

Abstract
Objectives
To study the percentage, suppressive function and plasticity of Treg in giant cell arteritis (GCA), and the effects of glucocorticoids and tocilizumab.
Methods
Blood samples were obtained from 40 controls and 43 GCA patients at baseline and after treatment with glucocorticoids + IV tocilizumab (n = 20) or glucocorticoids (n = 23). Treg percentage and phenotype were assessed by flow cytometry. Suppressive function of Treg was assessed by measuring their ability to inhibit effector T‐cell (Teff) proliferation and polarisation into Th1 and Th17 cells.
Results
Treg (CD4+CD25highFoxP3+) frequency in total CD4+ T cells was decreased in active GCA patients when compared to controls (2.5% vs. 4.7%, P < 0.001) and increased after treatment with tocilizumab but worsened after treatment with glucocorticoids alone. Treg lacking exon 2 of FoxP3 were increased in GCA patients when compared to controls (23% vs. 10% of total Treg, P = 0.0096) and normalised after treatment with tocilizumab + glucocorticoids but not glucocorticoids alone. In GCA patients, Treg were unable to control Teff proliferation and induced ˜50% increase in the amount of IL‐17+ Teff, which was improved after in vitro blockade of the IL‐6 pathway by tocilizumab.
Conclusion
This study reports quantitative and functional disruptions in the regulatory immune response of GCA patients and demonstrates that, unlike glucocorticoids, tocilizumab improves Treg immune response.
Keywords: giant cell arteritis, interleukin‐6, tocilizumab, Treg
In this study, we demonstrated quantitative and functional disruptions in the regulatory immune response of patients with giant cell arteritis, resulting in increased proliferation and Th17 polarization of effector T cells. In addition, our results suggest that, through a specific blockade of the IL‐6 pathway, tocilizumab associated with glucocorticoids restores a better quantitative and qualitative Treg immune response than glucocorticoids alone. GCA, giant cell arteritis. Teff, effector T‐cells; Th17, T helper‐17 cells; Treg, regulatory T‐cells.

Introduction
Giant cell arteritis (GCA) is a large vessel vasculitis that occurs in people older than 50 years and mainly affects the aorta and cranial arteries. The infiltration of T lymphocytes, macrophages and multinucleated giant cells into the wall of affected arteries, together with intimal hyperplasia and luminal obstruction, leads to ischaemic manifestations such as temporal headaches, jaw claudication, vision loss and stroke. Most patients with GCA present signs of systemic inflammation, including weight loss, fatigue and fever, along with an increase in the erythrocyte sedimentation rate (ESR) and C‐reactive protein (CRP) levels.1 Glucocorticoids (GC) are the cornerstone of GCA treatment, but are given for 1–2 years in average and even longer in case of relapse, which leads to the occurrence of GC‐related side effects in the majority of patients.2
Giant cell arteritis pathogenesis is not fully understood, but major progress has been made in the last few years. In particular, research has demonstrated the essential role of CD4+ T cells,3 which are characterised by an expansion of T helper‐1 (Th1) and Th17 cells in peripheral blood and affected arteries.4, 5, 6 Interferon‐γ (IFN‐γ) decreases at a slower rate than other cytokines, such as IL‐17 and IL‐6, after treatment with GC, which suggests that a chronic inflammatory process supported by IFN‐γ plays a major role in vascular remodelling.4, 7 By contrast, we previously demonstrated that the percentage of regulatory T cells (Treg) in the peripheral blood of GCA and polymyalgia rheumatica (PMR) patients was lower than in healthy controls.5 Interleukin‐6 (IL‐6), which is elevated in the serum of GCA patients,5, 8 maintains the balance between Th17 cells and Treg.9, 10 IL‐6 has therefore emerged as a promising therapeutic target in GCA, and subsequent trials with tocilizumab (TCZ), a humanised anti‐IL‐6 receptor antibody, found TCZ had a dramatic GC‐sparing effect.11, 12, 13 While both GC and TCZ are able to block the IL‐6 pathway, GC by decreasing IL‐6 production5, 8 and TCZ by blocking membranous and soluble receptors of IL‐6, it remains unclear why relapses are more frequent in patients treated with GC alone.12
In a recent paper, Miyabe et al. demonstrated that GCA patients had a Treg compartment enriched in IL‐17‐secreting Treg (Th17‐like Treg) that had an impaired suppressive capacity, which was mainly related to the expression of a hypofunctional isoform of FoxP3 lacking exon 2 (FoxP3Δ2).14 FoxP3 is indeed expressed as two isoforms in humans: a full‐length form and a form lacking exon 2, which is required for the antagonisation of RORγt and RORα by FoxP3 to control Th17 polarisation.15 Interestingly, TCZ can abrogate this population in the peripheral blood of GCA patients, but its capability to restore Treg suppressive ability, its effects on Treg plasticity and its ability to control Th17 polarisation are unknown.14
We therefore designed this study to investigate the compartment, phenotype, plasticity and suppressive functions of Treg in newly diagnosed GCA patients and to compare the effect of treatment with GC or GC + TCZ in these patients on Treg immune response.
Results
Population characteristics
Forty‐three newly diagnosed GCA patients were prospectively enrolled after providing written informed consent. The study was approved by the Institutional Review Board of Dijon University Hospital. All patients but one met ≥ 3/5 American College of Rheumatology criteria for the diagnosis of GCA.16 The remaining patient was 70 years old and had polymyalgia rheumatica (PMR) with constitutional symptoms, and PET‐CT showed aortitis and vasculitis of the subclavian, carotid and axillary arteries. Twenty GCA patients were treated with prednisone and 4 monthly infusions of IV TCZ (8 mg per kg per 4 weeks) from inclusion to visit of month 3, as previously reported.11 The 23 other GCA patients were treated with GC alone as stipulated in the French guidelines.17 Treatment allocation was not randomised. Patients were included from two distinct prospective trials (ClinicalTrials.gov: NCT01910038 and NCT02857192), which were conducted successively between 2014 and 2020. Blood samples were obtained at inclusion and after 3 months of treatment in both groups except for two patients (both in the GC‐alone group) who refused to undergo this second blood sampling. At baseline, none of the patients treated with GC alone had received prednisone before blood sampling, whereas 12 of 20 (60%) patients in the GC + TCZ group had received prednisone before blood sampling for a mean duration of 7.2 ± 1.5 days and at a mean dose of 26.4 ± 5.2 mg per day (P < 0.001; Table 1). Notably, TCZ was started after blood sampling of baseline in all patients of the GC + TCZ group. At blood sampling at 3 months, the mean prednisone dose was 16.0 ± 1.2 in patients treated with GC alone vs. 14.6 ± 0.7 in those treated with TCZ + GC (P = 0.344; Table 1).
Table 1.
Characteristics of the study population
| Healthy controls | GCA patients at diagnosis | GCA patients after 3 months of treatment | P‐valuea | |||||
|---|---|---|---|---|---|---|---|---|
| All | GC alone | GC + TCZ | All | GC alone | GC + TCZ | |||
| n = 40 | n = 43 | n = 23 | n = 20 | n = 41 | n = 21 | n = 20 | ||
| Age, mean ± SEM | 73.6 ± 1.9 | 74.9 ± 1.2 | 76.9 ± 1.6 | 72.6 ± 1.7 | 0.568 | |||
| Sex (H/F) | 16/24 | 14/29 | 9/14 | 5/15 | 0.481 | |||
| GCA characteristics, n (%) | ||||||||
| Weight loss | 33 (77) | 20 (87) | 13 (65) | 0 | 0 | 0 | ‐ | |
| Headache | 35 (81) | 18 (78) | 17 (85) | 0 | 0 | 0 | ‐ | |
| Jaw claudication | 15 (35) | 6 (23) | 9 (45) | 0 | 0 | 0 | ‐ | |
| Scalp tenderness | 18 (42) | 10 (44) | 8 (40) | 0 | 0 | 0 | ‐ | |
| Abnormal temporal artery | 25 (58) | 11 (48) | 14 (70) | 0 | 0 | 0 | ‐ | |
| Visual signs | 8 (19) | 5 (22) | 3 (15) | 0 | 0 | 0 | ‐ | |
| PMR | 18 (42) | 12 (52) | 6 (30) | 1 (2) | 1 (5) | 0 | ‐ | |
| Aortitisb | 15/37 (41) | 8/21 (38) | 7/16 (44) | 0 | 0 | 0 | ‐ | |
| Halo sign with US scan | 14/22 (64) | 8/10 (80) | 6/12 (50) | 0 | 0 | 0 | ‐ | |
| Positive TAB | 30/41 (73) | 13/22 (59) | 17/19 (89) | 0 | 0 | 0 | ‐ | |
| Biology, mean ± SEM | ||||||||
| Lymphocytes (G L−1) | 1.70 ± 0.01 | 1.46 ± 0.09 | 1.40 ± 0.13 | 1.53 ± 0.12 | 1.92 ± 0.14 | 1.63 ± 0.14 | 2.22 ± 0.23 | 0.077 |
| CD3+ (%) | 72 ± 1 | 75 ± 2 | 75 ± 2 | 75 ± 2 | 75 ± 1 | 72 ± 2 | 78 ± 2 | 0.090 |
| CD3+CD4+ (%) | 46 ± 2 | 54 ± 2 | 54 ± 3 | 53 ± 3 | 52 ± 2 | 51 ± 3 | 53 ± 3 | 0.008 |
| CD3+CD8+ (%) | 22 ± 1 | 19 ± 1 | 18 ± 2 | 20 ± 2 | 20 ± 2 | 19 ± 3 | 21 ± 2 | 0.126 |
| ESR (mm h−1) | 16.9 ± 1.6 | 84.6 ± 4.1 | 86.6 ± 5.1 | 82.3 ± 6.7 | 14.5 ± 2.4 | 24.6 ± 3.4 | 3.9 ± 1.0 | < 0.0001 |
| CRP (mg L−1) | 3.5 ± 0.3 | 80.7 ± 9.2 | 88.5 ± 11.9 | 71.6 ± 14.2 | 4.9 ± 1.2 | 7.7 ± 2.2 | 2.0 ± 0.3 | < 0.0001 |
| Fibrinogen (g L−1) | 3.4 ± 0.1 | 6.8 ± 0.2 | 6.8 ± 0.3 | 6.8 ± 0.3 | 3.0 ± 0.2 | 3.7 ± 0.2 | 2.2 ± 0.1 | < 0.0001 |
| Treatment | ||||||||
| On prednisone, n (%) | 0 | 12 (28) | 0 | 12 (60)c | 41 (100) | 21 (100) | 20 (100) | ‐ |
| Prednisone (mg per day), mean ± SEM | 0 | 12.3 ± 3.1 | 0 | 26.4 ± 5.2 | 15.3 ± 0.7 | 16.0 ± 1.2 | 14.6 ± 0.7 | ‐ |
PMR, polymyalgia rheumatica; SEM, standard error of the mean; TAB, temporal artery biopsy.
Healthy controls vs. all GCA patients at diagnosis.
Aortitis was defined as regular circumferential wall thickening ≥ 3 mm in the absence of calcification and/or significant atheroma on angio‐CT images, or a homogeneous vascular signal more intense than the liver on 18FDG‐PET images.
Prednisone was started from 7.2 ± 1.5 days.
Peripheral blood mononuclear cells (PBMCs) obtained from 40 age‐matched healthy volunteers were used as controls. Exclusion criteria for healthy volunteers were as follows: C‐reactive protein > 5 mg L−1, recent treatment with GC or immunosuppressants, history of cancer, recent acute or chronic infectious disease, and autoimmune disease.
A description of the included patients is provided in Table 1. There were no differences between groups regarding demographic, clinical or biological data at diagnosis. Importantly, age, which influences the number, phenotype and function of Treg,18 was comparable between controls and patients.
Treg percentage is decreased in the blood of GCA patients and increases after treatment with TCZ+ GC but not GC alone
First, we found a lower percentage of Treg (CD4+CD25highFoxP3+; Figure 1a) in total CD4+ T cells at GCA diagnosis than in healthy controls (2.3% vs. 4.7%, P < 0.001; Figure 1b). At baseline, the percentage of Treg was higher in patients subsequently treated with GC alone than in those subsequently treated with GC + TCZ (2.8% vs. 1.8%, P = 0.014). Actually, it was because of the fact that the percentage of Treg in the GC + TCZ group was twice as low in the 12 patients who had received prednisone for a few days at baseline, compared with the eight patients who were free of prednisone at baseline (1.3% vs. 2.6%, P = 0.026; Figure 1d). After 3 months of treatment with GC alone, Treg frequency continued to diminish (2.8% vs. 2.2%, P = 0.040), whereas it significantly increased after TCZ treatment (1.8% vs. 2.5%, P = 0.044) (Figure 1c).
Figure 1.
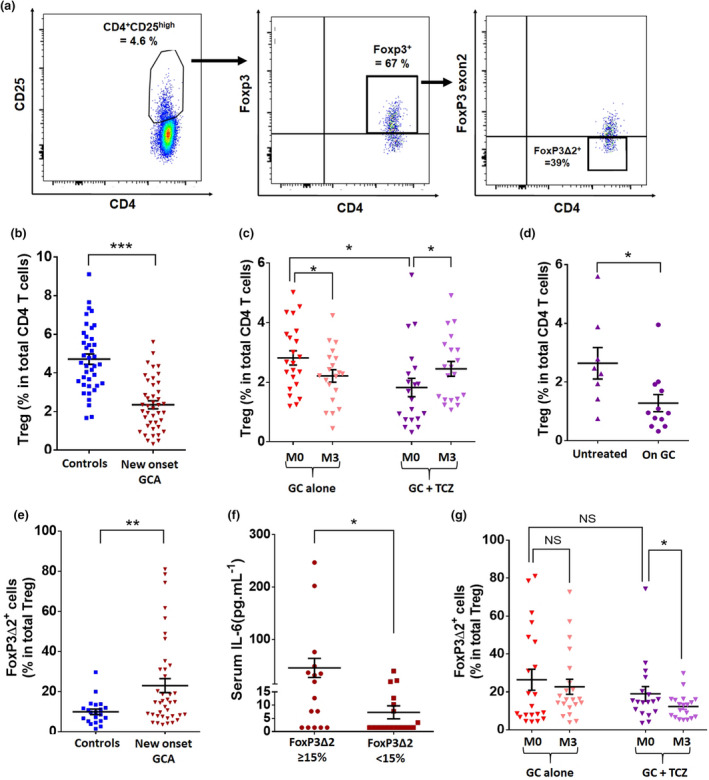
Decreased Treg frequency in GCA is corrected by treatment with TCZ + GC but not with GC. (a) Flow cytometric analysis of Treg cells. Treg are defined as CD4+CD25highFoxP3+. In this example of a new‐onset GCA patient, CD4+CD25high cells accounted for 4.6% of total CD4+ T cells. When gated on CD4+CD25high cells, 67% expressed FoxP3, so that Treg cells accounted for 3.08% of total CD4+ T cells. Among these Treg, 39% were deficient in exon 2 of FoxP3 (FoxP3Δ2 Treg). (b, c) Percentage of Treg in new‐onset GCA patients (n = 43) and controls (n = 40) (b) and their assessment at baseline (M0) and after treatment (M3): GC alone (n = 23) and GC and TCZ (n = 20). (d) Percentage of Treg at baseline in patients of the GC + TCZ group: at blood sampling, 12 had already received prednisone for a few days and eight were free of prednisone. (e) Percentage of Treg with an isoform of FoxP3 lacking exon 2 (FoxP3Δ2) in new‐onset GCA patients (n = 39) and controls (n = 21). (f) Serum IL‐6 (pg mL–1) in GCA patients depending on the percentage of circulating FoxP3Δ2 Treg at baseline (n = 34). (g) Percentage of FoxP3Δ2 Treg at baseline (M0) and after treatment (M3): GC alone (n = 21) and GC and TCZ (n = 18). Horizontal bars show the mean, error bars show the SEM, and P is the result of Student's t‐tests or paired Student's t‐tests, as appropriate. NS, not significant. *P < 0.05; **P < 0.01; and ***P < 0.001.
No differences were observed between groups regarding the level of expression of Helios or CD39 by circulating Treg (Supplementary figure 2). By contrast, the percentage of Treg expressing a spliced version of FoxP3 lacking exon 2 (FoxP3Δ2) in total Treg was increased in new‐onset GCA patients when compared to controls (23% vs. 10%, P = 0.0096; Figure 1e) and decreased after treatment with TCZ + GC (P = 0.049) but not GC alone (P = 0.788) (Figure 1g). Notably, the percentage of FoxP3Δ2 Treg at baseline was similar between patients subsequently treated with GC alone and those treated with GC + TCZ (26% vs. 19% in total Treg, P = 0.294; Figure 1g).
In GCA patients, serum IL‐6 at baseline was higher in patients who had an important increase in the percentage of circulating FoxP3Δ2 Treg (≥ 15% in total Treg) than in those in which the percentage of FoxP3Δ2 Treg was lower (< 15% in total Treg; 45.8 ± 18.3 vs. 7.3 ± 10.4 pg mL−1, P = 0.035; Figure 1f).
Treg proliferation is low and does not explain differences in the percentage of circulating Treg between groups
After isolation of circulating Treg (CD4+CD25high) and effector T cells (CD4+CD25low; Teff), we observed that the proliferation index of circulating Treg was low (1.2–1.4) in case of TCR activation with anti‐CD2, anti‐CD3 and anti‐CD28 microbeads, without any difference between Treg isolated from GCA patients (GCA Treg) and those isolated from healthy controls (control‐Treg). Furthermore, control Treg proliferation was not modified when cultured with control Teff, whereas the proliferative capacity of GCA Treg slightly increased when they were cultivated with GCA Teff (1.21 vs. 1.37, P = 0.001). This was not corrected in the presence of TCZ (1.37 vs. 1.45, P = 0.744) (Supplementary figure 3).
Treg ability to inhibit Teff proliferation is decreased in GCA patients and is improved by IL‐6 receptor blockade therapy
Figure 2 compares the ability of Treg from patients (GCA Treg) or controls (control‐Treg) to suppress Teff proliferation. The suppressive function of GCA Treg was strongly decreased when compared to control Treg (0.7% vs. 49.1%, P = 0.011). This defect significantly improved after in vitro blockade of IL‐6 signalling through treatment with TCZ (0.7% vs. 27.5%, P = 0.014). Furthermore, control Treg strongly inhibited the proliferation of GCA Teff (0.7% vs. 57.7%, P = 0.002). This suggests a functional defect of Treg rather than a lack of Treg or a resistance of GCA Teff to Treg inhibition (Figure 2a and b).
Figure 2.
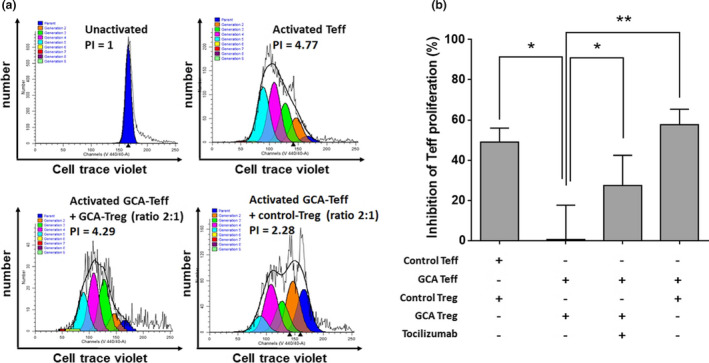
Ability of GCA Treg to inhibit Teff proliferation is altered in GCA and corrected by blockade of IL‐6 pathway with TCZ. (a) Proliferation of Teff (CD4+CFSE− cells) stimulated with anti‐CD2, anti‐CD3 and anti‐CD28 microbeads in the presence or not of GCA Treg or control Treg with a Teff/Treg ratio of 2:1. Proliferation index (PI) was calculated with ModFit LT software using CellTrace Violet incorporation. (b) Assessment of the inhibition of Teff proliferation by Treg: control Treg and control Teff (n = 15), GCA Treg and GCA Teff (n = 13), GCA Treg, GCA Teff and TCZ (5 µg mL−1) (n = 8) and control Treg and GCA Teff (n = 4). The percentage of inhibition was also calculated: 100 × (1‐(proliferation index of Teff cultured with Treg/proliferation index of Teff culture without Treg)). The higher this percentage is, the more the Treg are suppressive. Histograms show the mean ± SEM, and P is the result of Student's t‐tests or paired Student's t‐tests, as appropriate. NS, not significant. *P < 0.05 and **P < 0.01.
The same experiments were performed after in vivo treatment with GC or GC and TCZ, showing a trend towards increased functional activity of Treg after treatment, but with no clear difference between patients treated with GC and those treated with TCZ + GC (Supplementary figure 4a).
Treg increase Th17 polarisation of Teff in active GCA, which is corrected by TCZ but not GC alone
Figure 3 depicts Teff polarisation in the presence of TCZ and/or Treg from patients or controls. The percentage of IL‐17+ Teff and IFN‐γ+ Teff cells was not modified in the presence of control Treg (Figure 3c and d). However, GCA Treg triggered a ˜50% increase in the amount of IL‐17+ Teff (1.14% vs. 1.75%, P = 0.020). The ability of GCA Treg to induce Th17 polarisation was partially abrogated in the presence of TCZ (1.14% vs. 1.42%, P = 0.160). However, TCZ did not significantly decrease the percentage of IL‐17+ cells in Teff cultivated without Treg, suggesting that TCZ decreases Th17 polarisation through a restoration of Treg function rather than a direct effect on Teff differentiation. Furthermore, GCA Treg triggered a mild decrease in the amount of IFN‐γ+ Teff (7.94% vs. 6.36%, P = 0.019), which was amplified in the presence of TCZ (7.94 vs. 6.70, P = 0.002) (Figure 3c and d).
Figure 3.
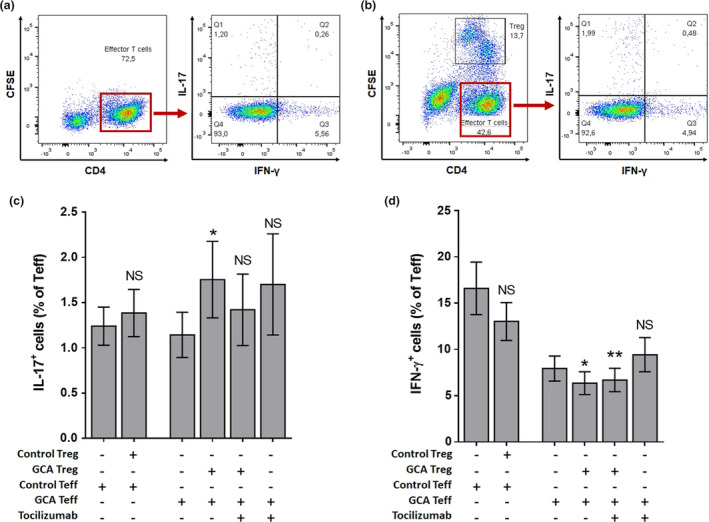
GCA Treg increase Teff polarisation in Th17 cells, which is attenuated by blockade of IL‐6 pathway with TCZ. (a, b) Flow cytometric study of the Teff (CD4+CFSE− cells) polarisation when Teff are cultivated alone (a) or in the presence of Treg (CD4+CFSE+) (Teff/Treg ratio = 2:1) (b). (c, d) Percentage of Th17 (CD4+IL‐17+) (c) and Th1 (CD4+IFN‐γ+) (d) in total Teff in the following conditions: control Teff (n = 14), control Treg and control Teff (n = 14), GCA Teff (n = 16), GCA Treg and GCA Teff (n = 16), GCA Teff and TCZ (5 µg mL−1) (n = 9) and GCA Treg, GCA Teff and TCZ (n = 9). Histograms show the mean ± SEM, and P is the result of ratio paired Student's t‐tests. NS, not significant. *P < 0.05 and **P < 0.01.
The effect of Treg on the polarisation of Teff after in vivo treatment of GCA patients is shown in Supplementary figure 4b and c. In patients treated with GC alone, IL‐17+ Teff induction in the presence of Treg was not modified, whereas it was decreased after 3 months of treatment with TCZ + GC (+87.6% vs. +6.04%, P = 0.028; Supplementary figure 4b). Regarding IFN‐γ+ Teff, no significant effect was observed, whatever the treatment group (Supplementary figure 4c).
Treg ability to produce IL‐17, but not IFN‐γ, is increased in GCA patients
Figure 4 shows the study of IL‐17 and IFN‐γ production by Treg (CD4+CFSE+) isolated from peripheral blood. We found that IL‐17 production by GCA Treg was more than threefold higher than control Treg (16.8% vs. 4.6%, P = 0.048). By contrast, co‐culture of Treg with autologous Teff did not significantly modify the production of IL‐17. In vitro treatment with TCZ tended to decrease IL‐17 production by Treg when cultured with Teff, but statistical significance was not reached (16.8% vs. 9.7%, P = 0.399; Figure 4b).
Figure 4.
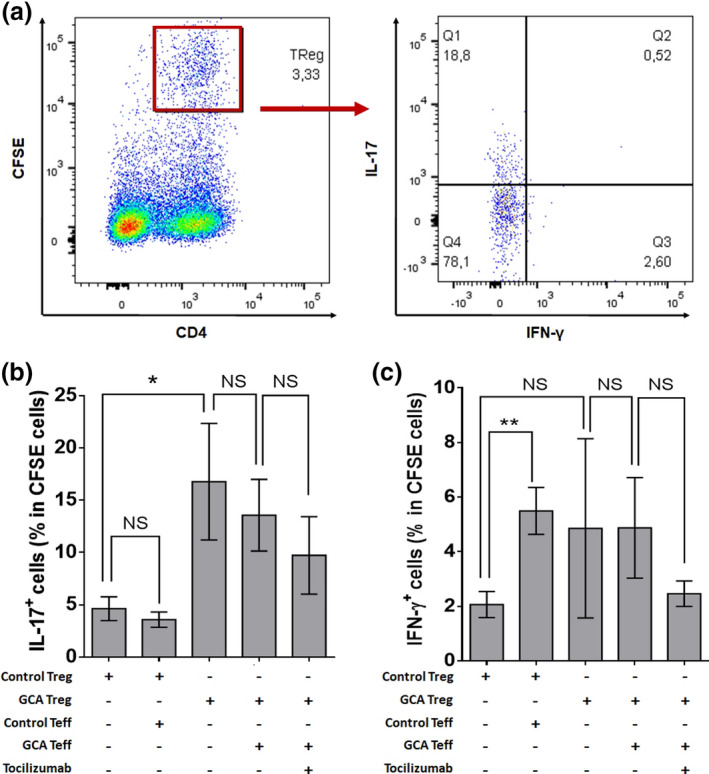
Assessment of the plasticity of circulating Treg. (a) Flow cytometric study of the Treg (CD4+CFSE+) production of IL‐17 and IFN‐γ. (b, c) Percentage of IL‐17+ Treg (b) and IFN‐γ+ Treg (c) when cultivated alone or in the presence of GCA or control Teff and/or TCZ (5 µg mL−1): control Treg (n = 14), control Treg and control Teff (n = 14), GCA Treg (n = 15), GCA Treg and GCA Teff (n = 16) and GCA Treg, GCA Teff and TCZ (n = 9). Histograms show the mean ± SEM, and P is the result of Student's t‐tests or paired Student's t‐tests, as appropriate. NS, not significant. *P < 0.05 and **P < 0.01.
In comparison with IL‐17, IFN‐γ production by Treg was dramatically lower. When cultured with control Teff, control Treg increased their level of expression of IFN‐γ while still remaining low (2.1% vs. 5.5%, P = 0.004). GCA Treg tended to produce more IFN‐γ than control‐Treg, but significance was not reached (2.1% vs. 4.9%, P = 0.423; Figure 4c).
FoxP3 and IL6 mRNA levels are decreased in ex vivo cultures of temporal arteries after treatment with dexamethasone but not tocilizumab
Figure 5 shows the assessment of IL6 and total FoxP3 mRNA expression in GCA temporal arteries cultivated for 5 days with or without TCZ or dexamethasone. While TCZ did not affect the expression of IL6 or FoxP3, dexamethasone triggered a significant decrease in the level of expression of both FoxP3 (P = 0.029) and IL6 (P = 0.005) (Figure 5a and b).
Figure 5.
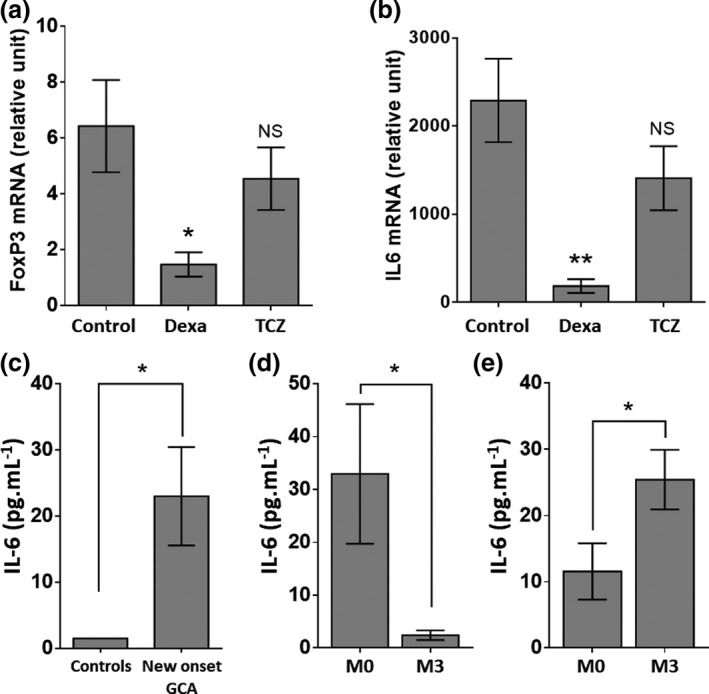
(a, b) FoxP3 (a) and IL‐6 (b) expressions in ex vivo cultures of temporal arteries (n = 13). Measures were performed after 5 days of culture in Matrigel with polyclonal IgG (control, n = 13), dexamethasone (0.5 µg mL−1, n = 7) or TCZ (10 µg mL−1, n = 10). RT‐PCR results are expressed in relative units with respect to GUSB expression (relative expression). (c–e) Assessment of serum IL‐6 in GCA patients (n = 43) and controls (n = 23) (c) and comparison of the IL‐6 concentration at baseline (M0) and after 3 months of treatment (M3): GC alone (n = 23) (d) and GC + TCZ (n = 20) (e). Histograms show the mean ± SEM, and P is the result of paired Student's t‐tests. NS, not significant. *P < 0.05 and **P < 0.01.
Serum IL‐6 is increased in active GCA patients and corrected by GC but not TCZ
Figure 5 shows that serum IL‐6 is increased in active GCA patients when compared to controls (23 vs. 1.5 pg mL−1, P = 0.039; Figure 5c). The levels were corrected after treatment with GC (P = 0.04; Figure 5d) but increased when patients received TCZ in addition to GC (P = 0.028; Figure 5e).
Discussion
This study aimed to investigate the frequency, function and plasticity of Treg in GCA and the effect of GC and TCZ, which are the two main approved treatments for this large vessel vasculitis.19 Even if Treg frequency was lower at baseline in patients who received GC + TCZ in comparison with GC alone, we confirmed, as previously demonstrated, that Treg frequency was severely decreased in the peripheral blood of newly diagnosed GCA patients5, 6 and that the level of circulating Treg improved after treatment with TCZ.10, 14 In addition and as suggested by previous studies,20, 21 our results demonstrated for the first time that GC worsened Treg deficiency, which may play a role in the higher frequency of relapse when patients are treated with GC alone in comparison with GC and TCZ.12
In this work, we also provide an in‐depth study of Treg function, specifically its ability to control T‐cell expansion and to modulate Th1 and Th17 cell polarisation, the latter having never been explored in GCA. We previously suggested that Treg might be functional in GCA, but our previous study included fewer patients, with a mix of GCA and PMR,5 so that our results were not consistent with a more recent study.14 In the present study, in which all included patients were affected by GCA, we clearly confirmed a defect in the functional activity of Treg. Furthermore, we demonstrated that this functional impairment in the control of Teff proliferation was related to Treg themselves and dependent on IL‐6, which supports the idea that TCZ can restore a functional Treg immune response by targeting Treg themselves rather than Teff.
Treg are essential in maintaining T‐cell homeostasis and control autoimmunity. Nevertheless, their effect on Th1 and Th17 polarisation has been rarely studied in human diseases. In rheumatoid arthritis, it was demonstrated that Treg increased Th17 differentiation, which was abrogated by TNF neutralisation (adalimumab) following a decreased production of IL‐6 by autologous monocytes.22 In the present study, we demonstrate for the first time a similar impairment of Treg biology in GCA since GCA Treg triggered an expansion of Th17 cells that was abrogated by the blockade of the IL‐6 pathway with TCZ. As it was the case in a previous study,14 the assessment of Treg function after in vivo treatment of GCA patients yielded less clear results, which can be explained by a smaller number of samples and/or by a less complete blockade of the IL‐6 pathway in vivo than in experiments in which TCZ was added in vitro. However, taken together, our results suggest that Treg may contribute to vascular inflammation by increasing IL‐17 production in active GCA, which can be resolved by treatment with TCZ but not GC alone.
Although plasticity between Treg and Th17 cells has been reported in the healthy state and in inflammatory diseases such as psoriasis, Crohn's disease and rheumatoid arthritis,23, 24, 25 it has not yet been studied in GCA. In the presence of IL‐6, Treg produce IL‐1726 and differentiate into Th17‐like Treg that have already been localised in GCA arteries.21 Th17‐like Treg are also known to have impaired suppression function and to exert a pathogenic role through the production of IL‐17.23 Interestingly, we observed that GCA Treg produced more than three times more IL‐17 than control‐Treg, suggesting that, in addition to their ability to induce a Th17 polarisation of Teff, Treg may be involved in vascular inflammation through their ability to differentiate into Th17‐like Treg. In contrast to what we observed in Teff, the environment had no impact on the ability of Treg to differentiate into Th17‐like Treg since co‐culture of Treg, Teff and TCZ did not induce a significant decrease in the percentage of Th17‐like Treg.
We hypothesise that the observed disruptions in Treg biology are mainly related to the expansion of FoxP3Δ2 Treg, which lacks exon 2 of FoxP3. FoxP3Δ2 Treg have indeed an increased ability to secrete IL‐17 and an impaired suppressive function, mainly related to the fact that the function of the missing exon 2 is to antagonise RORγt and RORα transcription factors and therefore to control Th17 polarisation.14, 15 In contrast to GC, TCZ decreased the FoxP3Δ2 Treg population in peripheral blood, which is consistent with the results of our in vitro functional study in which the blockade of the IL‐6 pathway with TCZ restored Treg's ability to inhibit the proliferation of Teff and their polarisation into Th17 cells. Along this line, the fact that we observed a significant increase in the level of serum IL‐6 in GCA patients having the highest percentage of circulating FoxP3Δ2 Treg strengthens the hypothesis that IL‐6 is involved in the expansion of these hypofunctional Treg.
In addition, the results obtained with ex vivo culture of temporal arteries showed that TCZ had no direct impact on the transcription of FoxP3 and IL6 genes, whereas GC triggered a decrease in the expression of both FoxP3 and IL6 mRNA. Along this line, and as previously demonstrated,5, 8, 11 serum IL‐6 decreased after treatment with GC, but it slightly increased after blockade of IL‐6R by TCZ. Altogether, these data suggest that the decrease in Treg in peripheral blood after treatment with GC may be related to a reduced transcription of FoxP3 mRNA, which may be induced by the blockade of the NF‐kB pathway by GC because this pathway upregulates FoxP3 after TCR engagement in T cells.27 By contrast, the blockade of the IL‐6 pathway by TCZ has no impact on IL6 and FoxP3 transcriptions but probably restores a normal splicing of FoxP3 mRNA and thus restores the expression of the exon 2 of FoxP3, decreases the production of IL‐17 by Treg and thus re‐establishes the Treg suppressive function making them able to inhibit Teff proliferation and their polarisation into Th17 cells.
Our results are strengthened by the homogeneity of the patients included in this study. All were newly diagnosed GCA patients, so the differences between groups were related to treatment and not biased by differences in the phenotypes of GCA patients (e.g. relapsers in the TCZ‐treated group compared with newly diagnosed patients in the GC‐treated group). Other notable strengths are the pairing of patients before and after treatment and the age matching of patients and controls, since ageing is an important factor modulating Treg immune response.18
Overall, our results reveal the existence of severe disruptions in the regulatory immune response of GCA. By promoting Th17 polarisation of Teff, GCA Treg may increase vascular inflammation rather than resolving it. Furthermore, our results suggest that, through a specific blockade of the IL‐6 pathway, TCZ restores a better quantitative and qualitative Treg immune response than GC. The GiACTA trial has demonstrated that the risk of relapse is decreased in patients receiving TCZ in addition to GC,12 and our study suggests that these observations could be related to the dichotomous effect of GC and TCZ on Treg immune response.
Methods
Cell preparation, antibodies and flow cytometric analysis
All analyses were performed on fresh PBMCs obtained by Ficoll gradient centrifugation. The following antibodies were used for flow cytometric analyses: anti‐CD4 PerCP, anti‐CD25 PE, anti‐FoxP3 Pacific Blue or Alexa Fluor 488 (clone 259D and clone 150D, the latter specifically detecting exon 2), anti‐Helios Pacific Blue, anti‐CD45RA eFluor 780 and anti‐CD39 Brilliant Violet 510 (Ozyme, Biolegend or Thermo Fisher Scientific, eBioscience, VILLEBON‐SUR‐YVETTE, France). Membrane staining was performed in 106 PBMCs. Cells were fixed and permeabilised (Fixation and Permeabilization Buffer; Thermo Fisher Scientific, eBioscience) before intracellular staining. Data were acquired on a BD Biosciences LSRII cytometer and analysed with FlowJo® v10 software.
Treg suppression assays
As presented in Supplementary figure 1, Treg (CD4+CD25high) and effector T cells (CD4+CD25low; Teff) were purified from fresh PBMCs by magnetic cell sorting following the manufacturers' instructions (CD4+CD25+ Treg Isolation Kit; Miltenyi Biotec, Paris, France). Teff and Treg were labelled with CellTrace Violet and CFSE (Thermo Fisher Scientific, Invitrogen, VILLEBON‐SUR‐YVETTE, France), respectively. T cells were activated with anti‐CD2/CD3/CD28 microbeads (Miltenyi Biotec) and cultured with or without Treg (Teff:Treg ratio = 2:1) or tocilizumab (5 µg mL−1). After 4 days, cells were activated with 0.1 µg mL−1 of phorbol 12‐myristate 13‐acetate (PMA) and 1 µg mL−1 of ionomycin (Sigma‐Aldrich, Saint‐Quentin‐Fallavier, France) in the presence of Brefeldin A (BD Golgi Plug; BD Bioscience, Le Pont‐de‐Claix, France) for 4 h and then fixed, permeabilised and stained with CD4‐PECy7, IL‐17‐PE and IFN‐γ‐APC. CellTrace Violet or CFSE incorporation and cytokine expression were analysed by flow cytometry to determine the proliferation index of Teff and Treg and the percentage of Th1 (CD4+IFN‐γ+) and Th17 (CD4+IL‐17+) cells among Teff (CD4+CFSE−) and Treg (CD4+CFSE+).
Cytokine assays
Serum IL‐6 concentration was measured by Luminex® (Thermo Fisher Scientific, eBioscience) following the manufacturer's instructions. Data were acquired and analysed on a Bio‐Plex® 200 System (Bio‐Rad, Marnes‐la‐Coquette, France).
Ex vivo culture of temporal arteries
Sections of 13 fresh temporal artery biopsy (TAB) specimens showing lesions of GCA were embedded in Matrigel® (BD Bioscience) and cultured as previously described.28 Briefly, surrounding tissue was carefully removed to keep the temporal artery before cutting regular sections of ∼1 mm of thickness. These sections were embedded in 25 µL of Matrigel® in a 24‐well plate (1 section/well and 2 sections/condition) and then covered by 1000 µL of RPMI 1640 medium (supplemented with 10% foetal bovine serum, L‐glutamine, amphotericin B and gentamycin) with or without tocilizumab (10 µg mL−1) or dexamethasone (0.5 µg mL−1). After 5 days, arterial sections were collected and homogenised in TRIzol reagent using a MINILYS homogeniser (Bertin Instruments®, Montigny‐le‐Bretonneux, France) before extraction of total mRNA.
RT‐PCR
Total RNA was extracted from cultured artery and cDNA obtained by reverse transcription employing a random hexamer priming kit (Thermo Fisher Scientific, Applied Bioscience, VILLEBON‐SUR‐YVETTE, France) in a final volume of 100 µL. Then, specific pre‐developed TaqMan probes from Thermo Fisher Scientific (TaqMan Gene Expression Assays) were used for PCR amplification of GUSB, FoxP3 and IL6 (Supplementary table 1). Fluorescence was detected with CFX96TM Real‐Time PCR Detection System, and results were analysed with CFX ManagerTM software (Bio‐Rad Laboratories). Gene expression was normalised to the expression of the endogenous control GUSB using the comparative ΔCt method. mRNA concentration was expressed in relative units with respect to GUSB expression (relative expression).
Statistics
Data are expressed as numbers (%) for categorical variables and means ± SEM for continuous variables. Chi‐squared tests or Fisher's exact tests were used to compare categorical variables, and unpaired or paired Student's t‐tests were performed to compare continuous variables, as appropriate. Statistical significance was set at P < 0.05 (two‐tailed). GraphPad Prism® was used for statistical analyses.
Conflict of Interest
Maxime Samson has received travel fees and personnel fees for symposium and boards from Roche–Chugaï, and consulting fees from AbbVie. Bernard Bonnotte serves as a board member for Roche–Chugaï. Catherine CREUZOT‐GARCHER has received a grant from Horus and Bayer, honoraria from Novartis and Bayer and serves as a medical advisory board member for Novartis, Bayer, Thea, Horus, Alcon and Allergan. Pierre‐Henry Gabrielle has received travel expenses from Allergan, Bayer and Novartis, and honoraria from Novartis and Bayer.
Author Contributions
Maxime Samson: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Software; Supervision; Validation; Visualization; Writing‐original draft. Hélène Greigert: Data curation; Formal analysis; Writing‐review & editing. Marion Ciudad: Conceptualization; Data curation; Methodology; Project administration; Writing‐review & editing. Claire Gerard: Data curation; Writing‐review & editing. Thibault Ghesquière: Data curation; Writing‐review & editing. Malika Trad: Data curation. Marc Corbera‐Bellalta: Formal analysis; Writing‐review & editing. Coraline Genet: Data curation; Writing‐review & editing. Sethi Ouandji: Formal analysis; Writing‐review & editing. Claudie Cladière: Data curation. Marine Thebault: Data curation. Kim Heang Ly: Data curation; Writing‐review & editing. Eric Liozon: Data curation; Writing‐review & editing. François Maurier: Data curation; Writing‐review & editing. Boris Bienvenu: Data curation; Writing‐review & editing. Benjamin Terrier: Data curation; Writing‐review & editing. Loïc Guillevin: Conceptualization; Data curation; Writing‐review & editing. Paris Charles: Data curation; Writing‐review & editing. Valérie Quipourt: Data curation; Writing‐review & editing. Hervé Devilliers: Conceptualization; Data curation; Formal analysis. Pierre‐Henry Gabrielle: Data curation; Writing‐review & editing. Catherine Creuzot‐Garcher: Data curation; Writing‐review & editing. Georges Tarris: Data curation; Writing‐review & editing. Laurent Martin: Data curation; Formal analysis; Writing‐review & editing. Philippe Saas: Resources; Writing‐original draft. Sylvain Audia: Conceptualization; Data curation; Formal analysis; Resources; Validation; Writing‐review & editing. Maria Cinta Cid: Data curation; Formal analysis; Writing‐review & editing. Bernard Bonnotte: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Software; Supervision; Validation; Visualization; Writing‐original draft.
Supporting information
Acknowledgments
We thank Sabine Berthier (Internal Medicine, Dijon), Nicolas Falvo (Internal Medicine, Dijon), Anne‐Laure Fauchais (Internal Medicine, Limoges), Alexandre Guilhem (Internal Medicine, Dijon), Vanessa Leguy (Internal Medicine, Dijon), Thibault Maillet (Internal Medicine, Dijon), Nadine Meaux Ruault (Internal Medicine, Besançon), Barbara Nicolas (Internal Medicine, Dijon) and Nathalie Vernier (Internal Medicine, Metz) for their participation in the study. We thank Anabelle Legrand, Arlette Hamman and Serge Monier from the Plateforme de Cytométrie, Institut Férératif de Recherche 100, Université de Bourgogne. We thank Alexandra Felin, Abderrahmane Bourredjem, Sandrine Daniel, Lydie Rossye and Marie‐Laure Humbert‐Asensio for their help in the management and the monitoring of the study. We thank the Direction de la Recherche Clinique et de l'Innovation du CHU de Dijon (DRCI) for the promotion and the management of the study, and Suzanne Rankin for reviewing the English. This work was supported by grants (MS) from the Fondation ARTHRITIS (2017‐2018), the Groupement Interrégional de Recherche Clinique et d'Innovation Est (GIRCI), Appel à Projet Jeunes Chercheurs 2014 and the French Ministry of Health (Programme Hospitalier de Recherche Clinique [PHRC] 2012) and registered with ClinicalTrials.gov, numbers NCT01910038 and NCT02857192.
References
- 1.Salvarani C, Cantini F, Hunder GG. Polymyalgia rheumatica and giant‐cell arteritis. Lancet 2008; 372: 234–245. [DOI] [PubMed] [Google Scholar]
- 2.Proven A, Gabriel SE, Orces Cet al. Glucocorticoid therapy in giant cell arteritis: duration and adverse outcomes. Arthritis Rheum 2003; 49: 703–708. [DOI] [PubMed] [Google Scholar]
- 3.Samson M, Corbera‐Bellalta M, Audia Set al. Recent advances in our understanding of giant cell arteritis pathogenesis. Autoimmun Rev 2017; 16: 833–844. [DOI] [PubMed] [Google Scholar]
- 4.Deng J, Younge BR, Olshen RAet al. Th17 and Th1 T‐cell responses in giant cell arteritis. Circulation 2010; 121: 906–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Samson M, Audia S, Fraszczak Jet al. Th1 and Th17 lymphocytes expressing CD161 are implicated in giant cell arteritis and polymyalgia rheumatica pathogenesis. Arthritis Rheum 2012; 64: 3788–3798. [DOI] [PubMed] [Google Scholar]
- 6.Terrier B, Geri G, Chaara Wet al. Interleukin‐21 modulates Th1 and Th17 responses in giant cell arteritis. Arthritis Rheum 2012; 64: 2001–2011. [DOI] [PubMed] [Google Scholar]
- 7.Corbera‐Bellalta M, Planas‐Rigol E, Lozano Eet al. Blocking interferon γ reduces expression of chemokines CXCL9, CXCL10 and CXCL11 and decreases macrophage infiltration in ex vivo cultured arteries from patients with giant cell arteritis. Ann Rheum Dis 2016; 75: 1177–1186. [DOI] [PubMed] [Google Scholar]
- 8.Roche NE, Fulbright JW, Wagner ADet al. Correlation of interleukin‐6 production and disease activity in polymyalgia rheumatica and giant cell arteritis. Arthritis Rheum 1993; 36: 1286–1294. [DOI] [PubMed] [Google Scholar]
- 9.Bettelli E, Carrier Y, Gao Wet al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006; 441: 235–238. [DOI] [PubMed] [Google Scholar]
- 10.Samson M, Audia S, Janikashvili Net al. Brief report: inhibition of interleukin‐6 function corrects Th17/Treg cell imbalance in patients with rheumatoid arthritis. Arthritis Rheum 2012; 64: 2499–2503. [DOI] [PubMed] [Google Scholar]
- 11.Samson M, Devilliers H, Ly KHet al. Tocilizumab as an add‐on therapy to glucocorticoids during the first 3months of treatment of Giant cell arteritis: a prospective study. Eur J Intern Med 2018; 57: 96–104. [DOI] [PubMed] [Google Scholar]
- 12.Stone JH, Tuckwell K, Dimonaco Set al. Trial of tocilizumab in giant‐cell arteritis. N Engl J Med 2017; 377: 317–328. [DOI] [PubMed] [Google Scholar]
- 13.Villiger PM, Adler S, Kuchen Set al. Tocilizumab for induction and maintenance of remission in giant cell arteritis: a phase 2, randomised, double‐blind, placebo‐controlled trial. Lancet 2016; 387: 1921–1927. [DOI] [PubMed] [Google Scholar]
- 14.Miyabe C, Miyabe Y, Strle Ket al. An expanded population of pathogenic regulatory T cells in giant cell arteritis is abrogated by IL‐6 blockade therapy. Ann Rheum Dis 2017; 76: 898–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Du J, Huang C, Zhou Bet al. Isoform‐specific inhibition of RORα‐mediated transcriptional activation by human FOXP3. J Immunol 2008; 180: 4785–4792. [DOI] [PubMed] [Google Scholar]
- 16.Hunder GG, Bloch DA, Michel BAet al. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum 1990; 33: 1122–1128. [DOI] [PubMed] [Google Scholar]
- 17.Bienvenu B, Ly KH, Lambert Met al. Management of giant cell arteritis: recommendations of the French Study Group for Large Vessel Vasculitis (GEFA). Rev Med Interne 2016; 37: 154–165. [DOI] [PubMed] [Google Scholar]
- 18.Salminen A. Activation of immunosuppressive network in the aging process. Ageing Res Rev 2020; 57: 100998. [DOI] [PubMed] [Google Scholar]
- 19.Hellmich B, Agueda A, Monti Set al. 2018 Update of the EULAR recommendations for the management of large vessel vasculitis. Ann Rheum Dis 2020; 79: 19–30. [DOI] [PubMed] [Google Scholar]
- 20.Cid MC, Campo E, Ercilla Get al. Immunohistochemical analysis of lymphoid and macrophage cell subsets and their immunologic activation markers in temporal arteritis. Influence of corticosteroid treatment. Arthritis Rheum 1989; 32: 884–893. [PubMed] [Google Scholar]
- 21.Espigol‐Frigole G, Corbera‐Bellalta M, Planas‐Rigol Eet al. Increased IL‐17A expression in temporal artery lesions is a predictor of sustained response to glucocorticoid treatment in patients with giant‐cell arteritis. Ann Rheum Dis 2013; 72: 1481–1487. [DOI] [PubMed] [Google Scholar]
- 22.Ehrenstein MR, Evans JG, Singh Aet al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti‐TNFα therapy. J Exp Med 2004; 200: 277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ren J, Li B. The functional stability of FOXP3 and RORγt in Treg and Th17 and their therapeutic applications. Adv Protein Chem Struct Biol 2017; 107: 155–189. [DOI] [PubMed] [Google Scholar]
- 24.Komatsu N, Okamoto K, Sawa Set al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med 2014; 20: 62–68. [DOI] [PubMed] [Google Scholar]
- 25.Miyao T, Floess S, Setoguchi Ret al. Plasticity of Foxp3+ T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity 2012; 36: 262–275. [DOI] [PubMed] [Google Scholar]
- 26.Yang XO, Nurieva R, Martinez GJet al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 2008; 29: 44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Long M, Park SG, Strickland Iet al. Nuclear factor‐kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity 2009; 31: 921–931. [DOI] [PubMed] [Google Scholar]
- 28.Corbera‐Bellalta M, Garcia‐Martinez A, Lozano Eet al. Changes in biomarkers after therapeutic intervention in temporal arteries cultured in Matrigel: a new model for preclinical studies in giant‐cell arteritis. Ann Rheum Dis 2014; 73: 616–623. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials