

Summary
In this study, methamphetamine (Meth)- and glutamate (Glu)-mediated intracellular Ca++ (Ca++i) signals were examined in real time in primary cortical neurons overexpressing an intracellular Ca++ probe, GCaMP5, by adeno-associated viral (AAV) serotype 1. Binding of Ca++ to GCaMP increased green fluorescence intensity in cells. Both Meth and Glu induced a rapid increase in Ca++i, which was blocked by MK801, suggesting that Meth enhanced Ca++i through Glu receptor in neurons. The Meth-mediated Ca++ signal was also blocked by Mg++, low Ca++ or the L-type Ca++ channel inhibitor nifedipine. The ryanodine receptor inhibitor dantrolene did not alter the initial Ca++ influx but partially reduced the peak of Ca++i. These data suggest that Meth enhanced Ca++ influx through membrane Ca++ channels, which then triggered the release of Ca++ from the endoplasmic reticulum in the cytosol. AAV-GCaMP5 was also injected to the parietal cortex of adult rats. Administration of Meth enhanced fluorescence in the ipsilateral cortex. Using immunohistochemistry, Meth-induced green fluorescence was found in the NeuN-containing cells in the cortex, suggesting that Meth increased Ca++ in neurons in vivo. In conclusion, we have used in vitro and in vivo techniques to demonstrate a rapid increase of Ca++i by Meth in cortical neurons through overexpression of GCaMP5. As Meth induces behavioral responses and neurotoxicity through Ca++i, modulation of Ca++i may be useful to reduce Meth-related reactions.
Keywords: Calcium, glutamate, magnesium, methamphetamine
Introduction
Methamphetamine (Meth) is a major substance of abuse worldwide. Meth induces psychoactive effect in human and experimental animals through dopamine (DA). Repeated administration of Meth induced behavioral sensitization, which is associated with increases in extracellular DA levels in the nucleus accumbens (NAc) and striatum (Fukakusa et al. 2008; Lan et al. 2009). High doses or long-term use of Meth causes neurodegeneration of dopaminergic (DA-ergic) terminals and Parkinson-like symptoms (Volkow et al. 2001; Luo et al. 2010; Yu et al. 2012). Meth also affects non-DA neurons, e.g. Meth alters gene expression in DA and non-DA neurons in the brain (Cadet et al. 2010; He et al. 2013). Apoptosis induced by systemic administration of Meth has been identified in non-DA-ergic brain areas (Deng et al. 2001; Shen et al. 2011).
Besides its indirect response through the release of catecholamines from synaptic terminals, Meth can modulate physiological reactions independent of catecholaminergic neurotransmitters (Kuczenski et al. 2007). For example, calcium (Ca++) is a major mediator for physiological responses of Meth or amphetamine (AM). Overexpression of Ca++/calmodulin-dependent protein kinase II in the NAc shell neurons potentiated AM-mediated locomotion and self-administration (Loweth et al. 2010). Meth-mediated behavioral sensitization was antagonized by gabapentin through inhibiting voltage-gated Ca++ channels (Kurokawa et al. 2011); the L-type Ca++ channel inhibitor nifedipine attenuated Meth-mediated place preference behavior (Suzuki et al. 1992). These data suggest that Meth alters behavioral responses through Ca++. The interaction of Meth and Ca++ has also been examined in cultured cells. Meth or AM increased intracellular Ca++ in fura-2 or fluo-4 AM-loaded PC12 cells (Kantor et al. 2004), EM4 cells (Goodwin et al. 2009), neonatal rat cardiomyocytes and HEK-293T cells overexpressing L-type Ca++ channel (Sugimoto et al. 2009). Depletion of intracellular Ca++ reduced AM-mediated DA release in the PC12 cells (Kantor et al. 2001). Nifedipine, but not ryanodine receptor (RyR) inhibitor, antagonized Meth-mediated Ca++ entry and beat rate in cultured cardiac cells (Sugimoto et al. 2009). Taken together, these data suggest that Meth alters intracellular Ca++ signaling through L-type Ca++ channel in non-neuronal cells. Limited studies have been conducted to examine the interactions of Meth and Ca++ in the central nervous system (CNS). High dose of Meth (5 mM) had no effects on Ca++ influx in rat hippocampal neuroprogenitor cells (NPCs) pre-loaded with fura-2. It is noted that a much higher dose of glutamate (Glu, 100 μM) was required to stimulate Ca++ influx in this study (Tian, Murrin & Zheng 2009a), suggesting that the NPCs may be less sensitive to Glu or Meth stimulation. In contrast to the responses in NPCs, Meth increased [Ca++]i in DA-ergic neurons isolated acutely from the ventral tegmental area of adult rat brains (Uramura et al. 2000). As DA can modulate Ca++ channels (Okada, Miyamoto & Toda 2003; Yasumoto et al. 2004), the Meth-induced [Ca++]i reaction is confounded by DA, released by Meth, in DA-ergic neurons. To our acknowledge, there are no published reports demonstrating the direct response of Meth on [Ca++]i in primary non-DA-ergic neuronal cultures.
Increasing number of evidence supports the interaction of Meth and glutamate (Glu) in the CNS (Hendrickson, Laurenzana & Owens 2006; Cadet et al. 2007). AM triggered Glu release in the NAc, prefrontal cortex (Reid, Hsu & Berger 1997; Del, Martinez & Mora 1998) and ventral tegmental area (Wolf et al. 2000). Memantine and dizcilpine (MK801), both N-methyl-d-aspartate receptor antagonists, attenuated Meth-mediated neurotoxicity (Boireau et al. 1995; Chipana et al. 2008). Similarly, MK801 antagonized AM-induced immediate early gene expression in striatal neurons and behavioral sensitization (Ohno, Yoshida & Watanabe 1994; Konradi, Leveque & Hyman 1996) as well as Meth-mediated DA overflow in rat striatum (Marshall, O’Dell & Weihmuller 1993; Finnegan & Taraska 1996), suggesting that Meth-mediated responses were inhibited by Glu antagonists.
Fura-2 or fluo-4 AM fluorescence has been widely used to detect intracellular Ca++. One study reported artifacts found in fura-2 intracellular Ca++ measurement (Kopp, Leech & Roe 2014). Recently, genetically encoded Ca++ indicators (GECIs) have been developed to monitor [Ca++]i in neurons, astrocytes or cardiac cells from Caenorhabditis elegans, flies, zebrafish and rodents (Mao et al. 2008; Tian et al. 2009b; Shigetomi, Kracun & Khakh 2010; Chung, Sun & Gabel 2013). These GECIs provide much faster response times to Ca++, which enables monitoring of rapid Ca++ transients following neuronal activity (Sun et al. 2013). GCaMP, a commonly used GECI probe, consists of a single circularly permuted green fluorescent protein (GFP), calmodulin (CaM) and M13 fragment from myosin light-chain kinase. Binding of Ca++ to CaM induces conformational changes in the GCaMP and results in increased fluorescence intensity in cells (Nakai, Ohkura & Imoto 2001). In this study, we employed GCaMP5 to cultured neuronal cells and brain parenchyma through adeno-associated viral (AAV) vector transduction (Akerboom et al. 2012). We demonstrated that Meth enhanced [Ca++]i in real time in neurons both in vivo and in vitro, which was antagonized by MK801, Mg++ or Ca++ channel blocker nifedipine.
Materials and Methods
Animals and materials
Adult female pregnant and male Sprague-Dawley rats (purchased from the BioLASCO, Taipei, Taiwan) were used in this study. Experimental procedures followed the guidelines of the ‘Principles of Laboratory Care’ (National Institutes of Health Publication No. 86–23, 1996) and were approved by the National Health Research Institutes (Taiwan) Animal Care and Use Committee. (+)Methamphetamine HCl, dantrolene, MK801, Glu and nifedipine were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA).
Construction, packaging and characterization of vectors
The cDNA encoding GCaMP5 (Akerboom et al. 2012) was PCR (polymerase chain reaction) amplified with linkered oligos from pCMV-GCaMP5G (Addgene #31788). The resulting vector pAAV-EF1a-GCaMP5 (Addgene #50964) was sequence verified and used in CsCl DNA preparation for packaging AAV serotype 1 vectors using modified triple transfection method (Howard et al. 2008) followed by affinity chromatography purification using AVB sepharose (GE Healthcare, Pittsburgh, PA, USA). Viruses were titered by qPCR using Taqman chemistry with primers and probe set to the EF1a promoter region. Titer for AAV1-EF1a-GCaMP5 was 1.7 × 1014 viral genomes (vg)/ml. Characterization of AAV vector is demonstrated in Supporting Information Fig. S1.
Primary cultures of rat cortical neurons and AAV transduction
Primary cultures were prepared from embryonic (E14–15) cortical tissues obtained from timed pregnant Sprague-Dawley rats. After removing the blood vessels and meninges, pooled cortices were trypsinized (0.05%, Invitrogen, Carlsbad, CA, USA) for 20 minutes at room temperature. After rinsing off trypsin with pre-warmed Dulbecco’s modified Eagle’s medium (Invitrogen), cells were dissociated by trituration, counted and plated into 96-well (3.0 × 104/well) cell culture plates pre-coated with polyethyleneimine (Sigma-Aldrich). The culture plating medium consisted of neurobasal medium supplemented with 2% heat-inactivated fetal bovine serum, 0.5 mM l-glutamine, 0.025 mM l-glutamate and 2% B27 (Invitrogen). Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air. The cultures were fed by exchanging 50% of media with feed media (neurobasal medium, Invitrogen) with 0.5 mM l-glutamate and 2% B27 with antioxidants supplement on days in vitro (DIV) 3. On DIV 5, 50% of media were removed and AAV-GCamP5 was added for 1 hour at 37°C. Cultures were fed with neurobasal media containing B27 supplement without antioxidants (Invitrogen) on DIV 7 and 10. Concentrations of Mg++ and Ca++ in the neurobasal media were 1.80 and 0.81 mM, respectively. Cultures were treated with reagents on DIV 10.
Real-time epifluorescence measurement of GCaMP5
Culture plates were placed on a motorized stage (Prior Scientific Inc., Fulbourn, Cambridge, UK) of a Nikon TE2000 inverted microscope (Nikon, Melville, NY, USA). Microscopic images were recorded through FITC filter from 1 minute before to 10 minutes after drug treatment at a rate of 2 frames/s. The intensity of intracellular green fluorescence of single cells (8–10 cells/well) was individually measured by the NIS Elements AR 3.2 Software (Nikon). The coordinates of images in culture plates were recorded for later image matching. Cultured cells were then fixed by 4% paraformaldehyde (PFA) for 1 hour at room temperature. The phenotype of imaged cells was verified using NeuN immunocytochemistry and matching images.
Immunocytochemistry
After removing 4% PFA solution, cells were washed with phosphate buffered saline (PBS). Fixed cells were treated with blocking solution [2% bovine serum albumin (BSA), 0.1% Triton X-100 (Sigma, St. Louis, MO, USA) and 5% goat serum in PBS] for 1 hour. The cells were incubated for 1 day at 4°C with a mouse monoclonal antibody against NeuN (1:500, Millipore, Billerica, MA, USA) and then rinsed three times with PBS. The bound primary antibody was visualized using Alexa Fluor 568 goat anti-mouse secondary (Invitrogen). Images were acquired using a monochrome camera Qi1-mc attached to Nikon TE2000-E inverted microscope.
In vivo delivery and imaging of GCaMP5 using the In Vivo Imaging System (IVIS)
Adult rats were anesthetized with chloral hydrate (400 mg/kg, i.p.). The animals were placed in a stereotaxic frame (Stoelting, Wood Dale, IL, USA), where a 10-μl Hamilton syringe (Stoelting) with a 30-gauge needle was used to stereotaxically deliver AAV1-GCaMP5 (2 μl of 5 × 109 vg/μl) into the left parietal cortex as previously described (Yu et al. 2013). The coordinates for the intracortical injection were 0.12 mm posterior to bregma, 2.1 mm lateral (left) to midline and 2.0 mm ventral to the brain surface. The rate of infusion (1 μl/ min) was adjusted by a microprocessor-controlled injector mounted to the stereotaxic frame (UMP4, World Precision Instruments, Sarasota, FL, USA). The needle remained in the brain for 2 minutes after the injection and then slowly removed. After recovery from anesthesia, animals were housed in their home cages. At 2 weeks after viral infection, animals were treated with Meth (5 mg/kg, i.p.) or vehicle (saline, i.p.). Brain tissues were harvested 30 minutes after administration of Meth or vehicle and immediately transferred to an IVIS Lumina 2 Imaging System chamber (Caliper Life Sciences, Cheshire, UK); GFP fluorescence images in whole brains were acquired using a charge-coupled device camera. The intensity of photon collected through IVIS was translated to false color images with strong fluorescence in yellow and was analyzed using Living ImageR Software Version 4.0 (PerkinElmer, Waltham, MA, USA).
Immunohistochemistry
Immediately after IVIS scanning, brain tissues were immersed in 4% PFA in phosphate buffer (PB; 0.1 M; pH 7.2) for 18–20 hours and transferred to 18% sucrose in 0.1 M PB for at least 16 hours. Serial sections of brains were cut at 30-μm thickness on a cryostat. Sections were rinsed with PB and were blocked with 4% BSA with 0.3% Triton X-100 in 0.1 M PB. Sections were then incubated with polyclonal anti-NeuN (1:100, Chemicon, Billerica, MA, USA) at 4°C for overnight. Sections were rinsed with 0.1 M PB and incubated in Alexa Fluor 568 secondary antibody solution (1:500, Molecular Probes, Eugene, OR, USA) and were mounted on slides and coverslipped. Control sections were incubated without primary antibody. Confocal analysis was performed using a Nikon D-ECLIPSE 80i microscope and EZ-C1 3.90 software as previously described (Yu et al. 2012).
Statistical analysis
Values are presented as means ± SEM. Unpaired t-test, two-way ANOVA and post hoc Newman–Keuls test were used for statistical analysis. A statistically significant difference was defined as P < 0.05.
Results
Characterization of Meth-mediated change in [Ca++]i in primary cortical neuronal culture
We first examined Meth-mediated change in [Ca++]i in neurons encoded with GCaMP5. Primary cortical neuronal culture cells were infected with AAV1-GCaMP5 on DIV 5. Intracellular Ca++, as indicated by the change in intracellular green fluorescence, was monitored on DIV 10. Different doses of Meth, ranging from 0.125 to 1 mM, were given to the cells. Meth dose-dependently increased the amplitude of intracellular fluorescence. The ED50 of Meth was close to 0.5 mM; this dose was chosen for the next experiment. As seen in the captured live images (Fig. 1a & Supporting Information Movie S1), Meth at 0.5 mM induced a rapid increase in intracellular Ca++. Peak fluorescence occurred around 20 seconds after treatment. Cells were fixed for NeuN immunostaining after treatment with Meth. Live image captured 26 seconds after Meth treatment was merged with immunocytochemical photomicrogram (Fig. 1a, merged). Cells sensitive to Meth were co-labeled by NeuN (Fig. 1a, far right, & Fig. 1d, left panel), suggesting a neuronal response of Meth. Meth-mediated intracellular fluorescence was conserved in the fixed cells when PFA was applied early after the peak reaction (Supporting Information Fig. S2), indicating an irreversible change of GCaMP5, after binding to Ca++, by PFA.
Figure 1.
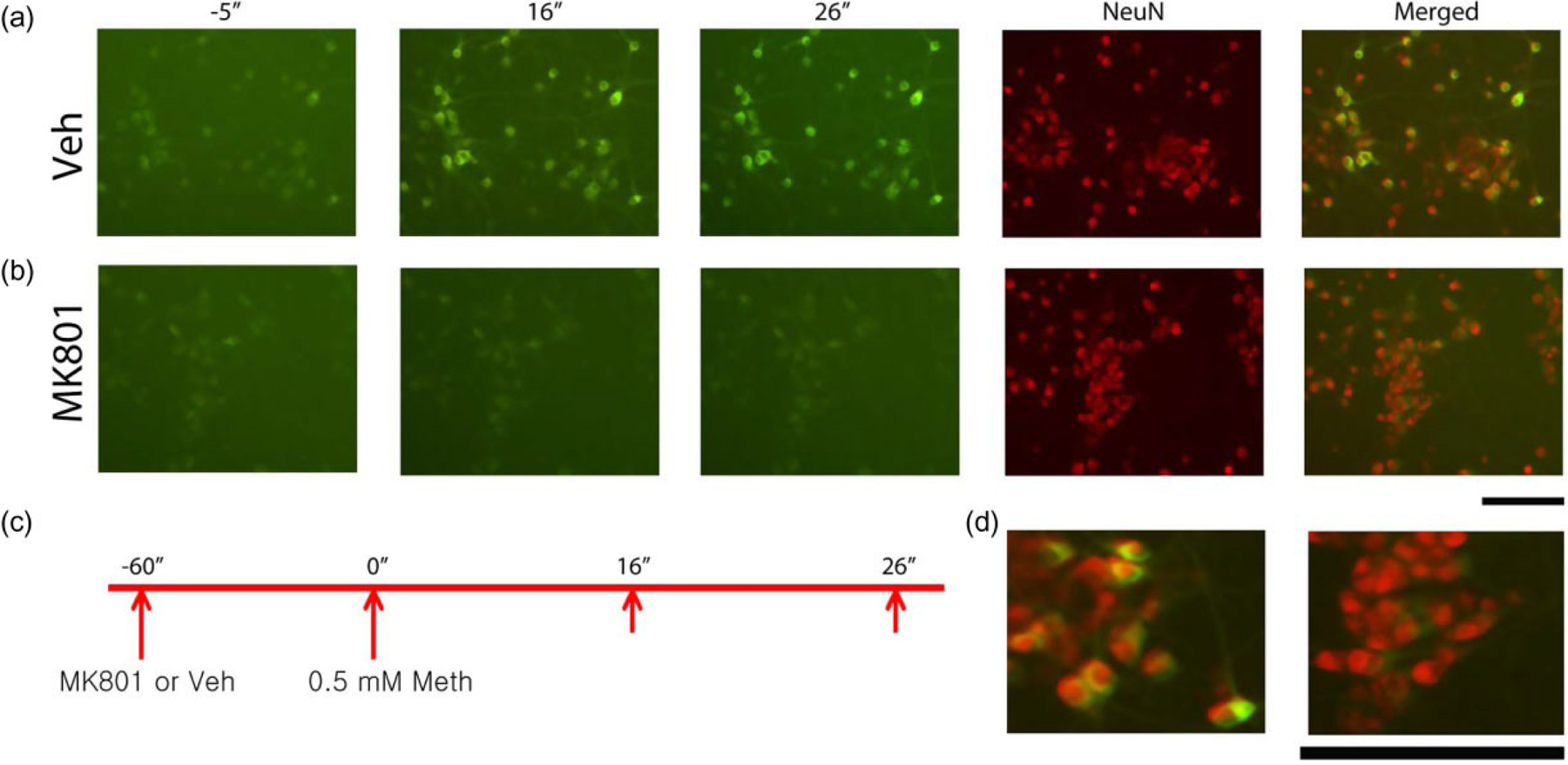
Methamphetamine (Meth) increased [Ca++]i in cultured primary cortical neurons overexpressing GCaMP5. (a) Real-time fluorescence images were taken 5ʺ before and 16ʺ and 26ʺ after Meth administration. Administration of Meth (0.5 mM) triggered a rapid and time-dependent increase in intracellular Ca++, as indicated by green fluorescence (left three panels). Peak fluorescence occurred 26 seconds after injection. Neuronal cells were identified by NeuN immunostaining (second panel from the far right). Meth enhanced green fluorescence only in NeuN (+) cells (right panel, merged). (d, Left) High magnification of merged photomicrograms indicates that Meth-mediated green fluorescence signal was found mainly in NeuN cells. (b) MK801 suppressed Meth-mediated intracellular Ca++ signals (left three panels). Most NeuN (+) cells did not express green fluorescence after Meth administration. (d, Right) High magnification of merged images indicates that pre-treatment with MK801 inhibited green fluorescence in NeuN cells. (c) Timeline indicates that primary cortical neurons were treated with vehicle or MK801 followed by Meth. Calibration: 50 μm
The dynamic intensity of intracellular GCaMP5 fluorescence of single cells was individually analyzed every second after subtracting the background noise. Eight to 15 cells/well were selected for this analysis. An example of real-time tracings of Meth (0.5 mM)-mediated rapid increase in intracellular fluorescence, pooled from 33 cells, is shown in Fig. 2. Meth at 0.5 mM induced a rapid increase in [Ca++]i.
Figure 2.
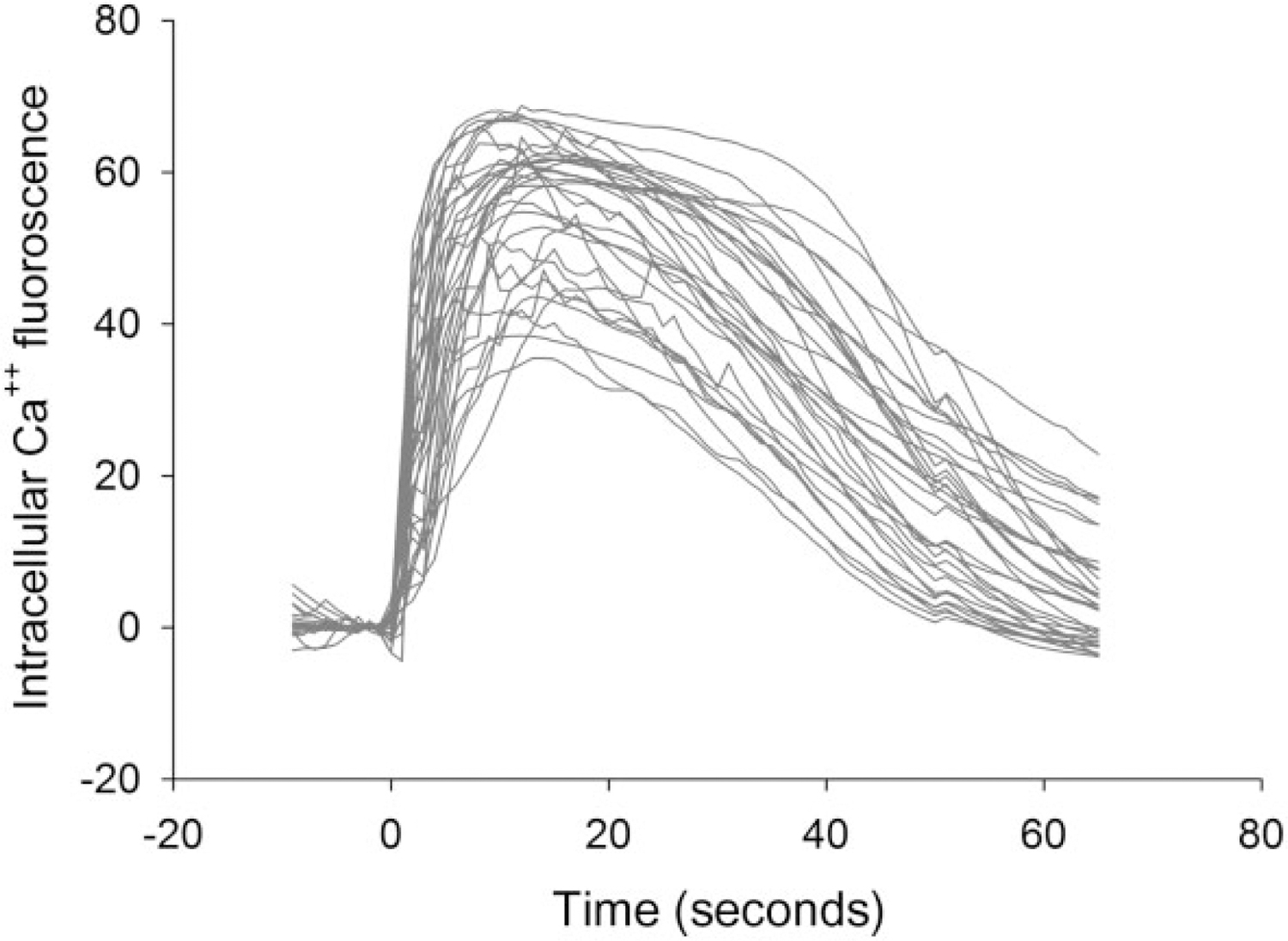
Real-time tracings of intracellular fluorescence of 33 neurons treated with methamphetamine (Meth) (0.5 mM)
Meth-mediated change in [Ca++]i is blocked by Glu antagonist MK801
We found that Glu, similar to Meth, also increased [Ca++]I in neurons encoded with GcaMP5. Different doses of Glu (7, 125 and 500 nM) were applied to the culture well. Glu dose-dependently increased the peak. ED50 of Glu was around 125 nM; this dose was chosen for the subsequent experiments. As seen in the captured live images (Fig. 3a, left 3 panels), administration of Glu (125 nM) time-dependently increased intracellular Ca++ in selective cells GCaMP5-labeled cells responsive to Glu co-expressed NeuN (Fig. 3a & d—left panel), suggesting a neuronal response of Glu.
Figure 3.
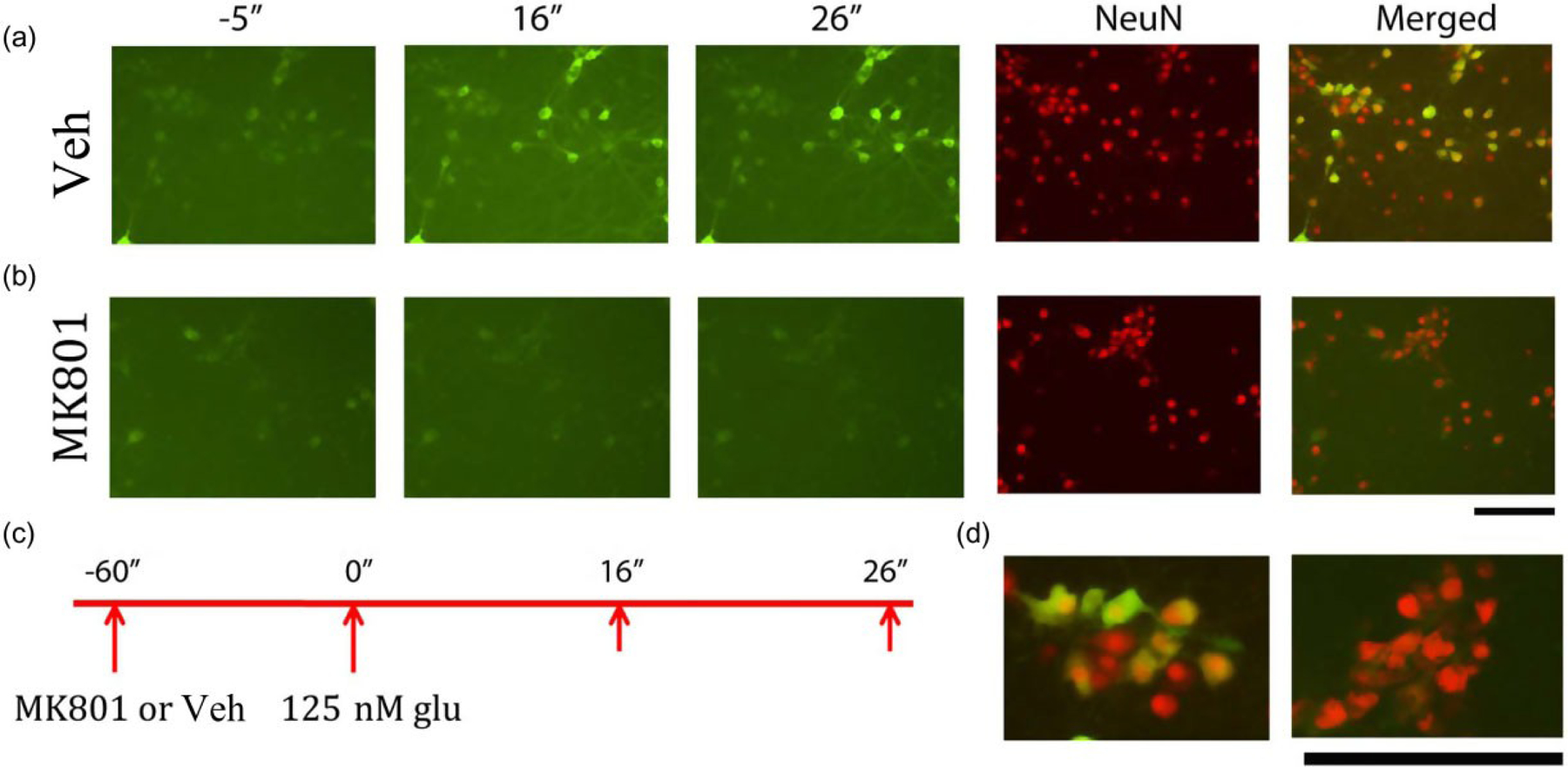
Increasing [Ca++]i by glutamate (Glu) using fluorescent live cell imaging in cultured primary cortical neurons overexpressed GCaMP5. (a) Real-time fluorescence images were taken 5″ before and 16″ and 26″ after Glu administration. Administration of Glu (top panels) triggered a rapid and time-dependent increase in intracellular Ca++, as indicated by green fluorescence (left three panels). Peak fluorescence occurred 26 seconds after injection. Neuronal cells were identified by NeuN immunostaining. Glu enhanced green fluorescence only in NeuN (+) cells (right panel, merged). MK801 suppressed Glu-mediated fluorescence signals (lower panel). Most NeuN (+) cells did not express green fluorescence after Glu administration (right and lower panels). (d) High magnification images of (a) (right panels). Green fluorescence signal was found in NeuN cells pre-treated with vehicle (left). Pre-treatment with MK801 inhibited green fluorescence in NeuN cells (right panel) (c) Timeline indicates that primary cortical neurons were treated with vehicle or MK801 followed by glutamate (125 nM). Calibration: 50 μm
MK801 (50 μM) was given to cells 1 minute before application of Glu (125 nM). A typical interaction of MK801 and Glu is shown in the captured live images (Fig. 3b). After treatment with MK801, Glu did not increase [Ca++]i in NeuN (+) cells (Fig. 3b versus Fig. 3a). At high magnification, most of NeuN (+) cells did not show green fluorescence, suggesting that Glu-mediated neuronal excitation was antagonized by MK801 (Fig. 3d, right panel).
MK801 also antagonized Meth-increased [Ca++]i. MK801 (50 μM) was given 1 minute before application of Meth (0.5 mM; Fig. 1d—right). MK801 reduced Meth-mediated Ca++i in the live images (Fig. 1b versus Fig. 1a; Supporting Information Movie S2). At high magnification, Meth did not induce Ca++-mediated fluorescence in NeuN (+) cells pre-treated with MK801 (Fig. 1d, right panel).
The intensity of fluorescence was further analyzed in 216 neurons, pre-treated with either saline or MK801. Glu (125 nM; Fig. 4a) and Meth (0.5 mM; Fig. 4b) significantly increased Ca++ fluorescence intensity (P < 0.01, one-way ANOVA). Pre-treatment with MK801 (50 μM) significantly antagonized both reactions (P < 0.01, two-way ANOVA + Newman–Keuls post hoc analysis; Fig. 4).
Figure 4:
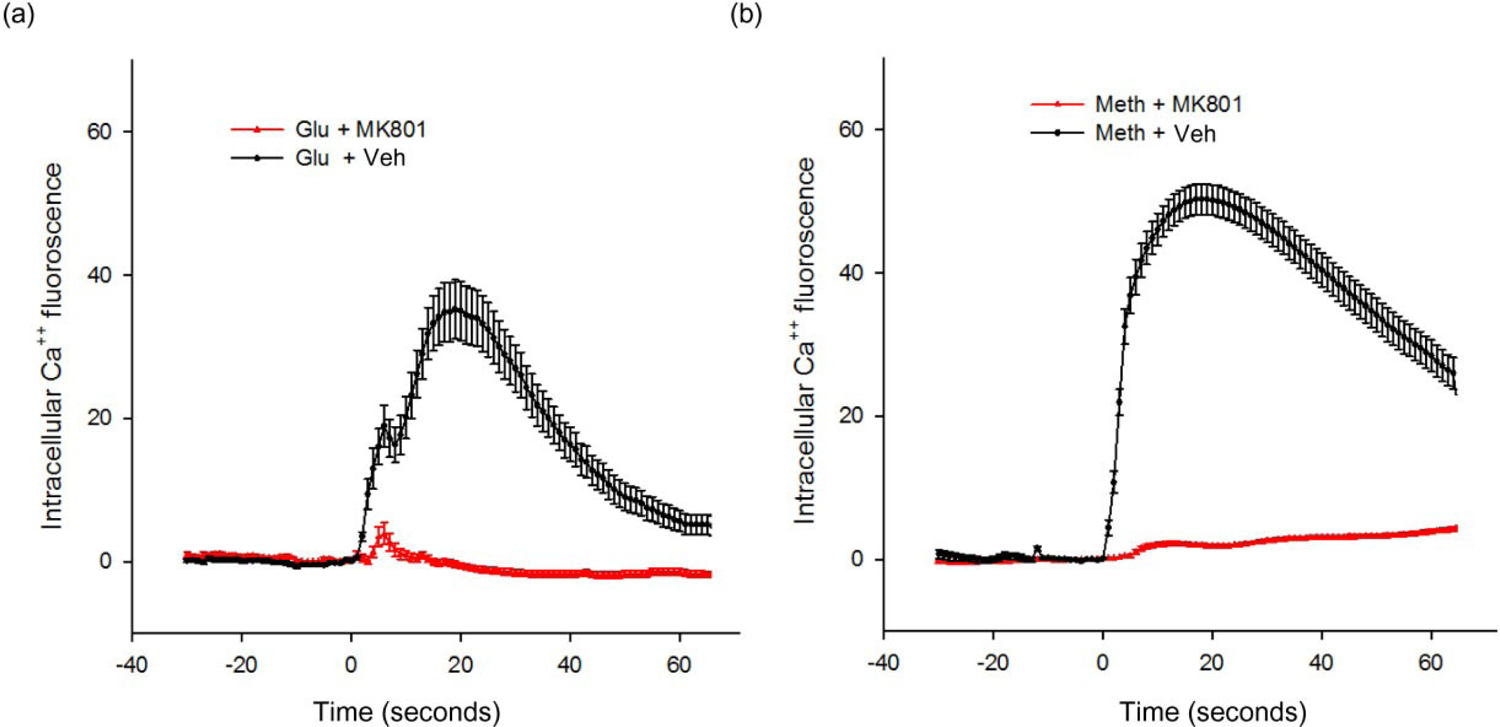
(a) MK801 antagonized glutamate (Glu)-mediated increase in [Ca++]i. MK801 (50 μM) was given to the culture wells 1 minute before application of Glu (125 nM at time 0). The intensity of fluorescence was analyzed in 216 neurons labeled by NeuN. Glu significantly increased Ca++ signal intensity, as compared to the response prior to Glu administration. Pre-treatment with MK801 significantly antagonized Glu-mediated increase in [Ca++]i (P < 0.001, two-way ANOVA). (b) MK801 antagonized methamphetamine (Meth)-mediated increase in [Ca++]i. MK801 (50 μM) was given to cells 1 minute before application of Meth (0.5 mM). MK801 significantly antagonized this response (P < 0.05, two-way ANOVA + Newman–Keuls post hoc analysis)
Mg++, nifedipine and low extracellular Ca++ altered Meth-mediated Ca++ signals
We examined the effects of Meth on primary neurons in low Ca++ medium. Cultured wells were washed with artificial cerebrospinal fluid (aCSF), with or without Ca++ (0.1 mM), on DIV 10 and then incubated for additional 20 minutes. Meth-induced intracellular green fluorescence from GCaMP5 was significantly reduced in aCSF containing Ca++ (0.1 mM) compared with the neural basal medium (Ca++ 1.8 mM; Fig. 5a versus Fig. 5b). Removing Ca++ in aCSF blocked the intracellular fluorescence induced by Meth (P < 0.01, two-way ANOVA; Fig. 5a). Mg++ (2 mM) was also added to the culture neurobasal medium containing 1.8 mM Ca++ and 0.81 mM Mg++ on DIV 10. Similar to the responses seen in the aCSF, exogenous Mg++ significantly suppressed intracellular fluorescence induced by Meth (P < 0.01, two-way ANOVA; Fig. 5b).
Figure 5:
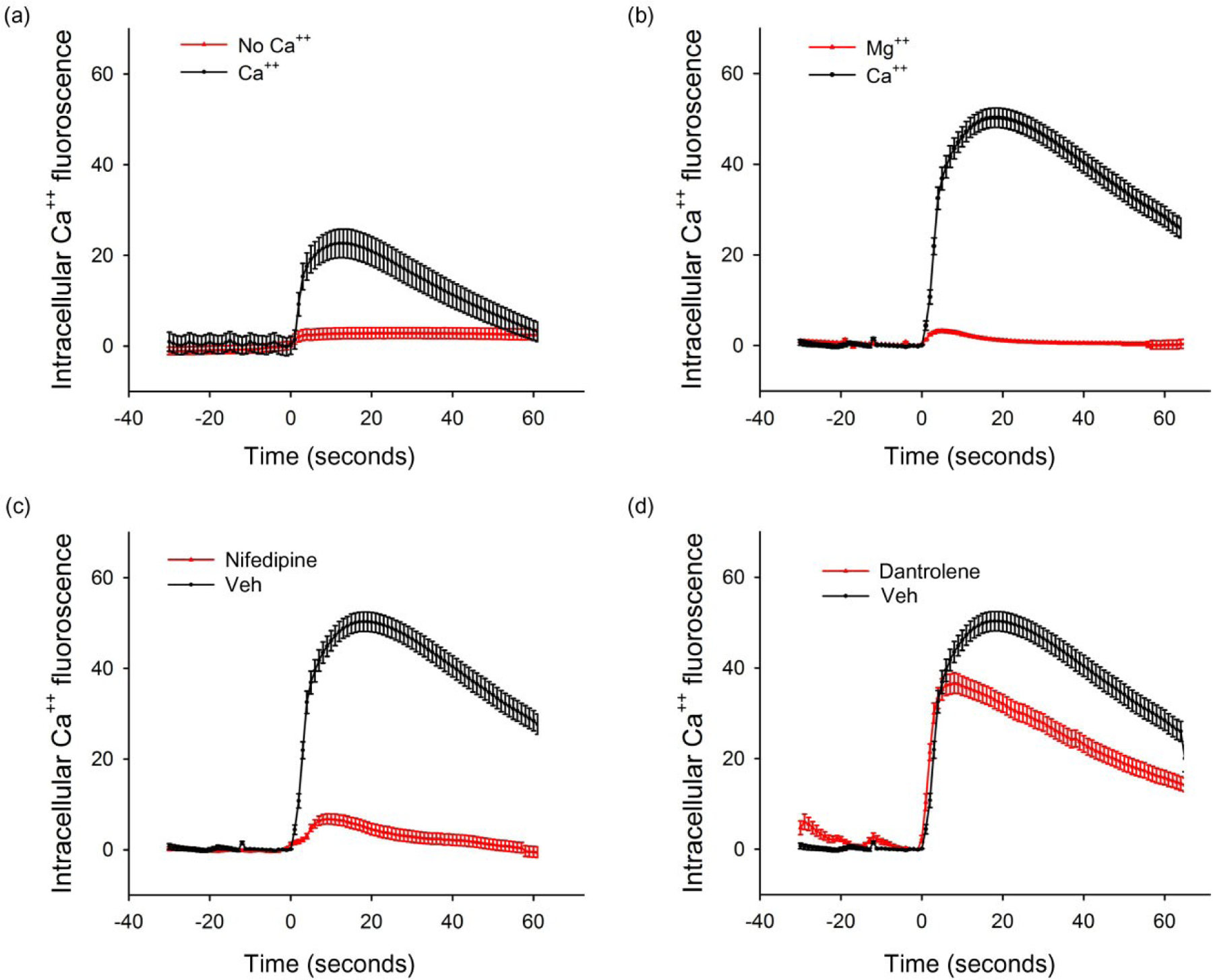
Mg++, nifedipine or low extracellular Ca++, blocked, while dantrolene partially antagonized, methamphetamine (Meth)-mediated increase in intracellular Ca++ signal in primary cortical neurons expressing GCaMP5. (a) Cultured primary cells expressing GCaMP5 were washed with artificial cerebrospinal fluid (aCSF) containing either Ca++ (0.1 mM) or no Ca++ on DIV 10. Administration of Meth (0.5 mM) increased intracellular fluorescence in cells incubated in aCSF medium with Ca++. Meth-mediated increase in intracellular Ca++ fluorescence was blocked by low Ca++. (B) Mg++ (2 mM) or vehicle (saline) was added to the culture neural basal medium containing 1.8 mM Ca++ on DIV 10. Meth induced a much higher intracellular Ca++ signal in neural basal medium (b) compared with aCSF (a). Treatment with Mg++ significantly suppressed Meth-mediated Ca++ influx into cytosol. (c) Nifedipine or vehicle was given to the culture medium 1 minute prior to Meth (0.5 mM). The amplitude of intracellular fluorescence induced by Meth was blocked by nifedipine. (d) Dantrolene did not alter the initial increase of Ca++ signal induced by Meth. The peak intensity of intracellular green fluorescence was, however, partially reduced by dantrolene
To examine the interaction with membrane Ca++ channels, culture medium was treated with nifedipine (5 μM) 1 minute prior to Meth (0.5 mM) on DIV 10. The amplitude of intracellular fluorescence induced by Meth was also markedly attenuated by nifedipine (P < 0.01, two-way ANOVA; Fig. 5c).
Dantrolene partially antagonized Meth-mediated increase in [Ca++]i in primary cortical neuronal culture
Cultured cells were treated with 300 nM dantrolene 1 minute prior to Meth. Dantrolene did not alter the initial increase of Ca++ signal induced by Meth. The slope of Ca++ influx (the rate of increase in intracellular fluorescence) was calculated in the first 5 seconds after delivery of Meth in 89 neurons. No significant difference was found between cells treated with dantrolene or vehicle (P = 0.43, t-test). However, dantrolene reduced the peak intensity of intracellular epifluorescence by 25% (P < 0.01, t-test; Fig. 5d), suggesting that dantrolene partially antagonized Meth-mediated increase in [Ca++]i.
Meth increased GCaMP5 fluorescence in vivo
AAV-GCaMP5 was administered to the parietal cortex in six adult rats. Fourteen days after viral delivery, animals were injected with either Meth or vehicle. Brains were scanned by IVIS 30 minutes after drug injection. Administration of Meth (5 mg/kg, i.p.), but not saline, significantly increased fluorescence in animals treated with AAV-GCaMP5 (P < 0.01, one-way ANOVA + post hoc Newman–Keuls test; Fig. 6). These data suggest that Meth enhanced [Ca++]i in the cerebral cortex in vivo.
Figure 6:
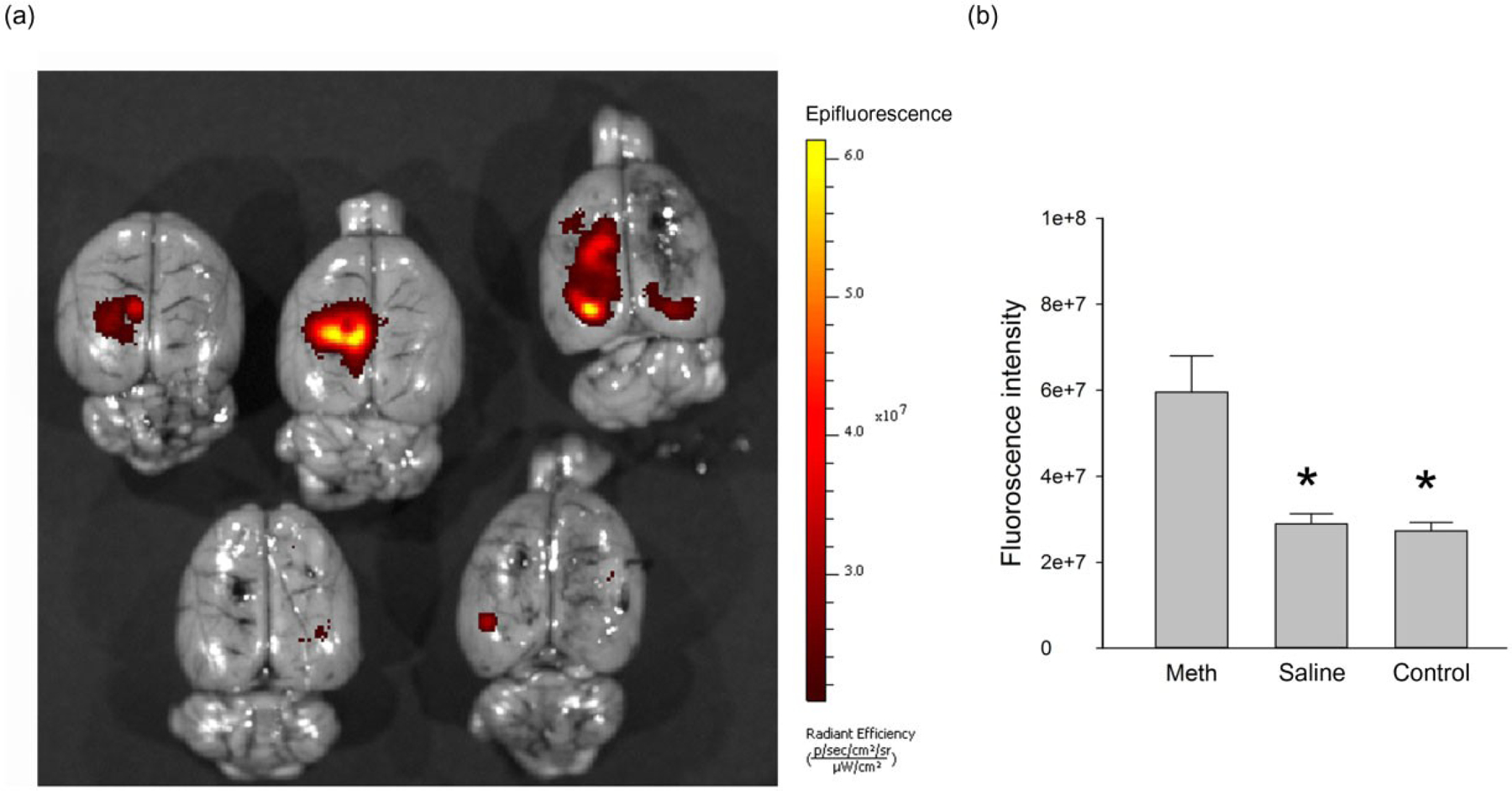
Representative IVIS (In Vivo Imaging System) scans from five brains indicates that methamphetamine (Meth) increased Ca++ signaling in animals pre-treated AAV-GCaMP5. AAV1-GCaMP5 was injected to the parietal cortex of adult rats 2 weeks prior to Meth or saline administration. (a) Brain tissues were collected for IVIS scanning 30 minutes after saline (lower two brains) or Meth (5 mg/kg, i.p., top three brains) administration. Meth administration increased green fluorescent protein (GFP) fluorescence in animal received Meth but not saline injection. (b) Fluorescent intensity analysis of selected area (0.15 cm2) near the injection site was performed in six rat brains using Living ImageR Software Version 4.0. Corresponding areas in the contralateral hemispheres were selected as controls. Meth significantly increased fluorescence in brain region receiving AAV-GCaMP5 injection (*P < 0.01, one-way ANOVA + post hoc Newman–Keuls test)
Immunohistochemistry
Adult rats receiving AAV-GCaMP5 were treated with saline or Meth (5 mg/kg, i.p.). Brains were collected and fixed 30–40 minutes after injection and were sectioned for immunohistochemistry. GCaMP5 fluorescence was found in the parietal cortex near the site of viral injection (Fig. 7a–h) and the needle track (Fig. 7j–k & m–o). The enhanced green fluorescence by Meth was localized mainly proximal to the site of viral injection (Fig. 7b versus Fig. 7f, Fig. 7i versus Fig. 7p). It could also be found near the needle track (Fig. 7j versus Fig. 7n). High-magnification confocal images indicated that these green fluorescence cells co-labeled with NeuN (Fig. 7d & h). These data suggest that Meth enhanced [Ca++]i in neurons of the cerebral cortex in vivo.
Figure 7:
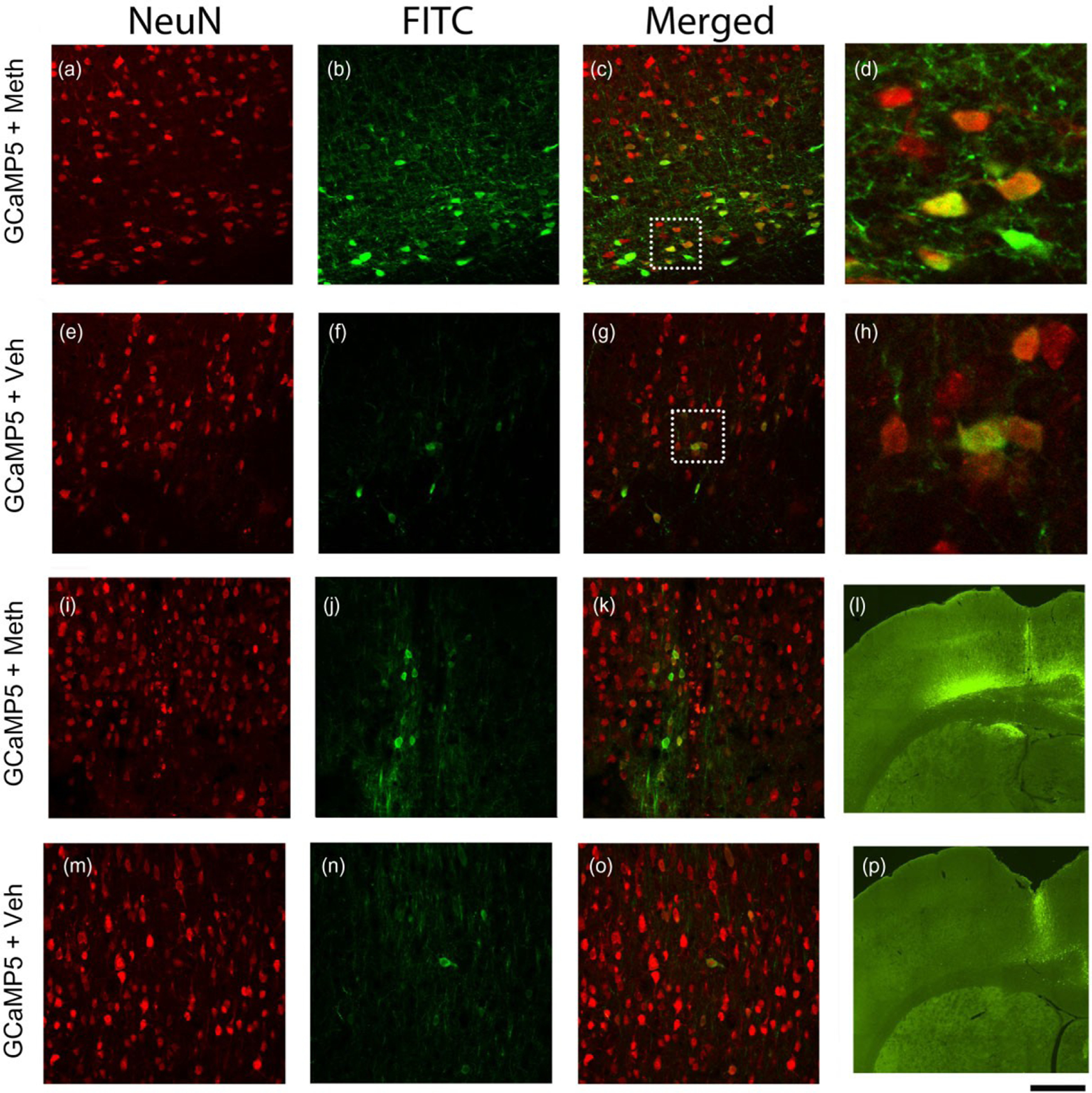
methamphetamine (Meth) increased Ca++-dependent green fluorescence of GCaMP5 in the cortical neurons from adult rat brains. Adult rats receiving AAV-GCaMP5 were treated with saline or Meth (5 mg/kg, i.p.). Brain tissues were fixed 30–40 minutes after injection and were later sectioned. Green fluorescence was found in the cerebral cortex near the site of injection (a–h) and needle track (i–k and m–o). Stronger green fluorescence was found in rats treated with Meth compared with vehicle (b versus f; j versus n; i versus p). Confocal images indicated that Meth enhanced green fluorescence in NeuN cells (c versus g; k versus o). (d & h) High magnification images taken from the injection sites (dotted squares in c, and g) indicate co-localization of green fluorescence and NeuN. (l) Meth enhanced green fluorescence in the needle track and injection site above the corpus callosum in the left parietal cortex. Less green fluorescence was found in another animal treated with saline (p). Scale bar: a–c, e–g, i–k, and m–o, 50 μm; d and h, 13 μm; l and p, 600 μm
Discussion
Calcium participates in many Meth- or AM-mediated physiological and pharmacological reactions. Calcium and L-type Ca++ channel are involved in the development of AM-induced behavioral sensitization (Uramura et al. 2000; Volkow et al. 2001). Meth-mediated place preference in mice was associated with acetylation of histone H3 following an increase in intracellular Ca++, which was also antagonized by L-type Ca++ blocker (Shibasaki et al. 2011). Nicardipine, given directly to the caudate putamen, inhibited Meth-mediated hyperactivity in rats (Hori et al. 1998). The calcium channel blocker verapamil reduced Meth-mediated apoptosis in the cerebellar neurons (Zhou, Liang & Li 2004). Isradipine, a dihydropyridine-class calcium channel antagonist, reduced Meth-mediated cognitive and physiological changes in human (Johnson, Ait-Daoud & Wells 2000). These data suggest a close interaction between Ca++ and Meth. In our study, we demonstrated a rapid increase in intracellular Ca++ in primary cortical cultured cells expressing GCaMP5 after Meth or Glu administration. Both Glu- and Meth-mediated Ca++i was antagonized by MK801. We also found that the antagonistic response of MK801 was revisable. The culture wells were washed twice with half media exchange after application of MK801 (i.e. exchange 50% culture media ×2 times). Meth-mediated excitation was partially recovered after wash, suggesting that the antagonistic response of MK801 was reversible. These Meth responsive cells expressed the neuronal marker NeuN. Our data support the notion that the interaction of Ca++i and Meth occurred in neurons through excitatory amino acids. In our control study, we found that Meth or Glu did not alter GFP fluorescence intensity in primary cortical cells overexpressing GFP by AAV1-GFP (data not shown), suggesting that these compounds did not alter the expression of fluorescence from GFP protein and the changes in fluorescence were due to the calcium binding property of GCaMP5 in primary cells.
Previous studies have demonstrated that DA can change [Ca++]i. For example, the D2 receptor antagonist raclopride induced a transient increase in Ca++i, which was antagonized by the L-type voltage-gated Ca++ channel antagonist nifedipine in primary neuronal cultures derived from fetal midbrain (Yasumoto et al. 2004). DA also modulated voltage-gated Ca++ channels through D2 receptor in primary olfactory neurons (Okada et al. 2003) and Ca++ current via D1 receptors in rat neostriatal neurons (Surmeier et al. 1995). As Meth activates DA receptors by releasing DA from nerve terminals, Meth-mediated [Ca++]i reaction can be confounded by its indirect reaction through DA. In our study, we examined Meth-mediated Ca++i in primary cortical neurons as neurons in the cerebral cortex do not contain DA biosynthetic enzyme tyrosine hydroxylase (Ungerstedt 1971). We demonstrated that Meth mediated Ca++i in the primary cortical neuronal culture, supporting a non-DA-ergic regulation of Ca++i by Meth.
The rise in cytosolic Ca++ can be derived from influx of extracellular Ca++ or release of Ca++ from the endoplasmic reticulum (ER). We examined the response of extracellular Ca++ by removing Ca++ in the aCSF medium. Lowering extracellular Ca++ reduced Meth-mediated cytosolic Ca++ signals from GCaMP5, suggesting that Meth enhanced the Ca++ influx from extracellular space to cytosol. We also increased extracellular Mg++ to the neural basal medium, which contained 1.8 mM Ca++. The exogenous Mg++ can compete with Ca++ for influx after Meth stimulation. Mg++, similar to MK801, is also a Glu receptor antagonist. We demonstrated that increasing Mg++ in medium abolished Meth-mediated increase in intracellular green fluorescence, suggesting that Mg++ effectively suppressed Ca++ entry induced by Meth and possibly its cascade of responses.
Nifedipine has been shown to attenuate Meth or AM-mediated Ca++ entry in non-neuronal cardiac cells (Sugimoto et al. 2009) or PC12 cells (Kantor et al. 2004). In this study, we examined the interaction of nifedipine and Meth in neuronal culture. We demonstrated that nifedipine, similar to MK801, antagonized Meth-mediated increase in Ca++i, suggesting that influx of Ca++ is mediated through L-type Ca++ channel in primary neuronal cells. It has been well documented that L-type Ca++ channel regulates excitatory amino acid-mediated cellular response (Rajadhyaksha et al. 1999; Cooper & White 2000). It is worthy to examine the common mechanisms between glutamate receptors and L-type Ca++ channels in Meth reaction.
Cytosolic Ca++ is also regulated by the released of Ca++ from the ER through RyR. Previous studies have shown that the RyR inhibitor ruthenium red did not antagonize Meth-enhanced [Ca++]i in cultured cardiomyocytes pre-loaded with fluo-4 AM (Sugimoto et al. 2009), suggesting a non-RyR reaction in non-neuronal cells. In this study, we examined the interaction of RyR and Meth in primary neuronal cells. Dantrolene, a clinically used RyR inhibitor, did not alter the influx of Ca++ after Meth administration as the slope of [Ca++]i at early time (i.e. 5 seconds) was not altered by dantrolene. However, dantrolene partially reduced the peak [Ca++]i by 25% 5 seconds after Meth administration, suggesting that Meth mobilized intracellular Ca++ from the ER after the influx of Ca++. As blocking Ca++ influx by Mg++ or Ca++ channel inhibitors completely abolished Meth-mediated [Ca++]i, the release of Ca++ from ER may be triggered by the initial influx of Ca++ through the membrane Ca++ channels. Our data also imply that dantrolene is a weak antagonist for Meth-mediated Ca++ responses.
Meth-activated intracellular Ca++ signals were also found in brain parenchyma in this study. We examined Ca++ and GCaMP5 interaction in brain tissue by IVIS analysis. Similar to the responses in neuronal culture, systemic injection of Meth increased fluorescence in brains pre-treated with AAV-GCaMP5. Weak fluorescence was found after saline injection. The enhanced fluorescence response was found 30 minutes after Meth injection, which correlated with behavioral responses of Meth reported previously. These data suggest that Ca++ entry may be a major target for Meth in behavioral responses in vivo. Using fluorescence microscopy and immunohistochemistry, fluorescence signal was found in the parietal cortex near the site of viral infection. Injection of saline induced a weak green fluorescence signal in the cerebral cortex, showing a regulated basal Ca++ signal from the infected cells. In contrast, Meth greatly enhanced green fluorescence in the brain cortex. The enhanced fluorescence by Meth co-localized primarily to NeuN (+) cells, indicating that our IVIS measurements represent a neuronal response. Taken together, our data suggest that Meth increased Ca++i mainly in neuronal cells in vivo.
Supplementary Material
Movie S1 Meth increased [Ca++]i in cultured primary cortical neurons overexpressing GCaMP5
Movie 2 MK801 suppressed Meth-mediated intracellular Ca++ signals in cultured primary cortical neurons overexpressing GCaMP5
SUPPORTING INFORMATION
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site:
Figure S1 Characterization of GCaMP5 from AAV in rat primary cortical neurons
Figure S2 Meth activated intracellular Ca++ fluorescence in PFA-fixed and non-fixed cells
Acknowledgements
The authors thank Doug Howard and Lowella Fortuno for their technical assistance (vector production and in vitro testing, respectively). This study was supported by the National Health Research Institutes, Taiwan.
Footnotes
Conflict of interest
All authors declare that they have no conflict of interest.
References
- Akerboom J, Chen TW, Wardill TJ, Tian L, Marvin JS, Mutlu S, Calderon NC, Esposti F, Borghuis BG, Sun XR, Gordus A, Orger MB, Portugues R, Engert F, Macklin JJ, Filosa A, Aggarwal A, Kerr RA, Takagi R, Kracun S, Shigetomi E, Khakh BS, Baier H, Lagnado L, Wang SS, Bargmann CI, Kimmel BE, Jayaraman V, Svoboda K, Kim DS, Schreiter ER, Looger LL (2012) Optimization of a GCaMP calcium indicator for neural activity imaging. J Neurosci 32:13819–13840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boireau A, Bordier F, Dubedat P, Doble A (1995) Methamphetamine and dopamine neurotoxicity: differential effects of agents interfering with glutamatergic transmission. Neurosci Lett 195:9–12. [DOI] [PubMed] [Google Scholar]
- Cadet JL, Brannock C, Krasnova IN, Ladenheim B, McCoy MT, Chou J, Lehrmann E, Wood WH, Becker KG, Wang Y (2010) Methamphetamine-induced dopamine-independent alterations in striatal gene expression in the 6-hydroxydopamine hemiparkinsonian rats. PLoS ONE 5:e15643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadet JL, Krasnova IN, Jayanthi S, Lyles J (2007) Neurotoxicity of substituted amphetamines: molecular and cellular mechanisms. Neurotox Res 11:183–202. [DOI] [PubMed] [Google Scholar]
- Chipana C, Torres I, Camarasa J, Pubill D, Escubedo E (2008) Memantine protects against amphetamine derivatives-induced neurotoxic damage in rodents. Neuropharmacology 54:1254–1263. [DOI] [PubMed] [Google Scholar]
- Chung SH, Sun L, Gabel CV (2013) In vivo neuronal calcium imaging in C. elegans. J Vis Exp 74:e50357. doi: 10.3791/50357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DC, White FJ (2000) L-type calcium channels modulate glutamate-driven bursting activity in the nucleus accumbens in vivo. Brain Res 880:212–218. [DOI] [PubMed] [Google Scholar]
- Del AA, Martinez R, Mora F (1998) Amphetamine increases extracellular concentrations of glutamate in the prefrontal cortex of the awake rat: a microdialysis study. Neurochem Res 23:1153–1158. [DOI] [PubMed] [Google Scholar]
- Deng X, Wang Y, Chou J, Cadet JL (2001) Methamphetamine causes widespread apoptosis in the mouse brain: evidence from using an improved TUNEL histochemical method. Mol Brain Res 93:64–69. [DOI] [PubMed] [Google Scholar]
- Finnegan KT, Taraska T (1996) Effects of glutamate antagonists on methamphetamine and 3,4-methylenedioxymetham-phetamine-induced striatal dopamine release in vivo. J Neurochem 66:1949–1958. [DOI] [PubMed] [Google Scholar]
- Fukakusa A, Nagai T, Mizoguchi H, Otsuka N, Kimura H, Kamei H, Kim HC, Nabeshima T, Takuma K, Yamada K (2008) Role of tissue plasminogen activator in the sensitization of methamphetamine-induced dopamine release in the nucleus accumbens. J Neurochem 105:436–444. [DOI] [PubMed] [Google Scholar]
- Goodwin JS, Larson GA, Swant J, Sen N, Javitch JA, Zahniser NR, De Felice LJ, Khoshbouei H (2009) Amphetamine and methamphetamine differentially affect dopamine transporters in vitro and in vivo. J Biol Chem 284:2978–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Yu S, Bae E, Shen H, Wang Y (2013) Methamphetamine alters reference gene expression in nigra and striatum of adult rat brain. Neurotoxicology 39:138–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson H, Laurenzana E, Owens SM (2006) Quantitative determination of total methamphetamine and active metabolites in rat tissue by liquid chromatography with tandem mass spectrometric detection. AAPS J 8:E709–E717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori Y, Takeda H, Tsuji M, Matsumiya T (1998) Differentiation of the inhibitory effects of calcium antagonists on abnormal behaviors induced by methamphetamine or phencyclidine. Pharmacology 56:165–174. [DOI] [PubMed] [Google Scholar]
- Howard DB, Powers K, Wang Y, Harvey BK (2008) Tropism and toxicity of adeno-associated viral vector serotypes 1,2,5,6,7, 8,9 in rat neurons and glia in vitro. Virology 372:24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BA, Ait-Daoud N, Wells LT (2000) Effects of isradipine, a dihydropyridine-class calcium channel antagonist, on D-methamphetamine-induced cognitive and physiological changes in humans. Neuropsychopharmacology 22:504–512. [DOI] [PubMed] [Google Scholar]
- Kantor L, Hewlett GH, Park YH, Richardson-Burns SM, Mellon MJ, Gnegy ME (2001) Protein kinase C and intracellular calcium are required for amphetamine-mediated dopamine release via the norepinephrine transporter in undifferentiated PC12 cells. J Pharmacol Exp Ther 297:1016–1024. [PubMed] [Google Scholar]
- Kantor L, Zhang M, Guptaroy B, Park YH, Gnegy ME (2004) Repeated amphetamine couples norepinephrine transporter and calcium channel activities in PC12 cells. J Pharmacol Exp Ther 311:1044–1051. [DOI] [PubMed] [Google Scholar]
- Konradi C, Leveque JC, Hyman SE (1996) Amphetamine and dopamine-induced immediate early gene expression in striatal neurons depends on postsynaptic NMDA receptors and calcium. J Neurosci 16:4231–4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp RF, Leech CA, Roe MW (2014) Resveratrol interferes with Fura-2 intracellular calcium measurements. J Fluoresc 24:279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczenski R, Everall IP, Crews L, Adame A, Grant I, Masliah E (2007) Escalating dose-multiple binge methamphetamine exposure results in degeneration of the neocortex and limbic system in the rat. Exp Neurol 207:42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa K, Shibasaki M, Mizuno K, Ohkuma S (2011) Gabapentin blocks methamphetamine-induced sensitization and conditioned place preference via inhibition of alpha(2)/ delta-1 subunits of the voltage-gated calcium channels. Neuroscience 176:328–335. [DOI] [PubMed] [Google Scholar]
- Lan KC, Chang AC, Liu SH, Ho IK, Lin-Shiau SY (2009) Enhancing effects of morphine on methamphetamine-induced reinforcing behavior and its association with dopamine release and metabolism in mice. J Neurochem 109:382–392. [DOI] [PubMed] [Google Scholar]
- Loweth JA, Singer BF, Baker LK, Wilke G, Inamine H, Bubula N, Alexander JK, Carlezon WA Jr., Neve RL, Vezina P (2010) Transient overexpression of alpha-Ca2+/calmodulin-dependent protein kinase II in the nucleus accumbens shell enhances behavioral responding to amphetamine. J Neurosci 30:939–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Wang Y, Kuang SY, Chiang YH, Hoffer BJ (2010) Decreased level of Nurr1 in heterozygous young adult mice leads to exacerbated acute and long-term toxicity after repeated methamphetamine exposure. PLoS ONE 5:e15193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao T, O’Connor DH, Scheuss V, Nakai J, Svoboda K (2008) Characterization and subcellular targeting of GCaMP-type genetically-encoded calcium indicators. PLoS ONE 3:e1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JF, O’Dell SJ, Weihmuller FB (1993) Dopamine-glutamate interactions in methamphetamine-induced neurotoxicity. J Neural Transm Gen Sect 91:241–254. [DOI] [PubMed] [Google Scholar]
- Nakai J, Ohkura M, Imoto K (2001) A high signal-to-noise Ca(2+) probe composed of a single green fluorescent protein. Nat Biotechnol 19:137–141. [DOI] [PubMed] [Google Scholar]
- Ohno M, Yoshida H, Watanabe S (1994) NMDA receptor-mediated expression of Fos protein in the rat striatum following methamphetamine administration: relation to behavioral sensitization. Brain Res 665:135–140. [DOI] [PubMed] [Google Scholar]
- Okada Y, Miyamoto T, Toda K (2003) Dopamine modulates a voltage-gated calcium channel in rat olfactory receptor neurons. Brain Res 968:248–255. [DOI] [PubMed] [Google Scholar]
- Rajadhyaksha A, Barczak A, Macias W, Leveque JC, Lewis SE, Konradi C (1999) L-type Ca(2+) channels are essential for glutamate-mediated CREB phosphorylation and c-fos gene expression in striatal neurons. J Neurosci 19:6348–6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid MS, Hsu K Jr., Berger SP (1997) Cocaine and amphetamine preferentially stimulate glutamate release in the limbic system: studies on the involvement of dopamine. Synapse 27:95–105. [DOI] [PubMed] [Google Scholar]
- Shen H, Luo Y, Yu SJ, Wang Y (2011) Enhanced neurodegeneration after a high dose of methamphetamine in adenosine A3 receptor null mutant mice. Neuroscience 194:170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibasaki M, Mizuno K, Kurokawa K, Ohkuma S (2011) L-type voltage-dependent calcium channels facilitate acetylation of histone H3 through PKCgamma phosphorylation in mice with methamphetamine-induced place preference. J Neurochem 118:1056–1066. [DOI] [PubMed] [Google Scholar]
- Shigetomi E, Kracun S, Khakh BS (2010) Monitoring astrocyte calcium microdomains with improved membrane targeted GCaMP reporters. Neuron Glia Biol 6:183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto K, Okamura K, Tanaka H, Takashima S, Ochi H, Yamamoto T, Matoba R (2009) Methamphetamine directly accelerates beating rate in cardiomyocytes by increasing Ca(2+) entry via L-type Ca(2+) channel. Biochem Biophys Res Commun 390:1214–1220. [DOI] [PubMed] [Google Scholar]
- Sun XR, Badura A, Pacheco DA, Lynch LA, Schneider ER, Taylor MP, Hogue IB, Enquist LW, Murthy M, Wang SS (2013) Fast GCaMPs for improved tracking of neuronal activity. Nat Commun 4:2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Bargas J, Hemmings HC Jr., Nairn AC, Greengard P (1995) Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron 14:385–397. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Shiozaki Y, Masukawa Y, Misawa M (1992) Effects of calcium antagonists on the cocaine- and methamphetamine-induced conditioned place preference. Arukoru Kenkyuto Yakubutsu Ison 27:81–90. [PubMed] [Google Scholar]
- Tian C, Murrin LC, Zheng JC (2009a) Mitochondrial fragmentation is involved in methamphetamine-induced cell death in rat hippocampal neural progenitor cells. PLoS ONE 4:e5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Hires SA, Mao T, Huber D, Chiappe ME, Chalasani SH, Petreanu L, Akerboom J, McKinney SA, Schreiter ER Bargmann CI, Jayaraman V, Svoboda K, Looger LL (2009b) Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat Methods 6:875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungerstedt U (1971) Stereotaxic mapping of the monoamine pathways in the rat brain. Acta Physiol Scand Suppl 367:1–48. [DOI] [PubMed] [Google Scholar]
- Uramura K, Yada T, Muroya S, Shioda S, Shiratani T, Takigawa M (2000) Methamphetamine induces cytosolic Ca2+ oscillations in the VTA dopamine neurons. Neuroreport 11:1057–1061. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Chang L, Wang GJ, Fowler JS, Franceschi D, Sedler M, Gatley SJ, Miller E, Hitzemann R, Ding YS, Logan J (2001) Loss of dopamine transporters in methamphetamine abusers recovers with protracted abstinence. J Neurosci 21:9414–9418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME, Xue CJ, Li Y, Wavak D (2000) Amphetamine increases glutamate efflux in the rat ventral tegmental area by a mechanism involving glutamate transporters and reactive oxygen species. J Neurochem 75:1634–1644. [DOI] [PubMed] [Google Scholar]
- Yasumoto F, Negishi T, Ishii Y, Kyuwa S, Kuroda Y, Yoshikawa Y (2004) Dopamine receptor 2 regulates L-type voltage-gated calcium channel in primary cultured mouse midbrain neural network. Cell Mol Neurobiol 24:877–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SJ, Airavaara M, Shen H, Chou J, Harvey BK, Wang Y (2012) Suppression of endogenous PPARγ increases vulnerability to methamphetamine -induced injury in mouse nigrostriatal dopaminergic pathway. Psychopharmacology (Berl) 221:479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SJ, Tseng K, Shen H, Harvey BK, Airavaara M, Wang Y (2013) Local administration of AAV-BDNF to subventricular zone induces functional recovery in stroke rats. PLoS ONE 8:e81750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JL, Liang JH, Li CL (2004) Inhibition of methamphetamine-induced apoptosis by the calcium channel blocker verapamil in rat cerebellar neurons. Beijing Da Xue Xue Bao 36:361–365. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1 Meth increased [Ca++]i in cultured primary cortical neurons overexpressing GCaMP5
Movie 2 MK801 suppressed Meth-mediated intracellular Ca++ signals in cultured primary cortical neurons overexpressing GCaMP5
SUPPORTING INFORMATION
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site:
Figure S1 Characterization of GCaMP5 from AAV in rat primary cortical neurons
Figure S2 Meth activated intracellular Ca++ fluorescence in PFA-fixed and non-fixed cells