

Abstract
Polyketide synthases (PKS) and nonribosomal peptide synthetases (NRPS) comprise biosynthetic pathways that provide access to diverse, often bioactive natural products. Metabolic engineering can improve production metrics to support characterization and drug-development studies, but often native hosts are difficult to genetically manipulate and/or culture. For this reason, heterologous expression is a common strategy for natural product discovery and characterization. Many bacteria have been developed to express heterologous biosynthetic gene clusters (BGCs) for producing polyketides and nonribosomal peptides. In this article, we describe tools for using Pseudomonas putida, a Gram-negative soil bacterium, as a heterologous host for producing natural products. Pseudomonads are known to produce many natural products, but P. putida production titers have been inconsistent in the literature and often low compared to other hosts. In recent years, synthetic biology tools for engineering P. putida have greatly improved, but their application towards production of natural products is limited. To demonstrate the potential of P. putida as a heterologous host, we introduced BGCs encoding the synthesis of prodigiosin and glidobactin A, two bioactive natural products synthesized from a combination of PKS and NRPS enzymology. Engineered strains exhibited robust production of both compounds after a single chromosomal integration of the corresponding BGC. Next, we took advantage of a set of genome-editing tools to increase titers by modifying transcription and translation of the BGCs and increasing the availability of auxiliary proteins required for PKS and NRPS activity. Lastly, we discovered genetic modifications to P. putida that affect natural product synthesis, including a strategy for removing a carbon sink that improves product titers. These efforts resulted in production strains capable of producing 1.1 g/L prodigiosin and 470 mg/L glidobactin A.
Keywords: Pseudomonas putida, heterologous expression, genome editing, polyketide, non-ribosomal peptide
1. Introduction
For most of the last century, natural products have been an essential source for the discovery of novel drugs for treating human diseases (Newman and Cragg, 2012). Natural products are secondary metabolites produced by living organisms that are often unnecessary for growth but provide advantages to the host organism in certain environments. These compounds evolved for specific interactions with biomolecules, and as a result, they often exhibit medically relevant properties, such as antibiotic or anti-cancer activities (Atanasov et al., 2021). Two of the most intriguing classes of natural products are polyketides and nonribosomal peptides. These compounds are often synthesized in an assembly line fashion by modular enzymes called type I polyketide synthases (PKS) and nonribosomal peptide synthetases (NRPS) (Keating and Walsh, 1999). Multiple enzymatic domains are encoded in single large peptides, and chemical diversity is achieved through the addition, deletion, and modification of individual domains (Vanner et al., 2013). The biosynthesis potential of these assembly-lines has long been touted as a panacea for accessing diverse molecular structures, but this potential remains mostly unrealized due to challenges in engineering enzymes to meet required synthesis metrics.
Bacteria, particularly Actinomycetes, are a rich source of natural products, including polyketides and nonribosomal peptides. The number of putative biosynthetic gene clusters (BGCs) has accelerated with the rise of next-generation sequencing (Niu, 2018), as has the number without a validated product (Rutledge and Challis, 2015). Connecting putative BGCs to purified compounds requires several challenges to be overcome. Putative BGC-containing hosts can be difficult to cultivate in a laboratory setting, and even if cultivation is achieved, the conditions required for inducing production of the desired natural product may be unknown (Ren et al., 2017). Alternatively, BGCs can be transferred to a heterologous host and expressed from modern synthetic biology vectors. This strategy by-passes growth and regulation problems associated with the native host by using a microorganism with well-characterized genetic tools that also grows robustly in laboratory conditions (Fu et al., 2012; Gaida et al., 2015; Pogorevc et al., 2019). Once a BGC is introduced into a heterologous host, novel compounds can be isolated for structure elucidation and titers can be improved with common metabolic engineering strategies to enable ex vivo bioactivity assays (Chen et al., 2017; Wang et al., 2019). Most heterologous BGC expression projects use Streptomyces species as hosts because actinomycetes are the most abundant source of bacterial natural products (Cook and Pfleger, 2019; Park et al., 2020). However, Streptomyces hosts are more difficult to genetically modify than the synthetic biology workhorse, Escherichia coli. Several groups have developed E. coli strains for polyketide and nonribosomal peptide production (D’Agostino and Gulder, 2018; Pfeifer et al., 2001). However, functional expression of these modular enzymes in E. coli is often difficult to achieve, so researchers have sought alternative hosts for producing heterologous polyketides and nonribosomal peptides (Bian et al., 2017; Liu et al., 2020; Mendez-Perez et al., 2011; Pogorevc et al., 2019).
Pseudomonas putida KT2440, a Gram-negative soil microbe, has become a popular metabolic engineering chassis in recent years, mostly due to its ability to utilize lignin-derived compounds and its tolerance to organic solvents and stresses associated with industrial-scale cultivations (Nikel and de Lorenzo, 2018; Niu et al., 2020). P. putida also has several traits that make it an attractive alternative host for producing natural products. Its genome has a relatively high GC content (61.5%) similar to that of actinomycete-derived natural product BGCs. P. putida natively expresses a promiscuous phosphopantetheinyl transferase (PPTase) capable of activating heterologous acyl- and peptidyl-carrier proteins, and it has excellent growth properties amenable to large-scale cultivations (Ankenbauer et al., 2020; Owen et al., 2011). There has also been an explosion in synthetic biology tools developed for P. putida, including genetic parts for expressing heterologous proteins and constructing genomic edits (Aparicio et al., 2018; Elmore et al., 2017; Martin-Pascual et al., 2021). Despite these advantages, P. putida has performed inconsistently in the literature as a heterologous host for producing polyketides and nonribosomal peptides, where it either achieves titers on par or greater than the native host (Li et al., 2010; Wenzel et al., 2005) or it performs poorly compared to alternative heterologous hosts (Chai et al., 2012; Wang et al., 2019). Most reports have investigated the effect of transcriptional control on heterologous BGC expression in P. putida (Domröse et al., 2017; Dudnik et al., 2013; Fu et al., 2008). Other variables that affect production titers, such as GC content of the BGC, translational efficiency, and specific activity of PKSs and NRPSs, have not been systematically explored in P. putida.
We selected prodigiosin and glidobactin A, two natural products natively produced by Gram-negative bacteria, to serve as model products for exploring factors that influence biosynthesis in P. putida. The BGCs for prodigiosin and glidobactin A biosynthesis both contain PKS and NRPS components, and both compounds possess medically relevant properties, such as anti-cancer activity (Danevčič et al., 2016; Han et al., 2005; Oka et al., 1988; Stein et al., 2012; Zhang et al., 2005). Prodigiosin is a tripyrrole secondary metabolite synthesized by many strains of Serratia marcescens (Williamson et al., 2006). It is a red pigment, providing an easily detectable phenotype for screening libraries for factors that increase prodigiosin production. The highest reported production titer of prodigiosin from a heterologous host is 150 mg/L from P. putida (Domröse et al., 2017). BGCs for glidobactin synthesis have been identified and characterized from Schlegelella brevitalea DSM7029 and Photorhabdus luminescens subsp. laumondii TT01 (Dudnik et al., 2013; Schellenberg et al., 2007). The two glidobactin BGCs differ in their GC content and in the presence of different genes required for biosynthesis, providing a case study that can reveal variables important for heterologous production in P. putida. The highest heterologous titer for this molecule was achieved after expression of the BGC from P. luminescens in Xenorhabdus doucetiae, which yielded 177 mg/L glidobactin A while production from expression in P. putida was not detected (Wang et al., 2019).
In this work, we designed a genome editing pipeline for integrating and expressing heterologous BGCs in P. putida. The BGCs responsible for prodigiosin (from S. marcescens ATCC274) and glidobactin (from S. brevitalea DSM7029 and P. luminescens subsp. laumondii TT01) biosynthesis serve as models for investigating the effects of transcription and translation on production titers. Focusing on glidobactin A biosynthesis, we compare strategies for improving the functional expression of modular PKSs and NRPSs. We also take advantage of the visible phenotype associated with prodigiosin production to devise a screen for discovering mutants with improved production titers of heterologous products. Through these efforts, we demonstrate that P. putida is capable of heterologous production titers in 100 mg - 1 g/L quantities from all three BGCs.
2. Results
2.1. Introducing heterologous BGCs into P. putida.
We initially focused on engineering a strain of P. putida to produce prodigiosin because the visible pigmentation of production cultures would allow for rapid detection of heterologous production from early production strains. The BGC for prodigiosin biosynthesis, designated pig, in S. marcescens ATCC274 is 20,948 bp in length and contains 14 genes expressed in a single operon (Harris et al., 2004) (Fig. 1a). Prodigiosin biosynthesis occurs as a bifurcated pathway where two precursors, 2-methyl-3-n-amyl-pyrrole (MAP) and 4-methoxy-2,2’-bipyrrole-5-carbaldehyde (MBC), are synthesized independently and then condensed in the final reaction that yields prodigiosin (Fig. 1b). MAP is derived from 2-octenoyl-ACP/-CoA, and MBC is derived from L-proline, malonyl-CoA, and L-serine. L-proline is incorporated into the pathway by a NRPS (PigG and PigI), followed by the incorporation of malonyl-CoA by a PKS (PigJ and PigH) (Williamson et al., 2006).
Fig. 1. Establishing a prodigiosin production strain.
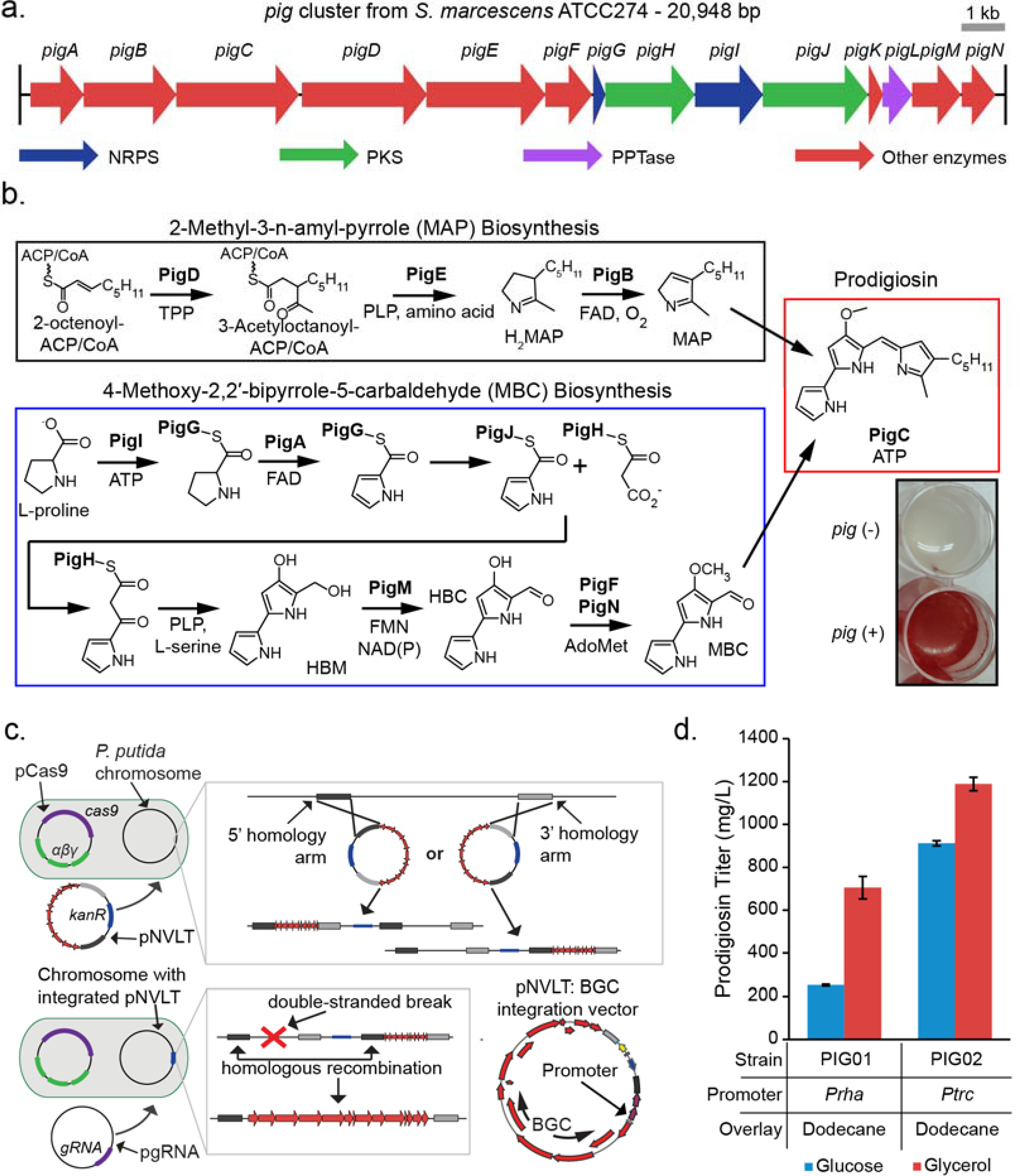
(a) Map of prodigiosin BGC from S. marcescens ATCC274. Colored arrows indicate the enzyme class according to the legend. (b) Prodigiosin biosynthesis begins with intermediates in fatty acid metabolism and L-proline. 2-octenoyl-CoA/ACP is converted to MAP by PigDEB. L-proline, serine, and malonyl-CoA are converted to MBC by PigIAJHMFN. The two intermediates are then fused by PigC to produce prodigiosin which generates a visible red color (inset) in culture. (c) Integration of BGCs into the chromosome of P. putida. A pNVLTv2 integration vector is introduced via conjugation into a strain of P. putida containing pCas9. The integration vector carries out a single-crossover recombination with the P. putida chromosome. The Cas9 counterselection is then enabled after electroporating pgRNA targeting the wild-type sequence to be replaced by the BGC. This selects for the double-crossover recombination of the integration vector and results in a markerless and stable integration of the BGC. (d) Prodigiosin production from cultures grown in glucose or glycerol-based media under control of Prha or Ptrc promoters. All cultures were supplied with dodecane as a product-sink. Error bars represent standard deviation, n = 3 biological replicates. Differences in values between all samples were found to be statistically significant (P<0.01) by the Student’s t-test.
Replicative vectors in P. putida can sometimes induce a growth defect, which can have a negative effect on heterologous production (Cook et al., 2018; Mi et al., 2016). Therefore, we sought to optimize a genome editing protocol for integrating large DNA constructs into the P. putida chromosome. We previously developed a set of vectors for constructing scarless deletions in P. putida KT2440 using Cas9-assisted homologous recombination (Cook et al., 2018). A replicative vector using the RK2 origin, pCas9, expressed Cas9 from Streptococcus pyogenes and the genes encoding the λRed system from λ bacteriophage, Exo, Beta, and Gam. The single guide RNA (sgRNA) was expressed from pgRNA, a replicative vector containing the BBR1 origin. The repair template was located in an integrative vector containing homology arms for the region to be deleted (Graf and Altenbuchner, 2011). The biggest difference between our needs and the existing genome editing method was the size of the required repair template, here containing a full BGC. Cloning BGCs can lead to large plasmid sizes and the introduction of genes that may be toxic to common E. coli cloning strains (Fu et al., 2012). Large plasmids are not as easily transformed by electroporation into P. putida, but conjugation is significantly more efficient (Fu et al., 2008). To address these issues, we edited the integrative vector to replace the high-copy ColE1 origin with the medium-copy p15A origin and add a functional origin of transfer (oriT) sequence for performing conjugations. Unnecessary genes in the original vector were also removed to reduce the plasmid size, resulting in the integrative vector pNVLTv2.
Next, we deleted the endogenous BGC for pyoverdine biosynthesis to simplify the baseline strain, create a “neutral” integration site, and eliminate potential analytical challenges (Choi et al., 2018; Gomez-Escribano and Bibb, 2011; Ravel and Cornelis, 2003). We had already created a strain from previous work with one gene deleted, (P. putida ΔpvdL), so we introduced homology arms for deleting the remaining three genes, pvdIJD, into pNVLTv2, resulting in pNVLTv2-pvdIJD. We designed a sgRNA plasmid targeting pvdI, pgRNAtet-pvdI, and successfully constructed the 24,920-bp deletion using the two-step Cas9-assisted protocol that our group has described previously, resulting in strain P. putida pvd-.
We selected two inducible promoter systems, the rha promoter (Prha, induced by L-rhamnose) and the trc promoter (Ptrc, induced by IPTG), to drive expression of heterologous BGCs at the pvdIJD locus. BGCs expressed as a single operon can be refactored for heterologous expression by replacing the native sequence directly upstream of the initial gene with a synthetic promoter and ribosome binding site (RBS) optimized for the heterologous host. Inducible promoters are ideal for these types of projects because they reduce BGC expression during cloning protocols and enable two-phase production cultures, where cultivation is separated into a growth stage and a production stage (Raj et al., 2020). Prha is tightly repressed by carbon catabolite repression in E. coli, but not in P. putida, making it an appropriate promoter for cloning BGCs in E. coli before transferring them into P. putida (Jeske and Altenbuchner, 2010). Ptrc is one the strongest promoters that we have characterized in P. putida, which allowed us to maximize expression of heterologous BGCs (Cook et al., 2018).
Experiments with each promoter linked to a fluorescent reporter construct confirmed that Ptrc has higher basal and maximum expression than Prha (Fig. S2a). Next, we attempted to construct pig integration vectors with both promoters using RecET direct cloning (Fu et al., 2012; Wang et al., 2016) in E. coli (Fig. 1c). We isolated correct clones with Prha only when transformation media was supplemented with glucose, conditions that silence Prha through catabolite repression. Consistently, we were unable to obtain a Ptrc version of the pig vector, presumably because leaky expression of the pig genes was toxic to E. coli. The expression construct including Prha and the pig BGC (23,247 bp total) was integrated into P. putida’s chromosome at the pvdIJD locus through the two-step protocol as described above, resulting in strain PIG01 (Fig. 1c). Upon addition of up to 0.5% (w/v) L-rhamnose to the growth media, cultures of PIG01 produced a visible red pigment characteristic of prodigiosin production (Fig. 1b).
2.2. Optimizing prodigiosin production.
Once we established prodigiosin production in P. putida, we then compared various media formulations and their effects on prodigiosin titers. Terrific Broth (TB) has been used previously to produce prodigiosin in P. putida cultures (Domröse et al., 2015). Alternatively, Riesenberg-Korz (RK) medium is a minimal medium developed for high-cell density cultures of E. coli, and variations of this medium have been used for the production of other products from P. putida (Lee et al., 2000; Riesenberg et al., 1991). After determining the optimum L-rhamnose concentration for induction (0.2% w/v), we compared prodigiosin production in rich media and minimal media (Fig. S2b). In non-baffled shake flask cultures of P. putida PIG01, prodigiosin titers were less than 100 mg/L in TB media (Fig. S3a). Prodigiosin is insoluble in water, so we added an organic overlay to provide a product sink, which greatly improved prodigiosin production in TB media to over 500 mg/L. Prodigiosin production in RK media with 2.5% glycerol reached approximately 500 mg/L without an organic overlay and over 600 mg/L with an overlay (Fig. S3a). Interestingly, using glucose as the carbon source in minimal media led to much lower prodigiosin titers of approximately 200 mg/L (Fig. 1d).
After establishing optimal media and cultivation conditions for strain PIG01, we sought to further improve prodigiosin production through transcriptional control. In lieu of cloning a pig integration construct with Ptrc, we designed a chromosomal integration vector for replacing Prha with Ptrc in PIG01, pNVLTv2-pvdIJD-Ptrc-pigA. Integration of this construct resulted in strain PIG02, which produced a strong red pigment in cultures supplemented with IPTG, and 1 mM was the optimal inducer concentration (Fig. S2c). Introducing a stronger promoter for pig expression improved prodigiosin production, as P. putida PIG02 generated higher prodigiosin titers than PIG01 (Fig. 1d). This strain also produced more prodigiosin on minimal media with glycerol than with glucose. PIG02 cultures grown in RK media with glycerol generated prodigiosin titers around 1.1 g/L, whereas production on glucose was approximately 900 mg/L.
2.3. Screening for mutants with improved prodigiosin biosynthesis.
To identify strategies for improving prodigiosin production, we devised a screen based on the appearance of P. putida colonies producing prodigiosin. Tn5 mutagenesis was used to create mutant libraries of P. putida PIG01 and PIG02, which normally have a light-pink phenotype on solid media with a low concentration of inducer (Fig. 2a). Libraries of these strains were plated on LB agar or minimal agar with glycerol, and individual colonies with a strong red color, indicating increased prodigiosin production, were isolated and sequenced to determine the location of the Tn5 insertion (Table S8). Surprisingly, the most common type of mutants were disruptions in components of the electron transport chain.
Fig. 2. Tn5 libraries highlight disruptions in electron transport chain.

(a) Generalized workflow for generating and screening mutant libraries for improved prodigiosin production. (b) Components of electron transport chain, highlighting role of the bo3 oxidase, Cyo. UQ – ubiquinones, UQH2 – ubiquinols, CIO – cyanide insensitive oxidase. Adapted from Ugidos et al. and Nikel et al. (c) Effect of deleting cyo operon on prodigiosin production on glucose and glycerol. Error bars represent standard deviation, n = 3 biological replicates. Differences between samples marked with asterisks were found to be statistically significant by the Student’s t-test (** = P<0.01).
The two operons containing Tn5 insertions, cyo and ccm, encode production of the bo3 type oxidase (Cyo) and cytochrome c maturation, respectively (Fig. 2b). Cyo is the primary terminal oxidase used during exponential growth and has been shown to be involved in gene regulation in P. putida (Morales et al., 2006). Mutants with disruptions in the ccm operon had a drastic growth defect on minimal media and did not grow on rich media. Disruptions in the cyo operon led to only a slight growth defect, so we investigated this operon’s effect on prodigiosin production (Fig. S5). The cyo operon contains five genes, cyoABCDE, and we deleted the region containing the RBS and all five genes in this operon in both the L-rhamnose- and IPTG-inducible prodigiosin producing strains (resulting in PIG07 and PIG08, respectively). Transcriptomic data available in the literature suggested that the cyo operon is upregulated during growth on glucose compared to glycerol (Nikel et al., 2014), so we compared prodigiosin production in RK media with glucose in addition to glycerol. No significant changes in production were observed from strains grown on glycerol, but PIG08 had improved production on glucose, making its performance equivalent to PIG02 on glycerol (Fig. 2c).
2.4. Heterologous expression of two glidobactin A BGCs.
The high titers of prodigiosin achieved from heterologous expression in P. putida were encouraging, but we also wanted to interrogate variables affecting the expression of PKSs and NRPSs with a canonical modular structure. Glidobactin A provided this opportunity, as its biosynthesis involves a hybrid PKS/NRPS that has three modules in total and is encoded by genes approximately 12.4 kb in length (Fig. 3a). The two homologous BGCs in S. brevitalea (glb) and P. luminescens (lum) both produce glidobactin A as the primary product (Bian et al., 2012; Schellenberg et al., 2007). Glidobactin A biosynthesis starts with the acylation of an L-threonine residue by GlbF/PLU1878p (Imker et al., 2010). The hybrid PKS/NRPS, GlbC/PLU1880p, then incorporates L-lysine, L-alanine, and malonyl-CoA before performing the final cyclization reaction (Wang et al., 2019). The most apparent difference between the two BGCs is that the glb and lum clusters have a GC content of 70% and 47%, respectively. The glb cluster also has three genes that are not present in the lum cluster: glbE, which encodes an MbtH-like protein (MLP); glbA, a transcriptional regulator; and glbH, which has been shown to be involved in the hydroxylation of a lysine residue that is incorporated into glidobactin A (Table S5) (Fu et al., 2012; Schellenberg et al., 2007). Unlike the prodigiosin BGC, neither glidobactin BGC contains a gene encoding a dedicated PPTase. Expressing each BGC afforded us the opportunity to assess several factors in heterologous production of polyketides and nonribosomal peptides: the effect of MLP expression on the activity of a heterologous NRPS, interactions between the host’s PPTase and heterologous PKSs and NRPSs, and the significance of similarities in phylogeny and GC content between P. putida and heterologous BGCs.
Fig. 3. Identifying glidobactin A as primary product from expression of glb and lum clusters.

(a) Gene maps of glidobactin BGCs from S. brevitalea DSM7029 and P. luminescens TT01. Colored arrows indicate the enzyme class according to the legend. The structure of glidobactin A is shown to the right. (b) Unique peak identified in HPLC analysis of extracts from glidobactin production strains. (c) LC-MS shows that the major peak has m/z value corresponding to glidobactin A + H+ (calculated m/z = 521.3334). (d) Effects of promoter and RBS strength on glidobactin production. All strains were grown in 25 mL of glycerol-based media in 250-mL non-baffled shake flasks. SD=Shine-Dalgarno sequence. Error bars represent standard deviation, n = 3 biological replicates. Differences in values between all samples were found to be statistically significant (P<0.01) by the Student’s t-test.
Using the same strategy applied to prodigiosin, both BGCs were integrated into the pvdIJD locus of P. putida pvd- with Prha directly upstream of the first gene (glbB, plu1881), resulting in the strains P. putida GLB01 and LUM01. Glidobactin A production from these two strains was initially assessed by HPLC; based on peak area, LUM01 produced 15-fold more product than GLB01 (Fig. 3b). We verified that the primary compound was glidobactin A using LC-MS and 1H-NMR (Fig. 3c, Fig. S9). Similar to prodigiosin, glidobactin A production was greater on glycerol compared to glucose (Fig. S8a). Before performing further modifications to strains GLB01 and LUM01, we confirmed by RT-PCR that both BGCs were fully transcribed in P. putida (Fig. S7). It has been shown that deleting the last gene in the glb cluster, glbH, does not abolish glidobactin A production but significantly reduces it (Schellenberg et al., 2007). The RT-PCR results demonstrated that glbH was transcribed, so we did not interrogate GlbH activity as a limiting step in glidobactin production in strain GLB01.
After determining the relative production titers between GLB01 and LUM01, we sought to improve production by modifying transcription and translation of the BGCs with the primary goal of determining why the glb cluster yielded less product than the lum cluster in P. putida. We continued to mirror our strategy with the pig cluster and swapped to the stronger promoter, Ptrc in both strains, resulting in strains GLB02 and LUM02. As expected, production of glidobactin A increased, with both strains producing approximately 2-fold more product (Fig. 3d). Production from GLB02 was still much lower than LUM02, so we investigated translation initiation as a potential factor. Using the RBS calculator (Farasat et al., 2014), we had designed the RBS sequences for the first gene in each BGC to have similar translation initiation rates (TIR) (glb: 13,000 au; lum: 9,000 au). Coincidentally, the Shine-Dalgarno sequences upstream of the BGCs in strains PIG01 and LUM01 were the same (AAGGAG), whereas GLB01 had a different sequence (GATTAG). When we changed the Shine-Dalgarno sequence for glb to AAGGAG in the RBS calculator, the predicted TIR increased to 43,000 au. Therefore, we constructed strain GLB03 by simultaneously inserting Ptrc and the AAGGAG Shine-Dalgarno sequence upstream of the glb cluster. Changing the Shine-Dalgarno sequence upstream of the glb cluster increased glidobactin A production by 2-fold compared to GLB02, which had Ptrc and the original RBS sequence, consistent with the increased RBS strength predicted in silico (Fig. 3d).
2.5. Improving glidobactin A production through MLP and PPTase overexpression.
Functional expression of large, multi-modular enzymes is often the greatest challenge in engineering natural product synthesis, so we identified the steps governed by PKSs and NRPSs as a possible bottleneck in glidobactin production. The presence of a cognate MLP in the glb cluster but not the lum cluster raised the question of whether MLP availability is affecting heterologous NRPS activity. NRPSs often require a complementary MLP for optimal solubility and activity, and homologs from different BGCs or strains are usually not interchangeable (Schomer and Thomas, 2017). Indeed, it has been demonstrated that soluble expression of the NRPS module, GlbF, in E. coli requires co-expression of its cognate MLP, GlbE (Imker et al., 2010). The genome of P. putida KT2440 contains one gene (PP_3808) encoding an MLP specific for the NRPSs responsible for pyoverdine biosynthesis. Deleting the gene encoding P. putida’s native MLP in strain GLB03 (resulting in GLB04) did not significantly change glidobactin A production (Fig. 4c). To increase MLP availability, we inserted a second copy of glbE with a constitutive promoter in place of P. putida’s native MLP, resulting in strain GLB05 (Fig. 4a). Overexpression of glbE resulted in greater than 2-fold increase in glidobactin A production compared to GLB03 and GLB04. In contrast, deleting PP_3808 or overexpressing glbE on a plasmid did not have a noticeable effect on glidobactin production for strain LUM01 (Fig. S8b,c).
Fig. 4. Improving functional expression of PKS/NRPS enzymes in glidobactin pathway.

(a) Strategies for overexpressing MLPs and PPTases in production strains. The MLP gene, glbE, was integrated in place of PP_3808, the gene encoding P. putida’s native MLP, along with a constitutive promoter from the Anderson promoter library. A constitutive promoter from the Anderson library was integrated upstream of PP_1183, the gene encoding P. putida’s native PPTase. The inducible promoter, Ptac, and sfp were integrated in place of pvdL. (b) Cas9-assisted ssDNA oligo recombination scheme for introducing ATG start codons in glbC/plu1880. To select for start codon mutations, an sgRNA targets the PAM sequence immediately downstream of the start codon. A mutagenic oligo contains mutations in the PAM and the start codon, and Cas9 activity selects against cells that don’t incorporate mutations from this oligo. PAM=protospacer adjacent motif. (c) and (d) Effects of MLP overexpression, PPtase overexpression, and start codon mutagenesis on glidobactin production strains. Charts are labeled and split into sections to emphasize genetic changes made (e.g. “PPTase” samples have modifications to PPTase expression). All cultures were in 25-mL of glycerol-based media in 250-mL non-baffled flasks. Error bars represent standard devitaion, n ≥ 3 biological replicates. Differences between samples marked with asterisks were found to be statistically significant by the Student’s t-test (n.s. = not significant, * = P<0.05, ** = P<0.01, *** = P<0.001).
After demonstrating that expression of glbE was non-optimal for glidobactin A production using only the glb cluster, we next investigated whether PPTase activity in P. putida could be limiting production. We initially modified the strains LUM01 and LUM02 by inserting a strong constitutive promoter upstream of PP_1183, resulting in the strains LUM03 and LUM04, respectively (Fig. 4a). Both strains saw an increase in production, and specifically, strain LUM04 had about 50% higher glidobactin A production compared to the parent strain, LUM02 (Fig. 4d). However, these strains had a slower growth rate compared to the parent strains, and when we measured glidobactin production from these strains in 3-mL cultures in tubes instead of 25-mL cultures in flasks, we found that they produced less glidobactin compared to the parent strains (Fig. S8d).
To avoid any deleterious effects on native metabolism from over-producing P. putida’s native PPTase, we introduced a heterologous PPTase as an orthogonal alternative. Sfp is a promiscuous PPTase from Bacillus subtilis that has enabled the production of functional PKS and NRPS enzymes in E. coli (Pfeifer et al., 2001). We designed an expression construct for integrating sfp into the pvdL locus with the tac promoter, enabling induction by the addition of IPTG in strains expressing LacI (Fig. 4a). Introducing this construct to the IPTG-inducible production strains, LUM02 and GLB05, resulted in LUM05 and GLB06. Co-expression of sfp improved glidobactin production by 30% in both strains (Fig. 4c,d). These strains also did not have a noticeable growth defect and did not have a significant drop in production in smaller cultures (Fig. S8d).
2.6. Modifying translation of hybrid PKS/NRPS via Cas9-assisted mutagenesis.
While analyzing GC content and codon usage of the BGCs described in this work, we noticed that the genes encoding the hybrid PKS/NRPS (glbC, plu1880) in both glidobactin clusters each used the alternative start codon, GTG, which lowers translation initiation rates compared to the canonical ATG start codon in prokaryotes, including P. putida (Elmore et al., 2021; Hecht et al., 2017). This realization provided the opportunity to further engineer glidobactin production through the simple strategy of generating a point mutation in the start codon of both genes. We hypothesized that this change would improve translation initiation for these genes and increase glidobactin A production.
To generate strains with these start codon mutations, we adapted our genome editing vectors for introducing point mutations via oligo recombineering. Wu and colleagues recently reported the generation of knockouts in P. putida using only one λ phage recombinase, Beta (Wu et al., 2019). We constructed a derivative of pCas9, pCas9-beta, and were able to construct small chromosomal deletions using 80-nt oligos (Table S6). However, it has been shown in the literature that P. putida can efficiently repair mismatches in its chromosome, and G to A changes are much more sensitive to mismatch repair compared to other mutations (Aparicio et al., 2020b). We accounted for this potential issue by incorporating a strategy developed by Aparicio et al. to temporally inhibit mismatch repair in P. putida. We constructed a second plasmid for oligo recombineering, pCas9-beta-mmr, which co-expresses beta and a defective mutant of mutL from P. putida, mutLE36K. Expression of the mutL mutant inhibits mismatch repair in P. putida, allowing for the generation of point mutations that would normally be repaired by the host.
We identified sgRNA targets 18–24 bp downstream of the start codon in glbC and plu1880 (Fig. 4b). The single-stranded DNA oligos (125 nt) designed for each BGC contained two point mutations: the desired G to A start codon mutation and a synonymous point mutation in the PAM site of the sgRNA target. Attempts to introduce these mutations with pCas9-beta resulted in most mutants having only the PAM mutation (Table S7). Repeating the recombineering protocols with pCas9-beta-mmr resolved this problem, and most mutants sequenced contained both the start codon and PAM mutations. We initially introduced these mutations into IPTG-inducible strains without sfp overexpression (LUM02 and GLB05), resulting in strains LUM06 and GLB07, which have the PAM mutation only (designated as SNP), and strains LUM07 and GLB08, which have both mutations (designated as ATG). The ATG derivatives of both strains produced about 20% more glidobactin A (Fig. 4c). Notably, GLB07 produced 20% less glidobactin A than GLB05, suggesting that the synonymous PAM mutation in glbC had a negative effect on expression, even though the mutated codon, TCG, is more common in P. putida than the native one, TCC. Next, we introduced the ATG mutation to strains containing sfp overexpression, resulting in strains LUM08 and GLB09. LUM08 did not have a significant increase in glidobactin A production compared to its parent strain, LUM05, but GLB09 produced 20% more glidobactin A than GLB06 (Fig. 4c,d).
The best glidobactin A production strain resulting from modifications mentioned above was LUM08, which had an approximately 3-fold increase in production compared to the initial production strain constructed with the lum cluster. GLB09 produced 30 times more glidobactin A than GLB01. After purifying a glidobactin A standard that we verified by 1H-NMR, we used LC-MS to determine the titer from LUM08 and GLB09, which we found to produce approximately 390 mg/L and 270 mg/L glidobactin A, respectively.
2.7. Engineering metabolism and gene regulation for improved natural product titers.
After establishing robust production of both prodigiosin and glidobactin A, we set out to identify metabolic engineering strategies that would improve production of both compounds. Despite differences in the PKS and NRPS components in prodigiosin and glidobactin biosynthesis, both compounds are synthesized from similar primary metabolites. Aided by the visible phenotype of prodigiosin production, we screened for metabolites that could be limiting prodigiosin production in P. putida. We hypothesized that results from these experiments could apply to improving glidobactin A production as well.
We grew cultures of P. putida PIG01 in RK media supplemented with compounds related to the metabolites directly incorporated into prodigiosin, including octanoic acid and amino acids (Fig. 5a). Octanoic acid was the only supplement that improved specific prodigiosin production, suggesting that the availability of 2-octenoyl-ACP/CoA could be limiting prodigiosin production in P. putida. This effect was only apparent when glycerol was the carbon source, and not on glucose (Fig. S3b). These results suggested to us that polyhydroxyalkanoate (PHA) biosynthesis was a potential carbon sink competing with the heterologous pathways for metabolites. Early reactions in both heterologous pathways incorporate an acyl-ACP or acyl-CoA into the natural product. P. putida synthesizes medium-chain-length PHAs (C8-C12) in order to store carbon and energy when an excess of carbon source is available (Prieto et al., 2016). Two PHA polymerases, PhaC-I and PhaC-II, are encoded in a single operon, phaC1ZC2, and incorporate 3-hydroxyalkanoates directly from fatty acid metabolism (Fig. 5b).
Fig. 5. Identification of fatty acyl precursors as a metabolic engineering target.

(a) Supplementing various precursors in minimal media (RK 2.5% glycerol) affects prodigiosin production. Cultures were grown in 3 mL media without dodecane overlay. (b) Metabolites from fatty acid metabolism that are incorporated into PHA, prodigiosin, and glidobactin A biosynthesis. P. putida incorporates mostly C8-C12 3-hydroxyalkanoates into PHAs. 2-octenoyl-ACP or CoA is incorporated into prodigiosin, and dodecanoyl-CoA or 2-dodecenoyl-CoA is incorporated into glidobactin A. (c) PHA composition from P. putida strains with and without ΔglpR and ΔphaC1ZC2 genotypes. Cultures were grown in RK media with glycerol and extractions were completed at 24h of growth. (d) Prodigiosin production from P. putida strains with and without ΔglpR and ΔphaC1ZC2 genotypes. Cultures were grown in 25 mL RK glycerol with dodecane overlay for 48h. (e) Effect of ΔglpR ΔphaC1ZC2 genotype on glidobactin A production. Cultures were grown in 25 mL RK glycerol for 48h. Error bars represent standard deviation, n = 3 biological replicates. Differences between samples marked with asterisks were found to be statistically significant by the Student’s t-test (n.s. = not significant, * = P<0.05, ** = P<0.01, *** = P<0.001).
We initially deleted phaC1ZC2 in strain PIG01, resulting in PIG03, which successfully abolished PHA biosynthesis from P. putida (Fig. 5c). However, PIG03 had lower prodigiosin production than the parent strain on RK media with glycerol (Fig. 5d). Based on reports in the literature, we had also identified glpR, which encodes the transcriptional regulator that represses the expression of genes involved in glycerol uptake and catabolism in P. putida (Fig. S4a) (Escapa et al., 2013). Escapa et al. found that deleting glpR decreased the length of lag phase during growth on glycerol as well as increased PHA production, so we hypothesized that this mutation could be beneficial for prodigiosin production as well. Deleting glpR alone in P. putida PIG01, resulting in PIG04, did not increase prodigiosin production, but introducing this mutation to PIG03, resulting in PIG05, slightly improved the prodigiosin titer to approximately 750 mg/L (Fig. 5d). In contrast, introducing the ΔglpR ΔphaC1ZC2 genotype to P. putida PIG02 did not improve production (Fig. S4b). However, measuring prodigiosin titers at earlier time points did reveal that strains with both deletions had increased initial specific productivity of prodigiosin compared to strains without (Fig. S4b). To test our hypothesis that metabolic modifications improving prodigiosin production can be applied to other natural products, we deleted glpR and phaC1ZC2 in LUM01, creating strain LUM09. This mutation had a more noticeable positive effect on glidobactin A production, resulting in a 30% increase in titer at 48h of growth as determined by HPLC (Fig. 5e). There was a similar benefit to production in the best glidobactin production strain, LUM08, resulting in a strain capable of producing 470 mg/L glidobactin A (LUM10).
3. Discussion
This work establishes several tools for rationally engineering natural product biosynthesis using P. putida as a heterologous host. After optimizing production through various genetic modifications, we achieved production titers greater than any previously reported values for heterologous production for both prodigiosin (1.1 g/L) and glidobactin A (470 mg/L) (Bian et al., 2014; Domröse et al., 2017; Wang et al., 2019). A major strength of our expression and production strategies is that all initial strains (i.e. strains with rha promoter and zero modifications to the BGC or host) were able to produce significant amounts of prodigiosin or glidobactin A, with titers that were on par or greater than values reported in the literature. Applying this approach to the heterologous expression of uncharacterized BGCs in P. putida could be a viable strategy for drug discovery. Combined with the simple metabolic background of P. putida, achieving functional expression of heterologous BGCs using a single integration provides a rapid workflow for building strains to analyze for novel compounds.
In cases where the initial heterologous production titers are lower than desired, there are multiple options for well-characterized promoters in P. putida (Lee et al., 2011). We have shown that in the case of chromosomal expression, increasing the promoter strength provides an expected increase in heterologous production (Fig. 1d, Fig. S2). In contrast, a recent paper that attempted to express the lum BGC in P. putida using T7 RNA polymerase (T7 RNAP) did not detect production of glidobactin A (Wang et al., 2019). Even though the T7 promoter is stronger than other bacterial promoters, transcription of long transcripts with T7 RNAP in P. putida may lead to poor mRNA stability because T7 RNAP, unlike bacterial RNAPs, is uncoupled from translation in bacteria (Fan et al., 2017; Iost et al., 1992; Iost and Dreyfus, 1995).
In contrast to transcription, translational control is rarely investigated in articles describing heterologous BGCs in bacteria. Here, we only tested two RBS sequences guided by in silico predictions for expressing the glb cluster (Fig. 3d). A systematic analysis of RBS libraries in future studies would reveal the extent to which translational control can affect heterologous expression of BGCs (Reis and Salis, 2020). Translation initiation of internal genes in heterologous BGCs also needs to be optimized, as demonstrated by the introduction of ATG start codons into the lum and glb clusters (Fig. 4b–d). With the availability of mutagenic recombineering techniques for P. putida, it would be feasible to optimize pathway expression by mutating the 5’ untranslated regions (UTRs) for individual genes (Aparicio et al., 2020a). Indeed, this strategy has been used recently to improve production of the natural product, violacein, in E. coli (Zhang et al., 2021).
GC content is often cited as a concern when designing heterologous BGCs, but the GC content of the two glidobactin BGCs did not appear to be a major factor in heterologous production. After accounting for issues in translation initiation and MLP expression in the glb cluster, glidobactin A production in P. putida was still higher with the low-GC lum cluster (47% GC) compared to the glb cluster (70% GC) (Fig. 4c,d). For comparison, the native BGC for pyoverdine biosynthesis in P. putida is 66% GC (Ravel and Cornelis, 2003). It is worth noting that P. putida and P. luminescens are both γ-proteobacteria while S. brevitalea belongs to the class β-proteobacteria, so the phylogenetic relationship between the native host and the heterologous host may have a greater influence on heterologous production than GC content and codon usage. Furthermore, codon optimization has been shown in the literature to be detrimental to heterologous gene expression in P. putida (Incha et al., 2020). For now, there is not enough knowledge on codon optimization for P. putida and other heterologous hosts to consistently rely on it for improving PKS and NRPS expression. Glidobactin A production with the lum cluster suggests that P. putida could be an alternative host for expressing BGCs from lower GC bacteria, such as cyanobacteria, which are becoming a more prevalent source for novel natural products (Blunt et al., 2018).
The results presented here also demonstrate how P. putida’s native metabolism affects heterologous production. For instance, media that elicited a strong carbon catabolite repression (CCR) response in P. putida, such as glucose-based or rich media (Kim et al., 2013), resulted in lower production titers compared to glycerol-based media (Fig. 1d, Fig. S8a). Bacteria use CCR to alter their metabolism to maximize growth rate and consumption of a preferred carbon source, which could prevent the diversion of metabolites needed for the production of a heterologous product (Stülke and Hillen, 1999). An alternative hypothesis is that the carbons in glycerol are more reduced than those in glucose and increase the availability of NAD(P)H for biosynthesis of the acyl-ACP/acyl-CoA substrates incorporated into the heterologous products (Villadsen et al., 2011). For example, P. putida has a higher composition of PHAs when grown on glycerol compared to glucose (Eggink et al., 1992), suggesting that P. putida has higher fluxes through fatty acid metabolism during growth on glycerol. Furthermore, the absence of the cyo operon in PIG08 improved prodigiosin production on glucose to be on par with production from PIG02 on glycerol (Fig. 2c). Inactivation of the cyo operon has been shown to alleviate CCR in P. putida (Dinamarca et al., 2002; Petruschka et al., 2001), and deleting a major component of oxidative phosphorylation could heavily impact the availability of reducing equivalents (Ebert et al., 2011). Improvements in prodigiosin production observed from Δcyo strains could be attributed to a partial disruption of CCR or an increased availability of NAD(P)H.
Another benefit to using P. putida as a heterologous host is that it is amenable to metabolic engineering for improved production of targeted molecules (Banerjee et al., 2020). Deleting PHA production in P. putida demonstrated that fatty acid metabolism can affect heterologous product titers and potentially established a general chassis strain for producing natural products containing fatty acid precursors (Fig. 5d,e). Strategies for overproducing fatty acids in P. putida have recently been reported in the literature and could be applied towards metabolic engineering efforts for natural products (Guss et al., 2021; Salvachúa et al., 2020). In addition, combinatorial engineering of enzymes in glidobactin A biosynthesis enabled the targeted production of derivatives with different chain lengths for the lipid tail, which can modulate the potency of glidobactin derivatives as an anti-cancer drug (Zhong et al., 2021). P. putida would be an ideal platform for furthering this work and engineering strains to produce novel derivatives of glidobactin A and other compounds.
4. Conclusions
The wealth of synthetic biology tools available to P. putida is its primary strength as a heterologous host. We generated multiple chromosomal deletions, insertions and point mutations in P. putida using a generalized genome editing toolkit. These methods enabled rational engineering of P. putida production strains to improve heterologous titers for both products, primarily through improving transcription and translation of the heterologous BGCs and overexpressing auxiliary proteins (MLPs and PPTases). Our efforts highlight simple and rational steps to improve heterologous production that could be applied to other heterologous hosts for improving expression and activity of PKSs and NRPSs. We also identified a carbon sink negatively affecting heterologous production and discovered a regulatory change required for improving productivity of prodigiosin and glidobactin A. As knowledge of metabolism and regulatory networks in P. putida continues to improve, increasing heterologous product titers from P. putida by engineering its native metabolism will be more common. In future studies, our methodology described here could be applied towards uncharacterized BGCs or enzymes engineered to synthesize natural product derivatives, establishing P. putida as a platform for producing novel drug candidates.
5. Materials and Methods
5.1. Plasmids, bacterial strains, and growth conditions.
The bacterial strains and plasmids used in this study are shown in Supplementary Table S1 and S2. Plasmid sequences are provided as supplementary material in Genbank format. Vectors constructed for various genome editing methods are available through Addgene. All E. coli strains were grown in LB medium at 37°C. P. putida KT2440 and its derivative strains were grown in LB medium at 30°C. LB medium was supplemented with kanamycin (50 μg/mL, Kan50), gentamycin (30 μg/mL, Gent30), tetracyline (10 μg/mL, LB Tet10 for E. coli, 25 μg/mL, Tet25 for P. putida), and irgasan (25 μg/mL, Irg25) when necessary. Serratia marcescens ATCC274 and Photorhabdus luminescens subsp. laumondii TT01 were grown in LB medium at 30°C. Schlegelella brevitalea sp. nov. DSM7029 was grown at 30°C on CY-Agar (3g/L casitone, 1.4g/L CaCl2 • 2H2O, 1.0g/L yeast extract, 15g/L agar) and in CYCG medium (6g/L casitone, 1.4g/L CaCl2 • 2H2O, 2.0g/L yeast extract, 25g/L glycerol) (Tu et al., 2016). All liquid cultures were shaken at 250 rpm during incubation.
For secondary metabolite production, P. putida was grown in RK medium (Riesenberg et al., 1991). The medium was prepared by mixing 13.3g potassium phosphate monobasic (KH2PO4), 4.0g ammonium phosphate (NH4)2PO4, 1.7g citric acid, 0.1g Fe(III) ammonium citrate, and 25g glycerol or 25g D-glucose to 800mL deionized/distilled water. To this solution, 10mL of sterile 100X RK batch trace minerals and 10mL of sterile 120g/L MgSO4 • 7H2O were added. The pH was adjusted to 6.7 with 5M NaOH and the volume was adjusted to 1L. The 100X trace minerals solution was prepared by adding 0.42g EDTA, 0.125g CoCl2 • 6H2O, 0.75g MnCl2 • 4H2O, 0.06g CuCl2, 0.15g H3BO3, 0.125g Na2(MoO4) • 2H2O, and 0.65g Zn(CH3COO)2 • 2H2O (zinc acetate) to 300mL deionized/distilled water and adjusting to a final solution volume of 500mL. All media components and the final media formulation were sterilized by filtration. RK medium was supplemented with kanamycin (25 μg/mL, Kan25) when necessary. Early production experiments also used Terrific Broth, supplemented with 5 g/L glycerol or 25 g/L glycerol.
5.2. Plasmid construction.
Most plasmids described in this work were constructed using Gibson assembly as described previously (Gibson et al., 2009). The plasmids pNVLTv2-pvdIJD-Prha-pig, pNVLTv2-pvdIJD-Prha-lum, and pNVLTv2-Prha-glb were constructed using RecET direct cloning (Fu et al., 2012; Wang et al., 2016). Genomic DNA was purified from S. marcescens, P. luminescens, and S. brevitalea using phenol:chloroform:isoamyl alcohol extraction as reported previously (Cook et al., 2018; Lee et al., 2006). Pure genomic DNA was prepared from 10mL of liquid culture, and the extraction protocol was scaled up accordingly. To “release” the BGC of interest from genomic DNA, 20μg of genomic DNA was digested with up to two restriction enzymes in 400-μL reactions and then purified by ethanol precipitation. Capture vectors were linearized by restriction digest in 100-μL reactions containing 2μg of plasmid DNA and then purified by gel extraction. Restriction digest reactions were incubated at 37°C for 2 hours. To prepare DNA for construction of pNVLTv2-pvdIJD-Prha-pig, S. marcescens genomic DNA was digested with AflII and the capture vector, pNVLTv2-pvdIJD-Prha-prepig, was digested with KpnI-HF. For pNVLTv2-pvdIJD-Prha-lum, P. luminescens genomic DNA was digested with PacI and MluI and the capture vector, pNVLTv2-pvdIJD-Prha-prelum, was digested with BsaI-HFv2. For pNVLTv2-pvdIJD-Prha-glb, S. brevitalea genomic DNA was digested with DraI and AvrII, and the capture vector, pNVLTv2-pvdIJD-Prha-preglb, was digested with BsaI-HFv2.
To prepare competent cells for RecET direct cloning, 5 mL of fresh LB were inoculated with 150 μL of overnight culture of E. coli GB05-dir. Cultures were incubated at 37°C for about 2 hours, and then induced with 100μL 20% (w/v) L-arabinose. Growth continued for another 45 minutes and then the cultures were placed on ice. For every aliquot of competent cells needed, 1mL of culture was centrifuged for 30 seconds at 11,000g and washed with 1mL 10% (v/v) glycerol. The samples were washed with 1mL 10% glycerol two more times, and then resuspended in 20μL 10% glycerol. At least 500ng of linearized vector and 5μg digested genomic DNA was added to each aliquot of competent cells to a total approximate volume of 50μL. The competent cells were then added to chilled 1-mm electroporation cuvettes and electroporated with a voltage of 1.8kV. After electroporation, cells were immediately mixed with 1mL SOC media and incubated at 37°C for 90 minutes. All of the recovered cells were plated on LB Kan25 supplemented with 0.5% (w/v) D-glucose. Plates were incubated overnight at 37°C.
5.3. Genome editing in P. putida.
For the 2-step λRed/Cas9 recombineering protocol, pNVLTv2 integration vectors were introduced into P. putida containing pCas9 by tri-parental conjugation, as described previously (Choi and Schweizer, 2006). The helper strain was E. coli HB101 containing pRK600 and the donor strain was E. coli DH5α or GB2005 containing pNVLTv2. Conjugations were plated on LB Gent30, Kan50, Irg25. Conjugants were inoculated in LB Gent30, Kan50 and grown overnight at 30°C. The next day, the λRed genes on pCas9 were induced by adding 0.6% w/v L-arabinose to the cultures and incubating for another 45 minutes. To prepare electrocompetent cells, 500 μL of culture was washed twice with 1mL 10% glycerol and resuspended in 50μL 10% glycerol. About 100ng pgRNAtet was added to each sample and the samples were added to chilled 1-mm electroporation cuvettes. Samples were electroporated with a voltage of 1.8kV and allowed to recover in 1 mL LB for 3 hours at 30°C. The recovered cells were selected on LB Gent30, Tet25 plates covered with aluminum foil to limit light exposure. Some electroporations were completed by washing and resuspending cells with 300 mM sucrose instead of 10% glycerol.
For oligo recombineering, P. putida containing pCas9-beta or pCas9-beta-mmr was used to prepare electrocompetent cells as described above. These cells were transformed with ~100 ng pgRNA plasmid DNA and 1 μL of 100 μM oligo by electroporation. Cells were recovered in 1mL LB for 3 hours and selected on LB Gent30, Kan50 or Gent30, Tet25. Transformants from all methods were screened for the desired knockout using colony PCR with primers flanking the gene of interest.
5.4. Prodigiosin production and extraction.
Prodigiosin production cultures were prepared by inoculating 25mL of fresh media to OD650 = 0.05 using overnight cultures. Pre-cultures were prepared in LB media and were washed with PBS before inoculation. Production cultures were incubated at 30°C and while shaking at 250 rpm until they reached an OD650 = 0.5. Prodigiosin production was then induced by adding either 0.2% (w/v) L-rhamnose or 1mM IPTG. For samples requiring a dodecane overlay, 10mL of dodecane was added at this time. The cultures were incubated at 30°C until up to 48 hours after inoculation.
At the end of cultivation, cultures were transferred to 50-mL conical tubes of a known weight. Samples were then centrifuged for 15 minutes at 3000g. If a dodecane overlay was used, then the upper dodecane layer was aspirated and transferred to a 15-mL conical tube of a known weight. The aqueous supernatant was decanted into a fresh 50-mL conical tube of known weight. The remaining pellet was resuspended in 10mL acidified ethanol (4% (v/v) 1M HCl in ethanol). The volume and weight of aqueous supernatant and dodecane was determined for each sample. The pellets resuspended in acidified ethanol were then centrifuged for 15 minutes at 3000g. The ethanol supernatant, the aqueous supernatant, and the dodecane were then diluted in acidified ethanol until the absorbance at 535nm (A535) for each sample was within the linear range of prodigiosin quantification. The molar extinction coefficient of prodigiosin (ε535 = 139,800 M−1cm−1, as determined previously) was used to quantify the concentration of prodigiosin in the dodecane overlay, the supernatant, and the pellet for each culture (Domröse et al., 2015). The total prodigiosin production was calculated from the recorded volumes and weights of the samples. The pellets were washed with 20mL sterile water and lyophilized overnight to determine the dry cell weight for each culture.
Smaller prodigiosin cultures without a dodecane overlay were analyzed for production by adding two volumes acidified ethanol directly to the culture. The extraction was mixed well by pipetting and 1 mL was collected. The samples were vortexed for 10 minutes at 1500 rpm and then centrifuged at 17,000g for 5 minutes. The supernatant was diluted in acidified ethanol and prodigiosin was quantified by absorbance at 535 nm.
5.5. Tn5 knockout library of prodigiosin producers.
Tn5 transposition was used to create libraries of random knockouts in the genomes of P. putida strains engineered to produce prodigiosin. The mini-transposon vector, pBAM1, was introduced into these strains via bi-parental conjugation. The donor strains were E. coli S17–1 λpir containing pBAM1 or E. coli BW29427 containing pBAM1 (Martínez-García et al., 2011) Transposon libraries were created in P. putida PIG01 and P. putida PIG02. The selection media for conjugation was LB Kan50 supplemented with 0.002–0.02% (w/v) L-rhamnose or RK Kan50 agar with 25g/L glycerol. In conjugations where E. coli S17–1 λpir was the donor strain, the selection media was supplemented with Irg25. The screens were designed so that the majority of transformants would appear light pink on the selection media, and colonies of interest were screened visually for an increase in pigmentation. Candidate colonies were re-streaked on selection media to confirm their altered phenotype. The transposon location for each mutant was determined by arbitrary priming PCR, as described previously (Das et al., 2005; Martínez-García et al., 2011).
5.6. Glidobactin A production and extraction.
Strains that were engineered to produce glidobactin A were cultivated in RK medium with glycerol or glucose. Production cultures were prepared by inoculating 25mL of fresh media to OD600 = 0.05. The cultures were incubated at 30°C while shaking at 250 rpm until they reached an OD600 = 0.5. Glidobactin A production was then induced with 0.2% (w/v) L-rhamnose or 1mM IPTG. The cultures continued to grow for up to 48 hours after inoculation.
Glidobactin A was extracted by adding 1 volume butanol to whole production cultures. After briefly shaking the culture/butanol mixtures by hand, solids were removed from the sides of the flask/tube using a spatula. The samples were shaken for 1 hour at 20C, creating a butanol-water emulsion. 1 mL of the emulsion was collected from each sample immediately after swirling by hand and then transferred to centrifuge tubes. Samples were left on ice for at least 10 minutes and centrifuged for 10 minutes at 10,000g and 4C. The butanol phase was transferred to a fresh tube and diluted 1:2 in butanol. This extract was filtered with a 0.22 μm nylon membrane in preparation for HPLC and LC-MS analysis. A second extraction protocol was initially used until we observed that glidobactin A was depositing on the inside of the glassware used for production cultures (Supplementary Methods).
5.7. HPLC and LC-MS analysis of glidobactin A.
HPLC analysis was completed with a Shimadzu HPLC system (Shimadzu Co., Columbia, MD, USA) equipped with a quaternary pump, autosampler, vacuum degasser, and fluorescence detector. HPLC separations were performed with an Agilent Eclipse Plus C18 column (2.1×50mm, with guard column). The injection volume was 2μL and the flow rate was 0.4mL/min. Samples were eluted with a gradient elution; mobile phase A was H2O with 0.1% formic acid and mobile phase B was 100% acetonitrile with 0.1% formic acid. For each injection, column was equilibrated with 95% mobile phase A and 5% mobile phase B for 1 minute, then switching over to 25% mobile phase B over 2 minutes. Mobile phase B was increased to 75% over 12 minutes, and then increased to 100% over 2 minutes. The solvent was held to 100% mobile phase B over the next 3 minutes before re-equilibrating the column in 95% mobile phase A.
Samples were analyzed by LC-MS with a Vanquish UPLC coupled via electrospray ionization operating in positive mode to a Q-Exactive orbitrap high-resolution mass spectrometer (ThermoScientific). Separation was conducted on a 2.1 × 100mm Acquity UHPLC BEH C18 column with 1.7μm particle size with a flow rate of 0.2ml/min. Solvent A was 95:5 H2O-acetonitrile with 0.1% formic acid. Solvent B was acetonitrile with 0.1% formic acid. The following gradient was used: 0 to 2 min, 0% B; 2 to 14 min, linear gradient from 0 to 100% B; 14 to 16 min, 100% B; 16 to 17 min, linear gradient from 100 to 0% B; 17 to 22 min, 0% B. The mass spectrometry parameters were: full MS-SIM (single ion monitoring) scanning between 450 and 600m/z at a resolution of 140000 full width at half maximum (FWHM), automatic control gain (ACG) target of 1e6, and maximum injection time (IT) of 40ms. The MAVEN software suite was used for data analysis (Clasquin et al., 2012; Melamud et al., 2010).
Supplementary Material
Acknowledgements
This study was supported by research grants from the National Science Foundation (MCB-1716594). TBC and MVV are recipients of the NIH Biotechnology Training Program Fellowships (NIGMS 5 T32 GM08349). MVV is the recipient of a NSF Graduate Research Fellowship (DGE- 1256259). The Bruker Avance III HD 600 NMR spectrometer was supported by grant NIH S10 OD012245. The authors would like to thank Víctor de Lorenzo for providing the plasmid pSEVA2514-rec2-mutLE36KPP and Francis A. Stewart for providing the strains E. coli GB2005 and E. coli GB05-dir. The authors would also like to thank William Cordell for his assistance in designing the glidobactin extraction protocol.
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ankenbauer A, Schäfer RA, Viegas SC, Pobre V, Voß B, Arraiano CM, Takors R, 2020. Pseudomonas putida KT2440 is naturally endowed to withstand industrial-scale stress conditions. Microb. Biotechnol 13, 1145–1161. 10.1111/1751-7915.13571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio T, de Lorenzo V, Martínez-García E, 2018. CRISPR/Cas9-Based Counterselection Boosts Recombineering Efficiency in Pseudomonas putida. Biotechnol. J 13, 1700161. 10.1002/biot.201700161 [DOI] [PubMed] [Google Scholar]
- Aparicio T, Nyerges A, Martínez-García E, de Lorenzo V, 2020a. High-Efficiency Multi-site Genomic Editing of Pseudomonas putida through Thermoinducible ssDNA Recombineering. iScience 23, 100946. 10.1016/j.isci.2020.100946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio T, Nyerges A, Nagy I, Pal C, Martínez-García E, Lorenzo V, 2020b. Mismatch repair hierarchy of Pseudomonas putida revealed by mutagenic ssDNA recombineering of the pyrF gene. Environ. Microbiol 22, 45–58. 10.1111/1462-2920.14814 [DOI] [PubMed] [Google Scholar]
- Atanasov AG, Zotchev SB, Dirsch VM, Supuran CT, 2021. Natural products in drug discovery: advances and opportunities. Nat. Rev. Drug Discov 20, 200–216. 10.1038/s41573-020-00114-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee D, Eng T, Lau AK, Sasaki Y, Wang B, Chen Y, Prahl J, Singan VR, Herbert RA, Liu Y, Tanjore D, Petzold CJ, Keasling JD, Mukhopadhyay A, 2020. Genome-scale metabolic rewiring improves titers rates and yields of the non-native product indigoidine at scale. Nat. Commun 11, 5385. 10.1038/s41467-020-19171-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian X, Huang F, Wang H, Klefisch T, Müller R, Zhang Y, 2014. Heterologous Production of Glidobactins/Luminmycins in Escherichia coli Nissle Containing the Glidobactin Biosynthetic Gene Cluster from Burkholderia DSM7029. ChemBioChem 15, 2221–2224. 10.1002/cbic.201402199 [DOI] [PubMed] [Google Scholar]
- Bian X, Plaza A, Zhang Y, Müller R, 2012. Luminmycins A–C, Cryptic Natural Products from Photorhabdus luminescens Identified by Heterologous Expression in Escherichia coli. J. Nat. Prod 75, 1652–1655. 10.1021/np300444e [DOI] [PubMed] [Google Scholar]
- Bian X, Tang B, Yu Y, Tu Q, Gross F, Wang H, Li A, Fu J, Shen Y, Li Y, Stewart AF, Zhao G, Ding X, Müller R, Zhang Y, 2017. Heterologous Production and Yield Improvement of Epothilones in Burkholderiales Strain DSM 7029. ACS Chem. Biol 12, 1805–1812. 10.1021/acschembio.7b00097 [DOI] [PubMed] [Google Scholar]
- Blunt JW, Carroll AR, Copp BR, Davis RA, Keyzers RA, Prinsep MR, 2018. Marine natural products. Nat. Prod. Rep 35, 8–53. 10.1039/C7NP00052A [DOI] [PubMed] [Google Scholar]
- Chai Y, Shan S, Weissman KJ, Hu S, Zhang Y, Müller R, 2012. Heterologous Expression and Genetic Engineering of the Tubulysin Biosynthetic Gene Cluster Using Red/ET Recombineering and Inactivation Mutagenesis. Chem. Biol 19, 361–371. 10.1016/j.chembiol.2012.01.007 [DOI] [PubMed] [Google Scholar]
- Chen R, Zhang Q, Tan B, Zheng L, Li H, Zhu Y, Zhang C, 2017. Genome Mining and Activation of a Silent PKS/NRPS Gene Cluster Direct the Production of Totopotensamides. Org. Lett 19, 5697–5700. 10.1021/acs.orglett.7b02878 [DOI] [PubMed] [Google Scholar]
- Choi K-H, Schweizer HP, 2006. mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nat. Protoc 1, 153–161. 10.1038/nprot.2006.24 [DOI] [PubMed] [Google Scholar]
- Choi KR, Cho JS, Cho IJ, Park D, Lee SY, 2018. Markerless gene knockout and integration to express heterologous biosynthetic gene clusters in Pseudomonas putida. Metab. Eng 47, 463–474. 10.1016/j.ymben.2018.05.003 [DOI] [PubMed] [Google Scholar]
- Clasquin MF, Melamud E, Rabinowitz JD, 2012. LC-MS Data Processing with MAVEN: A Metabolomic Analysis and Visualization Engine, in: Current Protocols in Bioinformatics. John Wiley & Sons, Inc., Hoboken, NJ, USA, pp. 14.11.1–14.11.23. 10.1002/0471250953.bi1411s37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook TB, Pfleger BF, 2019. Leveraging synthetic biology for producing bioactive polyketides and non-ribosomal peptides in bacterial heterologous hosts. Medchemcomm 10, 668–681. 10.1039/C9MD00055K [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook TB, Rand JM, Nurani W, Courtney DK, Liu SA, Pfleger BF, 2018. Genetic tools for reliable gene expression and recombineering in Pseudomonas putida. J. Ind. Microbiol. Biotechnol 45, 517–527. 10.1007/s10295-017-2001-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Agostino PM, Gulder TAM, 2018. Direct Pathway Cloning Combined with Sequence- and Ligation-Independent Cloning for Fast Biosynthetic Gene Cluster Refactoring and Heterologous Expression. ACS Synth. Biol 7, 1702–1708. 10.1021/acssynbio.8b00151 [DOI] [PubMed] [Google Scholar]
- Danevčič T, Borić Vezjak M, Zorec M, Stopar D, 2016. Prodigiosin - A Multifaceted Escherichia coli Antimicrobial Agent. PLoS One 11, e0162412. 10.1371/journal.pone.0162412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Noe JC, Paik S, Kitten T, 2005. An improved arbitrary primed PCR method for rapid characterization of transposon insertion sites. J. Microbiol. Methods 63, 89–94. 10.1016/j.mimet.2005.02.011 [DOI] [PubMed] [Google Scholar]
- Dinamarca MA, Ruiz-manzano A, Rojo F, 2002. Inactivation of Cytochrome o Ubiquinol Oxidase Relieves Catabolic Repression of the Pseudomonas putida GPo1 Alkane Degradation Pathway. J. Bacteriol 184, 3785–3793. 10.1128/JB.184.14.3785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domröse A, Klein AS, Hage-Hülsmann J, Thies S, Svensson V, Classen T, Pietruszka J, Jaeger K-E, Drepper T, Loeschcke A, 2015. Efficient recombinant production of prodigiosin in Pseudomonas putida. Front. Microbiol 6, 972. 10.3389/fmicb.2015.00972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domröse A, Weihmann R, Thies S, Jaeger K-E, Drepper T, Loeschcke A, 2017. Rapid generation of recombinant Pseudomonas putida secondary metabolite producers using yTREX. Synth. Syst. Biotechnol 2, 310–319. 10.1016/j.synbio.2017.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudnik A, Bigler L, Dudler R, 2013. Heterologous expression of a Photorhabdus luminescens syrbactin-like gene cluster results in production of the potent proteasome inhibitor glidobactin A. Microbiol. Res 168, 73–76. 10.1016/j.micres.2012.09.006 [DOI] [PubMed] [Google Scholar]
- Ebert BE, Kurth F, Grund M, Blank LM, Schmid A, 2011. Response of Pseudomonas putida KT2440 to Increased NADH and ATP Demand. Appl. Environ. Microbiol 77, 6597–6605. 10.1128/AEM.05588-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggink G, Waard P, Huijberts GNM, 1992. The role of fatty acid biosynthesis and degradation in the supply of substrates for poly(3-hydroxyalkanoate) formation in Pseudomonas putida. FEMS Microbiol. Lett 103, 159–163. 10.1111/j.1574-6968.1992.tb05833.x [DOI] [Google Scholar]
- Elmore JR, Dexter GN, Salvachúa D, Martinez-Baird J, Hatmaker EA, Huenemann JD, Klingeman DM, Peabody GL, Peterson DJ, Singer C, Beckham GT, Guss AM, 2021. Production of itaconic acid from alkali pretreated lignin by dynamic two stage bioconversion. Nat. Commun 12, 2261. 10.1038/s41467-021-22556-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore JR, Furches A, Wolff GN, Gorday K, Guss AM, 2017. Development of a high efficiency integration system and promoter library for rapid modification of Pseudomonas putida KT2440. Metab. Eng. Commun 5, 1–8. 10.1016/j.meteno.2017.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escapa IF, del Cerro C, García JL, Prieto MA, 2013. The role of GlpR repressor in Pseudomonas putida KT2440 growth and PHA production from glycerol. Environ. Microbiol 15, 93–110. 10.1111/j.1462-2920.2012.02790.x [DOI] [PubMed] [Google Scholar]
- Fan H, Conn AB, Williams PB, Diggs S, Hahm J, Gamper HB, Hou Y-M, O’Leary SE, Wang Y, Blaha GM, 2017. Transcription–translation coupling: direct interactions of RNA polymerase with ribosomes and ribosomal subunits. Nucleic Acids Res. 45, 11043–11055. 10.1093/nar/gkx719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farasat I, Kushwaha M, Collens J, Easterbrook M, Guido M, Salis HM, 2014. Efficient search, mapping, and optimization of multi-protein genetic systems in diverse bacteria. Mol. Syst. Biol 10, 731. 10.15252/msb.20134955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Bian X, Hu S, Wang H, Huang F, Seibert PM, Plaza A, Xia L, Müller R, Stewart a F., Zhang Y, 2012. Full-length RecE enhances linear-linear homologous recombination and facilitates direct cloning for bioprospecting. Nat. Biotechnol 30, 440–446. 10.1038/nbt.2183 [DOI] [PubMed] [Google Scholar]
- Fu J, Wenzel SC, Perlova O, Wang J, Gross F, Tang Z, Yin Y, Stewart AF, Müller R, Zhang Y, 2008. Efficient transfer of two large secondary metabolite pathway gene clusters into heterologous hosts by transposition. Nucleic Acids Res. 36, e113–e113. 10.1093/nar/gkn499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaida SM, Sandoval NR, Nicolaou SA, Chen Y, Venkataramanan KP, Papoutsakis ET, 2015. Expression of heterologous sigma factors enables functional screening of metagenomic and heterologous genomic libraries. Nat. Commun 6, 7045. 10.1038/ncomms8045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson D, Young L, Chuang R, 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. … 6, 12–17. 10.1038/NMETH.1318 [DOI] [PubMed] [Google Scholar]
- Gomez-Escribano JP, Bibb MJ, 2011. Engineering Streptomyces coelicolor for heterologous expression of secondary metabolite gene clusters. Microb. Biotechnol 4, 207–215. 10.1111/j.1751-7915.2010.00219.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf N, Altenbuchner J, 2011. Development of a Method for Markerless Gene Deletion in Pseudomonas putida. Appl. Environ. Microbiol 77, 5549–5552. 10.1128/AEM.05055-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guss AM, Elmore JR, Huenemann JD, 2021. Engineered microbes for conversion of organic compounds to medium chain length alcohols and methods of use. US 2021/024960 A1. [Google Scholar]
- Han S, Lee CW, Yoon YD, Kang JS, Lee KH, Yoon WK, Kim YK, Lee K, Park S, Kim HM, 2005. Effective prevention of lethal acute graft-versus-host disease by combined immunosuppressive therapy with prodigiosin and cyclosporine A. Biochem. Pharmacol 70, 1518–1526. 10.1016/j.bcp.2005.08.017 [DOI] [PubMed] [Google Scholar]
- Harris AKP, Williamson NR, Slater H, Cox A, Abbasi S, Foulds I, Simonsen HT, Leeper FJ, Salmond GPC, 2004. The Serratia gene cluster encoding biosynthesis of the red antibiotic, prodigiosin, shows species- and strain-dependent genome context variation. Microbiology 150, 3547–3560. 10.1099/mic.0.27222-0 [DOI] [PubMed] [Google Scholar]
- Hecht A, Glasgow J, Jaschke PR, Bawazer LA, Munson MS, Cochran JR, Endy D, Salit M, 2017. Measurements of translation initiation from all 64 codons in E. coli. Nucleic Acids Res. 45, 3615–3626. 10.1093/nar/gkx070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imker HJ, Krahn D, Clerc J, Kaiser M, Walsh CT, 2010. N-Acylation during Glidobactin Biosynthesis by the Tridomain Nonribosomal Peptide Synthetase Module GlbF. Chem. Biol 17, 1077–1083. 10.1016/j.chembiol.2010.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Incha MR, Thompson MG, Blake-Hedges JM, Liu Y, Pearson AN, Schmidt M, Gin JW, Petzold CJ, Deutschbauer AM, Keasling JD, 2020. Leveraging host metabolism for bisdemethoxycurcumin production in Pseudomonas putida. Metab. Eng. Commun 10, e00119. 10.1016/j.mec.2019.e00119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iost I, Dreyfus M, 1995. The stability of Escherichia coli lacZ mRNA depends upon the simultaneity of its synthesis and translation. EMBO J. 14, 3252–3261. 10.1002/j.1460-2075.1995.tb07328.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iost I, Guillerez J, Dreyfus M, 1992. Bacteriophage T7 RNA polymerase travels far ahead of ribosomes in vivo. J. Bacteriol 174, 619–622. 10.1128/JB.174.2.619-622.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeske M, Altenbuchner J, 2010. The Escherichia coli rhamnose promoter rhaP BAD is in Pseudomonas putida KT2440 independent of Crp–cAMP activation. Appl. Microbiol. Biotechnol 85, 1923–1933. 10.1007/s00253-009-2245-8 [DOI] [PubMed] [Google Scholar]
- Keating TA, Walsh CT, 1999. Initiation, elongation, and termination strategies in polyketide and polypeptide antibiotic biosynthesis. Curr. Opin. Chem. Biol 3, 598–606. 10.1016/S1367-5931(99)00015-0 [DOI] [PubMed] [Google Scholar]
- Kim J, Oliveros JC, Nikel PI, de Lorenzo V, Silva-Rocha R, 2013. Transcriptomic fingerprinting of Pseudomonas putida under alternative physiological regimes. Environ. Microbiol. Rep 5, 883–891. 10.1111/1758-2229.12090 [DOI] [PubMed] [Google Scholar]
- Lee CL, Ow DSW, Oh SKW, 2006. Quantitative real-time polymerase chain reaction for determination of plasmid copy number in bacteria. J. Microbiol. Methods 65, 258–267. 10.1016/j.mimet.2005.07.019 [DOI] [PubMed] [Google Scholar]
- Lee SY, Wong HH, Choi J, Lee SH, Lee SC, Han CS, 2000. Production of medium-chain-length polyhydroxyalkanoates by high-cell-density cultivation ofPseudomonas putida under phosphorus limitation. Biotechnol. Bioeng 68, 466–470. [DOI] [PubMed] [Google Scholar]
- Lee T, Krupa RA, Zhang F, Hajimorad M, Holtz WJ, Prasad N, Lee S, Keasling JD, 2011. BglBrick vectors and datasheets: A synthetic biology platform for gene expression. J. Biol. Eng 5, 12. 10.1186/1754-1611-5-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Weissman KJ, Müller R, 2010. Insights into Multienzyme Docking in Hybrid PKS-NRPS Megasynthetases Revealed by Heterologous Expression and Genetic Engineering. ChemBioChem 11, 1069–1075. 10.1002/cbic.201000103 [DOI] [PubMed] [Google Scholar]
- Liu X, Hua K, Liu D, Wu Z-L, Wang Y, Zhang H, Deng Z, Pfeifer BA, Jiang M, 2020. Heterologous Biosynthesis of Type II Polyketide Products Using E. coli. ACS Chem. Biol 15, 1177–1183. 10.1021/acschembio.9b00827 [DOI] [PubMed] [Google Scholar]
- Martin-Pascual M, Batianis C, Bruinsma L, Asin-Garcia E, Garcia-Morales L, Weusthuis RA, van Kranenburg R, Martins dos Santos VAP, 2021. A navigation guide of synthetic biology tools for Pseudomonas putida. Biotechnol. Adv 49, 107732. 10.1016/j.biotechadv.2021.107732 [DOI] [PubMed] [Google Scholar]
- Martínez-García E, Calles B, Arévalo-Rodríguez M, de Lorenzo V, 2011. pBAM1: an all-synthetic genetic tool for analysis and construction of complex bacterial phenotypes. BMC Microbiol. 11, 38. 10.1186/1471-2180-11-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamud E, Vastag L, Rabinowitz JD, 2010. Metabolomic Analysis and Visualization Engine for LC-MS Data. Anal. Chem 82, 9818–9826. 10.1021/ac1021166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez-Perez D, Begemann MB, Pfleger BF, 2011. Modular Synthase-Encoding Gene Involved in α-Olefin Biosynthesis in Synechococcus sp. Strain PCC 7002. Appl. Environ. Microbiol 77, 4264–4267. 10.1128/AEM.00467-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi J, Sydow A, Schempp F, Becher D, Schewe H, Schrader J, Buchhaupt M, 2016. Investigation of plasmid-induced growth defect in Pseudomonas putida. J. Biotechnol 231, 167–173. 10.1016/j.jbiotec.2016.06.001 [DOI] [PubMed] [Google Scholar]
- Morales G, Ugidos A, Rojo F, 2006. Inactivation of the Pseudomonas putida cytochrome o ubiquinol oxidase leads to a significant change in the transcriptome and to increased expression of the CIO and cbb3–1 terminal oxidases. Environ. Microbiol 8, 1764–1774. 10.1111/j.1462-2920.2006.01061.x [DOI] [PubMed] [Google Scholar]
- Newman DJ, Cragg GM, 2012. Natural Products As Sources of New Drugs over the 30 Years from 1981 to 2010. J. Nat. Prod 75, 311–335. 10.1021/np200906s [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikel PI, de Lorenzo V, 2018. Pseudomonas putida as a functional chassis for industrial biocatalysis: From native biochemistry to trans-metabolism. Metab. Eng 50, 142–155. 10.1016/j.ymben.2018.05.005 [DOI] [PubMed] [Google Scholar]
- Nikel PI, Kim J, de Lorenzo V, 2014. Metabolic and regulatory rearrangements underlying glycerol metabolism in Pseudomonas putida KT2440. Environ. Microbiol 16, 239–254. 10.1111/1462-2920.12224 [DOI] [PubMed] [Google Scholar]
- Niu G, 2018. Genomics-Driven Natural Product Discovery in Actinomycetes. Trends Biotechnol. 36, 238–241. 10.1016/j.tibtech.2017.10.009 [DOI] [PubMed] [Google Scholar]
- Niu W, Willett H, Mueller J, He X, Kramer L, Ma B, Guo J, 2020. Direct biosynthesis of adipic acid from lignin-derived aromatics using engineered Pseudomonas putida KT2440. Metab. Eng. 59, 151–161. 10.1016/j.ymben.2020.02.006 [DOI] [PubMed] [Google Scholar]
- Oka M, Nishiyama Y, Ohta S, Kamei H, Konishi M, Miyaki T, Oki T, Kawaguchi H, 1988. Glidobactins A, B and C, new antitumor antibiotics. I. Production, isolation, chemical properties and biological activity. J. Antibiot. (Tokyo) 41, 1331–1337. 10.7164/antibiotics.41.1331 [DOI] [PubMed] [Google Scholar]
- Owen JG, Copp JN, Ackerley DF, 2011. Rapid and flexible biochemical assays for evaluating 4’-phosphopantetheinyl transferase activity. Biochem. J 436, 709–717. 10.1042/BJ20110321 [DOI] [PubMed] [Google Scholar]
- Park D, Swayambhu G, Pfeifer BA, 2020. Heterologous biosynthesis as a platform for producing new generation natural products. Curr. Opin. Biotechnol 66, 123–130. 10.1016/j.copbio.2020.06.014 [DOI] [PubMed] [Google Scholar]
- Petruschka L, Burchhardt G, Müller C, Weihe C, Herrmann H, 2001. The cyo operon of Pseudomonas putida is involved in carbon catabolite repression of phenol degradation. Mol. Genet. Genomics 266, 199–206. 10.1007/s004380100539 [DOI] [PubMed] [Google Scholar]
- Pfeifer BA, Admiraal SJ, Gramajo H, Cane DE, Khosla C, 2001. Biosynthesis of Complex Polyketides in a Metabolically Engineered Strain of E. coli. Science (80-. ) 291, 1790–1792. 10.1126/science.1058092 [DOI] [PubMed] [Google Scholar]
- Pogorevc D, Tang Y, Hoffmann M, Zipf G, Bernauer HS, Popoff A, Steinmetz H, Wenzel SC, 2019. Biosynthesis and Heterologous Production of Argyrins. ACS Synth. Biol 8, 1121–1133. 10.1021/acssynbio.9b00023 [DOI] [PubMed] [Google Scholar]
- Prieto A, Escapa IF, Martínez V, Dinjaski N, Herencias C, de la Peña F, Tarazona N, Revelles O, 2016. A holistic view of polyhydroxyalkanoate metabolism in Pseudomonas putida. Environ. Microbiol 18, 341–357. 10.1111/1462-2920.12760 [DOI] [PubMed] [Google Scholar]
- Raj K, Venayak N, Mahadevan R, 2020. Novel two-stage processes for optimal chemical production in microbes. Metab. Eng 62, 186–197. 10.1016/j.ymben.2020.08.006 [DOI] [PubMed] [Google Scholar]
- Ravel J, Cornelis P, 2003. Genomics of pyoverdine-mediated iron uptake in pseudomonads. Trends Microbiol. 11, 195–200. 10.1016/S0966-842X(03)00076-3 [DOI] [PubMed] [Google Scholar]
- Reis AC, Salis HM, 2020. An Automated Model Test System for Systematic Development and Improvement of Gene Expression Models. ACS Synth. Biol 9, 3145–3156. 10.1021/acssynbio.0c00394 [DOI] [PubMed] [Google Scholar]
- Ren H, Wang B, Zhao H, 2017. Breaking the silence: new strategies for discovering novel natural products. Curr. Opin. Biotechnol 48, 21–27. 10.1016/j.copbio.2017.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riesenberg D, Schulz V, Knorre WA, Pohl H-D, Korz D, Sanders EA, Roß A, Deckwer W-D, 1991. High cell density cultivation of Escherichia coli at controlled specific growth rate. J. Biotechnol 20, 17–27. 10.1016/0168-1656(91)90032-Q [DOI] [PubMed] [Google Scholar]
- Rutledge PJ, Challis GL, 2015. Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat. Rev. Microbiol 13, 509–523. 10.1038/nrmicro3496 [DOI] [PubMed] [Google Scholar]
- Salvachúa D, Rydzak T, Auwae R, De Capite A, Black BA, Bouvier JT, Cleveland NS, Elmore JR, Furches A, Huenemann JD, Katahira R, Michener WE, Peterson DJ, Rohrer H, Vardon DR, Beckham GT, Guss AM, 2020. Metabolic engineering of Pseudomonas putida for increased polyhydroxyalkanoate production from lignin. Microb. Biotechnol 13, 290–298. 10.1111/1751-7915.13481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellenberg B, Bigler L, Dudler R, 2007. Identification of genes involved in the biosynthesis of the cytotoxic compound glidobactin from a soil bacterium. Environ. Microbiol 9, 1640–1650. 10.1111/j.1462-2920.2007.01278.x [DOI] [PubMed] [Google Scholar]
- Schomer RA, Thomas MG, 2017. Characterization of the Functional Variance in MbtH-like Protein Interactions with a Nonribosomal Peptide Synthetase. Biochemistry 56, 5380–5390. 10.1021/acs.biochem.7b00517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein ML, Beck P, Kaiser M, Dudler R, Becker CFW, Groll M, 2012. One-shot NMR analysis of microbial secretions identifies highly potent proteasome inhibitor. Proc. Natl. Acad. Sci 109, 18367–18371. 10.1073/pnas.1211423109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stülke J, Hillen W, 1999. Carbon catabolite repression in bacteria. Curr. Opin. Microbiol 2, 195–201. 10.1016/S1369-5274(99)80034-4 [DOI] [PubMed] [Google Scholar]
- Tu Q, Herrmann J, Hu S, Raju R, Bian X, Zhang Y, Müller R, 2016. Genetic engineering and heterologous expression of the disorazol biosynthetic gene cluster via Red/ET recombineering. Sci. Rep 6, 21066. 10.1038/srep21066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanner SA, Li X, Zvanych R, Torchia J, Sang J, Andrews DW, Magarvey NA, 2013. Chemical and biosynthetic evolution of the antimycin-type depsipeptides. Mol. Biosyst 9, 2712. 10.1039/c3mb70219g [DOI] [PubMed] [Google Scholar]
- Villadsen J, Nielsen J, Lidén G, 2011. Bioreaction engineering principles. Springer Science & Business Media. [Google Scholar]
- Wang G, Zhao Z, Ke J, Engel Y, Shi Y, Robinson D, Bingol K, Zhang Z, Bowen B, Louie K, Wang B, Evans R, Miyamoto Y, Cheng K, Kosina S, De Raad M, Silva L, Luhrs A, Lubbe A, Hoyt DW, Francavilla C, Otani H, Deutsch S, Washton NM, Rubin EM, Mouncey NJ, Visel A, Northen T, Cheng J, Bode HB, Yoshikuni Y, 2019. CRAGE enables rapid activation of biosynthetic gene clusters in undomesticated bacteria. Nat. Microbiol 4, 2498–2510. 10.1038/s41564-019-0573-8 [DOI] [PubMed] [Google Scholar]
- Wang H, Li Z, Jia R, Hou Y, Yin J, Bian X, Li A, Müller R, Stewart AF, Fu J, Zhang Y, 2016. RecET direct cloning and Redαβ recombineering of biosynthetic gene clusters, large operons or single genes for heterologous expression. Nat. Protoc 11, 1175–1190. 10.1038/nprot.2016.054 [DOI] [PubMed] [Google Scholar]
- Wenzel SC, Gross F, Zhang Y, Fu J, Stewart AF, Müller R, 2005. Heterologous Expression of a Myxobacterial Natural Products Assembly Line in Pseudomonads via Red/ET Recombineering. Chem. Biol 12, 349–356. 10.1016/j.chembiol.2004.12.012 [DOI] [PubMed] [Google Scholar]
- Williamson NR, Fineran PC, Leeper FJ, Salmond GPC, 2006. The biosynthesis and regulation of bacterial prodiginines. Nat. Rev. Microbiol 4, 887–899. 10.1038/nrmicro1531 [DOI] [PubMed] [Google Scholar]
- Wu Z, Chen Z, Gao X, Li J, Shang G, 2019. Combination of ssDNA recombineering and CRISPR-Cas9 for Pseudomonas putida KT2440 genome editing. Appl. Microbiol. Biotechnol 103, 2783–2795. 10.1007/s00253-019-09654-w [DOI] [PubMed] [Google Scholar]
- Zhang J, Shen Y, Liu J, Wei D, 2005. Antimetastatic effect of prodigiosin through inhibition of tumor invasion. Biochem. Pharmacol 69, 407–414. 10.1016/j.bcp.2004.08.037 [DOI] [PubMed] [Google Scholar]
- Zhang Yuyang, Chen H, Zhang Yao, Yin H, Zhou C, Wang Y, 2021. Direct RBS Engineering of the biosynthetic gene cluster for efficient productivity of violaceins in E. coli. Microb. Cell Fact 20, 38. 10.1186/s12934-021-01518-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, Diao X, Zhang N, Li F, Zhou H, Chen H, Bai X, Ren X, Zhang Y, Wu D, Bian X, 2021. Engineering and elucidation of the lipoinitiation process in nonribosomal peptide biosynthesis. Nat. Commun 12, 296. 10.1038/s41467-020-20548-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.