

Abstract
The research of extracellular vesicles (EVs) has boomed in the last decade, with the promise of them functioning as target‐directed drug delivery vehicles, able to modulate proliferation, migration, differentiation, and other properties of the recipient cell that are vital for health of the host organism. To enhance the ability of their targeted delivery, we employed an intrinsically overrepresented protein, CD81, to serve for recognition of the desired target antigen. Yeast libraries displaying mutant variants of the large extracellular loop of CD81 have been selected for binders to human placental laminin as an example target. Their specific interaction with laminin was confirmed in a mammalian display system. Derived sequences were reformatted to full‐length CD81 and expressed in EVs produced by HeLa cells. These EVs were examined for the presence of the recombinant protein and were shown to exhibit an enhanced uptake into laminin‐secreting mammalian cell lines. For the best candidate, the specificity of antigen interaction was demonstrated with a competition experiment. To our knowledge, this is the first example of harnessing an EV membrane protein as mediator of de novo target antigen recognition via in vitro molecular evolution, opening horizons to a broad range of applications in various therapeutic settings.
Keywords: antigen‐binding CD81, directed evolution, extracellular vesicle uptake, targeted extracellular vesicles, yeast display
Abbreviations
- BACE1
beta‐secretase 1
- BCA
bicinchoninic acid
- BSA
bovine serum albumin
- cel‐miR‐39
Caenorhabditis elegans micro‐RNA‐39Ct cycle threshold
- DMEM
Dulbecco's modified Eagle medium
- eGFP
enhanced green fluorescent protein
- EGFR
epithelial growth factor receptor
- EV
extracellular vesicle
- Fab
fragment antigen binding
- FACS
fluorescence‐activated cell sorting
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- FSC
forward scatter
- HA
hemagglutinin
- LEL
large extracellular loop
- MACS
magnetic‐activated cell sorting
- MES
2‐(N‐morpholino) ethanesulfonic acid
- MFI
mean fluorescence intensity
- miRNA
microRNA
- MWCO
molecular weight cut‐off
- NTA
nanoparticle tracking analysis
- PBS
phosphate buffered saline
- PBS‐T
PBS with 0.1% Tween‐20
- PCR
polymerase chain reaction
- PE
phycoerythrin
- PEG
polyethyleneglycol
- PEI
polyethyleneimine
- PVDF
polyvinylidene difluoride
- qPCR
Quantitative reverse transcription‐PCR in real time
- RIPA
radioimmunoprecipitation assay
- RT
room temperature
- SDS
sodium dodecylsulphate
- SEL
small extracellular loop
- siRNA
small interfering RNA
- SSC
side scatter
- TEM
transmission electron microscopy
- TM
midpoint of thermal denaturation
1. INTRODUCTION
In recent years, extracellular vesicles (EVs) have gained recognition as elaborate drug delivery vehicles (Yamamoto et al., 2019). These 50–150 nm large membrane enveloped structures, originating from the lamellae of the multivesicular body, have been discovered as cell‐to‐cell messengers, as their contents, composed of a cocktail of proteins, miRNAs and other non‐coding RNAs, originating from the producing cell, can efficiently be transferred to a recipient cell upon fusion with its cellular membrane (Yáñez‐Mó et al., 2015), by endocytosis and phagocytosis. EVs have been shown to play a vital role in intercellular communication in several biological situations crucial for sustaining homeostasis in tissues and organs (H. Rashed et al., 2017), aging and senescence (Terlecki‐Zaniewicz et al., 2019; Weilner et al., 2016), and further, to support processes such as regeneration after injury (Taverna et al., 2017) and limitation of tumorigenesis and malignant transformation (Wolfers et al., 2001). These properties have rendered them interesting for biotechnological applications, which has resulted in accelerated development of methods for their production, including optimization of source cells and respective culture media (Patel et al., 2018; Roura & Bayes‐Genis, 2019), improved purification processes (Zhang et al., 2019), standardization of protocols for characterization (Coumans et al., 2017) and stable storage of EV preparations (Jeyaram & Jay, 2018). It was soon accepted that exogenous loading of EVs with biologically diverse compounds, such as siRNAs, miRNAs, genes and toxins, can be applied for enhanced delivery of the desired substance into target cells (Batrakova & Kim, 2015). Sophisticated protocols have been devised to enable optimal enclosure of the drug of interest into the vesicles (Sutaria et al., 2017), employing a spectrum of strategies, including simple diffusion, lipofection, electroporation and sonication and endogenous loading methods related mostly to RNA‐cargo (overexpression in host cells (Hung & Leonard, 2015), extrusion of host cells (Lunavat et al., 2016), vexosome hybrid vehicles (Maguire et al., 2012) and introduction of EXOmotif to bias the miRNA loading into the EVs (Villarroya‐Beltri et al., 2013)).
On the other hand, targeted delivery of EVs has long been recognized as a feature that could crucially improve their biological effect. The membrane composition of the native EVs is unique as it exhibits substantial variation depending strongly on the cell of origin (Yoshioka et al., 2013) and assigning them a level of tropism for target cells (Hoshino et al., 2015). Nevertheless, the first experiments with EVs in vivo, especially those including intravenous applications, have shown that such agents could profit from improved specific delivery when their surface has been modified to enable the specific recognition of the target cells (Walker et al., 2019). Prominent examples featuring surface modification of EVs as beneficial for delivery of therapeutic compounds include overexpression of a surface‐bound peptide recognizing specifically the tumour marker EGFR, which led to a higher level of accumulation of modified vesicles in target cells, and in addition such particles loaded with let‐7a miRNA were able to inhibit tumour growth in mice (Ohno et al., 2013). Derivatization of an endogenously overrepresented EV surface protein to achieve enhanced targeted uptake was validated as a strategy with experiments where such particles were able to efficiently deliver anti‐BACE1 siRNA to the brain in mice (Alvarez‐Erviti et al., 2011). Targeted delivery could also be achieved via fusion with immunoglobulin‐based antigen‐binding molecules, such as single chain antibodies (Wang et al., 2018) and nanobodies (Kooijmans et al., 2016). Chemical strategies on the other hand involve ex vivo membrane modification, as presented for example for the ‘cloaking’ platform where streptavidin‐decorated EV surfaces can be endowed with a biotinylated targeting agent of choice (Antes et al., 2018). To sum up, derivatization of EV surface structures appears to be a viable strategy to enhance their targeted uptake, and the diverse spectrum of methods to achieve this goal is yet to be explored.
For this purpose, we have opted to leverage the innately overrepresented EV surface protein CD81, which is at the same time regarded as their hallmark surface marker, into an antigen‐recognition scaffold. This protein is a member of the tetraspanin family, which features four transmembrane segments (TM1–4) linked by a small intracellular loop, a small extracellular loop (SEL) and a large extracellular loop (LEL) (Levy et al., 1991). The crystallographic structure of a soluble form of the tetraspanin CD81 LEL domain is a five‐helix bundle stabilized by two disulfide bridges (Kitadokoro, 2001; Kitadokoro et al., 2001). The protein fold as well as its key structural features are conserved among tetraspanins, while a high degree of variability in its length and composition can be found in its central part that in CD81 comprises three helices (Seigneuret, 2006; Seigneuret et al., 2001) Previously, we have designed highly stabilized mutants of CD81 LEL via introduction of de novo disulfide bonds (Vogt et al., 2018). Two such CD81 LEL variants, the most stable one and the variant that exhibited reversible refolding after denaturation, were chosen to serve as scaffolds for libraries of antigen‐binding units, obtained by randomization of selected solvent‐exposed residues. The selection platforms were constructed using yeast display as a validated method of molecular evolution in vitro and were envisioned as a source of specific binders to an antigen of choice.
One of the most quickly expanding therapeutic areas that could profit from use of targeted EVs is regeneration of a variety of tissues, including bone (Inder et al., 2014; Qin et al., 2016), tendons (Wang et al., 2019), cartilage (Tan et al., 2020), skin (Terlecki‐Zaniewicz et al., 2019; Wu et al., 2018) and importantly neural tissue (Ching & Kingham, 2015; Dong et al., 2019), whose repair is complex depending on processes involving changes in Schwann cell phenotypes, the reprogramming of macrophages, and the reconstruction of the vascular network critically dependent on paracrine secretion (Cattin et al., 2015). Laminin can efficiently support the process of muscular regeneration (Riederer et al., 2015) and neural regeneration as it induces rapid and efficient adhesion of primary Schwann cells and vascular tube formation by endothelial cells (Hackethal et al., 2018). Therefore, we selected this protein as a target antigen for selection of specifically binding CD81 LEL mutants from a yeast library. To improve the discovery of hits that could function as recognition agents on the surface of EVs, we tested the antigen‐binding CD81 LEL variants in a mammalian display system and putative candidates were then expressed as full‐length CD81 in HeLa cells. To assess their novel functionality, internalization of such EVs into laminin‐secreting cell lines Huh‐7 and NCI‐N87 was compared with EVs derived from wild‐type CD81‐transfected production cell line, and further, their reactivity with several recombinant laminin isoforms was examined. The specificity of binding of laminin targeting EVs to their cognate antigen was tested under competitive conditions. Finally, the ability of laminin‐targeting EVs to transfer cel‐miR‐39 to Huh‐7 cell line was examined.
2. MATERIALS AND METHODS
2.1. Molecular design
The CD81 crystal structure PDB: 1G8Q was used as the basis of selection of solvent‐exposed residues that could upon randomization form a novel antigen binding site of about 600 Å2 in size. The first scaffold molecule was a CD81 LEL mutant that harbours two novel disulfide bonds, Ala134Cys/Lys144Cys and Val135Cys/Ser168Cys, and exhibits the midpoint of thermal transition (TM) of 109.4°C, 43°C above that of the wild‐type CD81 LEL. The library design named CD81LEL_2 involved randomization of 13 amino acid residues: 132–133, 136–139, 162–165, 167 and 171–172 (numbering as in PDB: 1G8Q). CD81LEL_L3 library design was based on the reversibly refolding CD81 LEL mutant, where the stabilizing mutations Ala130Cys/Ala146Cys and Val135Cys/Ser168Cys work additively to increase TM of CD81 LEL to 93.4°C, and render the mutated CD81 LEL the ability to reversibly refold when heated up to 110°C, unlike the wild‐type CD81 LEL. In this library, the amino acid residues at the positions 132–133, 136–141, 162–165, 167 and 171–172 were randomized to form a library of composite surfaces available for antigen binding.
2.2. Yeast display
2.2.1. Preparation of yeast display libraries
For validation of feasibility of yeast display of CD81 scaffold, CD81 LEL domain sequence (synthetized by Geneart, Thermo Fisher Scientific) was cloned into the pYD1 vector (Thermo Fisher Scientific) and transformed into S. cerevisiae EBY100 (Thermo Fisher Scientific) using the PEG3350/Li‐acetate/salmon sperm DNA protocol (Gietz & Schiestl, 2007). Selection, cultivation and induction of transformed colonies proceeded in S‐CAA media containing either 2% glucose (D) or 2% galactose/1% raffinose (G/R) as carbon source, according to published protocols (Chao et al., 2006). The recipient vector for the yeast display library was modified by deletions and insertions of restriction enzyme recognition sites using Quikchange Lightning Mutagenesis Kit (Agilent) to simplify linearization for use in gap‐repair driven homologous recombination. Oligonucleotides for yeast library construction were synthetized by ELLA Biotech. PCR recombination products encoding the randomized inserts were produced using in 100 μl aliquots using Q5 HiFi Polymerase MasterMix (New England Biolabs), 10 ng/μl template DNA encoding CD81 LEL, and 50 pmol of each of oligonucleotides L2for and EFrev for the library CD81LEL_L2, or 50 pmol of each of oligonucleotides L3for and EFrev (sequences of all oligos in the Table S1). The size of the yeast display libraries, which were produced each in two batches labelled A and B, was determined using dilution plating. To investigate the level of correctness, plasmid DNA was isolated from 10 μl of pelleted yeast cells using Zymoprep II kit (Zymo Research) and transformed to E. coli TOP10 using electroporation. The expression cassettes of CD81 LEL mutants were amplified using primers pyd forward and pyd reverse (Thermo Fisher Scientific) and sequenced using one of these primers.
2.2.2. Staining of yeast‐display libraries for FACS‐based phenotype control
For quality control, the yeast cells were induced in SG/RCAA medium with penicillin and streptomycin either for 48 h at 20°C or 24 h at 37°C. For staining, induced yeast cells were blocked in 2% BSA‐PBS solution for 30 min at RT at an OD600 of 1. Then they were resuspended into 100 μl‐aliquots and stained with anti‐Xpress antibody (R910‐25, Thermo Fisher Scientific) (1:1000) reactive with the N‐terminally positioned Xpress tag, and M38 antibody (10630D, Thermo Fisher Scientific) (1 μg/ml), which detects the properly folded CD81 LEL (Imai & Yoshie, 1993), in 2% BSA‐PBS for 1 h at RT. Cells were pelleted at 1000 × g, for 5 min at 4°C and resuspended in 2% BSA‐PBS with goat anti‐mouse (Fab’)2‐FITC conjugate (F‐2653, Sigma‐Aldrich), diluted 1:200. Samples stained with anti‐his‐tag‐Alexa Fluor 488 antibody (35351, QIAgen), diluted 1:200 in 2% BSA‐PBS and anti‐V5‐tag‐FITC antibody (MA1‐80281, Thermo Fisher Scientific), diluted 1:100 in 2% BSA‐PBS, were used to detect C‐terminally positioned tags to determine the percentage of read‐through sequences of the clones. The incubation time with fluorescent antibodies was 30 min on ice. The fluorescence of stained samples and unstained controls was determined using Guava easyCyte flow cytometer (Merck Millipore).
2.2.3. Selection of yeast display libraries with human placental laminin
Human placental laminin was purchased from Sigma‐Aldrich. For biotinylation, EZ‐Link Sulfo‐NHS‐LC‐LC‐biotin reagent (Thermo Fisher Scientific) was used in a 3:1 molar ratio, after dialysis of the antigen against 100‐fold volume of PBS overnight at 4°C. Incubation with biotinylation reagent proceeded for 1 h at RT with shaking. Unbound biotin was removed by dialysis against 100‐fold volume of PBS, using Snakeskin dialysis tubing with 10,000 Da MWCO (Thermo Fisher Scientific), at 4°C overnight with stirring.
Yeast display libraries L2A, L2B, L3A and L3B were cultured in SD‐CAA medium and the display of recombinant protein was induced by incubating them in SG/R‐CAA medium with shaking overnight at 37°C. For MACS, 109 induced yeast cells of each library were selected for binding to 1 μM biotinylated antigen and captured onto streptavidin microbeads (μMACS streptavidin kit, MACS Miltenyi), exactly according to manufacturer's instructions. In FACS selection rounds, the induced cell suspensions were diluted to 108 cells per ml 10% BSA‐PBS and labelled with 1 μM biotinylated laminin and streptavidin‐Alexa Fluor 647 (1:800) (S21374, Thermo Fisher Scientific) and anti‐V5‐FITC antibody (1:100) (MA1‐80281, Thermo Fisher Scientific) as secondary reagents. In all FACS‐assisted selection rounds, at least 20 times output of the previous sorting round was processed and 0.1% top anti‐V5‐antibody positive yeast cells were collected with a Sony SH8000 cell sorter. After visible enrichment, collected cells were plated out to characterize single yeast display clones. Yeast pools enriched for antigen binders entered an additional selection round using 200 nM antigen.
2.2.4. Analysis of antigen binding of CD81 LEL clones in mammalian cell display
The sequences of laminin binding clones were cloned between the SfiI and SalI restriction sites of the pDisplay vector (Thermo Fisher Scientific) that allows the expression of C‐terminally anchored protein of interest between N‐terminal HA‐tag and C‐terminal c‐myc‐tag. Plasmids encoding CD81 LEL mutants were introduced into 2‐ml‐cultures of HEK293‐6E cells (Canadian National Research Council) using PEI‐mediated transfection exactly according to manufacturer's instructions and the expression proceeded for 48 h on an orbital shaker at 180 rpm in humidified atmosphere with 5% CO2 at 37°C. After expression, the transfected cells were harvested at 300 × g, 10 min at 4°C, blocked for 30 min in 2% BSA‐PBS on ice. For the wild‐type CD81 LEL, the number of copies on the mammalian cell surface was determined using QIFIKIT (Agilent) and an anti‐c‐myc antibody 9E10 (Calbiochem) at 20 μg/ml, and the proper folding of the displayed CD81 LEL was confirmed with staining with M38 antibody (10630D, Thermo Fisher Scientific) at 10 μg/ml, followed by staining with anti‐mouse (Fab’)2‐FITC conjugate (F‐2653, Sigma‐Aldrich), diluted 1:200 in 2% BSA‐PBS. HEK cells expressing CD81 variants were stained with an anti‐c‐myc antibody (A‐14, sc789, Santa Cruz) at 10 μg/ml in 2% BSA‐PBS, for 30 min on ice. Binding was detected after incubation with anti‐rabbit IgG (H+L) ‐ Alexa Fluor 488 (A‐11034, Thermo Fisher Scientific), diluted 1:1000 in 2% BSA‐PBS, for 30 min on ice. Antigen reactivity was determined after incubation with 500 nM biotinylated human laminin and detection with streptavidin‐Alexa Fluor 647 (S21374, Thermo Fisher Scientific) at 1:1000 in 2% BSA‐PBS with Guava easyCyte flow cytometer (Merck Millipore).
2.3. EV preparation and characterization
2.3.1. Cell culture
HeLa cells (ATCC CCL‐2) were cultured at 37°C in 5% CO2 in RPMI 1640 medium (Sigma‐Aldrich), supplemented with 10% foetal bovine serum (FBS) and 4 mM L‐glutamine (Thermo Fisher Scientific) and used at passage 2–30. These cells were transduced with pBMN‐I‐GFP (Addgene)‐based constructs encoding full‐length CD81 variants with C‐terminal eGFP fusion (cloning details in File S1) using the retroviral Phoenix‐AMPHO transduction system. Phoenix‐AMPHO cells (ATCC CRL‐3213) were cultured in DMEM with 10% FBS and 4 mM L‐glutamine. Virus particle generation was performed by transfecting Phoenix‐AMPHO cells at 80 % confluency using jetPRIME (Polyplus Transfection) according to the manufacturer's brochure. Supernatants containing virus particles were filtered through a 0.45 μm PVDF filter and added to the HeLa cells. Transduced cells were sorted for strongly eGFP‐expressing variants with a Sony‐SH8000 sorter. To compare the expression of recombinant CD81‐eGFP variants, cells were harvested with trypsinization and the level of green fluorescence in a flow cytometry‐based assay was examined. Additionally, localization of the constructs was examined at cellular level using live‐cell microscopy (experimental details in File S1).
2.3.2. Isolation of extracellular vesicles
HeLa cells stably expressing CD81‐eGFP variants were cultured for 24–48 h. When cells reached a confluency of 75–85%, growth medium was discarded and replaced by OptiMEM reduced serum medium (Thermo Fisher Scientific), which was collected after 48 h when the cells were 95%–98% confluent with a viability over 95% as determined with Trypan blue exclusion method using the Vi‐CELL XR Counter (Beckman Coulter). The conditioned medium was centrifuged at 700 g (5804R; Eppendorf, Hamburg, Germany) for 5 min at 4°C to exclude cells and cellular debris. The supernatant was further centrifuged at 2000 × g (5804R; Eppendorf, Hamburg, Germany) for 10 min at 4°C to exclude larger agglomerated particles. After filtration through a 0.2‐μm PVDF‐filter, the supernatant was filled into polycarbonate 70‐ml 38 × 102 mm tubes (Beckman Coulter) to enrich EVs at 125,000 × g for 90 min at 4°C using a 45 Ti Rotor (Beckmann Coulter). The pellets were then re‐suspended in 1.5 ml of Live Cell Imaging Solution (Thermo Fisher Scientific), pH 7.4, per tube, and the ultracentrifugation step was repeated to obtain an EV pellet that was washed once, re‐suspended in 200–1000 μl of the same buffer and stored at ‐80°C until further analysis.
Where indicated, EV preparations were additionally treated: after ultracentrifugation, they were resuspended in 50 μl PBS and incubated for 45 min at 37°C with 550 μl of TrypLE‐Select agent (Thermo Fisher Scientific), to digest residual matrix that could contain laminin fragments and to reduce the EVs’ corona. The effect of TrypLE‐Select enzyme on the integrity of cell surface‐expressed CD81 was examined in comparison with 0.05% trypsin solution that is traditionally used (experimental details in File S1).
2.3.3. Nanoparticle tracking analysis (NTA)
The median, mean and mode sizes of extracellular vesicles as well as their concentration and size distribution were determined by nanoparticle tracking analysis on a ZetaView BASIC PMX‐120 (Particle Metrix GmbH). ZetaView software (version 8.05.11 SP4) was used to determine particle count, size parameters and distribution. The ZetaView device was calibrated with supplied standard beads. EV samples were diluted in PBS for further analysis. Particle measurements were captured and evaluated in three replicates.
2.3.4. Western blot
EV pellets isolated by ultracentrifugation were directly resuspended in RIPA buffer (50 mM Tris‐HCl, pH 8.0, with 150 mM sodium chloride, 1.0% Igepal CA‐630 (NP‐40), 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate), kept on ice, and vortexed five times every 5 min. HeLa cells were collected and the cell pellet was lysed with 100 μl of RIPA buffer, kept on ice, and vortexed five times every 5 min. The cell lysate was then spun at 12,000 × g for 10 min at 4°C and the supernatant was transferred to a new tube and kept on ice. Protein concentrations for the cell lysate supernatants and EV lysates were quantified using BCA assay (Thermo Fisher Scientific) according to manufacturer's instructions.
Thirty micrograms of cell lysate and 15 micrograms of EVs were mixed with buffer containing 0.5 M dithiothreitol, 0.4 M sodium carbonate (Na2CO3), 8% SDS, and 10% glycerol, and heated at 95°C for 10 min. The samples were loaded onto a NuPAGE Novex 4–12% Bis‐Tris Protein Gel (Invitrogen, Thermo Fisher Scientific) and run at 120 V in NuPAGE MES SDS running buffer (Invitrogen, Thermo Fisher Scientific) for 2 h. The proteins on the gel were transferred to a Trans‐Blot Turbo Mini PVDF Transfer for 7 min using the Trans Blot Turbo Blotting System (Bio‐RAD) apparatus. The membrane was blocked with 3% BSA in PBS with 0.1% Tween‐20 (PBS‐T, Sigma‐Aldrich), for 1 h with gentle shaking. After blocking, the membrane was incubated overnight at 4°C with primary antibody solution: anti‐syntenin at 1:1000 (TA504796, Origene, mouse monoclonal), anti‐TSG101 at 1:1000 (ab125011, Abcam, rabbit monoclonal), anti‐Alix at 1:2000 (ab117600, Abcam, mouse monoclonal), or anti‐calnexin at 1:1000 (ab22595, Abcam, rabbit polyclonal). The membrane was washed with PBS‐T three times for 5 min and incubated with the corresponding secondary antibody: donkey anti‐mouse‐IRDye 680RD at 1:10,000 (926‐68072, LI‐COR) or donkey anti‐rabbit‐ IRDye 800CW at 1:10,000 (925‐32213, LI‐COR), for 1 h at RT. Finally, the membrane was washed three times for 5 min with PBS‐T, twice for 5 min with PBS and visualized on the Odyssey infrared imaging system (LI‐COR) at 700 and 800 nm.
2.3.5. Transmission electron microscopy (TEM)
Samples of isolated EVs were pre‐diluted in Live Cell Imaging Solution (Thermo Fisher Scientific) and transferred to copper grids which were previously glow, discharged for 45 s and incubated for 1 min, followed by careful blotting. Uranyl formate solution (2% in 20 mM KOH) was delivered initially for 5 s, then discarded and used again for a 30‐s‐incubation. The staining solution was blotted and fully dried on air. Imaging was performed by using a Tecnai G2 Spirit electron microscope (FEI Company) operated at 120 kV and images were acquired by a CMOS 4k camera (TVIPS GmbH).
2.3.6. Uptake of targeting EVs
Hepatocarcinoma cell line Huh‐7 (JCRB0403, NIBIOHN) and gastric adenocarcinoma cell line NCI‐N87 (ATCC CRL‐5822) were maintained at 37°C in 5% CO2 in high glucose DMEM (Thermo Fisher Scientific), supplemented with 10% FBS (Sigma‐Aldrich), and 4 mM L‐glutamine (Thermo Fisher Scientific). Their secretion of laminin was visualized as described in the File S1. Per well, 1.5 × 105 Huh‐7 and 2 × 105 NCI‐N87 cells were seeded into 24 well‐plates and allowed to attach overnight. Medium was then replaced with 300 μl of OptiMEM reduced serum medium containing graded concentrations of 2.5, 1.25 and 0.75 × 1010 recombinant EVs harbouring CD81‐eGFP and the antigen‐targeting variants, and incubated for 4 h at 37°C. Cells were then detached with 0.1% trypsin‐EDTA (Thermo Fisher Scientific) for 5 min at 37°C, neutralized using 1x Trypsin Inhibitor (Thermo Fisher Scientific) and resuspended in 200 μl PBS. Flow cytometry was used to measure the uptake levels of eGFP‐positive EVs in that the MFI values of the main cell population, gated out in the FSC/SSC plot, were determined using a CytoFLEX S instrument (Beckman Coulter). The fluorescence of the untreated cells was subtracted from all readings. Fluorescence relative to the cells treated with wild‐type CD81 expressing EVs was calculated.
2.4. Transfer of cel‐miR‐39 by targeting EVs
2.4.1. Cel‐miR‐39 transfection
HeLa cells stably expressing CD81‐eGFP variants were reverse‐transfected with C. elegans specific cel‐miR‐39 precursor (AM17103, Thermo Fisher Scientific) using siPORT NeoFX transfection agent (AM4511, Thermo Fisher Scientific) according to the manufacturer's protocol. In brief, per well, 5 μl of siPORT NeoFX transfection agent and 7.5 μl of 10 μM pre‐cel‐miR‐39 were diluted in 100 μl of Opti‐MEM medium separately, then mixed and incubated for 10 min at room temperature. RNA/siPORT NeoFX Transfection Agent transfection complexes were dispensed into wells of a 6‐well‐culture plate containing 3 × 105 HeLa cells stably expressing CD81‐eGFP variants. These were incubated for 24 h at 37°C in 5% CO2, and then the medium was replaced with Opti‐MEM reduced serum medium and the incubation continued for 48 h. All the cel‐miR‐39 transfer studies were performed as before (Hill et al., 2020; Terlecki‐Zaniewicz et al., 2019).
2.4.2. EV isolation by ultrafiltration
The conditioned medium after 48‐h‐incubation was centrifuged at 700 × g for 5 min at 4°C to exclude cells and cellular debris. The supernatant was further centrifuged at 14,000 × g for 15 min at 4°C to exclude larger agglomerated particles. EVs in the conditioned medium were concentrated to 100 μl using Amicon Ultra‐0.5 centrifugal filter unit (UFC501096) at 14,000 × g at 4°C. EVs, secreted by 1 × 106 cells, were used for functional uptake studies in Huh‐7 cells. In brief, 3 × 105 Huh‐7 cells were seeded into 6‐well‐plates and allowed to attach overnight. Medium was then replaced with 1 ml of Opti‐MEM reduced serum medium containing recombinant EVs harbouring CD81‐eGFP or the antigen‐targeting variants produced by cel‐miR‐39‐transfected cells, and incubated for 24 h at 37°C. Where indicated, EV preparations were additionally treated with 1% Triton X‐100 and 38 μg/ml RNase A (Thermo Fisher Scientific) in PBS for 30 min at 37°C and afterwards washed using Amicon Ultra‐0.5 centrifugal filter unit (UFC501096) at 14,000 × g at 4°C, with the purpose to demonstrate that the cel‐miR‐39 is encapsulated in EVs.
2.4.3. RNA isolation
Transfected HeLa cells after EV harvest, and Huh‐7 cells after incubation with EVs carrying cel‐miR‐39, were lysed in TRIreagent (Sigma‐Aldrich) and small RNA isolation was performed using miRNeasy Mini kit (QIAgen) according to manufacturer's instructions. RNA concentration and quality were assessed using Nanodrop spectrometer (ND‐ONE‐W; Thermo Fisher Scientific).
2.4.4. cDNA synthesis
Ten ng of small RNA was used for cDNA synthesis using miRCURY LNA RT kit (QIAgen). cDNA was synthetized at 42°C for 60 min, followed by enzyme inactivation step for 5 min at 95°C. Specific synthesis reaction for UniSp6 used as a spike‐in was included in all samples as a control for enzyme activity.
2.4.5. Quantitative reverse transcription‐PCR in real time (qPCR)
miRNA qPCR analysis was performed using miRCURY LNA SYBR Green PCR kit (QIAgen) and LNA‐optimized primers. Primers used for qPCR are as follows: cel‐miR‐39, cel‐miR‐39‐3p miRCURY LNA miRNA PCR Assay (GeneGlobe ID: YP00203952; 339306; QIAgen); UniSp6, UniSp6 miRCURY LNA miRNA PCR Assay (GeneGlobe ID: YP00203954; 339306; QIAgen). All experiments were performed with a Rotor‐Gene Q cycler.
For all subsequent steps, the raw Ct values were normalized to total viable cell number for each sample. For better visualization, Ct values were further transformed to arbitrary units by assuming a Ct value of 45 to be 1 arbitrary unit (arbitrary units are presented as absolute values).
Statistical analysis was performed using one‐way ANOVA with Graph Pad Prism program, version 5.03.
2.4.6. Binding of recombinant EVs to laminin variants
Biotinylated recombinant laminin variants Lam521, 511, 421 and 332 and EHS‐laminin111 (Biolamina, Sundbyberg, Sweden) were used in bead‐based laminin‐binding assay. 2 × 1010 EVs were added to 4 × 103 CD9 Exosome Capture Beads (Immunostep) in a total volume of 80 μl 3% BSA‐PBS for 3 h at RT. Beads were collected with centrifugation at 3000 × g for 5 min at RT and washed once with 100 μl PBS. Five μg/ml of biotinylated laminin variants in 3% BSA‐PBS were added to the EV‐bead mixture and incubated for 30 min at RT. After pelleting and a washing step with 3% BSA‐PBS at 4°C, neutravidin‐PE (Thermo Fisher Scientific) (1:800) in 3% BSA‐PBS was added for 30 min on ice to detect EV‐bound laminin in a flow cytometry measurement performed on CytoFLEX S instrument (Beckman Coulter). Median fluorescence values of FSC/SSC gated population were expressed as multiple of readings obtained with wild‐type CD81 EVs after values obtained with the secondary reagent alone were subtracted.
2.4.7. Specificity of laminin binding of recombinant EVs
Five times 109 recombinant EVs harbouring wild‐type CD81‐eGFP or its laminin‐targeting variant L2ALU_1 were incubated with 6 × 103 CD9 Exosome Capture Beads (Immunostep) in 120 μl 3% BSA‐PBS for 3 h at RT. Beads were collected with centrifugation at 3000 × g for 5 min at RT and washed once with 100 μl PBS. Five μg/ml of biotinylated human placental laminin in 3% BSA‐PBS, and additionally 15 μg/ml of unlabelled laminin for competition in parallel samples were added to the EV‐bead mixture and incubated for 30 min at RT. After pelleting and a washing step with 3% BSA‐PBS at 4°C, neutravidin‐PE (Thermo Fisher Scientific) (1:800) in 3% BSA‐PBS was added for 30 min on ice to detect EV‐bound laminin in a flow cytometry measurement performed on CytoFLEX S instrument (Beckman Coulter).
3. RESULTS
3.1. Selection of CD81 LEL variants with antigen‐binding activity
Among the membrane‐overrepresented proteins considered markers for the EVs, CD81 has been prominent for a long time (Escola et al., 1998). Availability of several high‐resolution crystal structures has corroborated CD81 LEL as an independent folding unit (Cunha et al., 2018; Kitadokoro, 2001; Kitadokoro et al., 2001; Nelson et al., 2018). Previously, we have attempted to improve the biophysical properties of CD81 LEL and constructed mutants that exhibit in one case an extremely high level of thermostability and in one case reversible thermal denaturation (Vogt et al., 2018). As these should be more permissive for mutagenesis required to introduce a novel antigen binding site, we have identified amino acid residues that could form a novel antigen‐binding surface based on the above mutants. Addressed were the groups of residues that are distal from the cell membrane, to diminish the probability of steric occlusion upon the expression in the context of the full‐length CD81 on the surface of cells of EVs. Randomization of 13 amino acid residues in the library CD81LEL_L2 and 15 amino acid residues in CD81LEL_L3, which corresponds to 14.6% and 16.8% of the original CD81 LEL sequence, was hypothesized to lead to a postulated available antigen binding surface of about 600 Å2 (Figure 1). The decision to use yeast display for binder selection was based on efficient display of wild‐type CD81 LEL on the yeast surface, observed even after stress induction conditions at 37°C, where only properly folded proteins express well due to the stringent quality control system of yeast (Shusta et al., 2000). Over 70% of the cells were positive for binding of antibodies reactive with N‐terminal Xpress tag and C‐terminal c‐myc and his‐tags (Schematic in Figure 2a), and encouragingly a similar level of reactivity was observed with a structure‐dependent binding antibody M38 (Imai & Yoshie, 1993) (Figure 2b). The library sizes were determined to be 1.1 × 108 independent members for CD81LEL_L2 and 1.2 × 108 independent members for CD81LEL_L3, and the correctness of the library L2A was found to be 62.5%, L2B 87.5%, L3A 62.5% and L3B 100% as determined by single clone sequencing. The staining with C‐terminal tag‐reactive antibodies revealed at least 40% positive cells at stress induction conditions at 37°C, indicating that the libraries were suitable for antigen selection (Figure 2c).
FIGURE 1.
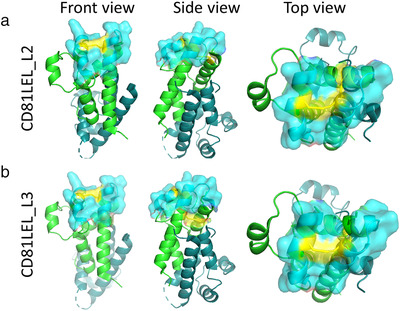
CD81 LEL library designs with de novo disulfide bridges (yellow) and mutated amino acid residues (cyan, surfaced) on protomer a (green), protomer b is in teal: (a) CD81LEL_L2; (b) CD81LEL_L3, front view (left panel), side view (centre panel) and side view (right panel). The figure is based on PDB: 1G8Q using The PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC
FIGURE 2.
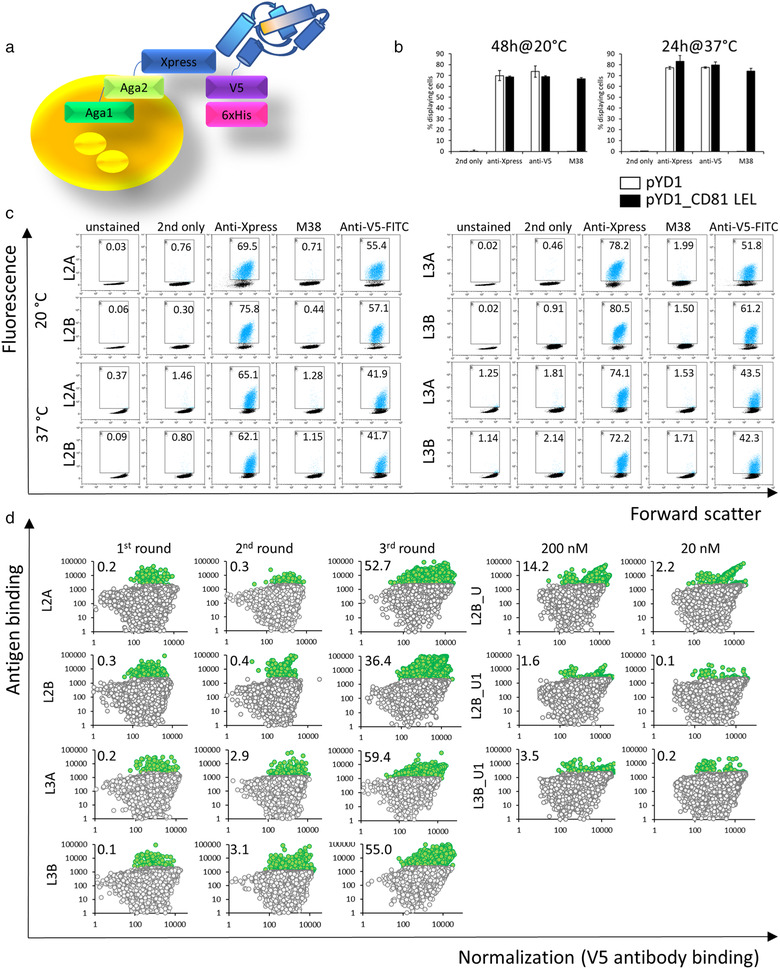
Yeast display of mutant CD81 LEL: (a) Schematic of yeast display; (b) Display of wild‐type CD81 LEL (black bars) in comparison with empty vector pYD1 (white bars), percentage of cells displaying N‐terminal Xpress tag, C‐terminal V‐tag and correctly folded CD81 LEL (M38‐antibody positive) are shown; (c) Quality control of libraries CD81LEL_L2 and CD81LEL_L3. Figures indicate the percentage of gated cells; (d) Results of sorting of CD81 LEL libraries for laminin binders. Figures indicate the percentage of antigen‐positive cells
Enrichment of antigen‐binding clones from all libraries was observed as a double‐positive population after MACS and two following FACS sorts and proceeded to a similar level for both library designs (Figure 2d). After the 3r sorting round different gates, U and U1, were set to improve the chance of discovery of unique clones: four such were identified from library 2 and four from library 3 (sequences in Table 1).
TABLE 1.
Amino acid sequences of laminin‐binding yeast clones
| LibraryAntigen concentration | AA residues | 132‐133 | 136‐139 | 162‐165 | 167 | 171‐172 |
|---|---|---|---|---|---|---|
| CD81LEL_L2 | Wild type | VDDD | LTAL | T | KN | |
| Clone | ||||||
| 1000 nM | L2ALU_1 | RE | NTFD | THSS | G | FF |
| L2ALU1_1 | MI | HRHY | PSST | P | QN | |
| L2BLU_1 | VF | FGGH | HPYK | K | QR | |
| L2BLU1_1 | WK | NSLQ | YLCA | Y | WP | |
| 200 nM | L1_1 | VF | FGGH | HPYK | K | QR |
| L1_13 | FI | FRRY | RNNE | A | AK |
| AA residues | 132‐133 | 136‐141 | 162‐165 | 167 | 171‐172 | |
|---|---|---|---|---|---|---|
| CD81LEL_L3 | Wild type | VDDDAN | LTAL | T | KN | |
| Clone | ||||||
| 1000 nM | L3ALU_1 | LW | SKRYKY | MGNL | E | IE |
| L3BLU_1 | RG | RDFTGS | KLRA | L | IK | |
| L3BLU_2 | KY | KQGDVE | VANM | R | PA | |
| L3BLU_4 | SW | NRKYEP | KANY | H | EM | |
| 200 nM | L1_17 | SW | NRKYEP | KANY | H | EM |
| L1_21 | RH | SGLSGR | IKHK | L | IK |
Enriched pools were then stained with decreasing concentrations of antigen and a double positive population was still discernible with 200 nM biotinylated laminin for sorts L2B_U, L2B_U1, and L3B_U1 (Figure 2d). Single colonies were sequenced and two new binder sequences were discovered (Table 1).
3.2. Laminin‐binding CD81 LEL clones in mammalian yeast display
To increase the likelihood of antigen binding of selected CD81 LEL candidates, when expressed as full‐length CD81 on the surface of EVs produced by mammalian cells and to reduce the frequency of false‐positive hits, we expressed the mutant LELs of candidate clones in the pDisplay system in HEK293‐6E cells (schematic in Figure 3a). With this system, correctly folded wild‐type CD81 LEL can be displayed on the cell surface (Figure 3b) at copy numbers of 20,000 molecules per cell, as determined using QIFIKIT and an anti‐c‐myc tag antibody. The level of display was comparable for the wild‐type CD81 LEL construct and putative laminin‐binding clones. For nine mutants of yeast‐derived CD81 LEL, reactivity with 0.5 μM laminin could be established while no signal above the threshold staining was observed with secondary reagent only (Figure 3c). The five best clones were expressed as full‐length CD81 variants on the surface on EVs.
FIGURE 3.
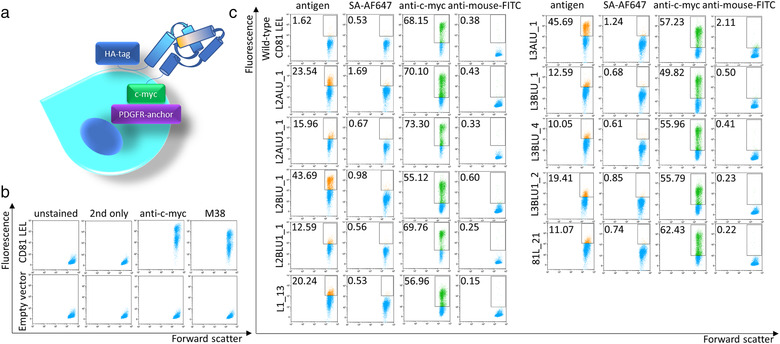
Screening of mutant CD81 LEL clones for laminin binding in mammalian expression system. (a) Schematic of surface display on HEK293 cells; (b) Display of wild‐type CD81 LEL; (c) Antigen binding and c‐myc epitope expression of selected laminin‐binding CD81 LEL variants. Figures indicate the percentage of gated cells. SA‐AF647: streptavidin‐Alexa Fluor 647
3.3. Production and characterization of recombinant EVs
Stable HeLa‐based cell lines, transduced with laminin‐binding CD81 LEL constructs cloned as full‐length CD81‐eGFP fusion, were found to express the mutant constructs at the same level as the analogous wild‐type variant and, similarly, the recombinant proteins predominantly localized to the cell membrane (Figure 4a and Figure S1). Each batch of isolated EVs (Schematic of EV preparation in Figure 4b) resulted in 1–2 × 1012 particles per ml with the median size between 100 and 125 nm as determined with NTA analysis, and there were no conspicuous differences between TrypLE‐Select treated and untreated EVs (Figure 4c and Figure S2A). Treatment of CD81‐expressing HeLa cells with TrypLE‐Select as applied here did not alter the surface expression of CD81 as opposed to 0.05% trypsin treatment (Figure S3). At least 40 % of the particles were eGFP‐fluorescent, indicating that they contained recombinant constructs (Figure 4d and Figure S2B). Importantly, a high yield of EV particles per cell was established for all mutants (Figure S2C). EV marker proteins syntenin, Alix and TSG101 were shown to be enriched in EV preparations in respect to HeLa cell lysate while calnexin, as a non‐EV protein, was absent (Figure 4e). TEM analysis confirmed the presence of the typical cup‐shaped structures (Figure 4f).
FIGURE 4.
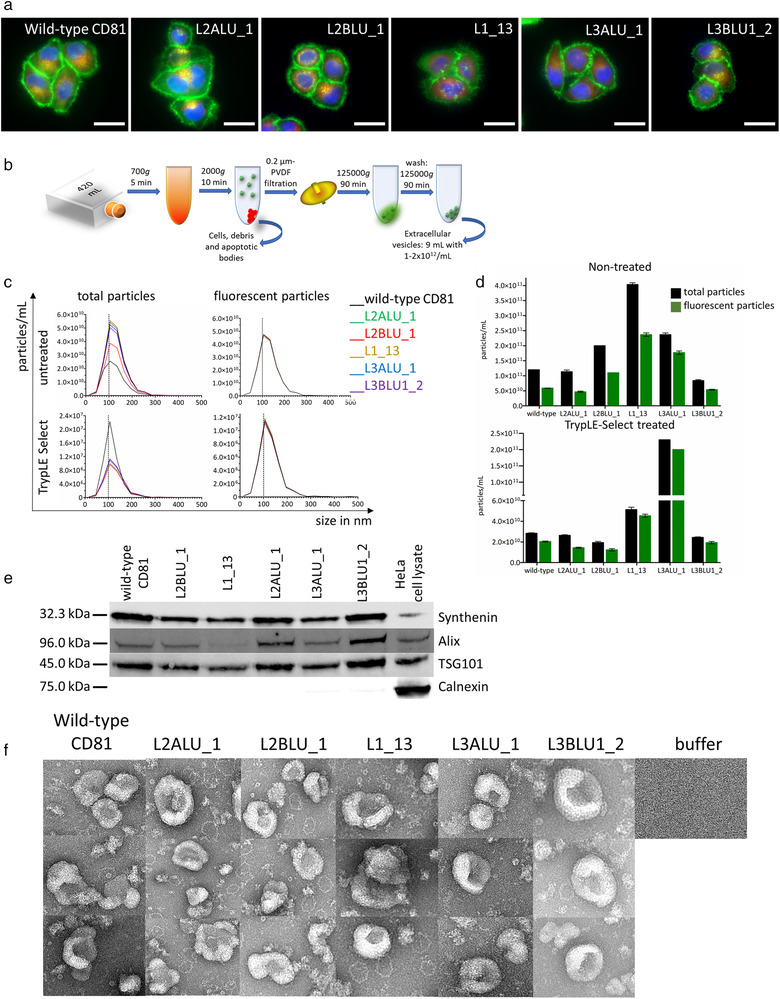
Production of EVs expressing antigen binding CD81 variants. (a) HeLa cells transformed with full‐length mutant CD81 analysed with live‐cell microscopy for distribution of recombinant CD81‐EGFP construct (green), Mitotracker (red) and Hoechst 33342 staining (blue) (bar represents 10 μm); (b) Scheme of EV enrichment; (c) Median size of isolated untreated and TrypLE‐Select‐ treated EVs produced by transformed cell lines (indicated in legend) for total and fluorescent particles; (d) Total (black bars) and recombinant EV (green bars) counts determined with NTA for untreated and TrypLE‐Select‐ treated EVs; (e) Western blot for EV marker proteins syntenin, Alix, TSG101, and the non‐EV marker calnexin; (f) TEM images of EVs isolated from wild‐type and mutant CD81‐transformed cells
3.4. Cellular uptake of recombinant EVs and specific binding to laminin
Laminin expression in Huh‐7 and NCI‐N87 cells was verified using live‐cell microscopy (Figure 5a and Figure S4). When Huh‐7 cells were treated with the highest tested concentration of EVs, most potent internalization was mediated by the clone L2ALU_1, where about five‐fold increase in fluorescence was measured in comparison with wild‐type CD81‐EV treated cells (Figure 5b). When the cells were exposed to TrypLE‐Select‐treated EVs, the difference amounted even to 9.9‐fold. Similar behaviour was observed for the clone L3ALU_1 where the fold change in fluorescence in respect to wild‐type increased from 1.9 to 8.4‐fold, and for L1_13 where 2.5‐ to 5.7‐fold increase could be recorded. For the other candidate binders, the increase in fluorescence was less prominent, 1.8‐ versus 2.7‐fold with TrypLE‐Select reagent treatment for the L2BLU_1 clone, and 1.2‐ and 1.7‐fold for the L3BLU1_2 clone. Dose‐dependent increase in fluorescence regarding the concentration of EVs used was observed for most assayed clones and conditions, but most prominently for TrypLE‐Select treated EVs expressing clones L2ALU_1, L3ALU_1 and L1_13.
FIGURE 5.

EVs expressing laminin‐binding CD81 internalize potently into laminin‐secreting cell lines. (a) Live‐cell microscopy images of Huh7 and NCI‐N87 cell lines visualizing laminin secretion (red), counterstained with anti‐vimentin (green) and Hoechst 33342 (blue) (bar represents 10 μm); (b) Internalization of laminin‐binding EVs into Huh‐7 and NCI‐N87 cell lines. For each clone, results of treatment with 2.5 × 1010 (dark colour), 1.25 × 1010 (medium dark colour) and 0.75 × 1010 (light colour) EVs are shown; (c) Binding of laminin isoforms (indicated in legend) to CD9‐bead captured recombinant EVs; (d) EVs with L2ALU_1 CD81 variant show specific binding of biotinylated laminin that can be outcompeted with excess of labelled antigen; (e) EV‐mediated transfer of cel‐miR‐39 to Huh‐7 cells. The levels of cel‐MiR‐39 are similar for EV‐producing HeLa cells transformed with wild‐type CD81 and L2ALU1 clone (left panel), but the expression in Huh‐7 cell line is significantly stronger after exposure L2ALU_1‐EVs (central panel). The levels of spike‐in RNA UniSp6 are similar for all cells and conditions (right panel). Individual measurements are presented as black bullets and 6 individual measurements were done for UniSp6 expression evaluation. Statistical analysis was done with one‐way ANOVA in GraphPad Prism version 5.03 (ns: P > 0.05; **: ≤ 0.01)
Most promising CD81 variants were also able to enhance the internalization into the NCI‐N87 cell line, 3.5‐ and 1.9‐fold increase in fluorescence signal could be measured for cells exposed to L2ALU_1 and L3ALU_1‐harboring EVs compared to wild‐type CD81 EVs. For this cell line, the effects with TrypLE‐Select treated EVs were stronger, with 17.5‐ and 16.8‐fold increase in fluorescence with clones L2ALU_1 and L3ALU_1. With this protocol a 9.6‐fold increase was observed with L1_13 clone and a 2.7‐fold increase with L2BLU_1 clone, while in both cases only background fluorescence levels were recorded when the EVs were not pretreated. The fluorescence of the cells treated with L3BLU1_2‐EV variant was at the level of wild‐type CD81 EV‐treated cells, regardless of the pre‐treatment protocol.
To corroborate the enhanced uptake of recombinant EVs, biological replicates of the preparations L2ALU_1 and L3ALU_1 were tested for the uptake into Huh‐7 and NCI‐N87 cells and the fold change in fluorescence in respect to wild‐type increased for about two‐ to three times. The enhanced EV internalization of TrypLE‐Select treated EVs was observed as a six‐ to eight‐fold increase in fluorescence compared to wild‐type EVs in both cell lines (Figure S5).
Recombinant EVs were tested for their reactivity with several variants of biotinylated laminin and the potently internalizing EVs with clone L2ALU_1 showed more than two‐fold increase in response when tested for reactivity with recombinant laminin variants with γ1 subunit, as well as a 1.5‐fold response with NHS‐laminin111, in comparison with wild‐type EVs (Figure 5c). EVs, displaying other clones, were within 1.5‐fold of the response of the wild‐type EVs.
To examine if the interaction of the recombinant EVs with the target antigen was specific, we performed a flow cytometry‐based assay where the EVs harbouring either wild‐type CD81‐GFP or the CD81 variant L2ALU_1 were first captured to anti‐CD9 coated beads and allowed to react with biotinylated human placental laminin and neutravidin‐PE. A higher level of fluorescence was seen for the L2ALU_1‐EVs than for wild‐type CD81‐EVs (Figure 5d). Specificity of interaction was tested in an experiment using competition with an excess of unlabelled laminin, which indeed reduced the amount of labelled laminin bound by L2ALU_1 clone.
3.5. EV‐packaged cel‐miR‐39 transfer
To assess the enhanced internalization of CD81 variants, HeLa cells stably expressing wild‐type CD81‐GFP or the CD81 variant L2ALU_1 were reversely transfected with C. elegans cel‐miR‐39 precursor (Terlecki‐Zaniewicz et al., 2019). Seventy‐two hours after transfection, EVs were isolated and used for Huh‐7 cells treatment. After 24 h of exposure, Huh‐7 small RNA was isolated and transfer of cel‐miR‐39 was analysed using qPCR. The expression of cel‐miR‐39 in EV‐producing HeLa cells was similar for wild‐type CD81‐transfected cells and L2ALU_1 variant. Remarkably, cel‐miR‐39 was detected in recipient Huh‐7 cells, and the levels were found to be significantly increased in Huh‐7 cells exposed to L2ALU_1‐EVs compared to wild‐type CD81 EVs, while levels of UniSp6 spike‐in were comparable for all cells and conditions (Figure 5e). Furthermore, the effect of the targeted EVs was lost when EVs were treated with Triton X‐100 and RNase A before exposing them to recipient Huh‐7 cells (Figure S6).
4. DISCUSSION
Stem cell therapies have been applied for the regeneration of a wide range of injured or lost tissues, but only recently it has been proposed that the therapeutic effects of stem cells largely depend on their paracrine activity and are mediated by growth factors and EVs (Azoidis et al., 2018). The advantage of applying EVs rather than growth factors is that they deliver a complex cargo of physiologically relevant proteins and nucleic acids and can even be loaded with exogenous bioactive substances. Comparing with cells, vesicles cannot replicate, do not change their phenotype and are more likely to reach the target tissue due to their small size. Specific targeting of EVs appears to be one of the challenging tasks to be accomplished before these efficient intercellular mediators can be widely used as therapeutic agents, and this has been approached with several strategies involving membrane surface decoration or alteration of their membrane composition by manipulation of their source cell (Murphy et al., 2019). We attempted here to enhance the interaction of extracellular vesicles with their target antigen by assigning one of the overrepresented EV‐membrane marker proteins, CD81, the ability of specific antigen recognition. The actual scaffold for directed evolution were thermally stabilized variants of the CD81 LEL with a high TM over 90°C, which is regarded prerequisite for randomization at a level required to achieve strong antigen binding. The amino acid residues, proposed for randomization, have solvent‐exposed side chains and should together form a confluent surface, which, in size, is comparable with antigen‐binding surfaces of clinically‐relevant alternative antigen‐binding scaffolds VHH that have already been shown to mediate high‐affinity antigen interaction, even though they are of smaller molecular surface areas and smaller diameters than conventional antibodies. The large extracellular loop of CD81 was well expressed in yeast display system, which triggered the choice of this platform. Furthermore, it has already served as a selection tool, not only for antibodies (Boder et al., 2012) and other immunoglobulin domain‐based binders, but also unrelated proteins of various architectures (Könning & Kolmar, 2018), such as Fibronectin 10th Type III domain (Koide et al., 1998), Sso7d (Gera et al., 2011) and Gp2 (Kruziki et al., 2015).
The yeast‐displayed libraries, based on the designed CD81 LEL scaffolds, were constructed with a high percentage of correct members, exhibited a high level of display of mutated proteins, and were rapidly enriched for the antigen of choice, human placental laminin. As the envisioned protocol for antigen‐binding EVs production involved the time‐consuming construction of stably transformed cell lines, a simple in‐between screening step of selected variants of CD81 LEL with mammalian surface display was introduced. Here the mutated LEL domains were expressed as C‐terminal fusions as opposed to the N‐terminal constellation in yeast display, and their expression was additionally under strict quality control of the mammalian folding machinery. Nevertheless, reactivity with the antigen could be corroborated for all variants enriched in yeast display, and the five, with the highest level of antigen binding, were expressed as GFP‐fused full‐length proteins in HeLa cells.
As the level of their expression as well as membrane‐localization was similar to the wild‐type CD81 construct, preparations of the derivatized EVs were compared for the ability to internalize into laminin‐secreting cell lines, Huh‐7 and NCI‐N87. Vesicles isolated from all host cell lines contained a high proportion of GFP‐positive entities and displayed similar uniform size distribution, with majority of the particles measuring 100 nm in diameter, which is typical for EVs. An increased uptake into two target laminin‐secreting cell lines was established for four out of the five selected EVs, and this feature was even more prominent after the pre‐treatment of EV preparation with TrypLE‐Select reagent. This protocol step was employed to digest away the laminin fragments present in the EV preparation and hence minimize the potential scavenging of the available binding sites, and additionally reduce the hydrodynamic radius of EVs by thinning of their corona zone (Skliar et al., 2018), which reportedly impedes their migration rate. The interaction of the most potently internalizing clone L2ALU_1 with laminin was shown to be specific upon competition with unlabelled laminin, and the potentiated reactivity with recombinant laminin variants 521, 511, 421, and EHS laminin111 indicated its specific binding to the γ1 laminin subunit. Importantly, the cel‐miR‐39 delivery into target cells was enhanced using L2ALU_1 derived EVs in comparison with wild‐type CD81 expressing EVs, corroborating the superior internalization of the target‐specific vesicles.
Laminins are major signalling and structural molecules of basement membranes and modulate several diverse cellular functions, such as maintaining tissue structure, adhesion and migration (Aumailley, 2013), but also cell polarity, survival and hormone signalling (Domogatskaya et al., 2012; Hohenester & Yurchenco, 2013; Xu et al., 2010). Laminin internalization could be mediated either by receptor‐independent mechanisms such as pinocytosis, or receptor‐dependent processes. Deficient endocytosis of laminins by dystroglycan results in a dysfunctional basement membrane, perturbed signalling from the endocytic compartment and affects laminin‐associated proteins, indispensable to normal cellular regulation (Leonoudakis et al., 2014). Another receptor connected with laminin endocytosis is the nonintegrin type 67 kDa laminin receptor (67LR). Laminin‐mediated internalization of this receptor can even be neuroprotective as it inhibits entry of β‐amyloid peptide into neurons, prevents its accumulation and decreases neurotoxicity (Gopalakrishna et al., 2018). There is accumulating evidence that EVs are bound to the molecules within the extracellular matrix, in part via ligand‐receptor interactions (Hoshino et al., 2015; Sung et al., 2015), and can in this form modulate cell migration and invasiveness (Mu et al., 2013) as well as mediate regulated delivery of molecular cargo (Clancy et al., 2015). Taken together, these facts point into a direction that EVs, with enhanced laminin binding, may efficiently support regenerative processes.
In summary, we have established a novel selectable antigen binding platform based on the CD81 LEL scaffold. The mutant laminin‐binding CD81 LEL sequences, derived from yeast display, were confirmed to interact with the cognate antigen, and the resulting EVs have indeed shown a higher level of internalization into laminin‐secreting cell lines, including the ability of potent cel‐miR‐39 transfer. In the view of the broad scope of applications for targeted EV‐based therapies, the advantage of the method presented here is that it can rapidly deliver binders to any antigen of choice, which can simply be ‘clicked’ into the full‐length CD81, recombinantly expressed on the EV surface, enabling specific EV‐mediated delivery to a large variety of cells and tissues.
CONFLICT OF INTEREST
J.G. is co‐founder and shareholder of Evercyte GmbH. The technology of antigen recognition via antigen binding marker proteins of extracellular vesicles of is covered in the patent application PCT/EP2019/071825.
Supporting information
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
ACKNOWLEDGEMENTS
We thank Rasmus Peter Thomson (Interdisciplinary Nanoscience Center, Aarhus University) for conceptualization and experimental support with electron microscopy. This project has been supported by the Austrian FFG‐COMET‐ Funding Program (grant to S.V.) and Evercyte GmbH. S.V. is a fellow of the international PhD programme “BioToP‐Biomolecular Technology of Proteins”, funded by the Austrian Science Fund (FWF) (W1224 to F.R. and J.G.). In addition, this project was supported by EQ‐BOKU VIBT GmbH and the BOKU Core Facility for Biomolecular and Cellular Analysis.
Vogt, S., Bobbili, M. R., Stadlmayr, G., Stadlbauer, K., Kjems, J., Rüker, F., Grillari, J., & Wozniak‐Knopp, G. (2021). An engineered CD81‐based combinatorial library for selecting recombinant binders to cell surface proteins: Laminin binding CD81 enhances cellular uptake of extracellular vesicles. Journal of Extracellular Vesicles, 10, e12139. 10.1002/jev2.12139
Stefan Vogt and Madhusudhan Reddy Bobbili contributed equally to this study.
DATA AVAILABILITY STATEMENT
The data supporting the findings of this study are available within the article and its supplementary materials.
REFERENCES
- Alvarez‐Erviti, L., Seow, Y., Yin, H., Betts, C., Lakhal, S., & Wood, M. J. A. (2011). Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nature Biotechnology, 29(4), 341–345. 10.1038/nbt.1807 [DOI] [PubMed] [Google Scholar]
- Antes, T. J., Middleton, R. C., Luther, K. M., Ijichi, T., Peck, K. A., Liu, W. J., Valle, J., Echavez, A. K., & Marbán, E. (2018). Targeting extracellular vesicles to injured tissue using membrane cloaking and surface display. Journal of Nanobiotechnology, 16(1), 61. 10.1186/s12951-018-0388-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aumailley, M. (2013). The laminin family. Cell Adhesion & Migration, 7(1), 48. 10.4161/CAM.22826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azoidis, I., Cox, S. C., & Davies, O. G. (2018). The role of extracellular vesicles in biomineralisation: Current perspective and application in regenerative medicine. Journal of Tissue Engineering, 9, 204173141881013. 10.1177/2041731418810130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batrakova, E. V., & Kim, M. S. (2015). Using exosomes, naturally‐equipped nanocarriers, for drug delivery. Journal of Controlled Release, 219, 396–405. 10.1016/j.jconrel.2015.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boder, E. T., Raeeszadeh‐Sarmazdeh, M., & Price, J. V. (2012). Engineering antibodies by yeast display. Archives of Biochemistry and Biophysics, 526(2), 99–106. 10.1016/j.abb.2012.03.009 [DOI] [PubMed] [Google Scholar]
- Cattin, A. ‐. L., Burden, J. J., Van Emmenis, L., Mackenzie, F. E., Hoving, J. J. A., Garcia Calavia, N., Guo, Y., Mclaughlin, M., Rosenberg, L. H., Quereda, V., Jamecna, D., Napoli, I., Parrinello, S., Enver, T., Ruhrberg, C., & Lloyd, A. C. (2015). Macrophage‐induced blood vessels guide Schwann cell‐mediated regeneration of peripheral nerves. Cell, 162(5), 1127–1139. 10.1016/j.cell.2015.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao, G., Lau, W. L., Hackel, B. J., Sazinsky, S. L., Lippow, S. M., & Wittrup, K. D. (2006).. Isolating and engineering human antibodies using yeast surface display. Nature Protocols, 1(2), 755–768. 10.1038/nprot.2006.94 [DOI] [PubMed] [Google Scholar]
- Ching, R., & Kingham, P. (2015). The role of exosomes in peripheral nerve regeneration. Neural Regeneration Research, 10(5), 743–747, 10.4103/1673-5374.156968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy, J. W., Sedgwick, A., Rosse, C., Muralidharan‐Chari, V., Raposo, G., Method, M., Chavrier, P., & D'souza‐Schorey, C. (2015). Regulated delivery of molecular cargo to invasive tumour‐derived microvesicles. Nature Communications, 6, 6919. 10.1038/ncomms7919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coumans, F. A.W., Brisson, A. R., Buzas, E. I., Dignat‐George, F., Drees, E. E.E., El‐Andaloussi, S., Emanueli, C., Gasecka, A., Hendrix, A., Hill, A. F., Lacroix, R., Lee, Y., Van Leeuwen, T. G., Mackman, N., Mäger, I., Nolan, J. P., Van Der Pol, E., Pegtel, D. M., Sahoo, S. ,… Nieuwland, R. (2017). Methodological guidelines to study extracellular vesicles. Circulation Research, 120(10), 1632–1648. 10.1161/CIRCRESAHA.117.309417 [DOI] [PubMed] [Google Scholar]
- Cunha, E. S., Sfriso, P., Rojas, A. L., Roversi, P., Hospital, A., Orozco, M., & Abrescia, N. G.A. (2018). Mechanism of structural tuning of the hepatitis C virus human cellular receptor CD81 large extracellular loop. Structure (London, England). 26(1), 181. 10.1016/j.str.2017.11.015 [DOI] [PubMed] [Google Scholar]
- Domogatskaya, A., Rodin, S., & Tryggvason, K. (2012). Functional diversity of laminins. Annual Review of Cell and Developmental Biology, 28(1), 523–553. 10.1146/annurev-cellbio-101011-155750 [DOI] [PubMed] [Google Scholar]
- Dong, R., Liu, Y., Yang, Y., Wang, H., Xu, Y., & Zhang, Z. (2019). MSC‐derived exosomes‐based therapy for peripheral nerve injury: a novel therapeutic strategy. BioMed Research International, 2019, 6458237. 10.1155/2019/6458237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escola, J.‐M., Kleijmeer, M. J., Stoorvogel, W., Griffith, J. M., Yoshie, O., & Geuze, H. J. (1998). Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B‐lymphocytes. Journal of Biological Chemistry, 273(32), 20121–20127. 10.1074/jbc.273.32.20121 [DOI] [PubMed] [Google Scholar]
- Gera, N., Hussain, M., Wright, R. C., & Rao, B. M. (2011). Highly stable binding proteins derived from the hyperthermophilic Sso7d scaffold. Journal of Molecular Biology, 409(4), 601–616. 10.1016/j.jmb.2011.04.020 [DOI] [PubMed] [Google Scholar]
- Gietz, R. D., & Schiestl, R. H. (2007).High‐efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nature Protocols, 2(1,) 31–34, 10.1038/nprot.2007.13 [DOI] [PubMed] [Google Scholar]
- Gopalakrishna, R., Gundimeda, U., Zhou, S., Bui, H., Davis, A., Mcneill, T., & Mack, W. (2018). Laminin‐1 induces endocytosis of 67KDa laminin receptor and protects Neuroscreen‐1 cells against death induced by serum withdrawal. Biochemical and Biophysical Research Communications, 495(1), 230–237. 10.1016/j.bbrc.2017.11.015 [DOI] [PubMed] [Google Scholar]
- H Rashed, M., Bayraktar, E. K., Helal, G., Abd‐Ellah, M., Amero, P., Chavez‐Reyes, A., & Rodriguez‐Aguayo, C. (2017). Exosomes: From garbage bins to promising therapeutic targets. International Journal of Molecular Sciences, 18(3), 538. 10.3390/ijms18030538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackethal, J., Schuh, C., & Hofer, A., Meixner, B., Hennerbichler, S., Redl, H., & Teuschl, A. H. (2018). Human placenta laminin‐111 as a multifunctional protein for tissue engineering and regenerative medicine. Advances in Experimental Medicine and Biology. 1077, 3–17. 10.1007/978-981-13-0947-2_1 [DOI] [PubMed] [Google Scholar]
- Hill, N., Michell, D. L., Ramirez‐Solano, M., Sheng, Q., Pusey, C., Vickers, K. C., & Woollard, K. J. (2020). Glomerular endothelial derived vesicles mediate podocyte dysfunction: A potential role for miRNA. Plos One, 15(3), e0224852. 10.1371/journal.pone.0224852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenester, E., & Yurchenco, P. D. (2013). Laminins in basement membrane assembly. Cell Adhesion & Migration, 7(1), 56–63. 10.4161/cam.21831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino, A., Costa‐Silva, B., Shen, T. ‐. L., Rodrigues, G., Hashimoto, A., Tesic Mark, M., Molina, H., Kohsaka, S., Di Giannatale, A., Ceder, S., Singh, S., Williams, C., Soplop, N., Uryu, K., Pharmer, L., King, T., Bojmar, L., Davies, A. E., Ararso, Y. … Lyden, D. (2015). Tumour exosome integrins determine organotropic metastasis. Nature, 527(7578), 329–335. 10.1038/nature15756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung, M. E., & Leonard, J. N. (2015). Stabilization of exosome‐targeting peptides via engineered glycosylation. Journal of Biological Chemistry, 290(13), 8166–8172. 10.1074/jbc.M114.621383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai, T., & Yoshie, O. (1993). C33 antigen and M38 antigen recognized by monoclonal antibodies inhibitory to syncytium formation by human T cell leukemia virus type 1 are both members of the transmembrane 4 superfamily and associate with each other and with CD4 or CD8 in T cells. Journal of Immunology, 151(11), 6470 LP – 6481. [PubMed] [Google Scholar]
- Inder, K. L., Ruelcke, J. E., Petelin, L., Moon, H., Choi, E., Rae, J., Blumenthal, A., Hutmacher, D., Saunders, N. A., Stow, J. L., Parton, R. G., & Hill, M. M. (2014). Cavin‐1/PTRF alters prostate cancer cell‐derived extracellular vesicle content and internalization to attenuate extracellular vesicle‐mediated osteoclastogenesis and osteoblast proliferation. Journal of extracellular vesicles, 3(1), 23784. 10.3402/jev.v3.23784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyaram, A., & Jay, S. M. (2018). Preservation and storage stability of extracellular vesicles for therapeutic applications. The Aaps Journal [Electronic Resource], 20(1), 1. 10.1208/s12248-017-0160-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitadokoro, K. (2001). CD81 extracellular domain 3D structure: Insight into the tetraspanin superfamily structural motifs. EMBO Journal, 20(1‐2), 12–18. 10.1093/emboj/20.1.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitadokoro, K., Galli, G., Petracca, R., Falugi, F., Grandi, G., & Bolognesi, M. (2001). Crystallization and preliminary crystallographic studies on the large extracellular domain of human CD81, a tetraspanin receptor for hepatitis C virus. Acta Crystallogr Sect D Biol Crystallogr, 57(Pt 1), 156–158, 10.1107/S0907444900015468 [DOI] [PubMed] [Google Scholar]
- Koide, A., Bailey, C. W., Huang, X., & Koide, S. (1998). The fibronectin type III domain as a scaffold for novel binding proteins. Journal of Molecular Biology. 284(4), 1141–1151, 10.1006/jmbi.1998.2238 [DOI] [PubMed] [Google Scholar]
- Könning, D., & Kolmar, H. (2018). Beyond antibody engineering: Directed evolution of alternative binding scaffolds and enzymes using yeast surface display. Microbial Cell Factories, 17(1), 32. 10.1186/s12934-018-0881-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooijmans, S. A. A., Aleza, C. G., Roffler, S. R., Van Solinge, W. W., Vader, P., & Schiffelers, R. M. (2016). Display of GPI‐anchored anti‐EGFR nanobodies on extracellular vesicles promotes tumour cell targeting. Journal of Extracellular Vesicles, 5(1), 31053. 10.3402/jev.v5.31053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruziki, M. A., Bhatnagar, S., Woldring, D. R., Duong, V. T., & Hackel, B. J. (2015). A 45‐amino‐acid scaffold mined from the PDB for high‐affinity ligand engineering. Chemistry & Biology, 22(7), 946–956. 10.1016/j.chembiol.2015.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonoudakis, D., Huang, G., Akhavan, A., Fata, J. E., Singh, M., Gray, J. W., & Muschler, J. L. (2014). Endocytic trafficking of laminin is controlled by dystroglycan and is disrupted in cancers. Journal of Cell Science, 127(Pt 22), 4894–4903. 10.1242/jcs.152728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy, S., Nguyen, V. Q., Andria, M. L., & Takahashi, S. (1991). Structure and membrane topology of TAPA‐1. Journal of Biological Chemistry, 266(22), 14597–14602. http://www.ncbi.nlm.nih.gov/pubmed/1860863 [PubMed] [Google Scholar]
- Lunavat, T. R., Jang, S. C., Nilsson, L., Park, H. T., Repiska, G., Lässer, C., Nilsson, J. A., Gho, Y. S., & Lötvall, J. (2016). RNAi delivery by exosome‐mimetic nanovesicles – Implications for targeting c‐Myc in cancer. Biomaterials, 102, 231–238. 10.1016/j.biomaterials.2016.06.024 [DOI] [PubMed] [Google Scholar]
- Maguire, C. A., Balaj, L., Sivaraman, S., Crommentuijn, M. H.w, Ericsson, M., Mincheva‐Nilsson, L., Baranov, V., Gianni, D., Tannous, B. A., Sena‐Esteves, M., Breakefield, X. O., & Skog, J. (2012). Microvesicle‐associated AAV vector as a novel gene delivery system. Molecular Therapy, 20(5), 960–971. 10.1038/mt.2011.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu, W., Rana, S., & Zöller, M. (2013). Host matrix modulation by tumor exosomes promotes motility and invasiveness. Neoplasia, 15(8), 875–IN4. 10.1593/neo.13786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy, D. E., De Jong, O. G., Brouwer, M., Wood, M. J., Lavieu, G., Schiffelers, R. M., & Vader, P. (2019). Extracellular vesicle‐based therapeutics: Natural versus engineered targeting and trafficking. Experimental & Molecular Medicine, 51(3), 32. 10.1038/s12276-019-0223-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, B., Adams, J., Kuglstatter, A., Li, Z., Harris, S. F., Liu, Y., Bohini, S., Ma, H., Klumpp, K., Gao, J., & Sidhu, S. S. (2018). Structure‐guided combinatorial engineering facilitates affinity and specificity optimization of anti‐CD81 antibodies. Journal of Molecular Biology, 430(14), 2139–2152. 10.1016/j.jmb.2018.05.018 [DOI] [PubMed] [Google Scholar]
- Ohno, S. ‐. I., Takanashi, M., Sudo, K., Ueda, S., Ishikawa, A., Matsuyama, N., Fujita, K., Mizutani, T., Ohgi, T., Ochiya, T., Gotoh, N., & Kuroda, M. (2013). Systemically injected exosomes targeted to EGFR deliver antitumor microrna to breast cancer cells. Molecular Therapy, 21(1), 185–191. 10.1038/mt.2012.180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, D. B., Santoro, M., Born, L. J., Fisher, J. P., & Jay, S. M. (2018). Towards rationally designed biomanufacturing of therapeutic extracellular vesicles: Impact of the bioproduction microenvironment. Biotechnology Advances, 36(8), 2051–2059. 10.1016/j.biotechadv.2018.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, Y., Sun, R., Wu, C., Wang, L., & Zhang, C. (2016). Exosome: A novel approach to stimulate bone regeneration through regulation of osteogenesis and angiogenesis. International Journal of Molecular Sciences, 17(5), 712. 10.3390/ijms17050712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riederer, I., Bonomo, A. C., Mouly, V., & Savino, W. (2015). Laminin therapy for the promotion of muscle regeneration. FEBS Letters, 589(22), 3449–3453. 10.1016/j.febslet.2015.10.004 [DOI] [PubMed] [Google Scholar]
- Roura, S., & Bayes‐Genis, A. (2019). Toward standardization of mesenchymal stromal cell‐derived extracellular vesicles for therapeutic use: A call for action. Proteomics, 19(1‐2), 1800397. 10.1002/pmic.201800397 [DOI] [PubMed] [Google Scholar]
- Seigneuret, M. (2006). Complete predicted three‐dimensional structure of the facilitator transmembrane protein and hepatitis C virus receptor CD81: Conserved and variable structural domains in the tetraspanin superfamily. Biophysical Journal, 90(1), 212–227. 10.1529/biophysj.105.069666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seigneuret, M., Delaguillaumie, A., Lagaudrière‐Gesbert, C., & Conjeaud, H. (2001). Structure of the tetraspanin main extracellular domain: A partially conserved fold with a structurally variable domain insertion. Journal of Biological Chemistry, 276(43), 40055–40064. 10.1074/jbc.M105557200 [DOI] [PubMed] [Google Scholar]
- Shusta, E. V., Holler, P. D., Kieke, M. C., Kranz, D. M., & Wittrup, K. D. (2000). Directed evolution of a stable scaffold for T‐cell receptor engineering. Nature Biotechnology, 18(7), 754–759. 10.1038/77325 [DOI] [PubMed] [Google Scholar]
- Skliar, M., Chernyshev, V. S., Belnap, D. M., Sergey, G. V., Al‐Hakami, S. M., Bernard, P. S., Stijleman, I. J., & Rachamadugu, R. (2018). Membrane proteins significantly restrict exosome mobility. Biochemical and Biophysical Research Communications, 501(4), 1055–1059. 10.1016/j.bbrc.2018.05.107 [DOI] [PubMed] [Google Scholar]
- Sung, B. H., Ketova, T., Hoshino, D., Zijlstra, A., Weaver, A. M. (2015). Directional cell movement through tissues is controlled by exosome secretion. Nature Communications. 6, 7164. 10.1038/ncomms8164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutaria, D. S., Badawi, M., Phelps, M. A., & Schmittgen, T. D. (2017). Achieving the promise of therapeutic extracellular vesicles: The devil is in details of therapeutic loading. Pharmaceutical Research, 34(5), 1053–1066. 10.1007/s11095-017-2123-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, S. H. S., Tjio, C. K. E., Wong, J. R. Y., Wong, K. L., Chew, J. R. J., Hui, J. H. P., & Toh, W. S. (2020). A systematic review of mesenchymal stem cell exosomes for cartilage repair and regeneration. Tissue Engineering. Part B, Reviews.27(1), 1–13, tEn.TEB.2019.0326. 10.1089/ten.TEB.2019.0326 [DOI] [PubMed] [Google Scholar]
- Taverna, S., Pucci, M., & Alessandro, R. (2017). Extracellular vesicles: Small bricks for tissue repair/regeneration. Annals of Translational Medicine, 5(4), 83–83. 10.21037/atm.2017.01.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terlecki‐Zaniewicz, L., Pils, V., Bobbili, M. R., Lämmermann, I., Perrotta, I., Grillenberger, T., Schwestka, J., Weiß, K., Pum, D., Arcalis, E., Schwingenschuh, S., Birngruber, T., Brandstetter, M., Heuser, T., Schosserer, M., Morizot, F., Mildner, M., Stöger, E., Tschachler, E. ,… Grillari, J. (2019). Extracellular vesicles in human skin: Cross‐talk from senescent fibroblasts to keratinocytes by miRNAs. Journal of Investigative Dermatology, 139(12), 2425–2436.e5.e5. 10.1016/j.jid.2019.05.015 [DOI] [PubMed] [Google Scholar]
- Villarroya‐Beltri, C., Gutiérrez‐Vázquez, C., Sánchez‐Cabo, F., Pérez‐Hernández, D., Vázquez, J., Martin‐Cofreces, N., Martinez‐Herrera, D. J., Pascual‐Montano, A., Mittelbrunn, M., & Sánchez‐Madrid, F. (2013). Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nature Communications, 4(1), 2980. 10.1038/ncomms3980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt, S., Stadlmayr, G., Stadlbauer, K., Sádio, F., Andorfer, P., Grillari, J., Rüker, F., & Wozniak‐Knopp, G. (2018). Stabilization of the CD81 large extracellular loop with De Novo disulfide bonds improves its amenability for peptide grafting. Pharmaceutics, 10(3), 138. 10.3390/pharmaceutics10030138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, S., Busatto, S., Pham, A., Tian, M., Suh, A., Carson, K., Quintero, A., Lafrence, M., Malik, H., Santana, M. X., & Wolfram, J. (2019). Extracellular vesicle‐based drug delivery systems for cancer treatment. Theranostics, 9(26), 8001–8017. 10.7150/thno.37097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. ‐. H., Forterre, A. V., Zhao, J., Frimannsson, D. O., Delcayre, A., Antes, T. J., Efron, B., Jeffrey, S. S., Pegram, M. D., & Matin, A. C. (2018). Anti‐HER2 scFv‐directed extracellular vesicle‐mediated mRNA‐based gene delivery inhibits growth of HER2‐positive human breast tumor xenografts by prodrug activation. Molecular Cancer Therapeutics. 17(5), 1133–1142, mOlcanther.0827.2017. 10.1158/1535-7163.MCT-17-0827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y., He, G., Guo, Y., Tang, H., Shi, Y., Bian, X., Zhu, M., Kang, X., Zhou, M., Lyu, J., Yang, M., Mu, M., Lai, F., Lu, K., Chen, W., Zhou, B., Zhang, J., & Tang, K. (2019). Exosomes from tendon stem cells promote injury tendon healing through balancing synthesis and degradation of the tendon extracellular matrix. Journal of Cellular and Molecular Medicine. 23(8), 5475–5485. 10.1111/jcmm.14430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weilner, S., Schraml, E., Wieser, M., Messner, P., Schneider, K., Wassermann, K., Micutkova, L., Fortschegger, K., Maier, A. B., Westendorp, R., Resch, H., Wolbank, S., Redl, H., Jansen‐Dürr, P., Pietschmann, P., Grillari‐Voglauer, R., & Grillari, J. (2016). Secreted microvesicular miR‐31 inhibits osteogenic differentiation of mesenchymal stem cells. Aging Cell, 15(4), 744–754. 10.1111/acel.12484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfers, J., Lozier, A., Raposo, G., Regnault, A., Théry, C., Masurier, C., Flament, C., Pouzieux, S., Faure, F., Tursz, T., Angevin, E., Amigorena, S., & Zitvogel, L. (2001). Tumor‐derived exosomes are a source of shared tumor rejection antigens for CTL cross‐priming. Nature Medicine, 7(3), 297–303. 10.1038/85438 [DOI] [PubMed] [Google Scholar]
- Wu, P., Zhang, B., Shi, H., Qian, H., & Xu, W. (2018). MSC‐exosome: A novel cell‐free therapy for cutaneous regeneration. Cytotherapy, 20(3), 291–301. 10.1016/j.jcyt.2017.11.002 [DOI] [PubMed] [Google Scholar]
- Xu, R., Spencer, V. A., Groesser, D. L., & Bissell, M. J. (2010). Laminin regulates PI3K basal localization and activation to sustain STAT5 activation. Cell Cycle, 9(21), 4315–4322. 10.4161/cc.9.21.13578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, T., Kosaka, N., & Ochiya, T. (2019). Latest advances in extracellular vesicles: From bench to bedside. Science and Technology of Advanced Materials, 20(1), 746–757. 10.1080/14686996.2019.1629835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yáñez‐Mó, M., Siljander, P. R.‐M., Andreu, Z., Bedina Zavec, A., Borràs, F. E., Buzas, E. I., Buzas, K., Casal, E., Cappello, F., Carvalho, J., Colás, E., Cordeiro‐Da Silva, A., Fais, S., Falcon‐Perez, J. M., Ghobrial, I. M., Giebel, B., Gimona, M., Graner, M., Gursel, I. ,… De Wever, O. (2015). Biological properties of extracellular vesicles and their physiological functions. Journal of Extracellular Vesicles, 4(2015), 1–60. 10.3402/jev.v4.27066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka, Y., Konishi, Y., Kosaka, N., Katsuda, T., Kato, T., Ochiya, T. (2013). Comparative marker analysis of extracellular vesicles in different human cancer types. Journal of Extracellular Vesicles. 2(1), 20424. 10.3402/jev.v2i0.20424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, P., Yeo, J. C., & Lim, C. T. (2019). Advances in technologies for purification and enrichment of extracellular vesicles. SLAS Technology Translating Life Sciences Innovation, 24(5), 477–488. 10.1177/2472630319846877 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Data Availability Statement
The data supporting the findings of this study are available within the article and its supplementary materials.