

Abstract
The objective of this study is to identify and analyze integrons and antibiotic resistance genes (ARGs) in samples collected from diverse sites in terrestrial Antarctica. Integrons were studied using two independent methods. One involved the construction and analysis of intI gene amplicon libraries. In addition, we sequenced 17 metagenomes of microbial mats and soil by high‐throughput sequencing and analyzed these data using the IntegronFinder program. As expected, the metagenomic analysis allowed for the identification of novel predicted intI integrases and gene cassettes (GCs), which mostly encode unknown functions. However, some intI genes are similar to sequences previously identified by amplicon library analysis in soil samples collected from non‐Antarctic sites. ARGs were analyzed in the metagenomes using ABRIcate with CARD database and verified if these genes could be classified as GCs by IntegronFinder. We identified 53 ARGs in 15 metagenomes, but only four were classified as GCs, one in MTG12 metagenome (Continental Antarctica), encoding an aminoglycoside‐modifying enzyme (AAC(6´)acetyltransferase) and the other three in CS1 metagenome (Maritime Antarctica). One of these genes encodes a class D β‐lactamase (blaOXA‐205) and the other two are located in the same contig. One is part of a gene encoding the first 76 amino acids of aminoglycoside adenyltransferase (aadA6), and the other is a qacG2 gene.
Keywords: antibiotic resistance, bioinformatics, horizontal gene transfer, microbial genomics
The objective of this study is to identify and analyze integrons and antibiotic resistance genes (ARGs) in samples collected from diverse sites in terrestrial Antarctica. The metagenomic analysis allowed us to identify novel predicted intI integrases and gene cassettes (GCs), which mostly encode unknown functions.
1. INTRODUCTION
Integrons are natural genetic platforms that incorporate, exchange, and express new genes. Through site‐specific recombination events, they integrate open reading frames ensuring their correct expression. All integrons share a common functional structure composed of a stable platform associated with a variable region of GCs that encode accessory functions (Cambray et al., 2010).
The stable region contains an integrase intI gene that codes for a site‐specific tyrosine recombinase (IntI). The enzyme catalyzes the insertion and excision of GCs (Stokes & Hall, 1989). This region also harbors a primary recombination site (attI) adjacent to intI where GCs are inserted (Collis et al., 1993), and a Pc promoter that regulates their expression. The Pc promoter is located in the coding region of the intI gene or between this gene and the attI site, and expression is oriented toward the site of integration (Collis & Hall, 1995; Jové et al., 2010). The variable region includes a gene array (GCs), each flanked by an attC site expressed under the control of Pc.
Integron studies were classically focused on isolates from clinical environments, using culture‐dependent methods. However, the incorporation of new sequencing methods allowed access to much more information. Metagenomic studies have allowed for the identification of new integrase sequences showing that there is large genetic diversity. In addition, metagenomic dataset analysis revealed that there is an extensive pool of GCs encoding functions required for environmental adaptation and others, mostly of unknown function (Holmes et al., 2003). The main obstacles to the identification of GCs are the limited tools available to recognize and predict the identification of attC sites (Cury et al., 2016). Some strategies were designed to recognize attC sites of “classical” integrons (Stokes et al., 2001), although this is difficult because they exhibit a relatively high diversity (Cury et al., 2016). This has even allowed for encryption of these sequences within protein‐coding sequences (Nivina et al., 2020).
Integrons are ancestral and usually stable structures that have played an essential role in bacterial genome evolution (Rowe‐Magnus and Mazel, 2001), due to their GCs shuffling properties in times of stress (Guerin et al., 2009). Considering the presence of an intI gene, integrons have been found in 9–17% of genomes available in databases (Boucher et al., 2007; Cambray et al., 2010; Cury et al., 2016). They are distributed throughout Gram‐negative bacteria, with the notable exception of α‐proteobacteria, and are more prevalent among γ‐proteobacteria (Cury et al., 2016).
Historically, integrons have been classified into two groups: chromosomal integrons (CIs) and mobile integrons (MIs), which are physically associated with transposons contained in conjugative plasmids (Mazel, 2006). Currently, CIs are termed as sedentary chromosomal integrons (SCIs), because MIs can also be found in chromosomes and the term could be confusing (Cury et al., 2016). SCIs share an evolutionary history, and their phylogeny reflects a pattern of vertical inheritance. For this reason, these integrons are not classified in classes such as MIs and are instead classified according to their host species (Cambray et al., 2011).
The first integrons were characterized at the end of the 1980s from a collection of bacterial isolates of clinical origin. It was then shown that integrons are commonly found in bacterial genomes. Analysis of integron diversity in natural environments indicates that they could play a fundamental role in the adaptation and evolution of these organisms. These studies were developed with samples from different environments and using different approaches. However, "extreme" environments like Antarctica have been relatively poorly explored (Ghaly et al., 2019; Gillings, 2014; Stokes et al., 2001).
The Antarctic convergence zone is classified into three biogeographical regions: Continental Antarctica, Maritime Antarctica, and Sub‐Antarctica (Convey, 2010). These three regions are differentiated mainly by their average annual temperatures, rainfall, and winds, and are also associated with characteristic biota. The Continental region is considered an especially inclement zone and includes the McMurdo Dry Valleys which is a polar desert. The highest rainfall is recorded in Maritime Antarctica (400–600 mm/year), with average annual temperatures ranging between 1℃ and −21℃. This region, with a less extreme climate than the Continental region, includes the NW of the Antarctic Peninsula and nearby islands. The Sub‐Antarctica region includes several islands located between 35°S and 60°S. The climate of these islands enables the establishment of breeding sites for birds and marine mammals.
Our objective was to identify and analyze integrons and ARGs in microbial mat and soil samples collected in different sites of the Antarctic continent. For the analysis of integrons, we used two culture‐independent methods. One of these strategies involved the construction and analysis of intI amplicon libraries from the soil. We also sequenced 17 metagenomes of microbial mats and soil by high‐throughput next generation sequencing (NGS) and analyzed these metagenomic data using the IntegronFinder program, which allows intI and GC identification.
As expected, these analyses allowed us to identify a broad spectrum of intI sequences. The vast majority were not related to mobile integrons integrase sequences, although some sequences were similar to others previously recovered in amplicon libraries from soil samples of non‐Antarctic environments. ARGs were analyzed in the 17 metagenomes using ABRicate with CARD database. We identified 53 ARGs by CARD, distributed in 15 metagenomes (except MTG3 and MTG4) and IntegronFinder identified three of these genes as GCs.
2. EXPERIMENTAL PROCEDURES
2.1. Construction of Inti amplicon library and DNA sequence analysis
Soil samples (Table 1) were collected in 50 mL sterile tubes and stored at −20ºC (except during transport, in which were kept refrigerated on ice) until processing in the laboratory (IIBCE, Montevideo). The two samples, identified CP2, and PD2 are described in Table 1. DNA extraction was done using the ZR Soil Microbe DNA Microprep kit (Zymo Research®, USA) following the manufacturer's instructions. Purified DNA was used as a template in PCR reactions for intI1 gene amplification (primers int1.F and int1.R) (484 bp) (Mazel et al., 2000). The protocol used for cloning PCR fragments was described previously by Antelo et al. (Antelo et al., 2015). Sequences obtained from Macrogen (Seoul, South Korea) (8 from PD2 and 14 from CP2) were analyzed following the protocol described by Antelo et al. (Antelo et al., 2015).
TABLE 1.
Sampling sites for amplicon libraries of intI genes (Mazel et al., 2000)
| Name of the sample | Campaign | Location | Site | Description |
|---|---|---|---|---|
| PD2 | April/2015 |
62°09´03´´S 58°56´27´´W |
Beach in front of Drake sea | Soil |
| CP2 | February/2016 | 62°14´35´´ S 58°39´99´´ W | Potter creek | Soil |
2.2. Sampling sites for NGS metagenomic analysis
Microbial mat samples (14 samples of ~ 20 g) were collected from different sites in Maritime and Continental Antarctica, representing a latitudinal gradient (Table 2). Microbial mats develop during Austral summer in glacier and snow melting currents. These communities are benthic microbial assemblages of high diversity, dominated by cyanobacteria (Callejas et al., 2018; Valdespino‐Castillo et al., 2018; Azziz et al., 2019). Soil samples (3 samples of ~ 60 g, from the upper portion of 10 cm deep, without snow cover) were collected on the Fildes peninsula during the Antarctic campaign of April 2015. Sampling sites included Antarctic Specially Protected Areas (ASPA) like Ardley Island (IA6; at ASPA 150), Halfthree‐Point (HTP2; at ASPA 125a), and sites next to the septic system chamber of BCAA (CS1).
TABLE 2.
Geographic location of sampling sites used for NGS metagenomics analysis
| Sample | Geographic reference and type of sample | Latitude | Longitude | Name of the contig_xxx |
|---|---|---|---|---|
| Drake(1) | King George Island (Fildes Peninsula, microbial mat) | 62°09’30’’ S | 58°56’31’’ W | MTG1_ |
| Espejo(2) | King George Island (Fildes Peninsula, microbial mat) | 62°09’59’’ S | 58°58’33’’ W | MTG2_ |
| HTP(3) | King George Island (Halfthree point, microbial mat) | 62°12’14’’ S | 58°57’16’’ W | MTG3_ |
| Pista(4) | King George Island (next to Chilean Airport, microbial mat) | 62° 10’0’’ S | 58° 58’34’’ W | MTG4_ |
| Carlini I(5) | King George Island (microbial mat) | 62° 14’35’’ S | 58° 40’39’’ W | MTG5_ |
| Carlini II(6) | King George Island (microbial mat) | 62° 14’34’’ S | 58° 40’26’’ W | MTG6_ |
| Esperanza(7) | Antarctic Peninsula (Trinity Peninsula, microbial mat) | 63°28’13’’ S | 57°12’3’’ W | MTG7_ |
| Primavera(8) | Antarctic Peninsula (microbial mat) | 64°09’22’’ S | 60°57’30’’ W | MTG8_ |
| San Martín(9) | Antarctic Peninsula (Fallieres Coast, microbial mat) | 68°07’45’’ S | 67°06’2’’ W | MTG9_ |
| B012‐2015–14(10) | McMurdo Dry Valleys (microbial mat) | 78°01’24’’ S | 163°55’03’’ E | MTG10_ |
| B012‐2015–15(11) | McMurdo Dry Valleys (microbial mat) | 78°01’23’’ S | 163°54’56’’ E | MTG11_ |
| B012‐2015–16(12) | McMurdo Dry Valleys (microbial mat) | 78°01’23’’ S | 163°54’07’’ E | MTG12_ |
| B012‐2015–16 Mid Good(13) | McMurdo Dry Valleys (microbial mat) | 78°01’30’’ S | 164°06’02’’ E | MTG13_ |
| B012‐2015–18(14) | McMurdo Dry Valleys (microbial mat) | 77°39’40’’ S | 163°05’31’’ E | MTG14_ |
| CS1(15) | King George Island (septic chamber next to BCAA, soil) | 62°11’35’’ S | 58°54’19’’ W | CS1_ |
| HTP2(16) | King George Island (Halfthree‐point, soil) | 62° 13’ 09’’S | 58° 57´ 09’´ W | HTP2_ |
| IA6(17) | King George Island (Ardley Island, soil) | 62° 12’ 03’’S | 58° 55’ 04‘’W | IA6_ |
2.3. Total DNA isolation and NGS metagenome sequencing
Metagenomic libraries were prepared with the Nextera DNA Flex library prep kit (Illumina, San Diego, CA) where fragments of total DNA (1 μg) were inserted into vectors and sequenced with whole‐genome sequencing technology (HiSeq2 × 150), at the Yale Center for Genomic Sciences. The data obtained for each metagenome had an average of 7.8 ± 2 Gb, which in total represented 109 Gb of sequenced DNA. The raw data passed through a quality filter and were assembled de novo using IDBA‐UD, with k‐mer sequences between 120 and 150 long (Peng et al., 2012). Metagenomes of soil samples were sequenced in Macrogen (Seoul, South Korea), using Illumina HiSeq 2500 technology.
2.4. Bioinformatic analysis of NGS total metagenomes
IntegronFinder (https://github.com/gem‐pasteur/IntegronFinder) (Cury et al., 2016) was used for the detection of integrons and their most distinctive components. The options used were –local_max, ‐‐linear, attC_evalue 1 and –func_annot. Results obtained were classified as complete integrons, including an integron‐integrase gene nearby attC site(s), In0 elements with just the integron‐integrase gene, and CALIN (Cassette Lacking Integrase), as clusters of attC sites without intI genes. Genomic contexts of the results obtained by IntegronFinder were annotated by RAST (Aziz et al., 2008). SnapGeneViewer 4.2.11 program was used to visualize the integron's genomic context.
Once the presence of an integrase gene was confirmed, the relative abundance of the contig bearing that gene was calculated. The software bowtie2 was used for mapping the filtered reads against all contigs of each metagenome. The number of reads mapping against the contigs bearing an integrase was divided by the length of the contig to obtain the RPK value. That value was normalized per million reads in the metagenome and the paired‐end nature of the reads was considered to express that value in fragments per kilobase of transcript per million reads mapped (FPKMs).
To identify conserved motifs characteristic of integron integrases, amino acid sequences selected from this study were aligned against IntI reference sequences (classes 1, 2, 3, 4, and 5) (GenBank A.N.: IntI1, AAQ16665; IntI2, AAT72891; IntI3, AAO32355; IntI4, AAC38424; IntI5, AF180939). Tyrosine recombinases XerC from Escherichia coli (P0A8P6) and XerD from E. coli (P0A8P8), XerC from Thiobacillus denitrificans (WP011313040), and XerD from T. denitrificans (WP018078134) were also included (Nunes‐Düby et al., 1998). For the alignment, Mafft program version 3.717 (Katoh et al., 2002) was used. Visualization and editing were done with Aliview program version 1.20 (Larsson, 2014). Representative integrases of selected organisms carrying chromosomal and super‐integrons were also included.
For the detection of total ARGs in the assembled metagenomes, ABRicate software (https://github.com/tseemann/abricate) was used along with the CARD database (https://card.mcmaster.ca/). This software runs a blast search which was filtered with 50% of coverage for the query sequence and 70% of identity for the alignment. Then, we verified if these genes could be identified as GCs using IntegronFinder.
2.5. Phylogenetic analysis of Antarctic integrases including Inti reference sequences
Phylogenetic analysis of IntI included 46 amino acid sequences of good quality (considering complete ORFs and not located at the edges of the contigs) identified by IntegronFinder and 15 nonredundant sequences recovered from intI amplicon libraries, obtained in this and a previous study (Antelo et al., 2015). The analysis also included 42 sequences of integrases from known organisms and XerC and XerD sequences of E. coli and T. denitrificans, which were used as external branches for the construction of the tree. Reference sequences were obtained from INTEGRALL—the Integron Database (Moura et al., 2009) (http://integrall.bio.ua.pt) and NCBI GenBank. All sequences were clustered to 90% of sequence identity using CD‐HIT (Fu et al., 2012) to remove redundancy. Analysis was done using NGPhylogeny.fr in the Advanced Workflow option, including PhyMLfor tree inference (Maximum‐likelihood method) (https://ngphylogeny.fr/workflows/advanced/). The tree was designed using Interactive Tree of Life (iTOL) v4 (Letunic & Bork, 2019).
3. RESULTS
3.1. Amplicon library analysis
King George Island (KGI) (25 de Mayo Island) in Maritime Antarctica is the largest island in the South Shetland archipelago. Amplicon libraries of intI genes were obtained from samples collected on this island (Table 1). Drake coast (PD2) and Potter Cove creek (CP2) contain a total of 22 IntI clones (8 in PD2, 14 inCP2). Some of these sequences were included for phylogenetic analysis discussed below. Similar to previous studies (Antelo et al., 2015), analysis of amplicon libraries indicated that the primers used, designed to amplify class 1 intI, are not specific for this class. This primer pair was designed using a limited database (Mazel et al., 2000) since at that time only sequences of clinical origin were available. However, we could identify 8 clones from CP2 highly similar (more than 99% identity in amino acid sequences) to the reference gene IntI1_AAQ16665 of clinical origin. The phylogenetic tree shows that this group of similar sequences also includes the gene previously identified in the Antarctic isolate IA12 (Antelo et al., 2018) and the intI1 gene from a complete integron in contig CS1_14211 (Figure 1). Finally, and according to the original design of this primer pair (Mazel et al., 2000), class 2 and class 3 intI genes were not amplified with this strategy.
FIGURE 1.
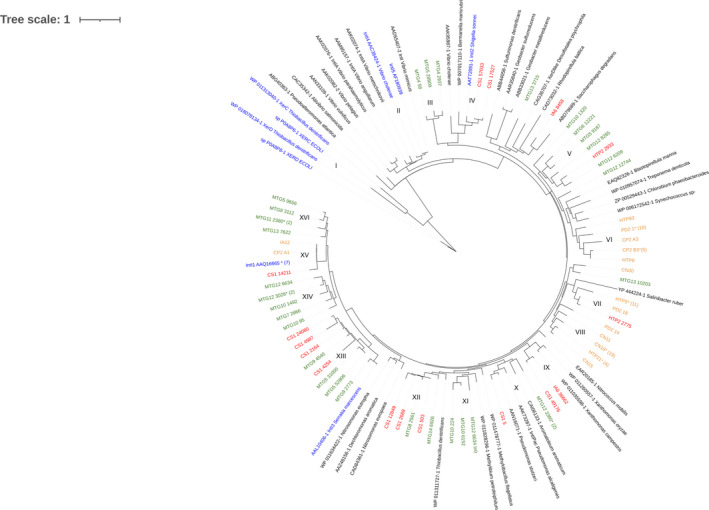
Phylogenetic tree of integrase protein sequences described in this work along with reference integrases. The different clusters are identified with Roman numbers. Classical reference sequences are written in blue (IntI1, IntI2, IntI3, IntI4, IntI5, XerC, and XercD sequences). Sequences recovered by NGS microbial mat metagenome analysis are written in green and by NGS soil metagenome analysis are written in red. Amplicon library sequences are written in orange. Sequences marked with * represent a cluster of sequences with more than 90% identity, the number of sequences in the cluster is shown between brackets
3.2. NGS metagenomic analysis
IntegronFinder was designed for the identification of integron elements, although the original analysis was restricted to complete bacterial genomes, avoiding problems associated with assemblies of metagenomes or draft genomes (Cury et al., 2016). In this case, we used IntegronFinder for metagenome analysis (Table 2), with the limitations related to the length of reads and contig assembly. The metagenomes allowed us to identify a large diversity of integrase genes and associated GCs (Table 3). Most intI genes exhibited similarities with environmental integrases described from different geographical regions (Figure 1) (Rodríguez‐Minguela et al., 2009). The relative quantification of integrase contigs consistently showed that those metagenomes with the highest richness of intI genes, related to the total number of predicted ORFs, were also those with the highest relative abundance of these genes assessed in fragments per Kilobase mapped (FPKM) (Table 3).
TABLE 3.
Identification of integron elements in Antarctic metagenomes using IntegronFinder
| Name of the sample | No of contigsa | Complete integrons | In0 | CALIN | FPKMb | |||
|---|---|---|---|---|---|---|---|---|
| Drake(1) | 14345 | 0 | 0 | 42 | ‐ | |||
| Espejo(2) | 11311 | 0 | 1 | 149 | ndc | |||
| HTP(3) | 10227 | 0 | 0 | 53 | ‐ | |||
| Pista(4) | 19554 | 0 | 2 | 43 | 0,0018257 | |||
| Carlini I(5) | 62551 | 3 | 4 | 119 |
0,002930121 |
|||
| Carlini II(6) | 47446 | 1 | 0 | 143 |
0,000534606 |
|||
| Esperanza(7) | 35849 | 0 | 1 | 104 | 0,002127371 | |||
| Primavera(8) | 53174 | 2 | 1 | 114 | 0,00293502 | |||
| San Martín(9) | 17447 | 1 | 1 | 44 | 0,004543001 | |||
| B012‐2015–14(10) | 18089 | 3 | 2 | 205 | 0,019193791 | |||
| B012‐2015–15(11) | 24919 | 1 | 0 | 94 | 0,000939058 | |||
| B012‐2015–16(12) | 23874 | 6 | 1 | 284 | 0,017899739 | |||
| B012‐2015–16 MidGood(13) | 23997 | 4 | 1 | 145 | 0,005845539 | |||
| B012‐2015–18(14) | 34862 | 1 | 0 | 70 | 0,001345873 | |||
| CS1(15) | 68030 | 10 | 3 | 283 | 0,007584958 | |||
| HTP2(16) | 28119 | 1 | 2 | 85 | 0,000933934 | |||
| IA6(17) | 54969 | 2 | 0 | 103 | 0,000127014 | |||
Number of contigs obtained from each metagenome.
Relative abundance of the contig bearing that intI gene.
Not determined
We could identify “Mobile Integron‐related” (MI‐related) intI genes phylogenetically related to class 2 (contigs CS1_17527 and CS1_57033, with more than 70% of amino acid sequence identity compared to AAT72891.1_IntI2), class 3 (contigs MTG5_31000, MTG5_52806, MTG9_2773, MTG9_4540, CS1_4254, CS1_2164, CS1_24080 and CS1_4987 with more than 70% of sequence identity compared to AAL10406.1_IntI3), and class 1 reference genes (CS1_14211, 88% identity to AAQ16665_IntI1). Using these strategies, amplicon library, and NGS analysis, we found that “MI‐related” intI genes represent about 21% of the total (13 from a total of 61). The analysis of the phylogenetic tree obtained with these recovered environmental sequences confirms that criteria for the classification of integrons should consider the high diversity of integrase sequences found in the environment.
3.3. Identification of inti genes and associated GCs
Table 3 summarizes results obtained with Integron Finder for analysis of these metagenomes. A total of 54 intI genes were found, 35 were classified as complete integrons including one intI gene and at least one attC site. The remaining 19 intI genes fell into the In0 group (empty integron) and the rest of the elements were classified as CALIN, a region containing a Cluster of attC sites Lacking INtegrase nearby. CALIN could be associated or not with a potential GC (2080 CALIN identified) (Table 3). IntegronFinder also annotates potential coding sequences (CDS) associated with these element classes. Then, a total of 3431 CDS were annotated within the region of 200 bp next to each integron element found in our dataset (intI and attC sites), including the 54 intI genes.
Alignment of IntI deduced amino acid sequences identified in this study with selected representative integrase sequences, allowed for recognition of conserved motifs characteristic of the tyrosine recombinase family. These motifs include four highly conserved residues (RHRY), Box I, Box II, Patches I, II, and III regions (Nunes‐Düby et al., 1998). The additional domain of around 35 aa, specific to integron integrases, was also identified (Messier & Roy, 2001; Nunes‐Düby et al., 1998). Essential amino acid residues for DNA binding and recombination processes are present in most sequences, suggesting that they represent functional enzymes (Messier & Roy, 2001; Macdonald et al., 2006) (Figure 2a).
FIGURE 2.

(a) Amino acid sequence alignment (SeaView) of IntIs recovered by amplicon library and NGS analysis. The first seven sequences were included as a reference. (b) Amino acid sequence alignment including the region with an additional domain, identified in some environmental IntIs
On the other hand, we found that three IntI sequences in NGS metagenomes from soil (CS1_12849, CS1_2689, CS1_503) and four from microbial mats (MTG5_28909, MTG8_2561, MTG10_95, and MTG14_6926) exhibited an additional extended domain of variable length, which was located after Patch III. This type of insertion was identified previously (Rodríguez‐Minguela et al., 2009) in environmental integrons recovered from diverse geographical regions. However, the implication of this type of insertion in integrase activity is unknown (Rodríguez‐Minguela et al., 2009) (Figure 2b).
3.4. Annotation of antibiotic resistance genes
Using the CARD database, ARGs were detected in most of the metagenomes analyzed (53 genes in 15 metagenomes) (Figure 3), which did not show a high richness of ARGs compared to other environmental samples (Van Goethem et al., 2018). However, in sample CS1, collected next to the septic chamber of BCAA we could identify 37 different ARGs. These ARGs were not identified as GCs by IntegronFinder, except blaOXA‐205 gene, present in contig CS1_503 (Appendix Figure A1a) as part of a complete integron and a fragment of aadA6 gene associated with a complete integron in CS1_14211 (Appendix Figure A1b). This integron also included a qacG2 gene cassette, identified by Blastn. In the same metagenome, we could find several determinants of macrolides resistance like mpH, mef, OPM, and erm. In terms of geographic distribution, dfrE (dihydrofolate reductase) and vatF (streptogramin A acetyltransferase) genes seem to be the most prevalent in the metagenomes analyzed, since they were detected in 8 and 7 different samples, respectively (Figure 3). According to this, dfrE, vatF, and macrolide resistance genes were not identified as GCs, although the length of contigs could be a limitation that does not allow us to affirm it with certainty.
FIGURE 3.
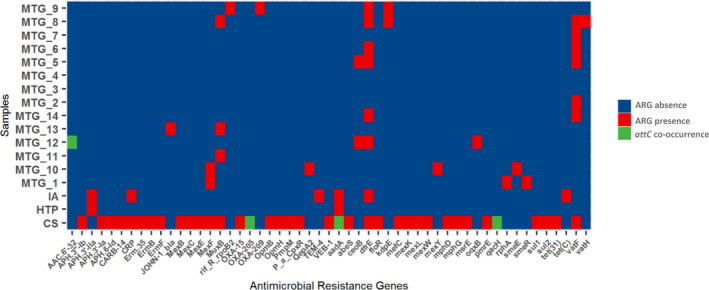
Heatmap of antimicrobial resistance genes in metagenomic assemblies and its association to attC sequences within the same contig
Finally, a second ARG located in MTG12 was identified in a region classified as CALIN by IntegronFinder (contig MTG12_18109), encoding an aminoglycoside‐modifying enzyme (100% identity to AAC(6´)‐acetyltransferase) (Figure A2).
3.5. Phylogenetic analysis of Inti sequences and description of their genomic context
This analysis included 61 IntI Antarctic sequences identified in this work and previous studies (Antelo et al., ,2015, 2018). We used 46 IntI sequences of good quality (considering complete ORFs and not located at the edges of the contigs), from those identified by NGS metagenomic analysis and 6 nonredundant IntIs recovered by amplicon library analysis obtained in this study. We also incorporated 9 Antarctic sequences obtained in previous studies (Antelo et al., 2015). In addition, 42 reference sequences and others recovered from the database were also included for analysis. IntI sequences identified in this study exhibited great diversity (less than 40% overall sequence identity at amino acid level) distributed throughout the tree in different groups. Likewise, several of our IntIs were not phylogenetically related to reference sequences and were grouped forming separate clades (Figure 1).
Previously published analyses showed that all IntI integrase sequences were separate from XerC and XerD recombinases, the closest group within the tyrosine recombinase family (Nunes‐Düby et al., 1998) (Group I, Figure 1). In addition, clades including integrases of class 1 (IntI1) (Group XV) and those with class 3 (IntI3) (Group XIII) are related to each other and distinct from the clade including class 2 (IntI2) (Group III), which form a separate group (Nemergut et al., 2008). Likewise, integrases from the Vibrio genus form a group separated from the rest of IntI integrases (Rowe‐Magnus and Mazel, 2001) (Group II). From a total of 61 IntI integron sequences analyzed in this study, only 13 sequences were related to mobile IntI (IntI1, GroupXV with 3 sequences; IntI2, Group III with 2 sequences; and IntI3, Group XIII with 8) with sequence identity higher than 70% at the amino acid level to reference IntI sequences.
3.6. Groups of sequences identified by phylogenetic analysis
XerC and XerD sequences, used as external branches, are identified as Group I, and reference sequences Int4 and Int5 are grouped with those of Vibrio metschnikovii and V. anguillarum, forming Group II. Reference IntI2 from Shigella sonnei of clinical origin belongs to Group III together with two IntI sequences of NGS metagenomes obtained from soil collected next to BCAA (CS1). Compared to IntI2, these two sequences exhibit an identity higher than 70%. Contig CS1_57033 contains one In0 and CS1_17527 has one attC site and one ORF that would encode a bacterioferritin (COG2193). The sequences related to IntI class 3 (Group XIII) form two subgroups, one includes MTG9_2773 and class 3 IntI reference sequence of Serratia marcescens. The other subgroup includes seven IntI sequences recovered from Antarctic microbial mat and soil samples (NGS metagenomes) (MTG5_52806, MTG5_31000, CS1_4254, MTG9_4540, CS1_2164, CS1_24080, CS1_4987). All these eight sequences exhibit identities greater than 70% compared to IntI3. Group XV includes 10 highly similar sequences that clustered with intI1 AAQ16665 as the reference sequence (more than 88% identity). This cluster includes eight almost identical sequences obtained by amplicon libraries from Potter creek soil (CP2) which we considered redundant and represented by CP2‐A1. This group also includes the IntI sequence identified in CS1 metagenome (CS1_14211) (88% sequence identity compared to IntI1) and IA12, identified previously in an Antarctic Enterobacteria isolate. Class 1 integrases appear to be almost identical with each other, which is consistent with that reported in the literature, which illustrates the great similarity exhibited by clinical class 1 integrases.
Groups X and XII are related. The first includes a reference sequence of Methylibium petroleiphilum (WP011828296) and three integrase sequences from Continental microbial mats from MTG10 and MTG12. The second one comprises sequences recovered from Maritime Antarctic soils (NGS metagenome) CS1_2689, CS1_12849, CS1_503, and MTG8_2561. Contig CS1_12849 has two attC sites and no associated ORFs, while CS1_503 contains three attC sites and two ORFs, one encodes, according to Resfam, a putative class D β‐lactamase (Resfam: RF0056) and the other a hypothetical protein (Figure A1a). The reference branch includes an IntI sequence of Thiobacillus denitrificans (WP_011311727).
Group X includes the IntI sequence of Pseudomonas stutzeri (AAN16071) and CS1_5 of soil (Maritime Antarctica), related to IntI from one unclassified bacterium of the Order Pseudomonadales (OHC29044.1) (Anantharaman et al., 2016). This group contains the longest integron identified in this study. CS1_5 has 15 attC sites and six ORFs (Appendix Figure A1c). The Integrons of the genus Pseudomonas are present only in a small number of species and may have been acquired through horizontal gene transfer at a late period in their evolutionary history. The number and composition of GCs vary between species; they can range from ten GCs (P. stutzeri) to more than 32 (P. alcaligenes) (Boucher et al., 2007). All ORFs identified in CS1_5 encode hypothetical proteins, except one ORF (385 bp) (not a GC) that would encode a penicillin acylase II (EC. 3.5.1.11). The number of attC sites (n = 15) present in the CS1_5 integron and its phylogenetic relationship with species of the genus Pseudomonas sp. that carry integrons in their chromosome suggest that it is a chromosomal integron.
4. DISCUSSION
In this study, we used IntegronFinder to identify genetic elements of integrons by NGS data analysis of Antarctic soil and microbial mat metagenomes collected from different sites on the continent. We also used samples of soil collected from other sites of Antarctica for the construction of amplicon libraries of intI integron‐integrase genes, using a pair of primers previously designed by Mazel et al. (2000). All recovered IntI deduced amino acid sequences were filtered by domain conservation analysis. Several novel results were obtained. First, although intI gene search tools were used in samples of different origins, the sequences obtained from each approach were different, according to the phylogenetic analysis (Figure 1). Only two groups included sequences obtained using both strategies. Group VII identified in the phylogenetic tree (Figure 1), contains two sequences, one obtained by NGS at Halfthree Point (HTP2_2775) and the other by amplicon library from a coastal area in the Drake sea (PD2_18), both from the Fildes peninsula (KGI). Besides, Group XV includes intI sequences obtained by amplicon libraries in this study (CP2) and one identified by NGS analysis of the CS1 metagenome. However, other groups identified in the phylogenetic tree contain sequences obtained from samples of diverse origins but using the same data collection strategy. For example, Group XVI contains sequences obtained from samples collected in KGI (MTG_5) and others in the Antarctic peninsula (MTG_8) and Mac Murdo Dry Valleys (MTG_11 and MTG_13), but all obtained by NGS data analysis.
As mentioned previously, we could identify “MI‐related” class 1 intI genes by NGS data analysis from one sample collected from one site exposed to human influence (CS1 metagenome) and by amplicon library with intI1 primers (Mazel et al., 2000), but identifying also, in this case, other diverse integrase genes. Previous studies have proposed that the relative abundance of “MI‐related” intI1 genes would be used as a proxy for anthropogenic pollution (Gillings et al., 2015; Zheng et al., 2020). These genetic platforms are usually linked to genes conferring resistance to antibiotics, disinfectants, and heavy metals, and they seem to be fixed by human selection. Mazel et al. (2000) designed this primer pair aimed at amplifying intI1 genes with relative specificity, avoiding the amplification of class 2 and 3 intI genes. Databases available at that time were relatively small and reduced to clinical isolates. Then, by including environmental samples as templates for amplification, it was later established that these primers had a lower specificity, being able to amplify integrase genes with a spectrum of diversity greater than expected (Antelo et al., 2015). However, we estimate that these primers have some specificity to “MI‐related” class 1 integrases, since “MI‐related” class 2 and 3 intI genes were not recovered by PCR, but two ORFs similar to class 2 intI genes were identified by NGS in CS1 metagenome and 8 ORFs similar to class 3 intI genes in CS1, MTG5 (next to Carlini base) and MTG9 (next to San Martín base) metagenomes.
The profile of integrase genes identified in these Antarctic communities is different from that found in samples of sewage water and beaches in a previous study (Fresia et al., 2019). In that work, the presence of integrase genes was also analyzed using IntegronFinder as a search tool. They found a high prevalence of “MI‐related” integrase genes in sewage (74%) and lower in beach (24%) samples. In that study, 90% of class 1, 2, and 3 “MI‐related” integrase sequences were contained in sewage samples and 61% of these integrons were associated with ARGs and characteristic attC sites. These resistance GCs, analyzed with Abricate using CARD and VFDB (Chen et al., 2005) as databases, were only identified in integrons from sewage samples.
In our case, using arbitrary thresholds, we could identify “MI‐related” integrase genes in 25% of samples (5 in a total of 20 samples), including soil and microbial mats. From this group, 32% of these genes were identified in the presumptive “contaminated” sample obtained from soil next to the septic chamber in BCAA (CS1). In this way, finding coherent identity thresholds for integrase sequence classification could be a good way to take into account the sequence diversity derived from metagenomic analysis. This could also be useful to compare with other works and gain insight into its distribution and occurrence in different environments (Roy, et al. 2021, Zhang et al., 2018). Also, ARGs were identified with ABRIcate using the CARD database. Although we used low identity and coverage thresholds, there could be a bias in the method used as we looked for ARGs using CARD database with mainly “clinical” resistance genes, which are no expected to be widely present in pristine Antarctic environments. In addition, Antarctic environmental communities could be a reservoir of non‐characterized antimicrobial resistance genes (Azziz et al., 2019). Only three GCs could be identified as ARGs using this tool, one in CS1 metagenome, encoding class D β‐lactamase (blaOXA‐205) in a complete integron associated with an IntI sequence similar to that of T. denitrificans. The second one was a fragment of a gene found also in the CS1 metagenome (76 amino acids), encoding an aminoglycoside adenyltransferase (aadA6). The other GC was located in a CALIN of MTG12 and encoded an aminoglycoside‐modifying enzyme (AAC(6´)‐acetyltransferase) (100% identity to AAC(6’)‐32). We also used the Resfams database with IntegronFinder to functionally characterize CDSs next to attC sites (in CALIN and complete integrons). With this strategy, IntegronFinder identified a total of 26 GCs as antibiotic resistance genes including blaOXA‐205, located in the complete integron of contig CS1_503. This gene was also identified using the ABRIcate‐CARD strategy. The rest of the resistance genes considered as potential GCs by IntegronFinder belong to CALIN elements, and they were not identified using the ABRIcate‐CARD strategy.
CS1 metagenome, sequenced from a sample of soil collected next to BCAA, showed the highest richness of ARGs (in relation to the total number of ORFs), which has also been reported in other studies (Rabbia et al., 2016; Jara et al., 2020). In addition, CS1 was the only one in which we were able to detect human fecal contamination using the reference genome of Crassphage phage as a biomarker (Dutilh et al., 2014). As previously shown, the high concentration of ARGs in the environment could be explained by human fecal contamination (Karkman et al., 2019). As expected, Antarctic scientific bases impact the surrounding environment. Searching for ARGs in this metagenome, we could find several macrolide resistance genes that involve different resistance mechanisms (Msr, pmr, Emr, mph, mef). Recent studies have shown that not only macrolide resistance genes but also low concentrations of macrolide antibiotics could be detected in water samples nearby Chilean, Russian, and Chinese scientific bases in King George Island (Hernández et al., 2019). These concentrations of antibiotics in the environment are not sufficient to promote selection for resistance genes. However, there is evidence from different studies that human presence in Antarctica is could lead to a release of macrolide resistance genes to the environment (Rabbia et al., 2016; Jara et al., 2020).
In relation to resistance genes, vat encodes a streptogramin A acetyltransferase which results in resistance to drugs of the group of streptogramins. These genes have been extensively identified, particularly in strains of Enterococcus spp. resistant to macrolides (Werner et al., 2002). However, there are no reports of the presence of the vatF gene in the Antarctic environment. Yuan et al. (2019) found macrolide resistance genes in soil samples, but all of them encoded efflux pumps rather than acetyltransferases. Similarly,Zaikova et al. (2019) found macrolides efflux pumps in 5 assembled genomes recovered from fossil microbial mats, but not vat genes. Also, Fitzpatrick & Walsh (2016) performed a meta‐analysis of metagenomes recovered from different environments, identifying 11 non‐efflux‐pump‐mediated genes that conferred resistance to macrolides in 23 metagenomes, but none in the metagenome of an Antarctic aquatic sample, included in the search. Therefore, the presence of vatF genes in 7 of our metagenomes is intriguing. Additional experiments should be performed to evaluate whether these genes are functional.
The family of dfr genes encodes dihydrofolate reductases that confer resistance to trimethoprim. In our study, we identified dfrE genes in 8 of the 17 Antarctic metagenomes analyzed. This variant of the dfr gene was previously identified in clinical isolates of Enterococcus faecalis (Coque et al., 1999). This gene is considered to be intrinsic in E. faecalis and confers resistance to trimethoprim in cells of E. coli carrying several copies of the gene. Trimethoprim resistance has been already found in Antarctic bacterial isolates (Rabbia et al., 2016), including variants of dfrA14 (Antelo et al., 2018) (associated with class 1 integron) and dfrA6 (Jara et al., 2020) (not determined the association to integrons). Finally, the presence of trimethoprim has been also confirmed in Antarctic sea samples (Hernández et al., 2019), a fact that could explain the occurrence of this gene in at least some of our metagenomes.
We could find several ARGs that are commonly found in the environment such as variants of aminoglycoside‐modifying enzymes AAC and APH. We could also detect other resistance genes that have been linked to clinical pathogens like blaVEB‐1. Another interesting gene is blaOXA‐209 which was first described in plasmids from isolates of Riemerella anatipestifer in diseased birds (Chen et al., 2012). This could have a link to the Antarctic environment as in this place most of the wildlife is composed of different species of birds. Birds have already been linked to the spread of ARGs in previous studies (Rabbia et al., 2016). We have also found several genes that act together like the Mex operon. Most of them were sequenced from the same sample and could be part of the same genome, although they were found in different contigs.
The sampled sites showed different antimicrobial resistance profiles. If we observe only the metagenomes sequenced from the Fildes peninsula, we could distinguish sites with a high richness of ARGs and others where we could not detect them at all. This suggests that the presence of “clinically‐derived” ARGs is patchy at the landscape and that there are certain factors that influence their presence. Sewage waters derived from human activity have already been described as one of these factors. In this work, the sewage impacted site could also be associated with the presence of integrons bearing antibiotic resistance GCs. In the rest of the samples, we could detect a wide diversity of integrases but not directly associated with known antimicrobial resistance functions. Therefore, the lack of association between ARGs and integrons in the samples analyzed supports the idea that evolutionary mechanisms that contribute to select resistance genes in a population are related to environmental pressures.
ETHICS STATEMENT
None required.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
Verónica Antelo: Conceptualization (lead); Data curation (lead); Formal analysis (lead); Investigation (lead); Methodology (lead); Resources (equal); Software (equal); Supervision (lead); Validation (lead); Visualization (equal); Writing‐original draft (equal); Writing‐review & editing (lead). Matías Giménez: Conceptualization (equal); Data curation (lead); Formal analysis (lead); Investigation (equal); Methodology (lead); Resources (equal); Software (lead); Supervision (equal); Validation (lead); Visualization (lead); Writing‐original draft (equal); Writing‐review & editing (lead). Gastón Azziz: Conceptualization (equal); Data curation (equal); Formal analysis (equal); Investigation (supporting); Methodology (equal); Software (equal); Supervision (supporting); Validation (equal); Visualization (supporting); Writing‐original draft (equal); Writing‐review & editing (equal). Patricia Valdespino: Data curation (supporting); Funding acquisition (equal); Methodology (equal); Project administration (equal); Resources (equal); Supervision (equal); Writing‐review & editing (equal). Luisa Falcón: Funding acquisition (lead); Investigation (equal); Project administration (lead); Resources (lead); Supervision (equal); Writing‐review & editing (equal). Lucas Ruberto: Resources (supporting); Walter MacCormack: Funding acquisition (supporting); Resources (equal). Didier Mazel: Formal analysis (equal); Writing‐review & editing (equal). Silvia Beatriz Batista: Conceptualization (lead); Formal analysis (lead); Funding acquisition (lead); Investigation (lead); Methodology (lead); Project administration (lead); Resources (lead); Software (supporting); Supervision (lead); Validation (lead); Visualization (equal); Writing‐original draft (lead); Writing‐review & editing (lead).
ACKNOWLEDGMENTS
We want to thank the Instituto Antártico Uruguayo and staff of the Base Científica Antártica Artigas for logistic support. Funding for this research was awarded through the PNUD‐URU program (project No. 13/001) and the AMEXCID International Cooperation program with Argentina. This work was also supported by PEDECIBA (Programa de Desarrollo de Ciencias Básicas) and ANII (Agencia Nacional de Investigación e Innovación). Technical support is acknowledged to O. Gaona (UNAM).
1.
FIGURE A1.
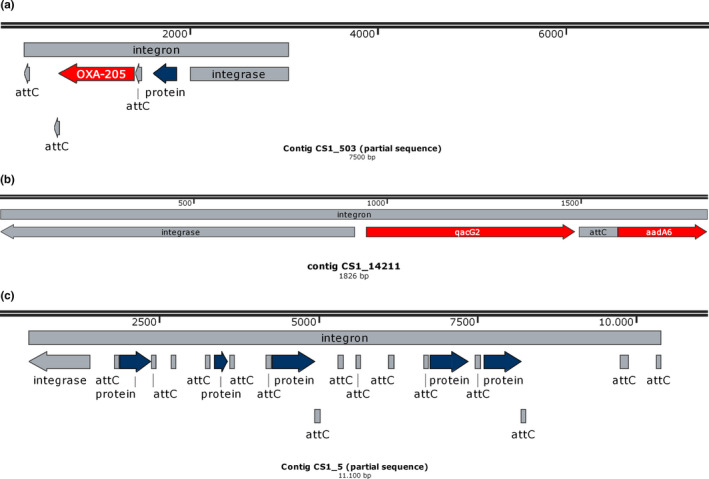
(a) Sequence map of integron detected in contig CS1_503. Only part of the contig is displayed. (b) Sequence map of integron detected in contig CS1_14211. The entire contig is displayed. (c) Sequence map of integron detected in contig CS1_5. No ARGs were detected in this integron. Only part of the contig is displayed
FIGURE A2.

Sequence map of CALIN detected in contig 18109 from metagenome 12. The entire sequence is displayed
Antelo, V., Giménez, M., Azziz, G., Valdespino‐Castillo, P., Falcón, L. I., Ruberto, L. A. M., Mac Cormack, W. P., Mazel, D., & Batista, S. (2021). Metagenomic strategies identify diverse integron‐integrase and antibiotic resistance genes in the Antarctic environment. MicrobiologyOpen, 10, e1219. 10.1002/mbo3.1219
Data Availability Statement
Contigs analyzed from Antarctic samples are deposited in NCBI BioProject PRJNA734336: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA734336 with accession numbers from MZ590516 to MZ590563.
REFERENCES
- Anantharaman, K., Brown, C. T., Hug, L. A., Sharon, I., Castelle, C. J., Probst, A. J., Thomas, B. C., Singh, A., Wilkins, M. J., Karaoz, U., Brodie, E. L., Williams, K. H., Hubbard, S. S., & Banfield, J. F. (2016). Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nature Communications, 7(1), 10.1038/ncomms13219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antelo, V., Guerout, A. M., Mazel, D., Romero, V., Sotelo‐Silveira, J., & Batista, S. (2018). Bacteria from Fildes Peninsula carry class 1 integrons and antibiotic resistance genes in conjugative plasmids. Antarctic Science, 30(1), 22–28. 10.1017/S0954102017000414 [DOI] [Google Scholar]
- Antelo, V., Romero, H., & Batista, S. (2015). Detection of integron integrase genes on King George Island, Antarctica. Advance Polar Science, 26, 30–37. [Google Scholar]
- Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., Formsma, K., Gerdes, S., Glass, E. M., Kubal, M., Meyer, F., Olsen, G. J., Olson, R., Osterman, A. L., Overbeek, R. A., McNeil, L. K., Paarmann, D., Paczian, T., Parrello, B., … Zagnitko, O. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics, 9, 75. 10.1186/1471-2164-9-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azziz, G., Giménez, M., Romero, H., Valdespino‐Castillo, P. M., Falcón, L. I., Ruberto, L. A. M., Mac Cormack, W. P., & Batista, S. (2019). Detection of presumed genes encoding beta‐lactamases by sequence based screening of metagenomes derived from Antarctic microbial mats. Frontiers of Environmental Science & Engineering, 13, 1–12. 10.1007/s11783-019-1128-1 [DOI] [Google Scholar]
- Boucher, Y., Labbate, M., Koenig, J. E., & Stokes, H. W. (2007). Integrons: Mobilizable platforms that promote genetic diversity in bacteria. Trends in Microbiology, 15, 301–309. 10.1016/j.tim.2007.05.004 [DOI] [PubMed] [Google Scholar]
- Callejas, C., Azziz, G., Souza, E. M., Gill, P. R., & Batista, S. (2018). Prokaryotic diversity in four microbial mats on the Fildes Peninsula, King George Island, maritime Antarctica. Polar Biology, 41(5), 935–943. 10.1007/s00300-018-2256-y [DOI] [Google Scholar]
- Cambray, G., Guerout, A.‐M., & Mazel, D. (2010). Integrons. Annual Review of Genetics, 44, 141–166. 10.1146/annurev-genet-102209-163504 [DOI] [PubMed] [Google Scholar]
- Cambray, G., Sanchez‐Alberola, N., Campoy, S., Guerin, É., Da Re, S., González‐Zorn, B., Ploy, M.‐C., Barbé, J., Mazel, D., & Erill, I. (2011). Prevalence of SOS‐mediated control of integron integrase expression as an adaptive trait of chromosomal and mobile integrons. Mobile DNA, 2, 6. 10.1186/1759-8753-2-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L., Yang, J., Yu, J., Yao, Z., Sun, L., Shen, Y., & Jin, Q. (2005). VFDB: A reference database for bacterial virulence factors. Nucleic Acids Research, 33(Database issue), D325–D328. 10.1093/nar/gki008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y.‐P., Lee, S.‐H., Chou, C.‐H., & Tsai, H.‐J. (2012). Detection of florfenicol resistance genes in Riemerella anatipestifer isolated from ducks and geese. Veterinary Microbiology, 154, 325–331. 10.1016/j.vetmic.2011.07.012 [DOI] [PubMed] [Google Scholar]
- Collis, C. M., Grammaticopoulos, G., Briton, J., Stokes, H. W., & Hall, R. M. (1993). Site‐specific insertion of gene cassettes into integrons. Molecular Microbiology, 9, 41–52. 10.1111/j.1365-2958.1993.tb01667.x [DOI] [PubMed] [Google Scholar]
- Collis, C. M., & Hall, R. M. (1995). Expression of antibiotic resistance genes in the integrated cassettes of integrons. Antimicrobial Agents and Chemotherapy, 39, 155–162. 10.1128/AAC.39.1.155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Convey, P. (2010). Terrestrial biodiversity in Antarctica ‐ recent advances and future challenges. Polar Science, 4, 135–147. 10.1016/j.polar.2010.03.003 [DOI] [Google Scholar]
- Coque, T. M., Singh, K. V., Weinstock, G. M., & Murray, B. E. (1999). Characterization of dihydrofolate reductase genes from trimethoprim‐ susceptible and trimethoprim‐resistant strains of Enterococcus faecalis. Antimicrobial Agents and Chemotherapy, 43, 141–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cury, J., Jové, T., Touchon, M., Néron, B., & Rocha, E. P. (2016). Identification and analysis of integrons and cassette arrays in bacterial genomes. Nucleic Acids Research, 44, 4539–4550. 10.1093/nar/gkw319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutilh, B. E., Cassman, N., McNair, K., Sanchez, S. E., Silva, G. G. Z., Boling, L., Barr, J. J., Speth, D. R., Seguritan, V., Aziz, R. K., Felts, B., Dinsdale, E. A., Mokili, J. L., & Edwards, R. A. (2014). ARTICLE A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nature Communications. 10.1038/ncomms5498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick, D., & Walsh, F. (2016). Antibiotic resistance genes across a wide variety of metagenomes. FEMS Microbiology Ecology, 92, 1–8. 10.1093/femsec/fiv168 [DOI] [PubMed] [Google Scholar]
- Fresia, P., Antelo, V., Salazar, C., Giménez, M., D’Alessandro, B., Afshinnekoo, E., Mason, C., Gonnet, G. H., & Iraola, G. (2019). Urban metagenomics uncover antibiotic resistance reservoirs in coastal beach and sewage waters. Microbiome, 7, 1–9. 10.1186/s40168-019-0648-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, L., Niu, B., Zhu, Z., Wu, S., & Li, W. (2012). CD‐HIT: Accelerated for clustering the next‐generation sequencing data. Bioinformatics, 28, 3150–3152. 10.1186/s40168-019-0648-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaly, T. M., Geoghegan, J. L., Alroy, J., & Gillings, M. R. (2019). High diversity and rapid spatial turnover of integron gene cassettes in soil. Environmental Microbiology, 21, 1567–1574. 10.1111/1462-2920.14551 [DOI] [PubMed] [Google Scholar]
- Gillings, M. R. (2014). Integrons: Past, present, and future. Microbiology and Molecular Biology Reviews, 78, 257–277. 10.1128/MMBR.00056-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillings, M. R., Gaze, W. H., Pruden, A., Smalla, K., Tiedje, J. M., & Zhu, Y.‐G. (2015). Using the class 1 integron‐integrase gene as a proxy for anthropogenic pollution. ISME, J9, 1269–1279. 10.1038/ismej.2014.226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerin, E., Cambray, G., Sanchez‐Alberola, N., Campoy, S., Erill, I., Da Re, S., Gonzalez‐Zorn, B., Barbe, J., Ploy, M.‐C., & Mazel, D. (2009). The SOS response controls integron recombination. Science, 324, 1034. 10.1126/science.1172914 [DOI] [PubMed] [Google Scholar]
- Hernández, F., Calısto‐Ulloa, N., Gómez‐Fuentes, C., Gómez, M., Ferrer, J., González‐Rocha, G., Bello‐Toledo, H., Botero‐Coy, A. M., Boıx, C., Ibáñez, M., & Montory, M. (2019). Occurrence of antibiotics and bacterial resistance in wastewater and sea water from the Antarctic. Journal of Hazardous Materials, 363, 447–456. 10.1016/j.jhazmat.2018.07.027 [DOI] [PubMed] [Google Scholar]
- Holmes, A. J., Gillings, M. R., Nield, B. S., Mabbutt, B. C., Nevalainen, K. M. H., & Stokes, H. W. (2003). The gene cassette metagenome is a basic resource for bacterial genome evolution. Environmental Microbiology, 5, 383–394. 10.1046/j.1462-2920.2003.00429.x [DOI] [PubMed] [Google Scholar]
- Jara, D., Bello‐Toledo, H., Domínguez, M., Cigarroa, C., Fernández, P., Vergara, L., Quezada‐Aguiluz, M., Opazo‐Capurro, A., Lima, C. A., & González‐Rocha, G. (2020). Antibiotic resistance in bacterial isolates from freshwater samples in Fildes Peninsula, King George Island, Antarctica. Scientific Reports, 10, 3145. 10.1038/s41598-020-60035-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jové, T., Da Re, S., Denis, F., Mazel, D., & Ploy, M.‐C. (2010). Inverse correlation between promoter strength and excision activity in class 1 integrons. PLoS Genetics, 6, e1000793. 10.1371/journal.pgen.1000793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkman, A., Pärnänen, K., & Larsson, D. G. J. (2019). Fecal pollution can explain antibiotic resistance gene abundances in anthropogenically impacted environments. Nature Communications, 10, 80. 10.1038/s41467-018-07992-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K., Misawa, K., Kuma, K., & Miyata, T. (2002). MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Research, 30, 3059–3066. 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson, A. (2014). AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics, 30, 3276–3278. 10.1093/bioinformatics/btu531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic, I., & Bork, P. (2019). Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Research, 47, W256–W259. 10.1093/nar/gkz239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald, D., Demarre, G., Bouvier, M., and Mazel, D. (2006). Structural basis for broad DNA specificity in integron recombination. 440. [DOI] [PubMed]
- Mazel, D. (2006). Integrons: agents of bacterial evolution. Nature Reviews Microbiology, 4, 608–620. 10.1038/nrmicro1462 [DOI] [PubMed] [Google Scholar]
- Mazel, D., Dychinco, B., Webb, V., & Davies, J. (2000). Antibiotic resistance in the ECOR collection:integrons and identification of a novel aad gene. Antimicrobial Agents and Chemotherapy, 44, 1568–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messier, N., & Roy, P. H. (2001). Integron integrases possess a unique additional domain necessary for activity. Journal of Bacteriology, 183, 6699–6706. 10.1128/JB.183.22.6699-6706.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moura, A., Soares, M., Pereira, C., Leitão, N., Henriques, I., & Correia, A. (2009). INTEGRALL: A database and search engine for integrons, integrases and gene cassettes. Bioinformatics, 25, 1096–1098. 10.1093/bioinformatics/btp105 [DOI] [PubMed] [Google Scholar]
- Nemergut, D. R., Robeson, M. S., Kysela, R. F., Martin, A. P., Schmidt, S. K., & Knight, R. (2008). Insights and inferences about integron evolution from genomic data. BMC Genomics, 9, 261. 10.1186/1471-2164-9-261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nivina, A., Grieb, M. S., Loot, C., Bikard, D., Cury, J., Shehata, L., Bernardes, J., & Mazel, D. (2020). Structure‐specific DNA recombination sites: Design, validation, and machine learning‐based refinement. Science Advances, 6(30). 10.1126/sciadv.aay2922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes‐Düby, S. E., Kwon, H. J., Tirumalai, R. S., Ellenberger, T., & Landy, A. (1998). Similarities and differences among 105 members of the Int family of site‐specific recombinases. Nucleic Acids Research, 26, 391–406. 10.1093/nar/26.2.391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, Y., Leung, H. C. M., Yiu, S. M., & Chin, F. Y. L. (2012). IDBA‐UD : a De Novo Assembler for single‐cell and Metagenomic Sequencing Data with Highly Uneven Depth., 28, 1420–1428. [DOI] [PubMed] [Google Scholar]
- Rabbia, V., Bello‐Toledo, H., Jiménez, S., Quezada, M., Domínguez, M., Vergara, L., Gómez‐Fuentes, C., Calisto‐Ulloa, N., González‐Acuña, D., López, J., & González‐Rocha, G. (2016b). Antibiotic resistance in Escherichia coli strains isolated from Antarctic bird feces, water from inside a wastewater treatment plant, and seawater samples collected in the Antarctic Treaty area. Polar Science, 10, 123–131. 10.1016/j.polar.2016.04.002 [DOI] [Google Scholar]
- Rodríguez‐Minguela, C. M., Apajalahti, J. H. A., Chai, B., Cole, J. R., & Tiedje, J. M. (2009). Worldwide prevalence of class 2 integrases outside the clinical setting is associated with human impact. Applied and Environment Microbiology, 75, 5100–5110. 10.1128/AEM.00133-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe‐Magnus, D. A., & Mazel, A. (2001). Integrons: Natural tools for bacterial genome evolution. Current Opinion in Microbiology, 4, 565–569. 10.1016/S1369-5274(00)00252-6 [DOI] [PubMed] [Google Scholar]
- Roy, P. H., Partridge, S. R., & Hall, R. M. (2021). Comment on ȘConserved phylogenetic distribution and limited antibiotic resistance of class 1 integrons revealed by assessing the bacterial genome and plasmid collectionȘ by A.N. Zhang et al. Microbiome, 9. 10.1186/s40168-020-00950-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes, H. W., & Hall, R. M. (1989). A novel family of potentially mobile DNA elements encoding site‐specific gene‐integration functions: Integrons. Molecular Microbiology, 3, 1669–1683. 10.1111/j.1365-2958.1989.tb00153.x [DOI] [PubMed] [Google Scholar]
- Stokes, H. W., Holmes, A. J., Nield, B. S., Holley, M. P., Nevalainen, K. M. H., Mabbutt, B. C., & Gillings, M. R. (2001). Gene cassette PCR: sequence‐independent recovery of entire genes from environmental DNA. Applied and Environment Microbiology, 67, 5240–5246. 10.1128/AEM.67.11.5240-5246.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdespino‐Castillo, P. M., Cerqueda‐García, D., Espinosa, A. C., Batista, S., Merino‐Ibarra, M., Taş, N., Alcántara‐Hernández, R. J., & Falcón, L. I. (2018). Microbial distribution and turnover in Antarctic microbial mats highlight the relevance of heterotrophic bacteria in low‐nutrient environments. FEMS Microbiology Ecology, 94(9), 10.1093/femsec/fiy129 [DOI] [PubMed] [Google Scholar]
- Van Goethem, M. W., Pierneef, R., Bezuidt, O. K. I., Van De Peer, Y., Cowan, D. A., & Makhalanyane, T. P. (2018). A reservoir of “historical” antibiotic resistance genes in remote pristine Antarctic soils. Microbiome, 6(1). 10.1186/s40168-018-0424-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner, G., Klare, I., & Witte, W. (2002). Molecular analysis of streptogramin resistance in enterococci. International Journal of Medical Microbiology, 292, 81–94. 10.1078/1438-4221-00194 [DOI] [PubMed] [Google Scholar]
- Yuan, K. E., Yu, K. E., Yang, R., Zhang, Q., Yang, Y., Chen, E., Lin, L., Luan, T., Chen, W., & Chen, B. (2019). Metagenomic characterization of antibiotic resistance genes in Antarctic soils. Ecotoxicology and Environmental Safety, 176, 300–308. 10.1016/j.ecoenv.2019.03.099 [DOI] [PubMed] [Google Scholar]
- Zaikova, E., Goerlitz, D. S., Tighe, S. W., Wagner, N. Y., Bai, Y. U., Hall, B. L., Bevilacqua, J. G., Weng, M. M., Samuels‐Fair, M. D., & Johnson, S. S. (2019). Antarctic relic microbial mat community revealed by metagenomics and metatranscriptomics. Front Ecol Evol, 7, 1. 10.3389/fevo.2019.00001 [DOI] [Google Scholar]
- Zhang, A. N., Li, L. G., Ma, L., Gillings, M. R., Tiedje, J. M., & Zhang, T. (2018). Conserved phylogenetic distribution and limited antibiotic resistance of class 1 integrons revealed by assessing the bacterial genome and plasmid collection. Environment International, 6, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, W., Huyan, J., Tian, Z., Zhang, Y., & Wen, X. (2020). Clinical class 1 integron‐integrase gene – a promising indicator to monitor the abundance and elimination of antibiotic resistance genes in an urban wastewater treatment plant. Environment International, 135, 105372. 10.1016/j.envint.2019.105372 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Contigs analyzed from Antarctic samples are deposited in NCBI BioProject PRJNA734336: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA734336 with accession numbers from MZ590516 to MZ590563.