

Abstract
Background:
Tolebrutinib is an oral, central nervous system–penetrant, irreversible inhibitor of Bruton’s tyrosine kinase, an enzyme expressed in B lymphocytes and myeloid cells (including microglia), which are major drivers of inflammation in multiple sclerosis (MS). The aim of this study was to determine the dose-response relationship between tolebrutinib and the reduction in new active brain lesions in patients with relapsing MS.
Methods:
This 16-week, phase 2b, randomised, placebo-controlled, crossover, dose-finding trial (NCT03889639) assessed tolebrutinib efficacy and safety in relapsing MS (relapsing-remitting MS or relapsing secondary progressive MS). A two-step randomisation process, conducted using an interactive voice/web response system, first subdivided the enrolled patients into two cohorts (1:1), and then each cohort (1:1:1:1) into four dosing arms (5, 15, 30, and 60 mg once daily). Cohort 1 received tolebrutinib for 12 weeks, then placebo for 4 weeks; in Cohort 2, the regimen was vice versa. Magnetic resonance imaging was performed every 4 weeks. The primary efficacy endpoint was the number of new gadolinium (Gd)-enhancing lesions detected after 12 weeks of tolebrutinib treatment (Cohort 1, week 12; Cohort 2, week 16), relative to the scan 4 weeks prior, compared to the lesions accumulated during 4 weeks of placebo run-in period in Cohort 2. Efficacy data were analysed on a modified intention-to-treat population, using a two-step multiple comparison procedure with modelling analysis.
Findings:
The study has been completed; it was conducted between May 14, 2019, and January 2, 2020. Of the 130 participants, assessed in 40 centres across 10 countries in Europe and North America (5 mg, n=33; 15 mg, n=32; 30 mg, n=33; 60 mg, n=32), 129 completed the treatment. At Week 12, there was a dose-dependent reduction in new Gd-enhancing lesions (mean ± standard deviation lesions/patient: placebo, 1·03 ± 2·50; 5 mg, 1·39 ± 3·20; 15 mg, 0·77 ± 1·48; 30 mg, 0·76 ± 3·31; 60 mg, 0·13 ± 0·43; p=0·03). One serious adverse event (hospitalisation due to MS relapse; 60 mg group) was reported. The most common non-serious adverse event was headache (all participants, 7% [9/130]; 5 mg, 3% [1/33]; 15 mg, 9% [3/32]; 30 mg, 3% [1/33]; 60 mg, 13% [4/32]). No safety-related discontinuations occurred.
Interpretation:
Twelve-week tolebrutinib treatment led to a dose-dependent reduction in new Gd-enhancing lesions (the 60-mg dose was most effective) and was well tolerated. Effective treatment for acute inflammation, combined with the potential to directly modulate the immune response within the central nervous system, provides a scientific rationale to pursue phase 3 clinical trials in relapsing and progressive forms of MS.
Funding:
Sanofi.
INTRODUCTION
Multiple sclerosis is an immune-mediated, inflammatory, demyelinating disorder leading to axon loss, neurological morbidity, and accumulating disability.1 Current immunomodulatory treatments can result in annualised relapse rates as low as 0·102–4; however, these results are offset by less impressive disability outcomes. Because existing treatments primarily affect peripheral adaptive immunity, we tested a new approach that combines peripheral and central nervous system (CNS) immunomodulation. Our goal was to simultaneously reduce acute and chronic neuroinflammation. The latter is thought to contribute to tissue loss and disability accumulation, and is the most significant unmet medical need for patients with MS.5–7
Bruton’s tyrosine kinase (BTK), a non-receptor tyrosine kinase expressed in most haematopoietic cells (excluding T cells and fully differentiated plasma cells), couples specific cell-surface receptors to downstream signalling pathways, linking immune stimulus to cellular activation.8–11 It is a critical signalling element in B lymphocytes and myeloid cells, including peripheral monocytes/macrophages and CNS-resident microglia.11 Thus, BTK inhibition was hypothesised to reduce the acute inflammation associated with contrast-enhancing lesions by modulating (rather than depleting) B lymphocytes.3,4,12–14 In addition, inhibiting BTK in the CNS could have beneficial effects on chronic lesions and meningeal inflammatory infiltrates.15 Such lesions contain microglia, perivascular or meningeal macrophages, and B-lineage cells,16,17 and have proven to be resistant to therapeutic intervention. Thus, targeting B and myeloid cells in the CNS and periphery may have a greater—and perhaps synergistic—impact on neuroinflammation and demyelination compared to current disease-modifying therapies.
Tolebrutinib is an orally available small molecule that irreversibly binds to and inhibits BTK. Phase 1 studies demonstrated pharmacologically relevant drug levels in cerebrospinal fluid. For example, a single oral administration of 120 mg in healthy volunteers resulted in the mean concentration in the cerebrospinal fluid of 4.1 nM after 2 hours, which is therapeutically relevant exposure based on cellular assays.18,19
This study aimed to establish a dose-response relationship for tolebrutinib in relapsing MS (RMS) using magnetic resonance imaging (MRI) measures of disease activity, which have been validated as being predictive of reduction in clinical relapse rates.20 It was further designed to determine the magnitude of effect on MRI lesions. We also pursued exploratory MRI measurements, such as “slowly evolving lesions”7 and “paramagnetic rim lesions”.21 These chronic lesions, resistant to approved therapies, are associated with activated microglia17,22 and are correlated to disability accumulation in patients with MS.23,24
The innovative crossover trial design was the first to use a 4-week placebo period rather than a dedicated placebo arm, in order to minimise exposure to placebo. It also ensured that all participants received active treatment for 12 weeks. Relative to traditional approaches, the multiple comparison procedure with modelling (MCP-Mod) analysis decreased the sample size necessary to establish a dose-response relationship. This design minimises the burdens and risks associated with trial participation.
METHODS
STUDY DESIGN AND PATIENTS
This was a randomised, placebo-controlled, double-blind, crossover, dose-ranging, phase 2b trial (ClinicalTrials.gov: NCT03889639; EudraCT: 2018-003927-12; WHO: U1111-1220-0572). Following screening, participants were randomised equally to one of four tolebrutinib dosing arms (5 mg, 15 mg, 30 mg, or 60 mg) within each of the two cohorts (for a total of eight treatment arms). At baseline, Cohort 1 initiated a 12-week tolebrutinib regimen and crossed over to placebo during the remaining 4 weeks of the trial; participants in Cohort 2 began with 4 weeks of placebo treatment and then crossed over to 12 weeks of tolebrutinib (figure S1, Supplementary Appendix, page 22). Although technically conducted as a crossover trial for blinding purposes, the 4-week placebo run-out from Cohort 1 was used for additional safety evaluation only, not for efficacy analyses.
Eligibility to participate in the trial included age 18–55 years and having a relapsing MS diagnosis (relapsing-remitting MS or relapsing secondary progressive MS) according to the 2013 clinical course revisions,25 as specified in the 2017 McDonald diagnostic criteria.26 (This is also consistent with the 2020 clarification of the 2013 clinical course descriptors.27) Participants had at least one relapse within the prior year, at least two relapses within the prior 2 years, or at least one active gadolinium (Gd)-enhancing brain lesion in the 6 months prior to screening. Exclusion criteria included diagnosis of primary progressive MS,26 diagnosis of secondary progressive MS without relapse, Expanded Disability Status Scale (EDSS) score >5·5 at screening, and relapses occurring within 30 days of randomisation (full exclusion criteria are provided in the Supplementary Appendix 2, page 6). For the purpose of this study, relapses were defined as acute, new neurological symptoms or worsening of previous neurological symptoms with an objective change on neurological examination. The symptoms had to be attributable to MS, at least 24 hours in duration, and be present at normal body temperature. An episode consistent with this definition, or an episode in which relapse could not be ruled out, required an EDSS assessment. Participants provided written informed consent, which was obtained during the screening period, from 4 weeks to 1 day before randomisation; therefore, prior to any study-related procedures.
The trial was conducted in accordance with the principles of the Declaration of Helsinki, the guidelines for Good Clinical Practice of the International Conference of Harmonisation, and additional local regulations. All procedures were approved by local institutional ethics review boards of participating sites.
RANDOMISATION AND MASKING
The randomisation list was created by Almac Clinical Technologies (San Francisco, CA, USA). Patients were randomised centrally, using an interactive voice/web response system, in a two-step procedure. In the first step, participants were randomised 1:1 to Cohort 1 or Cohort 2. In the second step, conducted within each cohort, participants were randomised 1:1:1:1 to one of the four tolebrutinib dosages (5 mg, 15 mg, 30 mg, or 60 mg). Participants and investigators were blinded for dose and tolebrutinib-placebo administration sequence. Identical-looking tablets were given for each tolebrutinib dose and placebo. Investigators, study team members, and study participants did not have access to unblinded data.
PROCEDURES
Double-blind study medication was administered orally once daily with or without food; participants were instructed to take the tablet under the same dietary conditions each day. MRI scans, including T2-weighted fluid-attenuation inversion recovery and Gd-enhanced T1-weighted scans, were obtained at screening and at scheduled trial visits every 4 weeks (±3 days) over 16 weeks. The screening MRI scans were to be performed as close to day 1 as possible, and were used in lieu of day 1 scans as comparators for assessment of changes at week 4. Additional assessments at each visit included pharmacokinetics (details are provided in the Supplementary Appendix 4, page 11). Gd-enhancing lesions were identified by one or more radiologists blinded to treatment assignment at an independent, central facility (NeuroRx, Montréal, Canada). New or enlarging T2 lesions were identified from sequential scans using a semi-automated process of joint time point segmentation, followed by visual inspection.28 In other words, the number of new or enlarging T2 lesions for each patient was determined by comparing scans at week 12 (Cohort 1) or week 16 (Cohort 2) with those obtained at the previous scan (week 8 for Cohort 1 and week 12 for Cohort 2). Analyses of slowly evolving lesions and paramagnetic rim lesions are detailed in Supplementary Appendix 6 (page 16). In brief, slowly evolving lesions were identified as contiguous areas of the baseline T2-lesion mask that showed constant and concentric local expansion from baseline to week 16, where local expansion is determined via Jacobian analysis.7 Paramagnetic rim lesions were manually identified on susceptibility-weighted images generated from six 3D-gradient echo-phase images (echo time ranging from 4·9–41 ms, resolution 0·8 x 0·8 x 0·8 mm).29 The schedule of study assessments, including efficacy, safety, and pharmacokinetics, is provided in Supplementary Appendix 4 (page 11). The details of safety laboratory assessments are summarised in Supplementary Appendix 5 (page 14).
OBJECTIVES AND ENDPOINTS
The primary objective was to determine the dose-response relationship between tolebrutinib and new active brain lesions detected using MRI. The primary efficacy endpoint was the number of new Gd-enhancing lesions detected on the scan performed after 12 weeks of tolebrutinib treatment (at week 12 for Cohort 1 and week 16 for Cohort 2), relative to the scan 4 weeks prior. Secondary endpoints were the number of new or enlarging T2 lesions detected on the same scan observed after 12 weeks of treatment, the total number of Gd-enhancing lesions after 12 weeks of tolebrutinib treatment (ie, all Gd-enhancing lesions on the scan at the end of 12 weeks of treatment), as well as adverse events (AEs), serious AEs, and AEs of special interest. Pharmacokinetics was a tertiary endpoint. In total, there were 15 prespecified exploratory endpoints and 23 prespecified MRI analyses; this report focuses on endpoints most pertinent to the recently initiated phase 3 trials of tolebrutinib in both relapsing (NCT04410978, NCT04410991) and progressive MS (NCT04411641, NCT04458051). Two exploratory endpoints of particular interest are reported here, the volume of slowly evolving lesions by dose arm, and the number of paramagnetic rim lesions in all active treatment arms combined in a subset of patients at sites with sufficient imaging capability. These endpoints are of considerable interest based on our working hypothesis that a brain-penetrant immunomodulator may be able to reduce these treatment-resistant lesions associated with neuroinflammation and provide benefit on disability progression in MS patients. A list of all exploratory endpoints is provided in Supplementary Appendix 7 (page 18).
STATISTICAL ANALYSIS
Based on the assumption that the rate of formation of new Gd-enhancing lesions in placebo-treated patients will remain constant over a period of 16 weeks, we used the 4-week placebo run-in data from Cohort 2, prior to the initiation of tolebrutinib treatment, as the comparator for week 12 (Cohort 1) or week 16 (Cohort 2) data in analyses for the primary and secondary efficacy endpoints. The 4-week placebo run-out from Cohort 1 was used for blinding purposes and additional safety evaluations.
A sample size of 120 participants, assuming a dropout rate of up to 15%, was estimated to provide ≥83% power to detect a maximum 85% reduction using a two-step multiple comparison procedure with modelling (MCP-Mod) technique process with six pre-defined dose-response curves (quadratic, linear, exponential, logistic, and two Emax With effective doses [ED50] of 10 mg and 30 mg), as described previously.30 The primary and secondary efficacy analyses were done on the modified intent-to-treat population, defined as all participants exposed to study intervention, in the treatment arm to which they were randomised. Analyses were based on pooled data from Cohorts 1 and 2 for each tolebrutinib dose, and dose response was evaluated using the two-step MCP-Mod with a negative logarithm transformation of the mean lesion count as input to the procedure. Step 1 tested for an efficacy signal versus the null hypothesis of a flat, non–dose-response curve. The primary endpoint was jointly evaluated for each of the six prespecified dose-response models with a contrast test that controlled the familywise error rate at two-sided alpha of 0·05. Step 2, undertaken only if Step 1 showed an efficacy signal, estimated a best-fit (smallest generalised Akaike information criterion) dose-response curve from the six dose-response models.
Mean lesion counts in each of the four dose arms at the end of 12-week tolebrutinib treatment and the end of 4-week placebo treatment were estimated using a negative binomial regression model with covariates of baseline Gd-enhancing T1 lesions (presence or absence; for new Gd-enhancing lesions and total number of Gd-enhancing lesions) and treatment arm. A generalised estimating equation approach was used to account for the potential correlation between the measurements in the 4-week placebo period and the subsequent tolebrutinib treatment period in Cohort 2 participants. Relative reductions with 95% confidence intervals in MRI lesions for each tolebrutinib dose versus placebo were estimated from the negative binomial model. There was no imputation of missing data.
Pharmacokinetic analyses included area under the curve (AUC0–24) measurements of parent drug concentrations (expressed as ng·hr/mL) by dose over 12 weeks of tolebrutinib treatment. The impact of fed status on tolebrutinib plasma exposure was determined for the full study population, imputing fed conditions for missing data. The relationship between AUC0–24 and MRI lesion count was also determined after 12 weeks of treatment as an exploratory analysis. Exploratory outcomes were analysed using descriptive statistics only.
Safety data were summarised by tolebrutinib dose for the overall population and reported over weeks 1–4 for both placebo and tolebrutinib treatment and over 12 weeks for tolebrutinib alone (ie, AEs observed between weeks 0–12 in Cohort 1 and weeks 4–16 in Cohort 2).
ROLE OF THE FUNDING SOURCE
The sponsor (Sanofi) designed the trial and was involved in data collection, analysis, and interpretation, along with manuscript drafting. The sponsor provided tolebrutinib and funded professional writing assistance. Safety was overseen by an independent data monitoring committee. The authors attest to adherence to trial protocol, along with the accuracy and completeness of the data reporting. The final version of the manuscript was approved by all authors, who had access to all the data in the study and accept responsibility for submitting this paper for publication.
RESULTS
The study was conducted across 40 centres in Europe and North America between March 27, 2019, and January 3, 2020. Out of 130 randomised participants (Cohort 1, n=64; Cohort 2, n=66), 129 (99%) successfully completed treatment (figure 1). One participant in the tolebrutinib 60-mg arm withdrew from treatment and follow-up due to unwillingness to comply with the protocol-mandated contraception requirement. Mean ± standard deviation (SD) exposure to tolebrutinib was 83 ± 2 days (5 mg), 83 ± 4 days (15 mg), 84 ± 2 days (30 mg), and 82 ± 7 days (60 mg). Exposure to placebo was 28 ± 2 days.
Figure 1: Participant disposition and study treatment exposure.
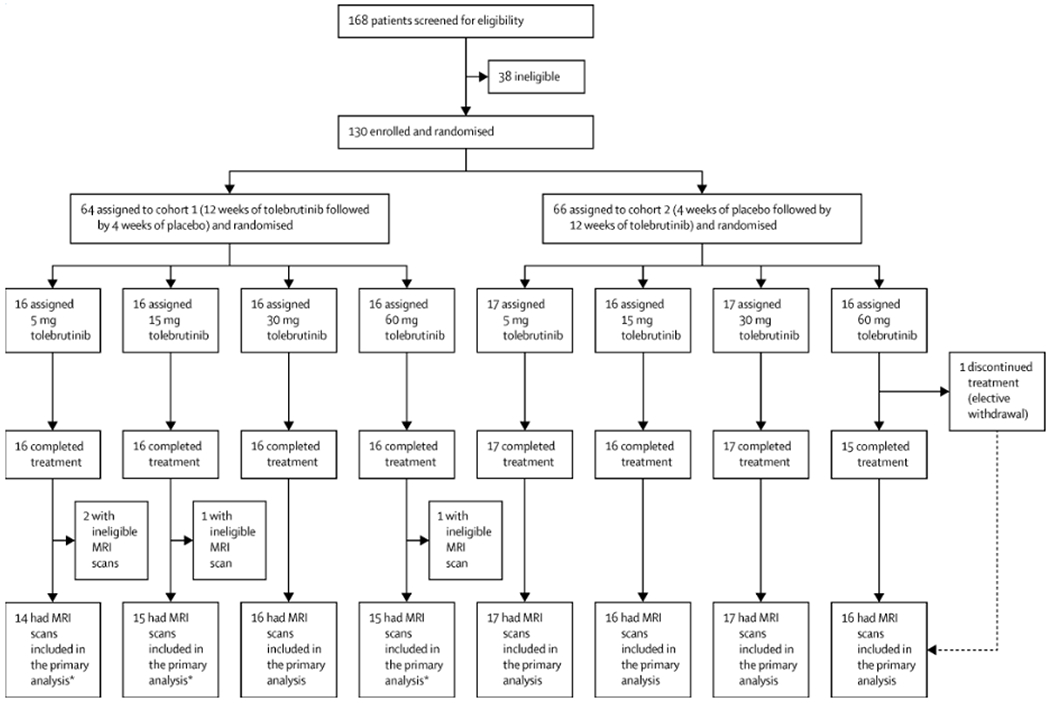
Participants were randomised in two steps: first (1:1) into two cohorts and then (1:1:1:1) into four dosage groups within each cohort. equally into one of eight treatment arms that were divided between two cohorts. Cohort 1 received tolebrutinib for 12 weeks beginning at baseline, followed by 4 weeks of placebo run-out in order to blind the treatment assignment and to provide additional safety data. Cohort 2 received placebo for 4 weeks as a run-in, followed by tolebrutinib for 12 weeks. Of the 130 participants randomised, 129 completed the treatment, and the participant with early treatment discontinuation still completed the study. For the primary analysis, an MRI assessment was excluded from analysis if the participant had received systemic corticosteroids within the 30 days prior to the MRI assessment date. Thus, MRI assessments from 126 of 130 participants were included in the primary analysis (Cohort 1: 5 mg, n=14; 15 mg, n=15; 30 mg, n=16; 60 mg, n=15. Cohort 2: 5 mg, n=17; 15 mg, n=16; 30 mg, n=17; 60 mg, n=16). R=randomisation.
Overall, 98% (128/130) of participants had diagnoses of relapsing-remitting MS and 2% (2/130) were diagnosed with secondary progressive MS with relapse. Thirty-five percent (44/130) of participants had Gd-enhancing lesions at baseline. Baseline characteristics were well balanced across treatment arms (table 1).
Table 1:
Demographic and disease characteristics of participants at baseline
|
|
||||||
|---|---|---|---|---|---|---|
| Tolebrutinib |
||||||
| All participants (N=130) | Placeboa (N=66) | 5 mg (n=33) | 15 mg (n=32) | 30 mg (n=33) | 60 mg (n=32) | |
| Age, years | 37 (10) | 36 (10) | 36 (10) | 36 (9) | 39 (10) | 37 (9) |
| Female, n (%) | 91 (70) | 46 (70) | 25 (76) | 21 (66) | 21 (64) | 24 (75) |
| Highly active disease, n (%) | 61 (47) | 29 (44) | 12 (36) | 19 (59) | 16 (48) | 14 (44) |
| RRMS,b n (%) | 128 (99) | 65 (99) | 33 (100) | 32 (100) | 32 (97) | 31 (97) |
| Time since initial relapse, years | 7·8 (7·4) | 7·7 (7·4) | 7·7 (7·8) | 8·0 (7·6) | 8·1 (7·8) | 7·3 (6·7) |
| Relapses in previous year | 1·2 (0·6) | 1·2 (0·5) | 1·2 (0·5) | 1·3 (0·6) | 1·3 (0·6) | 1·2 (0·4) |
| Relapses in previous 2 years | 1·7 (0·9) | 1·7 (0·7) | 1·7 (0·8) | 1·5 (0·8) | 1·8 (1·1) | 1·6 (0·9) |
| EDSS score | 2·5 (1·1) | 2·6 (1·1) | 2·5 (1·0) | 2·2 (1·0) | 2·6 (1·3) | 2·7 (1·2) |
| T2 hyperintense lesion volume, cm3 | 12·76 (11·31) | 12·95 (11·35) | 12·14 (9·86) | 11·74 (9·53) | 14·72 (13·80) | 12·41 (11·78) |
| T1 hypointense lesion volume, cm3 | 0·95 (1·62) | 1·07 (1·97) | 0·49 (0·52) | 1·02 (1·45) | 1·46 (2·11) | 0·85 (1·83) |
| Number of Gd-enhancing lesions | 1·8 (4·7)c | 2·2 (5·9) | 2·3 (5·9)d | 0·7 (1·8)e | 1·9 (4·9)d | 2·1 (4·9) |
| Participants with baseline Gd-enhancing lesions, n (%) | 44 (35)c | 25 (38) | 11 (34)d | 7 (23)e | 11 (34)d | 15 (47) |
Values are mean (SD) except where noted. Highly active disease was defined as 1 relapse in the year prior to screening and ≥1 Gd-enhancing lesion on MRI performed within 6 months prior to screening or ≥9 T2 lesions at baseline or ≥2 relapses in the year prior to screening.
Includes data from placebo-treated period of Cohort 2, which began tolebrutinib treatment at week 4 after the placebo run-in.
Two participants had a diagnosis of relapsing secondary progressive MS.
N=127.
N=32.
N=31.
EDSS=Expanded Disability Status Scale. Gd=gadolinium. MRI=magnetic resonance imaging. MS=multiple sclerosis. RRMS=relapsing-remitting MS. SD=standard deviation.
Using MCP-Mod methodology, the exponential model demonstrated a dose-response relationship between tolebrutinib and new Gd-enhancing lesions, which was used to reject the null hypothesis of a flat dose-response curve (test statistic 2·47; p=0·03; figure 2A). Maximal effect was observed with the 60-mg dose and corresponded to an adjusted relative reduction in new Gd-enhancing lesions of 85% (95% confidence interval [CI] 28–97%) versus placebo. The observed mean ± SD number of lesions was 0·13 ± 0·43 for tolebrutinib 60 mg versus 1·03 ± 2·50 for placebo (figure 2B). After 12 weeks of treatment, 90% (28/31) of participants who received tolebrutinib 60 mg had no new Gd-enhancing lesions, compared with 75% (44/59) of patients in the placebo arm (based on 4 weeks of placebo treatment).
Figure 2: Primary outcome: Number of new Gd-enhancing lesions on an MRI scan after 12 weeks of tolebrutinib treatment.
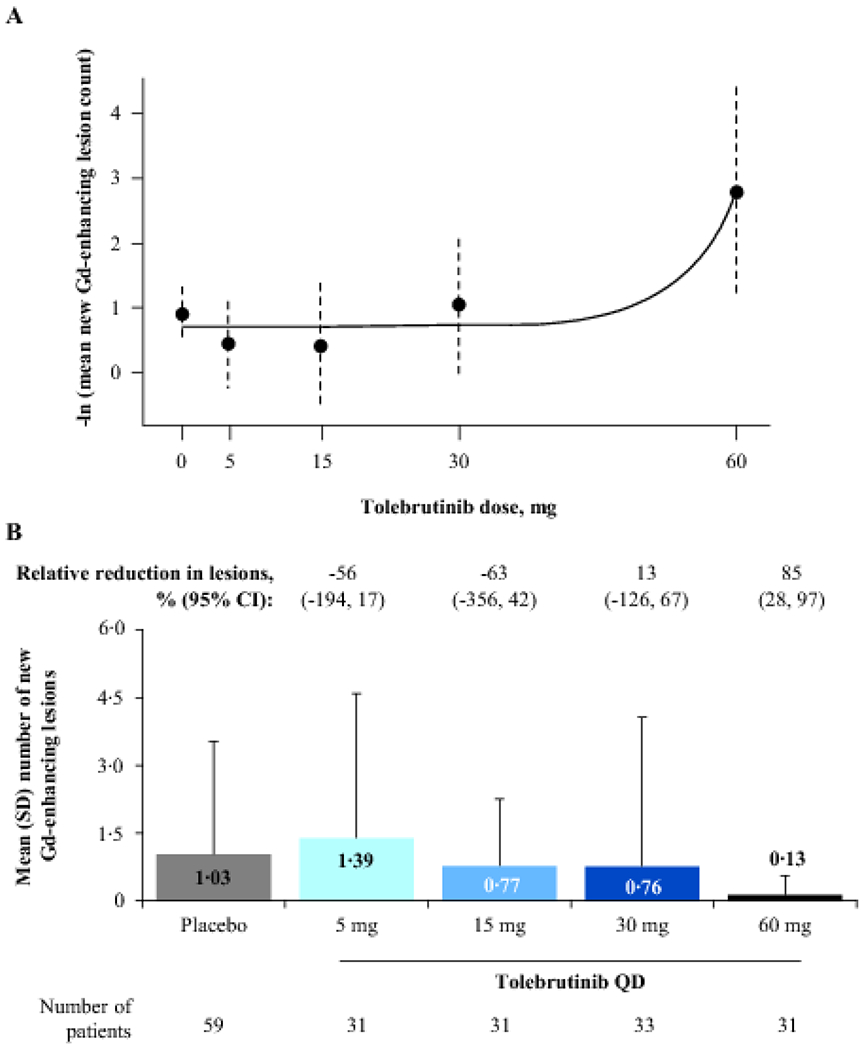
Panel A shows the estimated dose-response curve after applying the two-step MCP-Mod methodology; error bars represent 95% CIs. The exponential model was selected as the best fit based on the Akaike information criterion and was used to reject the null hypothesis (p=0·03). Panel B shows the mean (error bars: SD) number of new Gd-enhancing lesions per scan for pooled participants from Cohorts 1 and 2 at the end of 12 weeks of tolebrutinib treatment compared with a scan 4 weeks prior. In Cohort 2, the formation rate of new Gd-enhancing lesions over the 4-week placebo run-in period was assumed to be constant over the 16-week trial duration. Values are relative reduction in lesion count versus placebo, adjusted for presence or absence of baseline Gd-enhancing lesions, using a negative binomial model. CI=confidence interval. Gd=gadolinium. MCP-Mod=multiple comparison procedure-modelling. MRI=magnetic resonance imaging. QD=once daily. SD=standard deviation.
Analysis of secondary endpoints showed a dose-response relationship for the number of new or enlarging T2 lesions, with the linear model providing the best fit (test statistic 4·32; p<0·0001; figure 3A). Following 12 weeks of tolebrutinib treatment, a maximal effect was again observed with the 60-mg dose, with an 89% (95% CI 68–96%) relative reduction in the mean ± SD number of new or enlarging T2 lesions versus placebo (figure 3B: tolebrutinib 60 mg, 0·23 ± 0·62; placebo, 2·12 ± 5·16), and with 87% (27/31) of participants free of new or enlarging T2 lesions (placebo, 66% [39/59]: based on 4 weeks of placebo treatment). There was no significant dose response for the total number of Gd-enhancing lesions at the end of 12 weeks of treatment (figure 3C); therefore, dose-response modelling was not conducted for this outcome.
Figure 3: Secondary outcome: Number of new/enlarging T2 lesions and total number of Gd-enhancing lesions on an MRI scan after 12 weeks of tolebrutinib treatment.
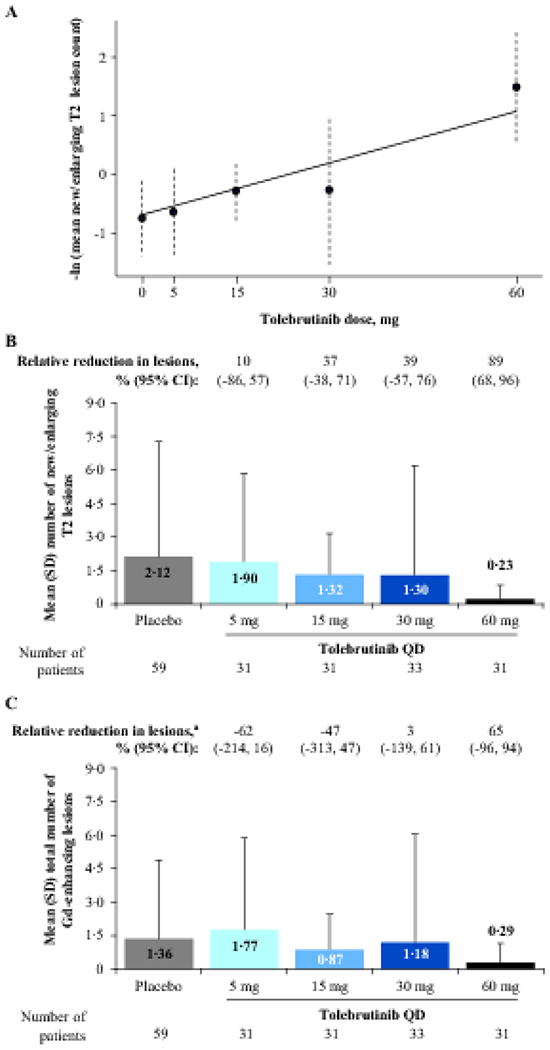
Panel A shows the estimated dose-response curve using MCP-Mod methodology for the number of new or enlarging T2 lesions; error bars represent 95% CIs. The linear model was selected as the best fit based on the Akaike information criterion and was used to reject the null hypothesis (p<0·0001). Panel B shows the mean (SD) number of new or enlarging T2 lesions for pooled participants from Cohorts 1 and 2 at the end of 12 weeks of tolebrutinib treatment. In Cohort 2, the formation rate of new Gd-enhancing lesions over the 4-week placebo run-in period was assumed to be constant over the 16-week trial duration. Relative reductions in lesions versus placebo were analysed using a negative binomial model. Panel C shows the total number of Gd-enhancing lesions at the end of 12 weeks of tolebrutinib treatment, for which the MCP-Mod procedure did not allow for rejection of the null hypothesis. aRelative reduction in lesions versus placebo, with adjustment for baseline presence or absence of Gd-enhancing lesion activity using a negative binomial model. CI=confidence interval. Gd=gadolinium. MCP-Mod=multiple comparison procedure-modelling. MRI=magnetic resonance imaging. QD=once daily. SD=standard deviation.
The pharmacokinetic assessment over 12 weeks of treatment detected an approximate 2·5-fold exposure increase between the 30- and 60-mg doses (mean AUC [coefficient of variation, %] 19·5 ng·h/mL [72%] versus 49·7 ng·h/mL [94%]; table S1, Supplementary Appendix, page 27). Meal status was well documented in 35% (135/389) of visits, in which 85% (115/135) included participants who have taken the treatment with food. Analysis comparing administration with or without food suggested an approximately two-fold higher plasma concentration of tolebrutinib when the 60-mg dose was administered with food (figure S3, Supplementary Appendix, page 24). To control for differences in drug exposure related to food intake, lesion counts were analysed as a function of AUC for each participant. Tolebrutinib exposures greater than 40 ng·hr/mL were associated with few new Gd-enhancing lesions and few new or enlarging T2 lesions (figure S4, Supplementary Appendix, page 25).
The median (interquartile range) total volume of slowly evolving lesions at week 16 was assessed as an exploratory analysis: 5 mg, 1·37 (3·61) cm3; 15 mg, 1·31 (2·59) cm3; 30 mg, 1·35 (2·46) mm3; 60 mg, 0·70 (2·31) cm3 (figure S5, Supplementary Appendix, page 26). Data from participants (n=32) evaluated in centres with susceptibility-weighted imaging capability show that 50% (16/32) of participants had paramagnetic rim lesions at baseline (figure S2, Supplementary Appendix, page 23).
The occurrence of AEs was similar across all tolebrutinib arms, with 50% (16/32) of participants treated with tolebrutinib 60 mg reporting at least one AE over 12 weeks (table 2). No AE led to treatment discontinuation or study discontinuation. Twelve AEs occurred in more than two participants during tolebrutinib treatment, of which headache (7% [9/130]), upper respiratory tract infection (5% [6/130]), and nasopharyngitis (4% [5/130]) were most frequent. An analysis of weeks 1–4 showed no serious AEs (table S2, Supplementary Appendix, page 28). During this 4-week period, AEs were reported by similar proportions of participants receiving tolebrutinib and placebo (13 [2/16] to 31% [5/16] tolebrutinib versus 35% [23/66] placebo), with headache being the most frequent AE across all treatment arms (n=5, four of whom received placebo). Over 12 weeks of tolebrutinib treatment, two participants had alanine aminotransferase (ALT) levels that exceeded three-fold the upper limit of normal (ULN; 30 mg, n=1; 60 mg, n=1). In the participant treated with the 30-mg dose, ALT was elevated at a single time point before returning to normal when measured 4 days later, with no interruption of treatment. The participant who received the 60-mg dose had a mild ALT elevation at screening and experienced an increase exceeding three-fold ULN at the week 4 visit at the end of the placebo run-in period. The patient initiated tolebrutinib treatment and ALT levels decreased gradually, reaching normal range at week 12, while remaining on treatment. Neither participant with elevated ALT discontinued tolebrutinib. Across doses, one serious AE was observed (severe MS relapse in a participant receiving 60-mg tolebrutinib in Cohort 1). Study drug treatment was uninterrupted. The participant recovered and completed the trial.
Table 2:
Secondary Outcome: Adverse Events During Tolebrutinib Treatment
| Tolebrutinib |
|||||
|---|---|---|---|---|---|
| Participants, n (%)a | All participants (N=130) | 5 mg (n=33) | 15 mg (n=32) | 30 mg (n=33) | 60 mg (n=32) |
| Any AE | 70 (54) | 19 (58) | 17 (53) | 18 (55) | 16 (50) |
| Severe AEb | 1 (1) | 0 | 0 | 0 | 1 (3) |
| Serious AEb | 1 (1) | 0 | 0 | 0 | 1 (3) |
| AE leading to death | 0 | 0 | 0 | 0 | 0 |
| AE leading to study discontinuation | 0 | 0 | 0 | 0 | 0 |
| Any AE leading to study treatment discontinuation | 0 | 0 | 0 | 0 | 0 |
| Any treatment-related AE | 17 (13) | 5 (15) | 1 (3) | 4 (12) | 7 (22) |
|
| |||||
| AEs occurring in >2 participants during 12 weeks of tolebrutinib treatment | |||||
| Headache | 9 (7) | 1 (3) | 3 (9) | 1 (3) | 4 (13) |
| Upper respiratory tract infection | 6 (5) | 2 (6) | 2 (6) | 1 (3) | 1 (3) |
| Nasopharyngitis | 5 (4) | 1 (3) | 0 | 1 (3) | 3 (9) |
| Back pain | 4 (3) | 1 (3) | 1 (3) | 2 (6) | 0 |
| Peripheral oedema | 4 (3) | 2 (6) | 0 | 0 | 2 (6) |
| Accidental overdose | 3 (2) | 0 | 0 | 0 | 3 (9) |
| Gastroenteritis | 3 (2) | 1 (3) | 0 | 0 | 2 (6) |
| Alanine aminotransferase increasedc | 3 (2) | 1 (3) | 0 | 1 (3) | 1 (3) |
| Respiratory tract infection | 3 (2) | 0 | 1 (3) | 1 (3) | 1 (3) |
| Muscle spasticity | 3 (2) | 0 | 0 | 1 (3) | 2 (6) |
| Oropharyngeal pain | 3 (2) | 1 (3) | 0 | 1 (3) | 1 (3) |
| Alopecia | 3 (2) | 1 (3) | 1 (3) | 0 | 1 (3) |
Percentage of participants with ≥1 event.
One participant from the tolebrutinib 60-mg arm experienced a serious AE, which was also determined to be severe; the AE was an MS relapse that was reported by an investigator due to hospitalisation for differential diagnosis.
Three participants had elevated alanine aminotransferase levels, 2 of whom (in the 30- and 60-mg arms) had levels >3 times the upper limit of normal.
AE=adverse event. MS=multiple sclerosis. See table S2 (Supplementary Appendix, page 27) for details about safety outcomes of tolebrutinib compared to placebo over weeks 1-4.
DISCUSSION
The primary objective of this trial was met, demonstrating a dose-related reduction in the number of new Gd-enhancing lesions after 12 weeks of tolebrutinib treatment. Our findings are consistent with phase 2 trials of disease-modifying therapies considered highly effective in MS.13,31 These results support continued development of tolebrutinib 60 mg in phase 3 trials with clinical endpoints, based on the well-established relationship between reductions of Gd-enhancing lesions and MS relapse rates.20,32 Due to the short treatment duration, clinical relapses and disability progression were not considered as primary or secondary endpoints in this trial.
Several pieces of data argue that the dose-response relationship was not based on the response to 60 mg only. First, the related secondary endpoint of new or enlarging T2 lesions was significant for all six dose-response models. Second, there was a steep decline in the number of Gd-enhancing lesions per scan with both the 30 mg and the 60 mg tolebrutinib dose, but the impact in the 30-mg group was masked by a single individual who had 19 Gd-enhancing lesions (figure S4, Supplementary Appendix, page 25). Finally, tolebrutinib plasma exposure was inversely related to lesion counts, and there were few new Gd-enhancing lesions in participants with high exposures.
Importantly, the exposure-response supports use of 60 mg as an appropriate dose for further evaluation of tolebrutinib in relapsing MS. Moreover, comparative pharmacokinetic analysis of the fed versus fasting states suggested maximal exposure was achieved when tolebrutinib was administered with food (figure S3, Supplementary Appendix, page 24). These results informed the dose selection for future MS trials: 60 mg once daily, taken with food.
Tolebrutinib was well tolerated, with no AEs leading to treatment withdrawal. Over 12 weeks of active treatment, 12 AEs were reported in more than two participants; headache was the most frequent. One serious (and severe) AE was reported, an MS relapse, in the tolebrutinib 60-mg arm. No other serious or severe AEs occurred over the study period. Our results suggest an acceptable safety and tolerability profile for tolebrutinib across all doses.
Imaging data demonstrated a clear and meaningful impact of tolebrutinib on acute inflammation based on the formation of new Gd-enhancing lesions. Approximately 50% of study participants at sites with susceptibility-weighted imaging capability had paramagnetic rim lesions: white matter lesions with ongoing myelin and axon destruction in association with activated microglia.21,33 These lesions can enlarge over time.23 The analysis of slowly evolving lesions7 in this study suggests that a longer course of treatment is likely necessary to assess whether BTK-driven modulation of CNS-resident microglia, infiltrating macrophages, and B lymphocytes trapped behind the blood-brain barrier would translate to clinical benefit related to slowing or halting of disability accumulation.
Of note, our findings are consistent with those of a phase 2 trial of evobrutinib, another orally administered BTK inhibitor, in patients with relapsing MS.4 However, differences in design between the two trials preclude direct comparison of efficacy.
Since the completion of this study, phase 3 studies in MS of several other BTK inhibitors have been initiated. Tolebrutinib and these other agents have distinct pharmacological profiles, differing in potency, selectivity, and CNS distribution. Based on CNS exposure and in vitro potency, tolebrutinib stands out as having the potential to achieve pharmacologically relevant exposures within this compartment. How that potential translates to clinical benefits will be evaluated in the ongoing phase 3 development program.
Limitations of this study include a relatively small sample size per dosing arm and a treatment period of only 12 weeks, which is too short to assess effects on clinical outcomes like relapses and disability. However, the objective here was to establish a safe and effective dose, which was accomplished. Longer monitoring of safety will improve our understanding of the risk-benefit profile, which will continue in the ongoing long-term safety study and four phase 3 studies underway in relapsing MS, primary progressive MS, and non-relapsing secondary progressive MS.
In summary, our study design with limited placebo exposure established an effect of tolebrutinib on MRI measures related to new lesion formation and identified an effective dose to test in phase 3 trials. Effective treatment for acute inflammation, combined with the potential to directly modulate the immune response within the central nervous system — known to be a key driver of MS clinical progression — provides scientific rationale to pursue phase 3 clinical trials in both relapsing and progressive MS.
Supplementary Material
RESEARCH IN CONTEXT.
Evidence before this study
PubMed was searched in May 2021 for clinical studies published in any language, using unrestricted dates and the terms “multiple sclerosis (MS) and Bruton’s tyrosine kinase (BTK)”, “Bruton’s tyrosine kinase inhibitor”, “chronic lesion”, or “slowly evolving lesion”. Accumulated evidence shows that chronic neuroinflammation, driven in part by B lymphocytes and activated microglia within the central nervous system, is a key contributor to disability accumulation in relapsing and progressive MS. BTK is a critical signalling element in B lymphocytes and myeloid cells. A phase 2 trial of evobrutinib, another BTK inhibitor, has provided proof of concept that targeting this enzyme in patients with MS can lead to improved clinical and magnetic resonance imaging outcomes. However, currently approved MS treatments act primarily outside the central nervous system. Tolebrutinib is an investigational BTK inhibitor that has been shown, in phase 1 studies, to penetrate the blood-brain barrier and reach pharmacologically relevant levels in the central nervous system.
Added value of this study
This phase 2 clinical trial is the first demonstration of tolebrutinib efficacy in patients with relapsing MS. Using a crossover trial design that minimised exposure to placebo, we showed a dose-dependent reduction of the number of gadolinium-enhancing brain lesions and new or enlarging T2 lesions after 12 weeks of treatment. Tolebrutinib also demonstrated a favourable safety profile.
Implications of all the available evidence
The results of this phase 2 study support investigation of tolebrutinib in phase 3 studies. In addition to the treatment effect demonstrated in relapsing MS, exploratory analysis of slowly evolving lesions raises the possibility that investigation of tolebrutinib in patients with progressive disease, in which chronic neuroinflammation is well documented, is warranted.
ACKNOWLEDGEMENTS
The authors and Sanofi thank the participants and their families for their involvement in the phase 2b trial. Critical review of the manuscript was provided by Svend S Geertsen, PhD, Bill Aschenbach, PhD, MBA, Alex L Lublin, PhD, Darren P Baker, PhD, and John C Reed, MD, PhD, of Sanofi. Statistical review was provided by Deborah Dukovic, MS, of Sanofi. Medical writing support was provided by Valerie P Zediak, PhD, and Vojislav Pejović, PhD, of Eloquent Scientific Solutions.
FUNDING
The tolebrutinib phase 2b trial was funded by Sanofi. Medical writing support for the development of this paper was funded by Sanofi.
DECLARATION OF INTERESTS
DS Reich is supported by the Intramural Research Program of NINDS (NIH) and has also received research support from Vertex. DL Arnold reports receiving consulting fees from Acorda Therapeutics, Biogen, Celgene, Genentech, GeNeuro, F Hoffmann-La Roche, Merck, Novartis, Roche, Sanofi, Teva, and Wave Life Science; financial support for research activities from Biogen Idec Canada Inc, Immunotec Inc, Novartis Canada Ltd, and Novartis Global Medical Affairs; and personal compensation from NeuroRx Research. P Vermersch reports receiving consulting and/or speaking fees and research support from Biogen, Celgene, Merck, Novartis, Roche, Sanofi, and Teva. A Bar-Or reports receiving consultancy fees and/or grant support from Biogen Idec, Genentech, GSK, Merck/EMD Serono, Novartis, Receptos, Roche, and Sanofi. RJ Fox reports receiving consulting fees from AB Science, Actelion, Biogen, Celgene, EMD Serono, Genentech, Immunic Therapeutics, Novartis, Sanofi, Teva, and TG Therapeutics and research support from Biogen and Novartis, and participating on advisory boards for Actelion, Biogen, Immunic Therapeutics, and Novartis. A Matta, T Turner, E Wallström, and X Zhang report being employees of Sanofi. M Mareţ and FA Khabirov do not have anything to disclose. A Traboulsee reports receiving compensation for consulting, serving on a scientific advisory board, speaking, or other activities from Biogen Idec, Roche, Sanofi, and Teva Innovation, along with grant and/or research support from Roche and Sanofi.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DATA SHARING STATEMENT
Qualified researchers may request access to patient-level data and related study documents including the clinical study report, study protocol with any amendments, blank case report form, statistical analysis plan, and dataset specifications. Patient-level data will be anonymised and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi’s data-sharing criteria, eligible studies, and process for requesting access can be found at https://www.clinicalstudydatarequest.com.
REFERENCES
- 1.Reich DS, Lucchinetti CF, Calabresi PA. Multiple Sclerosis. The New England journal of medicine 2018; 378(2): 169–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hauser SL, Bar-Or A, Cohen JA, et al. Ofatumumab versus Teriflunomide in Multiple Sclerosis. The New England journal of medicine 2020; 383(6): 546–57. [DOI] [PubMed] [Google Scholar]
- 3.Hauser SL, Bar-Or A, Comi G, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. The New England journal of medicine 2017; 376(3): 221–34. [DOI] [PubMed] [Google Scholar]
- 4.Montalban X, Arnold DL, Weber MS, et al. Placebo-controlled trial of an oral BTK inhibitor in multiple sclerosis. The New England journal of medicine 2019; 380(25): 2406–17. [DOI] [PubMed] [Google Scholar]
- 5.Bunai T, Terada T, Kono S, et al. Neuroinflammation following disease modifying therapy in multiple sclerosis: A pilot positron emission tomography study. J Neurol Sci 2018; 385: 30–3. [DOI] [PubMed] [Google Scholar]
- 6.Datta G, Colasanti A, Rabiner EA, et al. Neuroinflammation and its relationship to changes in brain volume and white matter lesions in multiple sclerosis. Brain 2017; 140(11): 2927–38. [DOI] [PubMed] [Google Scholar]
- 7.Elliott C, Wolinsky JS, Hauser SL, et al. Slowly expanding/evolving lesions as a magnetic resonance imaging marker of chronic active multiple sclerosis lesions. Multiple sclerosis (Houndmills, Basingstoke, England) 2019; 25(14): 1915–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hendriks RW. Drug discovery: New Btk inhibitor holds promise. Nature chemical biology 2011; 7(1): 4–5. [DOI] [PubMed] [Google Scholar]
- 9.Liang C, Tian D, Ren X, et al. The development of Bruton’s tyrosine kinase (BTK) inhibitors from 2012 to 2017: A mini-review. Eur J Med Chem 2018; 151: 315–26. [DOI] [PubMed] [Google Scholar]
- 10.Torke S, Pretzsch R, Hausler D, et al. Inhibition of Bruton’s tyrosine kinase interferes with pathogenic B-cell development in inflammatory CNS demyelinating disease. Acta Neuropathol 2020; 140(4): 535–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weber ANR, Bittner Z, Liu X, Dang TM, Radsak MP, Brunner C. Bruton’s Tyrosine Kinase: An Emerging Key Player in Innate Immunity. Front Immunol 2017; 8: 1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghajarzadeh M, Azimi A, Valizadeh Z, Sahraian MA, Mohammadifar M. Efficacy and safety of rituximab in treating patients with multiple sclerosis (MS): A systematic review and meta-analysis. Autoimmun Rev 2020; 19(8): 102585. [DOI] [PubMed] [Google Scholar]
- 13.Kappos L, Li D, Calabresi PA, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet (London, England) 2011; 378(9805): 1779–87. [DOI] [PubMed] [Google Scholar]
- 14.Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. The New England journal of medicine 2017; 376(3): 209–20. [DOI] [PubMed] [Google Scholar]
- 15.Keaney J, Gasser J, Gillet G, Scholz D, Kadiu I. Inhibition of Bruton’s Tyrosine Kinase Modulates Microglial Phagocytosis: Therapeutic Implications for Alzheimer’s Disease. J Neuroimmune Pharmacol 2019; 14(3): 448–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Magliozzi R, Howell O, Vora A, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2007; 130(Pt 4): 1089–104. [DOI] [PubMed] [Google Scholar]
- 17.Zrzavy T, Hametner S, Wimmer I, Butovsky O, Weiner HL, Lassmann H. Loss of ‘homeostatic’ microglia and patterns of their activation in active multiple sclerosis. Brain 2017; 140(7): 1900–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Francesco M, Wong M, LaStant J, et al. PRN2246, a potent and selective blood brain barrier penetrating BTK inhibitor, exhibits efficacy in central nervous system immunity. Multiple sclerosis (Houndmills, Basingstoke, England) 2017; 23(Issue 3_suppl): P989. [Google Scholar]
- 19.Smith P, Redfern J, Shu J, et al. Phase 1 clinical trial of PRN2246 (SAR442168), a covalent BTK inhibitor demonstrates safety, CNS exposure and therapeutic levels of BTK occupancy. Multiple sclerosis (Houndmills, Basingstoke, England) 2019; 25(Issue 1_suppl): P072. [Google Scholar]
- 20.Sormani MP, Bruzzi P. MRI lesions as a surrogate for relapses in multiple sclerosis: a meta-analysis of randomised trials. The Lancet Neurology 2013; 12(7): 669–76. [DOI] [PubMed] [Google Scholar]
- 21.Absinta M, Sati P, Schindler M, et al. Persistent 7-tesla phase rim predicts poor outcome in new multiple sclerosis patient lesions. J Clin Invest 2016; 126(7): 2597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jackle K, Zeis T, Schaeren-Wiemers N, et al. Molecular signature of slowly expanding lesions in progressive multiple sclerosis. Brain 2020; 143(7): 2073–88. [DOI] [PubMed] [Google Scholar]
- 23.Absinta M, Sati P, Masuzzo F, et al. Association of Chronic Active Multiple Sclerosis Lesions With Disability In Vivo. JAMA Neurol 2019; 76(12): 1474–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elliott C, Belachew S, Wolinsky JS, et al. Chronic white matter lesion activity predicts clinical progression in primary progressive multiple sclerosis. Brain 2019; 142(9): 2787–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology 2014; 83(3): 278–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. The Lancet Neurology 2018; 17(2): 162–73. [DOI] [PubMed] [Google Scholar]
- 27.Lublin FD, Coetzee T, Cohen JA, Marrie RA, Thompson AJ, International Advisory Committee on Clinical Trials in MS. The 2013 clinical course descriptors for multiple sclerosis: A clarification. Neurology 2020; 94(24): 1088–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elliott C, Arnold DL, Collins DL, Arbel T. Temporally consistent probabilistic detection of new multiple sclerosis lesions in brain MRI. IEEE Trans Med Imaging 2013; 32(8): 1490–503. [DOI] [PubMed] [Google Scholar]
- 29.Schofield MA, Zhu Y. Fast phase unwrapping algorithm for interferometric applications. Opt Lett 2003; 28(14): 1194–6. [DOI] [PubMed] [Google Scholar]
- 30.Selmaj K, Li DK, Hartung HP, et al. Siponimod for patients with relapsing-remitting multiple sclerosis (BOLD): an adaptive, dose-ranging, randomised, phase 2 study. The Lancet Neurology 2013; 12(8): 756–67. [DOI] [PubMed] [Google Scholar]
- 31.Miller DH, Khan OA, Sheremata WA, et al. A controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 2003; 348(1): 15–23. [DOI] [PubMed] [Google Scholar]
- 32.Sormani MP, Bonzano L, Roccatagliata L, Mancardi GL, Uccelli A, Bruzzi P. Surrogate endpoints for EDSS worsening in multiple sclerosis. A meta-analytic approach. Neurology 2010; 75(4): 302–9. [DOI] [PubMed] [Google Scholar]
- 33.Dal-Bianco A, Grabner G, Kronnerwetter C, et al. Slow expansion of multiple sclerosis iron rim lesions: pathology and 7 T magnetic resonance imaging. Acta Neuropathol 2017; 133(1): 25–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.