Huang C, Loumagne L, Tow C, Mackay C, Kato M, Prévost-Blondel A, Avril M-F, Nardin A, and Abastado J-P (2011). Chemotherapy induces intratumoral expression of chemokines in cutaneous melanoma, favoring T-cell infiltration and tumor control. Cancer Res. 71, 6997–7009. [DOI] [PubMed] [Google Scholar]
- Hsin LJ, Kao HK, Chen IH, Tsang NM, Hsu CL, Liu SC, Chang YS, and Chang KP (2013). Serum CXCL9 levels are associated with tumor progression and treatment outcome in patients with nasopharyngeal carcinoma. PLoS ONE 8, e80052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, and Bergers G (2019). Tumors vs. chronic wounds: an immune cell’s perspective. Front. Immunol 10, 2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idorn M, Skadborg SK, Kellermann L, Halldórsdóttir HR, Holmen Olofsson G, Met Ö, and Thor Straten P (2018). Chemokine receptor engineering of T cells with CXCR2 improves homing towards subcutaneous human melanomas in xenograft mouse model. OncoImmunology 7, e1450715–e1450715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen CS, Prokhnevska N, Master VA, Sanda MG, Carlisle JW, Bilen MA, Cardenas M, Wilkinson S, Lake R, Sowalsky AG, et al. (2019). An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 576, 465–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, Dyson KA, Grippin AJ, Deleyrolle LP, Zhang W, et al. (2019). CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat. Commun 10, 4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juric V, O’Sullivan C, Stefanutti E, Kovalenko M, Greenstein A, Barry-Hamilton V, Mikaelian I, Degenhardt J, Yue P, Smith V, and Mikels-Vigdal A (2018). MMP-9 inhibition promotes anti-tumor immunity through disruption of biochemical and physical barriers to T-cell trafficking to tumors. PLoS ONE 13, e0207255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalbasi A, Komar C, Tooker GM, Liu M, Lee JW, Gladney WL, Ben-Josef E, and Beatty GL (2017).Tumor-derived CCL2 mediates resistance to radiotherapy in pancreatic ductal adenocarcinoma. Clin. Cancer Res 23, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keklikoglou I, Cianciaruso C, Güç E, Squadrito ML, Spring LM, Tazzyman S, Lambein L, Poissonnier A, Ferraro GB, Baer C, et al. (2019). Chemotherapy elicits pro-metastatic extracellular vesicles in breast cancer models. Nat. Cell Biol 21, 190–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerdidani D, Chouvardas P, Arjo AR, Giopanou I, Ntaliarda G, Guo YA, Tsikitis M, Kazamias G, Potaris K, Stathopoulos GT, et al. (2019). Wnt1 silences chemokine genes in dendritic cells and induces adaptive immune resistance in lung adenocarcinoma. Nat. Commun 10, 1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim IS, Gao Y, Welte T, Wang H, Liu J, Janghorban M, Sheng K, Niu Y, Goldstein A, Zhao N, et al. (2019). Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat. Cell Biol 21, 1113–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin Y, Kobayashi S, Leader A, Amir E-D, Elefant N, Bigenwald C, Remark R, Sweeney R, Becker CD, Levine JH, et al. (2017). Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell 169, 750–765.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesch S, Blumenberg V, Stoiber S, Ogonek J, Cadilha B, Dantes Z, Rataj F, Dorman K, Lutz J, Karches C, et al. (2019). Arming T cells with C-X-C-motive receptor 6 enables adoptive T cell therapy of pancreatic cancer. Eur. J. Cancer 110, S25–S26. [Google Scholar]
- Li A, Herbst RH, Canner D, Schenkel JM, Smith OC, Kim JY, Hillman M, Bhutkar A, Cuoco MS, Rappazzo CG, et al. (2019). IL-33 signaling alters regulatory T cell diversity in support of tumor development. Cell Rep. 29, 2998–3008.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Zhu YH, Li Y, and Guan XY (2017). Identification of chemokine CXCL10 in tumor microenvironment by antibody array as a prognostic marker in hepatocellular carcinoma. Neoplasma 64, 778–786. [DOI] [PubMed] [Google Scholar]
- Lian J, and Luster AD (2015). Chemokine-guided cell positioning in the lymph node orchestrates the generation of adaptive immune responses. Curr. Opin. Cell Biol 36, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, Deng L, Hou Y, Meng X, Huang X, Rao E, Zheng W, Mauceri H, Mack M, Xu M, et al. (2017). Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat. Commun 8, 1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P, Li J, Wang G, Lan Z, Li J, et al. (2019). KRAS-IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell 35, 559–572.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber S, Reinartz S, Raifer H, Finkernagel F, Dreyer T, Bronger H, Jansen JM, Wagner U, Worzfeld T, Müller R, and Huber M (2018). Prognosis of ovarian cancer is associated with effector memory CD8+ T cell accumulation in ascites, CXCL9 levels and activation-triggered signal transduction in T cells. OncoImmunology 7, e1424672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JYH, Gerber SA, Murphy SP, and Lord EM (2014). Type I interferons induced by radiation therapy mediate recruitment and effector function of CD8(+) T cells. Cancer Immunol. Immunother 63, 259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Chen P, Xi D, Zhu H, and Gao Y (2017). ATF4 regulates CCL2 expression to promote endometrial cancer growth by controlling macrophage infiltration. Exp. Cell Res 360, 105–112. [DOI] [PubMed] [Google Scholar]
- Liu W, Liu Y, Fu Q, Zhou L, Chang Y, Xu L, Zhang W, and Xu J (2016). Elevated expression of IFN-inducible CXCR3 ligands predicts poor prognosis in patients with non-metastatic clear-cell renal cell carcinoma. Oncotarget 7, 13976–13983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugade AA, Sorensen EW, Gerber SA, Moran JP, Frelinger JG, and Lord EM (2008). Radiation-induced IFN-γ production within the tumor microenvironment influences antitumor immunity. J. Immunol. 180, 3132–3139. [DOI] [PubMed] [Google Scholar]
- Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, Portela Catani JP, Hannani D, Duret H, Steegh K, et al. (2013). Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 38, 729–741. [DOI] [PubMed] [Google Scholar]
- Ma Y, Mattarollo SR, Adjemian S, Yang H, Aymeric L, Hannani D, Portela Catani JP, Duret H, Teng MWL, Kepp O, et al. (2014). CCL2/CCR2-dependent recruitment of functional antigen-presenting cells into tumors upon chemotherapy. Cancer Res. 74, 436–445. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A, and Balkwill F (2008). Cancer-related inflammation. Nature 454, 436–444. [DOI] [PubMed] [Google Scholar]
- Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. (2018). TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura S, Wang B, Kawashima N, Braunstein S, Badura M, Cameron TO, Babb JS, Schneider RJ, Formenti SC, Dustin ML, and Demaria S (2008). Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J. Immunol 181, 3099–3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice NJ, McElrath MJ, Andersen-Nissen E, Frahm N, and Prlic M (2019). CXCR3 enables recruitment and site-specific bystander activation of memory CD8+ T cells. Nat. Commun 10, 4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikucki ME, Fisher DT, Matsuzaki J, Skitzki JJ, Gaulin NB, Muhitch JB, Ku AW, Frelinger JG, Odunsi K, Gajewski TF, et al. (2015). Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat. Commun 6, 7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, et al. (2019). Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol 20, 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlecnik B, Bindea G, Angell HK, Maby P, Angelova M, Tougeron D, Church SE, Lafontaine L, Fischer M, Fredriksen T, et al. (2016). Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity 44, 698–711. [DOI] [PubMed] [Google Scholar]
- Mlecnik B, Tosolini M, Charoentong P, Kirilovsky A, Bindea G, Berger A, Camus M, Gillard M, Bruneval P, Fridman WH, et al. (2010). Biomolecular network reconstruction identifies T-cell homing factors associated with survival in colorectal cancer. Gastroenterology 138, 1429–1440. [DOI] [PubMed] [Google Scholar]
- Mlecnik B, Van den Eynde M, Bindea G, Church SE, Vasaturo A, Fredriksen T, Lafontaine L, Haicheur N, Marliot F, Debetancourt D, et al. (2018). Comprehensive intrametastatic immune quantification and major impact of immunoscore on survival. J. Natl. Cancer Inst 110, 1. [DOI] [PubMed] [Google Scholar]
- Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, De Palma A, Mauri P, Monegal A, Rescigno M, et al. (2011). Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J. Exp. Med 208, 1949–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondini M, Loyher PL, Hamon P, Gerbé de Thoré M, Laviron M, Berthelot K, Clémenson C, Salomon BL, Combadière C, Deutsch E, and Boissonnas A (2019). CCR2-dependent recruitment of tregs and monocytes following radiotherapy is associated with TNFα-mediated resistance. Cancer Immunol. Res 7, 376–387. [DOI] [PubMed] [Google Scholar]
- Mortezaee K (2020). CXCL12/CXCR4 axis in the microenvironment of solid tumors: a critical mediator of metastasis. Life Sci. 249, 117534. [DOI] [PubMed] [Google Scholar]
- Mukaida N, Sasaki S, and Baba T (2014). Chemokines in cancer development and progression and their potential as targeting molecules for cancer treatment. Mediators Inflamm. 2014, 170381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarsheth N, Peng D, Kryczek I, Wu K, Li W, Zhao E, Zhao L, Wei S, Frankel T, Vatan L, et al. (2016). PRC2 epigenetically silences Th1-type chemokines to suppress effector T-cell trafficking in colon cancer. Cancer Res. 76, 275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarsheth N, Wicha MS, and Zou W (2017). Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol 17, 559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S, Van Keymeulen A, Brown D, Moers V, Lemaire S, et al. (2018). Identification of the tumour transition states occurring during EMT. Nature 556, 463–468. [DOI] [PubMed] [Google Scholar]
- Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, Sun Y, Zhao E, Vatan L, Szeliga W, et al. (2015). Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527, 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, Ye Y, Rabinovich BA, Liu C, Lou Y, Zhang M, Whittington M, Yang Y, Overwijk WW, Lizée G, and Hwu P (2010). Transduction of tumor-specific T cells with CXCR2 chemokine receptor improves migration to tumor and antitumor immune responses. Clin. Cancer Res 16, 5458–5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, et al. (2017). Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, Chudakov DM, and Rudensky AY (2016). Regulatory T cells exhibit distinct features in human breast cancer. Immunity 45, 1122–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L, Yu S, Yin C, Zhu B, Chen Z, Meng Z, and Wang P (2019). Plasma IFN-γ-inducible chemokines CXCL9 and CXCL10 correlate with survival and chemotherapeutic efficacy in advanced pancreatic ductal adenocarcinoma. Pancreatology 19, 340–345. [DOI] [PubMed] [Google Scholar]
- Rafat M, Aguilera TA, Vilalta M, Bronsart LL, Soto LA, von Eyben R, Golla MA, Ahrari Y, Melemenidis S, Afghahi A, et al. (2018). Macrophages promote circulating tumor cell-mediated local recurrence following radiotherapy in immunosuppressed patients. Cancer Res. 78, 4241–4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapp M, Grassmann S, Chaloupka M, Layritz P, Kruger S, Ormanns S, Rataj F, Janssen K-P, Endres S, Anz D, and Kobold S (2015). C-C chemokine receptor type-4 transduction of T cells enhances interaction with dendritic cells, tumor infiltration and therapeutic efficacy of adoptive T cell transfer. OncoImmunology 5, e1105428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapp M, Wintergerst MWM, Kunz WG, Vetter VK, Knott MML, Lisowski D, Haubner S, Moder S, Thaler R, Eiber S, et al. (2019). CCL22 controls immunity by promoting regulatory T cell communication with dendritic cells in lymph nodes. J. Exp. Med 216, 1170–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel A, Shorthouse D, Haas L, Hall BA, and Shields J (2016). Tumor-induced stromal reprogramming drives lymph node transformation. Nat. Immunol 17, 1118–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N, and Krummel MF (2016). Critical role for CD103(+)/CD141(+) dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell 30, 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, Daniel D, Hwang ES, Rugo HS, and Coussens LM (2014). Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 26, 623–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, et al. (2018). Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175, 998–1013.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon H, Remark R, Gnjatic S, and Merad M (2019). Host tissue determinants of tumour immunity. Nat. Rev. Cancer 19, 215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert L, Werba G, Tiwari S, Giao Ly NN, Alothman S, Alqunaibit D, Avanzi A, Barilla R, Daley D, Greco SH, et al. (2016). The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 532, 245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabaneh TB, Molodtsov AK, Steinberg SM, Zhang P, Torres GM, Mohamed GA, Boni A, Curiel TJ, Angeles CV, and Turk MJ (2018). Oncogenic BRAF(V600E) governs regulatory T-cell recruitment during melanoma tumorigenesis. Cancer Res. 78, 5038–5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma MD, Rodriguez PC, Koehn BH, Baban B, Cui Y, Guo G, Shimoda M, Pacholczyk R, Shi H, Lee EJ, et al. (2018). Activation of p53 in immature myeloid precursor cells controls differentiation into Ly6c+CD103+ monocytic antigen-presenting cells in tumors. Immunity 48, 91–106.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Schuster R, Stringer KF, Waltz SE, and Lentsch AB (2006). The Duffy antigen/receptor for chemokines (DARC) regulates prostate tumor growth. FASEB J. 20, 59–64. [DOI] [PubMed] [Google Scholar]
- Siddiqui I, Erreni M, van Brakel M, Debets R, and Allavena P (2016). Enhanced recruitment of genetically modified CX3CR1-positive human T cells into Fractalkine/CX3CL1 expressing tumors: importance of the chemokine gradient. J. Immunother. Cancer 4, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, et al. (2019). Intratumoral Tcf1+PD-1+CD8+ T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 50, 195–211.e10. [DOI] [PubMed] [Google Scholar]
- Singleton DC, and Harris AL (2012). Targeting the ATF4 pathway in cancer therapy. Expert Opin. Ther. Targets 16, 1189–1202. [DOI] [PubMed] [Google Scholar]
- Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remédios C, et al. (2014). Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med 20, 1301–1309. [DOI] [PubMed] [Google Scholar]
- Soucek L, Lawlor ER, Soto D, Shchors K, Swigart LB, and Evan GI (2007). Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat. Med 13, 1211–1218. [DOI] [PubMed] [Google Scholar]
- Spranger S, Bao R, and Gajewski TF (2015). Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235. [DOI] [PubMed] [Google Scholar]
- Spranger S, Dai D, Horton B, and Gajewski TF (2017). Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 31, 711–723.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, and Gajewski TF (2013). Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl. Med 5, 200ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, and Krek W (2003). Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature 425, 307–311. [DOI] [PubMed] [Google Scholar]
- Steinberg SM, Shabaneh TB, Zhang P, Martyanov V, Li Z, Malik BT, Wood TA, Boni A, Molodtsov A, Angeles CV, et al. (2017). Myeloid cells that impair immunotherapy are restored in melanomas with acquired resistance to BRAF inhibitors. Cancer Res. 77, 1599–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Sun J, Song B, Zhang L, Shao Q, Liu Y, Yuan D, Zhang Y, and Qu X (2016). Fucoidan inhibits CCL22 production through NF-κB pathway in M2 macrophages: a potential therapeutic strategy for cancer. Sci. Rep 6, 35855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Wang Y, Chlewicki LK, Zhang Y, Guo J, Liang W, Wang J, Wang X, and Fu YX (2016). Facilitating T cell infiltration in tumor microenvironment overcomes resistance to PD-L1 blockade. Cancer Cell 29, 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi K, and Karin M (2018). NF-κB, inflammation, immunity and cancer: coming of age. Nat. Rev. Immunol 18, 309–324. [DOI] [PubMed] [Google Scholar]
- Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, Sevillano M, Ibiza S, Carñellas A, Hernando-Momblona X, et al. (2018). TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 554, 538–543. [DOI] [PubMed] [Google Scholar]
- Thompson S, Martínez-Burgo B, Sepuru KM, Rajarathnam K, Kirby JA, Sheerin NS, and Ali S (2017). Regulation of chemokine function: the roles of GAG-binding and post-translational nitration. Int. J. Mol. Sci 18, 1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulvmar MH, Werth K, Braun A, Kelay P, Hub E, Eller K, Chan L, Lucas B, Novitzky-Basso I, Nakamura K, et al. (2014). The atypical chemokine receptor CCRL1 shapes functional CCL21 gradients in lymph nodes. Nat. Immunol 15, 623–630. [DOI] [PubMed] [Google Scholar]
- Vergani E, Di Guardo L, Dugo M, Rigoletto S,Tragni G, Ruggeri R, Perrone F, Tamborini E, Gloghini A, Arienti F, et al. (2016). Overcoming melanoma resistance to vemurafenib by targeting CCL2-induced miR-34a, miR-100 and miR-125b. Oncotarget 7, 4428–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetrano S, Borroni EM, Sarukhan A, Savino B, Bonecchi R, Correale C, Arena V, Fantini M, Roncalli M, Malesci A, et al. (2010). The lymphatic system controls intestinal inflammation and inflammation-associated Colon Cancer through the chemokine decoy receptor D6. Gut 59, 197–206. [DOI] [PubMed] [Google Scholar]
- Villablanca EJ, Raccosta L, Zhou D, Fontana R, Maggioni D, Negro A, Sanvito F, Ponzoni M, Valentinis B, Bregni M, et al. (2010). Tumor-mediated liver X receptor-alpha activation inhibits CC chemokine receptor-7 expression on dendritic cells and dampens antitumor responses. Nat. Med 16, 98–105. [DOI] [PubMed] [Google Scholar]
- Walens A, DiMarco AV, Lupo R, Kroger BR, Damrauer JS, and Alvarez JV (2019). CCL5 promotes breast cancer recurrence through macrophage recruitment in residual tumors. eLife 8, e43653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Yang L, Yu W, Wu Q, Lian J, Li F, Liu S, Li A, He Z, Liu J, et al. (2019a). Colorectal cancer cell-derived CCL20 recruits regulatory T cells to promote chemoresistance via FOXO1/CEBPB/NF-κB signaling. J. Immunother. Cancer 7, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Simons DL, Lu X, Tu TY, Solomon S, Wang R, Rosario A, Avalos C, Schmolze D, Yim J, et al. (2019b). Connecting blood and intratumoral Treg cell activity in predicting future relapse in breast cancer. Nat. Immunol 20, 1220–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing JB, Tanaka A, and Sakaguchi S (2019). Human FOXP3+ regulatory T cell heterogeneity and function in autoimmunity and cancer. Immunity 50, 302–316. [DOI] [PubMed] [Google Scholar]
- Wu Q, Zhou W, Yin S, Zhou Y, Chen T, Qian J, Su R, Hong L, Lu H, Zhang F, et al. (2019). Blocking triggering receptor expressed on myeloid cells-1-positive tumor-associated macrophages induced by hypoxia reverses immunosuppression and anti-programmed cell death ligand 1 resistance in liver cancer. Hepatology 70, 198–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Xu W, Wen Z, and Xiong S (2011). In situ prior proliferation of CD4+ CCR6+ regulatory T cells facilitated by TGF-β secreting DCs is crucial for their enrichment and suppression in tumor immunity. PLoS ONE 6, e20282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Yan C, Vilgelm AE, Chen S-C, Ayers GD, Johnson CA, and Richmond A (2020). Targeted deletion of CXCR2 in myeloid cells alters the tumor immune environment to improve antitumor immunity. Cancer Immunol. Res Published online November 11, 2020. 10.1158/2326-6066.CIR-20-0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelenay S, van der Veen AG, Böttcher JP, Snelgrove KJ, Rogers N, Acton SE, Chakravarty P, Girotti MR, Marais R, Quezada SA, et al. (2015). Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 162, 1257–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CY, Qi Y, Li XN, Yang Y, Liu DL, Zhao J, Zhu DY, Wu K, Zhou XD, and Zhao S (2015). The role of CCL20/CCR6 axis in recruiting Treg cells to tumor sites of NSCLC patients. Biomed. Pharmacother 69, 242–248. [DOI] [PubMed] [Google Scholar]
- Zhang Q, He Y, Luo N, Patel SJ, Han Y, Gao R, Modak M, Carotta S, Haslinger C, Kind D, et al. (2019). Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell 179, 829–845.e20. [DOI] [PubMed] [Google Scholar]