

Abstract
Half of patients with a ciliopathy syndrome remain unsolved after initial analysis of whole exome sequencing (WES) data, highlighting the need for improved variant filtering and annotation. By candidate gene curation of WES data, combined with homozygosity mapping, we detected a homozygous predicted synonymous allele in NPHP3 in 2 children with hepatorenal fibrocystic disease from a consanguineous family. Analyses on patient-derived RNA shows activation of a cryptic mid-exon splice donor leading to frameshift. Remarkably, the same rare variant was detected in 4 additional families with hepatorenal disease from UK, US and Saudi patient cohorts and in addition, another synonymous NPHP3 variant was identified in an unsolved case from the 100,000 Genomes dataset. We conclude that synonymous NPHP3 variants, not reported before and discarded by pathogenicity pipelines, solved several families with a ciliopathy. These findings prompt careful reassessment of synonymous variants, especially if they are rare and located in candidate genes.
Keywords: Next generation sequencing, RNA splicing, nephronophthisis, NPHP3, synonymous variant
Whole exome sequencing (WES) has become an accessible and cost-effective way of investigating inherited human diseases in routine clinical practice (Bamshad et al., 2011; Cameron-Christie et al., 2019; Groopman et al., 2019). Limited to protein coding regions of the genome (exome), which account for only ~2% of the genome, WES generates reduced amount of data, compared to whole genome sequencing (WGS), translating into decreased data storage costs as well as cheaper, quicker and easier data analysis. Indeed, variant annotation is generally more accurate in the protein coding regions of the genome because functional consequences are more readily assessable. Nevertheless, data generated from next generation sequencing (NGS) requires extensive pipeline analysis and automated filtering of variants or pathogenicity ranking remains challenging. Despite well-established workflows and software in place to process raw data (Jalali Sefid Dashti and Gamieldien, 2017), the typical European WGS in the 1K genome project has about 10,000 non-synonymous (1% singletons) variants (1000 Genomes Project Consortium et al., 2015), that need to be filtered down to a subset of variants relevant to the patient phenotype and pattern of inheritance. Crucially these final steps of variant prioritisation require biological and biomedical reasoning, demanding input from scientists and clinicians in addition to bioinformatics approaches. Besides filtering strategies, mutation detection rate by WES vastly depends on the underlying phenotype and the likelihood of a genetic aetiology (Mann et al., 2019). As a good example, ciliopathies are inherited disorders affecting approximately 1 in every 2000 individuals with autosomal dominant polycystic kidney disease (ADPKD) being by far the most common. Recessive forms often present with distinctive clinical features, classically combining nephronophthisis (NPHP) with specific extrarenal manifestations such as retinitis pigmentosa, skeletal manifestations or congenital hepatic fibrosis, not only allowing strong assumptions regarding a genetic origin but often narrowing it down to a handful of plausible candidate genes (Kagan et al., 2017). Yet, in familial cases of suspected nephronophthisis (NPHP), i.e., with high a priori likelihood of a genetic aetiology, WES detected a causative mutation in 63.3% of cases (Braun et al., 2016). This suggests that a significant number of cases remain unsolved, either because a novel gene is involved, the pathogenic variant is outside the coding regions or, because the pathogenicity of the underlying genetic variant is not readily assessable and requires in-depth curation.
Here we report an Omani pedigree where the parents were first cousins (Figure 1A), and in which WES was carried out in two siblings OM-1 (II.1) and OM-2 (II.4) with clinical features of a hepatorenal ciliopathy syndrome. Both presented with small echogenic kidneys with multiple cysts (Figure 1B), suggestive of nephronophthisis (NPHP) associated with congenital hepatic fibrosis and ultrasonographic signs of portal hypertension (Figure 1C & Supp. Figure S1). OM-1 had reached end stage kidney disease (ESKD) within 2 years of life (Table 1). Considering parental consanguinity, we applied initial standard WES filtering (using Qiagen Clinical Insight software) for rare (allele frequency (AF) <1% in any reference subpopulation) homozygous variants classified as pathogenic/likely pathogenic, truncating, predicted non-tolerated SNV or leading to splice site loss (see supplementary materials). This analysis revealed only 1 homozygous variant shared by the 2 siblings, a VUS in TSNARE1 (RefSeq NM_145003.5: c.761C>T; p.(Pro254Leu)), a gene implicated in synaptic vesicle exocytosis and polygenic risk for schizophrenia (Sleiman et al., 2013). Of note, no variants were detected in genes known to cause hepatorenal ciliopathies. Given the parental consanguinity, we next mapped the regions of homozygosity shared between OM-1 and OM-2 against 17 genes known or potentially associated with autosomal recessive cystic kidney disease and congenital hepatic fibrosis (Supp. Table S1) (Halbritter et al., 2013; Vilboux et al., 2017). We detected several, often non-overlapping regions of autozygosity in the 2 siblings, and only NPHP3 was located in the middle of a shared stretch of homozygosity (~3Mb) (Supp. Figure S2). By removing all pathogenicity filters and specifically looking for rare (AF<1%) homozygous variants in these candidate genes, we indeed identified 1 shared predicted synonymous VUS in NPHP3 (RefSeq NM_153240.5: c.2805C>T p.(Gly935Gly)) (Figure 1D) and recessive segregation of this variant was confirmed by Sanger sequencing (Figure 1A). The identified allele has not been reported before in cases of ciliopathy and is rare (gnomAD 2/251,374 alleles; no homozygous individuals). In line, we detected only 2 heterozygous adults without features of hepatorenal disease among ~200,000 individuals in the UK Biobank. Specialized in silico tools predict a possible impact on NPHP3 splicing (Table 1). Given its rarity, its location within a strong candidate gene NPHP3 in a shared region of homozygosity, the predicted effect on splicing and the absence of alternative genetic explanations, we first sought to look for additional patients with the identical allele.
Figure 1.
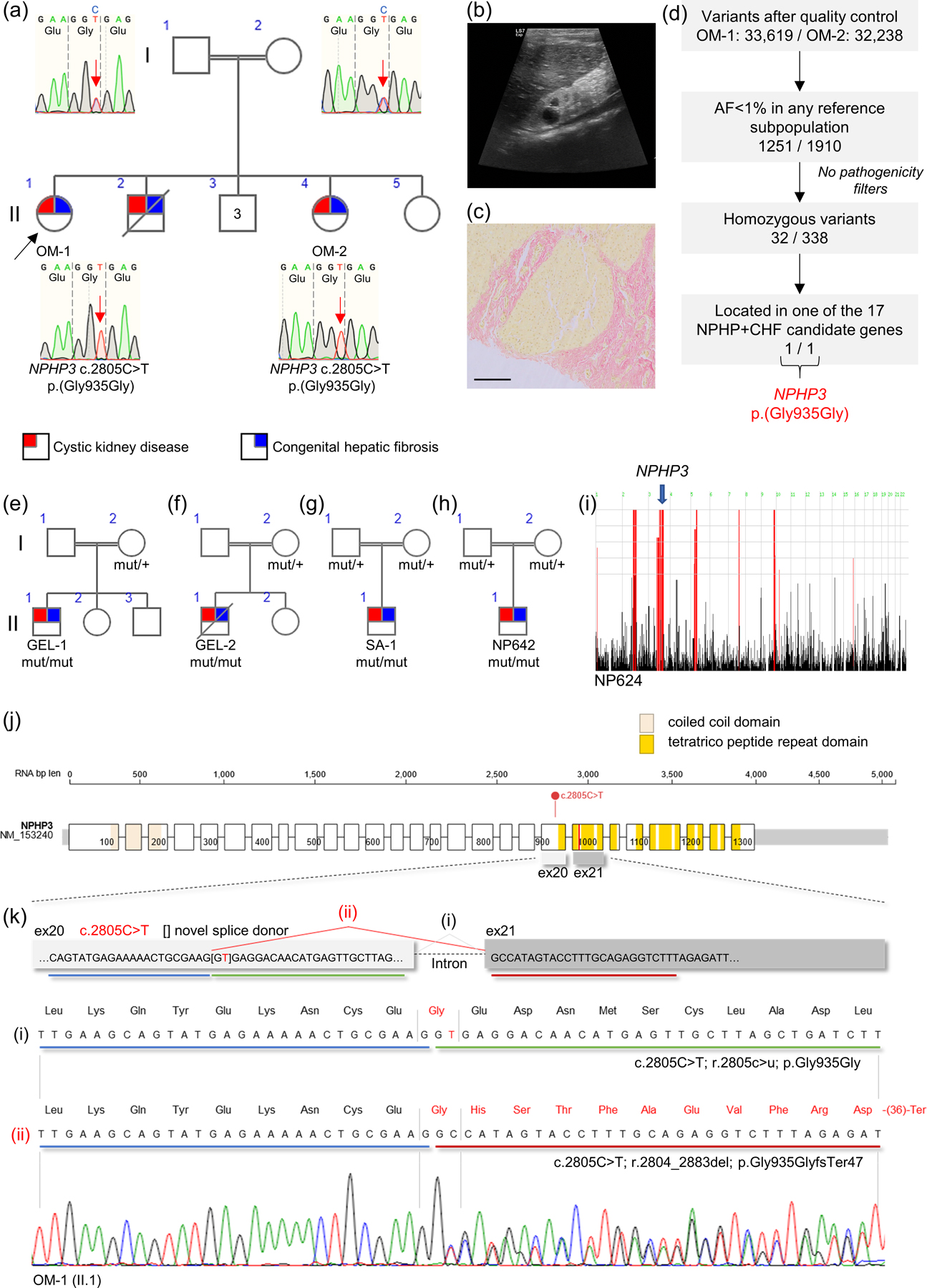
Consanguineous families with hepatorenal fibrocystic disease, NPHP3 variant identification and analysis of splicing defect
A. Index Omani family with genomic DNA Sanger chromatograms showing heterozygous and homozygous NPHP3 c.2805C>T change in parents and 2 patients, respectively. B. Left kidney ultrasonography from patient OM-1 (II.1) before reaching ESKD. Right and left kidneys were reduced in size (4.4 and 4.0 cm, respectively), with increased parenchymal echogenicity and loss of cortico-medullary differentiation. C. Van Gieson staining on liver biopsy from patient OM-1 (II.1) showing fibrous tissue around a hepatic nodule. Bar: 200μm. D. WES filtering strategy applied to identify NPHP3 variant. NPHP, nephronophthisis; CHF, congenital hepatic fibrosis. Additional families have been recruited within Genomics England 100,000 Genomes project (E, F), from a Saudi Arabia cohort (G) and from a US cohort (H). Segregation of the NPHP3 variant c.2805C>T is indicated by mut/+ for heterozygous individuals and mut/mut for homozygous patients. I. Genome-wide homozygosity plot for NP642. The genomic location of NPHP3 is indicated. Pedigrees were constructed and drawn using Progeny Free Online Pedigree Tool (Progeny Genetics LLC, Delray Beach, FL, www.progenygenetics.com). J. NPHP3 RNA (RefSeq NM_153240.5) and exon structure with UTR in grey and annotated with domains. Variant c.2805C>T mid-exon 20 is indicated. The red line indicates the predicted premature stop codon at amino acid position 982 (see below). Protein visualization using ProteinPaint (Zhou et al., 2016) and domain annotation with SMART (Schultz et al., 1998). K. Genomic map of NPHP3 exons 20 and 21 showing canonical splicing (i) and alternative splicing (ii) due to activation of a cryptic splice site mid-exon 20 in NPHP3 pre-mRNA harbouring c.2805C>T. Below is shown a Sanger sequence extract from RT-PCR (forward primer exon 19, reverse primer exon 21) performed on whole blood RNA from homozygous patient OM-1 (II.1). Sequence reveals presence of both canonically spliced (i) and alternatively spliced (ii) transcripts leading to shift in reading frame. The predicted amino acid sequence is above the nucleotide sequence. The full consequence of c.2805C>T is thus r.[2805c>u, 2804_2883del] p.[Gly935Gly, Gly935GlyfsTer47].
Table 1.
Clinical characteristics of patients and description of recurrent NPHP3 allele
| Patient ID | Gender | Ethnicity | Age at referral | Kidney disease | Additional clinical features | CKD stage | Parental consanguinity |
|---|---|---|---|---|---|---|---|
| OM-1 | F | Omani | 2y | Cystic kidney disease | Hypertension, CHF | 5 (ESKD at 2y) | Yes |
| OM-2 | F | Omani | 1y | Cystic kidney disease | Hypertension, CHF | 2 (at 3y) | Yes |
| GEL-1 | M | Not stated | at birth | Cystic kidney disease | DD, CHF, valvular cardiopathy, abnormal facial shape | 5 (ESKD at 1y) | Yes |
| GEL-2 | M | Not stated | at birth | Cystic kidney disease | DD, CHF, valvular cardiopathy, deceased at age 5y | 5 (ESKD at 2y) | Yes |
| SA-1 | M | Saudi Arabian | 2m | Cystic kidney disease | CHF | 5 (ESKD at 6y) | Yes |
| NP642 | M | Pakistani | 2y | Cystic kidney disease | Cholestasis | 5 (ESKD at 2y) | Yes |
| Gene name | Genomic coordinates | Nucleotide change | Predicted amino acid change | GERP RS | ACMG Classification | dbSNP ID | GnomAD | HSF impact prediction | MaxEnt Donor Site |
|---|---|---|---|---|---|---|---|---|---|
| NPHP3 | 3:132689152:G:A (GRCh38) | c.2805C>T | p.(Gly935Gly) | 1.72 | uncertain significance | rs1281725083 | 2/0/251374 | Activation of a cryptic splice donor site | 1.4 -> 9.16 +554.3% |
Transcript used: RefSeq NM_153240.5
Abbreviations: CHF, congenital hepatic fibrosis; DD, developmental delay; ESKD, end stage kidney disease; GERP, Genomic Evolutionary Rate Profiling – RS, rejected substitutions (number of substitutions expected assuming neutrality minus actual observed substitutions); GnomAD, Genome Aggregation Database; HSF, Human Splicing Finder; m, months; F, female; M, male; y, year
GERP version 2010 was accessed through Varsome (Kopanos et al., 2019). HSF impact predictions and MaxEnt Donor Site variation score were generated in 06/2021 through https://hsf.genomnis.com (Desmet et al., 2009) using search term ENST00000337331.10:c.2805C>T.
Searching the whole rare disease dataset (73,988 genomes) from the Genomics England 100,000 Genomes Project, we identified 2 probands with the identical homozygous change c.2805C>T in NPHP3 (GEL-1 & GEL-2) and phenotypes suggestive of a multisystem ciliopathy with features of hepatorenal fibrocystic disease (Figure 1E, 1F & Table 1). Homozygosity plots for GEL-1 & GEL-2 detected a large homozygous region on chromosome 3, including NPHP3, and in keeping with known parental consanguinity (Supp. Figure S3). In both cases, the unaffected mother is heterozygous for the NPHP3 allele (Supp. Figure S4A & S4B). Of note, both GEL-1 and GEL-2 are reported as genetically unsolved by the Genomics England analysis team as the predicted synonymous NPHP3 allele had been filtered out from variant tiering tables. A further two cases with NPHP-like disease associated with congenital liver disease were identified in patient databases from clinical collaborators. The first was identified in a cohort of WES data (~4500 whole exomes) from Saudi Arabia (SA-1) (Figure 1G) and the second one in a worldwide cohort of patients with inherited renal disease, including 800 patients with NPHP and related phenotypes (NP642) (Figure 1H & Table 1). In both consanguineous families, no alternative genetic diagnosis was detected. Noteworthy, the implication of NPHP3 in case NP642 is supported by a region of genome-wide homozygosity on chromosome 3 encompassing NPHP3 (Figure 1I).
The identified change c.2805C>T is located in the middle of NPHP3 exon 20 just before the C-terminal tetratrico peptide repeat domains (Figure 1J). In silico tools indicate a certain constraint for the cytosine at position 2805. Importantly, the thymine insertion creates a GT motif that is predicted to act as a cryptic alternative splice donor (Table 1). We evaluated the effect of this variant using whole blood-derived RNA from the index Omani family. We performed RT-PCR with primers targeted to exons 19 and 21 and sequenced the amplicon. In both heterozygous and homozygous individuals (Figure 1K & Supp. Figure S5), sequencing confirms the predicted alternative splicing event joining mid-exon 20 to exon 21. In addition, we also detected the canonically spliced RNA indicating that the effect on splicing is not 100%, at least not in blood. Altogether, the detected RNA sequence is compatible with the presence of both (i) canonically spliced mRNA leading to RNA substitution r.2805c>u and amino acid change p.Gly935Gly as well as (ii) alternatively spliced mRNA, out of phase (1 nucleotide from exon 20 and 2 nucleotides from exon 21) and leading to RNA deletion r.2804_2883del and frameshift p.Gly935GlyfsTer47 (Figure 1K). On gel electrophoresis, we confirm the presence of a shorter NPHP3 transcript, compatible with the expected alternative splicing, in unaffected heterozygotes and in homozygous patients but not in unrelated controls (Supp. Figure S6). The ratio of short to full-length transcripts is higher in homozygote’s blood RNA indicating a dosage effect on splicing.
Considering that a predicted synonymous variant discarded from several independent NGS pipelines and analyses provided a plausible genetic diagnosis in 5 families with this rare ciliopathy, we wondered whether filtering by rare predicted synonymous NPHP3 variants could solve additional cases in the 100,000 Genomes Project rare disease dataset (73,988 genomes). We did not detect other patients with rare homozygous NPHP3 synonymous variants and matching phenotypes but, when filtering for monoallelic variants, we identified a genetically unsolved 34-year-old female (GEL-3) presenting with ESKD in childhood and compatible with an autosomal recessive inheritance. This patient was compound heterozygous for a pathogenic NPHP3 nonsense allele (RefSeq NM_153240.5: c.1729C>T p.(Arg577Ter)) and a rare (UK Biobank exomes: 1/399,454 alleles) predicted synonymous NPHP3 allele (RefSeq NM_153240.5: c.3129T>C p.(Tyr1043Tyr)). The latter variant is situated 4 nucleotides from an intron-exon boundary and in silico tools predicted a possible effect on splicing (Supp. Table S2 & Supp. Figure S7) but unfortunately no patient RNA was available to test this hypothesis. Again, the rare synonymous variant was filtered out from the Genomics England tiering tables.
Synonymous variants occur frequently as they typically elude evolutionary constraint and may often be filtered out of automated lists of pathogenic variants. This is especially the case when in silico prediction scores based on amino acid changes such as SIFT and PolyPhen-2 determine these are benign alleles. Disease-causing predicted synonymous SNV have been reported in association with cystic kidney diseases including autosomal dominant polycystic kidney disease with variants identified in PKD1 and PKD2 (Claverie-Martin et al., 2015) and autosomal recessive polycystic kidney disease with variants identified in PKHD1 leading to aberrant splicing (Molinari et al., 2020). Finally, we have previously reported families with a clinical diagnosis of NPHP in whom a predicted synonymous SNV in NPHP3 was demonstrated to cause aberrant splicing, using RNA from urinary renal epithelial cells, (Molinari et al., 2018). In these cases, pathogenic synonymous variants have been detected because of investigator-led manual curation of disease-associated genes.
Mutations in NPHP3 are among the more common causes of infantile NPHP (Tory et al., 2009; Halbritter et al., 2013) and may also cause the perinatal lethal Meckel syndrome (Bergmann et al., 2008). Aside from the renal features which are commonly classified as NPHP and cystic kidney disease, NPHP3 causes liver phenotypes, typically congenital hepatic fibrosis (Olbrich et al., 2003). Remarkably, the predicted synonymous variants identified in this study were able to provide a plausible genetic diagnosis in 6 families suffering from a rare ciliopathy. The most recent and among the larger patient cohorts described 13 families with biallelic NPHP3 variants, without evidence for recurrent homozygous changes as we detected here (Tang et al., 2020). Among pathogenic truncating variants, the majority are located downstream of exon 21, suggesting indeed that the premature termination reported here is disease-causing (Chaki et al., 2011; Tang et al., 2020). Furthermore, exons 20 and 21 are present in all main NPHP3 transcripts (Olbrich et al., 2003) and no alternative splice junctions affecting exons 20 and 21 were detected in human RNA sequencing data from different tissues (https://www.gtexportal.org/home/). If translated, C-terminal truncation affects the tetratrico peptide repeat domain with potential functional importance in protein-protein interactions (Olbrich et al., 2003). Although we detected an allele-dosage effect on aberrant splicing, we still detected significant amounts of canonically spliced RNA (p.Gly935Gly) in homozygous patient blood. Considering the potential for cell-type and developmental stage differences in splicing, and in particular that splicing for NPHP3 markedly differed between whole blood and kidney (Molinari et al., 2018), we hypothesize that a predominant alternative splicing occurs in organs with disease manifestations. Unfortunately, we had no access to kidney or liver RNA and this constitutes a major limitation of this study. However, the identification of a total of 5 families sharing the same homozygous NPHP3 variant with overlapping phenotypes and the proof-of-principle demonstration that the identified variant indeed affects splicing strongly supports its pathogenicity.
The findings presented here and reported in literature show that rare predicted silent genetic variants are routinely discarded from NGS tiering algorithms, ignoring the functional consequences they can have on pre-mRNA splicing, and that some unexplained cases of ciliopathies may not be solved until a more refined analysis of any impact on splicing is performed to determine more accurately pathogenicity. It is estimated that cryptic splice mutations, i.e., outside of the essential GT and AG splice dinucleotides, are responsible for ~10% of pathogenic mutations in patients with rare genetic disorders (Jaganathan et al., 2019) and up to 62% of all pathogenic single-nucleotide variants are disrupting RNA splicing (Wai et al., 2020). There are a variety of tools that can be applied to this situation including SpliceAI (Jaganathan et al., 2019), MaxEntScan (Yeo and Burge, 2004) and Human Splicing Finder (Desmet et al., 2009; Moles-Fernández et al., 2018), with large variations in sensitivities and specificities for different types of splicing defects. A recent report from the UK Splicing and disease working group examined potential splicing-altering effects of 257 coding and non-coding VUS identified in a true-to-life clinical diagnostics context (mostly VUS in BRCA1/2 and FBN1) (Wai et al., 2020). Using RT-PCR or RNAseq on patient blood RNA, 33% of these VUS were associated with abnormal splicing. Interestingly, 13% of non–splice region variants (outside of the region defined as 3 exonic nucleotides and 8 intronic nucleotides from the exon-intron boundary) still significantly affected splicing. Despite the increasing accuracy of some bioinformatics prediction tools, all continue to show significant miscalling (Jaganathan et al., 2019; Wai et al., 2020). Therefore, one take home message from this study is that in silico tools should neither be relied on in isolation nor should pathogenic predictions represent a pre-requisite for seeking experimental evidence for altered splicing, especially for variants located outside of classical splice regions. The study concludes that experimental RNA analysis has the ability to produce clear results that help classify variant pathogenicity in relatively large cohorts and should be routinely considered to clarify VUS in unsolved genetic disease. However, further work and validation is needed to determine how best to incorporate experimental splicing analysis into clinical practice, how to report and standardize experimental evidence, the potential advantages of RNAseq over RT-PCR and how to translate insights on splicing from blood RNA to affected tissues (Wai et al., 2020).
Supplementary Material
Acknowledgements
We thank the affected individuals, their families, and their physicians who contributed to this study. EO is supported by an Early Postdoc Mobility Stipendium of the Swiss National Science Foundation (P2ZHP3_195181) and Kidney Research UK (Paed_RP_001_20180925). IAA was supported by the Ministry of Higher Education (Oman). EM is funded by Kidney Research UK (RP_006_20180227). MBG is funded by Kidney Research UK (ST_001_20171120) and the Northern Counties Kidney Research Fund. LP is funded by the Medical Research Council Discovery Medicine North Training Partnership. FH is the William E. Harmon Professor of Paediatrics and was supported by grants from the National Institutes of Health (DK068306). Part of sequencing and data processing was performed by the Yale Centers for Mendelian Genomics funded by the National Human Genome Research Institute (U54 HG006504). MAH is funded by Research Advisory Council at King Faisal Specialist Hospital and Research Centre (KFSHRC RAC# 2160 022), Riyadh, Saudi Arabia. JAS and CM are funded by Kidney Research UK and the Northern Counties Kidney Research Fund. This research was made possible through access to the data and findings generated by the 100,000 Genomes Project that is managed by Genomics England Limited (a wholly owned company of the Department of Health and Social Care). The 100,000 Genomes Project is funded by the National Institute for Health Research and NHS England. The Wellcome Trust, Cancer Research UK and the Medical Research Council have also funded research infrastructure. The 100,000 Genomes Project uses data provided by patients and collected by the National Health Service as part of their care and support. See supplemental information for consortium details. Furthermore, this research has been conducted using data from UK Biobank (project ID number 43879), a major biomedical database with online open access to researchers (www.ukbiobank.ac.uk). UK Biobank is generously supported by its founding funders the Wellcome Trust and UK Medical Research Council, as well as the Department of Health, Scottish Government, the Northwest Regional Development Agency, British Heart Foundation and Cancer Research UK. The organisation has over 150 dedicated members of staff, based in multiple locations across the UK.
Grant numbers:
EO: P2ZHP3_195181, Paed_RP_001_20180925; EM: RP_006_20180227; MBG: ST_001_20171120; FH: DK068306; MAH: KFSHRC RAC# 2160 022
Footnotes
Conflicts of Interest
The authors declare no conflict of interest.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials. Further phenotypic or sequencing data are available from the corresponding author (JAS), upon reasonable request. The identified genetic variant in NPHP3 has been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/ accession number SCV001478448).
References
- 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR. 2015. A global reference for human genetic variation. Nature 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J. 2011. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 12:745–755. [DOI] [PubMed] [Google Scholar]
- Bergmann C, Fliegauf M, Brüchle NO, Frank V, Olbrich H, Kirschner J, Schermer B, Schmedding I, Kispert A, Kränzlin B, Nürnberg G, Becker C, et al. 2008. Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia. Am J Hum Genet 82:959–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun DA, Schueler M, Halbritter J, Gee HY, Porath JD, Lawson JA, Airik R, Shril S, Allen SJ, Stein D, Al Kindy A, Beck BB, et al. 2016. Whole exome sequencing identifies causative mutations in the majority of consanguineous or familial cases with childhood-onset increased renal echogenicity. Kidney Int 89:468–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron-Christie S, Wolock CJ, Groopman E, Petrovski S, Kamalakaran S, Povysil G, Vitsios D, Zhang M, Fleckner J, March RE, Gelfman S, Marasa M, et al. 2019. Exome-Based Rare-Variant Analyses in CKD. J Am Soc Nephrol JASN 30:1109–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaki M, Hoefele J, Allen SJ, Ramaswami G, Janssen S, Bergmann C, Heckenlively JR, Otto EA, Hildebrandt F. 2011. Genotype-phenotype correlation in 440 patients with NPHP-related ciliopathies. Kidney Int 80:1239–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claverie-Martin F, Gonzalez-Paredes FJ, Ramos-Trujillo E. 2015. Splicing defects caused by exonic mutations in PKD1 as a new mechanism of pathogenesis in autosomal dominant polycystic kidney disease. RNA Biol 12:369–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmet F-O, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C. 2009. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 37:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groopman EE, Marasa M, Cameron-Christie S, Petrovski S, Aggarwal VS, Milo-Rasouly H, Li Y, Zhang J, Nestor J, Krithivasan P, Lam WY, Mitrotti A, et al. 2019. Diagnostic Utility of Exome Sequencing for Kidney Disease. N Engl J Med 380:142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbritter J, Porath JD, Diaz KA, Braun DA, Kohl S, Chaki M, Allen SJ, Soliman NA, Hildebrandt F, Otto EA, GPN Study Group. 2013. Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum Genet 132:865–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamosh A, Scott AF, Amberger J, Valle D, McKusick VA. 2000. Online Mendelian Inheritance in Man (OMIM). Hum Mutat 15:57–61. [DOI] [PubMed] [Google Scholar]
- Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, Kosmicki JA, Arbelaez J, Cui W, Schwartz GB, Chow ED, Kanterakis E, et al. 2019. Predicting Splicing from Primary Sequence with Deep Learning. Cell 176:535–548.e24. [DOI] [PubMed] [Google Scholar]
- Jalali Sefid Dashti M, Gamieldien J. 2017. A practical guide to filtering and prioritizing genetic variants. BioTechniques 62:18–30. [DOI] [PubMed] [Google Scholar]
- Kagan KO, Dufke A, Gembruch U. 2017. Renal cystic disease and associated ciliopathies. Curr Opin Obstet Gynecol 29:85–94. [DOI] [PubMed] [Google Scholar]
- Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, Massouras A. 2019. VarSome: the human genomic variant search engine. Bioinforma Oxf Engl 35:1978–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann N, Braun DA, Amann K, Tan W, Shril S, Connaughton DM, Nakayama M, Schneider R, Kitzler TM, AT van der Ven, Chen J, Ityel H, et al. 2019. Whole-Exome Sequencing Enables a Precision Medicine Approach for Kidney Transplant Recipients. J Am Soc Nephrol JASN 30:201–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moles-Fernández A, Duran-Lozano L, Montalban G, Bonache S, López-Perolio I, Menéndez M, Santamariña M, Behar R, Blanco A, Carrasco E, López-Fernández A, Stjepanovic N, et al. 2018. Computational Tools for Splicing Defect Prediction in Breast/Ovarian Cancer Genes: How Efficient Are They at Predicting RNA Alterations? Front Genet 9:366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinari E, Decker E, Mabillard H, Tellez J, Srivastava S, Raman S, Wood K, Kempf C, Alkanderi S, Ramsbottom SA, Miles CG, Johnson CA, et al. 2018. Human urine-derived renal epithelial cells provide insights into kidney-specific alternate splicing variants. Eur J Hum Genet EJHG 26:1791–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinari E, Srivastava S, Dewhurst RM, Sayer JA. 2020. Use of patient derived urine renal epithelial cells to confirm pathogenicity of PKHD1 alleles. BMC Nephrol 21:435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olbrich H, Fliegauf M, Hoefele J, Kispert A, Otto E, Volz A, Wolf MT, Sasmaz G, Trauer U, Reinhardt R, Sudbrak R, Antignac C, et al. 2003. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat Genet 34:455–459. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz J, Milpetz F, Bork P, Ponting CP. 1998. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci U S A 95:5857–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleiman P, Wang D, Glessner J, Hadley D, Gur RE, Cohen N, Li Q, Hakonarson H, Janssen-CHOP Neuropsychiatric Genomics Working Group. 2013. GWAS meta analysis identifies TSNARE1 as a novel Schizophrenia / Bipolar susceptibility locus. Sci Rep 3:3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Liu C, Liu X, Chen J, Fan X, Liu J, Ma D, Cao G, Chen Z, Xu D, Zhu Y, Jiang X, et al. 2020. Phenotype and genotype spectra of a Chinese cohort with nephronophthisis-related ciliopathy. J Med Genet. [DOI] [PubMed] [Google Scholar]
- Tory K, Rousset-Rouvière C, Gubler M-C, Morinière V, Pawtowski A, Becker C, Guyot C, Gié S, Frishberg Y, Nivet H, Deschênes G, Cochat P, et al. 2009. Mutations of NPHP2 and NPHP3 in infantile nephronophthisis. Kidney Int 75:839–847. [DOI] [PubMed] [Google Scholar]
- Vilboux T, Doherty DA, Glass IA, Parisi MA, Phelps IG, Cullinane AR, Zein W, Brooks BP, Heller T, Soldatos A, Oden NL, Yildirimli D, et al. 2017. Molecular genetic findings and clinical correlations in 100 patients with Joubert syndrome and related disorders prospectively evaluated at a single center. Genet Med Off J Am Coll Med Genet 19:875–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai HA, Lord J, Lyon M, Gunning A, Kelly H, Cibin P, Seaby EG, Spiers-Fitzgerald K, Lye J, Ellard S, Thomas NS, Bunyan DJ, et al. 2020. Blood RNA analysis can increase clinical diagnostic rate and resolve variants of uncertain significance. Genet Med 22:1005–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, Burge CB. 2004. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol J Comput Mol Cell Biol 11:377–394. [DOI] [PubMed] [Google Scholar]
- Zhou X, Edmonson MN, Wilkinson MR, Patel A, Wu G, Liu Y, Li Y, Zhang Z, Rusch MC, Parker M, Becksfort J, Downing JR, et al. 2016. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat Genet 48:4–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Web resources
Ensembl VEP: https://www.ensembl.org/info/docs/tools/vep/index.html
GnomAD v2.1.1: https://gnomad.broadinstitute.org/
GTEx: https://www.gtexportal.org/home/
The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS.
HomozygosityMapper: http://www.homozygositymapper.org/
Human Splicing Finder: https://hsf.genomnis.com/login (Desmet et al., 2009)
ImageJ: https://imagej.nih.gov/ij/ (Schneider et al., 2012)
Integrative Genomics Viewer: http://software.broadinstitute.org/software/igv/ (Robinson et al., 2011)
NCBI ClinVar: https://www.ncbi.nlm.nih.gov/clinvar/
NCBI Primer-BLAST: https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi
Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD), (Hamosh et al., 2000). World Wide Web URL: https://omim.org/
Pedigree tool: www.progenygenetics.com
ProteinPaint: https://pecan.stjude.cloud/proteinpaint (Zhou et al., 2016)
SMART: http://smart.embl-heidelberg.de/ (Schultz et al., 1998)
Varsome®: https://varsome.com/ (Kopanos et al., 2019)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.