

Abstract
Introduction and importance
MPL (myxoid pleomorphic liposarcoma) is an uncommon type of liposarcoma that affects mostly children and infants. Its aggressive behavior and tendency to recur warrant complete excision despite the challenges of troublesome locations.
Case presentation
A 12-month-old infant presented with an insidious onset of noisy breathing and respiratory distress not relieved by supplemental oxygen via face mask. Examination revealed dullness and decreased air entry on the left chest. Computed Tomographic (CT) scan showed a large solid mass occupying the left hemithorax and displacing the mediastinum to the right. Intraoperatively, a large solid mass arising from the left chest wall and attached to the fifth rib was seen. Histopathology of the resected mass showed myxoid pleomorphic liposarcoma which is non-reactive for MDM2 immunostain.
Clinical discussion
Unlike other liposarcomas, myxoid pleomorphic liposarcoma occurs in children, commonly in the chest. CT scan is the preferred imaging modality. Treatment is by complete excision where possible. Molecular studies like Fluorescent in-situ Hybridization (FISH) and Immunohistochemistry (IHC) is used for confirmation. It has a high propensity to metastasize and recurrence is expected. Chemotherapy and irradiation following complete resection decrease the disease recurrence.
Conclusion
Soft tissue malignancy must be considered in the differential diagnosis of a large intrathoracic tumor in an infant. FISH and IHC are essential for confirmation.
Keywords: Infant, Intrathoracic mass, MDM2, Myxoid pleomorphic liposarcoma
Highlights
-
•
Myxoid pleomorphic liposarcoma (MPL) is a rare form of liposarcoma usually occurring in children.
-
•
They are identified on radiological imaging in symptomatic children.
-
•
Histopathological and IHC studies helps to reach a definitive diagnosis.
1. Introduction
Liposarcomas are a group of malignancies in adults and have complex pathogenesis. Infants and children are less prone, except for MPL. It was first described by Alaggio et al. in 2009 as a distinctive subtype of liposarcoma that mainly affects a relatively young population and has been associated with Li-Fraumeni syndrome [1], [2]. Molecular studies with FISH or IHC for MDM2 amplification helps to confirm the diagnosis. This is a high-grade, aggressive tumor with a high tendency to recur [3]. Very few cases of MPL have been reported to date. Here, we describe a case of an intrathoracic MPL in a 12-month-old child confirmed by IHC. This case has been reported in line with SCARE criteria [4].
2. Case presentation
A 12 month-old-male child was brought to our hospital with a history of noisy breathing, nonproductive and intermittent cough for 2 weeks, and shortness of breath for a day. He was tachypneic, with chest indrawing. He required oxygen via a face mask to maintain saturation. His trachea was shifted to the right, and the air entry was decreased on the left chest. He had no fever, weight loss, or poor intake, no failure to thrive, and his past medical history was unremarkable with no family history of malignancy.
He had a hemoglobin of 9.9 g/dl [normal range = 14–18 g/dl], mean cell volume (MCV) of 71.3 femtolitres (fl) [normal range = 80–100 fl], a total count of 6930/ml3 (N: 30; L:65), serum iron at 26 μg/dl and a normal erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP). Peripheral blood smear showed normocytic normochromic cells. CT scan of the chest showed a large solid mass (with some cystic components) measuring 10.5 cm × 9.7 cm × 6.6 cm, occupying the entire left hemithorax, a collapsed left lung, with the heart and major vessels, shifted towards the right (Fig. 1).
Fig. 1.
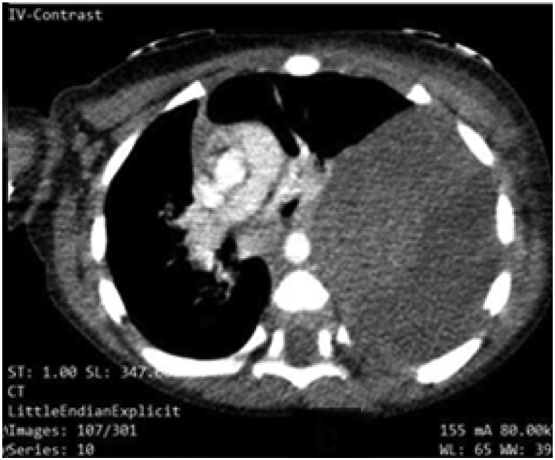
Huge, heterogeneously enhancing mass occupying the left hemithorax. Heart and the major vessels were displaced to the right.
Serum levels of uric acid, lactate dehydrogenase (LDH), alkaline phosphatase (ALP), alpha-fetoprotein (AFP), and beta-human chorionic gonadotropin (HCG) were normal. Ultrasound-guided biopsy from the mass revealed lipoblastoma as the closest differential.
The mass was excised via a left thoracotomy. The fifth rib, to which the tumor was adherent, was sub-totally excised, along with partial excision of the 4th and 6th ribs. The specimen, as received in the pathology lab, was in multiple fragments of variegated fleshy mass (Fig. 2). Histopathological examination showed tumor cells showing pleomorphic multi-vacuolated lipoblasts with scattered bizarre multinucleated hyperchromatic cells. Myxoid changes, calcifications, and foci of necrosis were also seen (Fig. 3). There were 4 mitoses in 10 high-power fields. The French Federation of Cancer Centers Sarcoma Group (FNCLCC) histological grading is Grade 2. The tumor involved the rib margin and diaphragmatic pleura; however, the bone was free. In IHC, the atypical cells were strongly immunoreactive for S100, CD34, P16, but MDM2, CDK-4, SMA, Desmin, and CD31 were non-reactive. Ki-67 proliferation index was 8–9%.
Fig. 2.

Resected specimen showed multiple pieces of variegated fleshy mass.
Fig. 3.

a) Anaplastic-looking tumor cells with bizarre pleomorphic nuclei. b) Bluish tinge of the stroma shows myxoid change with scattered lipoblasts. c) Focal area of necrosis. d) Lipoblasts showing indented, scalloped nuclei in a myxoid background.
After 6 weeks of convalescence, CT chest, whole-body bone scan and bone marrow biopsy were done, in preparation for adjuvant therapy. The CT did not show any recurrent or residual disease; the bone marrow and the bone scan, too, were normal (Fig. 4, Fig. 5). He has completed 4 cycles of chemotherapy with doxorubicin plus cyclophosphamide. Adjuvant radiation therapy is being planned. The child has tolerated treatment well and is asymptomatic after 5 months of surgery.
Fig. 4.

Preoperative chest x-ray (a) showed obliteration of the mediastinal structure which on CT scan (b) showed involvement of the whole left hemithorax.
Fig. 5.

Post-operative chest x-ray (a) and CT-scan (b) shows normal lung parenchyma.
3. Discussion
Liposarcoma is a malignancy of adipocytes. It accounts for approximately 15% of all soft tissue sarcomas and 75% of them occur in the lower extremities alone [5], [6]. Liposarcoma is rare in children, and it has no gender predilection. The common histological types of liposarcomas classified by the World Health Organization (WHO) are: well-differentiated, dedifferentiated, myxoid, and pleomorphic [7]. Myxoid Pleomorphic Liposarcoma has been recently added as a new entity in the recent fifth edition of WHO Classification of Soft Tissue and Bone Tumors [8].
MPL is an entity that shows the histological overlap between myxoid and pleomorphic liposarcoma. It is an extremely rare type of liposarcoma which occurs primarily in children and has a predilection for the axial region of the body, especially the mediastinum [2]. It is highly aggressive and has an increased risk of metastasis and recurrence [9].
MPL lacks fusion genes such as FUS-DDIT3 and EWSR1-DDIT3, usually present in a myxoid variant of liposarcoma. In addition, murine double minute 2 (MDM2) gene amplification, related to dedifferentiated liposarcoma and atypical lipomatous tumor, is also not detected in MPL [8]. Our case showed strong immunoreactivity for S100 and CD34, and MDM2 is non-reactive.
Intrathoracic MPL may present with clinical features suggesting invasion to the nearby structure, such as the superior vena cava, heart, trachea, pericardium, bronchi, and esophagus, causing symptoms like wheezing and shortness of breath, cough, tachycardia, and chest pain. However, there was no radiological or operative evidence of mediastinal invasion in our patient.
Radiological imaging shows nonspecific soft tissue mass displacing the adjacent organs with focal areas of hemorrhage and necrosis [10]. Fatty tissues are relatively decreased in the pleomorphic type of liposarcoma. MPLs have not been studied widely due to the rarity of these conditions, and no known characteristic, cytogenetic or molecular genetic abnormality associated with this disease has been found [9], [11].
MPLs are characterized by a high local recurrence, distant metastases, and a poor survival rate [2]. As in cases of other high-grade sarcomas, MPL has the propensity to metastasize to the lungs. Multimodality treatment comprises surgery, chemotherapy, and radiation therapy recommended for the treatment. Doxorubicin-based chemotherapy has shown good survival [12]. Age greater than 60 years, non-extremity lesions, deep-seated tumors, and large tumor size (>5 cm in diameter) are related to a bad prognosis [13].
4. Conclusion
Although rare, work-up of a huge intrathoracic mass in an infant should consider a possibility of myxoid pleomorphic liposarcoma. Appropriate clinical evaluation, imaging, and IHC studies help diagnose the condition.
Consent for publication
Written informed consent was obtained from the patient's father (due to his minor age) for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
Provenance and peer review
Not commissioned, externally peer-reviewed.
Ethical approval
Not required.
Funding
None.
Guarantor
Sansar Babu Tiwari accepts full responsibility for the work and/or the conduct of the study, has access to the data, and controls the decision to publish.
Research registration number
Not applicable.
CRediT authorship contribution statement
Sansar Babu Tiwari (SBT), Sumit Gami (SG), Ranjan Sapkota (RS) = Study concept, Data collection, and surgical therapy for the patient
Sansar Babu Tiwari (SBT), Sumit Gami (SG), Kamal Gautam (KG) = Writing - original draft preparation
Sujan Sharma (SS), Sansar Babu Tiwari (SBT) = Editing and writing
Shreya Shrivastav (SS), Ranjan Sapkota (RS) = Senior author and manuscript reviewer.
All the authors read and approved the final manuscript.
Declaration of competing interest
None.
Acknowledgment
None.
Contributor Information
Sansar Babu Tiwari, Email: sansartiwari@gmail.com.
Sujan Sharma, Email: sharmasujan@iom.edu.np.
References
- 1.Sinclair T.J., Thorson C.M., Alvarez E. Pleomorphic myxoid liposarcoma in an adolescent with Li-Fraumeni syndrome. Pediatr. Surg. Int. 2017;33:631–635. doi: 10.1007/s00383-017-4063-x. 2017/02/06. [DOI] [PubMed] [Google Scholar]
- 2.Zare S.Y., Leivo M., Fadare O. Recurrent pleomorphic myxoid liposarcoma in a patient with Li-Fraumeni syndrome. Int. J. Surg. Pathol. 2020;28:225–228. doi: 10.1177/1066896919878804. 2019/09/29. [DOI] [PubMed] [Google Scholar]
- 3.Francom C.R., Leoniak S.M., Lovell M.A. Head and neck pleomorphic myxoid liposarcoma in a child with Li-Fraumeni syndrome. Int. J. Pediatr. Otorhinolaryngol. 2019;123:191–194. doi: 10.1016/j.ijporl.2019.05.016. 2019/05/28. [DOI] [PubMed] [Google Scholar]
- 4.SCARE Group. Agha R.A., Franchi T., Sohrabi C., Mathew G., Kerwan A. The SCARE 2020 guideline: updating consensus Surgical CAse REport (SCARE) guidelines. Int. J. Surg. 2020;84:226–230. doi: 10.1016/j.ijsu.2020.10.034. [DOI] [PubMed] [Google Scholar]
- 5.Pui W.C., Ling W.H.Y., Najah M. Successful resection of a giant thoracic myxoid liposarcoma. Asian Cardiovasc. Thorac. Ann. 2018;26:410–412. doi: 10.1177/0218492318772763. [DOI] [PubMed] [Google Scholar]
- 6.Ma J., Zhang H.M., Zhang L.W. Primary mediastinal giant liposarcoma with smooth muscle and neural differentiation: a case report. Oncol. Lett. 2015;9:2667–2669. doi: 10.3892/ol.2015.3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.The WHO Classification of Tumours Editorial Board . 5th ed. IARC Press; Lyon: 2020. WHO Classification of Tumours Soft Tissue and Bone Tumours. [Google Scholar]
- 8.Suzuki K., Yasuda T., Watanabe K. Myxoid liposarcoma with cartilaginous differentiation showing DDIT3 rearrangement. Oncol. Lett. 2017;14:6789–6794. doi: 10.3892/ol.2017.7056. 2017/11/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alaggio R., Coffin C.M., Weiss S.W. Liposarcomas in young patients: a study of 82 cases occurring in patients younger than 22 years of age. Am. J. Surg. Pathol. 2009;33(5):645–658. doi: 10.1097/PAS.0b013e3181963c9c. [DOI] [PubMed] [Google Scholar]
- 10.Murphey M.D., Arcara L.K., Fanburg-Smith J. From the archives of the AFIP: imaging of musculoskeletal liposarcoma with radiologic-pathologic correlation. Radiographics. 2005 Sep-Oct;25(5):1371–1395. doi: 10.1148/rg.255055106. [DOI] [PubMed] [Google Scholar]
- 11.Hofvander J., Jo V.Y., Ghanei I. Comprehensive genetic analysis of a paediatric pleomorphic myxoid liposarcoma reveals near-haploidization and loss of the RB1 gene. Histopathology. 2016;69:141–147. doi: 10.1111/his.12913. 2015/12/10. [DOI] [PubMed] [Google Scholar]
- 12.Eilber F.C., Eilber F.R., Eckardt J. The impact of chemotherapy on the survival of patients with high-grade primary extremity liposarcoma. Ann. Surg. 2004;240:686–697. doi: 10.1097/01.sla.0000141710.74073.0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gebhard S., Coindre J.M., Michels J.J. Pleomorphic liposarcoma: clinicopathologic, immunohistochemical, and follow-up analysis of 63 cases: a study from the french Federation of Cancer Centers Sarcoma Group. Am. J. Surg. Pathol. 2002 May;26(5):601–616. doi: 10.1097/00000478-200205000-00006. [DOI] [PubMed] [Google Scholar]