

Abstract
Purpose:
Genomic alterations in DNA damage repair (DDR) genes other than BRCA may confer synthetic lethality with PARP inhibition in metastatic castration-resistant prostate cancer (mCRPC). To test this hypothesis, the phase 2 TRITON2 study of rucaparib included patients with mCRPC and deleterious non-BRCA DDR gene alterations.
Patients and Methods:
TRITON2 enrolled patients who had progressed on 1 to 2 lines of next-generation androgen receptor (AR)–directed therapy and 1 taxane-based chemotherapy for mCRPC. Key endpoints were investigator-assessed radiographic response per modified RECIST/PCWG3 and prostate-specific antigen (PSA) response (≥50% decrease from baseline).
Results:
TRITON2 enrolled 78 patients with a non-BRCA DDR gene alteration (ATM [n = 49], CDK12 [n = 15], CHEK2 [n = 12], and other DDR genes [n = 14]). Among patients evaluable for each endpoint, radiographic and PSA responses were observed in a limited number of patients with an alteration in ATM (2/19 [10.5%] and 2/49 [4.1%], respectively), CDK12 (0/10 [0%] and 1/15 [6.7%], respectively), or CHEK2 (1/9 [11.1%] and 2/12 [16.7%], respectively), including no radiographic or PSA responses in 11 patients with confirmed biallelic ATM loss or 11 patients with ATM germline mutations. Responses were observed in patients with alterations in the DDR genes PALB2, FANCA, BRIP1, and RAD51B.
Conclusions:
In this prospective, genomics-driven study of rucaparib in mCRPC, we found limited radiographic/PSA responses to PARP inhibition in men with alterations in ATM, CDK12, or CHEK2. However, patients with alterations in other DDR-associated genes (eg, PALB2) may benefit from PARP inhibition.
Keywords: DNA damage repair genes, metastatic castration-resistant prostate cancer, rucaparib
INTRODUCTION
Poly(ADP-ribose) polymerase (PARP) inhibitors are approved for use in BRCA1 and BRCA2 (BRCA)-mutant breast and ovarian cancers (1, 2), and have also shown evidence of activity in patients with BRCA-mutant metastatic castration-resistant prostate cancer (mCRPC), based on a well-described synthetically lethal effect (3). Confirmed radiographic and prostate-specific antigen (PSA) responses were observed in 25 (43.9%) of 57 and 51 (52.0%) of 98 patients with a deleterious BRCA alteration, respectively, in the phase 2 TRITON2 study (CO-338–052; NCT02952534) evaluating the PARP inhibitor rucaparib in men who progressed on an androgen receptor (AR)–directed therapy and chemotherapy (4). Olaparib and niraparib also have reported activity in patients with BRCA-mutant mCRPC (5–8). Rucaparib, olaparib, and niraparib have received Breakthrough Therapy designations from the U.S. Food and Drug Administration for the treatment of patients with BRCA-mutant mCRPC (9–11).
Beyond BRCA, deleterious alterations in other genes may also lead to DNA damage repair (DDR) deficiency and are hypothesized to render tumor cells sensitive to PARP inhibition. For example, ATM and CHEK2 are sensors of DNA damage, CDK12 is a positive regulator of BRCA genes, and PALB2 and FANCA interact with BRCA1 and/or BRCA2 during DNA repair (12–16). Clinical responses, defined variably as a radiographic response, or a decline in PSA or circulating tumor cell count, have been reported in mCRPC patients with alterations in ATM, CHEK2, FANCA, PALB2, or other non-BRCA DDR genes in studies evaluating olaparib (TOPARP-A [NCT01682772], TOPARP-B [NCT01682772] and niraparib (GALAHAD [NCT02854436]), and patients with alterations in these genes were included in a randomized study comparing olaparib to physician’s choice of abiraterone or enzalutamide (PROfound [NCT02987543]) (5, 6, 8, 17). Notably, the co-occurrence of alterations in different non-BRCA DDR genes, as seen in the TOPARP and PROfound studies (5, 6, 18), makes it difficult to attribute PARP inhibitor sensitivity to a given gene.
The available data provide a compelling rationale to explore PARP inhibition in men with mCRPC and a DDR gene alteration, and to define the set of DDR gene deficiencies that confer a synthetically lethal effect with PARP inhibitors. The TRITON2 study is investigating the PARP inhibitor rucaparib in patients with mCRPC associated with alterations in 15 DDR genes. Here, we report an ad hoc analysis of patients with deleterious alterations in non-BRCA DDR genes, including ATM, CDK12, CHEK2, FANCA, and PALB2.
METHODS
Study Design
TRITON2 is an open-label, phase 2 study evaluating the safety and efficacy of rucaparib in men with mCRPC associated with DDR deficiency. This trial is being conducted at 144 centers in 12 countries. Eligible patients at least 18 years old had histologically or cytologically confirmed adenocarcinoma or poorly differentiated carcinoma of the prostate that was metastatic and that had progressed on second–generation, AR–directed therapy (e.g., abiraterone acetate, enzalutamide, or apalutamide) for prostate cancer and 1 prior line of taxane-based chemotherapy for mCRPC. Disease progression on prior therapy was based on any of the following criteria: rise in prostate-specific antigen (PSA; minimum of 2 consecutive rising levels, with an interval of 1 week or more between each determination; the most recent screening measurement must have been greater than or equal to 2 ng/mL); transaxial imaging with new or progressive soft tissue masses on computed tomography (CT) or magnetic resonance imaging (MRI) as defined by Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST); or radionuclide bone scan with at least 2 new metastatic lesions. Patients were surgically or medically castrated, with serum testosterone levels of 50 ng/dL or less.
Patients with and without measurable visceral or nodal disease per RECIST criteria were eligible for the study; patients without measurable disease were required to have PSA levels of greater than 2 ng/mL on the most recent measurement. Patients had an Eastern Cooperative Oncology Group Performance Status of 0 or 1 and adequate organ function. Patients who had prior treatment with any PARP inhibitor, mitoxantrone, cyclophosphamide, or any platinum-based chemotherapy were excluded.
The study was approved by national or local institutional review boards and was carried out in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines of the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. Patients provided written informed consent before participation.
Procedures
Patients were screened for the presence of a deleterious somatic or germline alteration in BRCA1, BRCA2, ATM, BARD1, BRIP1, CDK12, CHEK2, FANCA, NBN, PALB2, RAD51, RAD51B, RAD51C, RAD51D, or RAD54L through central genomic testing of plasma or tumor tissue (archival or contemporaneous), or through local testing. Central testing was performed by Foundation Medicine, Inc (19, 20). To classify alterations as somatic or germline, whole blood samples were tested by Color Genomics using an assay validated to detect germline alterations (21, 22).
Patients received oral rucaparib 600 mg twice daily until confirmed radiographic disease progression as assessed by investigator based on modified RECIST and/or Prostate Cancer Clinical Trials Working Group 3 (PCWG3) criteria, unequivocal clinical disease progression, death, or other reason for discontinuation. Dose reductions were permitted if a patient had a grade 3 or greater or a persistent grade 2 adverse event (AE).
Tumor assessments (computed tomography/magnetic resonance imaging and bone scans) were performed every 8 weeks for 24 weeks, then every 12 weeks, and at treatment discontinuation. PSA was evaluated at baseline and every 4 weeks thereafter.
After discontinuation, all patients had a 28-day follow-up visit, and subsequent treatment and survival information was collected every 12 weeks from the last dose of rucaparib until death, loss to follow-up, or withdrawal of consent from study. For patients who discontinued for a reason other than disease progression, tumor assessments continued as above.
Outcomes
The proportion of patients achieving a confirmed complete or partial response per modified RECIST/PCWG3 as assessed by the investigator is reported for patients with measurable disease at baseline (hereafter, “evaluable patients”). The proportion of patients with a confirmed PSA response (50% or greater decrease from baseline confirmed by a second consecutive measurement at least 3 weeks later) is reported for patients with or without measurable disease at baseline. Additional outcomes reported here include clinical benefit rate (proportion of patients without radiographic progression per RECIST/PCWG3 criteria who were receiving treatment at a given time point [e.g., at 6 or 12 months]) and time to PSA progression.
The tumor responses were assessed by investigators per modified RECIST/PCWG3. Modified RECIST allows up to 10 target lesions, with a maximum of 5 per site, not including prostatic bed or bone lesions; MRI is allowed. For PSA response (≥ 50% decrease from baseline confirmed by a second consecutive measurement at least 3 weeks later), measurements were performed by local laboratories. Early rises in PSA (before 12 weeks following the first dose of study drug) were not considered in determining PSA progression.
The clinical benefit rate was calculated as the number of patients without radiographic progression who were on study through the given time interval divided by the number of patients who had the given amount of follow-up time. Time to PSA progression was defined as the time from the first dose of rucaparib to the date that a 25% or greater increase and absolute increase of at least 2 ng/mL above the nadir (or baseline if there was no PSA decline after baseline) in PSA was measured, plus 1 day.
Safety was assessed by monitoring AEs and vital signs, laboratory testing, and physical examination.
Statistical Analysis
For this ad hoc analysis, data are reported for patients with a non-BRCA DDR gene alteration enrolled in TRITON2 as of Jan 4, 2019, using a visit cutoff date of April 29, 2019; therefore, patients in this analysis had a minimum of 16 weeks of follow-up, allowing for at least 2 radiographic tumor assessments and for confirmation of response. The safety population included all patients who received at least 1 dose of study treatment.
Time to PSA progression was summarized using Kaplan-Meier methodology. Statistical analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC, USA).
RESULTS
Patients and Genomic Characteristics
Between Feb 15, 2017, and Jan 4, 2019, 78 patients were enrolled based on a non-BRCA DDR gene alteration identified during screening. Patients were allowed to submit multiple tissue and plasma samples for genomic testing. The majority of patients had results from 1 genomic test (44 [56.4%] patients), with the remainder having results from 2 (31 [39.7%] patients) or 3 (3 [3.8%] patients; Supplementary Table). Patients were grouped by gene alteration (ATM, CDK12, CHEK2, and other DDR gene), and patients with co-occurring alterations were included in multiple gene groups. For example, a patient with co-occurring CDK12 and CHEK2 alterations was included in both the CDK12 and CHEK2 groups.
Including cases of co-occurring alterations identified prior to enrolment or in samples tested after enrolment, 49 patients had an ATM alteration, 15 patients had a CDK12 alteration, and 12 patients had a CHEK2 alteration (Fig. 1; Supplementary Table). Among 14 patients with other DDR gene alterations, 4 had a FANCA alteration, 4 had an NBN alteration, 2 had a BRIP1 alteration, 2 had a PALB2 alteration, and 1 each had a RAD51, RAD51B, or RAD54L alteration (Fig. 1). Among 49 patients with an ATM alteration, 11 (22.4%) had a germline alteration; the most frequent type of ATM alteration was a frameshift mutation (36.1% of alterations; Supplementary Table and Supplementary Figs. S1 and S2). Zygosity could be determined for 17 (34.7%) patients with an ATM alteration, 11 of whom had a biallelic alteration and 6 had a monoallelic alteration. Among the 15 patients with CDK12 alterations, 7 (47%) had 2 distinct CDK12 alterations presumed to result in biallelic inactivation, and an additional 2 (13%) had loss of heterozygosity of the other allele. We could not confirm that these alterations were somatic due to the absence of CDK12 from our germline panel, but CDK12 alterations have not been reported to occur in the germline (16). Of the 12 patients with CHEK2 alterations, 4 were germline and 8 somatic, and biallelic inactivation was identified in 3 of 4 evaluable cases, 2 of which were germline (Figure 2, Supplementary Table).
Figure 1.
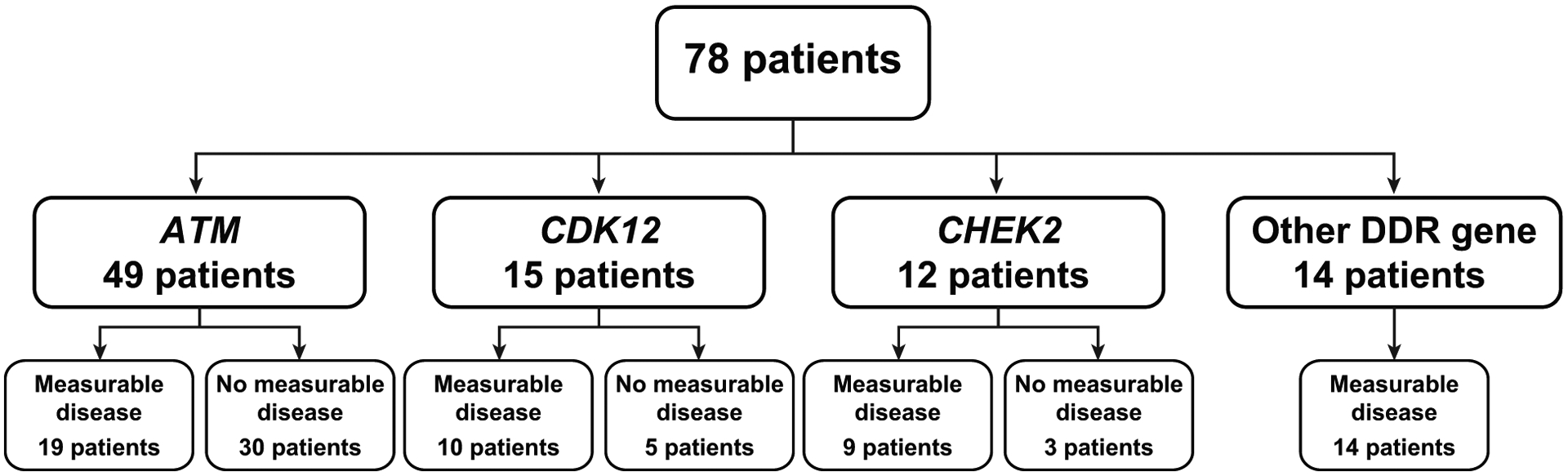
Overview of analysis groups for patients from TRITON2 with non-BRCA DDR gene alterations. Groups are not mutually exclusive and contain patients with co-occurring DDR gene alterations (see Supplementary Table). The “other DDR gene” group includes patients with an alteration in FANCA (n = 4), NBN (n = 4), BRIP1 (n = 2), PALB2 (n = 2), RAD51 (n = 1), RAD51B (n = 1), and/or RAD54L (n = 1). Abbreviation: DDR, DNA damage repair.
Figure 2.
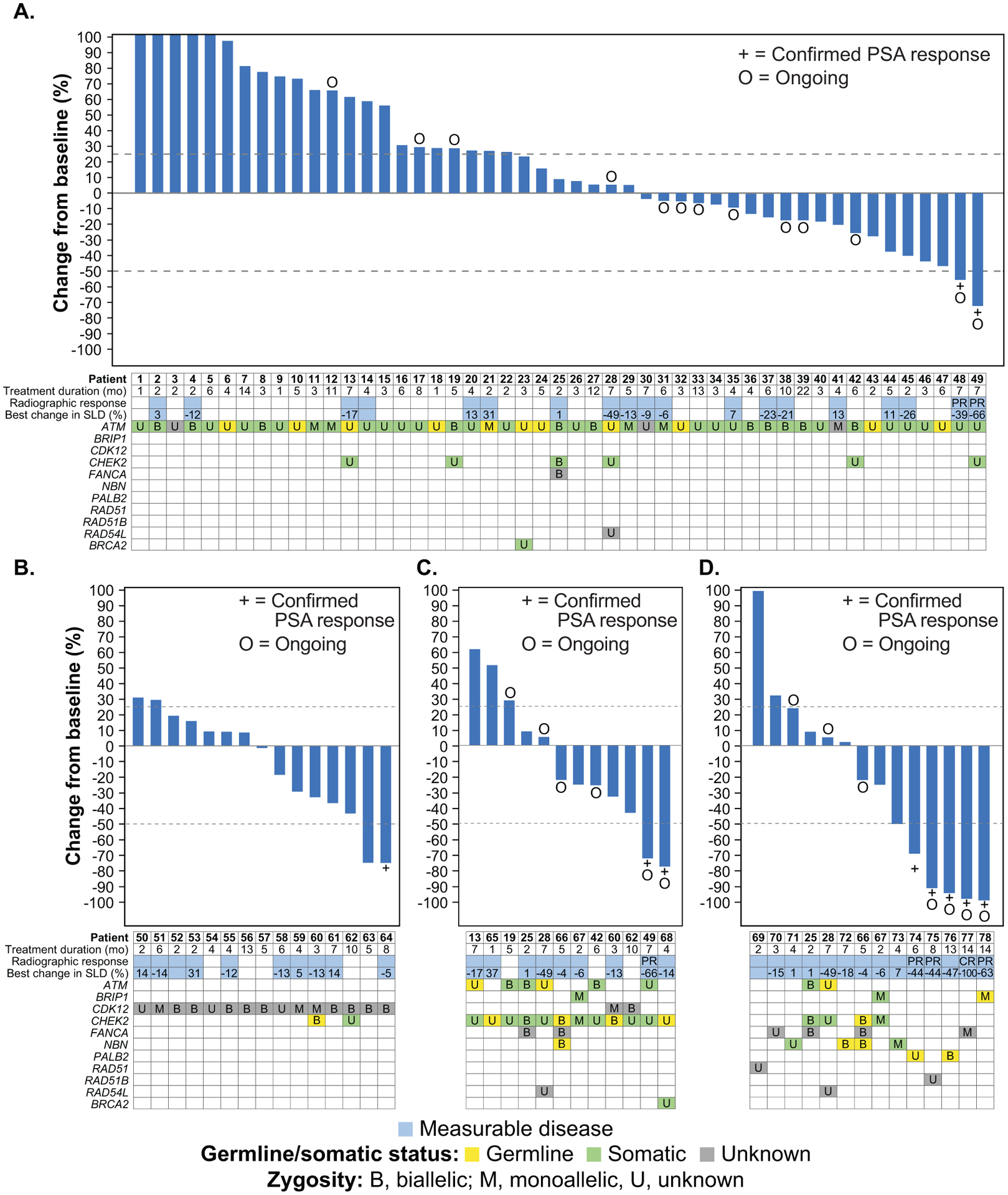
Best change from baseline in PSA in patients with an ATM alteration (A), CDK12 alteration (B), CHEK2 alteration (C), or other DDR gene alteration (D). PSA increases for patients 1–5 were 319%, 142%, 126%, 109%, and 106%; bars were capped at 100% for visual clarity. Patients 55, 56, 57, 61, 62, 63, and 64 had 2 distinct CDK12 alterations identified through tissue and/or plasma testing and were considered to have biallelic loss. Abbreviations: CR, complete response; PR, partial response; PSA, prostate-specific antigen; SLD, sum of the longest diameter.
Patients with an ATM alteration generally had lower Gleason scores at diagnosis and less measurable disease at study entry than those in the other groups (Table 1). At the visit cutoff date, patients still receiving treatment included 13 (26.5%) with an ATM alteration, 6 (50.0%) with a CHEK2 alteration, and 7 (50.0%) with another DDR gene alteration.
Table 1:
Baseline patient demographics and disease characteristics
| DDR gene group | ||||
|---|---|---|---|---|
| ATM (n = 49) | CDK12 (n = 15) | CHEK2 (n = 12) | Othera (n = 14) | |
| Age, median (IQR), years | 73.0 (68.0–77.0) | 64.0 (56.0–72.0) | 71.5 (64.5–75.0) | 66.5 (61.0–72.0) |
| Race, n (%) | ||||
| White | 35 (71.4) | 6 (40.0) | 6 (50.0) | 11 (78.6) |
| Black or African American | 3 (6.1) | 2 (13.3) | 1 (8.3) | 0 |
| Unknown | 11 (22.4) | 7 (46.7) | 5 (41.7) | 3 (21.4) |
| ECOG PS, n (%) | ||||
| 0 | 21 (42.9) | 7 (46.7) | 5 (41.7) | 2 (14.3) |
| 1 | 27 (55.1) | 8 (53.3) | 7 (58.3) | 11 (78.6) |
| ≥ 2 | 1 (2.0) | 0 | 0 | 1 (7.1) |
| Baseline PSA, median (IQR), ng/mL | 108.7 (19.7–286.8) | 72.7 (37.2–615.0) | 119.9 (33.2–256.5) | 98.8 (33.2–255.8) |
| Baseline alkaline phosphatase, median (IQR), U/L | 102.0 (72.0–198.0) | 107.0 (78.0–172.0) | 88.5 (66.5–164.5) | 84.5 (72.0–126.0) |
| Baseline albumin, median (IQR), g/L | 38.6 (36.0–41.0) | 39.0 (35.0–44.0) | 37.0 (34.5–40.0) | 38.5 (35.0–42.0) |
| Baseline lactate dehydrogenase, median (IQR), ukat/L | 3.9 (3.2–6.3) | 4.4 (2.8–6.7) | 4.2 (3.6–7.3) | 4.4 (3.3–7.2) |
| Gleason score ≥ 8 at diagnosis, n (%) | 20 (40.8) | 14 (93.3) | 6 (50.0) | 8 (57.1) |
| No. prior CRPC therapies, median (IQR)b | 2 (2–3) | 3 (2–4) | 3 (2–4) | 2 (2–3) |
| Prior therapies, n (%)c | ||||
| Abiraterone | 34 (69.4) | 11 (73.3) | 11 (91.7) | 12 (85.7) |
| Enzalutamide | 36 (73.5) | 14 (93.3) | 9 (75.0) | 7 (50.0) |
| Abiraterone and enzalutamide | 23 (46.9) | 10 (66.7) | 8 (66.7) | 5 (35.7) |
| Docetaxel | 46 (93.9) | 13 (86.7) | 12 (100) | 13 (92.9) |
| Cabazitaxel | 4 (8.2) | 2 (13.3) | 0 | 2 (14.3) |
| Sipuleucel-T | 6 (12.2) | 1 (6.7) | 1 (8.3) | 0 |
| Radium-223 | 11 (22.4) | 2 (13.3) | 3 (25.0) | 1 (7.1) |
| Measurable disease status and type (per investigator), n (%) | ||||
| Measurable disease | 19 (38.8) | 10 (66.7) | 9 (75.0) | 14 (100) |
| Only measurable nodal disease | 9 (47.4) | 6 (60.0) | 8 (88.9) | 9 (64.3) |
| Measurable visceral ± nodal disease | 10 (52.6) | 4 (40.0) | 1 (11.1) | 5 (35.7) |
| Nonmeasurable disease | 30 (61.2) | 5 (33.3) | 3 (25.0) | 0 |
| Bone-only disease | 25 (83.3) | 2 (40.0) | 1 (33.3) | 0 |
| Other non-measurable disease | 5 (16.7) | 3 (60.0) | 2 (66.7) | 0 |
Visit cutoff date: April 29, 2019.
Includes patients with an alteration in FANCA (n = 4), NBN (n = 4), BRIP1 (n = 2), PALB2 (n = 2), RAD51 (n = 1), RAD51B (n = 1), and/or RAD54L (n = 1).
Does not include luteinizing hormone–releasing hormone analogues, first-generation antiandrogens, hormones, corticosteroids, bone-targeted agents, hematopoietic growth factors, or docetaxel given for hormone-sensitive disease.
Categories are not mutually exclusive.
Abbreviations: CRPC, castration-resistant prostate cancer; DDR, DNA damage repair; ECOG PS, Eastern Cooperative Oncology Group Performance Status; IQR, interquartile range; PSA, prostate-specific antigen.
Efficacy Outcomes
ATM Cohort
Among 19 evaluable patients with an ATM alteration, 2 (10.5%) had confirmed partial radiographic responses, including 1 with a co-occurring CHEK2 alteration (Table 2, Fig. 2A). Both patients had ongoing responses (1.8+ and 2.0+ months from the date of radiographic response) at the time of the visit cutoff (Fig. 3A). A confirmed PSA response was observed in 2 (4.1%) of 49 overall patients with an ATM alteration (Table 2 and Supplementary Fig. S3A), occurring in the same 2 patients who achieved a radiographic response. The 6- and 12-month clinical benefit rate for patients with an ATM alteration with or without measurable disease was 28.6% (12 of 42 patients) and 16.7% (3 of 18 patients) (Table 2).
Table 2:
Response by DDR gene alteration
| By DDR gene group | ||||
|---|---|---|---|---|
| ATM (n = 49) | CDK12 (n = 15) | CHEK2 (n = 12) | Othera (n = 14) | |
| Confirmed investigator-assessed objective responseb | 2/19 (10.5) [1.3–33.1] | 0/10 (0) [0.0–30.8] | 1/9 (11.1) [0.3–48.2] | 4/14 (28.6) [8.4–58.1] |
| Complete response | 0/19 (0.0) | 0/10 (0) | 0/9 (0) | 1/14 (7.1) |
| Partial response | 2/19 (10.5) | 0/10 (0) | 1/9 (11.1) | 3/14 (21.4) |
| Stable disease | 9/19 (47.4) | 6/10 (60.0) | 6/9 (66.7) | 8/14 (57.1) |
| Progressive disease | 7/19 (36.8) | 3/10 (30.0) | 2/9 (22.2) | 1/14 (7.1) |
| Not evaluable | 1/19 (5.3) | 1/10 (10.0) | 0/9 (0) | 1/14 (7.1) |
| 6-month clinical benefit ratec | 12/42 (28.6) [15.7–44.6] | 3/15 (20.0) [4.3–48.1] | 3/8 (37.5) [8.5–75.5] | 6/11 (54.5) [23.4–83.3] |
| 12-month clinical benefit rated | 3/18 (16.7) [3.6–41.4] | 1/14 (7.1) [0.2–33.9] | 0/5 (0) [0.0–52.2] | 3/8 (37.5) [8.5–75.5] |
| Confirmed PSA responsee | 2/49 (4.1) [0.5–14.0] | 1/15 (6.7) [0.2–31.9] | 2/12 (16.7) [2.1–48.4] | 5/14 (35.7) [12.8–64.9] |
| Median time to PSA progression, mo (95% CI) | 3.1 (2.8–4.6) | 3.2 (2.8–4.6) | 7.4 (2.8–7.4) | 11.1 (3.0-NR) |
Visit cutoff date: April 29, 2019. Data are n/N (%) [95% CI] unless stated otherwise.
Includes patients with an alteration in FANCA (n = 4), NBN (n = 4), BRIP1 (n = 2), PALB2 (n = 2), RAD51 (n = 1), RAD51B (n = 1), and/ or RAD54L (n = 1).
Per modified RECIST/PCWG3 criteria; includes patients who had measurable disease at baseline per the investigator and ≥ 16 weeks of follow-up.
Proportion of patients without radiographic progression per RECIST/PCWG3 criteria who were ongoing with treatment at 6 months.
Proportion of patients without radiographic progression per RECIST/PCWG3 criteria who were ongoing with treatment at 12 months.
Defined as ≥ 50% reduction in PSA from baseline; includes patients who had ≥ 16 weeks of follow-up.
Abbreviations: CI, confidence interval; NR, not reached; PCWG3, Prostate Cancer Clinical Trials Working Group 3; PSA, prostate-specific antigen; RECIST, Response Evaluation Criteria In Solid Tumors version 1.1.
Figure 3.
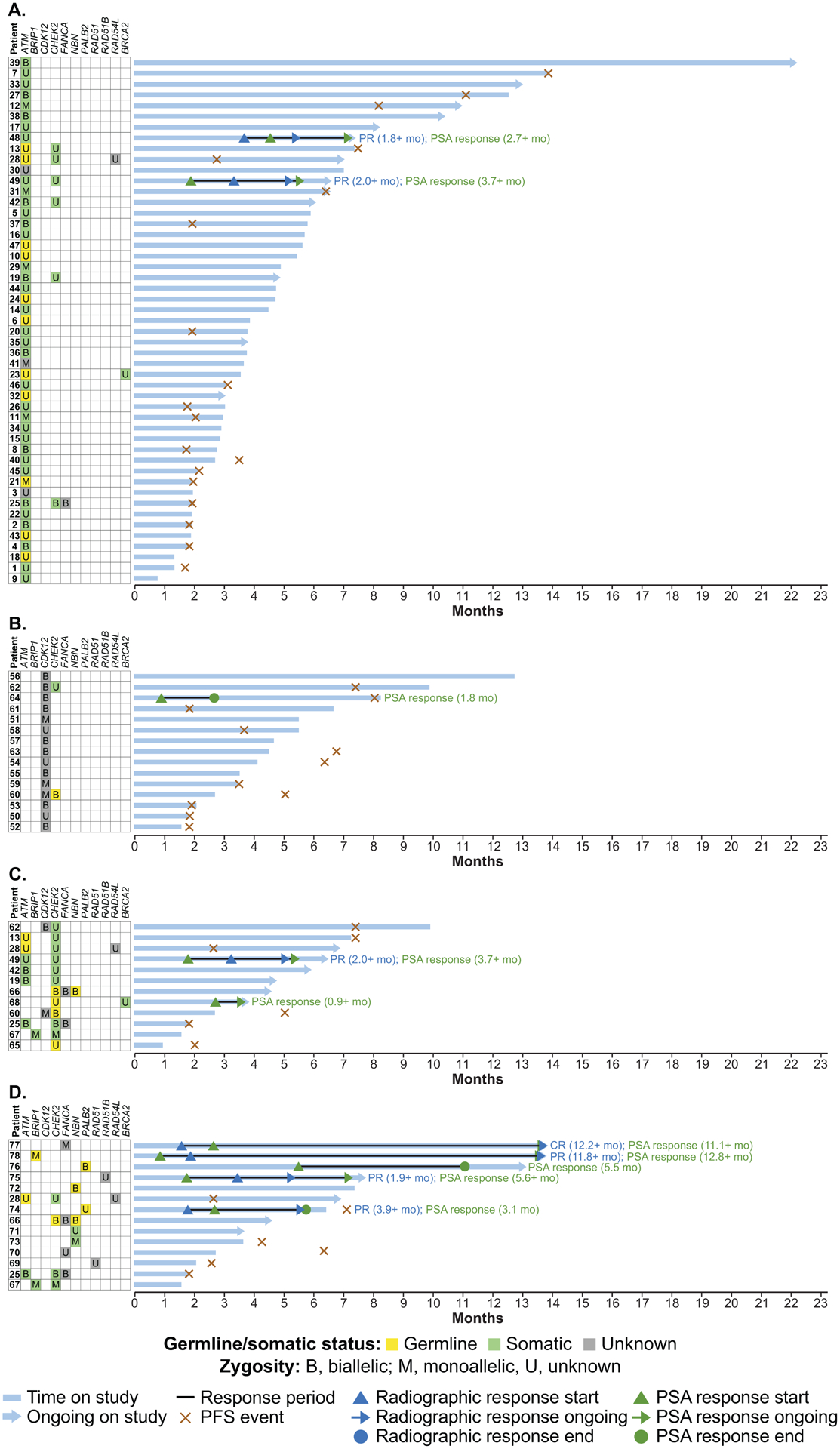
Treatment duration in patients with an ATM alteration (A), CDK12 alteration (B), CHEK2 alteration (C), or other DDR gene alteration (D). Patients 55, 56, 57, 61, 62, 63, and 64 had 2 distinct CDK12 alterations identified through tissue and/or plasma testing and were considered to have biallelic loss. Abbreviations: CR, complete response; DDR, DNA damage repair; PFS, progression-free survival; PR, partial response; PSA, prostate-specific antigen
The 2 patients who had radiographic and PSA responses had somatic ATM alterations detected through cell-free DNA (cfDNA) screening, so the possibility of homozygous loss of another DDR gene (e.g., BRCA2 or PALB2) cannot be ruled out. Archival tissue was not available for these patients to assess ATM alteration zygosity. However, none of the 11 patients with known biallelic alterations in ATM or 11 patients with germline alterations in ATM had a radiographic or PSA response.
CDK12 Cohort
No confirmed radiographic responses were observed among the 10 evaluable patients with a CDK12 alteration (Table 2). Among 15 overall patients with a CDK12 alteration, 1 (6.7%) had a confirmed PSA response lasting 1.8 months (Table 2, Fig. 3B, and Supplementary Fig. S3B). The 6- and 12-month clinical benefit rate in the CDK12 cohort was 20.0% (3 of 15 patients) and 7.1% (1 of 14 patients) (Table 2).
The patient with a PSA response had 2 distinct CDK12 alterations that are presumed to be biallelic and were identified in both cfDNA and tissue (Supplementary Table and Supplementary Fig. S2B).
CHEK2 Cohort
Among 9 evaluable patients with a CHEK2 alteration, 1 (11.1%) with a co-occurring ATM alteration had a confirmed partial radiographic response, as previously described (Table 2). A confirmed PSA response was observed in 2 (16.7%) of 12 overall patients with a CHEK2 alteration, 1 of whom was the patient with a radiographic response. Both were still on study as of the visit cutoff date and had ongoing PSA responses (3.7+ and 0.9+ months; Fig. 3C and Supplementary Fig. S3C). The 6-month clinical benefit rate for patients with a CHEK2 alteration was 37.5% (3 of 8 patients); no patients with a CHEK2 alteration were still receiving treatment at 12 months (Table 2).
CHEK2 alterations for both patients with PSA responses were identified through cfDNA screening; 1 was somatic and the other was germline. The PSA responder with a germline CHEK2 alteration (patient 68) was found to have a somatic BRCA2 alteration through tissue analysis conducted after study enrolment (Supplementary Table).
Other DDR Gene Cohort
Of the 14 evaluable patients with other DDR gene alterations, 4 (28.6%) had a confirmed radiographic response and 5 (35.7%) had a PSA response (Table 2, Fig. 3D). Both patients with a PALB2 alteration had PSA responses, and 1 patient also had a partial radiographic response that was ongoing for 3.9+ months as of the visit cutoff; the other patient had a 47% reduction in tumor volume but had not undergone a follow-up scan for confirmation of the response as of the visit cutoff. Of 4 patients with a FANCA alteration, 1 patient with a monoallelic truncating alteration had complete radiographic and PSA responses that were ongoing at the time of the visit cutoff. One of 2 patients with a BRIP1 alteration had a partial radiographic and a PSA response, both ongoing.
The patient with a RAD51B alteration had a partial radiographic and a PSA response, both ongoing.
Safety
The safety population included 78 patients with a non-BRCA DDR gene alteration who received at least 1 dose of rucaparib. At the visit cutoff date, the median (range) treatment duration was 4.3 (0.7–22.1) months.
A treatment-emergent AE (TEAE) of any grade occurred in 76 (97.4%) patients, the most frequent (reported in at least 20% of patients) were asthenia or fatigue, nausea, decreased appetite, anemia, constipation, vomiting, and diarrhea (Table 3). Among all 39 (50.0%) patients with grade 3 or higher TEAEs, anemia, asthenia or fatigue, and thrombocytopenia were the most frequent (Table 3). No TEAEs of myelodysplastic syndrome or acute myeloid leukemia were reported.
Table 3:
Most common (10% or more of patients) TEAEs of any grade in the safety population
| Overall (n = 78) | ||
|---|---|---|
| Any grade | Grade ≥ 3 | |
| Asthenia or fatigue | 41 (52.6) | 7 (9.0) |
| Nausea | 35 (44.9) | 1 (1.3) |
| Decreased appetite | 27 (34.6) | 1 (1.3) |
| Anemiaa | 24 (30.8) | 12 (15.4) |
| Constipation | 18 (23.1) | 0 |
| Vomiting | 18 (23.1) | 1 (1.3) |
| Diarrhea | 17 (21.8) | 1 (1.3) |
| ALT or AST increased | 14 (17.9) | 2 (2.6) |
| Back pain | 14 (17.9) | 2 (2.6) |
| Edema, peripheral | 14 (17.9) | 0 |
| Dizziness | 13 (16.7) | 0 |
| Weight decreased | 13 (16.7) | 0 |
| Blood creatinine increased | 12 (15.4) | 0 |
| Dysgeusia | 10 (12.8) | 0 |
| Thrombocytopeniab | 10 (12.8) | 4 (5.1) |
| Arthralgia | 9 (11.5) | 1 (1.3) |
| Cough | 8 (10.3) | 0 |
| Hematuria | 8 (10.3) | 1 (1.3) |
| Pain in extremity | 8 (10.3) | 0 |
Visit cutoff date: April 29, 2019. Data are n (%).
Includes preferred terms of anemia and decreased hemoglobin.
Includes preferred terms of thrombocytopenia and decreased platelets.
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; TEAE, treatment-emergent adverse event.
Treatment interruption due to a TEAE occurred in 34 (43.6%) patients, most commonly due to asthenia or fatigue (10 [12.8%] patients) and anemia (8 [10.3%]). Dose reduction due to a TEAE occurred in 21 (26.9%) patients, most commonly due to asthenia or fatigue (6 [7.7%] patients), anemia (3 [3.8%]), and thrombocytopenia (3 [3.8%]).
Four (5.1%) patients discontinued because of a TEAE (excluding disease progression), 1 each due to asthenia or fatigue, decreased appetite, hematuria, and postoperative respiratory failure. One (1.5%) death due to intestinal ischemia was reported and was considered unrelated to rucaparib by the investigator.
DISCUSSION
PARP inhibitors have shown dramatic responses in heavily pretreated mCRPC patients with BRCA gene alterations; however, the utility of alterations in non-BRCA DDR genes as biomarkers for PARP inhibitor sensitivity is not well established. This report describes 78 patients with mCRPC and prospectively identified non-BRCA DDR gene alterations treated with the PARP inhibitor rucaparib. The safety profile of rucaparib in these patients was consistent with that in patients with ovarian cancer and other solid tumors (23, 24). A limited number of radiographic and PSA responses was observed in patients with ATM, CDK12, or CHEK2 gene alterations, whereas responses were observed in patients with alterations in other DDR genes less frequently altered in mCRPC such as PALB2, BRIP1, FANCA, and RAD51B. These results suggest there are non-BRCA biomarkers associated with PARP inhibitor activity in mCRPC that warrant further investigation.
Radiographic and PSA responses in the ATM cohort of this study were observed in 10.5% and 4.1% of patients evaluable for each endpoint, respectively. The ATM cohort included patients with frameshift mutations, truncating mutations, homozygous deletions, and rearrangements (Supplementary Figs. S1 and S2A) expected to result in complete loss of function of that allele, whereas few patients (11.5%) had missense alterations predicted to be oncogenic. The ATM cohort also included patients with confirmed germline ATM alterations and patients with biallelic loss of ATM (Fig. 2A). Despite the various types and locations of ATM alterations observed, there was no clear association between response and the characteristics of the genomic alterations. These results suggest that ATM deficiency alone is not sufficient to result in synthetic lethality with PARP inhibition in mCRPC. In the TOPARP-A study, which did not enroll patients based on biomarker selection, although PSA and/or circulating tumor cell responses were observed in 4 patients with an ATM alteration, none of these patients had a radiographic response. In TOPARP-B, RECIST and PSA responses were observed in 8.3% and 5.3% of patients with an ATM alteration (6). Similarly, among 23 men with mCRPC who received olaparib, no PSA responses were observed in 6 patients with an ATM alteration, compared to 13 (76.4%) of 17 patients with a BRCA alteration (25). In PROfound, median radiographic PFS was similar in patients with an ATM alteration who received olaparib and those who received enzalutamide or abiraterone (8). In our study, although only a limited number of patients with an ATM alteration had a radiographic or PSA response, most patients had a best response of stable disease. These data, as well as the 6- and 12-month clinical benefit rates, suggest that rucaparib may provide, at best, disease stabilization in patients with an ATM alteration, although it should be noted that TRITON2 was not designed to assess this outcome. Detailed results from randomized studies such as PROfound or the phase 3 TRITON3 study of rucaparib (NCT02975934) are needed to more fully evaluate the effect of PARP inhibition in this patient subgroup while controlling for a potential prognostic effect of ATM alterations. The apparent lower sensitivity to PARP inhibitors of tumors harboring an ATM alteration may be due to ATM’s role as a sensor of DNA damage rather than a mediator of homologous recombination repair (15).
Most patients with CDK12 alterations enrolled in TRITON2 had a best response of stable disease and only 1 patient had a PSA response; as a result, enrolment of patients with a CDK12 alteration into TRITON2 was halted per protocol. CDK12 plays a role in the elongation phase of transcription, and studies suggest that inactivation of CDK12 can reduce DDR gene expression and sensitize tumor cells to PARP inhibition (26–28). However, a recent study reported that tumors with biallelic inactivation of CDK12 have distinct genomic and transcriptional profiles compared with DDR-deficient tumors (16). In TOPARP-B, no radiographic or PSA responses were observed in the 20 patients with a CDK12 alteration (6). Similarly, in a recent retrospective study, none of 11 men with CDK12-altered advanced prostate cancer had a radiographic or PSA response to PARP inhibitor treatment (29). In PROfound, median radiographic PFS in patients with a CDK12 alteration was longer with olaparib than with enzalutamide or abiraterone, although the statistical significance of this finding is unclear (8). Together, the genomics and clinical data suggest that CDK12 deficiency does not reliably predict response to PARP inhibitors.
We identified 1 confirmed radiographic response in the CHEK2 cohort, and 2 patients had a PSA response. However, CHEK2 alterations alone do not appear sufficient to render tumor cells responsive to PARP inhibitor treatment as co-occurring alterations in other DDR genes were often observed in TRITON2 patients with a CHEK2 alteration. In particular, both patients with CHEK2 alterations that responded were enrolled from plasma testing, and subsequent tissue testing in 1 patient revealed the presence of a somatic BRCA2 alteration.
Among patients with alterations in other DDR genes, patients with alterations in PALB2, FANCA, BRIP1, and RAD51B had both confirmed radiographic and PSA responses. The duration of response was up to a year in some of these patients, highlighting the durability of the responses. These data are consistent with reports of PARP inhibitor activity in patients with mCRPC (5, 6, 30, 31). For example, radiographic and/or PSA responses were observed in 57% of patients (4 of 7) with PALB2 alterations in TOPARP-B (6). Additionally, reversion mutations in PALB2 have been detected in a patient with mCRPC who progressed on olaparib treatment, providing strong evidence that the original mutation sensitized the tumor to PARP inhibitor treatment (31). Although it is challenging to draw conclusions because of the small sample size, these data strongly support continued examination of patients with alterations in this subset of DDR genes to determine if the alterations are robustly associated with PARP inhibitor sensitivity.
In this manuscript, we describe a large population of patients with mCRPC who were prospectively enrolled based on a deleterious gene alteration in a non-BRCA DDR gene, enabling analysis of the impact of PARP inhibitor treatment on these relatively infrequent gene alterations. Additionally, many patients had genomic test results from 2 or 3 assays, helping to overcome some of the limitations inherent to both tissue and plasma profiling and allowing for a more comprehensive, real-world evaluation of genomic alterations. Furthermore, we present results grouped by presence of at least 1 alteration in a given DDR gene to enable better assessment of the predictive utility of a specific DDR gene alteration.
One limitation of this study is the small sample size for the CDK12, CHEK2, and other non-BRCA DDR gene cohorts. Additionally, tissue and plasma test results were not available for all patients. Thus, a complete evaluation of co-occurring alterations cannot be performed, and tumor zygosity could be determined only for a subset of patients with central tissue test results. Loss of heterozygosity may occur more frequently with certain gene alterations than others, calling into question if a lack of response is related to alteration zygosity or if loss of the gene is not truly synthetically lethal with PARP inhibition. Although biallelic DDR gene alterations were identified among these cohorts without apparent association with response, overall the number of biallelic alterations observed for each gene was small. Further research to add upon these findings with greater numbers is warranted.
Although PARP inhibitor treatment has demonstrated radiographic and PSA responses in patients with BRCA alterations (4–8), the data presented here offer compelling evidence that response to PARP inhibitors is limited in men with mCRPC harboring an ATM, CDK12, or CHEK2 alteration. These data provide an optimistic outlook for treatment of prostate cancer with other DDR gene alterations, such as PALB2. Further work is needed to determine if PARP inhibitors provide disease stabilization in patients with an ATM or CDK12 alteration or whether there are other genomic characteristics for patients who achieve a response, and to better define the subset of other DDR gene alterations that confer sensitivity to PARP inhibitors in the setting of mCRPC.
Supplementary Material
Statement of Translational Relevance.
Deleterious alterations in BRCA1, BRCA2, and other DNA damage repair genes are thought to sensitize tumor cells to poly(ADP-ribose) polymerase (PARP) inhibition. In this analysis of patients with metastatic castration-resistant prostate cancer from TRITON2, loss of function alterations in ATM, CDK12, or CHEK2 alone did not significantly confer synthetic lethality with the PARP inhibitor rucaparib, as measured by objective radiographic and prostate-specific antigen responses. Responses were observed, however, in a number of patients with alterations in FANCA, PALB2, BRIP1, or RAD51B, genes associated with homologous recombination repair; these promising results warrant further studies. This analysis expands current understanding of predictive genomic biomarkers for treatment with a PARP inhibitor in patients with prostate cancer, but as well in patients with other malignancies, including breast, pancreatic, and ovarian cancer, where these biomarkers are also being explored.
Acknowledgments
Medical writing and editorial support funded by Clovis Oncology were provided by Nathan Yardley and Frederique H. Evans of Ashfield Healthcare Communications.
Financial Support
TRITON2 was designed by the funder and the first/last authors (W. Abida and S. Chowdhury). This article was written by the authors, with medical writing and copy editing support paid for by the funder. Data were collected by the investigators, analyzed by the funder, and interpreted by all authors. All authors had full access to all trial data and had final responsibility for the decision to submit for publication.
Disclosure of Potential Conflicts of Interest
W. Abida has served in a consulting or advisory role for Clovis Oncology, Janssen, MORE Health, and ORIC Pharmaceuticals; has received financial support for travel and/or accommodation from Clovis Oncology and ORIC Pharmaceuticals; and his institution has received research funding from Clovis Oncology, AstraZeneca, Prostate Cancer Foundation, and Zenith Epigenetics.
D. Campbell has nothing to disclose.
A. Patnaik has served in a consulting or advisory role for Exelixis, Janssen, and Jounce Therapeutics; has received honoraria from Clovis Oncology, Merck, Prime Inc, and Roche; and has received research funding from Clovis Oncology, Bristol-Myers Squibb, and GlaxoSmithKline.
J. Shapiro has served in a consulting or advisory role for Amgen, Astellas, Ipsen, Merck, and Roche and received financial support for travel from Amgen and Merck.
B. Sautois has served a consulting or advisory role for Clovis Oncology, Astellas, Janssen, and Sanofi and received financial support for travel and/or accommodation from Janssen.
N.J. Vogelzang has served in a consulting or advisory role for Clovis Oncology, Astellas, AstraZeneca, Bayer, Caris, Eisai, Janssen, Merck, Pfizer, and Sanofi; holds stock options from Caris; and serves as an editor for Up-To-Date.
R. McDermott has served in a consulting or advisory role for Bayer, Janssen, and Pfizer; has received support for travel and/or accommodation from Celgene, Janssen, and Pfizer; and has received research funding related to clinical trials from Clovis Oncology, Amgen, Bayer, Bristol-Myers Squibb, and Merck.
J. Zhang has served in a consulting or advisory role and/or on speakers bureaus for Clovis Oncology, AstraZeneca, and Sanofi.
J.M. Piulats has served in a consulting or advisory role for Clovis Oncology, Janssen, Pfizer, Merck, Bristol-Myers Squibb, VCN biosciences, Astellas, Roche-Genentech, BeiGene, Sanofi, ImmunoCore, and Novartis; has received financial support for travel and/or accommodation from Janssen and Roche-Genentech; has received research funding from Bristol-Myers Squibb, AstraZeneca, Janssen, Incyte, Pfizer, and Merck.
K. Fizazi has received personal fees from Clovis Oncology, Amgen, Astellas, AstraZeneca, Bayer, CureVac, ESSA, Janssen, Orion Pharma, Roche-Genentech, and Sanofi.
A.S. Merseburger has served as a principal investigator or subinvestigator for clinical studies by Clovis Oncology, Astellas, Bayer, GlaxoSmithKline, Ipsen, Janssen, Novartis, Pfizer, Teva, and Wyeth; served as a speaker for Astellas, Bayer, GlaxoSmithKline, Hexal, Ipsen, Janssen, Novartis, Sanofi Aventis, and Teva; served on an advisory board for Clovis Oncology, Astellas, Bayer, Ipsen, Janssen, Novartis, Pfizer, and Teva; and received research grants from Wyeth.
C.S. Higano has served in a consulting or advisory role for Clovis Oncology, Aptevo, Asana, Astellas, Bayer, Blue Earth Diagnostics, Churchill Pharmaceuticals, Dendreon, Endocyte, Ferring, Hinova, Janssen, Myriad, Orion, and Pfizer; and has received research funding from Clovis Oncology, Aptevo, Aragon Pharma, Astellas, AstraZeneca, Bayer, Dendreon, eFFECTOR Therapeutics, Emergent, Ferring, Genentech, Hoffman-LaRoche, Medivation, and Pfizer.
L.E. Krieger has served in a consulting and advisory role and/or on speakers bureaus for Clovis Oncology, Astellas, Janssen, Pfizer, Merck Sharpe & Dohme, AstraZeneca, Ipsen, Roche, Bristol-Myers Squibb, and Bayer; and has received financial support for travel and accommodation from Astellas, Ipsen, Merck Sharp & Dohme, AstraZeneca and Janssen.
C.J. Ryan has received research support from Clovis Oncology and has served as a consultant for AstraZeneca and Bayer.
F.Y. Feng has served in a consulting or advisory role for Bayer, Blue Earth Diagnostics, Celgene, Dendreon, EMD Serono, Ferring, Genentech, Janssen, Astellas, Myovant Sciences, and Nutcraker Therapeutics; has received honoraria from Clovis Oncology; and is a founding member with ownership interests of PFS Genomics.
A.D. Simmons, A. Loehr, D. Despain, M. Dowson, F. Green, S.P. Watkins, and T. Golsorkhi are employees of Clovis Oncology and may own stock or have stock options in that company.
S. Chowdhury has served in a consulting or advisory role and/or on speakers bureaus for Clovis Oncology, Astellas, BeiGene, Janssen, and Pfizer; received honoraria from GlaxoSmithKline; and received financial support for travel and accommodation from Clovis Oncology.
All other authors have nothing to disclose.
References
- 1.Cook SA, Tinker AV. PARP inhibitors and the evolving landscape of ovarian cancer management: a review. BioDrugs 2019;33:255–73. [DOI] [PubMed] [Google Scholar]
- 2.Keung MYT, Wu Y, Vadgama JV. PARP inhibitors as a therapeutic agent for homologous recombination deficiency in breast cancers. J Clin Med 2019;8:E435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lord CJ, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017;355:1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abida W, Campbell D, Patnaik A, Sautois B, Shapiro J, Vogelzang NJ, et al. Preliminary results from the TRITON2 study of rucaparib in patients (pts) with DNA damage repair (DDR)-deficient metastatic castration-resistant prostate cancer (mCRPC): Updated analyses. Ann Oncol 2019;30:abst 846PD. [Google Scholar]
- 5.Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med 2015;373:1697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mateo J, Porta N, Bianchini D, McGovern U, Elliott T, Jones R, et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol 2020;21:162–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith MR, Sandhu SK, Kelly WK, Scher HI, Efstathiou E, Lara P, et al. Phase II study of niraparib in patients with metastatic castration-resistant prostate cancer (mCRPC) and biallelic DNA-repair gene defects (DRD): Preliminary results of GALAHAD. J Clin Oncol 2019;37:abst 202. [Google Scholar]
- 8.Hussain M, Mateo J, Fizazi K, Saad F, Shore ND, Sandhu S, et al. PROfound: Phase III study of olaparib versus enzalutamide or abiraterone for metastatic castration-resistant prostate cancer (mCRPC) with homologous recombination repair (HRR) gene alterations. Ann Oncol 2019;30:abst LBA12. [Google Scholar]
- 9.Clovis Oncology, Inc. 2018. Clovis Oncology receives Breakthrough Therapy designation for Rubraca® (rucaparib) for treatment of BRCA1/2-mutated metastatic castration resistant prostate cancer (mCRPC). <https://ir.clovisoncology.com/investors-and-news/news-releases/press-release-details/2018/Clovis-Oncology-Receives-Breakthrough-Therapy-Designation-for-Rubraca-rucaparib-for-Treatment-of-BRCA12-Mutated-Metastatic-Castration-Resistant-Prostate-Cancer-mCRPC/default.aspx>. AccessedFebruary 5, 2020.
- 10.AstraZeneca PLC. 2016. Lynparza™ (olaparib) granted Breakthrough Therapy designation by US FDA for treatment of BRCA1/2 or ATM gene mutated metastatic Castration Resistant Prostate Cancer. <https://www.astrazeneca.com/media-centre/press-releases/2016/Lynparza-Olaparib-granted-Breakthrough-Therapy-Designation-by-US-FDA-for-treatment-of-BRCA1-2-or-ATM-gene-mutated-metastatic-Castration-Resistant-Prostate-Cancer-28012016.html#>. AccessedFebruary 5, 2020.
- 11.Janssen Pharmaceuticals, Inc. 2019. Janssen announces U.S. FDA Breakthrough Therapy designation granted for niraparib for the treatment of metastatic castration-resistant prostate cancer. <https://www.janssen.com/us/sites/www_janssen_com_usa/files/janssen_announces_us_fda_breakthrough_therapy_designation_granted_for_niraparib.pdf>. AccessedFebruary 5, 2020.
- 12.Zhang F, Fan Q, Ren K, Andreassen PR. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol Cancer Res 2009;7:1110–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Folias A, Matkovic M, Bruun D, Reid S, Hejna J, Grompe M, et al. BRCA1 interacts directly with the Fanconi anemia protein FANCA. Hum Mol Genet 2002;11:2591–7. [DOI] [PubMed] [Google Scholar]
- 14.Zannini L, Delia D, Buscemi G. CHK2 kinase in the DNA damage response and beyond. J Mol Cell Biol 2014;6:442–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weigelt B, Bi R, Kumar R, Blecua P, Mandelker DL, Geyer FC, et al. The landscape of somatic genetic alterations in breast cancers from ATM germline mutation carriers. J Natl Cancer Inst 2018;110:1030–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu YM, Cieslik M, Lonigro RJ, Vats P, Reimers MA, Cao X, et al. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell 2018;173:1770–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith MR, Sandhu SK, Kelly WK, Scher HI, Efstathiou E, Lara PN, et al. Pre-specified interim analysis of GALAHAD: A phase II study of niraparib in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) and biallelic DNA-repair gene defects (DRD). Ann Oncol 2019;30:abst LBA50. [Google Scholar]
- 18.de Bono JS, Fizazi K, Saad F, Shore N, Sandhu SK, Mehra N, et al. Central, prospective detection of homologous recombination repair gene mutations (HRRm) in tumour tissue from >4000 men with metastatic castration-resistant prostate cancer (mCRPC) screened for the PROfound study. Ann Oncol 2019;30:abst 847PD. [Google Scholar]
- 19.Clark TA, Chung JH, Kennedy M, Hughes JD, Chennagiri N, Lieber DS, et al. Analytical validation of a hybrid capture–based next-generation sequencing clinical assay for genomic profiling of cell-free circulating tumor DNA. J Mol Diagn 2018;20:686–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013;31:1023–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crawford B, Adams SB, Sittler T, van den Akker J, Chan S, Leitner O, et al. Multi-gene panel testing for hereditary cancer predisposition in unsolved high-risk breast and ovarian cancer patients. Breast Cancer Res Treat 2017;163:383–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neben CL, Zimmer AD, Stedden W, van den Akker J, O’Connor R, Chan RC, et al. Multi-gene panel testing of 23,179 individuals for hereditary cancer risk identifies pathogenic variant carriers missed by current genetic testing guidelines. J Mol Diagn 2019;21:646–57. [DOI] [PubMed] [Google Scholar]
- 23.Rubraca (rucaparib) tablets [prescribing information]. Boulder, CO: Clovis Oncology, Inc.; 2018. [Google Scholar]
- 24.Rubraca (rucaparib) tablets [summary of product characteristics]. Swords, Ireland: Clovis Oncology Ireland Ltd.; 2019. [Google Scholar]
- 25.Marshall CH, Sokolova AO, McNatty AL, Cheng HH, Eisenberger MA, Bryce AH, et al. Differential response to olaparib treatment among men with metastatic castration-resistant prostate cancer harboring BRCA1 or BRCA2 versus ATM mutations. Eur Urol 2019;76:452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joshi PM, Sutor SL, Huntoon CJ, Karnitz LM. Ovarian cancer-associated mutations disable catalytic activity of CDK12, a kinase that promotes homologous recombination repair and resistance to cisplatin and poly(ADP-ribose) polymerase inhibitors. 2014;289:9247–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ekumi KM, Paculova H, Lenasi T, Pospichalova V, Bösken CA, Rybarikova J, et al. Ovarian carcinoma CDK12 mutations misregulate expression of DNA repair genes via deficient formation and function of the Cdk12/CycK complex. Nucleic Acids Res 2015;43:2575–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bajrami I, Frankum JR, Konde A, Miller RE, Rehman FL, Brough R, et al. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res 2014;74:287–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Antonarakis ES, Velho PI, Agarwal N, Santos VS, Maughan BL, Pili R, et al. CDK12-altered prostate cancer: Clinical features and therapeutic outcomes to standard systemic therapies, PARP inhibitors, and PD1 inhibitors. Ann Oncol 2019;30:abst 845PD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horak P, Weischenfeldt J, von Amsberg G, Beyer B, Schütte A, Uhrig S, et al. Response to olaparib in a PALB2 germline mutated prostate cancer and genetic events associated with resistance. Cold Spring Harbor Mol Case Stud 2019;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goodall J, Mateo J, Yuan W, Mossop H, Porta N, Miranda S, et al. Circulating cell-free DNA to guide prostate cancer treatment with PARP inhibition. Cancer Discovery 2017;7:1006–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.