

Abstract
The CLN3 gene was identified over two decades ago, but the primary function of the CLN3 protein remains unknown. Recessive inheritance of loss of function mutations in CLN3 are responsible for juvenile neuronal ceroid lipofuscinosis (Batten disease, or CLN3 disease), a fatal childhood onset neurodegenerative disease causing vision loss, seizures, progressive dementia, motor function loss and premature death. CLN3 is a multipass transmembrane protein that primarily localizes to endosomes and lysosomes. Defects in endocytosis, autophagy, and lysosomal function are common findings in CLN3-deficiency model systems. However, the molecular mechanisms underlying these defects have not yet been fully elucidated. In this mini-review, we will summarize the current understanding of the CLN3 protein interaction network and discuss how this knowledge is starting to delineate the molecular pathogenesis of CLN3 disease. Accumulating evidence strongly points towards CLN3 playing a role in regulation of the cytoskeleton and cytoskeletal associated proteins to tether cellular membranes, regulation of membrane complexes such as channels/transporters, and modulating the function of small GTPases to effectively mediate vesicular movement and membrane dynamics.
Keywords: CLN3, cytoskeleton, actin, ion channels, intracellular trafficking, endocytosis, autophagy
1. Introduction
The neuronal ceroid lipofuscinoses (NCLs, Batten disease) are a group of inherited metabolic disorders that primarily lead to childhood onset neurodegeneration, with clinical symptoms that include seizures, vision loss, progressive loss of cognitive and motor function, and premature death [1]. A main pathological feature is the progressive accumulation of autofluorescent lysosomal storage material in cells within and outside of the central nervous system (CNS) [1]. Originally subclassified by age of onset, thirteen genetic NCL subtypes have now been defined [2]; therefore each subtype is now referred to by its gene nomenclature [3]. Autosomal recessive CLN2 and CLN3 disease (originally classified as classical late-infantile and juvenile NCL, respectively) are the most common subtypes in most demographic regions [1]. While there are significant efforts to develop enzyme replacement and gene therapies for some forms of NCL [4, 5], and a CNS-directed enzyme replacement therapy has been approved for CLN2 disease [6], patient care is limited to palliative treatment in most forms of NCL.
CLN3 disease is usually first recognized with new onset of vision loss between four and ten years of age. Progressively declining cognitive and motor function, emergence of seizures, and behavioral and psychiatric disturbances are typical in the progression of CLN3 disease, and most patients succumb to the disease in their late teens or early twenties [7, 8]. Linkage mapping led to the identification of the CLN3 gene in 1995, which localizes to chromosome 16p11.2 [9]. The CLN3 protein is a multipass transmembrane protein with 6 membrane spanning domains, with the N- and C-termini oriented facing the cytosol [10, 11]. The genetics of CLN3 disease and biochemical features of the CLN3 protein have been extensively reviewed elsewhere [2, 12–16]. The purpose of this mini-review is to highlight recent advances delineating molecular pathways in which CLN3 likely functions, and we will discuss how the loss of CLN3 function in these pathways may impact cellular physiology in CLN3 disease.
2. Cellular phenotypes in CLN3-deficiency models
The identification of the CLN3 gene facilitated substantial efforts towards the creation of model systems to investigate CLN3 cell biology and the consequences of loss of CLN3 function on the molecular, cellular and whole organism levels. The application of numerous overexpression, knock-out and targeted mutation strategies to generate disease models has given rise to a diverse collection of tools for this important area of CLN3 disease research. These include lower organism models, such as yeast [17–19], Caenorhabditis elegans [20], Dictyostelium discoideum [21], Drosophila melanogaster [22], and zebrafish [23], as well as mammalian models, such as mice [24–28] and a mini pig model that is in development [29]. Reprogramming and gene editing technologies have also led to the establishment of a number of human in vitro stem cell model systems [30–36]. For a selection of comprehensive reviews covering models for CLN3 disease research, see [37–42].
The CLN3 protein has been reported to localize to numerous subcellular sites including endoplasmic reticulum (ER), Golgi, mitochondria, plasma membrane, endosomes and lysosomes [43–57]. Some of the reported localization results may have been influenced by systems that utilized overexpressed and/or fusion tags on the CLN3 protein or were influenced by non-specific antibodies, as discussed in [13, 58]. Nevertheless, an overwhelming majority of those studies support that CLN3 traffics through the secretory pathway following its synthesis, ultimately being targeted to endosomes and lysosomes possibly via the plasma membrane.
Consistent with the trafficking route of CLN3 and its primary localization to endosomes and lysosomes, a robust body of evidence has documented abnormalities in the secretory, plasma membrane and endolysosomal systems in CLN3 deficiency models. These include alterations in lysosomal pH [59–61], Ca2+ homeostasis and signaling [21, 62–65], defects in osmoregulation [66–68], changes in lipid microdomain properties [69, 70], altered endocytosis and trafficking of cell surface receptors [52, 71–74], disrupted sorting of lysosomal-targeted proteins [52, 56, 75], and defects in autophagy [76–79]. In the following sections, we will discuss how these abnormalities may be explained by an emerging picture regarding CLN3 protein interactions within its trafficking pathway.
3. Recent insights into pathomechanisms from protein-protein interaction (PPI) studies
CLN3 regulation of ion channels/transporters
Ion channels and transporters are integral membrane proteins that play roles in a wide array of biological activities, such as regulation of vascular function, cardiac function, renal function, and synaptic transmission [80]. The well-documented role that ion channel and ion transporter dysfunction plays in human diseases, such as epilepsy [81], cardiovascular disease [82, 83], kidney disease [84, 85], and neurodegenerative disease [86–89], underscores their importance in human biology.
Ion flux plays a critical role in endolysosomal regulation and function [90]. As noted above, a number of studies have demonstrated that loss of CLN3 leads to ion imbalance. This includes abnormal lysosomal pH regulation [60, 61, 91] (although normal lysosomal pH has also been reported in some CLN3 disease cell models [92]), altered Ca2+ homeostasis and signaling [21, 62–65], and altered biometals homeostasis [93]. While it has not been demonstrated that CLN3 directly functions to transport ions, several possible molecular mechanisms by which CLN3 may impact ion transport and signaling have emerged from PPI studies.
CLN3’s role in calsenilin-mediated modulation of K+ channels and Ca2+ dependent cell death
One of the first proteins shown to interact with CLN3 was calsenilin [94]. Calsenilin, also named KChIP3 (K+ Channel Interacting Protein) and DREAM, is a neuronal Ca2+ sensor that binds and regulates ion channels, including Kv4 channels, voltage dependent Ca2+ channels, and the Inositol trisphosphate 3 receptor (IP3R) (reviewed in [95]). Calsenilin also binds presenilins 1 and 2 to enhance gamma secretase activity and programmed cell death [96, 97], and in the nucleus binds downstream regulatory element (DRE) containing genes (hence the pseudonym DREAM) to repress transcription [98]. In a search for interactors of calsenilin using a classical yeast two-hybrid approach, Chang et al., pulled out a fragment of CLN3 and later validated the interaction at the endogenous level in SH-SY5Y human neuroblastoma cells, also further mapping the interaction to the C-terminus of CLN3 [94]. The interaction of CLN3 with calsenilin was Ca2+ dependent, with a reduced interaction in the presence of Ca2+. Moreover, Chang et al suggested that the interaction of CLN3 and calsenilin conferred anti-apoptotic properties to SH-SY5Y cells because in the absence of CLN3, these cells were more sensitive to Ca2+-dependent cell death mediated by calsenilin, and overexpression of CLN3 protected against Ca2+-dependent cell death [94].
More recently, Seifert et al confirmed the ability of CLN3 to exert an effect on calsenilin-mediated processes. In this 2020 study, the authors demonstrated that CLN3 could modulate calsenilin/KChIP3-mediated Kv4.2 function [99]. In HEK293 cells, overexpression of CLN3 reduced the amount of calsenilin/KChIP3 bound to the Kv4.2 channel. Furthermore, in a co-expression paradigm where co-expression of calsenilin/KChIP3 and Kv4.2 in HEK293 cells modulates Kv4.2 current properties, the addition of CLN3 co-expression dampened the effect of calsenilin/KChIP3 on the Kv4.2 current properties. Seifert et al. speculated that CLN3, by virtue of its binding interaction with calsenilin/KChIP3, interferes with KChIP3 association with Kv4.2 channels to negatively regulate their function [99]. Furthermore, this effect was Ca2+ dependent, whereby the KChIP3-Kv4.2 interaction and KChIP3-induced effects on Kv4.2 channel properties were stabilized by the presence of Ca2+. The demonstration that a disease-associated missense variant and a C-terminal deletion version of CLN3 had reduced or no effect on KChIP3-mediated modulation of Kv4.2 suggested this modulation of Kv4.2 channels may have some disease relevance. However, an important caveat of these studies was that Seifert et al did not observe a direct interaction between CLN3 and calsenilin/KChIP3 [99], in contrast to the findings of Chang et al. This discrepancy was most likely due to the use of differently tagged versions of the overexpressed proteins, as discussed by Seifert et al in their report [99]. Most notably Seifert et al used GFP-tagged CLN3 in their co-immunoprecipitation analysis, while Chang et al used untagged CLN3 [94]. It is well documented that tags can sometimes interfere with efficient CLN3 folding and trafficking [16, 46].
Taken together the findings from Chang et al and Seifert et al suggest that CLN3 functions as a direct modulator of Ca2+-dependent calsenilin function in both ion channel regulation and Ca2+-dependent cell death (Figure 1). In addition to the need for further delineation of the precise molecular mechanisms by which CLN3 modulates calsenilin-mediated function, many important questions remain to be answered surrounding these key discoveries in the study of CLN3 protein function: Does CLN3 also modulate other calsenilin-mediated processes, such as presenilin/γ-secretase cleavage of amyloid precursor protein (APP) and nuclear translocation and transcriptional repression? What is the role of the calsenilin activities that CLN3 modulates in the CLN3 neurodegenerative disease process? Notably, overexpression of wildtype CLN3 (but not disease-associated mutant CLN3) has been reported to interfere with gamma-secretase mediated cleavage of amyloid precursor protein (APP) in HEK293 cells, which is a presenilin-dependent process [100, 101]. Notch2-associated changes have also been reported in the cerebellum of Cln3-deficient mice [102], and Notch signaling was inhibited in a CLN3 overexpression Drosophila model [22]; presenilin is a key regulator of notch signaling [103, 104]. These observations indicate investigation of a potential role for CLN3 in calsenilin-mediated presenilin function is warranted.
Figure 1. Hypothetical model depicting CLN3-regulation of calsenilin function.
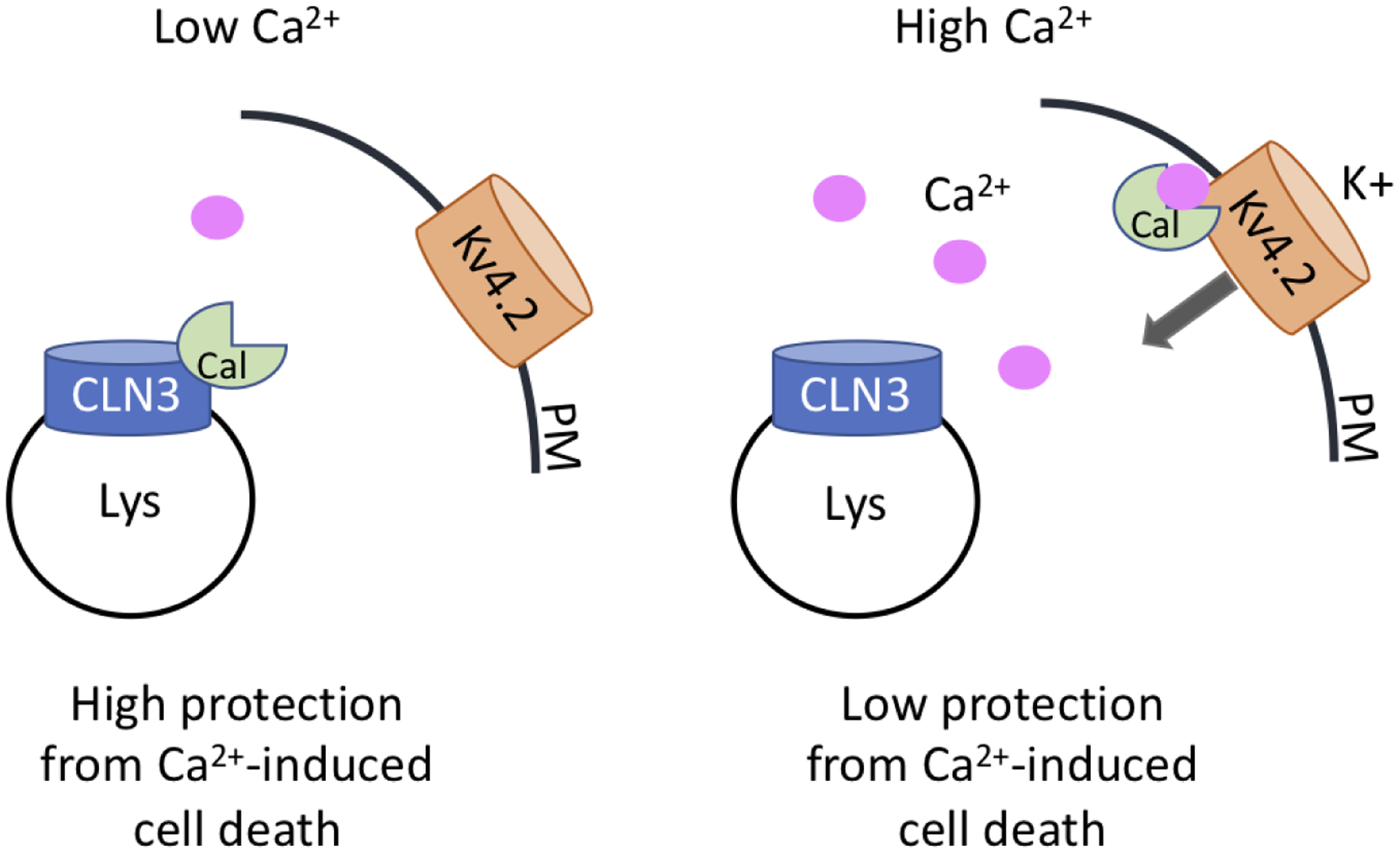
Under low Ca2+ conditions, CLN3 can bind calsenilin which is protective from Ca2+-induced cell death and calsenilin regulation of Kv4.2. In high Ca2+ conditions, calsenilin binds Ca2+ and dissociates from CLN3, where it is then able to interact with other proteins like Kv4.2 to influence their trafficking and/or function. A pool of CLN3 localized at the plasma membrane (PM) (not shown) may alternatively be involved in Kv4.2 channel regulation. Cal=calsenilin, Lys=lysosome
CLN3 association with plasma membrane Na,K-ATPase and the plasma-membrane associated cytoskeletal protein, β-fodrin
In another yeast 2-hybrid screening approach, using an N-terminus CLN3 bait, an interaction with the plasma membrane-associated Na,K-ATPase (both the ß1 and ß2 isoforms) was uncovered [105]. In this study, co-immunoprecipitation of overexpressed full length CLN3 validated the novel yeast 2-hybrid interaction. Interestingly, the cytoskeletal protein ß-fodrin (or ß-II-spectrin) and GRP78/BiP also co-immunoprecipitated with the CLN3-Na,K-ATPase complex [105]; both ß-fodrin and GRP78 are previously known interactors of Na,K-ATPase, and ß-fodrin was also identified as a candidate interactor in the CLN3 screen [105]. Na,K-ATPase ion transport function is important for maintaining membrane potential and for regulation of Ca2+ homeostasis. A function beyond ion transport has also been more recently established; Na,K-ATPase also plays a key role as a cellular signaling receptor in a number of important signaling pathways including Src and MEK signal transduction pathways [106, 107].
Uusi-Rauva and colleagues examined the plasmalemmal pumping activity of the Na,K-ATPase in CLN3-deficient primary mouse neurons and saw no appreciable difference compared to wildtype primary mouse neurons [105]. However, a difference in the relative amount of plasma membrane associated Na,K-ATPase and in its rate of endocytosis following ouabain treatment (which blocks the activity of the Na,K-ATPase pump) significantly differed in the wild-type versus CLN3-deficient primary mouse neurons. As a result, the authors speculated that CLN3 may modulate endocytosis of Na,K-ATPase through regulation of the fodrin cytoskeleton, a process that may also involve Grp78/BiP. Consistent with this hypothesis was an altered fodrin cytoskeleton in Cln3 knockout mouse brain and in CLN3 patient fibroblasts [105]. Grp78/BiP, a well-known chaperone of the endoplasmic reticulum (ER)[108, 109], has also been found to be associated with the plasma membrane [110], and a potential role for Grp78/BiP in oubain-induced endocytosis of the Na,K-ATPase has been suggested [111].
Role of CLN3 in regulating the actin cytoskeleton
In addition to the interaction of CLN3 with ß-fodrin already discussed here, CLN3 has also been reported to interact with the cytoskeletal associated protein myosin-II-b [112]. Using a modified yeast 2-hybrid approach (CytoTrap yeast 2-hybrid) and a C-terminal CLN3 bait, Getty et al identified an interaction with myosin-II-b that was verified with myc-tagged overexpressed CLN3 in NIH/3T3 fibroblasts [112]. Myosin-II-b is one of three different isoforms of non-muscle myosin heavy chain, which is an important regulator of the actin cytoskeleton, functioning in cell polarity, cell migration, synaptogenesis and synaptic plasticity (reviewed in [113]); myosin-II-b is the main non-muscle myosin II isoform in neuronal synapses [114].
A functional interaction of CLN3 and myosin-II-b was further supported by observations in Cln3 knock-out mouse embryonic fibroblasts (MEFs), where wound healing was significantly compromised compared to wild-type MEFs, similar to the effect seen when wild-type MEFs were treated with the non-muscle myosin inhibitor, blebbistatin [112]. Moreover, blebbistatin treatment did not alter the wound healing in the Cln3 knock-out MEFs. Interestingly, the authors further analyzed the effect of chloroquine treatment on wound healing given CLN3 is primarily an endolysosomal protein. Disruption of lysosomal function by chloroquine significantly delayed cell migration following scratch wounding of the monolayer of wild-type MEFs, while it completely ablated cell migration following scratch wounding of the monolayer of Cln3 knock-out MEFs, indicating that lysosomal activity and CLN3 were required for proper MEF cell migration in the wound healing assay [112].
Thus, CLN3 may serve as a tether between the endolysosomal membrane and the actin cytoskeleton by binding interaction with ß-fodrin and myosin-II-b [105, 112]. Notably, disruption of the actin cytoskeleton and defects in cell migration in CLN3 deficiency models have also been reported by other groups, lending additional support to this possible role for CLN3 [65, 72].
Owing to its involvement in synaptic function and plasticity, non-muscle myosin II plays a role in a wide range of neurologic disorders including neurodevelopmental disorders and neurodegenerative disease (reviewed in [113, 115]). Non-muscle myosin II is also expressed in glia where it plays a role in neuroinflammation and myelination (reviewed in [115]). Hence, the signaling pathways that modulate non-muscle myosin II function are under investigation for their potential as therapeutic targets for neurologic disease, including PAK and ROCK inhibitors (reviewed in [113, 116–118]. Notably, in a proteomics analysis of presynaptic-enriched synaptosomes isolated from Cln3 knock-out mouse brain, the “Signaling by Rho family GTPases” pathway in IPA analysis was significantly upregulated as compared to wild-type control synaptosomes, which included the ROCK2 protein [119]. Hypothesizing this may reflect synaptic degeneration, the authors analyzed whether ROCK2 inhibition by RNAi knockdown or pharmacological inhibition using fasudil would alter CLN3-induced neurodegeneration in a Drosophila eye model. Crossing CLN3-overexpressing flies, which display a small eye phenotype due to degeneration of photoreceptors [22], with two independent ROCK2 ortholog RNAi fly lines significantly rescued the CLN3-induced small eye degenerative phenotype [119]. Treatment with ROCK inhibitor, fasudil, was less effective but also significantly dampened the CLN3 overexpression induced degeneration [119]. While further studies are required to determine the potential for targeting ROCK signaling in CLN3 disease, these findings nevertheless provide additional support for a CLN3 role in mediating actin cytoskeleton dynamics.
Role of CLN3 in endocytosis
Several studies have linked CLN3 to various forms of endocytosis [52, 69, 71, 72]. Endocytosis is an important cellular process that can internalize molecules, down-regulate cell surface proteins and modulate cell growth and movement [120, 121]. Three major subtypes of endocytosis have been identified; clathrin-mediated endocytosis (CME), clathrin-independent endocytosis (CIE), and macropinocytosis (fluid-phase endocytosis). CME is the most well studied, and as the name implies, requires the formation of clathrin-coated vesicles at the plasma membrane (PM) [122, 123]. CIE is mediated by a variety of mechanism including caveolae [124]. The formation of caveolae requires lipids such as cholesterol and sphingolipids, along with proteins called caveolins [125]. In cells lacking CLN3, caveolae dependent endocytosis was defective [69]. This was most likely due to the lack of proper sorting of caveolin from the Golgi to the PM, highlighting a potential role of CLN3 at the Golgi.
Although actin is required for all forms of endocytosis, it plays a particularly important role in macropinocytosis (Reviewed in [126]). In this form of endocytosis, actin polymerization results in the formation of membrane ruffles which form large vacuoles, leading to the internalization of extracellular fluids and molecules [127]. CDC42, a member of the Rho family of small GTPases, is a regulator of actin polymerization, and plays crucial roles in both CIE and macropinocytosis [128, 129]. During macropinocytosis, GTP-loaded CDCD42 is recruited to specific regions of the PM via an interaction with phosphatidylinositol (3,4,5)-trisphosphate (PI(3,4,5)P3), which drives actin polymerization and the formation of filopodia near the forming macropinocytosis invagination [130]. In order for CDC42 to cycle to a GDP bound form, the GAP ARHGAP21 (also known as ARHGAP10) is recruited to the site. This recruitment is mediated by the small GTPase Arf1 [131]. It is important to point out that CDC42 must cycle between GDP and GTP bound states in order to function. Although depletion of ARHGAP21 results in more GTP-loaded CDC42 with increased actin polymerization, it also leads to reduced macropinocytosis [131]. CLN3 deficient murine brain microvascular endothelial cells (MBEC) had defects in macropinocytosis. This was due to an elevated amount of GTP-loaded CDC42 [72]. Further work demonstrated that the increase in GTP-loaded CDC42 was due to a decrease in PM localized ARHGAP21. Furthermore, it was demonstrated that ARF1 GTP loading was reduced in CLN3 deficient MBEC cells [72]. As Arf1 is required to recruit ARHGAP21 to the PM, this explains the observed increase in GTP loaded CDC42. How CLN3 regulates Arf1 GTP loading is yet to be determined. One possibility is that CLN3 is required to recruit the GEF responsible for Arf1 activation at the PM, although the identity of that protein remains to be elucidated. Alternatively, CLN3 may act as a scaffold, regulating the spatiotemporal interaction of Arf1 recruited ARHGAP21 with CDC42.
Role of CLN3 in regulating Rab GTPases
Small GTPases are small molecular switches that regulate various cellular functions. Five families of small GTPase are expressed in mammalian cells; Ras, Rho, Rab, Arf and Ran (Reviewed in [132]). Each family is associated with specific cellular functions including cell growth, differentiation and survival (Ras), cytoskeleton organization and vesicular trafficking (Rho), regulation of vesicular trafficking (Rab and Arf), and control of nucleocytoplasmic transport (Ran). Small GTPases function by interacting with downstream effectors proteins. A key regulation of effector protein binding is accomplished by turning on the small GTPase by binding to GTP. Activation is modulated by members of the guanine nucleotide exchange factor (GEF), which loads GTP onto the small GTPase. In the majority of cases, activated small GTPases are membrane bound. GTP loaded small GTPases can then bind an effector protein to mediate function. On the other hand, GTPase-activating proteins (GAPs) can facilitate the hydrolization of GTP to GDP, which turns the small GTPase off, and returns it to the cytosol (Reviewed in [133]).
At the trans-Golgi Network (TGN), the lysosomal sorting receptors (LSRs) are responsible for the sorting and trafficking of soluble lysosomal resident proteins (cargo) such as cathepsin D and prosaposin to lysosomes [134, 135]. This is accomplished via the interaction of the cytosolic tail of the receptor with adaptor protein-1 (AP-1), which subsequently binds clathrin (Figure 2). The spatiotemporal recruitment of AP-1 to the membranes of the TGN is regulated by the small GTPase Arf1, which both stabilizes and opens AP-1 for membrane binding and interaction with the LSR. The monomeric clathrin adaptors, the Golgi-localized, gamma adaptin ear-containing, ARF-binding proteins (GGAs), have also been implicated in this process. CLN3 has been implicated in membrane trafficking, but the molecular details have not been well studied [19, 52, 56]. In cells depleted of CLN3, export from the TGN of CI-MPR was significantly delayed, resulting in defective lysosomal enzyme function [56]. Further supporting a role for CLN3 in sorting soluble lysosomal cargo proteins, an unbiased screen using a proteomics approach demonstrated reduced levels of several soluble lysosomal proteins in cells lacking functional CLN3 [92]. The mechanism behind the delayed exit of CI-MPR from the TGN has not been elucidated. CLN3 can interact with Rab1a, a small GTPase localized to the Golgi apparatus [136]. Rab1a is implicated in the recruitment of Golgi-specific brefeldin A-resistance guanine nucleotide exchange factor 1 (GBF1), a GEF for Arf1 [137]. The recruitment of GBF1 leads to the recruitment of the GGA family of adaptors [138]. Alternatively, through a GEF cascade with Brefeldin A-inhibited guanine nucleotide-exchange protein 1 (BIG1), the recruitment of GFB1 can also recruit AP-1. Further investigation will be required to determine the molecular role of CLN3 in sorting at the TGN.
Figure 2. Schematic representation of cellular pathways modulated by CLN3.
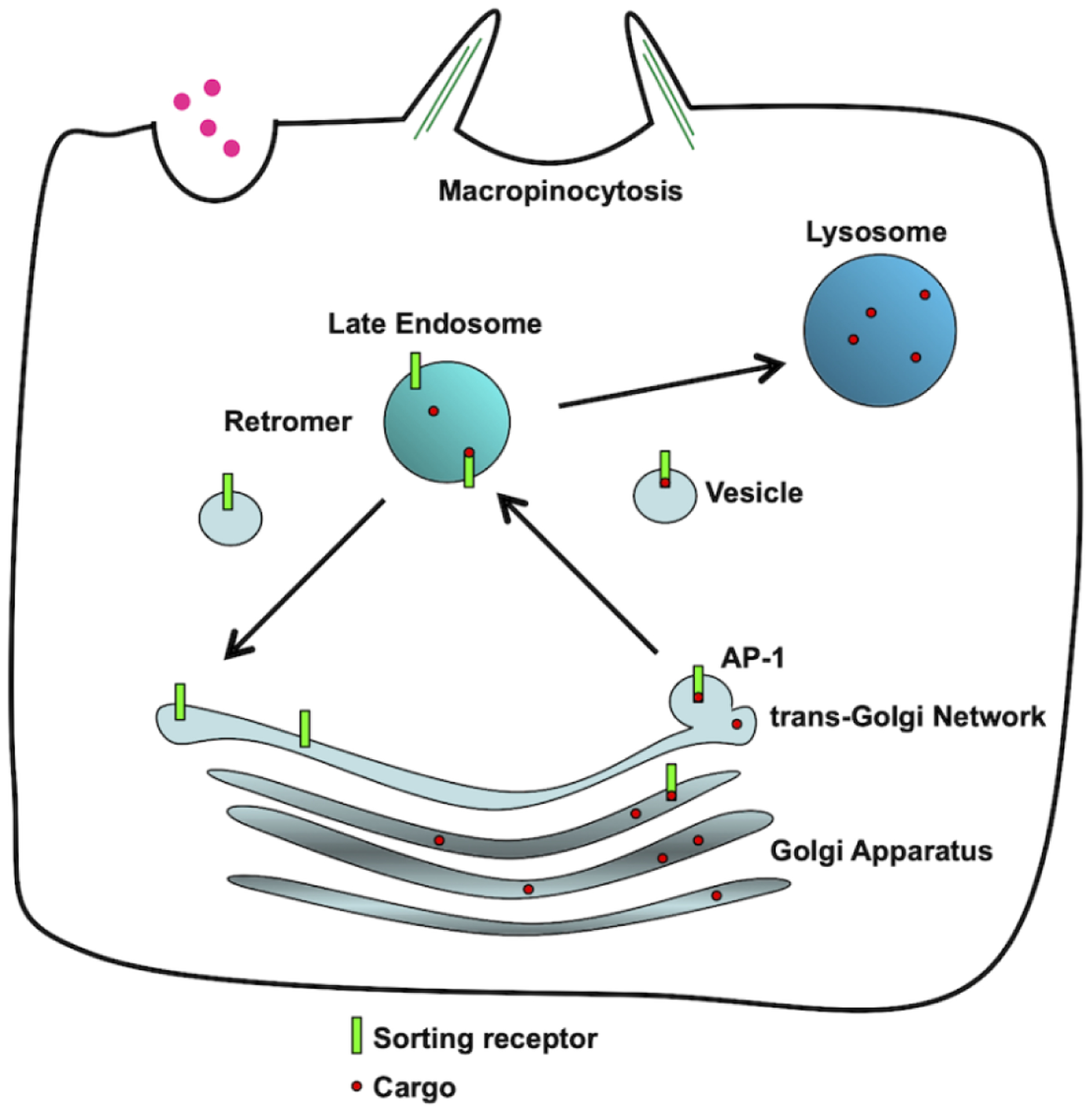
CLN3 can regulate Golgi export by potentially regulating AP-1 recruitment, receptor retrieval from late endosomes with retromer, and macropinocytosis by regulating the actin cytoskeleton.
Of the small GTPase family, the Rab family comprises the largest number, with 60 encoded in the mammalian genome (Reviewed in [139]). Rab GTPases play key roles in the formation, movement and tethering of trafficking vesicles at various cellular locations. CLN3 was shown to interact with Rab7A, a small GTPase localized to late endosomes [136, 140]. Rab7A performs many key functions including regulating endosome-to-Trans Golgi network (TGN) trafficking, the degradation of endocytic cargo and the fusion of endolysosomes with autophagosomes [141–144]. These various functions require that Rab7A interact with various effector proteins. Modulation of effector binding is at least partially regulated by the GEF Mon1/Ccz1, which is thought to activate Rab7A resulting in the membrane recruitment of Rab7A to late endosomes where it can interact with an effector protein [145, 146]. Termination of effector binding is mediated by the GAPs TBC1D2 [147], TBC1D5 [143, 148] and TBC1D15 [149, 150], which are able to shut off Rab7A, returning it to the cytosol.
Retromer is a cytosolic trimeric protein complex composed of Vacuolar protein sorting (VPS) 26, VPS29 and VPS35 [151, 152]. Dysfunction of retromer has been implicated in a variety of neurodegenerative diseases including Alzheimer’s disease [153], Parkinson’s disease [73, 154] and Amyotrophic lateral sclerosis (ALS) [155]. More recent work has shown that retromer dysfunction also occurs in various forms of NCL, including CLN3 and CLN5 [136, 156]. Retromer is recruited to late endosomes, where it interacts with the cytosolic tail of CI-MPR and sortilin [151, 157] (Figure 3). This interaction is required to sort the LSRs out of the late endosome, and return them to the TGN [151, 152]. In cells lacking functional retromer, the LSRs remain trapped in late endosomes and are eventually degraded in lysosomes. GTP-loaded Rab7A is required to recruit retromer to late endosomes, a step necessary for efficient retromer binding to the LSRs [142, 143]. In HeLa cells lacking CLN3, the Rab7A/retromer and retromer/sortilin interactions are significantly weakened [136]. This is not due to lack of activation of Rab7A, as it is still membrane bound, suggesting that its GTP loading is occurring. CLN3 appears to function as a scaffold protein, ensuring efficient interactions between Rab7A with retromer, and retromer with sortilin [136]. It is noteworthy to point out that other NCL proteins, including CLN6, CLN7 and CLN8, are also required to efficiently sort soluble lysosomal cargo proteins, suggesting a possible common disease mechanism across multiple forms of NCL [158–160].
Figure 3. Retrieval of the lysosomal sorting receptors requires CLN3.
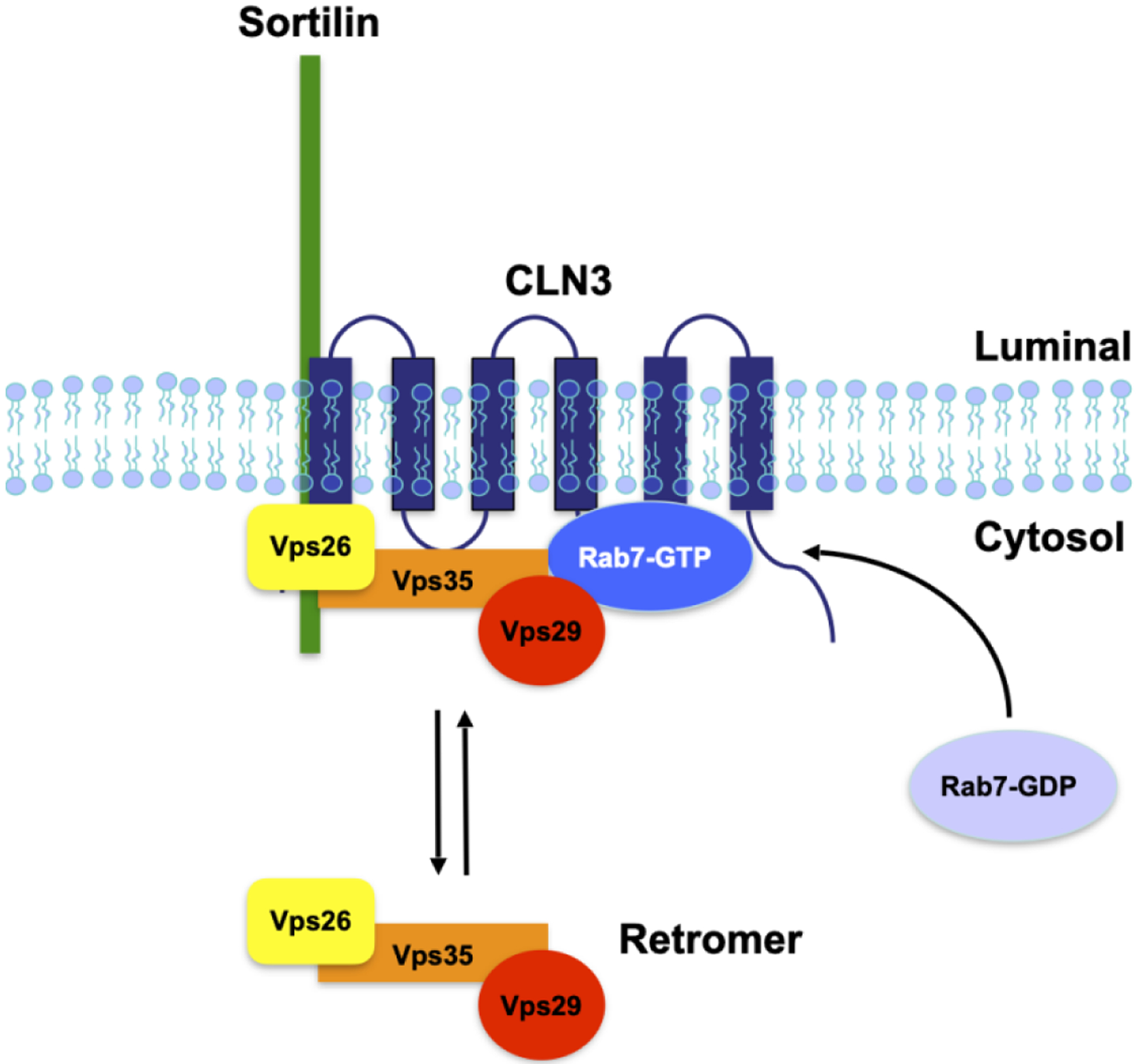
CLN3 coordinates the efficient interactions between Rab7, retromer and the lysosomal sorting receptor, sortilin at late endosomes. In cells lacking functional CLN3, these interactions are deficient, resulting in the lysosomal degradation of the receptors
Autophagy is known to be dysfunctional in various CLN3 model systems [76, 78, 79]. Lack of autophagosome/lysosome fusion has previously been observed in CLN3-deficient mouse cerebellar cells [63]. The lack of autophagosome/lysosome fusion could be due to dysfunctional fusion machinery, or due to lack of movement of organelles favouring fusion. Rab7A can regulate both these processes by interacting with different effector proteins. Pleckstrin homology domain-containing family M member 1 (PLEKHM1) is a Rab7A effector that functions as a tethering factor, mediating the fusion of late endosomes with lysosomes and autophagosomes with lysosomes [161]. Rab-interacting lysosomal protein (RILP) is also a Rab7A effector and plays a role in both fusion and minus end transport of lysosomes and autophagosomes [162–164]. In cells lacking CLN3, the Rab7A/PLEKHM1 interaction is disrupted, resulting is significant delays in EGFR degradation. However, the Rab7A/RILP interaction was not affected [136]. Although dysfunction in retromer activity resulting in defective lysosomal function could affect autophagy, lack of lysosome/autophagosome fusion in CLN3-deficient cells, due to decreased PLEKHM1 function, could as well.
The functional regulation of small GTPases by CLN3 appears to be emerging as a significant role for this integral membrane protein. It will be interesting to determine if CLN3 regulates other small GTPases implicated in other cellular pathways. Does CLN3 function mainly as a scaffold protein, or does it regulate signal via other mechanisms? Further work will be required to determine its roles.
CLN3 in lysosomal positioning
In various models of CLN3 disease, the localization of lysosomes within cells is affected. In HeLa cells overexpressing CLN3 bearing a disease-causing mutation, CLN3E295K, lysosomes were more compact, and localized towards the perinuclear region. This suggested a role for CLN3 in moving lysosomes in a plus end direction, towards the periphery [140]. In order to determine how CLN3 could be implicated in organelle positioning, a co-immunoprecipitation strategy was used. CLN3 was found to interact with RILP, the dynactin subunit p150Glued, dynein and the kinesin-2 subunit KIF3A [140]. The first three proteins form a complex required for minus-end transport (towards the peri nuclear space) of lysosomes and autophagosomes, while kinesin-2 has been shown to function in plus-end transport (transport towards the periphery) of lysosomes [162–165]. On the other hand, mouse cerebellar cells harbouring the most common disease causing mutation, a ~1-kb deletion (Cln3Δex7/8 cerebellar cells), lysosomes were more dispersed to the cell periphery [52], a finding consistent with the observation that lysosomes were more distal from the cell bodies in CLN3-deficient iPSC-derived neurons from CLN3 patients [166]. How can these seemingly opposing results be explained? In the HeLa cell experiments, wild-type and mutant CLN3 was expressed in cells containing endogenous CLN3. On the other hand, the mouse cerebellar cells and CLN3-patient iPSC-derived neurons lack wild-type CLN3 and could only express mutant CLN3 from the endogenous locus, and most likely at much reduced levels due to non-sense mediated decay of the mutant Cln3/CLN3 mRNA [167]. Additionally, the experiments involved systems with different CLN3 mutations. It is possible that the mutations affect interactions with motor proteins differently. As the plus and minus end motors often co-purify with lysosomes together, the different CLN3 mutations could affect these interactions differently[168]. Finally, the observed differences could be due to the nature of the cells themselves. Organelle position is much more crucial and regulated in neurons compared to other cell types.
4. Molecular discoveries shed light on possible impacts on differing cell types
Function of CLN3 in cells of the CNS: Glial Cells
The CNS contains four types of glial cells, microglia, astrocytes, oligodendrocytes and progenitor cells. These cells were once thought to function as a glue, keeping the brain together, but have been shown to play major roles in the CNS and have been implicated in a variety of diseases.
Microglia are the resident immune cells of the brain and perform several functions including cytokine release and phagocytosis [169]. Microglia dysfunction has been associated with neurodegenerative diseases including Alzheimer’s disease, and it has been reported that microglia can clear Aβ fragments via macropinocytosis [170], Microglia dysfunction has also been reported in rare lysosomal diseases such as Niemann-Pick type C and CLN3 disease [171, 172]. How could defects in CLN3 function affect these microglia processes? As these cells play important roles in phagocytosis and macropinocytosis, defects in CLN3 function could severely affect these processes. Although phagocytosis in microglia has not been shown to be affected in models of CLN3 disease, this process is affected in other cell types such as retinal pigment epithelium cells [36]. Neuroinflammation occurs in several neurodegenerative diseases, including CLN3 disease [173]. Microglia play an important role in mediating inflammation through many mechanisms, although the most studied is the NLRP3 inflammasome complex. This complex senses external and internal stimuli, which leads to the activation of caspase-1, resulting in the activation of pro-inflammatory cytokines and rapid cell death [174]. This complex can be degraded via autophagy, which enables cells to down regulate inflammation. Indeed, following activation, components of the inflammasome are ubiquitinated and interact with p62 [174]. It has been shown that microglia isolated from CLN3-deficient mice are more primed towards inflammasome activation and caspase-1 activation, compared to wild-type microglia [171]. Since lysosomes in CLN3-deficient cells are dysfunctional affecting autophagy, inflammation associated with CLN3 disease could be due to prolonged inflammasome signaling. In CLN3 disease, two possible functions of microglia could be impaired, resulting in neuroinflammation.
Astrocytes are the most abundant glia cells in the CNS and perform a variety of functions including ion homeostasis [175]. Along with microglia, astrocytes play an important role in maintaining neuronal health, and their dysfunction has been associated with lysosomal diseases [176]. Astrocytosis and morphological changes in astrocytic end feet have been reported in CLN3-deficient mouse brain [177–179]. A recent study also found significant impairments in the function of astrocytes derived from the brain of CLN3-deficient mice. In this study, the authors demonstrated that these astrocytes had defects in their actin cytoskeleton, and impairments in their calcium signaling abilities, roles associated with CLN3 function [65]. An additional study also reported abnormalities in calcium signaling and metabolism in astrocytes isolated from CLN3-deficient mice [64]. Furthermore, using a co-culture system of mouse-derived astrocytes and cortical neurons, Parviainen et al showed that astrocytes derived from CLN3-deficient mice had a negative impact on the survival and shape of neurons from both wild-type and CLN3-deficient mice [65]. This points to the role of glia, and particularly astrocytes, in the neurodegeneration observed in CLN3 disease. From a molecular view, the role of CLN3 in modelling the actin cytoskeleton and its role in modulating ion channels and lysosomal targeting could explain the phenotypes observed in the mouse studies.
Neurons
Neurons are polarized cells that contain a cell body (soma), dendrites and an axon. In certain cases, the axon can extend up to 1 meter in length. Each region contains a subset of organelles. The sorting of organelles into these distinct regions is highly regulated, as is the transport of organelles in axons. For example, lysosomes are positioned throughout the soma and along axons. However, those lysosomes are not all the same. Lysosomes in the soma are more acidic, contain more degradative enzymes, and have the ability to degrade material much more that more peripheral lysosomes [180]. CLN3 could play a role in this polarized sorting by favouring minus or plus end transport of lysosomes in neurons. Consistent with this hypothesis, overexpressed CLN3 was more abundantly associated with retrograde moving, Rab7-positive vesicles (versus anterograde moving vesicles) in primary hippocampal mouse neurons [57]. As already discussed previously, CLN3 binds to both plus end and minus end motors, and in mouse cerebellar cells harbouring a disease-causing mutation of CLN3, and in iPSC-derived neurons from CLN3 patients, lysosomal positioning was affected [52, 166]. In neurons, autophagosome formation occurs in the distal axons, which are then transported in a minus-end direction in order to fuse to lysosomes [181]. Rab7A has been implicated in the movement of endolysosomes in both directions and is required for autophagosome/lysosome fusion [161, 182]. Combined with potential defects in lysosomal positioning, and autophagosome/lysosome fusion, and the need to move proteins and organelles over greater distances, neurons could be more affected by CLN3 mutations than other cell types. CLN3 has also been shown to localize to vesicles in neuronal synaptic regions, where it may play a role in regulation of synaptic activity [183], consistent with a growing number of reports documenting alterations in neurodevelopment and/or neurotransmission in CLN3 disease models [34, 119, 184–188]. Thus, CLN3-deficient neurons may be especially vulnerable to dysfunctioning lysosomal and synaptic pathways, which, combined with functional defects in glial cells, may explain the prominent impact CLN3 disease has on nervous system function.
Function of CLN3 in cells outside of the CNS
Although CLN3 disease primarily affects nervous system function, the CLN3 gene is ubiquitously expressed, and indeed lysosomal storage pathology is found in many cell types, including those found in peripheral organ systems [1, 9]. In fact, an impact on cardiac function has more recently been recognized as a clinically significant component of CLN3 disease, particularly in later disease stages [189, 190]. Intriguingly, relatively high levels of CLN3 expression (compared to brain) are seen in the muscle, kidney and digestive system in multiple organisms [9, 20, 57, 66].
Epithelial cells are of particular interest for understanding CLN3 function, as a number of studies have shown abnormalities in epithelial cells in various organs due to CLN3 deficiency. These include the retinal pigment epithelium, principal cells of the kidney, and clear cells of the epidydimis of the male reproductive tract [36, 66, 191]. An important functional role for CLN3 in epithelial cells, which, like neurons, are a polarized cell type, would be consistent with CLN3’s role in regulation of phagocytosis and endocytosis as well as in osmoregulation, membrane organization and regulation of the actin cytoskeleton, all of which may make epithelial cells vulnerable to CLN3 deficiency. Consistent with this notion, it has recently been suggested that a reduction in phagocytosis of photoreceptor outer segments by the retinal pigment epithelium may significantly contribute to the vision loss in CLN3 disease [36].
Vascular endothelial cells have also been shown to express high levels of CLN3 in mouse studies [27, 69]. In the brain, these cells are a central component of the blood-brain-barrier, which is compromised in Cln3 loss of function mouse models [69, 72, 192]. As discussed previously in this review, MBEC cells lacking CLN3 displayed defects in micropinocytosis, as well as defects in membrane microdomain fluidity and an abnormal response to osmotic stress, which were at least partly attributed to misregulated Arf1-CDC42 signaling and actin-driven processes [69, 72].
Further work to better understand the impact of CLN3 deficiency in cells outside of the CNS, together with ongoing studies aimed at establishing a more comprehensive CLN3 disease natural history will contribute important knowledge towards a broader understanding of CLN3 function and CLN3 disease pathogenesis.
5. Conclusions
Through a number of protein-protein interaction (PPIs) studies using different techniques (summarized in Table 1), the interactome of CLN3 has provided clues to its function. Emerging themes of CLN3 function include the regulation of cytoskeletal/cytoskeletal associated proteins (actin cytoskeleton and dynein motor-RILP) to tether cellular membranes, regulation of membrane complexes such as channels/transporters, and modulating the function of small GTPases to effectively mediate vesicular movement and membrane dynamics (such as fission, fusion, chemotaxis/cell migration). As technology develops, we have access to novel tools to learn more about the function of CLN3. For instance, proximity labelling techniques such as BioID (proximity-dependent biotin identification) and APEX (engineered ascorbate peroxidase) have been developed that have several advantages over traditional methods including detection of interactions with proteins expressed in their native environments, in live cells, and detection of transient interactions [193]. By engineering the tag into the endogenous locus using CRISPR/Cas9 technology, CLN3 interactomes using these methods could detect PPIs without over expressing CLN3, and with CLN3 localized to the membrane. This could result in new interacting partners missed in previous unbiased screens and identify new roles for CLN3. Further studies focusing on other interacting partners already identified, or to be identified, could shed further light on CLN3 function. A more robust understanding of CLN3, its interacting partners and function should lead to the identification of novel therapeutic targets, and possible treatments for children affected by CLN3 disease.
Table 1. Summary of CLN3 interactions.
The table shows known interacting partners of CLN3, and the method that demonstrated the interaction. For information on yeast 2-hybrid, please see [195]. Co-immunoprecipitation (co-IP). Bioluminescence resonance energy transfer (BRET) is a method to detect protein-protein interactions in live cells, in real time and with proteins expressed in their native environment. For more information, please refer to [196]. Several additional reports of hits in CLN3 PPI screens or of fragment binding interactions are not included in the table, given the limited validation done on those hits (see references [71, 197, 198]).
| Interacting Protein | Method | Reference |
|---|---|---|
| Calsenilin | Yeast 2-Hybrid, co-IP | Chang et al., 2007 [94] |
| Na,K-ATPase | Yeast 2-Hybrid, co-IP | Uusi-Rauva et al., 2008 [105] |
| β-Fodrin | Yeast 2-Hybrid, co-IP | Uusi-Rauva et al., 2008 [105] |
| GRP78/BiP | co-IP | Uusi-Rauva et al., 2008 [105] |
| myosin-II-b | Cytotrap Yeast 2-Hybrid, co-IP | Getty et al., 2011 [112] |
| SBDS | Cytotrap Yeast 2-Hybrid, co-IP | Vitiello et al., 2010 [194] |
| Rab1A | BRET | Yasa et al., 2020 [136] |
| Vps26A | BRET, co-IP | Yasa et al., 2020 [136] |
| Rab7A | co-IP, GST pulldown BRET, co-IP |
Uusi-Rauva et al., 2012 [140] Yasa et al., 2020 [136] |
| sortilin | BRET, co-IP | Yasa et al., 2020 [136] |
| RILP | co-IP | Uusi-Rauva et al., 2012 [140] |
| p150Glued (Dynactin subunit) | co-IP | Uusi-Rauva et al., 2012 [140] |
| Dynein | co-IP | Uusi-Rauva et al., 2012 [140] |
| KIF3A (Kinesin-2 subunit) | co-IP | Uusi-Rauva et al., 2012 [140] |
SBDS=Shwachman-Bodian-Diamond syndrome protein
Highlights.
Autosomal recessive mutations in CLN3 lead to CLN3 disease
CLN3 disease is a fatal neurodegenerative disease that primarily affects children
CLN3 is a membrane protein localized to the secretory and endolysosomal pathways
Loss of CLN3 function leads to defective autophagy and lysosomal function
CLN3 is implicated in regulating the cytoskeleton, ion channels, and trafficking
Acknowledgements
SL acknowledges funding from the Joint Programme in Neurodegenerative Diseases (Neuronode), the Canadian Institutes for Health Research (ENG-155186), and the Canadian Foundation for Innovation (35258). SC acknowledges funding from the National Institutes of Neurological Disorders and Stroke (R56NS113891), NCL Stiftung, and from Dr. Sandra Nusinoff Lehrman and Stephen Lehrman.
Funding source declaration:
Work included in this mini-review was funded by the Joint Programme in Neurodegenerative Diseases (Neuronode), the Canadian Institutes for Health Research (ENG-155186), and the Canadian Foundation for Innovation (35258) to SL. SC receives funding from the National Institutes of Neurological Disorders and Stroke (R56NS113891).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none
References
- [1].Mole SE, Williams RE, Goebel HH, The Neuronal Ceroid Lipofuscinoses (Batten Disease), Second Edition ed., Oxford University Press, Oxford, 2011. [Google Scholar]
- [2].Mole SE, Cotman SL, Genetics of the neuronal ceroid lipofuscinoses (Batten disease), Biochim Biophys Acta 1852(10 Pt B) (2015) 2237–41. 10.1016/j.bbadis.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Williams RE, Mole SE, New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses, Neurology 79(2) (2012) 183–91. https://doi.org/79/2/183 [pii] 10.1212/WNL.0b013e31825f0547. [DOI] [PubMed] [Google Scholar]
- [4].Johnson TB, Cain JT, White KA, Ramirez-Montealegre D, Pearce DA, Weimer JM, Therapeutic landscape for Batten disease: current treatments and future prospects, Nat Rev Neurol 15(3) (2019) 161–178. 10.1038/s41582-019-0138-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kohlschutter A, Schulz A, Bartsch U, Storch S, Current and Emerging Treatment Strategies for Neuronal Ceroid Lipofuscinoses, CNS Drugs 33(4) (2019) 315–325. 10.1007/s40263-019-00620-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Schulz A, Ajayi T, Specchio N, de Los Reyes E, Gissen P, Ballon D, Dyke JP, Cahan H, Slasor P, Jacoby D, Kohlschutter A, Group CLNS, Study of Intraventricular Cerliponase Alfa for CLN2 Disease, N Engl J Med 378(20) (2018) 1898–1907. 10.1056/NEJMoa1712649. [DOI] [PubMed] [Google Scholar]
- [7].Marshall FJ, de Blieck EA, Mink JW, Dure L, Adams H, Messing S, Rothberg PG, Levy E, McDonough T, DeYoung J, Wang M, Ramirez-Montealegre D, Kwon JM, Pearce DA, A clinical rating scale for Batten disease: reliable and relevant for clinical trials, Neurology 65(2) (2005) 275–9. [DOI] [PubMed] [Google Scholar]
- [8].Ostergaard JR, Juvenile neuronal ceroid lipofuscinosis (Batten disease): current insights, Degener Neurol Neuromuscul Dis 6 (2016) 73–83. 10.2147/DNND.S111967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].International Batten Disease Consortium, Isolation of a novel gene underlying Batten disease, CLN3. The International Batten Disease Consortium, Cell 82(6) (1995) 949–57. [DOI] [PubMed] [Google Scholar]
- [10].Nugent T, Mole SE, Jones DT, The transmembrane topology of Batten disease protein CLN3 determined by consensus computational prediction constrained by experimental data, FEBS Letters 582(7) (2008) 1019–24. https://doi.org/S0014-5793(08)00164-6 [pii] 10.1016/j.febslet.2008.02.049. [DOI] [PubMed] [Google Scholar]
- [11].Ratajczak E, Petcherski A, Ramos-Moreno J, Ruonala MO, FRET-assisted determination of CLN3 membrane topology, PLoS ONE 9(7) (2014) e102593. 10.1371/journal.pone.0102593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kousi M, Lehesjoki AE, Mole SE, Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses, Human Mutation 33(1) (2012) 42–63. 10.1002/humu.21624. [DOI] [PubMed] [Google Scholar]
- [13].Phillips SN, Benedict JW, Weimer JM, Pearce DA, CLN3, the protein associated with Batten disease: structure, function and localization, Journal of Neuroscience Research 79 (2005) 573–583. [DOI] [PubMed] [Google Scholar]
- [14].Carcel-Trullols J, Kovacs AD, Pearce DA, Cell biology of the NCL proteins: What they do and don’t do, Biochim Biophys Acta 1852(10 Pt B) (2015) 2242–55. 10.1016/j.bbadis.2015.04.027. [DOI] [PubMed] [Google Scholar]
- [15].Cotman SL, Staropoli JF, The juvenile Batten disease protein, CLN3, and its role in regulating anterograde and retrograde post-Golgi trafficking, Clin Lipidol 7(1) (2012) 79–91. 10.2217/clp.11.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mirza M, Vainshtein A, DiRonza A, Chandrachud U, Haslett LJ, Palmieri M, Storch S, Groh J, Dobzinski N, Napolitano G, Schmidtke C, Kerkovich DM, The CLN3 gene and protein: What we know, Mol Genet Genomic Med 7(12) (2019) e859. 10.1002/mgg3.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Pearce DA, Sherman F, BTN1, a yeast gene corresponding to the human gene responsible for Batten’s disease, is not essential for viability, mitochondrial function, or degradation of mitochondrial ATP synthase, Yeast 13(8) (1997) 691–7. [DOI] [PubMed] [Google Scholar]
- [18].Pearce DA, Sherman F, A yeast model for the study of Batten disease, Proceedings of the National Academy of Sciences of the United States of America 95(12) (1998) 6915–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Codlin S, Haines RL, Mole SE, btn1 affects endocytosis, polarization of sterol-rich membrane domains and polarized growth in Schizosaccharomyces pombe, Traffic 9(6) (2008) 936–50. https://doi.org/TRA735 [pii] 10.1111/j.1600-0854.2008.00735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].de Voer G, van der Bent P, Rodrigues AJ, van Ommen GJ, Peters DJ, Taschner PE, Deletion of the Caenorhabditis elegans homologues of the CLN3 gene, involved in human juvenile neuronal ceroid lipofuscinosis, causes a mild progeric phenotype, J Inherit Metab Dis 28(6) (2005) 1065–80. [DOI] [PubMed] [Google Scholar]
- [21].Huber RJ, Myre MA, Cotman SL, Loss of Cln3 function in the social amoeba Dictyostelium discoideum causes pleiotropic effects that are rescued by human CLN3, PLoS ONE 9(10) (2014) e110544. 10.1371/journal.pone.0110544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tuxworth RI, Vivancos V, O’Hare MB, Tear G, Interactions between the juvenile Batten disease gene, CLN3, and the Notch and JNK signalling pathways, Human molecular genetics 18(4) (2009) 667–78. https://doi.org/ddn396 [pii] 10.1093/hmg/ddn396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wager K, Zdebik AA, Fu S, Cooper JD, Harvey RJ, Russell C, Neurodegeneration and Epilepsy in a Zebrafish Model of CLN3 Disease (Batten Disease), PLoS ONE 11(6) (2016) e0157365. 10.1371/journal.pone.0157365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mitchison HM, Bernard DJ, Greene ND, Cooper JD, Junaid MA, Pullarkat RK, de Vos N, Breuning MH, Owens JW, Mobley WC, Gardiner RM, Lake BD, Taschner PE, Nussbaum RL, Targeted disruption of the Cln3 gene provides a mouse model for Batten disease. The Batten Mouse Model Consortium [corrected], Neurobiol Dis 6(5) (1999) 321–34. 10.1006/nbdi.1999.0267S096999619990267X [pii]. [DOI] [PubMed] [Google Scholar]
- [25].Katz ML, Johnson GS, Mouse gene knockout models for the CLN2 and CLN3 forms of ceroid lipofuscinosis, European Journal of Paediatric Neurology 5Suppl A (2001) 109–14. [DOI] [PubMed] [Google Scholar]
- [26].Cotman SL, Vrbanac V, Lebel LA, Lee RL, Johnson KA, Donahue LR, Teed AM, Antonellis K, Bronson RT, Lerner TJ, MacDonald ME, Cln3(Deltaex7/8) knock-in mice with the common JNCL mutation exhibit progressive neurologic disease that begins before birth, Hum Mol Genet 11(22) (2002) 2709–21. [DOI] [PubMed] [Google Scholar]
- [27].Eliason SL, Stein CS, Mao Q, Tecedor L, Ding SL, Gaines DM, Davidson BL, A knock-in reporter model of Batten disease, J Neurosci 27(37) (2007) 9826–34. https://doi.org/27/37/9826 [pii] 10.1523/JNEUROSCI.1710-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Langin L, Johnson TB, Kovacs AD, Pearce DA, Weimer JM, A tailored Cln3(Q352X) mouse model for testing therapeutic interventions in CLN3 Batten disease, Sci Rep 10(1) (2020) 10591. 10.1038/s41598-020-67478-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Johnson TB, Sturdevant DA, White KA, Drack AV, Bhattarai S, Rogers C, Cooper JD, Pearce DA, Weimer JM, Characterization of a novel porcine model of CLN3-Batten disease, Molecular Genetics & Metabolism 126(2) (2019) S81. [Google Scholar]
- [30].Lojewski X, Staropoli JF, Biswas-Legrand S, Simas AM, Haliw L, Selig MK, Coppel SH, Goss KA, Petcherski A, Chandrachud U, Sheridan SD, Lucente D, Sims KB, Gusella JF, Sondhi D, Crystal RG, Reinhardt P, Sterneckert J, Scholer H, Haggarty SJ, Storch A, Hermann A, Cotman SL, Human iPSC models of neuronal ceroid lipofuscinosis capture distinct effects of TPP1 and CLN3 mutations on the endocytic pathway, Human molecular genetics 23(8) (2014) 2005–22. https://doi.org/ddt596 [pii] 10.1093/hmg/ddt596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhang X, Zhang D, Chen SC, Lamey T, Thompson JA, McLaren T, De Roach JN, Chen FK, McLenachan S, Generation of an induced pluripotent stem cell line from a patient with non-syndromic CLN3-associated retinal degeneration and a coisogenic control line, Stem Cell Res 29 (2018) 245–249. 10.1016/j.scr.2018.04.014. [DOI] [PubMed] [Google Scholar]
- [32].Wiley LA, Burnight ER, Drack AV, Banach BB, Ochoa D, Cranston CM, Madumba RA, East JS, Mullins RF, Stone EM, Tucker BA, Using Patient-Specific Induced Pluripotent Stem Cells and Wild-Type Mice to Develop a Gene Augmentation-Based Strategy to Treat CLN3-Associated Retinal Degeneration, Hum Gene Ther 27(10) (2016) 835–846. 10.1089/hum.2016.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Burnight ER, Bohrer LR, Giacalone JC, Klaahsen DL, Daggett HT, East JS, Madumba RA, Worthington KS, Mullins RF, Stone EM, Tucker BA, Wiley LA, CRISPR-Cas9-Mediated Correction of the 1.02 kb Common Deletion in CLN3 in Induced Pluripotent Stem Cells from Patients with Batten Disease, CRISPR J 1 (2018) 75–87. 10.1089/crispr.2017.0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gomez-Giro G, Arias-Fuenzalida J, Jarazo J, Zeuschner D, Ali M, Possemis N, Bolognin S, Halder R, Jager C, Kuper WFE, van Hasselt PM, Zaehres H, Del Sol A, van der Putten H, Scholer HR, Schwamborn JC, Synapse alterations precede neuronal damage and storage pathology in a human cerebral organoid model of CLN3-juvenile neuronal ceroid lipofuscinosis, Acta Neuropathol Commun 7(1) (2019) 222. 10.1186/s40478-019-0871-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kinarivala N, Morsy A, Patel R, Carmona AV, Sajib MS, Raut S, Mikelis CM, Al-Ahmad A, Trippier PC, An iPSC-Derived Neuron Model of CLN3 Disease Facilitates Small Molecule Phenotypic Screening, ACS Pharmacol Transl Sci 3(5) (2020) 931–947. 10.1021/acsptsci.0c00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Tang C, Han J, Dalvi S, Manian K, Winschel L, Volland S, Soto CA, Galloway CA, Spencer W, Roll M, Milliner C, Bonilha VL, Johnson TB, Latchney L, Weimer JM, Augustine EF, Mink JW, Gullapalli VK, Chung M, Williams DS, Singh R, A human model of Batten disease shows role of CLN3 in phagocytosis at the photoreceptor-RPE interface, Commun Biol 4(1) (2021) 161. 10.1038/s42003-021-01682-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Cooper JD, Russell C, Mitchison HM, Progress towards understanding disease mechanisms in small vertebrate models of neuronal ceroid lipofuscinosis, Biochim Biophys Acta 1762(10) (2006) 873–89. [DOI] [PubMed] [Google Scholar]
- [38].Shacka JJ, Mouse models of neuronal ceroid lipofuscinoses: useful pre-clinical tools to delineate disease pathophysiology and validate therapeutics, Brain Res Bull 88(1) (2012) 43–57. 10.1016/j.brainresbull.2012.03.003. [DOI] [PubMed] [Google Scholar]
- [39].Bond M, Holthaus SM, Tammen I, Tear G, Russell C, Use of model organisms for the study of neuronal ceroid lipofuscinosis, Biochim Biophys Acta (2013). https://doi.org/S0925-4439(13)00013-6 [pii] 10.1016/j.bbadis.2013.01.009. [DOI] [PubMed] [Google Scholar]
- [40].Faller KM, Gutierrez-Quintana R, Mohammed A, Rahim AA, Tuxworth RI, Wager K, Bond M, The neuronal ceroid lipofuscinoses: Opportunities from model systems, Biochim Biophys Acta 1852(10 Pt B) (2015) 2267–78. 10.1016/j.bbadis.2015.04.022. [DOI] [PubMed] [Google Scholar]
- [41].Huber RJ, Hughes SM, Liu W, Morgan A, Tuxworth RI, Russell C, The contribution of multicellular model organisms to neuronal ceroid lipofuscinosis research, Biochim Biophys Acta Mol Basis Dis 1866(9) (2020) 165614. 10.1016/j.bbadis.2019.165614. [DOI] [PubMed] [Google Scholar]
- [42].Minnis CJ, Thornton CD, FitzPatrick LM, McKay TR, Cellular models of Batten disease, Biochim Biophys Acta Mol Basis Dis 1866(9) (2020) 165559. 10.1016/j.bbadis.2019.165559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Katz ML, Gao CL, Prabhakaram M, Shibuya H, Liu PC, Johnson GS, Immunochemical localization of the Batten disease (CLN3) protein in retina, Investigative Ophthalmology & Visual Science 38(11) (1997) 2375–86. [PubMed] [Google Scholar]
- [44].Jarvela I, Sainio M, Rantamaki T, Olkkonen VM, Carpen O, Peltonen L, Jalanko A, Biosynthesis and intracellular targeting of the CLN3 protein defective in Batten disease, Human molecular genetics 7(1) (1998) 85–90. [DOI] [PubMed] [Google Scholar]
- [45].Jarvela I, Lehtovirta M, Tikkanen R, Kyttala A, Jalanko A, Defective intracellular transport of CLN3 is the molecular basis of Batten disease (JNCL). [erratum appears in Hum Mol Genet 1999 Aug;8(8):1585], Human molecular genetics 8(6) (1999) 1091–8. [DOI] [PubMed] [Google Scholar]
- [46].Haskell RE, Derksen TA, Davidson BL, Intracellular trafficking of the JNCL protein CLN3, Molecular Genetics & Metabolism 66(4) (1999) 253–60. [DOI] [PubMed] [Google Scholar]
- [47].Golabek AA, Kaczmarski W, Kida E, Kaczmarski A, Michalewski MP, Wisniewski KE, Expression studies of CLN3 protein (battenin) in fusion with the green fluorescent protein in mammalian cells in vitro, Molecular Genetics & Metabolism 66(4) (1999) 277–82. [DOI] [PubMed] [Google Scholar]
- [48].Vesa J, Peltonen L, Mutated genes in juvenile and variant late infantile neuronal ceroid lipofuscinoses encode lysosomal proteins, Current Molecular Medicine 2(5) (2002) 439–44. [DOI] [PubMed] [Google Scholar]
- [49].Ezaki J, Takeda-Ezaki M, Koike M, Ohsawa Y, Taka H, Mineki R, Murayama K, Uchiyama Y, Ueno T, Kominami E, Characterization of Cln3p, the gene product responsible for juvenile neuronal ceroid lipofuscinosis, as a lysosomal integral membrane glycoprotein, Journal of neurochemistry 87(5) (2003) 1296–308. [DOI] [PubMed] [Google Scholar]
- [50].Mao Q, Xia H, Davidson BL, Intracellular trafficking of CLN3, the protein underlying the childhood neurodegenerative disease, Batten disease, FEBS Letters 555(2) (2003) 351–7. [DOI] [PubMed] [Google Scholar]
- [51].Storch S, Pohl S, Braulke T, A dileucine motif and a cluster of acidic amino acids in the second cytoplasmic domain of the batten disease-related CLN3 protein are required for efficient lysosomal targeting, Journal of Biological Chemistry 279(51) (2004) 53625–34. https://doi.org/M410930200 [pii] 10.1074/jbc.M410930200. [DOI] [PubMed] [Google Scholar]
- [52].Fossale E, Wolf P, Espinola JA, Lubicz-Nawrocka T, Teed AM, Gao H, Rigamonti D, Cattaneo E, MacDonald ME, Cotman SL, Membrane trafficking and mitochondrial abnormalities precede subunit c deposition in a cerebellar cell model of juvenile neuronal ceroid lipofuscinosis, BMC Neurosci 5 (2004) 57. https://doi.org/1471-2202-5-57 [pii] 10.1186/1471-2202-5-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kyttala A, Ihrke G, Vesa J, Schell MJ, Luzio JP, Two motifs target Batten disease protein CLN3 to lysosomes in transfectedd nonneuronal and neuronal cells, Molecular Biology of the Cell 15 (2004) 1313–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kyttala A, Yliannala K, Schu P, Jalanko A, Luzio JP, AP-1 and AP-3 facilitate lysosomal targeting of Batten disease protein CLN3 via its dileucine motif, Journal of Biological Chemistry 280(11) (2005) 10277–83. https://doi.org/M411862200 [pii] 10.1074/jbc.M411862200. [DOI] [PubMed] [Google Scholar]
- [55].Kitzmuller C, Haines RL, Codlin S, Cutler DF, Mole SE, A function retained by the common mutant CLN3 protein is responsible for the late onset of juvenile neuronal ceroid lipofuscinosis, Human molecular genetics 17(2) (2008) 303–12. https://doi.org/ddm306 [pii] 10.1093/hmg/ddm306. [DOI] [PubMed] [Google Scholar]
- [56].Metcalf DJ, Calvi AA, Seaman M, Mitchison HM, Cutler DF, Loss of the Batten disease gene CLN3 prevents exit from the TGN of the mannose 6-phosphate receptor, Traffic 9(11) (2008) 1905–14. https://doi.org/TRA807 [pii] 10.1111/j.1600-0854.2008.00807.x. [DOI] [PubMed] [Google Scholar]
- [57].Oetjen S, Kuhl D, Hermey G, Revisiting the neuronal localization and trafficking of CLN3 in juvenile neuronal ceroid lipofuscinosis, J Neurochem 139(3) (2016) 456–470. 10.1111/jnc.13744. [DOI] [PubMed] [Google Scholar]
- [58].Pearce DA, Localization and processing of CLN3, the protein associated to Batten disease: where is it and what does it do?, Journal of Neuroscience Research 59(1) (2000) 19–23. [PubMed] [Google Scholar]
- [59].Pearce DA, Nosel SA, Sherman F, Studies of pH regulation by Btn1p, the yeast homolog of human Cln3p, Molecular Genetics & Metabolism 66(4) (1999) 320–3. [DOI] [PubMed] [Google Scholar]
- [60].Gachet Y, Codlin S, Hyams JS, Mole SE, btn1, the Schizosaccharomyces pombe homologue of the human Batten disease gene CLN3, regulates vacuole homeostasis, Journal of Cell Science 118(Pt 23) (2005) 5525–36. https://doi.org/jcs.02656 [pii] 10.1242/jcs.02656. [DOI] [PubMed] [Google Scholar]
- [61].Holopainen JM, Saarikoski J, Kinnunen PK, Jarvela I, Elevated lysosomal pH in neuronal ceroid lipofuscinoses (NCLs), European Journal of Biochemistry 268(22) (2001) 5851–6. [DOI] [PubMed] [Google Scholar]
- [62].An Haack K, Narayan SB, Li H, Warnock A, Tan L, Bennett MJ, Screening for calcium channel modulators in CLN3 siRNA knock down SH-SY5Y neuroblastoma cells reveals a significant decrease of intracellular calcium levels by selected L-type calcium channel blockers, Biochim Biophys Acta 1810(2) (2011) 186–91. https://doi.org/S0304-4165(10)00220-5 [pii] 10.1016/j.bbagen.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Chandrachud U, Walker MW, Simas AM, Heetveld S, Petcherski A, Klein M, Oh H, Wolf P, Zhao WN, Norton S, Haggarty SJ, Lloyd-Evans E, Cotman SL, Unbiased Cell-based Screening in a Neuronal Cell Model of Batten Disease Highlights an Interaction between Ca2+ Homeostasis, Autophagy, and CLN3 Protein Function, J Biol Chem 290(23) (2015) 14361–80. 10.1074/jbc.M114.621706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Bosch ME, Kielian T, Astrocytes in juvenile neuronal ceroid lipofuscinosis (CLN3) display metabolic and calcium signaling abnormalities, J Neurochem 148(5) (2019) 612–624. 10.1111/jnc.14545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Parviainen L, Dihanich S, Anderson GW, Wong AM, Brooks HR, Abeti R, Rezaie P, Lalli G, Pope S, Heales SJ, Mitchison HM, Williams BP, Cooper JD, Glial cells are functionally impaired in juvenile neuronal ceroid lipofuscinosis and detrimental to neurons, Acta Neuropathol Commun 5(1) (2017) 74. 10.1186/s40478-017-0476-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Stein CS, Yancey PH, Martins I, Sigmund RD, Stokes JB, Davidson BL, Osmoregulation of ceroid neuronal lipofuscinosis type 3 in the renal medulla, Am J Physiol Cell Physiol 298(6) (2010) C1388–400. 10.1152/ajpcell.00272.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Getty A, Kovacs AD, Lengyel-Nelson T, Cardillo A, Hof C, Chan CH, Pearce DA, Osmotic stress changes the expression and subcellular localization of the Batten disease protein CLN3, PLoS ONE 8(6) (2013) e66203. 10.1371/journal.pone.0066203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Mathavarajah S, McLaren MD, Huber RJ, Cln3 function is linked to osmoregulation in a Dictyostelium model of Batten disease, Biochim Biophys Acta Mol Basis Dis 1864(11) (2018) 3559–3573. 10.1016/j.bbadis.2018.08.013. [DOI] [PubMed] [Google Scholar]
- [69].Tecedor L, Stein CS, Schultz ML, Farwanah H, Sandhoff K, Davidson BL, CLN3 loss disturbs membrane microdomain properties and protein transport in brain endothelial cells, J Neurosci 33(46) (2013) 18065–79. https://doi.org/33/46/18065 [pii] 10.1523/JNEUROSCI.0498-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Rusyn E, Mousallem T, Persaud-Sawin DA, Miller S, Boustany RM, CLN3p impacts galactosylceramide transport, raft morphology, and lipid content, Pediatric Research 63(6) (2008) 625–31. 10.1203/PDR.0b013e31816fdc17. [DOI] [PubMed] [Google Scholar]
- [71].Luiro K, Yliannala K, Ahtiainen L, Maunu H, Jarvela I, Kyttala A, Jalanko A, Interconnections of CLN3, Hook1 and Rab proteins link Batten disease to defects in the endocytic pathway, Human molecular genetics 13(23) (2004) 3017–27. [DOI] [PubMed] [Google Scholar]
- [72].Schultz ML, Tecedor L, Stein CS, Stamnes MA, Davidson BL, CLN3 deficient cells display defects in the ARF1-Cdc42 pathway and actin-dependent events, PLoS ONE 9(5) (2014) e96647. 10.1371/journal.pone.0096647 PONE-D-14–09456 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Vilarino-Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, Soto-Ortolaza AI, Cobb SA, Wilhoite GJ, Bacon JA, Behrouz B, Melrose HL, Hentati E, Puschmann A, Evans DM, Conibear E, Wasserman WW, Aasly JO, Burkhard PR, Djaldetti R, Ghika J, Hentati F, Krygowska-Wajs A, Lynch T, Melamed E, Rajput A, Rajput AH, Solida A, Wu RM, Uitti RJ, Wszolek ZK, Vingerhoets F, Farrer MJ, VPS35 mutations in Parkinson disease, Am J Hum Genet 89(1) (2011) 162–7. 10.1016/j.ajhg.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Kovacs AD, Hof C, Pearce DA, Abnormally increased surface expression of AMPA receptors in the cerebellum, cortex and striatum of Cln3(−/−) mice, Neurosci Lett 607 (2015) 29–34. 10.1016/j.neulet.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Codlin S, Mole SE, pombe S btn1, the orthologue of the Batten disease gene CLN3, is required for vacuole protein sorting of Cpy1p and Golgi exit of Vps10p, J Cell Sci 122(Pt 8) (2009) 1163–73. https://doi.org/jcs.038323 [pii] 10.1242/jcs.038323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Cao Y, Espinola JA, Fossale E, Massey AC, Cuervo AM, MacDonald ME, Cotman SL, Autophagy is disrupted in a knock-in mouse model of juvenile neuronal ceroid lipofuscinosis, J Biol Chem 281(29) (2006) 20483–93. https://doi.org/M602180200 [pii] 10.1074/jbc.M602180200. [DOI] [PubMed] [Google Scholar]
- [77].Chang JW, Choi H, Cotman SL, Jung YK, Lithium rescues the impaired autophagy process in CbCln3(Deltaex7/8/Deltaex7/8) cerebellar cells and reduces neuronal vulnerability to cell death via IMPase inhibition, J Neurochem 116(4) (2011) 659–668. 10.1111/j.1471-4159.2010.07158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Vidal-Donet JM, Carcel-Trullols J, Casanova B, Aguado C, Knecht E, Alterations in ROS activity and lysosomal pH account for distinct patterns of macroautophagy in LINCL and JNCL fibroblasts, PLoS ONE 8(2) (2013) e55526. 10.1371/journal.pone.0055526 PONE-D-12–26637 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Wavre-Shapton ST, Calvi AA, Turmaine M, Seabre MC, Cutler DF, Futter CE, Mitchison HM, Photoreceptor phagosome processing defects and disturbed autophagy in retinal pigment epithelium of Cln3Deltaex1-6 mice modelling juvenile neuronal ceroid lipofuscinosis (Batten disease), Hum Mol Genet (2015). 10.1093/hmg/ddv406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Bagal SK, Brown AD, Cox PJ, Omoto K, Owen RM, Pryde DC, Sidders B, Skerratt SE, Stevens EB, Storer RI, Swain NA, Ion channels as therapeutic targets: a drug discovery perspective, J Med Chem 56(3) (2013) 593–624. 10.1021/jm3011433. [DOI] [PubMed] [Google Scholar]
- [81].Menezes LFS, Sabia EF Junior, Tibery DV, Carneiro LDA, Schwartz EF, Epilepsy-Related Voltage-Gated Sodium Channelopathies: A Review, Front Pharmacol 11 (2020) 1276. 10.3389/fphar.2020.01276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Nabauer M, Kaab S, Potassium channel down-regulation in heart failure, Cardiovasc Res 37(2) (1998) 324–34. 10.1016/s0008-6363(97)00274-5. [DOI] [PubMed] [Google Scholar]
- [83].Weisbrod D, Small and Intermediate Calcium Activated Potassium Channels in the Heart: Role and Strategies in the Treatment of Cardiovascular Diseases, Front Physiol 11 (2020) 590534. 10.3389/fphys.2020.590534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Gianesello L, Del Prete D, Ceol M, Priante G, Calo LA, Anglani F, From protein uptake to Dent disease: An overview of the CLCN5 gene, Gene 747 (2020) 144662. 10.1016/j.gene.2020.144662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Soleimani M, Xu J, SLC26 chloride/base exchangers in the kidney in health and disease, Semin Nephrol 26(5) (2006) 375–85. 10.1016/j.semnephrol.2006.07.005. [DOI] [PubMed] [Google Scholar]
- [86].Lee K, Jo YY, Chung G, Jung JH, Kim YH, Park CK, Functional Importance of Transient Receptor Potential (TRP) Channels in Neurological Disorders, Front Cell Dev Biol 9 (2021) 611773. 10.3389/fcell.2021.611773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Bose S, He H, Stauber T, Neurodegeneration Upon Dysfunction of Endosomal/Lysosomal CLC Chloride Transporters, Front Cell Dev Biol 9 (2021) 639231. 10.3389/fcell.2021.639231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Grimm C, Butz E, Chen CC, Wahl-Schott C, Biel M, From mucolipidosis type IV to Ebola: TRPML and two-pore channels at the crossroads of endo-lysosomal trafficking and disease, Cell Calcium 67 (2017) 148–155. 10.1016/j.ceca.2017.04.003. [DOI] [PubMed] [Google Scholar]
- [89].Hwang SM, Lee JY, Park CK, Kim YH, The Role of TRP Channels and PMCA in Brain Disorders: Intracellular Calcium and pH Homeostasis, Front Cell Dev Biol 9 (2021) 584388. 10.3389/fcell.2021.584388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Chadwick SR, Grinstein S, Freeman SA, From the inside out: Ion fluxes at the centre of endocytic traffic, Curr Opin Cell Biol 71 (2021) 77–86. 10.1016/j.ceb.2021.02.006. [DOI] [PubMed] [Google Scholar]
- [91].Pearce DA, Ferea T, Nosel SA, Das B, Sherman F, Action of BTN1, the yeast orthologue of the gene mutated in Batten disease, Nat Genet 22(1) (1999) 55–8. 10.1038/8861. [DOI] [PubMed] [Google Scholar]
- [92].Schmidtke C, Tiede S, Thelen M, Kakela R, Jabs S, Makrypidi G, Sylvester M, Schweizer M, Braren I, Brocke-Ahmadinejad N, Cotman SL, Schulz A, Gieselmann V, Braulke T, Lysosomal proteome analysis reveals that CLN3-defective cells have multiple enzyme deficiencies associated with changes in intracellular trafficking, J Biol Chem 294(24) (2019) 9592–9604. 10.1074/jbc.RA119.008852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Grubman A, Pollari E, Duncan C, Caragounis A, Blom T, Volitakis I, Wong A, Cooper J, Crouch PJ, Koistinaho J, Jalanko A, White AR, Kanninen KM, Deregulation of biometal homeostasis: the missing link for neuronal ceroid lipofuscinoses?, Metallomics 6(4) (2014) 932–43. [DOI] [PubMed] [Google Scholar]
- [94].Chang JW, Choi H, Kim HJ, Jo DG, Jeon YJ, Noh JY, Park WJ, Jung YK, Neuronal vulnerability of CLN3 deletion to calcium-induced cytotoxicity is mediated by calsenilin, Hum Mol Genet 16(3) (2007) 317–26. https://doi.org/ddl466 [pii] 10.1093/hmg/ddl466. [DOI] [PubMed] [Google Scholar]
- [95].Bahring R, Kv channel-interacting proteins as neuronal and non-neuronal calcium sensors, Channels (Austin) 12(1) (2018) 187–200. 10.1080/19336950.2018.1491243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Jo DG, Chang JW, Hong HS, Mook-Jung I, Jung YK, Contribution of presenilin/gamma-secretase to calsenilin-mediated apoptosis, Biochem Biophys Res Commun 305(1) (2003) 62–6. 10.1016/s0006-291x(03)00688-0. [DOI] [PubMed] [Google Scholar]
- [97].Jo DG, Lee JY, Hong YM, Song S, Mook-Jung I, Koh JY, Jung YK, Induction of proapoptotic calsenilin/DREAM/KChIP3 in Alzheimer’s disease and cultured neurons after amyloid-beta exposure, J Neurochem 88(3) (2004) 604–11. 10.1111/j.1471-4159.2004.02159.x. [DOI] [PubMed] [Google Scholar]
- [98].Carrion AM, Link WA, Ledo F, Mellstrom B, Naranjo JR, DREAM is a Ca2+-regulated transcriptional repressor, Nature 398(6722) (1999) 80–4. 10.1038/18044. [DOI] [PubMed] [Google Scholar]
- [99].Seifert C, Storch S, Bahring R, Modulation of Kv4.2/KChIP3 interaction by the ceroid lipofuscinosis neuronal 3 protein CLN3, J Biol Chem 295(34) (2020) 12099–12110. 10.1074/jbc.RA120.013828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Golabek AA, Kida E, Walus M, Kaczmarski W, Michalewski M, Wisniewski KE, CLN3 protein regulates lysosomal pH and alters intracellular processing of Alzheimer’s amyloid-beta protein precursor and cathepsin D in human cells, Molecular Genetics & Metabolism 70(3) (2000) 203–13. [DOI] [PubMed] [Google Scholar]
- [101].Golabek AA, Kida E, Walus M, Kaczmarski W, Wujek P, Wisniewski KE, CLN3 disease process: missense point mutations and protein depletion in vitro, European Journal of Paediatric Neurology 5Suppl A (2001) 81–8. [DOI] [PubMed] [Google Scholar]
- [102].Weimer JM, Benedict JW, Getty AL, Pontikis CC, Lim MJ, Cooper JD, Pearce DA, Cerebellar defects in a mouse model of juvenile neuronal ceroid lipofuscinosis, Brain Research 1266 (2009) 93–107. https://doi.org/S0006-8993(09)00291-1 [pii] 10.1016/j.brainres.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Guner G, Lichtenthaler SF, The substrate repertoire of gamma-secretase/presenilin, Semin Cell Dev Biol 105 (2020) 27–42. 10.1016/j.semcdb.2020.05.019. [DOI] [PubMed] [Google Scholar]
- [104].Wolfe MS, Unraveling the complexity of gamma-secretase, Semin Cell Dev Biol 105 (2020) 3–11. 10.1016/j.semcdb.2020.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Uusi-Rauva K, Luiro K, Tanhuanpaa K, Kopra O, Martin-Vasallo P, Kyttala A, Jalanko A, Novel interactions of CLN3 protein link Batten disease to dysregulation of fodrin-Na+, K+ ATPase complex, Exp Cell Res 314(15) (2008) 2895–905. https://doi.org/S0014-4827(08)00243-7 [pii] 10.1016/j.yexcr.2008.06.016. [DOI] [PubMed] [Google Scholar]
- [106].Xie Z, Askari A, Na(+)/K(+)-ATPase as a signal transducer, Eur J Biochem 269(10) (2002) 2434–9. 10.1046/j.1432-1033.2002.02910.x. [DOI] [PubMed] [Google Scholar]
- [107].Xie Z, Cai T, Na+-K+--ATPase-mediated signal transduction: from protein interaction to cellular function, Mol Interv 3(3) (2003) 157–68. 10.1124/mi.3.3.157. [DOI] [PubMed] [Google Scholar]
- [108].Haas IG, Wabl M, Immunoglobulin heavy chain binding protein, Nature 306(5941) (1983) 387–9. 10.1038/306387a0. [DOI] [PubMed] [Google Scholar]
- [109].Casas C, GRP78 at the Centre of the Stage in Cancer and Neuroprotection, Front Neurosci 11 (2017) 177. 10.3389/fnins.2017.00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Tsai YL, Zhang Y, Tseng CC, Stanciauskas R, Pinaud F, Lee AS, Characterization and mechanism of stress-induced translocation of 78-kilodalton glucose-regulated protein (GRP78) to the cell surface, J Biol Chem 290(13) (2015) 8049–64. 10.1074/jbc.M114.618736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Kesiry R, Liu J, GRP78/BIP is involved in ouabain-induced endocytosis of the Na/K-ATPase in LLC-PK1 cells, Front Biosci 10 (2005) 2045–55. 10.2741/1680. [DOI] [PubMed] [Google Scholar]
- [112].Getty AL, Benedict JW, Pearce DA, A novel interaction of CLN3 with nonmuscle myosin-IIB and defects in cell motility of Cln3(−/−) cells, Exp Cell Res 317(1) (2011) 51–69. https://doi.org/S0014-4827(10)00434-9 [pii] 10.1016/j.yexcr.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Newell-Litwa KA, Horwitz R, Lamers ML, Non-muscle myosin II in disease: mechanisms and therapeutic opportunities, Dis Model Mech 8(12) (2015) 1495–515. 10.1242/dmm.022103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Ryu J, Liu L, Wong TP, Wu DC, Burette A, Weinberg R, Wang YT, Sheng M, A critical role for myosin IIb in dendritic spine morphology and synaptic function, Neuron 49(2) (2006) 175–82. 10.1016/j.neuron.2005.12.017. [DOI] [PubMed] [Google Scholar]
- [115].Javier-Torrent M, Saura CA, Conventional and Non-Conventional Roles of Non-Muscle Myosin II-Actin in Neuronal Development and Degeneration, Cells 9(9) (2020). 10.3390/cells9091926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Aguilar BJ, Zhu Y, Lu Q, Rho GTPases as therapeutic targets in Alzheimer’s disease, Alzheimers Res Ther 9(1) (2017) 97. 10.1186/s13195-017-0320-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Koch JC, Tatenhorst L, Roser AE, Saal KA, Tonges L, Lingor P, ROCK inhibition in models of neurodegeneration and its potential for clinical translation, Pharmacol Ther 189 (2018) 1–21. 10.1016/j.pharmthera.2018.03.008. [DOI] [PubMed] [Google Scholar]
- [118].Yan Y, Yu J, Gao Y, Kumar G, Guo M, Zhao Y, Fang Q, Zhang H, Yu J, Jiang Y, Zhang HT, Ma CG, Therapeutic potentials of the Rho kinase inhibitor Fasudil in experimental autoimmune encephalomyelitis and the related mechanisms, Metab Brain Dis 34(2) (2019) 377–384. 10.1007/s11011-018-0355-7. [DOI] [PubMed] [Google Scholar]
- [119].Llavero Hurtado M, Fuller HR, Wong AMS, Eaton SL, Gillingwater TH, Pennetta G, Cooper JD, Wishart TM, Proteomic mapping of differentially vulnerable pre-synaptic populations identifies regulators of neuronal stability in vivo, Sci Rep 7(1) (2017) 12412. 10.1038/s41598-017-12603-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Doherty GJ, McMahon HT, Mechanisms of endocytosis, Annual review of biochemistry 78 (2009) 857–902. 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- [121].Mayor S, Parton RG, Donaldson JG, Clathrin-independent pathways of endocytosis, Cold Spring Harbor perspectives in biology 6(6) (2014). 10.1101/cshperspect.a016758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Kaksonen M, Roux A, Mechanisms of clathrin-mediated endocytosis, Nature reviews. Molecular cell biology 19(5) (2018) 313–326. 10.1038/nrm.2017.132. [DOI] [PubMed] [Google Scholar]
- [123].McMahon HT, Boucrot E, Molecular mechanism and physiological functions of clathrin-mediated endocytosis, Nature reviews. Molecular cell biology 12(8) (2011) 517–33. 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- [124].Johannes L, Parton RG, Bassereau P, Mayor S, Building endocytic pits without clathrin, Nature reviews. Molecular cell biology 16(5) (2015) 311–21. 10.1038/nrm3968. [DOI] [PubMed] [Google Scholar]
- [125].Parton RG, Caveolae: Structure, Function, and Relationship to Disease, Annual review of cell and developmental biology 34 (2018) 111–136. 10.1146/annurevcellbio-100617-062737. [DOI] [PubMed] [Google Scholar]
- [126].Hinze C, Boucrot E, Local actin polymerization during endocytic carrier formation, Biochemical Society transactions 46(3) (2018) 565–576. 10.1042/BST20170355. [DOI] [PubMed] [Google Scholar]
- [127].Mercer J, Helenius A, Gulping rather than sipping: macropinocytosis as a way of virus entry, Current opinion in microbiology 15(4) (2012) 490–9. 10.1016/j.mib.2012.05.016. [DOI] [PubMed] [Google Scholar]
- [128].Chadda R, Howes MT, Plowman SJ, Hancock JF, Parton RG, Mayor S, Cholesterol-sensitive Cdc42 activation regulates actin polymerization for endocytosis via the GEEC pathway, Traffic 8(6) (2007) 702–17. 10.1111/j.1600-0854.2007.00565.x. [DOI] [PubMed] [Google Scholar]
- [129].Nevins AK, Thurmond DC, Caveolin-1 functions as a novel Cdc42 guanine nucleotide dissociation inhibitor in pancreatic beta-cells, J Biol Chem 281(28) (2006) 18961–72. 10.1074/jbc.M603604200. [DOI] [PubMed] [Google Scholar]
- [130].Basquin C, Trichet M, Vihinen H, Malarde V, Lagache T, Ripoll L, Jokitalo E, Olivo-Marin JC, Gautreau A, Sauvonnet N, Membrane protrusion powers clathrin-independent endocytosis of interleukin-2 receptor, EMBO J 34(16) (2015) 2147–61. 10.15252/embj.201490788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Kumari S, Mayor S, ARF1 is directly involved in dynamin-independent endocytosis, Nature cell biology 10(1) (2008) 30–41. 10.1038/ncb1666. [DOI] [PubMed] [Google Scholar]
- [132].Rojas AM, Fuentes G, Rausell A, Valencia A, The Ras protein superfamily: evolutionary tree and role of conserved amino acids, The Journal of cell biology 196(2) (2012) 189–201. 10.1083/jcb.201103008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Cherfils J, Zeghouf M, Regulation of small GTPases by GEFs, GAPs, and GDIs, Physiological reviews 93(1) (2013) 269–309. 10.1152/physrev.00003.2012. [DOI] [PubMed] [Google Scholar]
- [134].Lefrancois S, Zeng J, Hassan AJ, Canuel M, Morales CR, The lysosomal trafficking of sphingolipid activator proteins (SAPs) is mediated by sortilin, Embo J 22(24) (2003) 6430–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Pohlmann R, Boeker MW, von Figura K, The two mannose 6-phosphate receptors transport distinct complements of lysosomal proteins, J Biol Chem 270(45) (1995) 27311–8. 10.1074/jbc.270.45.27311. [DOI] [PubMed] [Google Scholar]
- [136].Yasa S, Modica G, Sauvageau E, Kaleem A, Hermey G, Lefrancois S, CLN3 regulates endosomal function by modulating Rab7A-effector interactions, J Cell Sci 133(6) (2020). 10.1242/jcs.234047. [DOI] [PubMed] [Google Scholar]
- [137].Dumaresq-Doiron K, Savard MF, Akam S, Costantino S, Lefrancois S, The phosphatidylinositol 4-kinase PI4KIIIalpha is required for the recruitment of GBF1 to Golgi membranes, Journal of cell science 123(Pt 13) (2010) 2273–80. https://doi.org/jcs.055798 [pii] 10.1242/jcs.055798. [DOI] [PubMed] [Google Scholar]
- [138].Lefrancois S, McCormick PJ, The Arf GEF GBF1 Is Required for GGA Recruitment to Golgi Membranes, Traffic 8(10) (2007) 1440–1451. [DOI] [PubMed] [Google Scholar]
- [139].Hutagalung AH, Novick PJ, Role of Rab GTPases in membrane traffic and cell physiology, Physiological reviews 91(1) (2011) 119–49. https://doi.org/91/1/119 [pii] 10.1152/physrev.00059.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Uusi-Rauva K, Kyttala A, van der Kant R, Vesa J, Tanhuanpaa K, Neefjes J, Olkkonen VM, Jalanko A, Neuronal ceroid lipofuscinosis protein CLN3 interacts with motor proteins and modifies location of late endosomal compartments, Cell Mol Life Sci 69(12) (2012) 2075–89. 10.1007/s00018-011-0913-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Gutierrez MG, Munafo DB, Beron W, Colombo MI, Rab7 is required for the normal progression of the autophagic pathway in mammalian cells, Journal of cell science 117(Pt 13) (2004) 2687–97. 10.1242/jcs.01114. [DOI] [PubMed] [Google Scholar]
- [142].Rojas R, van Vlijmen T, Mardones GA, Prabhu Y, Rojas AL, Mohammed S, Heck AJ, Raposo G, van der Sluijs P, Bonifacino JS, Regulation of retromer recruitment to endosomes by sequential action of Rab5 and Rab7, The Journal of cell biology 183(3) (2008) 513–26. https://doi.org/jcb.200804048 [pii] 10.1083/jcb.200804048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Seaman MN, Harbour ME, Tattersall D, Read E, Bright N, Membrane recruitment of the cargo-selective retromer subcomplex is catalysed by the small GTPase Rab7 and inhibited by the Rab-GAP TBC1D5, Journal of cell science 122(Pt 14) (2009) 2371–82. https://doi.org/jcs.048686 [pii] 10.1242/jcs.048686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Vanlandingham PA, Ceresa BP, Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration, J Biol Chem 284(18) (2009) 12110–24. 10.1074/jbc.M809277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Kinchen JM, Ravichandran KS, Identification of two evolutionarily conserved genes regulating processing of engulfed apoptotic cells, Nature 464(7289) (2010) 778–82. https://doi.org/nature08853 [pii] 10.1038/nature08853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Nordmann M, Cabrera M, Perz A, Brocker C, Ostrowicz C, Engelbrecht-Vandre S, Ungermann C, The Mon1-Ccz1 complex is the GEF of the late endosomal Rab7 homolog Ypt7, Curr Biol 20(18) (2010) 1654–9. https://doi.org/S0960-9822(10)00990-5 [pii] 10.1016/j.cub.2010.08.002. [DOI] [PubMed] [Google Scholar]
- [147].Frasa MA, Maximiano FC, Smolarczyk K, Francis RE, Betson ME, Lozano E, Goldenring J, Seabra MC, Rak A, Ahmadian MR, Braga VM, Armus is a Rac1 effector that inactivates Rab7 and regulates E-cadherin degradation, Curr Biol 20(3) (2010) 198–208. 10.1016/j.cub.2009.12.053. [DOI] [PubMed] [Google Scholar]
- [148].Seaman MNJ, Mukadam AS, Breusegem SY, Inhibition of TBC1D5 activates Rab7a and can enhance the function of the retromer cargo-selective complex, Journal of cell science 131(12) (2018). 10.1242/jcs.217398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Peralta ER, Martin BC, Edinger AL, Differential effects of TBC1D15 and mammalian Vps39 on Rab7 activation state, lysosomal morphology, and growth factor dependence, J Biol Chem 285(22) (2010) 16814–21. https://doi.org/M110.111633 [pii] 10.1074/jbc.M110.111633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Yamano K, Fogel AI, Wang C, van der Bliek AM, Youle RJ, Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy, Elife 3 (2014) e01612. 10.7554/eLife.01612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Arighi CN, Hartnell LM, Aguilar RC, Haft CR, Bonifacino JS, Role of the mammalian retromer in sorting of the cation-independent mannose 6-phosphate receptor, The Journal of cell biology 165(1) (2004) 123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Seaman MN, Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer, The Journal of cell biology 165(1) (2004) 111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Small SA, Kent K, Pierce A, Leung C, Kang MS, Okada H, Honig L, Vonsattel JP, Kim TW, Model-guided microarray implicates the retromer complex in Alzheimer’s disease, Ann Neurol 58(6) (2005) 909–19. 10.1002/ana.20667. [DOI] [PubMed] [Google Scholar]
- [154].Zimprich A, Benet-Pages A, Struhal W, Graf E, Eck SH, Offman MN, Haubenberger D, Spielberger S, Schulte EC, Lichtner P, Rossle SC, Klopp N, Wolf E, Seppi K, Pirker W, Presslauer S, Mollenhauer B, Katzenschlager R, Foki T, Hotzy C, Reinthaler E, Harutyunyan A, Kralovics R, Peters A, Zimprich F, Brucke T, Poewe W, Auff E, Trenkwalder C, Rost B, Ransmayr G, Winkelmann J, Meitinger T, Strom TM, A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease, Am J Hum Genet 89(1) (2011) 168–75. 10.1016/j.ajhg.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Muzio L, Sirtori R, Gornati D, Eleuteri S, Fossaghi A, Brancaccio D, Manzoni L, Ottoboni L, Feo L, Quattrini A, Mastrangelo E, Sorrentino L, Scalone E, Comi G, Marinelli L, Riva N, Milani M, Seneci P, Martino G, Retromer stabilization results in neuroprotection in a model of Amyotrophic Lateral Sclerosis, Nature communications 11(1) (2020) 3848. 10.1038/s41467-020-17524-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Mamo A, Jules F, Dumaresq-Doiron K, Costantino S, Lefrancois S, The role of ceroid lipofuscinosis neuronal protein 5 (CLN5) in endosomal sorting, Mol Cell Biol 32(10) (2012) 1855–66. https://doi.org/MCB.06726-11 [pii] 10.1128/MCB.06726-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Canuel M, Lefrancois S, Zeng J, Morales CR, AP-1 and retromer play opposite roles in the trafficking of sortilin between the Golgi apparatus and the lysosomes, Biochemical and biophysical research communications 366(3) (2008) 724–30. [DOI] [PubMed] [Google Scholar]
- [158].di Ronza A, Bajaj L, Sharma J, Sanagasetti D, Lotfi P, Adamski CJ, Collette J, Palmieri M, Amawi A, Popp L, Chang KT, Meschini MC, Leung HE, Segatori L, Simonati A, Sifers RN, Santorelli FM, Sardiello M, CLN8 is an endoplasmic reticulum cargo receptor that regulates lysosome biogenesis, Nat Cell Biol 20(12) (2018) 1370–1377. 10.1038/s41556-018-0228-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Danyukova T, Ariunbat K, Thelen M, Brocke-Ahmadinejad N, Mole SE, Storch S, Loss of CLN7 results in depletion of soluble lysosomal proteins and impaired mTOR reactivation, Hum Mol Genet 27(10) (2018) 1711–1722. 10.1093/hmg/ddy076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Bajaj L, Sharma J, di Ronza A, Zhang P, Eblimit A, Pal R, Roman D, Collette JR, Booth C, Chang KT, Sifers RN, Jung SY, Weimer JM, Chen R, Schekman RW, Sardiello M, A CLN6-CLN8 complex recruits lysosomal enzymes at the ER for Golgi transfer, J Clin Invest 130(8) (2020) 4118–4132. 10.1172/JCI130955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].McEwan DG, Popovic D, Gubas A, Terawaki S, Suzuki H, Stadel D, Coxon FP, Miranda de Stegmann D, Bhogaraju S, Maddi K, Kirchof A, Gatti E, Helfrich MH, Wakatsuki S, Behrends C, Pierre P, Dikic I, PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins, Molecular cell 57(1) (2015) 39–54. 10.1016/j.molcel.2014.11.006. [DOI] [PubMed] [Google Scholar]
- [162].van der Kant R, Fish A, Janssen L, Janssen H, Krom S, Ho N, Brummelkamp T, Carette J, Rocha N, Neefjes J, Late endosomal transport and tethering are coupled processes controlled by RILP and the cholesterol sensor ORP1L, Journal of cell science 126(Pt 15) (2013) 3462–74. 10.1242/jcs.129270. [DOI] [PubMed] [Google Scholar]
- [163].Wijdeven RH, Janssen H, Nahidiazar L, Janssen L, Jalink K, Berlin I, Neefjes J, Cholesterol and ORP1L-mediated ER contact sites control autophagosome transport and fusion with the endocytic pathway, Nature communications 7 (2016) 11808. 10.1038/ncomms11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Khobrekar NV, Quintremil S, Dantas TJ, Vallee RB, The Dynein Adaptor RILP Controls Neuronal Autophagosome Biogenesis, Transport, and Clearance, Dev Cell 53(2) (2020) 141–153 e4. 10.1016/j.devcel.2020.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Brown CL, Maier KC, Stauber T, Ginkel LM, Wordeman L, Vernos I, Schroer TA, Kinesin-2 is a motor for late endosomes and lysosomes, Traffic 6(12) (2005) 1114–24. 10.1111/j.1600-0854.2005.00347.x. [DOI] [PubMed] [Google Scholar]
- [166].Lojewski X, Staropoli JF, Biswas-Legrand S, Simas AM, Haliw L, Selig MK, Coppel SH, Goss KA, Petcherski A, Chandrachud U, Sheridan SD, Lucente D, Sims KB, Gusella JF, Sondhi D, Crystal RG, Reinhardt P, Sterneckert J, Scholer H, Haggarty SJ, Storch A, Hermann A, Cotman SL, Human iPSC models of neuronal ceroid lipofuscinosis capture distinct effects of TPP1 and CLN3 mutations on the endocytic pathway, Hum Mol Genet 23(8) (2014) 2005–22. 10.1093/hmg/ddt596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Miller JN, Chan CH, Pearce DA, The role of nonsense-mediated decay in neuronal ceroid lipofuscinosis, Hum Mol Genet 22(13) (2013) 2723–34. https://doi.org/ddt120 [pii] 10.1093/hmg/ddt120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Hendricks AG, Perlson E, Ross JL, Schroeder HW 3rd, Tokito M, Holzbaur EL, Motor coordination via a tug-of-war mechanism drives bidirectional vesicle transport, Curr Biol 20(8) (2010) 697–702. 10.1016/j.cub.2010.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Kettenmann H, Hanisch UK, Noda M, Verkhratsky A, Physiology of microglia, Physiological reviews 91(2) (2011) 461–553. 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- [170].Mandrekar S, Jiang Q, Lee CY, Koenigsknecht-Talboo J, Holtzman DM, Landreth GE, Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis, The Journal of neuroscience : the official journal of the Society for Neuroscience 29(13) (2009) 4252–62. 10.1523/JNEUROSCI.5572-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Xiong J, Kielian T, Microglia in juvenile neuronal ceroid lipofuscinosis are primed toward a pro-inflammatory phenotype, J Neurochem 127(2) (2013) 245–58. 10.1111/jnc.12385. [DOI] [PubMed] [Google Scholar]
- [172].Colombo A, Dinkel L, Muller SA, Sebastian Monasor L, Schifferer M, Cantuti-Castelvetri L, Konig J, Vidatic L, Bremova-Ertl T, Lieberman AP, Hecimovic S, Simons M, Lichtenthaler SF, Strupp M, Schneider SA, Tahirovic S, Loss of NPC1 enhances phagocytic uptake and impairs lipid trafficking in microglia, Nature communications 12(1) (2021) 1158. 10.1038/s41467-021-21428-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Dolisca SB, Mehta M, Pearce DA, Mink JW, Maria BL, Batten disease: clinical aspects, molecular mechanisms, translational science, and future directions, Journal of child neurology 28(9) (2013) 1074–100. 10.1177/0883073813493665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A, Kehrl JH, Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction, Nature immunology 13(3) (2012) 255–63. 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Kimelberg HK, Functions of mature mammalian astrocytes: a current view, The Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry 16(1) (2010) 79–106. 10.1177/1073858409342593. [DOI] [PubMed] [Google Scholar]
- [176].Chen G, Li HM, Chen YR, Gu XS, Duan S, Decreased estradiol release from astrocytes contributes to the neurodegeneration in a mouse model of Niemann-Pick disease type C, Glia 55(15) (2007) 1509–18. 10.1002/glia.20563. [DOI] [PubMed] [Google Scholar]
- [177].Pontikis CC, Cotman SL, MacDonald ME, Cooper JD, Thalamocortical neuron loss and localized astrocytosis in the Cln3Deltaex7/8 knock-in mouse model of Batten disease, Neurobiol Dis 20(3) (2005) 823–36. https://doi.org/S0969-9961(05)00153-1 [pii] 10.1016/j.nbd.2005.05.018. [DOI] [PubMed] [Google Scholar]
- [178].Bosch ME, Aldrich A, Fallet R, Odvody J, Burkovetskaya M, Schuberth K, Fitzgerald JA, Foust KD, Kielian T, Self-Complementary AAV9 Gene Delivery Partially Corrects Pathology Associated with Juvenile Neuronal Ceroid Lipofuscinosis (CLN3), J Neurosci 36(37) (2016) 9669–82. 10.1523/JNEUROSCI.1635-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Schultz ML, Tecedor L, Lysenko E, Ramachandran S, Stein CS, Davidson BL, Modulating membrane fluidity corrects Batten disease phenotypes in vitro and in vivo, Neurobiol Dis 115 (2018) 182–193. 10.1016/j.nbd.2018.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Gowrishankar S, Yuan P, Wu Y, Schrag M, Paradise S, Grutzendler J, De Camilli P, Ferguson SM, Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques, Proceedings of the National Academy of Sciences of the United States of America 112(28) (2015) E3699–708. 10.1073/pnas.1510329112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Maday S, Holzbaur EL, Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway, Dev Cell 30(1) (2014) 71–85. 10.1016/j.devcel.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Raiborg C, Wenzel EM, Pedersen NM, Olsvik H, Schink KO, Schultz SW, Vietri M, Nisi V, Bucci C, Brech A, Johansen T, Stenmark H, Repeated ER-endosome contacts promote endosome translocation and neurite outgrowth, Nature 520(7546) (2015) 234–8. 10.1038/nature14359. [DOI] [PubMed] [Google Scholar]
- [183].Luiro K, Kopra O, Lehtovirta M, Jalanko A, CLN3 protein is targeted to neuronal synapses but excluded from synaptic vesicles: new clues to Batten disease, Hum Mol Genet 10(19) (2001) 2123–31. [DOI] [PubMed] [Google Scholar]
- [184].Ahrens-Nicklas RC, Tecedor L, Hall AF, Lysenko E, Cohen AS, Davidson BL, Marsh ED, Neuronal network dysfunction precedes storage and neurodegeneration in a lysosomal storage disorder, JCI Insight 4(21) (2019). 10.1172/jci.insight.131961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Song JW, Misgeld T, Kang H, Knecht S, Lu J, Cao Y, Cotman SL, Bishop DL, Lichtman JW, Lysosomal activity associated with developmental axon pruning, J Neurosci 28(36) (2008) 8993–9001. https://doi.org/28/36/8993 [pii] 10.1523/JNEUROSCI.0720-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Luiro K, Kopra O, Blom T, Gentile M, Mitchison HM, Hovatta I, Tornquist K, Jalanko A, Batten disease (JNCL) is linked to disturbances in mitochondrial, cytoskeletal, and synaptic compartments, J Neurosci Res 84(5) (2006) 1124–38. 10.1002/jnr.21015. [DOI] [PubMed] [Google Scholar]
- [187].Burkovetskaya M, Karpuk N, Kielian T, Age-dependent alterations in neuronal activity in the hippocampus and visual cortex in a mouse model of Juvenile Neuronal Ceroid Lipofuscinosis (CLN3), Neurobiol Dis 100 (2016) 19–29. 10.1016/j.nbd.2016.12.022. [DOI] [PubMed] [Google Scholar]
- [188].Grunewald B, Lange MD, Werner C, O’Leary A, Weishaupt A, Popp S, Pearce DA, Wiendl H, Reif A, Pape HC, Toyka KV, Sommer C, Geis C, Defective synaptic transmission causes disease signs in a mouse model of juvenile neuronal ceroid lipofuscinosis, Elife 6 (2017). 10.7554/eLife.28685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Ostergaard JR, Rasmussen TB, Molgaard H, Cardiac involvement in juvenile neuronal ceroid lipofuscinosis (Batten disease), Neurology 76(14) (2011) 1245–51. https://doi.org/76/14/1245 [pii] 10.1212/WNL.0b013e31821435bd. [DOI] [PubMed] [Google Scholar]
- [190].Rietdorf K, Coode EE, Schulz A, Wibbeler E, Bootman MD, Ostergaard JR, Cardiac pathology in neuronal ceroid lipofuscinoses (NCL): More than a mere co-morbidity, Biochim Biophys Acta Mol Basis Dis 1866(9) (2020) 165643. 10.1016/j.bbadis.2019.165643. [DOI] [PubMed] [Google Scholar]
- [191].Staropoli JF, Haliw L, Biswas S, Garrett L, Holter SM, Becker L, Skosyrski S, Da Silva-Buttkus P, Calzada-Wack J, Neff F, Rathkolb B, Rozman J, Schrewe A, Adler T, Puk O, Sun M, Favor J, Racz I, Bekeredjian R, Busch DH, Graw J, Klingenspor M, Klopstock T, Wolf E, Wurst W, Zimmer A, Lopez E, Harati H, Hill E, Krause DS, Guide J, Dragileva E, Gale E, Wheeler VC, Boustany RM, Brown DE, Breton S, Ruether K, Gailus-Durner V, Fuchs H, de Angelis MH, Cotman SL, Large-scale phenotyping of an accurate genetic mouse model of JNCL identifies novel early pathology outside the central nervous system, PLoS ONE 7(6) (2012) e38310. 10.1371/journal.pone.0038310 PONE-D-12–05357 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Lim MJ, Alexander N, Benedict JW, Chattopadhyay S, Shemilt SJ, Guerin CJ, Cooper JD, Pearce DA, IgG entry and deposition are components of the neuroimmune response in Batten disease, Neurobiol Dis 25(2) (2007) 239–51. 10.1016/j.nbd.2006.09.005. [DOI] [PubMed] [Google Scholar]
- [193].Roux KJ, Kim DI, Raida M, Burke B, A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells, J Cell Biol 196(6) (2012) 801–10. 10.1083/jcb.201112098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Vitiello SP, Benedict JW, Padilla-Lopez S, Pearce DA, Interaction between Sdo1p and Btn1p in the Saccharomyces cerevisiae model for Batten disease, Human molecular genetics 19(5) (2010) 931–42. https://doi.org/ddp560 [pii] 10.1093/hmg/ddp560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Paiano A, Margiotta A, De Luca M, Bucci C, Yeast Two-Hybrid Assay to Identify Interacting Proteins, Current protocols in protein science 95(1) (2019) e70. 10.1002/cpps.70. [DOI] [PubMed] [Google Scholar]
- [196].Sauvageau E, Lefrancois S, A beginner’s guide to bioluminescence resonance energy transfer (BRET), The Biochemist 41(6) (2019) 36–40. 10.1042/bio04106036. [DOI] [Google Scholar]
- [197].Scifo E, Szwajda A, Debski J, Uusi-Rauva K, Kesti T, Dadlez M, Gingras AC, Tyynela J, Baumann MH, Jalanko A, Lalowski M, Drafting the CLN3 protein interactome in SH-SY5Y human neuroblastoma cells: a label-free quantitative proteomics approach, J Proteome Res 12(5) (2013) 2101–15. 10.1021/pr301125k. [DOI] [PubMed] [Google Scholar]
- [198].Behrends C, Sowa ME, Gygi SP, Harper JW, Network organization of the human autophagy system, Nature 466(7302) (2010) 68–76. https://doi.org/nature09204 [pii] 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]