

Abstract
Circulating tumor DNA (ctDNA) sequencing is being rapidly adopted in precision oncology, but the accuracy, sensitivity, and reproducibility of ctDNA assays is poorly understood. Here we report the findings of a multi-site, cross-platform evaluation of the analytical performance of five industry-leading ctDNA assays. We evaluated each stage of the ctDNA sequencing workflow with simulations, synthetic DNA spike-in experiments, and proficiency testing on standardized cell line–derived reference samples. Above 0.5% variant allele frequency, ctDNA mutations were detected with high sensitivity, precision and reproducibility by all five assays, whereas below this limit detection became unreliable and varied widely between assays, especially when input material was limited. Missed mutations (false-negatives) were more common than erroneous candidates (false-positives), indicating that the reliable sampling of rare ctDNA fragments is the key challenge for ctDNA assays. This comprehensive evaluation of the analytical performance of ctDNA assays serves to inform best-practice guidelines and provides a resource for precision oncology.
Editorial summary:
Reliable detection of mutations below 0.5% variant allele frequency remains a key challenge for circulating tumor DNA sequencing assays.
INTRODUCTON
Cancer cells undergoing apoptosis or necrosis release fragments of DNA into the circulatory system1,2. These circulating tumor DNA (ctDNA) fragments may harbor somatic mutations from their tumor of origin, and their abundance correlates with tumor size and stage3,4. Accordingly, ctDNA can act as an accessible biomarker to inform cancer detection, molecular stratification, therapeutic monitoring and post-treatment surveillance5–9.
Assays that measure ctDNA have several advantages over tumor-tissue biopsies (see Supplementary Table 1). The collection of ctDNA is fast, cheap, minimally invasive, and can be performed serially to monitor tumor evolution or response to therapy. In theory, ctDNA can provide a representative cross-section of heterogeneous tumors and multi-focal disease. Moreover, whilst a tumor-tissue biopsy cannot be performed without prior knowledge of the tumor, ctDNA assays can identify evidence of unknown lesions, thereby enabling detection of minimal residual disease following treatment or even cancer screening in healthy populations5–9.
These advantages are best realized via the unbiased analysis of ctDNA by next-generation sequencing (NGS) and, on this basis, ctDNA sequencing is being rapidly adopted in precision oncology. However, ctDNA sequencing assays face major technical challenges. Cell-free DNA exists as small fragments (~160 bp) at low concentrations (typically < 10 ng, or < 3000 genome copies, per mL of plasma in cancer patients)10. Furthermore, only a small fraction of cell-free DNA is tumor-derived (commonly < 1% of alleles in circulation, but sometimes as low as < 0.01%)10. The detection of rare somatic mutations from such limited input material is highly challenging.
CtDNA sequencing assays are also affected by a range of experimental variables and artifacts. Extensive PCR amplification is typically required to generate an NGS library from the small quantity of cell-free DNA available, as well as further targeted enrichment of informative cancer genes (by hybrid-capture or amplicon methods)10. The small size of cell-free DNA fragments can inhibit target enrichment, reduce alignability to the human reference genome and prevent the resolution of complex loci or mutations. These variables further exacerbate the quantitative biases and sequencing errors that affect all NGS experiments11.
In spite of these challenges, ctDNA assays of increasing resolution have been developed12–24. With clinical adoption already underway, it is critical for the community to understand the sensitivity, accuracy and reproducibility of ctDNA assays, and the variables that impact analytical performance. Discordant results between alternative assays or parallel ctDNA and tumor-biopsy tests have been reported25–27. Accordingly, a joint review by the American Society of Clinical Oncology and College of American Pathologists recently identified the pressing need for proficiency testing using standardized samples to assess the analytical validity of ctDNA assays and enable unbiased comparisons between different technology platforms and laboratories28.
Here we report the findings of a multi-lab, cross-platform evaluation of analytical performance among NGS-based ctDNA assays carried out as part of the FDA-led Sequencing Quality Control Phase 2 (SEQC2) project, or the fourth phase of the MAQC consortia. The Oncopanel Sequencing Working Group – comprising academic, industry, government and regulatory stakeholders – tested the performance of five leading ctDNA assays across twelve participating clinical and research facilities. We employed simulated and synthetic experiments, as well as rigorous proficiency testing on contrived human ctDNA reference materials to measure the impact of variables at each step within the ctDNA sequencing workflow. The study assesses the analytical validity of ctDNA assays for potential clinical applications and informs best practice guidelines (see Box 1).
Box 1. Summary of concepts, findings & recommendations.
| Issue | Findings | Outlook & recommendations |
|---|---|---|
| Mutation frequency | Mutations present above ~0.5% VAF were detected with high sensitivity and reproducibility by all participating ctDNA assays but performances were generally suboptimal below this level and variable between assays (Fig. 4). | A key challenge in ongoing development of ctDNA sequencing assays is to improve detection sensitivity for low-frequency mutations (< 0.5% VAF). |
| Coverage depth & heterogeneity | Fragment-depth was a critical variable in ctDNA assays, with high coverage essential for sensitive detection of low-frequency mutations (Fig. 3,4). In addition to depth, even coverage across target regions was important to ensure high sensitivity and reproducibility (Fig. S3). | Improvements to the efficiency/stability of capture enrichment, NGS library conversion and amplification may yield increased coverage depth and decreased heterogeneity, leading to improved performance and robustness. |
| DNA input quantity | Increasing DNA input quantity generally improved fragment-depth, sensitivity and reproducibility (Fig. 5). | Limited availability of cell-free DNA is a challenge for clinical translation of ctDNA assays. Input material may be increased via improvements to efficiency of plasma-DNA extractions, increasing the volume of patient blood draws (when feasible) or obtaining cell-free DNA from other body fluids (e.g., urine, stool, CSF, etc.) for relevant cancers. |
| UMIs | Unique molecular identifiers (UMIs) enabled effective consensus error correction, minimizing the detection of false-positives (Table 2). | Wherever possible, UMIs should be employed for consensus error correction in ctDNA sequencing assays. |
| Inter-laboratory variation | Participating assays were robust to technical variables between test labs – from plasma extraction to sequencing workflow stages – and were impacted largely by random, rather than systematic variation (Fig. 4, Fig. S6). | Robustness to technical variables is essential for clinical implementation of ctDNA sequencing assays. |
| Random sampling | The detection of low-frequency mutations (VAF < 0.5%) by random sampling poses an inherent statistical challenge, even when high fragment-depth is available (Fig. 1). | Novel strategies for the enrichment of ctDNA fragments over non-cancerous cell-free DNA (e.g., by fragment size selection) and alternative signals, such as ctDNA methylation profiles, may help overcome limitations of random sampling. |
| Targeted enrichment method | Performance was broadly comparable between participating amplicon and hybrid-capture assays, with sensitivity and robustness largely determined by the fragment-depth achieved, not the method of enrichment (Fig. 6). | Amplicon methods can enable sensitive, cost effective detection of ctDNA mutations in single genes or mutation hotspots but small panel sizes limit their suitability for unbiased surveillance (e.g., for tumor evolution profiling). |
| Exon edge-effect | In hybrid-capture sequencing, mutations in exon edge regions were detected with lower sensitivity than central regions, due to lower coverage (Fig. 1,2). | Increasing the size of captured flanking regions around exons during panel design may alleviate this exon edge-effect. |
| Sequence context | Mutations in challenging genome sequence contexts, such as high/low GC-content, low sequence complexity or suboptimal alignability, were detected with lower sensitivity (Fig. 1,2). | Some of these effects may be alleviated by increasing capture-probe density in challenging regions or via improvements to NGS library preparations. |
| Reference standards | Reproducibility measurements provided a useful but imperfect proxy for analytical performance that is not dependent on the availability of a reference sample and annotation (Fig. 4). | Well-characterized reference standards can directly measure analytic performance characteristics in absence of confounding biological variables and are a useful tool for comparing ctDNA assays. |
RESULTS
Evaluating ctDNA assays with simulated sequencing data
The detection of ctDNA fragments occurs by random sampling from a background of non-cancerous cell-free DNA. To investigate the analytical variables that govern this process, in the absence of confounding experimental variables, we initially generated simulated NGS libraries that emulate targeted analysis of cell-free DNA by hybrid-capture sequencing (see Methods).
We created simulated libraries from 155 cancer genes, covered at ~9,000-fold depth by ~160 bp sequence fragments (Fig. S1a). This level of coverage can be theoretically obtained from a routine patient blood-draw, given that cell-free DNA occurs at up to ~3,000 genome copies per mL of plasma (see Supplementary Table 2). The cancer genes harbored 2,356 simulated somatic mutations (one COSMIC SNV per exon) that were represented at known variant allele frequencies (VAFs) ranging from 5% to 0.1% (Fig. S1a). We then performed an in silico hybrid-capture enrichment step to obtain typical convex coverage profiles over targeted exons, resulting in a final 8,252-fold median fragment-depth at mutation sites (Fig. S1a,b). An example is shown for the oncogene MET (Fig. 1a).
Figure 1. Evaluating ctDNA assays with simulated sequencing data.
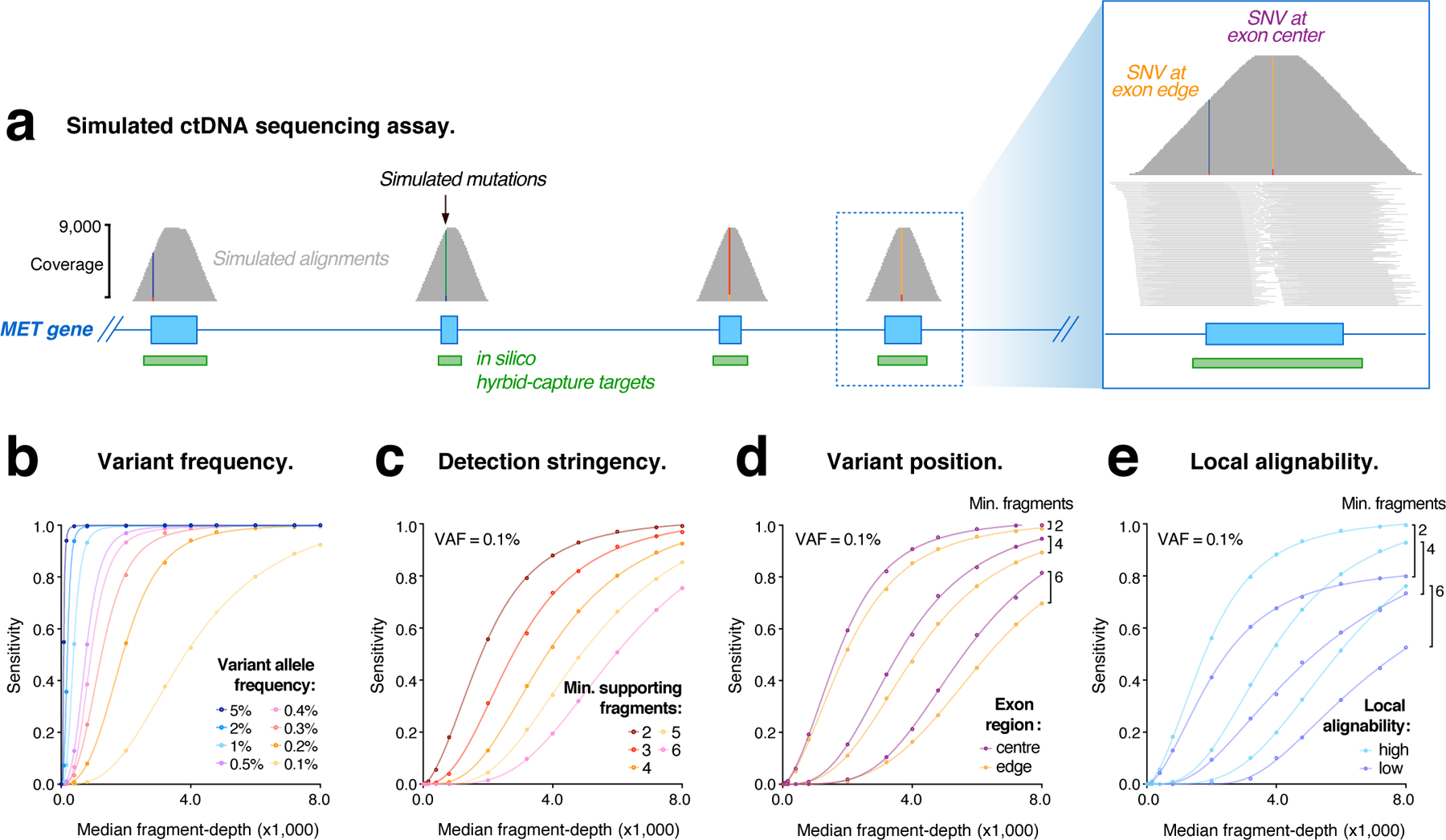
(a) Genome browser view showing coverage of simulated sequencing fragments within the MET oncogene, with single nucleotide variants (SNVs) represented in each exon. Inset (right) shows the distribution of fragment coverage within a single coding exon, illustrating the convex coverage profile that results from in silico capture enrichment and causes lower fragment-depth among mutations in edge regions. (b–e) Curves modelling the relationship between simulated library depth (median fragment-depth) and detection sensitivity for simulated mutations under various conditions: (b) shows mutations represented at different frequencies (0.1–5% VAF), with ≥ 4 supporting fragments required for detection; (c) mutations at VAF = 0.1%, with different levels of detection stringency applied (≥ 2–6 supporting fragments); (d) mutations within exon edge regions (< 20bp from exon boundary), compared to central regions (> 50bp from exon boundary); (e) mutations in regions of sub-optimal alignability (low), compared to optimal regions (high).
We used these simulated libraries to evaluate the impact of coverage on the detection of ctDNA mutations. Due to random sampling, the number of sequence fragments containing a given mutation follows a Poisson distribution, with a median fragment count that is proportional to the product of VAF and global fragment-depth (Fig. S1c). At maximum depth, >99% of mutations were detected by at least two independent fragments (Fig. S1d). However, any decrease in coverage or increase in detection stringency (i.e., >2 supporting fragments) caused a reduction in sensitivity.
To model these relationships, we incrementally adjusted the simulated alignment coverage and detection stringency (see Methods). For low-frequency mutations (VAF < 0.5%), coverage had a pronounced impact on detection sensitivity, with this relationship modelled by a sigmoidal function (Fig. 1b; Fig. S1d). The stringency imposed during mutation detection similarly impacted sensitivity for low-frequency mutations (Fig. 1c; Fig. S1d). By contrast, mutations at higher frequencies (VAF > 0.5%) were detected with maximum sensitivity even at relatively low fragment-depth and high stringencies (Fig. 1b,c; Fig. S1d). These analyses illustrate the inherent challenge of reliably detecting low-frequency ctDNA mutations by random sampling.
The enrichment of DNA fragments by hybrid-capture results in heterogeneous coverage across targeted exons. Even in the absence of hybridization biases, we found the detection of ctDNA mutations was similarly heterogeneous: since mutations in the edge regions of exons had lower fragment-depth than central mutations, detection sensitivity was up to 10% lower among edge mutations (Fig. 1d; Fig. S1e,f). Given that many pathogenic mutations occur within exon edge regions, especially at splice-site positions where coverage is lowest29, this edge-effect is a relevant consideration when designing hybrid-capture panels for ctDNA sequencing.
The short fragment length of cell-free DNA can cause erroneous or ambiguous alignment to the human reference genome. We found that ~5% of exons analyzed (118 of 2,356) had sub-optimal alignability, with these exons exhibiting lower fragment-depth and the mutations they harbored being detected with reduced sensitivity (Fig. 1e; Fig. S1f). This effect hindered the detection of mutations in notable gene families, such as the RAS family (KRAS, NRAS, HRAS)30. The impact of local alignability and exon position were most pronounced when evaluating low-VAF mutations, and when depth or stringency were limiting (Fig. 1d,e). These results demonstrate that, even in the absence of experimental variables, genomic context can have an influence on the detection of ctDNA mutations.
Evaluating ctDNA assays using synthetic DNA controls (sequins)
We next evaluated the detection of ctDNA mutations using synthetic DNA controls known as ‘sequins’. Sequins are synthetic DNA sequences that emulate natural human genes and mutations, and recapitulate many of the technical biases that impact their analysis by NGS31,32 (Fig. 2a).
Figure 2. Evaluating ctDNA assays with sequins.
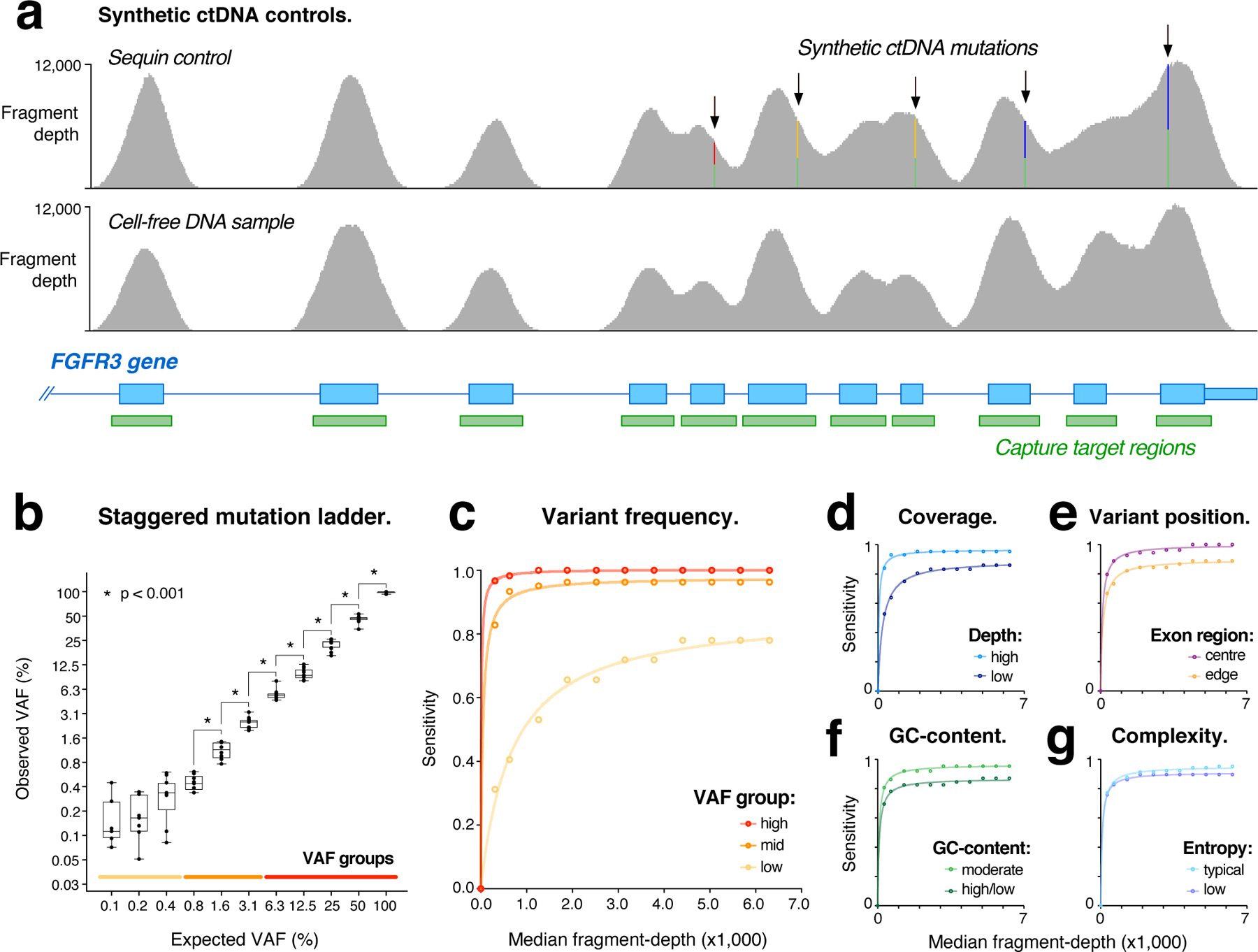
(a) Genome browser view showing fragment coverage within a synthetic sequin control (upper) representing the oncogene FGFR3, harboring multiple synthetic mutations at VAF = 50%. For comparison, coverage is also shown within the natural FGFR3 gene (lower) obtained from the accompanying human sample. (b) Scatter-box plots show observed vs expected variant allele frequencies (VAFs) for synthetic sequin mutations (n = 134), which are represented in two-fold VAF increments from 0.1%−100%. Asterisks indicate significant differences in measured VAFs between increments (two-sided t-test; p < 0.001; n > 8 data points per bin). Boxes show median ± range (whisker) and interquartile range (box). Colored lines indicate high, mid and low VAF groups used in c. (c–g) Curves modelling the relationship between library depth (median-fragment depth) and detection sensitivity for synthetic sequin mutations under various conditions: (c) shows mutations within different VAF groups, indicated on lower axis of b; (d) mutations with high fragment-depth (> 5000-fold), compared to low fragment-depth (< 3000-fold); (e) mutations within exon edge regions (< 20bp from exon boundary), compared to central regions (> 50 bp from exon boundary); (f) mutations in regions of high or low GC-content (< 40% / > 60%), compared to moderate regions; (g) mutations in regions of low sequence entropy (< 1.9), compared to typical regions.
We assembled a mixture of sequin controls representing 134 recurrent and/or clinically actionable somatic mutations within the functional domains of 87 cancer-related genes (Supplementary Data 1). Sequins representing wild-type and mutant alleles for each gene were combined in precise ratios to form a staggered reference ladder spanning a wide range of VAF levels (from 0.1% to 100%; Fig. 2b). The sequin mixture was fragmented and size-selected to emulate cell-free DNA fragments (Fig. S2a), then added at ~0.2% fractional abundance to human mock cell-free DNA samples (described below). These combined samples were then analyzed by targeted hybrid-capture sequencing (see Methods). In total, 119 kb of synthetic sequence was captured and analyzed.
We evaluated the detection of synthetic mutations encoded by the sequin mixture. After calibrating sequins to match the coverage of their accompanying human sample (6311-fold median fragment-depth; Fig. S2b), 125/134 synthetic mutations were detected (sensitivity = 0.93). To assess the impact of coverage on sensitivity, we repeated variant detection across a range of down-sampled libraries. Decreasing coverage had a strong impact on the detection of low-frequency mutations (VAF < 0.5%), whilst mutations at intermediate (0.5–5%) and high (> 5%) frequencies were detected with high sensitivity even at low fragment-depths (0.96–1.00; Fig. 2c). We observed little difference in sensitivity for single nucleotide variants (SNVs) and small insertions/deletions (1–16 bp; Fig. S2c), suggesting variant frequency had a larger effect than variant type.
In addition to global coverage depth, performance is also influenced by coverage heterogeneity, resulting from regional variation in hybridization kinetics, PCR amplification and library conversion efficiency. Sequin mutations ranged over 300-fold in fragment-depth (12,080-fold to 36-fold), with sequin coverage profiles closely resembling corresponding genes in their accompanying human sample (r2 = 0.89; Fig 2a; Fig. S2d,e). Such heterogeneity had a strong effect on variant detection: mutations in regions of high coverage (> 5000-fold) were detected with up to 30% higher sensitivity than regions of low coverage (< 3000-fold; Fig. 2d).
To elucidate the underlying determinants of this heterogeneity, synthetic variants were stratified according to genomic context (see Methods). We observed reductions in detection sensitivity for mutations in (i) exon edge regions (Fig. 2e), (ii) regions of high or low GC-content (Fig. 2f) and (iii) regions of low sequence complexity (Fig. 2g), identifying these as likely contributing variables. Many pathogenic mutations exist in such challenging genomic contexts. For example, low coverage was obtained within the GC-rich TERT promoter region, obscuring detection of the synthetic c.−57A>C mutation33, represented at 1.5% VAF in this region (Fig. S2f).
Changes observed in the abundance of ctDNA fragments in patient plasma may indicate tumor progression, response or resistance to therapy, or disease relapse9. It is therefore essential that in addition to detecting ctDNA mutations, ctDNA assays can accurately measure their frequency. At maximum fragment-depth, two-fold magnitude changes in ctDNA abundance could be reliably resolved down to a VAF of 0.8% but were inaccurate below this level (Fig. 2b). Resolution was also impacted by fragment-depth, with decreasing coverage eroding the lower limit of quantitative accuracy (Fig. S2g). This demonstrates the difficulty of accurately identifying changes in the abundance of low-frequency ctDNA mutations during patient treatment.
Multi-site, cross-platform proficiency study
We next undertook a large-scale proficiency study utilizing a set of contrived reference DNA samples that are described in detail in a companion article34. Briefly, genomic DNA extracted from ten diverse human cancer cell-lines was pooled at equal abundance to create a mock cancer sample (Sample A; Fig. 3a). This pooled sample, as well as each individual cell-line, was genotyped to establish a set of ~40,000 ‘known variants’ and ~10.2 Mb of ‘known negatives’ (positions that matched the human reference genome in every cell line) within the exonic coding regions. Together, these form a reference annotation against which diagnostic performance was subsequently evaluated.
Figure 3. Structure of cross-platform ctDNA sequencing proficiency study.
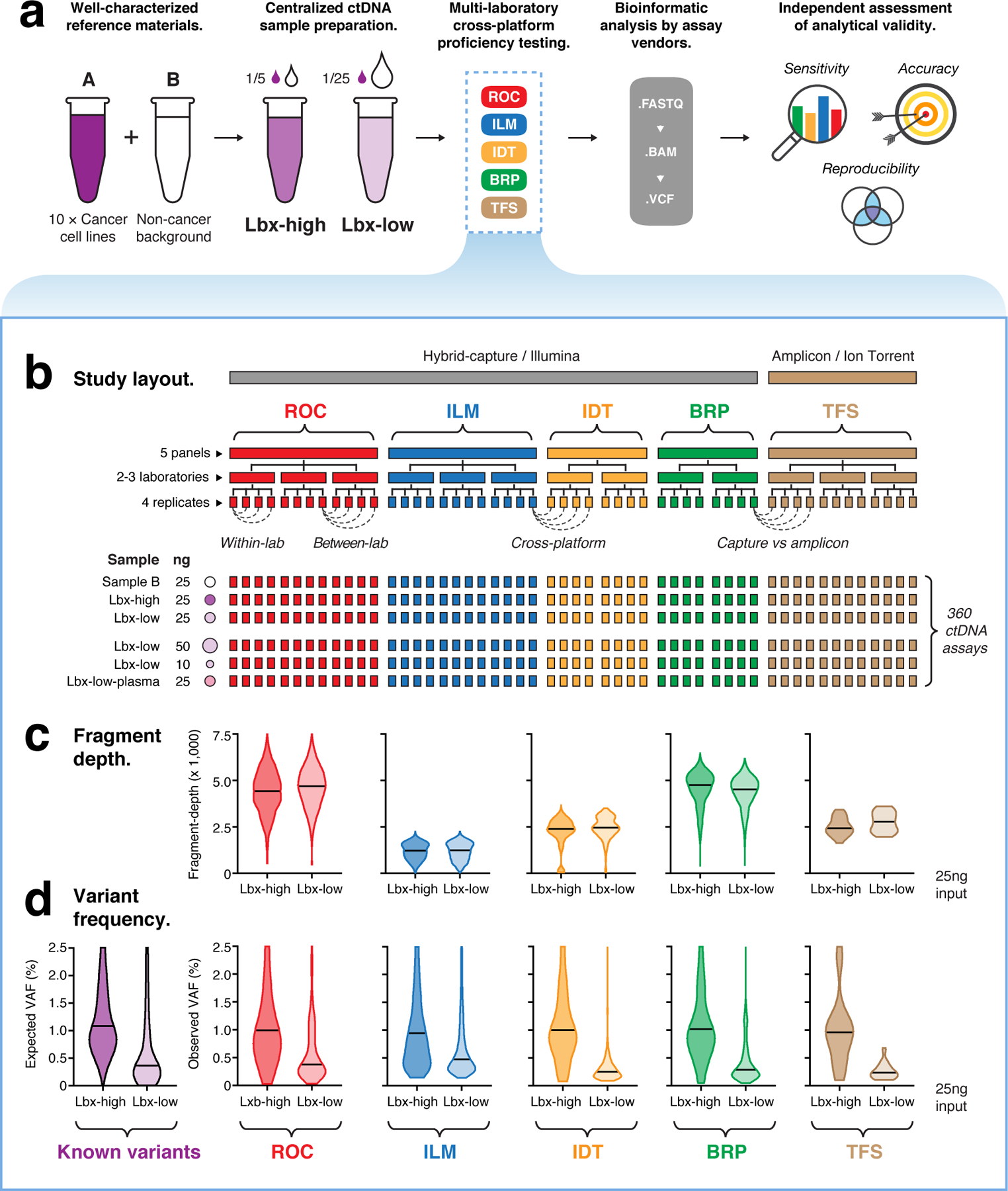
(a) Schematic overview of the proficiency study. Briefly, contrived mock cell-free DNA samples (Lbx-high, Lbx-low) were administered to 12 test labs, where they were analyzed by one or more participating ctDNA sequencing assays (ROC, ILM, IDT, BRP, TFS; see Supplementary Table 3). Bioinformatic analysis was performed by the relevant assay vendor, using their custom pipelines. Results were then submitted for analytical evaluation by an independent team. (b) Schematic overview of the proficiency testing scheme. Each participating ctDNA assay was performed at two or three independent test labs, with four technical replicates per lab generated for each test sample. Each of Lbx-high, Lbx-low and Sample B were analyzed at a fixed 25 ng input amount, and Lbx-low was additionally analyzed at 10 ng and 50 ng input amounts, and at 25ng input following extraction from a synthetic plasma solution (Lbx-low-plasma). In total, 360 ctDNA assays were evaluated. (c; upper) Violin plots show coverage distributions (unique fragment-depth) for Lbx-high and Lbx-low (25 ng input) replicates in each participating assay. (c; lower) Distribution of variants allele frequency (VAF) for on-target variant candidates in Lbx-high and Lbx-low (25 ng input). For comparison, expected VAF distributions for known variants in Lbx-high and Lbx-low are also shown (lower left).
To emulate the range of VAFs typically encountered in ctDNA assays, Sample A was combined at known ratios with DNA extracted from a non-cancer background cell-line (Sample B) to create two further reference samples: Lbx-high (20% A / 80% B) and Lbx-low (4% A / 96% B; Fig. 3a). These samples were enzymatically sheared, and size selected to form DNA fragment-size distributions of ~160–180 bp (see Methods).
These mock cell-free DNA samples were administered to twelve independent laboratory sites, across the United States, United Kingdom, China and Australia (Fig. 3a). Each laboratory performed one or more participating ctDNA sequencing assay (Supplementary Table 3), which included hybrid-capture assays from Roche Sequencing Solutions (ROC), Illumina (ILM), Integrated DNA Technologies (IDT) and Burning Rock Dx (BRP), and an amplicon sequencing panel from Thermo Fisher Scientific (TFS). All sequencing was performed using Illumina instruments (NovaSeq or NextSeq), with the exception of the TFS amplicon assay, which was sequenced using Thermo Fisher Scientific’s IonTorrent instrument (Fig. 3b).
Each participating assay was performed at 2–3 independent test labs, with four technical replicates per lab for each mock ctDNA sample, at a fixed DNA input amount (25 ng). In addition, Lbx-low was analyzed with increased (50 ng) and decreased (10 ng) input amounts, to investigate the impact of cell-free DNA input quantity. To assess the impact of technical variables during plasma DNA extraction, Lbx-low was also analyzed following extraction from a synthetic plasma solution, with extractions performed independently at each test lab (Fig. 3b).
Each sequencing library was then analyzed by the relevant ctDNA assay vendor. Bioinformatic analysis was not standardized across the study, with each vendor instead employing an internal analysis pipeline, and providing a final set of variant candidates for centralized evaluation by an independent team (Fig. 3a). Together, the proficiency study encompassed 360 ctDNA assays, and constitutes the most comprehensive evaluation of analytical performance in ctDNA sequencing to date (Supplementary Data 2).
Coverage depth & heterogeneity
We evaluated coverage depth, which is considered a key variable in ctDNA sequencing10. We observed substantial differences in coverage between different assays, with median unique fragment-depth ranging from ~4,700-fold (BRP, ROC) to ~1,200-fold (ILM; at 25ng input; Fig. 3c). Given DNA input quantities were standardized, these differences reflect the capacity of each assay to exhaustively profile the unique DNA fragments within the input sample and may have a relevant impact on assay performance. Assuming 25ng input equates to ~7,500 genome equivalent copies, estimated molecular recovery rates ranged from ~63% in ROC to 17% in ILM (Fig. 3c; Supplementary Tables 2,4). The TFS amplicon assay achieved comparable fragment-depth to participating hybrid-capture assays (Fig. 3c).
As observed above, multiple technical variables can result in uneven coverage across target regions. After normalizing for overall depth, assays were distinguished by clear differences in coverage heterogeneity (Fig. S3a,b). For example, although the BRP and ROC assays achieved similar median fragment-depths, BRP showed lower heterogeneity than ROC across their respective target regions (normalized IQR = 0.23 vs 0.35; Fig. S3a) and at matched sites present on both hybrid-capture panels (normalized IQR = 0.20 vs 0.46; Fig. S3b).
Analytical sensitivity
We next evaluated the sensitivity of hybrid-capture ctDNA assays by measuring the fraction of on-target known variants that were detected in each library (see Methods; Supplementary Data 2). Known variants were detected with superior sensitivity in Lbx-high compared to Lbx-low for all assays, reflecting their higher frequency in the former sample (Fig. 3d, Fig. S3c,d, Fig. S4a,b). Indeed, all assays were highly sensitive for known variants at high (VAF > 2.5%; 0.99–1.00) and intermediate (0.5–2.5%; 0.96–1.00) frequencies but showed progressively weaker sensitivity for variants at lower frequencies (VAF < 0.5%), with significant variation observed between assays (0.1–0.5%; 0.39–0.83; Fig. 4a). The most sensitive assays (IDT, BRP) achieved sensitivity > 0.90 for variants with 0.3–0.5% VAF, however, no assays reached this mark for variants with 0.2–0.3% or 0.1–0.2% VAF (Fig. 4a).
Figure 4. Comparison of performance between hybrid-capture ctDNA assays at 25ng input.
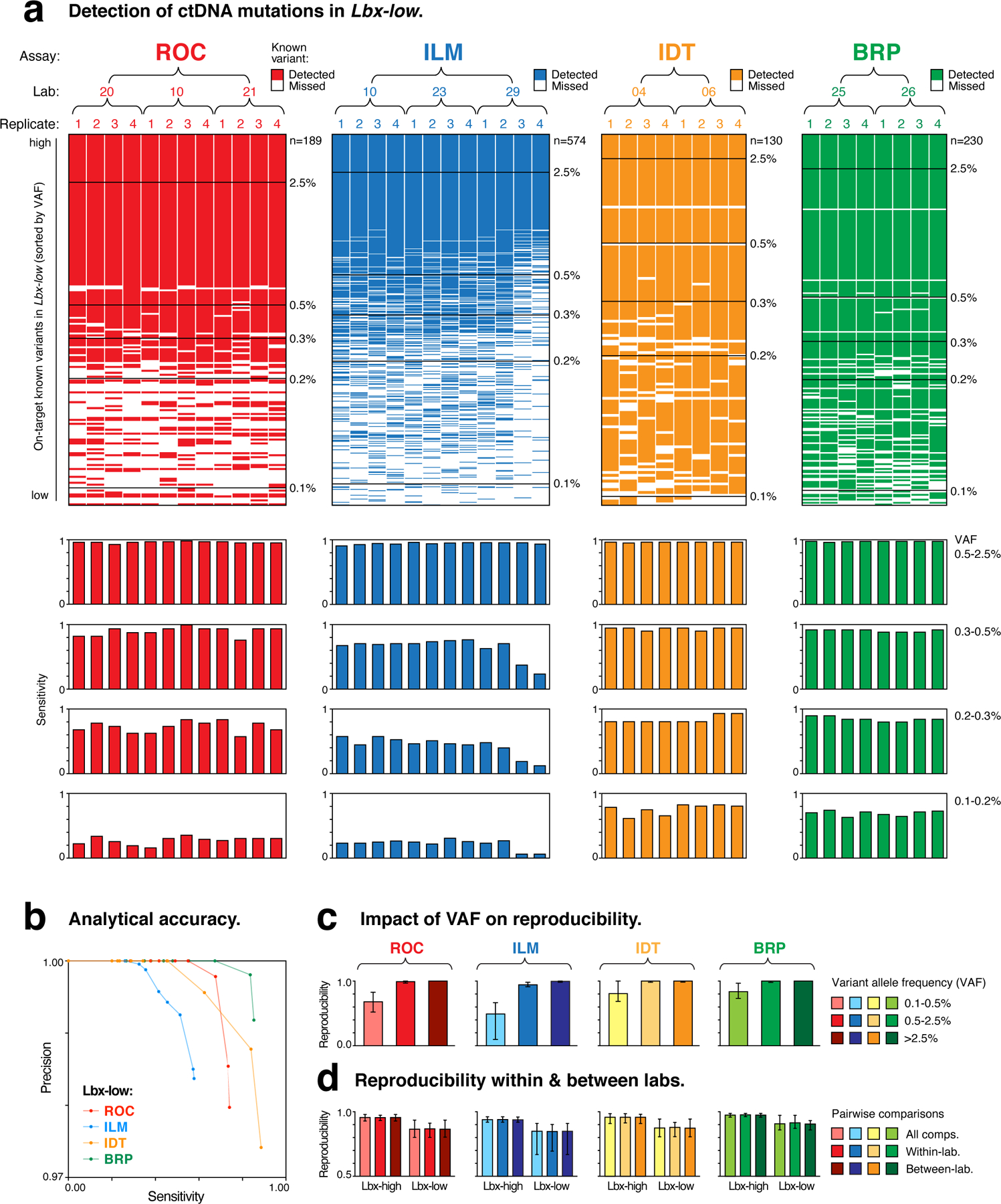
(a; upper) Ordered heatmaps show the detection of known variants (rows) in ctDNA assay replicates (columns). All on-target variants for a given assay are shown. Variants are sorted by expected variant allele frequency (VAF) in descending order, and replicates are arranged hierarchically by assay type, test lab and replicate number. Heatmaps show results for Lbx-low at 25ng input and equivalent heatmaps for Lbx-high are shown in Fig. S4a. (a; lower) Aligned below each heatmap column, bar charts indicate the sensitivity of variant detection in each replicate. Sensitivity is reported separately for known variants in the following VAF ranges: 2.5–0.5%, 0.5–0.3%, 0.3–0.2%, 0.2–0.1%, with measurements taken from both Lbx-high (high- and mid-VAF) and Lbx-low (low-VAF). (b) Precision-recall curves compare diagnostic performance of participating ctDNA assays for Lbx-low (25ng input; VAF range 2.5–0.1%). Equivalent curves for Lbx-high are shown in Fig. S4c. (c,d) Bar charts show pairwise reproducibility scores for participating assays (n = 132 for ROC, ILM; n = 56 for IDT, BRP; median ± range): (c) reproducibility is reported separately for variant candidates at high, mid and low frequency (as above); (d) reproducibility is reported separately for all within-lab and between-lab pairwise comparisons. Note that due to its smaller panel size and VAF distribution of on-target variants the TFS amplicon sequencing assay is not included in these analyses.
As demonstrated above, high coverage is essential for reliable sampling of rare ctDNA mutations. Consistent with this, differences in assay sensitivity partially reflected the differences observed between assays in coverage depth. For example, the high depth achieved by BRP enabled sensitive detection for variants as low as 0.3–0.5% VAF (0.89–0.93), whereas the ILM assay exhibited lower fragment-depth and lower sensitivity at this level (0.24–0.77; Fig. 3c, Fig. 4a). However, coverage depth alone was not necessarily a good predictor of sensitivity, with IDT achieving superior sensitivity to ROC despite its lower fragment-depth (Fig. 3c; Fig. 4a). This result is likely attributed, at least in part, to the lower coverage heterogeneity in the IDT assay (Fig. S3a,b), emphasizing the importance of achieving even coverage, in addition to overall depth.
Sensitivity is also influenced by bioinformatic variables. For example, among BRP assays, there were several known variants that were missed by every replicate despite other variants of similar or lower VAF being reproducibly detected (Fig. 4a). This suggests the BRP analysis pipeline was highly stringent, with strict filtering of variant candidates slightly reducing the sensitivity of variant detection that was achieved (Fig. 3c; Fig. 4a).
Analytical accuracy
We next identified false-positive (FP) variant candidates that were erroneously detected at known negative positions by each assay (see Methods; Supplementary Data 2). With all assays utilizing unique molecular identifiers (UMIs) to correct sequencing errors35, FPs were relatively rare, ranging from a mean of 1.65 to 5.3 FP candidates per replicate (at 25ng input; Supplementary Table 5). After accounting for panel sizes, BRP exhibited the lowest FP rate (0.03 FP/kb) and IDT the highest (0.07 FP/kb), however, given the small number of FPs, the differences between assays were not statistically significant (p > 0.05). Erroneous variant candidates occurred almost exclusively at low frequency (VAF < 0.5%; Supplementary Table 5).
To compare the accuracy of the participating ctDNA assays we generated precision-recall curves36, ranking known variants and FPs according to their observed VAFs. For Lbx-low samples at 25 ng input, BRP was the most accurate assay, with roughly equivalent sensitivity but superior precision to IDT (Fig. 4b, Fig. S4c). While this analysis enables useful cross-platform comparisons, it should be noted that precision is strongly influenced by the mutational burden of the sample under analysis (i.e., the number of positives available for detection), and should not be taken to indicate the inherent precision of the participating ctDNA assays. Overall, despite the differences observed in fragment-depth and sensitivity among the different ctDNA assays, FP rates were modest and broadly similar. Therefore, sensitivity, rather than precision was the major determinant of overall analytical performance.
Reproducibility between assay replicates
We next evaluated reproducibility by comparing the outcomes of replicate assay within and between labs (see Methods). We defined reproducibility as the fraction of variant candidates shared between any pair of replicates, with all possible pairwise comparisons considered.
Similar to sensitivity, reproducibility was generally high (0.99–1.00 and 0.95–1.00) for variants at high (>2.5%) and intermediate (0.5–2.5%) VAF, respectively, but was relatively low and differed widely between panels for low-frequency variants (0.1–0.5%; 0.58–0.83; Fig. 4c; Fig. S4d). Once again, coverage was a relevant variable, but was not alone sufficient to explain differences in reproducibility between assays, with IDT achieving relatively high reproducibility despite its lower fragment-depth (Fig. 3c; Fig. 4c). The results highlight the difficulty of reproducibly detecting low-frequency ctDNA mutations, and suggest that sensitivity is the major determinant of assay reproducibility. Within known positions, we found that FPs constituted only a small minority (< 10%) of the discordant variant candidates between any given pair of replicates (Fig. S4e), although we note that this fraction may be larger in samples with lower mutational burden than the reference samples analyzed here.
The concordance between reproducibility and sensitivity indicates that measurements of reproducibility can provide a useful proxy for diagnostic performance that is not dependent on the availability of a reference annotation. However, we noted that, whereas FNs are guaranteed to reduce assay sensitivity, this is not necessarily true for reproducibility, where systematic FNs (i.e., known variants missed in every replicate) are not penalized. In this case, reproducibility would slightly over-estimate the performance of the BRP assay, in which systematic FNs were most common (Fig. 4a,c). This emphasizes the value of well-characterized reference samples that can directly measure diagnostic performance37.
Finally, despite the wide variation between samples and panels, we observed no significant differences in reproducibility across within- and between-lab comparisons (Fig. 4d). This implies that all assays were robust to technical variables between facilities, and were impacted largely by random, rather than systematic variation.
Impact of cell-free DNA input quantity & plasma extraction
The quantity of cell-free DNA retrieved from a patient blood-draw is typically small, and this can be a major limitation for ctDNA assays10. To assess the impact of cell-free DNA input quantity, we next measured the performance of each hybrid-capture assay with high (50 ng), medium (25 ng) and low (10 ng) amounts of input DNA (Lbx-low; see Methods).
Coverage depth scaled linearly with input quantity for a given assay but varied widely between assays (Fig. 5a). Low input (10 ng) ILM assays did not reach the minimum coverage requirements for analysis, so were excluded from subsequent evaluation (Fig. 5a).
Figure 5. Impact of cell-free DNA input quantity (Lbx-low) on hybrid-capture ctDNA assay performance.
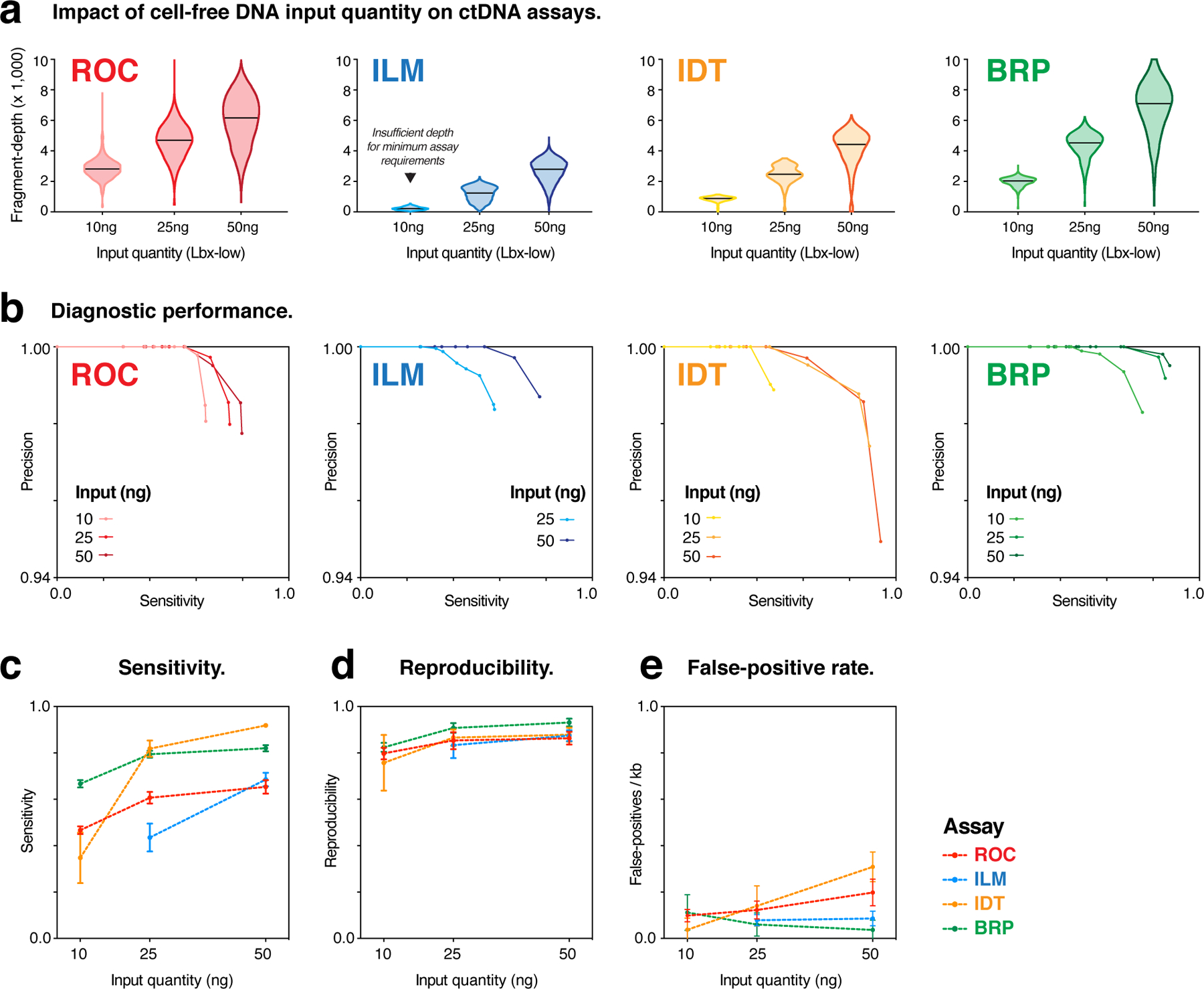
(a) Violin plots show coverage distributions (unique fragment-depth) for Lbx-low replicates at 10ng, 25ng and 50ng input amounts for hybrid-capture ctDNA assays. Note that 10ng ILM assays did not reach minimum coverage requirements, so were excluded from subsequent analysis (b) Precision-recall curves compare diagnostic performance of participating ctDNA assays for Lbx-low at each input amount above (VAF range 2.5–0.1%). (c-e) Curves showing the relationship between cell-free DNA input quantity (Lbx-low) and variant detection sensitivity (c), pairwise reproducibility (d) and false-positive rates (FPs/kb; e) for each participating ctDNA assay profiling low frequency variants (VAF range 0.5–0.1%). Error bars are mean ± 95% CI . Note that, due to its smaller panel size and VAF distribution of on-target variants, the TFS amplicon sequencing assay is not included in these analyses.
The increasing fragment-depth afforded by 25 ng input, compared to 10 ng, resulted in substantial improvements in sensitivity, reproducibility and overall diagnostic performance for all assays, particularly for low-frequency variants (Fig. 5b–e; Fig. S5a,b). However, some assays (BRP, ROC) showed minimal further improvement with the addition of 50 ng input (Fig. 5b–e; Fig. S5a,b). The extent to which performance varied over the range of input quantities tested indicates the robustness of each assay to the variable cell-free DNA input amounts encountered in the clinic. Overall, the greater fragment-depth achieved by an assay at a given input level, the more robust that assay was to variation in input quantity, with BRP being the most stable (Fig. 5b–e).
We also evaluated the impact of cell-free DNA extraction, with each laboratory independently performing multiple extractions on DNA from Lbx-low suspended in synthetic plasma solutions (Lbx-low-plasma). Extraction efficiencies ranged from mean 33% (TFS) to 55% (BRP; Supplementary Data 3), with the DNA retrieved being subsequently quantified and analyzed at 25ng input quantities (see Methods). In general, we observed no significant difference in sensitivity, FP-rates or overall accuracy between Lbx-low and Lbx-low-plasma (Fig. S6a–c). Pairwise reproducibility was equivalent for Lbx-low and Lbx-low-plasma replicates, as well as for the pairwise comparison of Lbx-low to Lbx-low-plasma replicates (Fig. S6d). Finally, just as for Lbx-low, there was no difference in pairwise reproducibility across within-lab and between-lab comparisons for Lbx-low-plasma replicates (Fig. S6e). These results indicate that all participating ctDNA assays were relatively robust to technical variables between test labs at the plasma-DNA extraction stage.
Comparison of TFS amplicon assay to hybrid-capture panels
Amplicon sequencing methods enable targeted analysis of cancer mutation hotspots and can be applied in ctDNA analysis. We next compared the Thermo Fisher Scientific Oncomine cfDNA assay (TFS) to the other participating ctDNA-assays, which all use hybrid-capture enrichment (Fig. 3a,b). The TFS target regions (~1.9 kb) encompass driver-mutation hotspots within 11 cancer genes (Supplementary Table 3), such as BRAF p.V600E, a highly recurrent melanoma mutation38 that is one of the known variants in our analysis (Fig. 6a). With these target regions almost entirely contained within the ROC, ILM and BRP capture panels, we were able to perform direct comparisons of sensitivity, accuracy and reproducibility within this clinically relevant window (see Methods).
Figure 6. Evaluation of TFS amplicon sequencing assay.
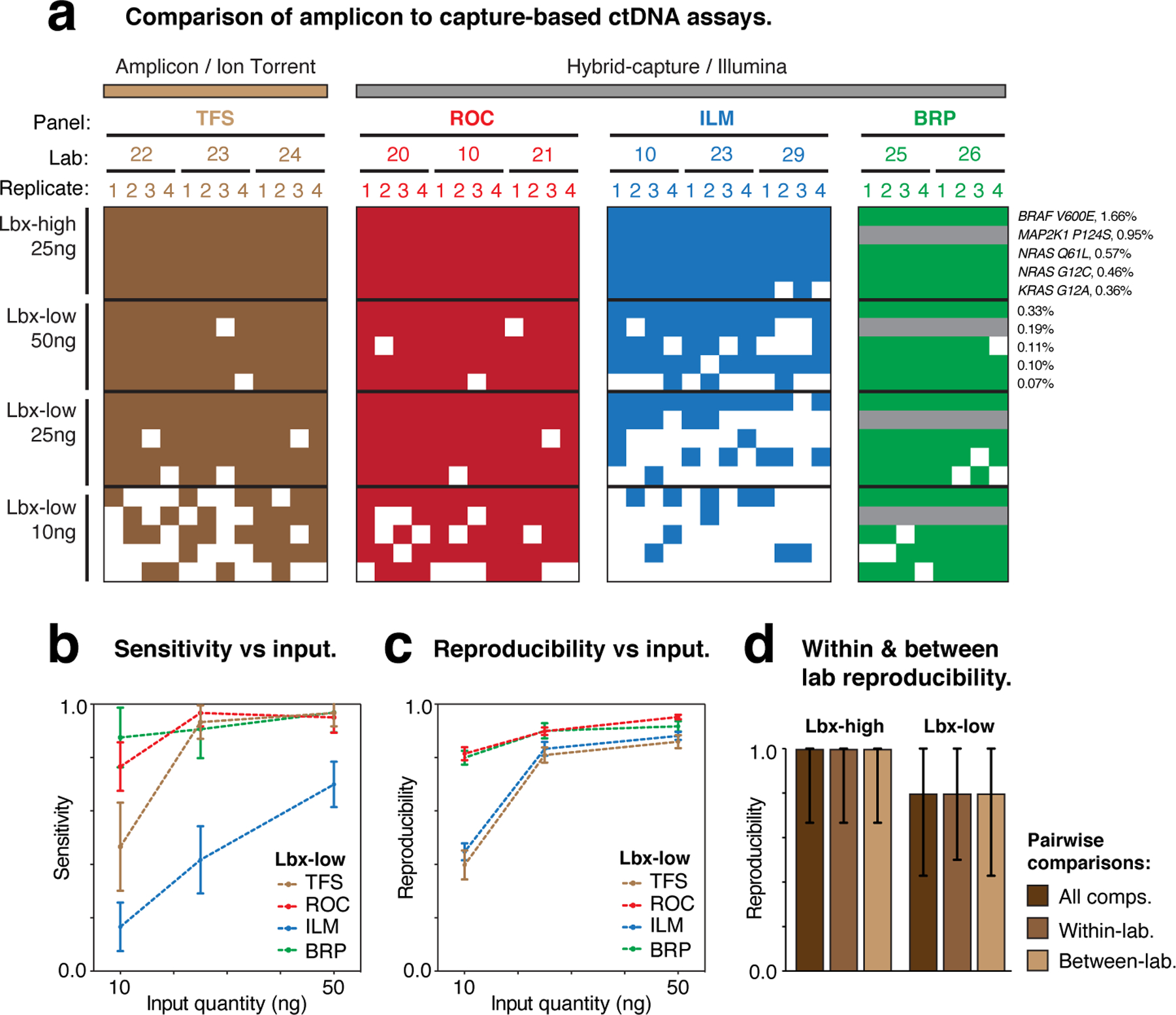
(a) Heatmaps show the detection of known variants (rows) in ctDNA assay replicates (columns). Variants are sorted by expected variant allele frequency (VAF) in descending order for each sample/input quantity (Lbx-high 25ng, Lbx-low 10–50ng), and replicates are arranged hierarchically by assay type, test lab and replicate number. Grey rows indicate where known variant was not within the target regions for a given assay. (b,c) Curves showing the relationship between cell-free DNA input quantity (Lbx-low) and variant detection sensitivity (b) and pairwise reproducibility (c). (d) Bar charts show pairwise reproducibility scores for participating assays (n = 132 for ROC, ILM TFS; n = 56 for BRP; median ± range). Reproducibility is reported separately for all pairwise comparisons in Lbx-high and Lbx-low and separately for all within-lab and between-lab comparisons. Note that the IDT hybrid-capture assay is not included in these comparisons because this panel had limited overlap with TFS amplicon target regions.
Overall, performance was similar between the TFS amplicon assay and hybrid-capture assays. TFS showed perfect detection sensitivity for on-target known variants in Lbx-high, and achieved equivalent sensitivity to ROC/BRP for Lbx-low, when high input was used (25 or 50 ng; Fig. 6a,b). Due to its lower fragment-depth (Fig. 3c), TFS suffered a larger reduction in sensitivity than ROC/BRP when input quantity was restricted (10 ng), but still outperformed ILM, the hybrid-capture assay with the lowest fragment-depth (Fig. 6a,b). With this loss of sensitivity, BRAF p.V600E (VAF = 0.33%) was missed in half of all TFS replicates at 10 ng, whereas it was detected with perfect reliability at 25 and 50 ng (Fig. 6a). TFS and ILM also showed poor reproducibility at low input levels, compared to ROC and BRP (Fig. 6c). Finally, as we observed for hybrid-capture panels (Fig. 4d), TFS showed no difference in reproducibility between within- and between-lab comparisons (Fig. 6d).
To further evaluate the detection of low-frequency mutations, TFS test sites also analyzed synthetic DNA control (AcroMetrix Oncology Hotspot Control) containing 15 known cancer mutations that overlapped TFS hotspot regions (out of 521 in total; 50ng input; see Supplementary Methods). This enabled more robust measurement of diagnostic performance for low frequency variants, with all 15 mutations being present at ~0.1% VAF. Low-frequency mutations were detected with relatively high sensitivity (0.86–1.0; Fig. S7a). A number of false-positives were also detected, however, these were almost entirely excluded by applying a minimum detection threshold of VAF > 0.05% (Fig. S7a,b). Accordingly, a strong improvement in reproducibility (median 0.65 vs 0.94) was observed when applying this filter, at a relatively small cost to sensitivity (median 0.96 vs 0.90; Fig. S7c).
Overall, these results indicate comparable performance between the TFS amplicon sequencing assay and participating hybrid-capture based assays for the detection of SNVs. Indeed, we found the performance of a given assay, and especially its robustness to reductions in input quantity, was largely determined by the fragment-depth achieved, not the method of target enrichment or sequencing.
DISCUSSION
The ability to diagnose and monitor cancer through ctDNA sequencing promises to revolutionize clinical oncology5–9. Accordingly, there is considerable interest and investment in the ongoing development of NGS-based ctDNA assays39. Yet the reliable detection of trace amounts of fragmented ctDNA from a routine blood-draw remains a major technical challenge10. Government, regulatory and clinical organizations have therefore called for thorough analytical evaluation of ctDNA assays, in order to define diagnostic limits, assess reproducibility and identify key experimental variables that impact performance28.
This study begins to address these unmet needs and to the best of our knowledge provides the first large-scale assessment of analytical performance among industry-leading ctDNA assays. We report that mutations represented above ~0.5% VAF could be detected with high sensitivity, accuracy and reproducibility by all participating assays. However, variant detection was generally unreliable and variable between assays for mutations lower than ~0.5% VAF. This was primarily driven by the proportion of known variants that missed detection (i.e., lack of sensitivity) due to stochastic sampling, in agreement with our initial simulated experiments. False-positives were a less significant source of discordant results, with UMIs used effectively to minimize errors in all assays. Cell-free DNA input quantity was a key variable, with increasing input leading to improved fragment-depth, sensitivity and reproducibility.
Previous studies have reported discordant results between alternative assays or parallel ctDNA and tumor-biopsy tests, although the underlying causes and extent to which this resulted from biological, rather than technical, factors were unclear25–27 (see below). We also observed discordant results between vendors, labs and assay replicates. However, this was limited to low-frequency mutations (VAF < 0.5%), and largely reflected the limitations of stochastic sampling rather than technical biases or errors. In fact, we found that participating assays were generally robust to technical variables between test labs, from plasma extraction to sequencing workflow stages.
The performance characteristics of the assays evaluated here were broadly similar to what has been reported by several ctDNA sequencing providers (based on internal testing) that did not participate in this study. During validation of the Guardant360 CDx™ hybrid-capture assay, variants were detected with high sensitivity (~94%) at VAF ≥ 0.4%, declining to ~64% among variants with VAF ranging from 0.05%–0.25%23. FoundationACT™ showed ~99% sensitivity for SNVs with VAF > 0.5%, ~95% for 0.25%–0.5% VAF and ~70% for 0.125–0.25% VAF13. MSK-ACCESS™ showed ~98% sensitivity for SNVs with VAF > 0.5%, declining to ~74% for 0.1%–0.5% VAF12. Validation of the amplicon-based InVisionFirst™ assay, suggested this may have superior LOD to the hybrid-capture assays above, with ~99% sensitivity for SNVs as low as 0.25–0.35% VAF24. Consistent with our findings, all of these providers also reported low false-positive rates. Although direct comparisons between studies that used different test samples and DNA input quantities must be treated with caution, it appears generally true that the sensitive detection of ctDNA mutations below ~0.5% VAF is a major challenge.
While the accurate detection of mutations > 0.5% VAF and robustness to technical variables among ctDNA assays is cause for optimism, data arising during early clinical implementation of several tests highlight necessity for reliable detection of low-frequency mutations. A survey of > 1,000 plasma samples from cancer patients tested with Guardant360 CDx™ found that half of all detected SNVs occurred below ~0.5% VAF and a quarter below ~0.2% VAF40. Among 859 patients tested with FoundationACT™, half of all variants detected had VAFs below ~1.3% and a third below ~0.5%13. Among 435 patients tested with MSK-ACCESS™, >5% of all mutations detected were missed by NGS but identified by more sensitive genotyping methods, with these having a median VAF of ~0.08%12. These results, which are generally based on the analysis of patients with advanced disease, demonstrate the tendency for ctDNA mutations to be represented at very low frequencies. Moreover, given the likelihood that many low-frequency variants missed detection, the median VAFs reported in these studies represent upper-bound estimates.
The analytical performance characteristics of a given ctDNA assay determine its potential suitability for specific applications in research and clinical oncology, with different assays being suited to different purposes. For example, higher sensitivity and precision and lower LOD are required for molecular characterization of early-stage vs late-stage cancer, due to the lower mutational burden and abundance of ctDNA fragments in circulation8,9.
That mutations above ~0.5% VAF were accurately detected indicates that the participating ctDNA assays may be suitable for molecular stratification and profiling tumor evolution in advanced cancer patients, where informative mutations are commonly detected with VAFs ranging from ~1–10%14,18–20. Given that variants were also accurately quantified above an LOQ of ~0.8% VAF, ctDNA sequencing appears suitable to monitor frequencies over time and in response to therapeutic intervention14. Characterization of early-stage, localized disease with ctDNA sequencing requires accurate detection of mutations with VAFs ranging from ~0.1–1% VAF, although we note that this is likely to vary between cancer types and individual patients3,4,41,42. In patients with Non-Small-Cell Lung Cancer, for example, a tumor ~10 cm3 in volume yields ctDNA fragments at ~0.1% VAF, on average, in the plasma3. Therefore, while further improvement is required to ensure reliable detection of low frequency mutations, suitability for early-stage cancer appears within reach of current ctDNA sequencing assays.
The ability to detect and monitor post-surgical minimal residual disease (MRD) via ctDNA sequencing is an application with great potential utility8,9. However, MRD monitoring demands highly sensitive detection of mutations with VAFs ranging from ~0.1–0.01% to reliably predict disease relapse3,5,43,44. Given the participating assays in our study were generally unreliable for mutations with VAFs ~10-fold higher than this, substantial further development is required for use in monitoring MRD, and targeted analysis of known mutations by droplet digital PCR (ddPCR; or related approaches) remains the most promising strategy for this application.
The breadth a ctDNA assay’s target regions and the types of mutations detected are also a relevant consideration. For example, large hybrid-capture panels like the ILM panel tested here (154 target genes, ~500 kb) are ideal for unbiased genomic characterization in advanced metastatic disease, but are not cost effective for targeted monitoring of known driver mutations during and after therapy. Amplicon methods like the TFS panel tested here (11 target genes, ~1.9 kb) enable more affordable, focused analysis of mutation hotspots but their small panel sizes limit suitability for unbiased genomic surveillance. Limited ability to detect mutations types beyond SNVs and small indels is a further drawback of amplicon-based approaches.
The use of ctDNA sequencing for early cancer detection demands unbiased surveillance of broad target regions with high sensitivity and low LOD. Moreover, given the low prior probabilities involved in screening healthy subjects, false-positive rates must be exquisitely low for an assay to achieve clinical utility7. Given these requirements, considerable improvements to existing assays, as well as continued depreciation of sequencing costs, will be needed for this much-anticipated application to approach feasibility.
Given the discussions above, improved sensitivity for mutations below ~0.5% VAF should be a priority for the ongoing development of ctDNA assays. Optimizations to the efficiency of plasma-DNA extractions, target capture and NGS library preparations may yield incremental improvements in coverage and, thereby, in the detection of low-frequency mutations. Such advances are critical to ensure the robustness of ctDNA assays to the variable cell-free DNA input quantities encountered in the clinic10. However, participating assays showed diminishing returns with increasing sample input and/or fragment-depth and remained unable to exhaustively identify low-frequency mutations.
This deficit in sensitivity reflects the difficulty of reproducibly detecting rare ctDNA fragments by random sampling of amplified fragments; an inherent statistical challenge that may not be fully overcome simply by increasing global fragment-depth. Substantial additional improvements to ctDNA assays may therefore require further innovations, such as the selective enrichment of ctDNA fragments over the background of non-cancerous cell-free DNA fragments, potentially by exploiting discrepancies in fragment size45,46 or methylation status47.
CtDNA assay performance will also benefit from ongoing improvement in the detection of cancer mutations beyond SNVs and small indels. Translocations and other structural variants48,49, copy number alterations50 and microsatellite instability51 can be informative cancer biomarkers, but their detection is challenging due to the small fragment sizes and amplification biases in ctDNA assays. Further assay development and proficiency testing on these features is therefore required.
The reliability of ctDNA sequencing assays is commonly assessed by measuring concordance between alternative assays or assay replicates27, parallel ctDNA and tumor-biopsy tests25,26,40, or orthogonal analysis of plasma samples with non-NGS based techniques, such is ddPCR23. These approaches have been applied across large cohorts of clinical specimens to inform assay development and validation12,23,40.
While highly useful, several caveats must be acknowledged. As noted earlier, concordance measurements between assays do not penalize systematic errors, such as mutations that are missed by multiple assays or replicates, and may therefore over-estimate performance. The comparison of plasma and tumor-tissue biopsies is also confounded by an array of biological factors, such as tumor type, stage, morphology and heterogeneity. For example, a mutation detected in a tumor-tissue biopsy but not with matched ctDNA sequencing might be truly absent from the plasma, due to restricted local circulation at its site of origin. Therefore, discordance is not necessarily indicative of poor analytical performance for either test. While, orthogonal analysis of a plasma sample with ddPCR can reliably determine whether detected mutations are true, it is not practical with this targeted approach to assess all invariant sites across a large hybrid-capture sequencing panel to rule out potential false-negatives28.
Arguably the most relevant limitation in using bona fide cell-free DNA samples for analytical validation experiments, is the inability to standardise across different assays, sites & replicates. Patient plasma samples vary widely in mutational burden and cell-free DNA yields, with no single sample representing the full diversity of mutation types and frequencies that should be considered in rigorous proficiency testing. The use of different clinical cohorts, cell-free DNA input quantities and test materials makes it difficult to draw reliable comparisons between validation studies performed by different ctDNA sequencing providers12,13,23,24.
To avoid these caveats, we utilised a combination of synthetic DNA controls (sequins31,32, AcroMetrix) and cell-line derived reference samples (Lbx-high, Lbx-low)34 to evaluate the analytical performance of ctDNA assays. This approach allows: (i) appropriate numbers, types and frequencies of known mutations to be analyzed; (ii) unambiguous classification or true/false-positives/negatives across large target regions; (iii) standardization of samples and input quantities between sites, assays and assay replicates and; (iv) absence of confounding biological variables.
However, there are also limitations to this approach. The enzymatic process by which samples were fragmented does not perfectly emulate the fragmentation of natural cell-free DNA, on which the participating assays have been optimized. It is therefore possible that the efficiency of fragment capture and library conversion for these contrived samples may be lower/higher for any given kit than for natural cell-free DNA. Moreover, we cannot be certain that the kinetics of fragmentation are equivalent between all genome regions, potentially resulting in some sites being over/under-represented or represented by fragments of atypical sizes. While we deliberately devised reference samples with a high mutational burden to improve the power of performance measurements, this means that measures of assay precision are inflated and should not be interpreted as realistic measures of performance on clinical samples (instead, they are useful for making technical comparisons between assays, given common reference samples were tested).
Contrived reference samples can never fully recapitulate the many nuanced biological factors that influence the potential utility of ctDNA sequencing assays in real clinical contexts. For example, the ability to distinguish informative ctDNA mutations from a background of benign variants in cell-free DNA, generated during clonal haematopoiesis52, is a challenge that cannot be evaluated using synthetic reference materials. Therefore, while they are an ideal substrate on which to assess the analytical performance characteristics of ctDNA assays in the absence of confounding biological variables, contrived samples alone cannot be used to determine clinical thresholds (e.g., LOD & LOB), which must account for, rather than exclude, such variables. Importantly, clinical performance cannot exceed analytical performance and analytical validity is a pre-requisite for clinical validity and clinical utility, with these properties requiring demonstration in clinical trials employing assays that have achieved analytical validity28.
This study advances the community’s understanding of analytical performance characteristics in ctDNA sequencing, outlines a set of best-practice guidelines (Box 1), and constitutes astep toward the ultimate goal of establishing clinical utility for precision oncology. Moreover, the study establishes a unique set of reference materials, annotations and an analytic framework for standardized proficiency testing on ctDNA assays. While ongoing studies are required to establish the potential clinical validity and utility of ctDNA assays, the SEQC2 Oncopanel Sequencing Working Group has helped lay the foundation for such future work.
METHODS
Simulated ctDNA sequencing assays
To model the parameters of ctDNA sequencing assays, we generated simulated NGS libraries that emulate targeted analysis of cell-free DNA by hybrid-capture sequencing. Simulated reads were created using wgsim (v1.9; https://github.com/lh3/wgsim), with mutation and error rates set to zero (-e 0 -R 0 -r 0 -X 0). Paired-end (2 × 150 bp) read-fragments were generated, with a mean fragment size of 160 bp and a standard deviation of 15 bp (−1 150 −2 150 -d 160 -s 15). Read-fragments were simulated uniformly over 155 cancer-related loci on a generic gene panel for oncology applications (Roche NimbleGen; For Research Use Only, Not for Diagnostic Procedures), based either on the hg38 reference sequence or a modified hg38 sequence containing one Cosmic SNV per exon (n = 2,356; selected randomly and inserted using gatk FastaAlternateReferenceMaker (v3.8)53.
Reads simulated from reference and mutant sequences were combined in precise ratios to create eight independent simulated libraries in which all SNVs were represented at a specified VAF level (5%, 2%, 1%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%), with genes covered uniformly at ~9,000-fold fragment-depth. Simulated read-fragments were aligned to hg38 using bwa mem (v0.7.16)54. In silico capture enrichment was performed by intersecting aligned read-fragments with the capture targets BED file and retaining only fragments with ≥ 60 bp overlap to a target region. This process creates convex coverage profiles over targeted exons that resemble typical coverage profiles obtained during hybrid-capture sequencing (Fig. S1a). These libraries were then down-sampled (gatk DownsampleSam) to create additional libraries with incremental reductions in fragment-depth.
Simulated SNVs were then detected using VarScan (v2.4.3)55 and detection sensitivity (TPs/(TPs+FNs)) was calculated for SNVs within each library, across a range of detection stringency levels (i.e., the minimum number of supporting fragments for a SNV to be called). To measure the effect of variant position, simulated SNVs were parsed based on their distance to the nearest exon boundary (‘edge regions’ < 20 bp; ‘central regions’ > 50 bp). To measure the effect of local alignability, SNVs were parsed based on the alignability of their occupied exon sequence. Exons were considered to have ‘sub-optimal’ alignability if ≥ 5% of overlying alignments had MapQ = 0.
Manufacture and sequencing of synthetic DNA controls (sequins)
We evaluated the detection of ctDNA mutations using synthetic DNA controls known as ‘sequins’ (www.sequinstandards.com). Synthetic controls were manufactured in a purpose-built facility at the Garvan Institute of Medical Research (Sydney, Australia). Detailed descriptions of the design, manufacture and experimental validation of sequins have been published previously31,32.
For this study a custom sequin mixture for ctDNA sequencing experiments was created, encompassing 354 individual sequins ranging in size from ~1–6 kb for a combined ~757 kb of synthetic DNA sequence in total. This mixture provides synthetic ‘chiral’ representations of relevant exons/domains within 87 cancer-related genes and includes 134 synthetic mutations (Supplementary Data 1).
As described previously, sequin sequences were initially synthesized and validated by a commercial vendor (Thermo Fisher Scientific, GeneArt). Within a purpose-built manufacturing facility, synthetic sequences were amplified by bacterial culture, excised by restriction enzyme digest, quantified by UV fluorometry (Thermo Fisher Scientific Qubit) and combined using a liquid-handling robot (Eppendorf epMotion). By combining synthetic molecules representing reference and variant alleles in precise ratios, synthetic mutations were represented across a wide range of VAF levels, ranging from 100% to 0.1% in two-fold increments. This staggered reference ladder allows detection sensitivity and quantitative accuracy to be assessed at different VAF levels.
To emulate the fragmentation of cell-free DNA, the synthetic sequin mixture was enzymatically sheared using NEBNext dsDNA Fragmentase in 10X Fragmentase Reaction Buffer for 30 minutes at 37°C. The reaction was terminated by addition of 0.5M EDTA and the resulting fragments were purified using double sided SPRI size selection. 0.65X Agencourt AMPure XP beads were used to exclude fragments >250 bp, while 1.8X Agencourt AMPure XP beads were used to enrich fragments < 250 bp and the purified DNA was visualized with an Agilent TapeStation. This neat, fragmented sequin mixture was validated by NGS, using a Nextera XT DNA library prep kit and sequenced on an Illumina MiSeq.
Following validation, the fragmented sequin mixture was spiked into mock human ctDNA reference samples (Lbx-high, Lbx-low; described below) at ~0.2% fractional concentration. These combined samples were analyzed by hybrid-capture sequencing, using a custom oncology panel (Roche NimbleGen; For Research Use Only, Not for Diagnostic Procedures) targeting the 87 human cancer genes that were represented by sequin controls, as well as the sequin controls themselves (this approach is described in detail elsewhere32). In total, 119 kb of synthetic sequence was captured and analyzed.
NGS libraries were prepared with a KAPA LTP Library Preparation Kit (Illumina platform KR0453 – v6.17), in conjunction with IDT xGen dual index adaptors, according to the manufacturer’s protocol with 10 cycles of PCR amplification. Capture enrichment was performed according to an established protocol (Roche Double Capture Technical Note, August 2012). Purified libraries were quantified on an Agilent TapeStation and sequenced on an Illumina NovaSeq (S1 flow cell).
Sequin bioinformatics analysis
Targeted NGS libraries containing reads from mock human ctDNA samples spiked with synthetic sequin controls were initially trimmed using TrimGalore (https://github.com/FelixKrueger/TrimGalore) then processed using the purpose-built anaquin toolkit for sequin analysis, via a workflow that is described in detail elsewhere31. Briefly, sample-derived and sequin-derived reads were separated, and sequin reads were reversed in orientation (anaquin split). Sample and sequin reads were aligned separately to the hg38 reference genome using bwa mem (v0.7.16). Off-target reads were excluded and PCR duplicates collapsed using gatk MarkDuplicates (v4.0). Sequin-derived alignments were then calibrated to equivalent coverage depth to accompanying sample alignments within matched genome regions (anaquin calibrate). VarScan (v2.4.3) was then used to call variants (SNVs and indels) within on-target sequin regions, with a minimum of three supporting read-fragments required for detection. Anaquin somatic was used to evaluate the detection of sequin variants.
Sequin libraries were then incrementally down-sampled (gatk DownsampleSam) and variant detection was repeated across a range of depreciating fragment-depths. Detection sensitivity (the fraction of known sequin variants detected) was calculated in each down-sampled library to generate curves that model the relationship between sensitivity and fragment-depth, with sequin variants parsed into pre-defined VAF bins (< 0.5%, 0.5–5%, > 5%). Sequin variants were also parsed according to: (i) fragment depth (high fragment-depth > 5000-fold; low fragment-depth < 3000-fold), (ii) distance to the nearest exon boundary (edge regions < 20 bp; central regions > 50 bp). (iii) GC-content within a 120 bp local window. GC-content was calculated using bedtools nuc (v2.25; high > 60%, low < 40%). (iv) Sequence complexity within a local 120 bp window. Complexity was calculated using SeqComplex (https://github.com/caballero/SeqComplex), with windows showing entropy scores < 1.9 considered to have low complexity.
Preparation of human ctDNA reference samples
The cross-platform ctDNA sequencing proficiency study from the present manuscript utilized a set of contrived reference DNA samples that are described in detail in a companion article34. Briefly, Sample A comprised genomic DNA extracted from ten diverse cancer cell-lines (Agilent UHRR cell lines) and Sample B is non-cancerous genomic DNA (Agilent Male Control DNA). Sample A and Sample B were combined to create two further reference samples: Lbx-high (20% A / 80% B) and Lbx-low (4% A / 96% B). Note that Lbx-high and Lbx-low are also referred to as Sample D and Sample E (prior to fragmentation) in the accompanying article describing the preparation of reference samples34.
Aliquots of these samples, as well as Sample A and Sample B, were enzymatically fragmented, using the KAPA Frag kit (KAPA Biosystems) according to the manufacturer’s instructions. Samples dissolved in TE buffer were first purified with the Agencourt AmPure XP Kit at a 3:1 bead to sample volumetric ratio to remove EDTA. Purified DNA samples (5 μg) in 10 mM Tris-HCl pH 8.0 (35 μL) were mixed with KAPA Frag Enzyme (10 μL) and 10X KAPA Frag Buffer (5 μL) on ice and incubated at 37°C for 25 minutes. After fragmentation, DNA samples were purified with Agencourt AmPure XP beads again, as described above.
Size selection of fragmented DNA (5 µg / well) was performed on a Pippin Prep instrument (Sage Science) using 3% agarose gel cassette (Sage Science # CDP3010) with the range from BP start (110) to the BP end (190) to achieve an average fragment length of ~165 bp. Post size-selection DNA samples were characterized using an Agilent Bioanalyzer 2100 with DNA high sensitivity kit (Agilent Technologies, Inc.). Typically, a yield of 6–8% was obtained. Samples were quantified and dissolved in Tris buffer (10 mM Tris, pH 8.0) at 5 ng/μL for storage and distribution.
The plasma DNA sample (Lbx-low-plasma) was prepared with a DNA concentration at 40 ng/mL in synthetic plasma (Horizon Discovery, United Kingdom) and two aliquots of 8 mL were shipped to each test site in two 10 mL tubes. Test labs did not assess the concentration of DNA samples except the plasma sample to avoid any site-to-site variation that could be introduced by independent quantification. Qubit dsDNA HS (High Sensitivity) Assay Kit Q32851 (Thermo Fisher Scientific) was used for DNA sample quantification for all ctDNA samples and also mandated at each test site for quantification after DNA extraction from synthetic plasma. An SOP was developed and distributed to all test sites for plasma sample quantification including a step of Qubit dsDNA HS assay calibration with distributed ctDNA samples as standards.
Multi-site ctDNA proficiency testing
Each testing laboratory performed one or more participating ctDNA sequencing assays (five in total; Supplementary Table 3), according to the vendor’s instructions. Sequencing was performed using Illumina (NovaSeq 6000, NextSeq 500) or Thermo Fisher Scientific IonTorrent instruments (Supplementary Table 3). Sequencing information is supplied in Supplementary Table 6. Detailed experimental procedures for each assay are provided in Supplementary Methods.
Each participating assay was performed in 2–3 independent labs, with four technical replicates per lab for each mock ctDNA sample, at a fixed DNA input amount (25 ng). In addition, Lbx-low was analyzed with increased (50 ng) and decreased (10 ng) input amounts. Each test lab also performed four independent plasma-DNA extractions on the provided Lbx-low-plasma sample (2.5 mL per replicate) and analyzed the extracted DNA at a fixed DNA input amount (25 ng per replicate).
All sequencing libraries were then administered to the relevant ctDNA assay vendor for blind analysis. Bioinformatic analysis was not standardized across the study, with each vendor instead employing an internal analysis pipeline and providing a final set of variant candidates for centralized evaluation by an independent team at the Garvan Institute of Medical Research and the US FDA National Centre for Toxicological Research. All participating assays utilized Unique Molecular Identifiers (UMIs) to collapse duplicate read-pairs into consensus fragments. Comparisons of assay yields/depths throughout the study are based on unique fragment-depth, rather than raw read or alignment counts. Detailed information about the bioinformatics pipeline employed by each vendor is provided in Supplementary Methods.
Evaluation of results
Pre-processing variant candidates
Assay vendors provided a single independent VCF file containing candidate variants called in each assay replicate, at each test lab. The following pre-processing steps were used to ensure direct comparability between all call-sets. Where relevant, candidates marked with filter flags by the vendor bioinformatics pipeline, or indicated as VAF = 0, were excluded. Multi-allelic variant sites were broken into multiple individual variants using bcftools norm (v1.9). Complex and/or multi-nucleotide variants were broken into their simplest individual components using RTG-tools (https://github.com/RealTimeGenomics/rtg-tools) vcfdecompose (v3.10.1) with the --break-mnps --break-indels parameters set to TRUE and gatk LeftAlignAndTrimVariants (v4.0.11) was used to ensure consistent representation of indels.
Sensitivity and accuracy
To measure the sensitivity of participating ctDNA assays we compared each set of variant candidates to the reference annotation described in34. RTG-tools vcfeval (v3.10.1) was used to compare the VCF file for each assay replicate to the set of ‘known variants’ in Lbx-high/Lbx-low, and the set of known germline variants in the non-cancer background Sample B. These comparisons were restricted to the intersection of all ‘known positions’ with the on-target reportable regions provided by the relevant assay vendor. All candidates within known positions were classified as true-positive (TP) or false-positive (FP). Sensitivity was defined as the number of TPs in a given replicate divided by the number of on-target known variants for the relevant panel, and was calculated both globally and within pre-defined VAF bins (0.1–0.5%, 0.5–2.5%, >2.5%). FP-rates were defined as the number of FPs in a given replicate divided by the size of the on-target known negative positions for the relevant panel, thereby accounting for differences in panel size. Precision-recall curves were generated by incrementally varying the minimum VAF threshold (below which candidates are excluded) from 0% to 100% and re-calculating sensitivity (TP/(TP+FN)) and precision (TP/(TP+FP)) at each increment.
Reproducibility
To measure the reproducibility of participating ctDNA assays we performed reciprocal pairwise comparisons between variant call-sets for all replicates of a given assay/sample/input. Comparisons were performed using RTG-tools vcfeval (v3.10.1) and were restricted to the capture target regions provided by the relevant assay vendor. For a given pair of replicates, reproducibility was defined as the fraction of total variant candidates that were concordant between call-sets, with all possible pairwise comparisons being performed. Reproducibility was calculated globally, across within-lab and between-lab comparisons, and within pre-defined VAF bins (0.1–0.5%, 0.5–2.5%, >2.5%). When calculating reproducibility within VAF bins, candidates in a given bin in the first sample were compared to the whole call-set for the second sample, and vice versa, in order to avoid bin-edge effects.
Supplementary Material
ACKNOWLEDGEMENTS
All SEQC2 participants freely donated their time, reagents, and computing resources for the completion and analysis of this project. We thank our expert colleague Prof. Sarah-Jane Dawson for providing useful feedback during manuscript preparation. We acknowledge the following funding sources: NHMRC grants APP1108254 & APP1114016 (to T.R.M.), BAA grant HHSF223201510172C (to D.J.Jr), Shanghai Municipal Science and Technology Major Project grant 2017SHZDZX01 (to L.S.), the National Natural Science Foundation of China grant 31720103909 (to L.S.), MRFF grant MRF1173594, Cancer Institute NSW Early Career Fellowship 2018/ECF013 and philanthropic support from The Kinghorn Foundation (to I.W.D). The contents of the published materials are solely the responsibility of the administering institution, a participating institution or individual authors, and they do not reflect the views of any funding body listed above.
Footnotes
CODE AVAILABILITY
Variant call-sets for each ctDNA sequencing assay were generated by internal bioinformatics pipelines by each assay vendor. While these pipelines are not open source, detailed descriptions and relevant software version numbers are provided in the Supplementary Methods section. All data plots were generated using R (version 3.5 or later) or GraphPad Prism (version 8).
REPORTING SUMMARY
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Suggestion: Reliable detection of mutations below 0.5% variant allele frequency remains a key challenge for circulating tumor DNA sequencing assays.
DISCLAIMERS & COMPETING INTERESTS
This research includes contributions from, and was reviewed by, the FDA and the NIH. This work has been approved for publication by these agencies, but it does not necessarily reflect official agency policy. Certain commercial materials and equipment are identified in order to adequately specify experimental procedures. In no case does such identification imply recommendation or endorsement by the FDA or the NIH, nor does it imply that the items identified are necessarily the best available for purpose. The Garvan Institute of Medical Research has filed patent applications on synthetic controls for genomics. The authors declare no other competing financial interests.
DATA AVAILABILITY
Descriptive data about individual ctDNA assays are provided in Supplementary Data 2. Descriptive data about individual variants, including their detection status in each ctDNA assay, are provided in variant classification tables within the Source Data Excel file. These tables were used to generate variant detection heatmaps and other data plots. Raw sequencing data has been deposited to the NCBI Bioproject PRJNA677999. Variant calls generated by each assay vendor (in VCF format) and panel region files (in BED format) can be accessed at the following link: https://figshare.com/projects/SEQC2_Onco-panel_Sequencing_Working_Group_-_Liquid_Biopsy_Study/94523
REFERENCES
- 1.Leon SA, Shapiro B, Sklaroff DM & Yaros MJ Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res 37, 646–650 (1977). [PubMed] [Google Scholar]
- 2.Stroun M, Anker P, Lyautey J, Lederrey C & Maurice PA Isolation and characterization of DNA from the plasma of cancer patients. Eur J Cancer Clin Oncol 23, 707–712 (1987). [DOI] [PubMed] [Google Scholar]
- 3.Abbosh C et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 545, 446–451 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bettegowda C et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 6, 224ra24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abbosh C, Birkbak NJ & Swanton C Early stage NSCLC - challenges to implementing ctDNA-based screening and MRD detection. Nat Rev Clin Oncol 15, 577–586 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Aggarwal C et al. Strategies for the successful implementation of plasma-based NSCLC genotyping in clinical practice. Nat Rev Clin Oncol (2020). [DOI] [PubMed]
- 7.Aravanis AM, Lee M & Klausner RD Next-Generation Sequencing of Circulating Tumor DNA for Early Cancer Detection. Cell 168, 571–574 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Siravegna G, Marsoni S, Siena S & Bardelli A Integrating liquid biopsies into the management of cancer. Nat Rev Clin Oncol 14, 531–548 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Wan JCM et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer 17, 223–238 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Volckmar A-L et al. A field guide for cancer diagnostics using cell-free DNA: From principles to practice and clinical applications. Genes Chromosomes Cancer 57, 123–139 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Ross MG et al. Characterizing and measuring bias in sequence data. Genome Biol. 14, R51 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brannon AR et al. Enhanced specificity of high sensitivity somatic variant profiling in cell-free DNA via paired normal sequencing: design, validation, and clinical experience of the MSK-ACCESS liquid biopsy assay. bioRxiv 2020.06.27.175471 (2020).
- 13.Clark TA et al. Analytical Validation of a Hybrid Capture–Based Next-Generation Sequencing Clinical Assay for Genomic Profiling of Cell-Free Circulating Tumor DNA. J Mol Diagn 20, 686–702 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dawson S-J et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med 368, 1199–1209 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Forshew T et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med 4, 136ra68 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Kinde I, Wu J, Papadopoulos N, Kinzler KW & Vogelstein B Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A 108, 9530–9535 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klein EA et al. Development of a comprehensive cell-free DNA (cfDNA) assay for early detection of multiple tumor types: The Circulating Cell-free Genome Atlas (CCGA) study. J Clin Oncol 36, 12021–12021 (2018). [Google Scholar]
- 18.Miller AM et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature 565, 654–658 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murtaza M et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 497, 108–112 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Murtaza M et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat Commun 6, 8760 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Newman AM et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med 20, 548–554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newman AM et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol 34, 547–555 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Odegaard JI et al. Validation of a Plasma-Based Comprehensive Cancer Genotyping Assay Utilizing Orthogonal Tissue- and Plasma-Based Methodologies. Clin Cancer Res 24, 3539 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Plagnol V et al. Analytical validation of a next generation sequencing liquid biopsy assay for high sensitivity broad molecular profiling. PLoS One 13, e0193802 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuderer NM et al. Comparison of 2 commercially available next-generation sequencing platforms in oncology. JAMA Oncol 3, 996–998 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stetson D et al. Orthogonal comparison of four plasma NGS tests with tumor suggests technical factors are a major source of assay discordance. JCO Precision Oncology 1–9 (2019). [DOI] [PubMed]
- 27.Torga G & Pienta KJ Patient-paired sample congruence between 2 commercial liquid biopsy tests. JAMA Oncol 4, 868–870 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Merker JD et al. Circulating tumor DNA analysis in patients with cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J Clin Oncol 36, 1631–1641 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Shiraishi Y et al. A comprehensive characterization of cis-acting splicing-associated variants in human cancer. Genome Res. 28, 1111–1125 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bos JL The ras gene family and human carcinogenesis. Mutat Res 195, 255–271 (1988). [DOI] [PubMed] [Google Scholar]
- 31.Blackburn J et al. Use of synthetic DNA spike-in controls (sequins) for human genome sequencing. Nat Protoc 14, 2119–2151 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Deveson IW et al. Chiral DNA sequences as commutable controls for clinical genomics. Nat Commun 1–13 (2019). [DOI] [PMC free article] [PubMed]
- 33.Horn S et al. TERT promoter mutations in familial and sporadic melanoma. Science 339, 959–961 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Jones W et al. A Verified Genomic Reference Sample for Assessing Performance of Cancer Panels Detecting Small Variants of Low Allele Frequency. Genome Biology (2021). 10.1186/s13059-021-02316-z. [DOI] [PMC free article] [PubMed]
- 35.Fu GK, Hu J, Wang P-H & Fodor SPA Counting individual DNA molecules by the stochastic attachment of diverse labels. Proc Natl Acad Sci U S A 108, 9026–9031 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saito T & Rehmsmeier M The precision-recall plot is more informative than the ROC plot when evaluating binary classifiers on imbalanced datasets. PLoS One 10, e0118432 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hardwick SA, Deveson IW & Mercer TR Reference standards for next-generation sequencing. Nat Rev Genet 18, 473–484 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Hodis E et al. A landscape of driver mutations in melanoma. Cell 150, 251–263 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheridan C Investors keep the faith in cancer liquid biopsies. Nat Biotechnol 37, 972–974 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Lanman RB et al. Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell-free circulating tumor DNA. PLoS One 10, e0140712 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phallen J et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci Transl Med 9, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cohen JD et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 359, 926–930 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tie J et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med 8, 346ra92 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Diehl F et al. Circulating mutant DNA to assess tumor dynamics. Nat Med 14, 985–990 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mouliere F et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci Transl Med 10, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Underhill HR et al. Fragment Length of Circulating Tumor DNA. PLoS Genet 12, e1006162 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shen SY et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 563, 579–583 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Kim Y-W et al. Monitoring circulating tumor DNA by analyzing personalized cancer-specific rearrangements to detect recurrence in gastric cancer. Experimental & Molecular Medicine 51, 1–10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klega K et al. Detection of somatic structural variants enables quantification and characterization of circulating tumor DNA in children with solid tumors. JCO Precision Oncology 2018, 10.1200/PO.17.00285 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peng H et al. CNV detection from circulating tumor DNA in late stage non-small cell lung cancer patients. Genes (Basel) 10, 926 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cai Z et al. Detection of microsatellite instability from circulating tumor DNA by targeted deep sequencing. J Mol Diagn 22, 860–870 (2020). [DOI] [PubMed] [Google Scholar]
- 52.Hu Y et al. False-positive plasma genotyping due to clonal hematopoiesis. Clin Cancer Res 24, 4437–4443 (2018). [DOI] [PubMed] [Google Scholar]
- 53.McKenna A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li H & Durbin R Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koboldt DC et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22, 568–576 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Descriptive data about individual ctDNA assays are provided in Supplementary Data 2. Descriptive data about individual variants, including their detection status in each ctDNA assay, are provided in variant classification tables within the Source Data Excel file. These tables were used to generate variant detection heatmaps and other data plots. Raw sequencing data has been deposited to the NCBI Bioproject PRJNA677999. Variant calls generated by each assay vendor (in VCF format) and panel region files (in BED format) can be accessed at the following link: https://figshare.com/projects/SEQC2_Onco-panel_Sequencing_Working_Group_-_Liquid_Biopsy_Study/94523