

Abstract
Objectives:
Metformin is widely used for the treatment of type 2 diabetes mellitus and found to have a crucial rule in the induction of apoptosis in several cancer types including pancreatic cell carcinoma, epithelial ovarian cancer, breast cancer, and renal cell carcinoma. In this study, we propose to explore the potential role of metformin as an adjuvant of irinotecan to target colorectal cancer (CRC) cell lines, exploring the effects underlying the anticancer properties of metformin on CRC cell lines.
Methods:
HCT116 and SW480 cell lines were treated with metformin, irinotecan and their combination. The effect of metformin on cell viability was evaluated using MTT assay. Flow cytometry technique was used to analyze apoptosis and cell cycle progression. While, detection of protein expression was analyzed by Western blot.
Results:
Metformin was found to inhibit growth in both HCT1116 and SW480 cell lines. On combination with irinotecan, it has been revealed that metformin sensitized CRC cells to irinotecan-induced cytotoxicity. Flow cytometry analysis showed that metformin did not induce apoptosis, but blocked cell cycle in G1 and S phases. This blockage was accompanied by decreased cyclin E and Cdk2 levels and increased p21 level.
Conclusion:
Combination of metformin with irinotecan may be an effective treatment strategy for targeting colorectal cancer that are resistant to irinotecan monotherapy.
Keywords: Colorectal cancer, cell cycle arrest, irinotecan, metformin, P21
Introduction
Irinotecan (CPT-11) is a widely used anticancer agent to treat colorectal cancer. It exerts its action through inhibition of topoisomerase I enzyme which leads to DNA strands breakage and ultimately cell death.[1,2] Irinotecan is a known water soluble derivative of natural alkaloid camptothecin (CPT). CPT is characterized by its selective binding to the cleavage complex, which is formed by both cleaved DNA and topoisomerase I.[3]
Initially, irinotecan was approved for the treatment of advanced colorectal cancer as second-line therapy. However, its combination with leucovorin and 5-Fluorouracil is being used recently as first-line option.[4] Irinotecan-acquired resistance has emerged for several reasons, such as intracellular level decrease and reduction in drug uptake, mutations in topoisomerase I that resulted in its structural change and topoisomerase I expression decrease.[5]
Diabetes mellitus, especially type 2, is considered a risk factor for CRC development.[6-8] Increased level of insulin (hyperinsulinemia) and insulin-like growth factor 1(IGF-1) enhance the proliferation and growth of colon cells, already existing transformed cells and finally development of CRC.[9] This mitogenic pathway of insulin can be enhanced by decreased level of IGF-1 binding protein, and subsequently, increased level of free IGF that plays an important role in enhancing cell proliferation and DNA synthesis with subsequent decrease in apoptosis.[10] For this reason, treatment that raises insulin blood level might contribute in the development of cancerous cells.
Another suggested mechanism that contributes to the emergence of CRC chemotherapy-resistance is through the dysregulation of p53 intracellular pathway. P53 is a tumor-suppression gene that helps in the prevention of tumor formation.[11]
Metformin is a widely used agent for the treatment of diabetes from biguanide group.[12] It decreases liver glucose production and activates insulin receptors.[13] Several modalities have been suggested to explain the antiproliferative effect of metformin. First, inhibition of complex 1 pathway inside the mitochondria results in the activation of adenosine monophosphate-activated protein kinase (AMPK) that causes protein synthesis downregulation and protecting the cell from apoptosis.[14] Second, inhibition of mammalian target of rapamycin (mTOR) as a consequence of AMPK activation ends up with inhibition of protein synthesis and cell proliferation.[15] Third, ameliorating the effect of hyperinsulinemia and IGF on cell proliferation and, consequently, the risk of colorectal cancer development.[16] Fourth, inhibition of the precancerous aberrant crypt foci (ACF) formation.[17] Finally, induction of G0/G1 phase cell cycle arrest and S phase arrest several cancerous cell lines.[15]
When combined with sorafenib in thyroid carcinoma cell line, metformin 5 mM induced both apoptosis and G1 phase cell cycle arrest.[18] Moreover, the addition of metformin to 5-flourouracil in esophageal adenocarcinoma cell lines sensitized them to the cytotoxic effect of 5-flourouracil and induced apoptosis.[19] In addition, metformin showed a synergistic effect by enhancing imatinib cytotoxicity on CRC HCT19 and by causing cell cycle arrest at S/G2 phase.[20]
However, no previous studies have investigated the effect of using metformin to induce irinotecan sensitivity in irinotecan-resistance CRC cells.
In this study, we investigated the antiproliferative and apoptotic potential of both Irinotecan and metformin in human colorectal cancer cells, in addition to evaluation the effect of metformin on irinotecan-induced cytotoxicity.
Methods
Cell culture
Human colorectal cancer cell lines (HCT116, SW480) were kindly donated by Dr Rick Throne (Newcastle, Australia). Cell lines were cultured in T-75 flasks as monolayer in Dulbecco’s Modified Eagle’s Medium (DMEM) high glucose with L Glutamine containing 10% fetal calf serum (FCS), 1% amphotericin B (250 μg/ml), and 1% penicillin/streptomycin (5000U penicillin/ml and 5000 streptomycin/ml). Cells were incubated in a humidified atmosphere of 95% air with 5% CO2 at 37°C. The cells were passed and harvested when they were 90% confluent.
Cell viability assay
To assess the antiproliferative effect of metformin on human CRC cell lines, MTT assay (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was used. Briefly, cells were seeded in 1 × 104 cell per well in flat- bottomed 96-well plate and left overnight until cells were attached. Next day, cells were pretreated with either different concentration of irinotecan alone or metformin or mixture of metformin and irinotecan. Media in the first column were replaced with fresh media at the same day of treatment and used as control.
Cells then were labeled with MTT according to manufacturer’s instruction and resulting formazan was solubilized with DMSO (Dimethyl Sulfoxide). Absorbance was read in a microplate reader at 540 nm.
Calculating percentage of viable cells
Cell viability was calculated by dividing mean absorbance of treated wells over mean absorbance of untreated control wells using this formula:

Apoptosis assay by flow cytometry (FACS)
Flow cytometry (FCM) analysis using propidium iodide (PI) was used to evaluate the apoptosis by measuring cellular DNA content and analyze cell cycle phases by measuring percentage of cells in G(1), S, and G(2)/M phases.[21] For both apoptosis detection and cell cycle analysis, cells were seeded in 15 × 104 cells per well in duplicate in 24-well plate and left overnight until cells were attached. Next day, cells were pretreated with metformin at concentration of 20 mM alone, irinotecan at concentration of 5 μM, and combination of irinotecan and metformin for 24 h. Fresh media were used as control.
PI stain was solubilized with PBS (Phosphate Buffered Saline) and was added to each well, plates then incubated for 30 min. The stained cells were added to the pellet that was formed by centrifuging the treated cells. After that cells in the centrifuge tube were incubated for 24 h and protected from light using aluminum foil. Finally, cells were analyzed by flow cytometry. Percentage of apoptotic cells and cells in each phase of the cell cycle were determined.
Analysis of protein expression by Western blotting
To detect the presence or absence of protein in treated cells, Western blotting (immunoblotting) was used. Cells were seeded in 45 × 104 cells per well in triplicate in 6-well plate, and left overnight to allow cell attachment. In the next day, cells were pretreated with metformin at concentration of 20 mM or treated with irinotecan at concentration of 5 μM for 24 h. Treated cells with metformin and irinotecan were divided into three groups and left for 24, 36, and 48 h.
To harvest HCT116 and SW480 cells for Western blotting, cells were washed with PBS. A lysis buffer was used for this harvesting. The cell lysate was centrifuged at 3000 rpm (round per minute) for 1 h, and the supernatants that contain proteins were used. The lysate protein content was determined using the Bradford dye assay. A solution that contains 2 μL lysate protein, 800 μL distilled water, and 200 μL Bradford dye was prepared. On binding with protein, this dye will form a stable unprotonated form (blue). The absorbance with spectrophotometer at 495 nm was measured. Standard curve was used to determine the total lysate protein content in cell samples.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was used to separate proteins according to their size. After that, proteins were transferred from SDS gel to a nitrocellulose membrane, and the membranes were blocked for 1 h using the blocking buffer that contains 5% BSA (Bovine serum albumin) in TBST (Tween Tris Buffer Saline).
The membranes were incubated overnight with primary antibodies at 4°C: Rabbit anti -GRP-78 monoclonal antibody, mouse anti-p21 monoclonal antibody, mouse anti-cyclin E monoclonal antibody, mouse anti-cyclin A monoclonal antibody, mouse anti-Cdk2 monoclonal antibody, and mouse anti-Cdk1 monoclonal antibody. Then, the membranes were washed for 5 min with wash buffer (TTBS). GAPDH monoclonal antibody was used as loading control antibody.
After that, the membranes were incubated with the secondary antibody at room temperature for 1 h on a platform shaker. Finally, the bands were visualized.
Statistical analysis
GraphPad Prism statistical software version 5.0 was used to conduct all descriptive statistical analysis. ANOVA and post hoc Tukey’s tests were used for such statistical analysis. For all results, differences were considered significant if P-value was <0.05 (P < 0.05).
Results
1. Metformin inhibited CRC cells growth and sensitized CRC cells to irinotecan-induced cytotoxicity
To assess the antiproliferative activity of metformin in human CRC cells; HCT116 and SW480 cell lines were treated with metformin at 20 mM for 72 h, then cell viability was evaluated using MTT assay. Results in Table 1 indicate that Metformin induced cell growth inhibition in both cell lines. It is also noted that irinotecan did not induce cytotoxic effect against both CRC cells when used at concentrations that ranged between 1 and 7.5 μM. However, only SW480 cells were partially sensitive to irinotecan when used at 10 μM.
Table 1.
Percentage of cell viability for HCT116 and SW480 after using several concentrations of Irinotecan and Metformin
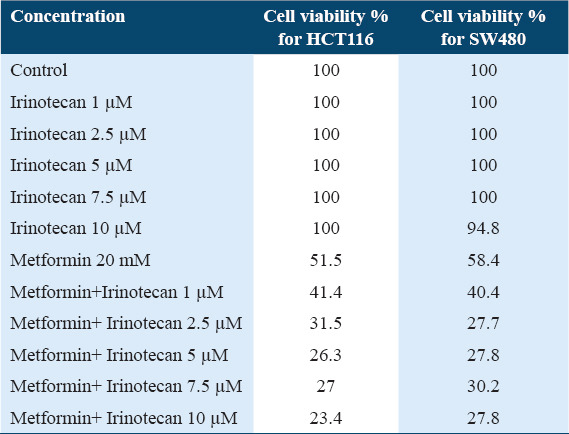
In addition, results in Table 1 reveal that metformin significantly sensitized CRC cells to irinotecan-induced growth inhibition in a dose-dependent way. Lower cells viability was noticed with higher irinotecan concentration.
We found that cell lines that were pretreated with the combination of metformin and irinotecan had significantly lower percentage of cell viability when compared with metformin-only, irinotecan-only, or control cell lines [Figure 1].
Figure 1.
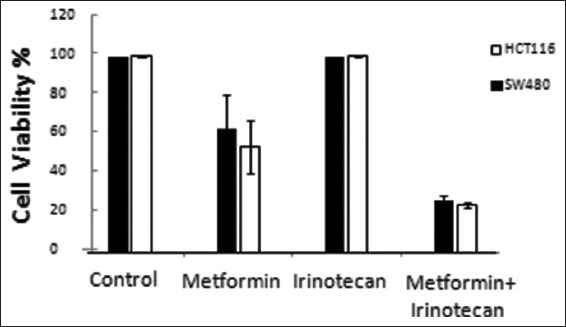
Percentage of viable cells for HCT116 and SW480 after treatment with Metformin 20 mM, Irinotecan 5μM, combination of Metformin 20 mM and Irinotecan 5 μM compared with control “with no treatment” (* On comparison of cell viability % in HCT116 and SW480 cell lines treated with Metformin, and combination of Metformin+Irinotecan with control, differences were statistically considered significant, P < 0.05)
2. Metformin sensitized CRC cells to irinotecan by induction of cell cycle arrest
To study the apoptotic effect of metformin and irinotecan, cell lines were pretreated with metformin, irinotecan, and combination of metformin and irinotecan. Apoptotic cells were measured by flow cytometry. When compared with control, pretreated cell lines with metformin showed higher percentage of apoptotic cells. However, the differences in percentages between control and different pretreated cell lines were not statistically significant [Figure 2].
Figure 2.
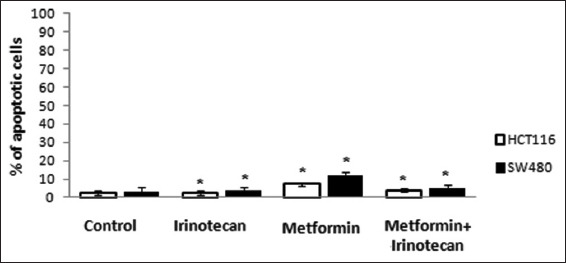
Percentage of apoptotic cells by flow cytometry. (*On comparison of % of apoptotic cells in HCT116 and SW480 cell lines treated with metformin, irinotecan and combination of metformin+irinotecan with control, differences were statistically considered non-significant)
The percentage of cells in Sub-G1 phase which represents dead cells or DNA fragmentation that assessed by flow cytometry showed that neither metformin alone nor in combination with Irinotecan induced apoptosis. By quantification of cells present in Sub-G1, G1, S, and G2/M phase, it was found that metformin when combined with irinotecan induced G0/G1and S phases cell cycle arrest in both HCT116 and SW480 cell lines by observing that cells’ percentage in G0/G1phase increased normalized to the control (from 28.4% to 45.02% in HCT116, and from 35.08% to 43.66% in SW480), and in S phase (from 19.48% to 38.22% in HCT116, and from 21.3% to 48.40% in SW480) as illustrated in Table 2, Figures 3 and 4. Results also indicated that irinotecan exerts its action by blocking the cell cycle at S phase (increase in cell population in S phase accompanied by reduction in the distribution of cells in other phases).
Table 2.
Effect of metformin on cell cycle progression in HCT116 and SW480 treated cells. Flow cytometry analysis of HCT116 and SW480 cells treated with DMSO (control), metformin, metformin+ irinotecan, and irinotecan for G0/G1, S and G2/M phase quantification
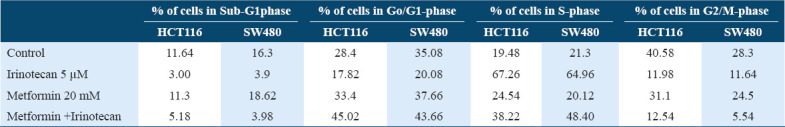
Figure 3.
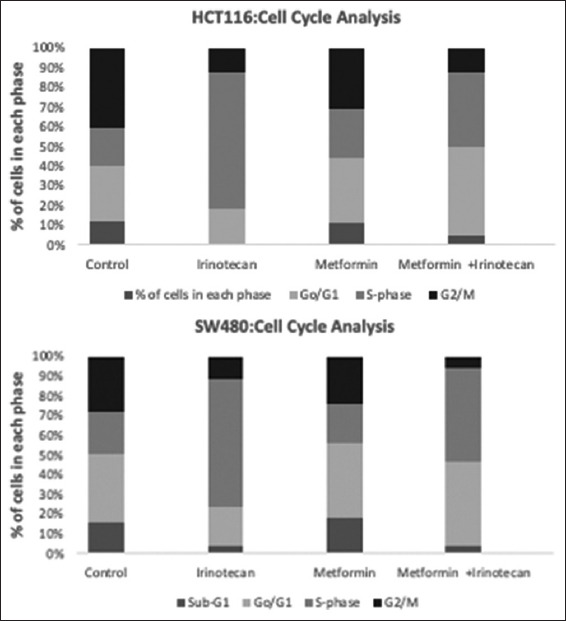
Cell cycle analysis by flow cytometry. Metformin inhibited cell cycle progression at G1 phase. Colorectal cancer cell lines, HCT116, and SW480, were treated with metformin, Irinotecan and combination of metformin and irinotecan normalized to the control
Figure 4.
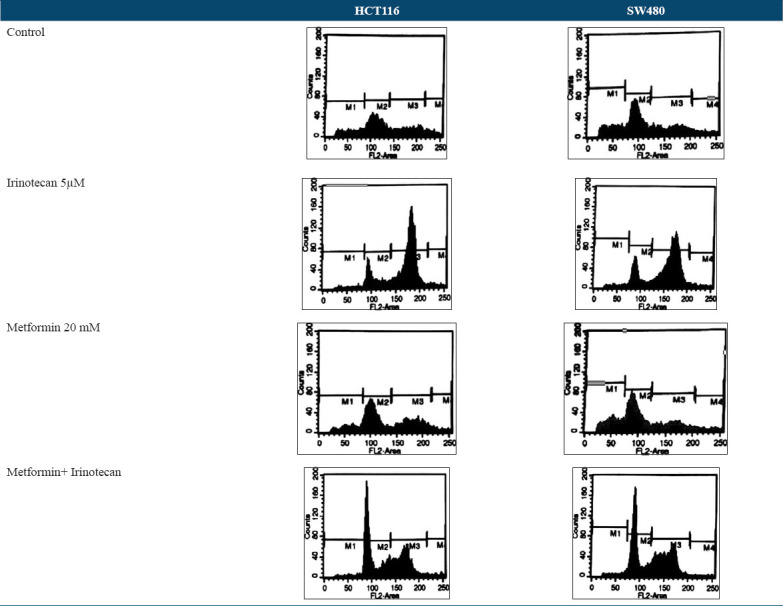
DNA cell cycle that was analyzed by propidium iodide and measured by flow cytometry. M1 indicates Sub-G1, M2 indicates G0/G1, M3 indicates S, and M4 indicates G2/M phases. Accumulation of cells in G0/G1 phase when they were treated with combination of metformin and irinotecan means that cell cycle block occurred here
3. Metformin downregulated Cdk2, cyclin E and upregulated P21
Metformin treatment increased cells percentage in G1 phase (from 28.4% to 45.02% in HCT116, and from 35.08% to 43.66% in SW480), and in S phase (from 19.48% to 38.22% in HCT116, and from 21.3% to 48.40% in SW480), this increase suggested that metformin inhibited cell cycle in G1 and S phase.
To examine the effect of irinotecan and metformin on regulation of Cdk2, cyclin E, and P21 expression, we used Western blot analysis to assess the expression levels in different cell lines.
In our experiment, expression of p21 protein was found to be upregulated in pretreated cell lines with Metformin, Irinotecan, and combination of metformin and Irinotecan. On the other hand, the expression of Cdk2, cyclin E, Cdk1, and cyclin A was downregulated as shown in Figures 5 and 6. A broad range of marker ladder was used as a loading control.
Figure 5.
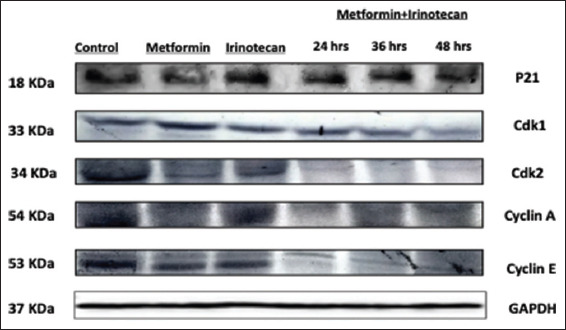
Western blot analysis of P21, Cdk1, Cdk2, Cyclin E, Cyclin A, and GRP-78 expression level in SW480 cell line
Figure 6.
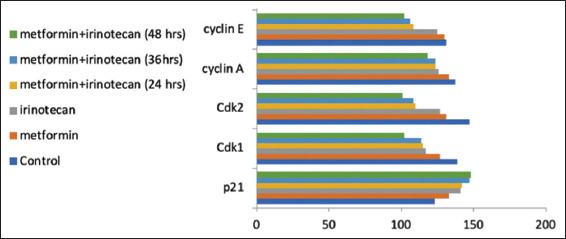
Protein bands intensity in Western blot experiment
Discussion
Metformin is a widely used drug in the treatment of type 2 diabetes mellitus. Recently, metformin was recognized to have potential anti-tumorigenic properties due to its antiproliferative effect on cancer cells in vitro.[22,23] Several epidemiological studies showed patients with type 2 diabetes who were treated with metformin had lower incidence and mortality of cancer.[24-27]
Our study aimed to explore the effect of metformin on irinotecan-induced cytotoxicity in CRC cell lines. Our findings proved three main outcomes; first, metformin inhibited the growth of both CRC cell lines HCT116 and SW480 by 42–50% when compared with the control. Mogavero et al. results on HT29, HCT116, and HCT116 p53−/− CRC cell lines demonstrated a decrease in proliferation when treated with metformin 5 mM for 72 h by 25–30% which is close to our findings.[28] The results of evaluating the chemo-sensitizing effect of metformin on resistant epithelial ovarian cancer (EOC) cell lines support our findings by reducing significantly EOC cells viability up to 40%.[29]
Although our selected CRC cell lines in this study were resistant to irinotecan, where HCT116 is null of p53 gene, while SW480 is known to have a mutant p53 gene, the pretreatment with metformin sensitized both cell lines to irinotecan. In a dose-dependent pattern of killing, metformin with irinotecan reduced cells viability by 60% with the least concentration of irinotecan used in our study and up to 75% with 10 μM of irinotecan. This shows the important role of metformin to induce cancer cells sensitivity and reduce risk of treatment resistance in patients who are resistant to irinotecan.
With the limited data and studies of using metformin to sensitize cell lines to irinotecan, our results showed consistent findings with Kunthur et al. where they indicated in their study that using metformin alone at 1 mmol/l concentration, or in sequential design in combination with either irinotecan (5 μM), 5-flourouracil (20 μM), or oxaliplatin (5 μM) on two human colon cancer cell lines (HT29 and HCT116) decreased cell viability by 35–70% in metformin groups and 40–55% in the combination groups.[30] The findings from testing metformin ability to sensitize resistant A2780 cell lines to paclitaxel[29] confirm our conclusion that metformin has an intrinsic mechanism to sensitize many cell lines to different anti-cancer agents; including irinotecan. Sorafenib was shown to express additional growth inhibition when combined with metformin on HTh74 anaplastic thyroid carcinoma by reducing cell viability up to 80%.[18]
Finally, metformin when combined with irinotecan did not induce apoptosis in both CRC cell lines yet induced cell cycle arrest through blocking the cell cycle progression at G1 and S phases. This is verified by increased cells percentage in G1 phase from 28.4% to 45.02% in HCT116 and from 35.08% to 43.66% in SW480, while in S phase from 19.48% to 38.22% in HCT116, and from 21.3% to 48.40% in SW480. The absence of apoptosis can be explained by the p53-null/mutant cell lines that were used in our study. This block at S phase is expected for irinotecan and is consistent with the findings when irinotecan was tested on p53-negative Caco-2 and p53-positive CW2 human colorectal cancer cell in concentrations ranging from 0.3 to 30 μmol/l for 24–48 h. Irinotecan never induced apoptosis in both cell lines, but increased the proportions of cells at S and G2/M phases with subsequent decrease in G1 phase cells count.[31]
In our study, HCT116 cell lines when treated with metformin showed partial cell cycle arrest at G0/G1 phase. Although, metformin was found to significantly induce G0/G1 cell cycle arrest in different cancerous cell lines.[28,32-36] However, other studies found partial induction of cell cycle arrest at G0/G1 phases by metformin.[37] We suggest that metformin might have other pathways to induce cell cycle arrest that are not mediated by p53 gene, as our HCT116 is a p53-null cell line. This suggestion is supported by recent evidence which showed that metformin induce cell cycle arrest by signaling pathways, the AMP-activated protein kinase (AMPK) pathway, and the phosphatidylinositol 3-kinase (PI3K). The activation of AMPK pathway will lead to inactivation of mTOR pathway and ultimately cell death.[36,38-40]
Cell cycle arrest, as induced by metformin and irinotecan, was accompanied with the downregulation of cell cycle regulators Cdk2 and cyclin E and upregulation of p21, which is consistent with its role in CDKs-cyclin complex inhibition and blockage of cell cycle.[28,32] Several (CDKs) mediate the progression of cell cycle.[41] In mammalian cells, the passage through the G1 phase is regulated by sequential actions of D-and E-type cyclins in combination with CDK4/6 and CDK2, respectively.[42] Cyclin D binds with CDK4 or CDK6 and the complex enters the nucleus, then this complex is activated by phosphorylation by CDK activating kinase (CAK). The activity of this complex drives cells to the S-phase as long as the growth factors is present.[43] Cyclin E is the key regulator to overcome the Restriction point at the G1-to-S transition phase. P21 protein is a cyclin dependent kinases inhibitor,[44] that is required for cell cycle progression. The p21 gene is usually controlled by p53 protein.[45] As a result, p21 may promote p53-dependent cell cycle arrest or apoptosis. We expect that the upregulation of p21 in our study mediated the induction of cell cycle arrest by the combination of metformin and irinotecan.
The strength of our study emerges from its uniqueness in examining the role of metformin in sensitizing two different CRC cell lines to irinotecan. To the best of our knowledge, there are no studies that measured the effect of metformin with irinotecan on these cell lines that are resistant to irinotecan. Moreover, we combined metformin with different concentrations of irinotecan (1, 2.5, 5, 7.5, and 10 μM) and established a dose-dependent inhibition of CRC cell lines. This will serve as a starting point for future research to determine the optimal concentration of irinotecan to avoid its toxic cumulative effect. However, our study has some limitations. We used metformin in one concentration only (20 mM), rather than several concentrations. In addition, the mechanism of metformin-induced cell cycle arrest was not examined in details.
Conclusion
We conclude that combining metformin with irinotecan would be beneficial in inhibiting the growth of cancerous cells that are resistant to irinotecan. We recommend to investigate the underlying mechanism that mediates the metformin and irinotecan killing of cells when combined together using in vivo cell lines which would be a starting point to move this research from benchside to bedside.
Authors’ Declaration Statements
We confirm that the manuscript has been read and approved by all named authors and that there are no other persons who satisfied the criteria for authorship but are not listed. We further confirm that the order of authors listed in the manuscript has been approved by all of us. We confirm that we have given due consideration to the protection of intellectual property associated with this work and that there are no impediments to publication, including the timing of publication, with respect to intellectual property.
Ethics approval and consent to participate
Not applicable (no human subjects participated in this research).
Availability of data and material
Data sharing is not applicable to this article as no datasets were generated or analyzed during this study.
Competing Interests
Authors have no competing interest to declare.
Funding Statement
This work was supported by the Deanship of Research at Jordan University of Science and Technology.
Authors’ Contributions
EK and NM were responsible for the conception and design of this study. EK, NM, and MA were responsible for running the experimental work. EK, NM, and WI analyzed and interpreted the results. All authors were responsible for writing the first draft and revising the manuscript critically for important intellectual content.
Acknowledgment
We acknowledge Deanship of Research at Jordan University of Science and Technology for funding our research, and Dr. Rick Throne (Newcastle, Australia) for providing us with both CRC cell lines.
ORCID link of the submitting author: https://orcid.org/0000-0002-7569-0275
References
- 1.Hammond WA, Swaika A, Mody K. Pharmacologic resistance in colorectal cancer:A review. Ther Adv Med Oncol. 2016;8:57–84. doi: 10.1177/1758834015614530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Temmink OH, Hoebe EK, Fukushima M, Peters GJ. Irinotecan-induced cytotoxicity to colon cancer cells in vitro is stimulated by pre-incubation with trifluorothymidine. Eur J Cancer. 2007;43:175–83. doi: 10.1016/j.ejca.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 3.Pommier Y. Camptothecins and topoisomerase I:A foot in the door. Targeting the genome beyond topoisomerase I with camptothecins and novel anticancer drugs:Importance of DNA replication, repair and cell cycle checkpoints. Curr Med Chem Anticancer Agents. 2004;4:429–34. doi: 10.2174/1568011043352777. [DOI] [PubMed] [Google Scholar]
- 4.Schilling G, Lipp R, Hegewisch-Becker S, Hossfeld DK. UFT/leucovorin plus weekly irinotecan in advanced or metastatic colorectal cancer. Oncology (Williston Park) 2000;14(Suppl 9):38–40. [PubMed] [Google Scholar]
- 5.Xu Y, Villalona-Calero MA. Irinotecan:Mechanisms of tumor resistance and novel strategies for modulating its activity. Ann Oncol. 2002;13:1841–51. doi: 10.1093/annonc/mdf337. [DOI] [PubMed] [Google Scholar]
- 6.Cossor FI, Adams-Campbell LL, Chlebowski RT, Gunter MJ, Johnson K, Martell RE, et al. Diabetes, metformin use, and colorectal cancer survival in postmenopausal women. Cancer Epidemiol. 2013;37:742–9. doi: 10.1016/j.canep.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ross CM. Re:Prospective study of adult onset diabetes mellitus (Type 2) and risk of colorectal cancer in women. J Natl Cancer Inst. 1999;91:1334. doi: 10.1093/jnci/91.15.1334. [DOI] [PubMed] [Google Scholar]
- 8.Buysschaert M, Sadikot S. Diabetes and cancer:A 2013 synopsis. Diabetes Metab Syndr. 2013;7:247–50. doi: 10.1016/j.dsx.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 9.Berster JM, Goke B. Type 2 diabetes mellitus as risk factor for colorectal cancer. Arch Phys Biochem. 2008;114:84–98. doi: 10.1080/13813450802008455. [DOI] [PubMed] [Google Scholar]
- 10.Erbach M, Mehnert H, Schnell O. Diabetes and the risk for colorectal cancer. J Diabetes Complications. 2012;26:50–5. doi: 10.1016/j.jdiacomp.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 11.Li XL, Zhou J, Chen ZR, Chng WJ. P53 mutations in colorectal cancer-molecular pathogenesis and pharmacological reactivation. World J Gastroenterol. 2015;21:84–93. doi: 10.3748/wjg.v21.i1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee JH, Jeon SM, Hong SP, Cheon JH, Kim TI, Kim WH. Metformin use is associated with a decreased incidence of colorectal adenomas in diabetic patients with previous colorectal cancer. Dig Liver Dis. 2012;44:1042–7. doi: 10.1016/j.dld.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 13.Pollak M. Metformin and other biguanides in oncology:Advancing the research agenda. Cancer Prev Res (Phila) 2010;3:1060–5. doi: 10.1158/1940-6207.CAPR-10-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dowling RJ, Goodwin PJ, Stambolic V. Understanding the benefit of metformin use in cancer treatment. BMC Med. 2011;9:33. doi: 10.1186/1741-7015-9-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ben Sahra I, Le Marchand-Brustel Y, Tanti JF, Bost F. Metformin in cancer therapy:A new perspective for an old antidiabetic drug? Mol Cancer Ther. 2010;9:1092–9. doi: 10.1158/1535-7163.MCT-09-1186. [DOI] [PubMed] [Google Scholar]
- 16.Zhang ZJ, Zheng ZJ, Kan H, Song Y, Cui W, Zhao G, et al. Reduced risk of colorectal cancer with metformin therapy in patients with Type 2 diabetes:A meta-analysis. Diabetes Care. 2011;34:2323–8. doi: 10.2337/dc11-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hosono K, Endo H, Takahashi H, Sugiyama M, Sakai E, Uchiyama T, et al. Metformin suppresses colorectal aberrant crypt foci in a short-term clinical trial. Cancer Prev Res (Phila) 2010;3:1077–83. doi: 10.1158/1940-6207.CAPR-10-0186. [DOI] [PubMed] [Google Scholar]
- 18.Chen G, Nicula D, Renko K, Derwahl M. Synergistic anti-proliferative effect of metformin and sorafenib on growth of anaplastic thyroid cancer cells and their stem cells. Oncol Rep. 2015;33:1994–2000. doi: 10.3892/or.2015.3805. [DOI] [PubMed] [Google Scholar]
- 19.Honjo S, Ajani JA, Scott AW, Chen Q, Skinner HD, Stroehlein J, et al. Metformin sensitizes chemotherapy by targeting cancer stem cells and the mTOR pathway in esophageal cancer. Int J Oncol. 2014;45:567–74. doi: 10.3892/ijo.2014.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jaeryun L, Deokbae P, Youngki L. Metformin synergistically potentiates the antitumor effects of imatinib in colorectal cancer cells. Dev Reprod. 2017;21:139–50. doi: 10.12717/DR.2017.21.2.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Riccardi C, Nicoletti I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protoc. 2006;1:1458–61. doi: 10.1038/nprot.2006.238. [DOI] [PubMed] [Google Scholar]
- 22.Martin-Castillo B, Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. Metformin and cancer:Doses, mechanisms and the dandelion and hormetic phenomena. Cell Cycle. 2010;9:1057–64. doi: 10.4161/cc.9.6.10994. [DOI] [PubMed] [Google Scholar]
- 23.Del Barco S, Vazquez-Martin A, Cufí S, Oliveras-Ferraros C, Bosch-Barrera J, Joven J, et al. Metformin:Multi-faceted protection against cancer. Oncotarget. 2011;2:896–917. doi: 10.18632/oncotarget.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang P, Li H, Tan X, Chen L, Wang S. Association of metformin use with cancer incidence and mortality:A meta-analysis. Cancer Epidemiol. 2013;37:207–18. doi: 10.1016/j.canep.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 25.Noto H, Goto A, Tsujimoto T, Noda M. Cancer risk in diabetic patients treated with metformin:A systematic review and meta-analysis. PLoS One. 2012;7:e33411. doi: 10.1371/journal.pone.0033411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer:A cohort study among people with Type 2 diabetes. Diabetes Care. 2009;32:1620–5. doi: 10.2337/dc08-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–5. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mogavero A, Maiorana MV, Zanutto S, Varinelli L, Bozzi F, Belfiore A, et al. Metformin transiently inhibits colorectal cancer cell proliferation as a result of either AMPK activation or increased ROS production. Sci Rep. 2017;7:15992. doi: 10.1038/s41598-017-16149-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dos Santos Guimaraes I, Ladislau-Magescky T, Tessarollo NG, Dos Santos DZ, Gimba ER, Sternberg C, et al. Chemosensitizing effects of metformin on cisplatin- and paclitaxel-resistant ovarian cancer cell lines. Pharmacol Rep. 2018;70:409–17. doi: 10.1016/j.pharep.2017.11.007. [DOI] [PubMed] [Google Scholar]
- 30.Kunthur A, Aldwairi A, Simmen F, Govindarajan R. Effect of metformin alone and in combination with 5-fluorouracil (5FU), oxaliplatin (O) and irinotecan (I) on human colon cancer cell lines. J Clin Onco. 2011;29(Suppl 15):e13041. [Google Scholar]
- 31.Kaku Y, Tsuchiya A, Kanno T, Nishizaki T. Irinotecan induces cell cycle arrest, but not apoptosis or necrosis, in Caco-2 and CW2 colorectal cancer cell lines. Pharmacology. 2015;95:154–9. doi: 10.1159/000381029. [DOI] [PubMed] [Google Scholar]
- 32.Miyoshi H, Kato K, Iwama H, Maeda E, Sakamoto T, Fujita K, et al. Effect of the anti-diabetic drug metformin in hepatocellular carcinoma in vitro and in vivo. Int J Oncol. 2014;45:322–32. doi: 10.3892/ijo.2014.2419. [DOI] [PubMed] [Google Scholar]
- 33.Siregar Y, Widyawati T, Hasibuan PA, Rangkuti IY. Metformin inhibits growth of breast cancer cell T47 through decreasing expression of protein P53, BCL2 and cyclin D1. J Med Res Innov. 2019;3:e000164. [Google Scholar]
- 34.Pasha M, Sivaraman SK, Frantz R, Agouni A, Munusamy S. Metformin induces different responses in clear cell renal cell carcinoma Caki cell lines. Biomolecules. 2019;9:113. doi: 10.3390/biom9030113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cantrell LA, Zhou C, Mendivil A, Malloy KM, Gehrig PA, Bae-Jump VL. Metformin is a potent inhibitor of endometrial cancer cell proliferation-implications for a novel treatment strategy. Gynecol Oncol. 2010;116:92–8. doi: 10.1016/j.ygyno.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Savarino V, Marabotto E, Zentilin P, Furnari M, Bodini G, De Maria C, et al. Proton pump inhibitors:Use and misuse in the clinical setting. Exp Rev Clin Pharmacol. 2018;11:1123–34. doi: 10.1080/17512433.2018.1531703. [DOI] [PubMed] [Google Scholar]
- 37.Alimova IN, Liu B, Fan Z, Edgerton SM, Dillon T, Lind SE, et al. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle. 2009;8:909–15. doi: 10.4161/cc.8.6.7933. [DOI] [PubMed] [Google Scholar]
- 38.Zhao B, Luo J, Yu T, Zhou L, Lv H, Shang P. Anticancer mechanisms of metformin:A review of the current evidence. Life Sci. 2020;254:117717. doi: 10.1016/j.lfs.2020.117717. [DOI] [PubMed] [Google Scholar]
- 39.Chen YH, Yang SF, Yang CK, Tsai HD, Chen TH, Chou MC, et al. Metformin induces apoptosis and inhibits migration by activating the AMPK/p53 axis and suppressing PI3K/AKT signaling in human cervical cancer cells. Mol Med Rep. 2021;23:88. doi: 10.3892/mmr.2020.11725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen YC, Li H, Wang J. Mechanisms of metformin inhibiting cancer invasion and migration. Am J Transl Res. 2020;12:4885–901. [PMC free article] [PubMed] [Google Scholar]
- 41.Morgan DO. Cyclin-dependent kinases:Engines, clocks, and microprocessors. Annu Rev Cell Dev Biol. 1997;13:261–91. doi: 10.1146/annurev.cellbio.13.1.261. [DOI] [PubMed] [Google Scholar]
- 42.Sherr CJ. D-type cyclins. Trends Biochem Sci. 1995;20:187–90. doi: 10.1016/s0968-0004(00)89005-2. [DOI] [PubMed] [Google Scholar]
- 43.Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12:2245–62. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- 44.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–16. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 45.El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–25. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during this study.