

Abstract
The genetic causes of atrial fibrillation (AF) with slow conduction are unknown. Eight kindreds with familial AF and slow conduction, including a family affected by early onset AF, heart block and incompletely penetrant non-ischemic dilated cardiomyopathy (DCM) underwent whole exome sequencing. A known pathogenic mutation in the desmin (DES) gene resulting in p.S13F substitution (NM_001927.3:c.38C>T) at a PKC phosphorylation site was identified in all four members of the kindred with early-onset AF and heart block, while only two developed DCM. Higher penetrance for AF and heart block prompted a genetic screening for DES modifier(s). A deleterious mutation in the phosphodiesterase-4D-interacting-protein (PDE4DIP) gene resulting in p.A123T substitution (NM_001002811:c.367G>A) was identified that segregated with early onset AF, heart block and the DES mutation. Three additional novel deleterious PDE4DIP mutations were identified in four other unrelated kindreds. Characterization of PDE4DIPA123T in vitro suggested impaired compartmentalization of PKA and PDE4D characterized by reduced colocalization with PDE4D, increased cAMP activation leading to higher PKA phosphorylation of the β2-adrenergic-receptor, and decreased PKA phosphorylation of desmin after isoproterenol stimulation.
Our findings identify PDE4DIP as a novel gene for slow AF and unravel its epistatic interaction with DES mutations in development of conduction disease and arrhythmia.
Keywords: Desmin, DES, PDE4DIP, myomegalin, early onset atrial fibrillation, slow conduction, slow ventricular response, heart block, cardiomyopathy, ENG
Graphical Abstract
1. Introduction
Atrial fibrillation (AF) is the most common cardiac arrhythmia in the general population, with an estimated lifetime incidence of 7% to 26%. It is associated with major cardiovascular complications, including stroke, tachyarrhythmia and heart failure (Go et al., 2013). While AF typically leads to a rapid ventricular response in the majority of patients, a subset of AF is associated with a slow heart rate (Amat-y-Leon et al., 1974; R. K. Kumar, Saxena, & Talwar, 1991; Yamashita, Murakawa, Ajiki, & Omata, 1997).
AF has a major genetic component with a mode of inheritance of a complex trait and in rare cases is inherited as a single gene disorder. More than 44 disease-causing genes with rare damaging mutations have been identified in monogenic forms of familial AF. In addition, over 95 genetic loci have been identified by genome wide association studies of AF (Alzahrani et al., 2018). The pathophysiology of the distinct clinical entity characterized by AF and slowing of conduction along the electrical conduction system is poorly understood. Conduction disease leading to early onset heart block may occur at the time of birth with an incidence of 1 in 15,000-25,000 live births (Costedoat-Chalumeau, Georgin-Lavialle, Amoura, & Piette, 2005) and is often associated with maternal anti-Ro/SSA and/or anti-La/SSB autoantibodies, and is less commonly due to congenital syphilis, rheumatic fever or diphtheria infection (Michaelsson, Riesenfeld, & Jonzon, 1997). Progressive conduction system disease, which may lead to heart block is heritable and has been associated with mutations in the genes encoding cardiac ion channels (Baruteau, Probst, & Abriel, 2015). Other forms of conduction system disease develop later in childhood or early adult life and are associated with dilated cardiomyopathy (Moak et al., 2001; Udink ten Cate et al., 2001) or AF but its etiology remains poorly understood and are likely caused by different genetic mutations. However, the genetic causes of AF with slow ventricular response are vastly unknown.
We describe the genetic screening of a multiplex family with several family members affected by early onset AF and heart block requiring pacemaker implantation in the fourth decade of life. A few affected family members developed non-ischemic dilated cardiomyopathy (DCM) later in life. The genetic causes of these traits were investigated using whole exome sequencing (WES). A deleterious DES mutation was identified that explained the incompletely penetrant cardiomyopathy but not the highly penetrant slow atrial fibrillation. A search for genetic modifiers led to the identification of a novel mutation in PDE4DIP. The genetic screening of seven independent kindreds with AF and conduction disease by WES led to the identification of three additional novel PDE4DIP mutations that segregated with the disease. The PDE4DIP mutation in the larger family was characterized in vitro. These studies identified PDE4DIP mutations as genetic modifiers for the DES mutation and as risk alleles for AF with slow conduction.
2. Materials and Methods
Study Subjects
Patients with early onset familial AF, including a multiplex family from Lebanon with early onset familial AF, cardiomyopathy, and heart block requiring pacemaker placement were referred to the Yale Cardiovascular Genetics clinic for genetic screening. All other kindreds had isolated slow AF with no structural heart disease. The protocol was approved by the institutional review board at Yale University School of Medicine. Informed consent to participate in the study and to undergo genetic sequencing was obtained from all patients. Detailed clinical information, including laboratory data and clinical imaging were collected. Genomic DNA was extracted from peripheral blood leukocytes and sent for exome sequencing. Family history was obtained from the index cases, and pedigrees were constructed based on self-reported phenotypes.
Whole Exome Sequencing and Targeted Sequence Capture
Genomic DNA was captured on exomes at the W.M. Keck Facility at Yale University using Roche NimbleGen 2.1M Human Exome Array, as described earlier (Keramati et al., 2014). In brief, DNA libraries were prepared and sequenced on the Illumina Genome Analyzer, followed by image analysis and base calling. Sequences were aligned against human reference genome (UCSC Genome Browser hg19) and processed using MAQ program SAMtools. SAMtools was also used for the single-nucleotide variant detection and filtering against the reference genome as described earlier. Filters were applied against published databases.
Exome analysis
Segregation analysis was performed on all three phenotypes of interest in the Lebanese family, which may exhibit epistatic interaction in development of cardiomyopathy, heart block and AF. A computer script was designed for variant annotation based on the novelty, conservation, tissue expression and their effect on protein function. They were considered non-conservative if the substituted amino acid was conserved in all species. Filtering criteria included frequency (<1/1000), and splice site location (−2 to +2 intron exon boundary). Synonymous, intronic variants, and variants with low quality were filtered out. Variants that were predicted to be benign by two in silico tools (SIFT, and Polyphen2) as well as by other laboratories were filtered out early in the analysis. CADD score was additionally used to predict pathogenicity and variants with a score of less than 10 were excluded. Regions prone to sequencing errors were screened then all the related duplications and deletions were filtered out. Also, all variants in known AF-associated genes were screened for allele frequencies <1% in the Gnomad database. Lastly, intolerance score and phyloP 46-way score were used as a measure of conservation. The GTEx portal was used to filter out genes without cardiac tissue expression.
Familial AF kindreds and mutation burden analysis
The candidate gene lists segregating with AF and heart block were further narrowed down by screening an additional exome dataset of 7 kindreds with AF and conduction disease. The same filtering criteria for exome analysis detailed above was used for the identified genetic variants.
Exome data derived from 158 ethnically matched individuals without atrial fibrillation or cardiomyopathy served as the control dataset. Data from 7 additional kindreds with atrial fibrillation and conduction disease, constituted the ‘cases’ dataset. A gene-based burden analysis was carried out, comparing the aggregate burden of rare, protein-altering variants. Fisher exact test was used for statistical analysis to evaluate the mutation burden of PDE4DIP.
Cloning and transfection of PDE4DIP
The PDE4DIP NM_001002811 transcript was obtained in a GFP-tagged ORF CMV plasmid from ORIGENE (plasmid code RG215787). The PDE4DIP transgene was then sub-cloned into an mCherry-tagged lentiviral plasmid (pLVX-mCherry-N1) and a point mutation was induced using site directed mutagenesis resulting in an alanine to threonine substitution at amino acid 123. The subcloning and mutagenesis were outsourced to SGI-DNA service (now known as Codex DNA, Inc). The wild type and mutant PDE4DIP- pLVX-mCherry-N1 plasmid sequences were verified using sanger sequencing (primers: 5’TGCTAAACTGTATGTCTGGCTCT3’ and 5’ TCTTGCTTGCCGCTATTTGC3’). Human embryonic kidney (HEK) cell line 293T was transfected with the mutant PDE4DIP-mCherry plasmid, wild type PDE4DIP-mCherry plasmid and empty pLVX-mCherry-N1 control plasmid using the Lipofectamine transfection kit. Transfection efficiency was verified by percentage m-cherry expression on fluorescent microscopy (>50%). Along with the above-mentioned plasmids, the cells were co-transfected with the fluorescence resonance energy transfer (FRET) sensor plasmids and incubated on 35mm microwell dishes with 14mm glass coverslip for 3 days to achieve 60-70% confluency.
In vitro Fluorescence Resonance Energy Transfer (FRET) imaging of cAMP signaling
Intracellular cAMP imaging was done using FRET sensors. A fourth generation Epac-based FRET sensor (mTurquoise2Δ-Epac (CD, ΔDEP, Q270E)-tdcp173Venus (Epac-SH187)) was used as previously described (Klarenbeek, Goedhart, van Batenburg, Groenewald, & Jalink, 2015). This sensor consists of a full-length cAMP-binding Rap-1 activating protein Epac, which is sandwiched between the donor and acceptor fluorescent proteins with larger conformational change, and FRET change compared to sensors with partial Epac. Fourth generation of cAMP sensors have superior photo-stability, dynamic range and signal-to-noise ratios. The design is based on mTurquoise2 as a bleaching-resistant donor, and a tandem of two cp173Venus fluorophores as acceptors.
FRET was quantified using 3 cube ratiometric imaging with analysis in MATLAB using previously developed custom software (A. Kumar et al., 2016). Images were acquired on an inverted Nikon Eclipse Ti widefield microscope equipped with a cooled charged-coupled device Cool SNAP HQ2 camera using a 20x 0.75 NA objective at 37°C. For each time point and field, three sequential images were acquired with the following filter combinations: donor (mTurquoise2) channel with 460/20 (excitation filter-ex), T455lp (dichroic mirror-di) and 500/22 (emission filter-em); FRET channel with 460/20 (ex), T455lp (di) and 435/30 (em); and acceptor (Venus) channel with 492/18 (ex), T515lp (di) and 535/30 (em) filter combinations. For analysis, donor leakage was determined from 293T cells transfected with mTurquoise2 and acceptor cross excitation was obtained from Vinculin-Venus transfected cells. All three FRET images (mTurquoise2, Venus, FRET) were background subtracted and filtered by three-point smoothening. FRET maps and pixel-wise FRET index was calculated as:
FRET index = [FRET channel – x(Donor channel) – y(Acceptor channel)]/[Acceptor channel]
Where x is the leakage co-efficient and y is the cross-excitation fraction. Masks for each cell were generated by thresholding mCherry-PDE4DIP wild type and mutant positive cells using the mTurquoise images. Mean FRET index was calculated as the average FRET signal for all of the mCherry positive cells in the field of view. A total of 18-20 fields were analyzed per group (from two independent experiments). For each field, FRET was measured at baseline and at 4 minutes after stimulation with 1uM isoproterenol (cAMP activator). Additional images were acquired 4 minutes after stimulation with 25 uM of Forskolin (strong cAMP activator) under live cell wide field microscopy. Statistical comparisons were performed in Graphpad Prism version 8. A two-way ANOVA with Sidak’s post hoc was used with statistical significance set at p<0.05.
Immunohistochemistry
The C2C12 cells were transfected with lipofectamine-3000 reagent. The lipofectamine:DNA complexes were prepared according to the manufacturer’s protocol (ThermoFisher, lipofectamine 3000). 4 mg of plasmid (control-mCherry, PDE4DIP-WT-mCherry or PDE4DIP-mut-mCherry) was used to make lipofectamine: DNA complexes for 105 cells. The C2C12 cells were trypsinized, resuspended in media (DMEM+10%FBS, without antibiotics). The single cell suspension was added to the lipofectamine:DNA mix at a concentration of 105 cells/well for a 6-well dish. The cells were plated on a 6-well plate and allowed to adhere overnight. Next day, the media was changed to DMEM+10%FBS, with antibiotics. The following day, the cells were treated with either DMSO or isoproterenol at the concentration of 1μM for 8 minutes and processed for either immunohistochemistry or western blot.
For immunohistochemistry, the cells were washed with 1XPBS, and fixed with 4% formaldehyde overnight at 4°C. The cells were permeabilized with 1XPBS-0.1%Trition, 3X, 5 minutes each, followed by blocking in 1XPBS-0.1%Trition+10%FBS for 1 hour and overnight incubation in primary antibody at 3 μgm/ml for PDE4D, catalog# PA521590, Lot #VH3049391A or 20 μgm /ml for phospho-desmin at serine 60 antibody (p-desmin Ser60), ThermoFisher, Catalog# PA5-38837. This was followed by washes in 1XPBS-0.1%Trition, 3X, 5 minutes each, and overnight incubation in secondary antibody with conjugated Alexa-488 (ThermoFisher, goat anti-rabbit Catalog#A32731). The cells were washed again in 1XPBS-0.1%Trition, 3X, 5 minutes each, mounted in ProlongGold Anti-fade solution, and imaged with 63X oil immersion objective on SP8 confocal microscope.
Images were acquired with the following filter combinations: mCherry-PDE4DIP red channel with 580/610 (excitation/emission) and PDE4D green channel with 488/525 (excitation/emission filter). Similarly, the p-desmin immunostaining was acquired with green channel 488/525 (excitation/emission filter). The PDE4D and mCherry-PDE4DIP co-localization was measured as percentage of double positive particles per cell divided by the total number of red (mCherry) particles per cell. The results were analyzed using one-way ANOVA and Tukey’s post hoc test. Semi-quantitative measurement of PDE4D and p-desmin protein expression was done via mean fluorescence intensity weighted by fluorescent area of visualized cells using ImageJ software. The results were analyzed using one-way ANOVA and Tukey’s post hoc test. For all the immunohistochemistry experiments 3 biological replicates were performed per condition.
Western Blotting
The C2C12 cells were transfected with empty vector, wild type or mutant PDE4DIP plasmids (control-mCherry, PDE4DIP-WT-mCherry or PDE4DIP-mut-mCherry) using lipofectamine 3000 as mentioned above and cultured in a 100 mm plates. At day 3, cells reached 70% confluency and plasmid expression was verified by percentage mCherry fluorescence (>50%) using confocal microscopy. Cells were either treated with DMSO or stimulated with isoproterenol (1 mM) for 8 minutes and harvested in Cell Lysis buffer with fresh protease and phosphatase inhibitors. Cells were quickly collected, placed in liquid nitrogen and subsequently prepared for western blot analysis. The western blot was carried out using standard procedures to quantify phosphorylation changes in the beta-2-adrenergic receptor. The same amounts of lysates (10mg) were loaded on all the gels in all the experiments and antibodies were used at a concentration of 1 μg/ml. Ponceau S stain was used for checking loading across different samples. The following antibodies were used: p-beta adrenergic receptor Ser355, 356 (Catalog # PA538403, ThermoFisher), p-beta adrenergic receptor Ser346 (Catalog#Ab192821, Abcam), total beta-adrenergic receptor (Catalog#Ab182136, Abcam), and HRP conjugated beta-actin (Catalog#5125, Cell Signalling). These experiments were done in triplicates with two sets of biological replicates with identical results. The Signal intensity ± standard error of means was quantified using ImageJ and the significance was calculated using student’s t-test.
Results
3.1. Pathogenic desmin (DES) mutation and its association with cardiac traits
The index case of the Lebanese family was a 48-year-old man (III-2, figure 1A) who presented with syncope and was diagnosed with heart block. Interestingly, his underlying cardiac rhythm at the time was AF. No reversible factors were identified for the AF or heart block. He underwent a permanent pacemaker placement and has been doing well to this date at age 54 without cardiomyopathy. Examination of the remaining family revealed very similar presentations in his 52-year-old brother (III-1, figure 1A) and maternal uncle (II-2) who developed heart block with AF in their early 40 years of age, necessitating a permanent pacemaker with a major difference that they both developed non-ischemic dilated cardiomyopathy (DCM) also in their mid-40 years of age. The mother (II-1) also had similar presentations with early onset heart block and AF but no cardiomyopathy. A 60-year-old maternal aunt (II-6) had AF at 57 years of age but no cardiomyopathy or conduction disease. In sum, there were 4 living family members with heart block and early onset AF, with only 2 of them having DCM, one of whom died shortly after the ascertainment.
Figure 1.
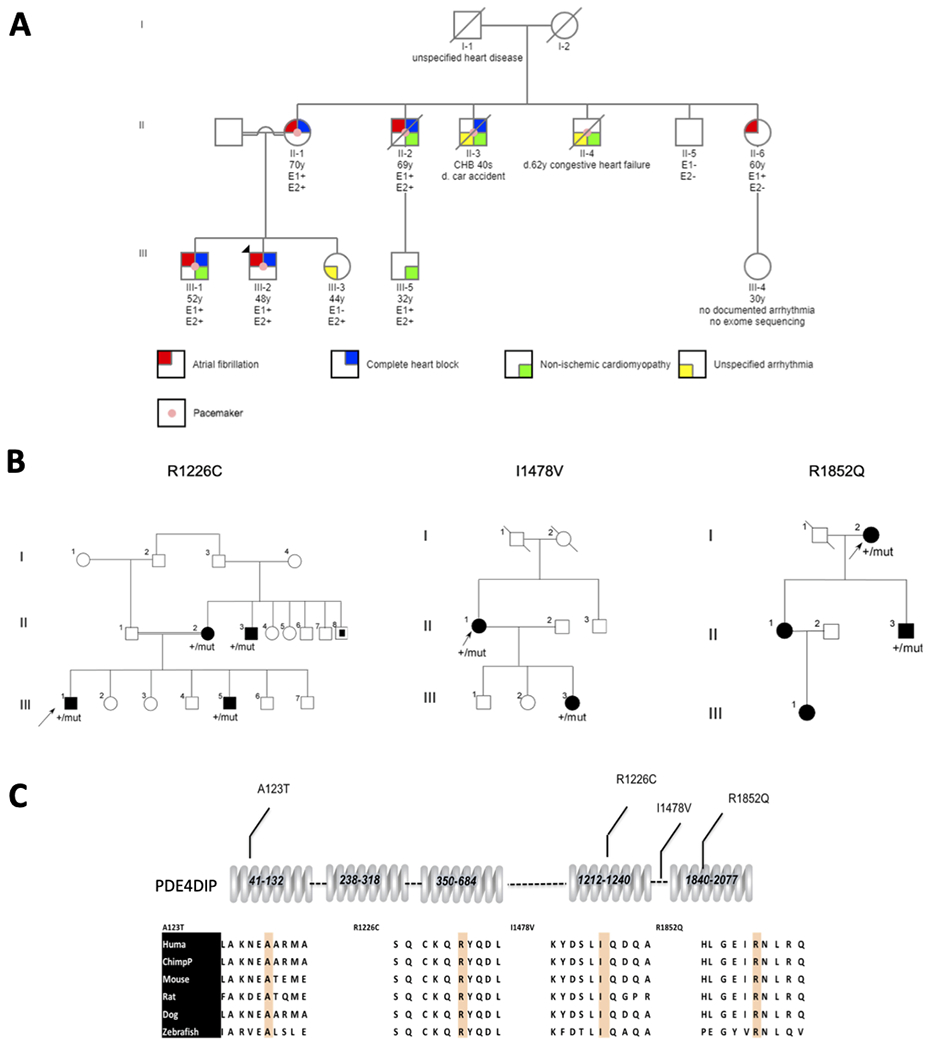
(A) Pedigree of the multiplex Lebanese family with atrial fibrillation, heart block and non-ischemic cardiomyopathy. Each trait is shown in different color as indicated. The index case (III-2) is shown with an arrow. E1: carrier status for p.S13F mutation in desmin and E2: carrier status for p.A123T in PDE4DIP. All patients underwent exome sequencing, and the mutations were confirmed by sanger sequencing. Circles represent females; squares represent males. Symbols with slash through them indicate deceased subjects. (B) Pedigrees with early onset atrial fibrillation and slow ventricular response and their corresponding PDE4DIP mutations. The index cases are indicated by arrows. Individuals with slow AF are indicated by black symbols; unaffected individuals are shown as unfilled symbols. (C) Segments of the PDE4DIP protein sequences flanking the substituted amino acids in the kindreds with slow AF are shown from diverse vertebrate species. As demonstrated, all substitutions in the familial AF kindreds occurred at evolutionarily conserved sites.
Of note, there were 2 other maternal uncles who had died prior to the ascertainment; one of them had known heart block (II-3) and had died in a car accident at 40 years of age, whereas the other (II-4) had an implantable cardiac defibrillator device for his heart failure and died at age 62 from end stage heart failure. While they carried the diagnosis of unspecified arrhythmia, a history AF could not be verified due to absent medical records. Most recently, one young family member (III-5) developed DCM with left bundle branch block.
All four subjects with early onset AF harbored a known pathogenic mutation in the DES gene (NC_000002.11:g.220283222C>T) that resulted in a serine to phenylalanine substitution at codon 13 (p.S13F). A healthy 65-year-old maternal uncle (II-5) who underwent exome sequencing was negative for the DES mutation (figure 1A).
Interestingly, the DES mutation leading to p.S13F substitution had been previously reported in 8 other families affecting a total of 45 patients (Supp. Table S1) (Bergman et al., 2007; Pica, Kathirvel, Pramono, Lai, & Yee, 2008; van Tintelen et al., 2009). On average, 38% of patients harboring the p.S13F substitution developed heart block requiring pacemaker implantation at an average age of 41 years, and 38% of patients developed cardiomyopathy (mostly dilated and arrhythmogenic right ventricular cardiomyopathy) at an average age of 44.6 years, but only 11% of patients had AF in their disease course (Supp. Table S1). Furthermore, 31% had mild proximal and distal skeletal myopathies (Supp. Table S1). Members of the Lebanese family did not have evidence of significant skeletal myopathies. A more comprehensive review of all known mutations throughout the DES gene indicated that on average 49% develop cardiomyopathy (DCM, arrhythmogenic cardiomyopathy, restrictive cardiomyopathy, and non-compaction cardiomyopathy), 36% develop conduction disease requiring pacemaker placement, and a minority (<10%) develop AF (Brodehl, Gaertner-Rommel, & Milting, 2018; Brodehl et al., 2019; Marakhonov et al., 2019; Protonotarios et al., 2020; van Spaendonck-Zwarts et al., 2011). While the incidence of cardiomyopathy in the Lebanese pedigree (50%) was similar to that reported in the literature, the incidence of heart block (67%) requiring pacemaker and AF (83%) were much greater.
3.2. Epistatic interaction between DES and PDE4DIP mutations
The remaining mutations were filtered as described in the methods to remove common variants and genes not expressed in the heart and performed segregation analyses of the remaining genes. This revealed a mutation in the PDE4DIP gene that segregated in all 4 affected family members with early onset heart block and AF. The PDE4DIP mutation (NM_001002811:c.367G>A) results in a non-conservative alanine to threonine substitution at amino acid 123 (p.A123T). The presence of PDE4DIP mutation in all family members with very early onset AF and heart block suggested an epistatic interaction with the DES gene, resulting in increased penetrance of these two traits. Another member of the family (II-6), who was found to be a carrier of the DES mutation had only developed late onset AF at age 57 but has neither DCM nor conduction disease (figure 1A). One young unaffected 32-year-old member of the kindred who has been considered as too young to develop disease (III-5) was found to be a carrier of both mutations DES and PDE4DIP mutations and is being closely followed by a local cardiologist.
PDE4DIP is an anchoring protein that interacts with both phosphodiesterase 4D (PDE4D) and cAMP dependent protein kinase A (PKA) (Dodge et al., 2001) and several other proteins to form a multiprotein complex that plays an important role in targeting signaling processes to subcellular locations. PDE4D hydrolyzes cAMP and regulates its levels within cardiac myocytes where it also complexes with proteins mediating sympathetic signals to heart, including β-adrenergic receptors (Mongillo et al., 2004; Perry et al., 2002; Xiang et al., 2005).
Additionally, novel deleterious heterozygous variant of the ENG gene (NC_000009.11:g.130578055G>A) causing a p.A628V substitution co-segregated with cardiomyopathy. The ENG gene encodes a membrane glycoprotein primarily expressed in the vascular endothelium and myocardium and has been previously identified as a modifier gene for hypertrophic cardiomyopathy (HCM) in patients with pathogenic MYH7 mutations, resulting in more profound myocardial fibrosis (Frustaci, Lanfranchi, Bellin, & Chimenti, 2012).
3.3. PDE4DIP Mutations Among Patients with early onset AF and mutation burden analysis
We subsequently screened 7 kindreds with AF and slow ventricular response suggestive of conduction disease using WES. All variants with allele frequencies greater than 1/1,000 or considered as benign by PolyPhen and Sift, including a number of variants in PDE4DIP were filtered. There were 3 independent novel deleterious nonconservative mutations in PDE4DIP gene that segregated with the disease in 4 kindreds (table 1, figures 1B–C). A gene-based burden analysis was carried out, comparing the aggregate burden of rare, protein-altering variants in each gene between WES variants of all cases compared to 158 age and ethnically matched controls. Statistical analysis showed significantly higher burden of mutations in PDE4DIP gene in cases vs. controls (adjusted p value <0.05). No other variants AF or heart block genes were identified in the 7 kindreds.
Table 1.
Rare damaging PDE4DIP mutations identified in independent kindreds with familial slow AF. Het: heterozygous, Hom: homozygous, PolyPhen 2: Polymorphism Phenotyping v2 software.
| PDE4DIP Gene Mutation | Amino acid substitution | Het/Hom | Prediction by PolyPhen 2 | 1,000 Genomes | gnomAD minor allele frequency | EXAC frequency |
|---|---|---|---|---|---|---|
|
NC_000001.10:g.144931342C>T NM_001002811:c.367G>A |
A123T | Het | Damaging | Novel | 0.00002386 | 2.47e−05 |
|
NC_000001.10:g.144877255T>C NM_001198834.1:c.4432A>G |
I1478V | Het | Damaging | Novel | 0.0003792 | 4.71e−04 |
|
NC_000001.10:g.144866687C>T NM_001198834.1:c.5555G>A |
R1852Q | Het | Damaging | Novel | 0.0002832 | 5.27e−04 |
|
NC_000001.10:g.144881520G>A NM_001198834.1:c.3676C>T |
R1226C | Het | Damaging | Novel | 0.0001874 | 5.77e−05 |
3.4. Functional characterization of the PDE4DIP mutation
We first examined whether the p.A123T substitution alters the interaction between PDE4D and PDE4DIP, using immunofluorescence staining and specific antibody against PDE4D. Wildtype and mutant PDE4DIP and an empty mCherry-tagged plasmid were expressed in C2C12 cells and the expression was verified by positive mCherry fluorescence. Careful examination showed that upon isoproterenol stimulation, PDE4D and wildtype PDE4DIP show increased colocalization but the co-localization of PDE4D and PDE4DIPpA123T was dramatically reduced (figure 2A–C). Furthermore, there was a trend towards reduced PDE4D levels in the mutant compared to wild type, which could be suggestive of decreased protein stability and possibly increased turnover due to decreased colocalization with PDE4DIP (figure 2D).
Figure 2.
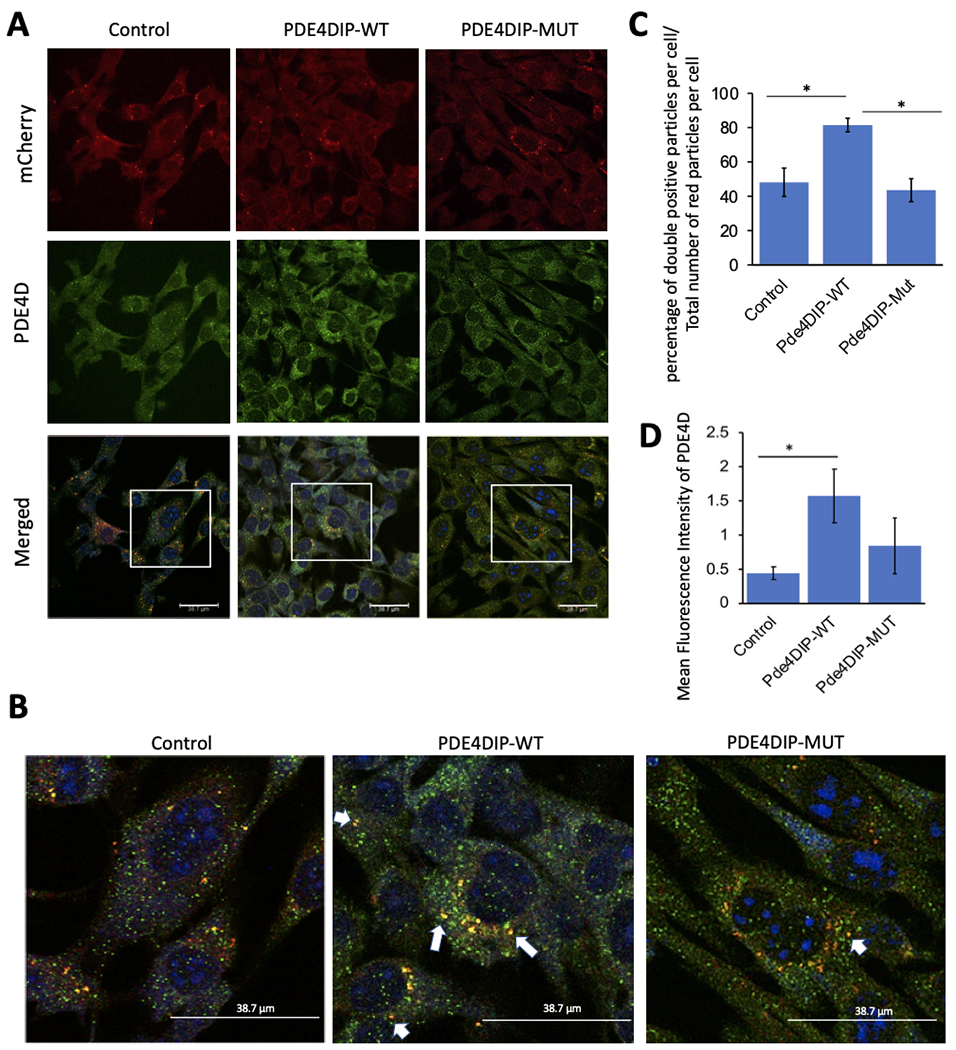
Immunofluorescent images of C2C12 cells expressing mCherry-tagged wildtype (WT) and mutant (mut p.A123T) PDE4DIP or mCherry-empty plasmid control after isoproterenol exposure. mCherry-PDE4DIP is shown in red, PDE4D is in green, all scale bars are 38.7 μm (A). Magnified merged images depict the colocalization of PDE4DIP and PDE4D in yellow color, which is higher in the WT cells, examples labelled with white arrows (B). PDE4D and mCherry-PDE4DIP co-localization was measured as percentage of double positive (yellow) particles per cell divided by the total number of red (mCherry) particles per cell and shown as bar graph with standard error of mean (C). PDE4D expression was quantified using mean fluorescence intensity of PDE4D antibody within cells (D). The “*” indicates significant difference (p-value <0.05).
To assess how the altered interaction between PDE4DIPA123T and PDE4D affects cAMP levels, cells were transfected with a plasmid either containing the PDE4DIPA123T or wild type PDE4DIP. Since cAMP activation occurs at compartmental level within the cells, the effect of PDE4DIPA123T on intracellular cAMP signaling was explored using a FRET-based sensor (figure 3A). Isoproterenol was used to stimulate the endogenous beta-2 adrenergic receptor (β2AR). The FRET sensor imaging showed increased cAMP levels (decreased FRET) in mutant compared to wildtype transfected cells in response to isoproterenol stimulation (figure 3B–E).
Figure 3.
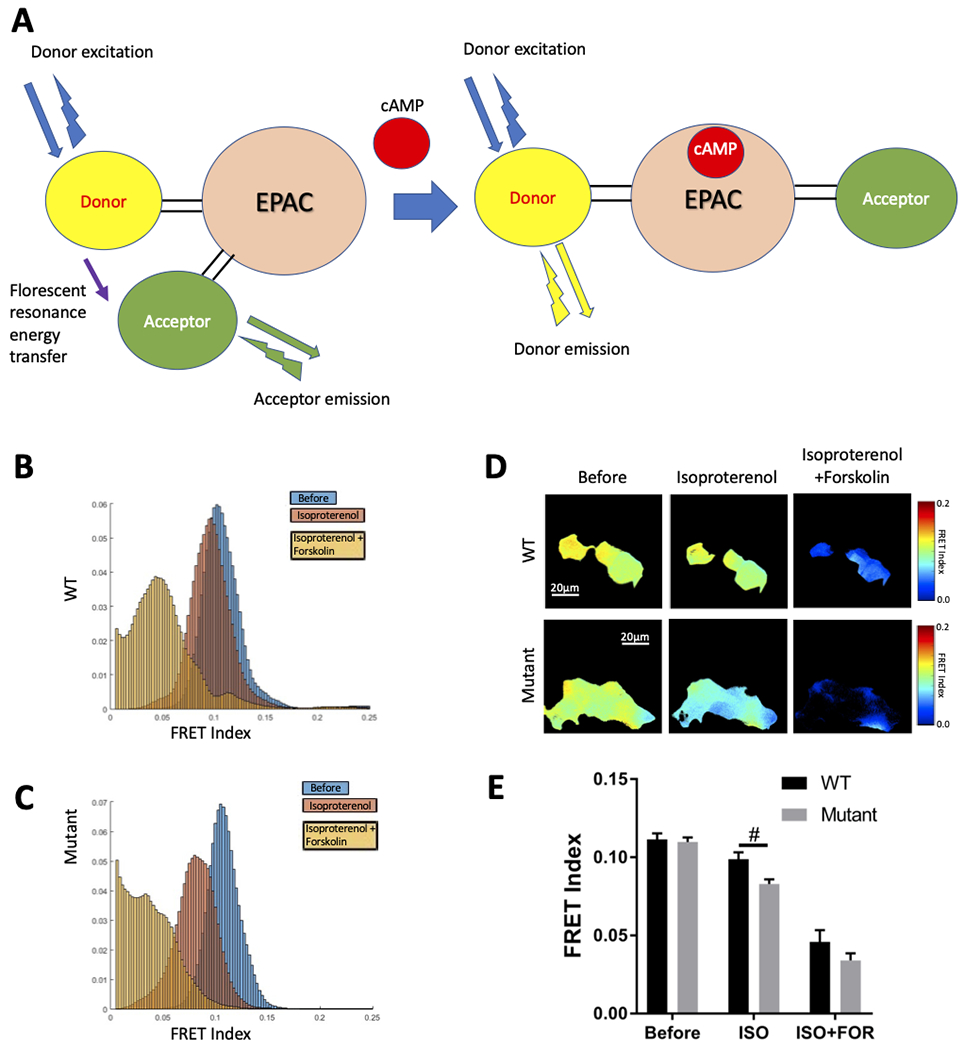
Fluorescence resonance energy transfer (FRET) imaging for intracellular cAMP. Schematic of the FRET molecular biosensor used in this study for cAMP detection, at baseline in the absence of cAMP the donor excitation allows resonance energy transfer to the acceptor arm that is in close proximity and that emits energy at its own wavelength (high FRET condition) whereas in the presence of cAMP the donor and acceptor arms are further apart in distance and orientation hence the donor emits light at its own wavelength (low FRET) (A). Images of Isoproterenol (ISO 1uM) and Forskolin (FOR 25uM) induced cAMP activation in 293T cells expressing wild type (WT) and mutant PDE4DIP were obtained under live confocal microscopy at baseline and at 4 minutes after stimulation with 1uM isoproterenol. Further imaging was done 4 minutes after stimulation with 25 uM of forskolin. The FRET indices are shown for WT (B) and mutant (C) proteins under different stimulations (color coded as blue: before stimulation, red: stimulation with isoproterenol and orange after stimulation with forskolin). The heat maps on the right show the upper and lower limits of the FRET ranges in the colored keys for WT and mutant cells under different stimulations, scale bar of 20 μm (D). FRET index shows a significant increase of cAMP activity (decreased FRET) in cells transfected by the mutant (p.A123T) compared to wild type PDE4DIP after stimulation with Isoproterenol (ISO 1uM), bar graph results with field average (mean +/− standard error of mean) reflect quantification of 18-20 fields from two independent experiments per group (E), “#” indicates statistically significant difference with p-value<0.05.
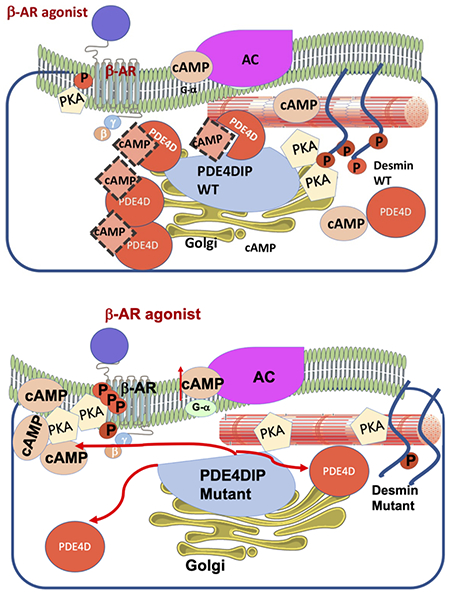
Examination of the PKA and the and G protein-coupled receptor serine/threonine kinases (GRKs) phosphorylation sites on the β2AR revealed an increase in PKA mediated phosphorylation at residue Serine-346 (figure 4A , 4C and Supp. Figure S2) but lower β2AR phosphorylation at the GRK residues (Serine-355 and 356) in cells expressing PDE4DIPA123T compared to wild type PDE4DIP (figure 4B, 4D and Supp. Figure S3) after isoproterenol stimulation that was measured as the ratio of phosphorylated to total β2AR signal. Of note, at baseline cells transfected with wild type PDE4DIP had increased PKA and GRK phosphorylation of the β2AR compared to control and mutant transfected cells. Examination of the PKA phosphorylation of desmin by immunofluorescent microscopy revealed decreased phosphorylation at its serine 60 site in PDE4DIPA123T cells (figure 5). The reduced p-desmin in the mutants suggests that PDE4DIPA123T mutation causes loss of compartmentalization of both PDE4D and PKA, resulting in increased PKA phosphorylation of β2AR but reduced phosphorylation of desmin. These findings are highly relevant since inactivation of sarcoplasmic PDE4D and altered desmin phosphorylation have both been linked to cardiomyopathy and arrhythmias (Beca et al., 2011; Lehnart et al., 2005; Rainer et al., 2018).
Figure 4.
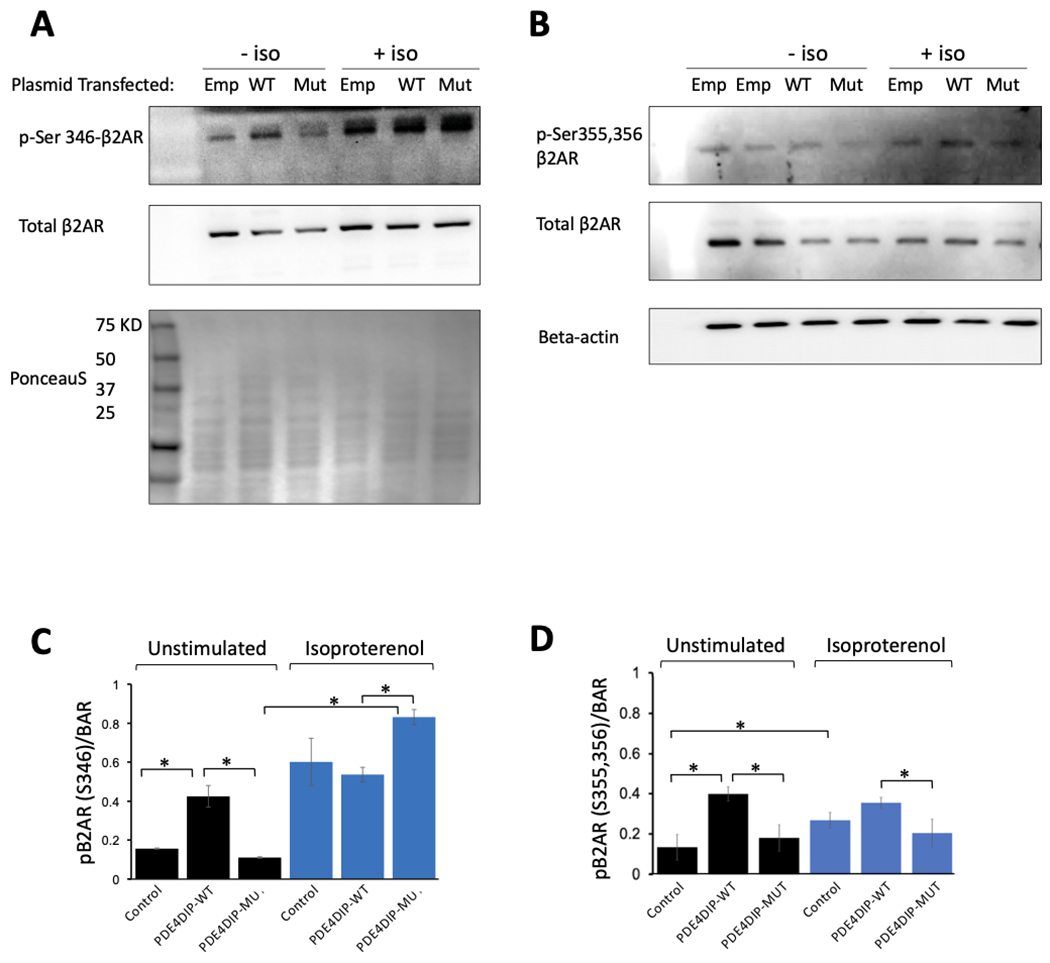
Western blot analysis of β 2 adrenergic receptor (β2AR) phosphorylation. Panel (A) shows phosphorylation at the PKA site (p-Ser 346) in the β2AR before (−iso) and 8 minutes after 1uM of isoproterenol stimulation (+iso) in C2C12 cells transfected with mCherry-empty plasmid, or wild type mCherry-PDE4DIP (WT) plasmid, or mutant mCherry-PDE4DIP (mut) plasmid, also shown is the corresponding total β2AR and a PonceauS protein stain as a loading control. Panel (B) shows phosphorylation at the GRK sites (p-Ser 355,356) in the β2AR before (−iso) and 8 minutes after 1uM of isoproterenol stimulation (+iso), and the corresponding total β2AR, also shown is beta-actin as an additional protein loading control, of note all the western blots (A&B) were loaded with the identical amount of lysate. Panel (C) shows the ratio of phosphorylated β2AR at the PKA site (Ser346) to the total β2AR. There is increased PKA phosphorylation at baseline in the wild type PDE4DIP compared to the control and the mutant however, after isoproterenol stimulation, there is a much higher levels of PKA mediated phosphorylation of the β2AR in the mutant compared to wild type PDE4DIP transfected cells, there is also an increase in PKA phosphorylation of the β2AR in the control cells. Panel (D) shows the ratio of phosphorylated β2AR at the GRK sites (Ser355,356) to the total β2AR. There is increased PKA phosphorylation at baseline in the wild type PDE4DIP compared to the control and the mutant, however after isoproterenol stimulation the GRK phosphorylation remains lower in the mutant compared to wild type transfected cells, there is also an increase in GRK phosphorylation of the β2AR in the control cells. The “*” indicates statistically significant difference (p-value <0.05).
Figure 5.
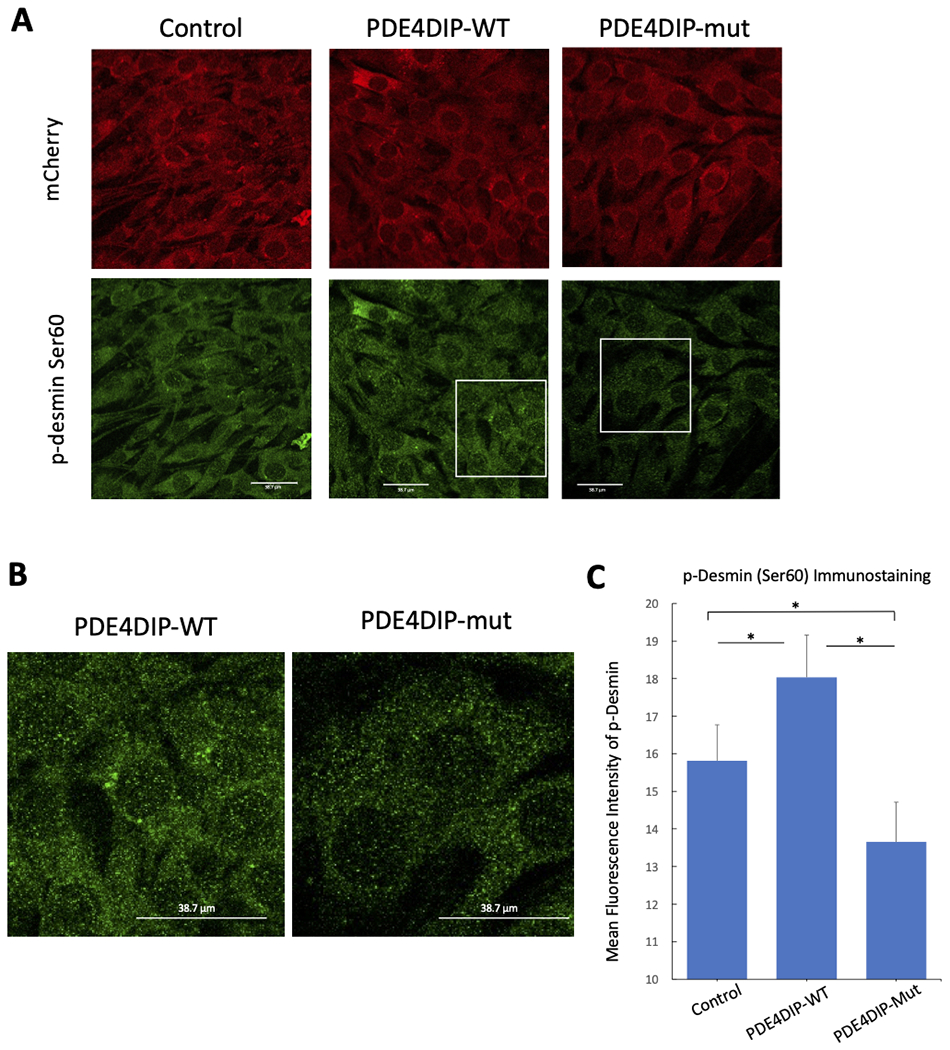
Reduction of total phospho-desmin (Ser60) in C2C12 cells expressing the mutant (p.A123T) compared to wildtype PDE4DIP. Panel A shows immunofluorescent images of C2C12 cells expressing mCherry-empty plasmid, wild type mCherry-PDE4DIP (WT) and mutant mCherry-PDE4DIP (mut p.A123T) plasmids. mCherry is shown in red and p-desmin (Ser60) is shown in green after isoproterenol exposure, all scale bars are 38.7 μm. Panel B shows magnified images of p-desmin (Ser60) in cells expressing PDE4DIP-WT and PDE4DIP-mut. Panel C shows p-desmin (Ser60) quantified using mean fluorescence intensity of p-desmin antibody within cells. The “*” indicates significant difference (p-value <0.05).
4. Discussion
In this manuscript we describe the identification of PDE4DIP as the genetic modifier of DES. Desmin is a type III intermediate filament protein that plays a pivotal role in the electromechanical functioning of cardiomyocytes (Mado et al., 2019). It connects the Z discs, which are at the center of the contractile unit, to costameres, desmosomes, nucleus and mitochondria and plays a pivotal role in the contractile function of cardiomyocytes. Mutations in DES are associated with autosomal dominant dilated cardiomyopathy with incomplete penetrance. Over 40 pathogenic DES mutations have been causally linked to myofibrillar myopathies involving skeletal myopathies, non-ischemic cardiomyopathies and cardiac conduction disease (van Tintelen et al., 2009). The p.S13F substitution occurs within a highly conserved motif in the non-helical head domain of the protein that is shared by type III and IV intermediate filaments (Bar, Strelkov, Sjoberg, Aebi, & Herrmann, 2004; Pica et al., 2008). This substitution eliminates serine at a known phosphorylation site for protein kinase C, thus interfering with the protein organization and likely leading to deleterious desmin aggregation and disruption of the cytoskeletal network (Brodehl et al., 2012; Kitamura et al., 1989). Although desmin is widely known as a skeletal muscle protein it is highly expressed within heart and within the conduction system of the heart and the pulmonary vein myocardial sleeves that are foci of origin of AF (Kugler et al., 2018). Accordingly, the mutations have pleotropic effects with cardiomyopathy being the most prominent trait, followed by cardiac conduction disease and much less commonly AF (van Spaendonck-Zwarts et al., 2011). The genetic modifiers of DES have not been identified. Interestingly, six members of the Lebanese kindred who were carriers of the pathological desmin p.S13F substitution had slightly higher incidence of DCM compared to the literature (50% vs 38% in published data), but considerably higher incidence of early onset AF (83% vs 11% in published data) and heart block requiring a pacemaker (67% vs 38% in published data) (Supp. Table S1) (Bergman et al., 2007; Pica et al., 2008; van Tintelen et al., 2009). The high incidence of AF and heart block suggested epistatic interactions between the DES mutation and other genetic mutations and provided an exceptional opportunity to search for genetic modifiers.
Our study identified the PDE4DIPpA123T mutation as a genetic modifier of DES that increases the penetrance of heart block and early onset AF in DES mutation carriers. Also known as myomegalin, PDE4DIP is highly expressed in cardiac atrial and ventricular tissue (Supp. Figure S1). It is a large gene located on the long arm of chromosome 1 and contains 64 exons coding for a 2,362 amino acid protein that was first isolated in 2001 in a study of cardiac genes (Soejima et al., 2001) with several splicing variants. The protein is composed of alpha-helical and coiled-coil structures and is also heavily expressed in skeletal muscles (Supp. Figure S1). Within myocytes, PDE4DIP is predominantly localized in the Golgi compartment, in proximity to the cytoskeletal apparatus and in the nucleus (Bouguenina et al., 2017; Soejima et al., 2001; Wang, Zhang, & Qi, 2014; Wu et al., 2016). It is an A-kinase-anchoring protein that is involved in the assembly of the cAMP dependent protein kinase A (PKA)/phosphodiesterase 4D (PDE4D) cAMP signaling module in a multiprotein complex (Dodge et al., 2001). Co-compartmentalization of both PKA and PDE4D is critical for sustained specificity of adrenergic signaling to subcellular locations, contractility of cardiomyocytes and timely termination of the second messenger response (Fink et al., 2001).
PDE4D hydrolyzes cAMP and regulates its levels within cardiac myocytes where it also complexes with proteins mediating sympathetic signals to the heart, including β-adrenergic receptors (Mongillo et al., 2004; Perry et al., 2002; Xiang et al., 2005). The activity of PDE4D has been also localized to the transverse (T) tubule/sarcoplasmic reticulum (SR) junctional space thus mediating cAMP/calcium homeostasis that is involved in excitation-contraction coupling (Mika, Richter, & Conti, 2015; Mongillo et al., 2004; Zaccolo & Pozzan, 2002). Of note, loss of function of PDE4D is associated with cardiomyopathy and AF (Jorgensen, Yasmeen, Iversen, & Kruuse, 2015).
We further established an association between multiple rare non-conservative PDE4DIP mutations with large effects in four out of seven unrelated families referred for early onset familial AF and slow conduction without structural heart disease. The functional significance of these mutations was established by the combined statistical power, high conservation of the mutated bases, and in vitro functional studies.
The in vitro characterization of PDE4DIPpA123T suggested a gain of function mutation leading to increased PKA activity. This effect is likely due to the disruption of the spatial-temporal activity of PDE4D, which is normally anchored to PDE4DIP and showed reduced colocalization with PDE4DIPpA123T. Consequently, there was increased cAMP activation by PDE4DIPpA123T vs. wildtype PDE4DIP in response to isoproterenol as assayed by FRET imaging. Under normal circumstances, the rise of cAMP in response to isoproterenol triggers PDE4D activation as well as desensitization of the G-protein coupled β2AR receptor via phosphorylation by PKA and G protein-coupled receptor serine/threonine kinases (GRKs) at distinct phosphorylation sites (Xin, Tran, Richter, Clark, & Rich, 2008). PKA mediated phosphorylation of the β2AR at Ser345/346 by PKA causes a switch from stimulatory Gs to inhibitory Gi protein (Martin, Whalen, Zamah, Pierce, & Lefkowitz, 2004; Zamah, Delahunty, Luttrell, & Lefkowitz, 2002). On the other hand, prolonged β2AR stimulation results in its GRK mediated phosphorylation at Ser 355/356, its binding to β-arrestin, termination of G protein-mediated signaling, and facilitated receptor endocytosis resulting in receptor desensitization (Nobles et al., 2011; Shenoy & Lefkowitz, 2005). In addition, PDE4D binds to β-arrestin and is subsequently recruited to the plasma membrane to regulate PKA mediated phosphorylation of β2AR.(Houslay & Baillie, 2005; Perry et al., 2002; Willoughby et al., 2007). Interestingly, there was more pronounced phosphorylation of the β2AR at the PKA residue (Ser 346) in the mutant versus control cells while the phosphorylation at the GRK residues (Ser355-356) was reduced. The former may underlie the mechanism of slow heart rate in patients with AF.
Like all A-kinase-anchoring proteins, PDE4DIP binds to the regulatory subunit of PKA and anchors it to the N-terminal region of cMyBPC, whereby it mediates cMyBPC phosphorylation and plays an important role in regulation of cardiac contractility. The PDE4DIP colocalization with cMyBPC and cTNI has been shown to increase upon β adrenergic activation and play a role in their stability (Uys et al., 2011). Desmin is also known to be a phosphorylation target for cAMP-dependent kinases (Gard & Lazarides, 1982; O’Connor, Gard, & Lazarides, 1981). Specifically, PKA phosphorylation of desmin regulates its function and assembly (Inagaki et al., 1988; Tao & Ip, 1991). Interestingly, our studies in C2C12 cells showed that p-desmin was lower in the mutant PDE4DIP compared to wildtype PDE4DIP after β adrenergic activation. This suggests that PDE4DIP directs PKA to desmin, but PDE4DIPpA123T induced loss of compartmentalization of both PDE4D and PKA leads to PKA phosphorylation of β2AR but reduced phosphorylation of the desmin, which has also lost a PKC phosphorylation site by the p.S13F mutation. Phosphorylation changes in desmin are shown to alter its assembly and trigger accumulation of toxic preamyloid oligomers in acquired heart failure (Rainer et al., 2018). Thus, the perturbed compartmentalization of PDE4DIPpA123T and further reduction in desmin phosphorylation is a mechanistic link to the modifier effect of the PDE4DIP variant on the mutant desmin, leading to increased penetrance of heart block and AF in desmin p.S13F carriers.
It is noteworthy that a common nonsynonymous variant (rs1778155) of the PDE4DIP gene, resulting in arginine for histidine substitution at codon 1761 has been associated with ischemic stroke in NHLBI Exome Sequence Project (odds ratio: 2.15; p-value: 2.63 × 10−8) (Auer et al., 2015). This disorder is believed to be associated with AF. Thus, our findings may provide a molecular link between ischemic stroke and slow AF, which is often subclinical due to the absence of tachycardia and palpitations.
Our success in identifying the disease gene and its modifier was largely due to our systematic study of outlier subjects with well-characterized disease and the segregation analysis of the rare variants. The investigated kindreds in our study all had early onset AF with slow conduction and demonstrated an autosomal dominant pattern of inheritance. This approach is particularly superior to case–control association studies for identification of disease-causing rare variants of large genes with high mutation burden such as PDE4DIP. It is, however, noteworthy that not all identified segregating PDE4DIP mutations may have sufficient power to be independently disease causing; this is particularly true for variants with higher allele frequencies.
One limitation of our study is that it does not examine the effect of the PDE4DIP mutation on the potassium channels and ryanodine receptor (RyR2). PDE4D has been shown to regulate the slow delayed rectifier potassium current (Terrenoire, Houslay, Baillie, & Kass, 2009) and RyR2 (Lehnart et al., 2005; Mika, Richter, Westenbroek, Catterall, & Conti, 2014). Its inhibition leads to increased PKA activation of the slow delayed rectifier potassium current (Terrenoire et al., 2009), a mechanism that may underlie the AF (Sampson et al., 2008). It remains to be determined if PDE4DIP mutation generate a functional substrate for AF in the atrium by altering the potassium current. In addition, a Mendelian randomization study examining link between rs1778155 variant, AF, and stroke could support the potential link between PDE4DIP common variant and the risk for atrial fibrillation and should be a topic for future investigations.
5. Conclusion and Future Prospects
Desmin mutations have pleotropic effects and often exhibit incomplete penetrance, but modifier genes that explain this behavior had not been identified. In this study, we show epistatic interaction between DES and PDE4DIP mutations. In addition, we find increased mutation burden of the PDE4DIP gene in kindreds with slow AF. The in vitro characterization of one of the mutations showed impaired spatial regulation of cAMP/PKA, causing altered phosphorylation of several proteins that play important roles in cardiac contractile function and conduction. In summary, our study has implicated PDE4DIP in maintenance of cardiac function and as a potential target for development of novel drugs for treatment of arrhythmias and cardiomyopathies.
Supplementary Material
Acknowledgements:
We would like to thank the participating families. The authors would like to thank the NHLBI GO Exome Sequencing Project and its ongoing studies which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926) and the Heart GO Sequencing Project (HL-103010)67. We would like to also thank Ms. Joanna Kucharczak for her help with this study. This study was supported by the grant to study the Mendelian Disorders and R01HL094784-05 grants (to A.M.).
Grant numbers:
This study was supported by the grant to study the Mendelian Disorders and R01HL094784-05 grants (to A.M.).
Footnotes
The authors have no conflicts of interest to report.
Data Availability Statement:
Data available on request due to privacy/ethical restrictions.
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions. The variant data has been uploaded to ClinVar and can be accessed on this link: https://www.ncbi.nlm.nih.gov/clinvar/?term=SUB9925062
References:
- Alzahrani Z, Ornelas-Loredo A, Darbar SD, Farooqui A, Mol D, Chalazan B, … Darbar D, (2018). Association Between Family History and Early-Onset Atrial Fibrillation Across Racial and Ethnic Groups. JAMA Netw Open, 1(5), e182497. doi: 10.1001/jamanetworkopen.2018.2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amat-y-Leon F, Racki AJ, Denes P, Ten Eick RE, Singer DH, Bharati S, … Rosen KM, (1974). Familial atrial dysrhythmia with A-V block. Intracellular microelectrode, clinical electrophysiologic, and morphologic observations. Circulation, 50(6), 1097–1104. doi: 10.1161/01.cir.50.6.1097 [DOI] [PubMed] [Google Scholar]
- Auer PL, Nalls M, Meschia JF, Worrall BB, Longstreth WT Jr., Seshadri S, … Blood Institute Exome Sequencing, P. (2015). Rare and Coding Region Genetic Variants Associated With Risk of Ischemic Stroke: The NHLBI Exome Sequence Project. JAMA Neurol, 72(7), 781–788. doi: 10.1001/jamaneurol.2015.0582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar H, Strelkov SV, Sjoberg G, Aebi U, & Herrmann H, (2004). The biology of desmin filaments: how do mutations affect their structure, assembly, and organisation? J Struct Biol, 148(2), 137–152. doi: 10.1016/j.jsb.2004.04.003 [DOI] [PubMed] [Google Scholar]
- Baruteau AE, Probst V, & Abriel H, (2015). Inherited progressive cardiac conduction disorders. Curr Opin Cardiol, 30(1), 33–39. doi: 10.1097/HCO.0000000000000134 [DOI] [PubMed] [Google Scholar]
- Beca S, Helli PB, Simpson JA, Zhao D, Farman GP, Jones P, … Backx PH. (2011). Phosphodiesterase 4D regulates baseline sarcoplasmic reticulum Ca2+ release and cardiac contractility, independently of L-type Ca2+ current. Circ Res, 109(9), 1024–1030. doi: 10.1161/CIRCRESAHA.111.250464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman JE, Veenstra-Knol HE, van Essen AJ, van Ravenswaaij CM, den Dunnen WF, van den Wijngaard A, & van Tintelen JP, (2007). Two related Dutch families with a clinically variable presentation of cardioskeletal myopathy caused by a novel S13F mutation in the desmin gene. Eur J Med Genet, 50(5), 355–366. doi: 10.1016/j.ejmg.2007.06.003 [DOI] [PubMed] [Google Scholar]
- Bouguenina H, Salaun D, Mangon A, Muller L, Baudelet E, Camoin L, … Badache A, (2017). EB1-binding-myomegalin protein complex promotes centrosomal microtubules functions. Proc Natl Acad Sci U S A, 114(50), E10687–E10696. doi: 10.1073/pnas.1705682114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodehl A, Gaertner-Rommel A, & Milting H, (2018). Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys Rev, 10(4), 983–1006. doi: 10.1007/s12551-018-0429-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodehl A, Hedde PN, Dieding M, Fatima A, Walhorn V, Gayda S, … Milting H, (2012). Dual color photoactivation localization microscopy of cardiomyopathy-associated desmin mutants. J Biol Chem, 287(19), 16047–16057. doi: 10.1074/jbc.M111.313841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodehl A, Pour Hakimi SA, Stanasiuk C, Ratnavadivel S, Hendig D, Gaertner A, … Milting H, (2019). Restrictive Cardiomyopathy is Caused by a Novel Homozygous Desmin (DES) Mutation p.Y122H Leading to a Severe Filament Assembly Defect. Genes (Basel), 10(11). doi: 10.3390/genes10110918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costedoat-Chalumeau N, Georgin-Lavialle S, Amoura Z, & Piette JC, (2005). Anti-SSA/Ro and anti-SSB/La antibody-mediated congenital heart block. Lupus, 14(9), 660–664. doi: 10.1191/0961203305lu2195oa [DOI] [PubMed] [Google Scholar]
- Dodge KL, Khouangsathiene S, Kapiloff MS, Mouton R, Hill EV, Houslay MD, … Scott JD, (2001). mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. EMBO J, 20(8), 1921–1930. doi: 10.1093/emboj/20.8.1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink MA, Zakhary DR, Mackey JA, Desnoyer RW, Apperson-Hansen C, Damron DS, & Bond M, (2001). AKAP-mediated targeting of protein kinase a regulates contractility in cardiac myocytes. Circ Res, 88(3), 291–297. doi: 10.1161/01.res.88.3.291 [DOI] [PubMed] [Google Scholar]
- Frustaci A, Lanfranchi G, Bellin M, & Chimenti C, (2012). Coronary telangiectasia associated with hypertrophic cardiomyopathy. Eur J Heart Fail, 14(12), 1332–1337. doi: 10.1093/eurjhf/hfs125 [DOI] [PubMed] [Google Scholar]
- Gard DL, & Lazarides E, (1982). Analysis of desmin and vimentin phosphopeptides in cultured avian myogenic cells and their modulation by 8-bromo-adenosine 3′,5′-cyclic monophosphate. Proc Natl Acad Sci U S A, 79(22), 6912–6916. doi: 10.1073/pnas.79.22.6912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, … Stroke Statistics, S. (2013). Heart disease and stroke statistics--2013 update: a report from the American Heart Association. Circulation, 127(1), e6–e245. doi: 10.1161/CIR.0b013e31828124ad [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houslay MD, & Baillie GS, (2005). Beta-arrestin-recruited phosphodiesterase-4 desensitizes the AKAP79/PKA-mediated switching of beta2-adrenoceptor signalling to activation of ERK. Biochem Soc Trans, 33(Pt 6), 1333–1336. doi: 10.1042/BST20051333 [DOI] [PubMed] [Google Scholar]
- Inagaki M, Gonda Y, Matsuyama M, Nishizawa K, Nishi Y, & Sato C, (1988). Intermediate filament reconstitution in vitro. The role of phosphorylation on the assembly-disassembly of desmin. J Biol Chem, 263(12), 5970–5978. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/2833525 [PubMed] [Google Scholar]
- Jorgensen C, Yasmeen S, Iversen HK, & Kruuse C, (2015). Phosphodiesterase4D (PDE4D)--A risk factor for atrial fibrillation and stroke? J Neurol Sci, 359(1-2), 266–274. doi: 10.1016/j.jns.2015.11.010 [DOI] [PubMed] [Google Scholar]
- Keramati AR, Fathzadeh M, Go GW, Singh R, Choi M, Faramarzi S, … Mani A, (2014). A form of the metabolic syndrome associated with mutations in DYRK1B. N Engl J Med, 370(20), 1909–1919. doi: 10.1056/NEJMoa1301824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura S, Ando S, Shibata M, Tanabe K, Sato C, & Inagaki M, (1989). Protein kinase C phosphorylation of desmin at four serine residues within the non-alpha-helical head domain. J Biol Chem, 264(10), 5674–5678. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/2494168 [PubMed] [Google Scholar]
- Klarenbeek J, Goedhart J, van Batenburg A, Groenewald D, & Jalink K, (2015). Fourth-generation epac-based FRET sensors for cAMP feature exceptional brightness, photostability and dynamic range: characterization of dedicated sensors for FLIM, for ratiometry and with high affinity. PLoS One, 10(4), e0122513. doi: 10.1371/journal.pone.0122513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugler S, Nagy N, Racz G, Tokes AM, Dorogi B, & Nemeskeri A, (2018). Presence of cardiomyocytes exhibiting Purkinje-type morphology and prominent connexin45 immunoreactivity in the myocardial sleeves of cardiac veins. Heart Rhythm, 15(2), 258–264. doi: 10.1016/j.hrthm.2017.09.044 [DOI] [PubMed] [Google Scholar]
- Kumar A, Ouyang M, Van den Dries K, McGhee EJ, Tanaka K, Anderson MD, … Schwartz MA, (2016). Talin tension sensor reveals novel features of focal adhesion force transmission and mechanosensitivity. J Cell Biol, 213(3), 371–383. doi: 10.1083/jcb.201510012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar RK, Saxena A, & Talwar KK, (1991). Lone atrial fibrillation with complete heart block in a child. Int J Cardiol, 30(3), 349–351. doi: 10.1016/0167-5273(91)90014-g [DOI] [PubMed] [Google Scholar]
- Lehnart SE, Wehrens XH, Reiken S, Warrier S, Belevych AE, Harvey RD, … Marks AR. (2005). Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell, 123(1), 25–35. doi: 10.1016/j.cell.2005.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mado K, Chekulayev V, Shevchuk I, Puurand M, Tepp K, & Kaambre T, (2019). On the role of tubulin, plectin, desmin, and vimentin in the regulation of mitochondrial energy fluxes in muscle cells. Am J Physiol Cell Physiol, 316(5), C657–C667. doi: 10.1152/ajpcell.00303.2018 [DOI] [PubMed] [Google Scholar]
- Marakhonov AV, Brodehl A, Myasnikov RP, Sparber PA, Kiseleva AV, Kulikova OV, … Skoblov MY. (2019). Noncompaction cardiomyopathy is caused by a novel in-frame desmin (DES) deletion mutation within the 1A coiled-coil rod segment leading to a severe filament assembly defect. Hum Mutat, 40(6), 734–741. doi: 10.1002/humu.23747 [DOI] [PubMed] [Google Scholar]
- Martin NP, Whalen EJ, Zamah MA, Pierce KL, & Lefkowitz RJ, (2004). PKA-mediated phosphorylation of the beta1-adrenergic receptor promotes Gs/Gi switching. Cell Signal, 16(12), 1397–1403. doi: 10.1016/j.cellsig.2004.05.002 [DOI] [PubMed] [Google Scholar]
- Michaelsson M, Riesenfeld T, & Jonzon A, (1997). Natural history of congenital complete atrioventricular block. Pacing Clin Electrophysiol, 20(8 Pt 2), 2098–2101. doi: 10.1111/j.1540-8159.1997.tb03636.x [DOI] [PubMed] [Google Scholar]
- Mika D, Richter W, & Conti M, (2015). A CaMKII/PDE4D negative feedback regulates cAMP signaling. Proc Natl Acad Sci U S A, 112(7), 2023–2028. doi: 10.1073/pnas.1419992112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mika D, Richter W, Westenbroek RE, Catterall WA, & Conti M, (2014). PDE4B mediates local feedback regulation of beta(1)-adrenergic cAMP signaling in a sarcolemmal compartment of cardiac myocytes. J Cell Sci, 127(Pt 5), 1033–1042. doi: 10.1242/jcs.140251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moak JP, Barron KS, Hougen TJ, Wiles HB, Balaji S, Sreeram N, … Buyon JP. (2001). Congenital heart block: development of late-onset cardiomyopathy, a previously underappreciated sequela. J Am Coll Cardiol, 37(1), 238–242. doi: 10.1016/s0735-1097(00)01048-2 [DOI] [PubMed] [Google Scholar]
- Mongillo M, McSorley T, Evellin S, Sood A, Lissandron V, Terrin A, … Zaccolo M, (2004). Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ Res, 95(1), 67–75. doi: 10.1161/01.RES.0000134629.84732.11 [DOI] [PubMed] [Google Scholar]
- Nobles KN, Xiao K, Ahn S, Shukla AK, Lam CM, Rajagopal S, … Lefkowitz RJ. (2011). Distinct phosphorylation sites on the beta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin. Sci Signal, 4(185), ra51. doi: 10.1126/scisignal.2001707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor CM, Gard DL, & Lazarides E, (1981). Phosphorylation of intermediate filament proteins by cAMP-dependent protein kinases. Cell, 23(1), 135–143. doi: 10.1016/0092-8674(81)90278-6 [DOI] [PubMed] [Google Scholar]
- Perry SJ, Baillie GS, Kohout TA, McPhee I, Magiera MM, Ang KL, … Lefkowitz RJ. (2002). Targeting of cyclic AMP degradation to beta 2-adrenergic receptors by beta-arrestins. Science, 298(5594), 834–836. doi: 10.1126/science.1074683 [DOI] [PubMed] [Google Scholar]
- Pica EC, Kathirvel P, Pramono ZA, Lai PS, & Yee WC, (2008). Characterization of a novel S13F desmin mutation associated with desmin myopathy and heart block in a Chinese family. Neuromuscul Disord, 18(2), 178–182. doi: 10.1016/j.nmd.2007.09.011 [DOI] [PubMed] [Google Scholar]
- Protonotarios A, Brodehl A, Asimaki A, Jager J, Quinn E, Stanasiuk C, … Lopes LR. (2020). The Novel Desmin Variant p.Leu115Ile Is Associated With a Unique Form of Biventricular Arrhythmogenic Cardiomyopathy. Can J Cardiol. doi: 10.1016/j.cjca.2020.11.017 [DOI] [PubMed] [Google Scholar]
- Rainer PP, Dong P, Sorge M, Fert-Bober J, Holewinski RJ, Wang Y, … Agnetti G, (2018). Desmin Phosphorylation Triggers Preamyloid Oligomers Formation and Myocyte Dysfunction in Acquired Heart Failure. Circ Res, 122(10), e75–e83. doi: 10.1161/CIRCRESAHA.117.312082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson KJ, Terrenoire C, Cervantes DO, Kaba RA, Peters NS, & Kass RS, (2008). Adrenergic regulation of a key cardiac potassium channel can contribute to atrial fibrillation: evidence from an I Ks transgenic mouse. J Physiol, 586(2), 627–637. doi: 10.1113/jphysiol.2007.141333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, & Lefkowitz RJ, (2005). Seven-transmembrane receptor signaling through beta-arrestin. Sci STKE, 2005(308), cm10. doi: 10.1126/stke.2005/308/cm10 [DOI] [PubMed] [Google Scholar]
- Soejima H, Kawamoto S, Akai J, Miyoshi O, Arai Y, Morohka T, … Mukai T, (2001). Isolation of novel heart-specific genes using the BodyMap database. Genomics, 74(1), 115–120. doi: 10.1006/geno.2001.6527 [DOI] [PubMed] [Google Scholar]
- Tao JX, & Ip W, (1991). Site-specific antibodies block kinase A phosphorylation of desmin in vitro and inhibit incorporation of myoblasts into myotubes. Cell Motil Cytoskeleton, 19(2), 109–120. doi: 10.1002/cm.970190206 [DOI] [PubMed] [Google Scholar]
- Terrenoire C, Houslay MD, Baillie GS, & Kass RS, (2009). The cardiac IKs potassium channel macromolecular complex includes the phosphodiesterase PDE4D3. J Biol Chem, 284(14), 9140–9146. doi: 10.1074/jbc.M805366200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udink ten Cate FE, Breur JM, Cohen MI, Boramanand N, Kapusta L, Crosson JE, … Meijboom EJ. (2001). Dilated cardiomyopathy in isolated congenital complete atrioventricular block: early and long-term risk in children. J Am Coll Cardiol, 37(4), 1129–1134. doi: 10.1016/s0735-1097(00)01209-2 [DOI] [PubMed] [Google Scholar]
- Uys GM, Ramburan A, Loos B, Kinnear CJ, Korkie LJ, Mouton J, … Moolman-Smook JC. (2011). Myomegalin is a novel A-kinase anchoring protein involved in the phosphorylation of cardiac myosin binding protein C. BMC Cell Biol, 12, 18. doi: 10.1186/1471-2121-12-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Spaendonck-Zwarts KY, van Hessem L, Jongbloed JD, de Walle HE, Capetanaki Y, van der Kooi AJ, … van Tintelen JP. (2011). Desmin-related myopathy. Clin Genet, 80(4), 354–366. doi: 10.1111/j.1399-0004.2010.01512.x [DOI] [PubMed] [Google Scholar]
- van Tintelen JP, Van Gelder IC, Asimaki A, Suurmeijer AJ, Wiesfeld AC, Jongbloed JD, … van den Berg MP. (2009). Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart Rhythm, 6(11), 1574–1583. doi: 10.1016/j.hrthm.2009.07.041 [DOI] [PubMed] [Google Scholar]
- Wang Z, Zhang C, & Qi RZ, (2014). A newly identified myomegalin isoform functions in Golgi microtubule organization and ER-Golgi transport. J Cell Sci, 127(Pt 22), 4904–4917. doi: 10.1242/jcs.155408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willoughby D, Baillie GS, Lynch MJ, Ciruela A, Houslay MD, & Cooper DM, (2007). Dynamic regulation, desensitization, and cross-talk in discrete subcellular microdomains during beta2-adrenoceptor and prostanoid receptor cAMP signaling. J Biol Chem, 282(47), 34235–34249. doi: 10.1074/jbc.M706765200 [DOI] [PubMed] [Google Scholar]
- Wu J, de Heus C, Liu Q, Bouchet BP, Noordstra I, Jiang K, … Akhmanova A, (2016). Molecular Pathway of Microtubule Organization at the Golgi Apparatus. Dev Cell, 39(1), 44–60. doi: 10.1016/j.devcel.2016.08.009 [DOI] [PubMed] [Google Scholar]
- Xiang Y, Naro F, Zoudilova M, Jin SL, Conti M, & Kobilka B, (2005). Phosphodiesterase 4D is required for beta2 adrenoceptor subtype-specific signaling in cardiac myocytes. Proc Natl Acad Sci U S A, 102(3), 909–914. doi: 10.1073/pnas.0405263102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin W, Tran TM, Richter W, Clark RB, & Rich TC, (2008). Roles of GRK and PDE4 activities in the regulation of beta2 adrenergic signaling. J Gen Physiol, 131(4), 349–364. doi: 10.1085/jgp.200709881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Murakawa Y, Ajiki K, & Omata M, (1997). Incidence of induced atrial fibrillation/flutter in complete atrioventricular block. A concept of ‘atrial-malfunctioning’ atrio-hisian block. Circulation, 95(3), 650–654. doi: 10.1161/01.cir.95.3.650 [DOI] [PubMed] [Google Scholar]
- Zaccolo M, & Pozzan T, (2002). Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science, 295(5560), 1711–1715. doi: 10.1126/science.1069982 [DOI] [PubMed] [Google Scholar]
- Zamah AM, Delahunty M, Luttrell LM, & Lefkowitz RJ, (2002). Protein kinase A-mediated phosphorylation of the beta 2-adrenergic receptor regulates its coupling to Gs and Gi. Demonstration in a reconstituted system. J Biol Chem, 277(34), 31249–31256. doi: 10.1074/jbc.M202753200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data available on request due to privacy/ethical restrictions.
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions. The variant data has been uploaded to ClinVar and can be accessed on this link: https://www.ncbi.nlm.nih.gov/clinvar/?term=SUB9925062