

Abstract
Susceptibility to atrial fibrillation (AF) is determined by well-recognized risk factors such as diabetes or hypertension, emerging risk factors such as sleep apnea or inflammation, and increasingly well-defined genetic variants. As discussed in detail in a companion article in this series, studies in families and in large populations have identified multiple genetic loci, specific genes, and specific variants increasing susceptibility to AF. Since it is becoming increasingly inexpensive to obtain genotype data and indeed whole genome sequence data, the question then becomes to define whether using emerging new genetics knowledge can improve care for patients both prior to and after development of AF. Examples of improvements in care could include identifying patents at increased risk for AF (and thus deploying increased surveillance or even low-risk preventive therapies should these be available), identifying patient subsets in whom specific therapies are likely to be effective or ineffective or in whom the driving biology could motivate the development of new mechanism-based therapies, or identifying an underlying susceptibility to comorbid cardiovascular disease. While current guidelines for the care of patents with AF do not recommend routine genetic testing,1 this rapidly increasing knowledge base suggests that testing may now or very soon have a place in the management of select patients. The opportunity is to generate, validate, and deploy clinical predictors (including family history) of AF risk, to assess the utility of incorporating genomic variants into those predictors, and to identify and validate interventions such as wearable or implantable device-based monitoring ultimately to intervene in patients with AF before they present with catastrophic complications like heart failure or stroke.
Subject Terms: Atrial Fibrillation, Genetics
Keywords: Atrial fibrillation, genetics, human, clinical
INTRODUCTION
Susceptibility to atrial fibrillation (AF) is determined by well-recognized risk factors such as diabetes or hypertension, emerging risk factors such as sleep apnea or inflammation, and increasingly well-defined genetic variants. As discussed in detail in a companion article in this series, studies in families and in large populations have identified multiple genetic loci, specific genes, and specific variants increasing susceptibility to AF. Since it is becoming increasingly inexpensive to obtain genotype data and indeed whole genome sequence data, the question then becomes to define whether using emerging new genetics knowledge can improve care for patients both prior to and after development of AF. Examples of improvements in care could include identifying patents at increased risk for AF (and thus deploying increased surveillance or even low-risk preventive therapies should these be available), identifying patient subsets in whom specific therapies are likely to be effective or ineffective or in whom the driving biology could motivate the development of new mechanism-based therapies, or identifying an underlying susceptibility to comorbid cardiovascular disease.
While current guidelines for the care of patents with AF do not recommend routine genetic testing,1 this rapidly increasing knowledge base suggests that testing may now or very soon have a place in the management of select patients. The opportunity is to generate, validate, and deploy clinical predictors (including family history) of AF risk, to assess the utility of incorporating genomic variants into those predictors, and to identify and validate interventions such as wearable or implantable device-based monitoring ultimately to intervene in patients with AF before they present with catastrophic complications like heart failure or stroke.
Genomic variation
It is convenient to classify genomic variants increasing susceptibility to any complex disease by their effect size.2 At one end of this spectrum are typical rare single gene Mendelian conditions with very large effect sizes (Supplementary Figure). These cannot be common across broad populations (otherwise, many would have the disease) and have therefore usually been identified in families or in closed populations. As described below, associations have been more recently described between rare genetic variants and common traits like AF using whole exome or genome sequencing in very large population subgroups. At the other end of the effect size spectrum are common variants, usually identified using the genome-wide association study (GWAS) paradigm, that individually confer very modest increased risk but may be combined to create polygenic risk scores (PRS) to identify patients with clinically-meaningful increased risk.
COMMON GENETIC VARIATION IN AF
Finding common variants mediating AF risk
GWAS interrogates hundreds of thousands of common variants across the genome to identify associations between loci, genes, and individual variants and a trait of interest.2 “Common” is generally defined as a minor allele frequency greater than ~5%. The method uses single nucleotide variants (SNVs) that “tag” haplotype blocks, which are genetic data that are co-inherited; the result is that the SNVs with the strongest association with the trait being interrogated are not necessarily those with the biologic activity that underlies the association, nor is the nearest gene necessarily the one whose function mediates the biology. Unraveling new biology from GWAS results continues to present a major opportunity and challenge to genome science.
The first GWAS were expensive and studied small patient populations (thousands) and few variants (less than a million). Initial GWAS results for common traits and conditions like AF,3 coronary artery disease (CAD),4–6 or the QT interval7 defined specific loci at which variants modulate risk. These findings, in turn, have been used to define specific risk alleles and underlying biology, as described in companion articles in this series. One additional hope of these early studies was that risk alleles could be used to identify specific patients at elevated disease risk, and indeed soon after initial GWAS results genetic testing companies began to deliver risk allele information to patients.8 Further, individual SNVs were studied for their effects across clinically-related traits. For example, a very early signal examining variability in the QT interval was at chromosome 1 near NOS1AP,7 and subsequent studies have suggested that the risk allele modifies penetrance in the congenital long QT syndrome,9–11 risk for amiodarone-related long QT syndrome,12 and risk for sudden death in the broad population.13 However, the hazard ratio for these effects is modest, generally 1.5 or less.
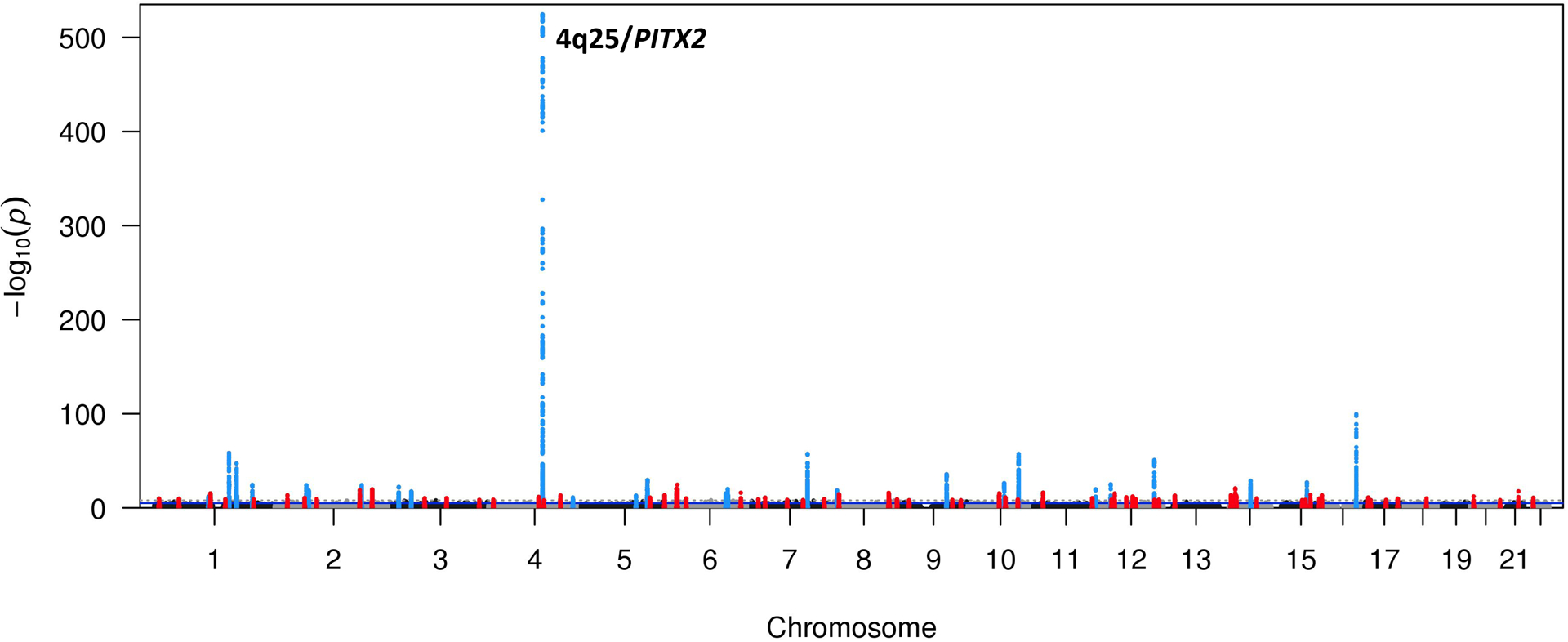
The initial GWAS for AF risk, which studied 550 cases unselected for any other risk factor, identified variants at chromosome 4q25 near PITX2 as risk alleles,3 and subsequent increasingly larger and ancestrally diverse patient sets have identified dozens of loci at which SNVs confer risk for AF.14 Unlike the situation in many other common diseases or traits, the AF GWAS result is unusual in that the 4q25 locus carries much smaller P values (10−550) and larger effect sizes, odds ratio ~1.7-fold per allele, than other loci (Figure 1). Importantly, genetic association studies have identified multiple independent risk loci at 4q25, and interestingly these seem to vary across ancestries;14 one likely interpretation is that each locus independently regulates expression of target gene(s), possibly PITX2. A major outcome of AF GWAS to date has been to define novel AF susceptibility pathways and these fall into 3 broad categories: electrical signaling, contractile function, and cardiac development.14
Figure 1. Results from a recent AF GWAS.
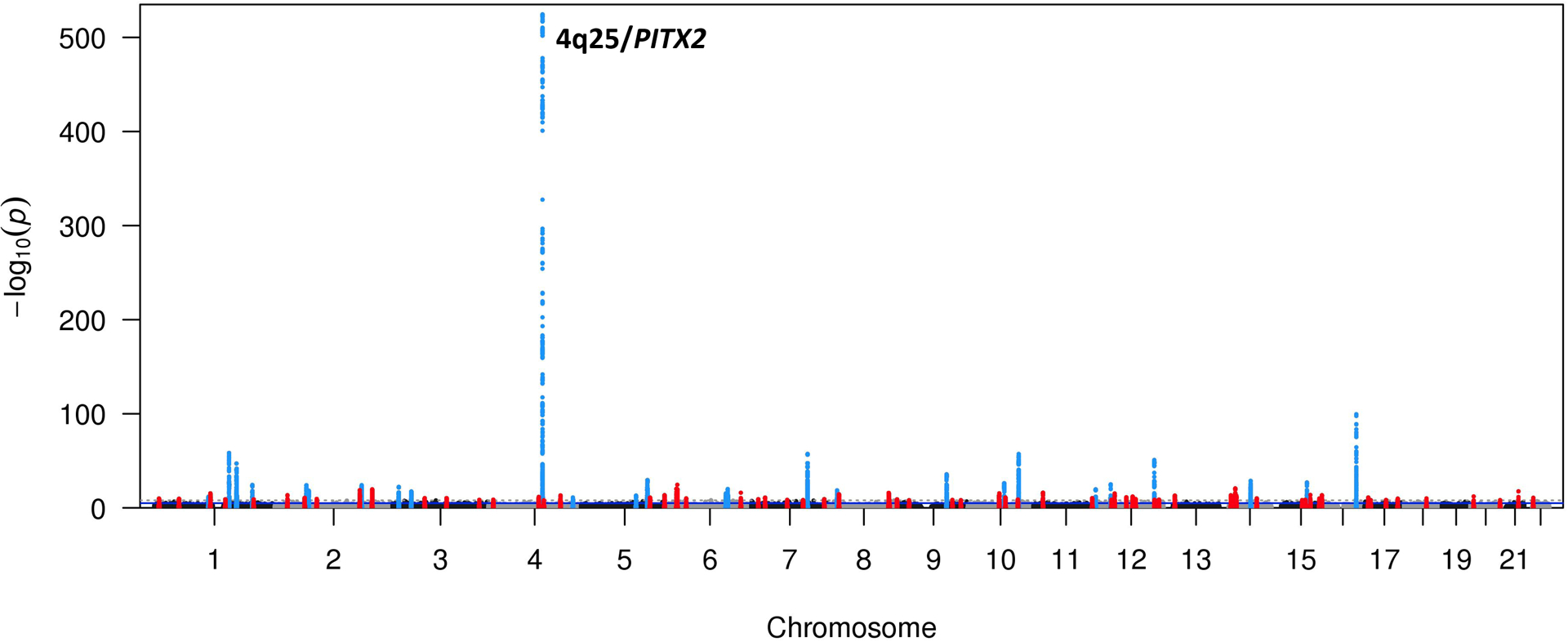
The 4q25 AF susceptibility locus is one of the most significant genetic associations detected for any phenotype with a P-value equal to 3.4×10−155. 22 other loci at which common variants are associated with AF at the genome-wide significance level (P<5 × 10−8) are shown. Figure courtesy of Patrick Ellinor.14
Predicting AF – role of common variants
One major challenge to the field is to identify individuals at high risk for AF before they present with arrhythmia-mediated heart failure15 or stroke. Epidemiological studies such as the Framingham Heart Study (FHS) have proven invaluable in identifying AF risk factors such a hypertension or diabetes. Further, early reports from the founders of modern electrocardiography,16, 17 studies in FHS,18 as well as in patients seen for AF at tertiary care institutions,19, 20 have identified a positive family history of AF as a risk factor. GWAS results can also be used to estimate the heritability of common traits; for AF in the UK Biobank, a very large middle-aged, predominantly European ancestry population, this estimate is ~22%.21 Importantly, most of this risk was attributable to common SNVs, but only a minority of those SNVs were located near known risk loci for arrhythmias (including AF) or cardiomyopathy. Taken together, these data support a role for multiple genetic factors in determining AF risk, and suggest further studies may be useful to further delineate the mechanisms underlying this shared risk.
The first study to use GWAS-identified SNVs to attempt to predict AF risk found that adding SNVs at three GWAS loci, near PITX2, KCNN3, and ZFHX3, did not improve prediction of AF beyond conventional risk factors in FHS.22 Another early study identified 3 independent risk SNVs at chromosome 4q25, and reported that a few individuals (<2%) carried 5 or 6 risk variants, and were at >5-fold relative risk for AF (Figure 2);23 a similar result was later replicated in both European and Japanese ancestry subjects.24 These initial studies suggested that combining multiple risk SNVs into a polygenic risk score (PRS) might enhance the ability of genetic variation to predict AF. In an initial study, scores based on 11 to 719 common SNVs were developed. Hazard ratios comparing highest versus lowest risk quartiles were significantly greater than 1: the hazard ratios for incident AF varied from 1.28 (for 719 SNVs) to 1.67 (for 25 SNVs), and a 127 SNV score predicted those with cardioembolic stroke.25 A study in the UK biobank dataset used a 6,730,541-SNV PRS to identify 6.1% of the population at >3-fold risk for prevalent and incident AF.26 The authors also studied PRS for four other common diseases (coronary artery disease, breast cancer, inflammatory bowel disease, and type 2 diabetes), and reported that in each case a small group at >3-fold risk could be identified, that this risk was similar to or greater than that for monogenic diseases conferring risk (such familial hypercholesterolemia for CAD), and that therefore it is worth considering incorporating PRS into clinical decision making. The optimal number of SNVs to be incorporated into a PRS has not yet been established.
Figure 2. The evolution of polygenic risk scores for AF.
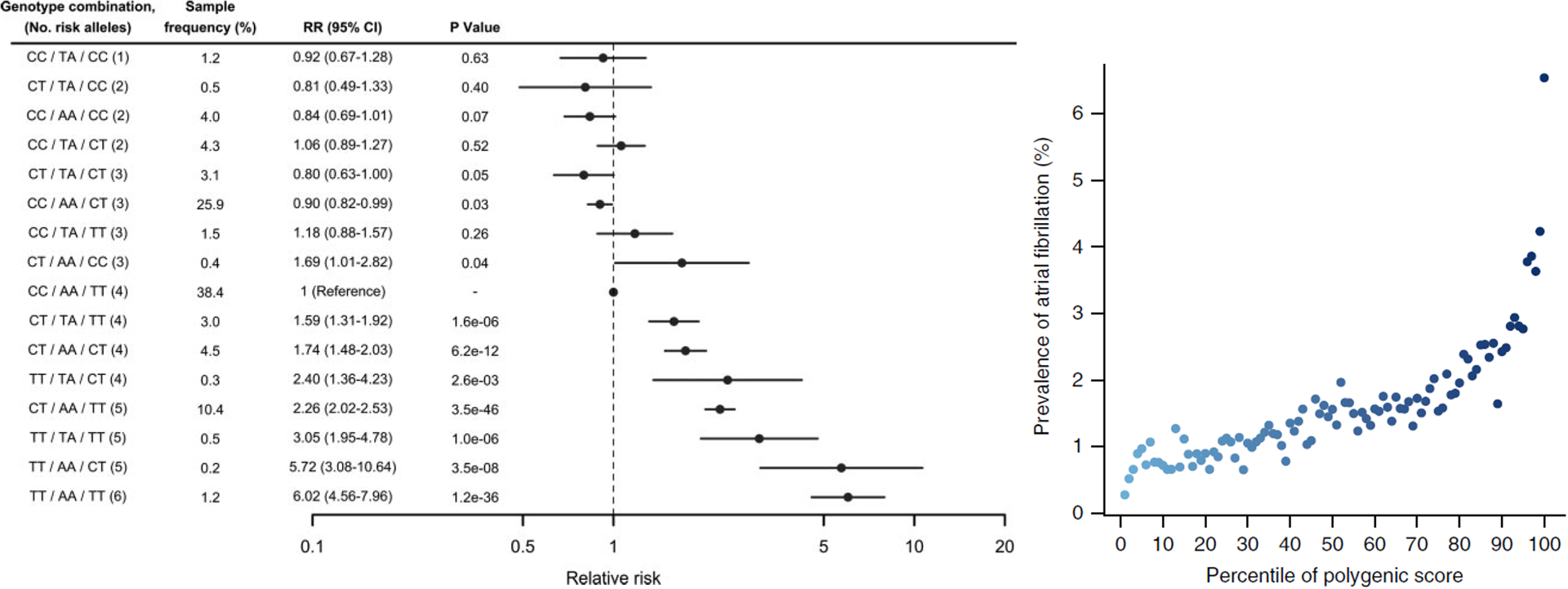
Early experience with PRS in AF used 3 SNVs from the 4q25 locus (Panel A) compared to a recent PRS in AF that used 6,730,541 SNVs (Panel B). Panel A: Reproduced with permission from Circulation. 2010. 122(10): 976–984. Copyright © 2010 American Heart Association. All rights reserved.23 Panel B: Reproduced with permission from Nature Genetics. 2018. 50(9): 1219–1224. Copyright © 2018 Nature Publishing Group. All rights reserved
PERSONALIZATION OF CARE: ROLE OF COMMON VARIATION
An understanding of the genetic basis of AF susceptibility has motivated studies to predict not only who will develop AF, but also clinical outcomes such as stroke or arrhythmia recurrence after AF ablation discussed below. Initial studies in this area focused on individual common variants that were selected from candidate genes based on known biology; for example, common variants in ACE (angiotensin converting enzyme)27 and IL6R (interleukin-6 receptor)28 were examined for an association with AF ablation outcomes due to their possible roles in fibrosis and inflammation. Subsequently, candidate SNVs such as the chromosome 4q25 susceptibility alleles were examined.29–32 Today, with the development of PRS to estimate the combined effect of thousands of common variants, the association between overall common variant susceptibility to AF and AF outcomes is underway.
Predicting Stroke
The goal of preventing strokes through early diagnosis of AF is a major driving force behind AF genetics research. Adding to the potential for clinical application in this area, GWAS to define the genetic associations with stroke discovered that common variants associated with AF (4q25/PITX2, 16q22/ZFHX3) were also among the strongest associations with cardioembolic stroke.33, 34 In an early iteration of polygenic risk modeling for AF, a genetic risk score for AF derived from 12 known AF susceptibility alleles was examined for association with stroke in the Malmo Diet and Cancer Study. Among 27,471 participants, 1,495 participants experienced incident stroke and individuals in the highest genetic risk quintile for AF had a 23% higher risk for stroke compared to the lowest genetic risk quintile.35 More recently, using a variety of PRS for AF derived from as many as 719 SNVs, polygenic risk for AF was calculated in 18,919 individuals and found to confer up to a 2.25-fold increase in risk of cardioembolic stroke in the highest versus lowest AF genetic risk quartiles.25 While this sounds highly promising, there has been concern that the addition of PRS provides only a small improvement in AF (or stroke) risk prediction beyond clinical only models. This was addressed in a more recent study by Weng et al. by using a new PRS derived from a larger discovery cohort and by focusing on long-term rather than short-term risk for AF; the results provided further support for an AF PRS to reclassify patient’s risk for AF.36
Predicting outcomes after AF Ablation
Originally recommended for patients with paroxysmal AF and few comorbidities, AF ablation has become an increasingly common therapy for AF as its use has expanded to patients with almost any clinical and genetic risk profile.37 However, the procedural approach is largely the same across patient subgroups despite a multitude of potentially different AF mechanisms.37 All patients receive pulmonary vein (PV) isolation and the decision of whether or how to modify the atrial substrate or ablate non-PV triggers is a source of general disagreement.37 Accordingly, 20–40% of patients have residual AF requiring continuation of antiarrhythmic medications or repeat ablation38, which must be weighed along with the risk of procedural complications and cost when considering a patient for AF ablation. Taken together, AF ablation is a treatment for which a personalized approach, potentially based on a clear understanding of mechanism and of underlying genetics, could be useful.
One of the earliest candidate SNV analyses detected an association between AF risk alleles at the chromosome 4q25/PITX2 locus and increased odds of recurrence39, and this result has been replicated in 3 European ancestry cohorts.40, 41 In populations of Asian-ancestry, an initial report by Choi et al. in 2015 detected no association with recurrence in a multicenter cohort from the Korean AF Network,32 but subsequently 3 separate reports from Chinese and Japanese AF ablation cohorts detected an association with increased risk of recurrence.42–44 Most recently, a meta-analysis of seven qualifying studies of both European and Asian ancestries reported the AF risk allele at rs2200733 (4q25/PITX2) conferred a 45% increased risk of AF recurrence following ablation.45 Of note, there was no association with recurrence for common variants at the other top AF susceptibility loci (1q21/KCNN2 and 16q22/ZFHX3).
Experimental data suggest that PITX2 regulates development of the pulmonary vein myocardial sleeve and/or increased automaticity of left atrial cardiomyocytes, both of which may predispose to arrhythmia mechanisms poorly addressed by PV isolation.46 Thus, the association of 4q25 SNVs with ablation outcomes motivated other studies that investigated candidate SNVs associated with AF mechanisms thought to respond poorly to PV isolation; these included genes associated with cardiac fibrosis and inflammation (e.g. ACE, IL6R).27, 28
The absolute difference in ablation outcomes between carriers and non-carriers of individual variants is too small to be used to guide clinical management decisions, such as selecting patients for AF ablation. A recent study evaluated a 929-SNV PRS for AF in 4,276 patients from 10 AF ablation centers (unpublished data). At baseline, patients with higher AF genetic susceptibility were younger and had fewer comorbidities; however, there was no association with recurrence after ablation. These results suggest that genetic susceptibility to AF and to recurrence after ablation are genetically different phenotypes, and so a PRS predictor of AF recurrence after ablation may be best derived from studies of ablation response, and not AF susceptibility in the general population.
In summary, the current state of this field is left with evidence that some common variants are associated with increased risk of recurrence, however the effect size for individual SNVs is small and the optimal panel of SNVs to derive a polygenic risk score is yet to be determined. Future studies that integrate data from GWAS of AF in the general population into mechanistic pathways using various bioinformatic approaches could be used to enhance the predictive power of PRS.
Predicting other outcomes
Carriers of the common Arg389Gly SNV in the β1 adrenergic receptor gene (ADRB1) were found to require lower doses of rate control medications (beta-blockers and calcium channel blockers) to achieve target heart rates in AF.47 This SNV had previously been reported to predict a beneficial effect of the beta-blocker bucindolol in patients with heart failure,48 and reanalysis of data from that trial found that bucindolol also reduced incident AF in Arg389 carriers.49 This prompted a large, multicenter genotype-guided clinical drug trial randomizing patients with Arg389Arg ADRB1 to bucindolol versus metoprolol.50 While the study found no difference between treatment groups, it is noteworthy as the first large randomized controlled pharmacogenomic trial for rhythm-control in AF.51
Outcomes with antiarrhythmic drug therapy and cardioversion were also explored for an association with the top 3 AF susceptibility loci at 1q21, 4q25, and 16q22. One study reported that a 4q25 SNV (rs10033464) was associated with higher odds of favorable response to antiarrhythmic drug therapy as measured by remaining on the same antiarrhythmic drug for 6 months with a corresponding reduction in symptomatic AF burden (OR 4.7 in the discovery cohort and 1.5 in the validation cohort).52 A different common SNV at 4q25 (rs2200733) has also been associated with AF recurrence following cardioversion, conferring a 2.4-fold higher risk of recurrence (HR 2.4, 95% CI: 1.4–4.1, P=0.001).53 Taken together, these studies suggest 4q25 is statistically associated with response to medical rhythm-control therapies, but it remains unknown whether the effect size is clinically significant. Interestingly, 4q25 SNVs have also been associated with an increased risk for sudden death.54
Other biomarkers
Biomarkers often derived from transcriptomic, metabolomic, or proteomic studies may also be useful in predicting AF,55, 56 and will be discussed in further detail in other sections of this series. Both natriuretic peptides57–60 and C-reactive protein60 have been proposed as predictors of AF. One appeal of candidate biomarkers is that they may be “downstream” of genomic predictors and thus may capture both environment and genomic variation. A challenge is to validate such newer biomarkers and to assess the extent to which they provide predictive value beyond other markers like family history, clinical variables and genomics. A further difficulty is that a candidate biomarker may not have been measured in the large numbers of patients (often from epidemiologic cohorts) required for validation. One proposed approach to this problem has been to develop “virtual” biomarkers: a candidate biomarker is measured in an epidemiologic cohort such as FHS and a multi-SNV instrument is then developed to predict the biomarker in any set that has longitudinal data and dense genotype data, even if the biomarker itself has not been measured.61, 62 One application of this approach was to assess associations between predicted plasma concentrations of Thyroid Stimulating Hormone (TSH) and EHR-based phenotypes in subjects without recognized thyroid disease (Figure 3). In most such subjects, TSH has not been measured but a multi-SNV “virtual” TSH derived from FHS identified associations between AF and low TSH.63
Figure 3. Leveraging genetics for biomarker research.
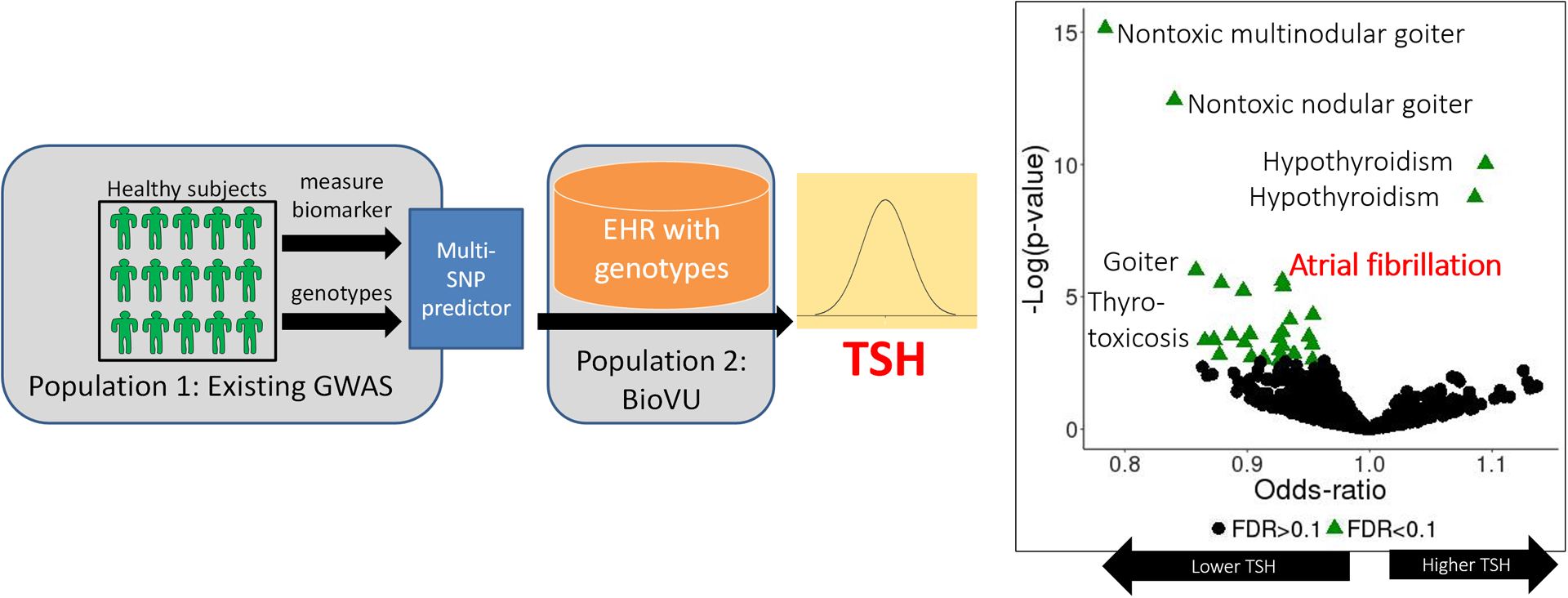
Using a population that has both GWAS data and a biomarker measured (e.g. TSH), a PRS can be derived to predict that biomarker’s value in any separate population with GWAS data. Using this “virtual biomarker” approach, a PRS for TSH was shown to be associated with thyroid disorders and AF. Reproduced with permission from JAMA. 2019. 4(2):136–143. Copyright © 2019 American Medical Association. All rights reserved.63
Measured biomarkers associated with genetic markers have been used to help establish causative pathways of AF using the Mendelian Randomization approach. If genetic markers, which are randomly distributed in a natural setting, are associated with an outcome such as AF, and if these association are dependent only on the measured biomarker levels, then this is powerful evidence of a causative pathway. This approach has been used to validate the causative relationship between thyroid function and AF64.
PERSONALIZATION OF CARE: ROLE OF RARE VARIATION
Finding rare variants mediating AF risk
The strongest evidence that a rare variant mediates a human trait comes from linkage analysis of large kindreds in which individuals from multiple generations can be unambiguously classified as affected or unaffected. While a number of reports have described multi-generation families with a high incidence of AF, only a few have successfully used conventional linkage analysis to identify causative genetic variants in these families, often with other phenotypes like heart failure or conduction system disease (Supplementary Table I)65–72 One possible explanation for the failure to identify causative variants in many of these kindreds is that AF is common and has variable, age-related penetrance, so even in affected families some individuals may have AF without carrying the family’s AF-causing variant, while variant carriers may not necessarily develop AF. Some affected families may have other manifestations, such as sudden death in childhood73 or heart failure (e.g. laminopathies74), which can help assign phenotype. Further, AF is increasingly recognized as an initial manifestation of a well-described cardiomyopathy or channelopathy as discussed in further detail below.
An alternative to family studies is to search for rare variants by sequencing likely candidate genes (e.g. those encoding ion channels or contractile proteins) in patients presenting with AF. This approach has the greatest appeal in families in which multiple members present with early onset of AF (eoAF) and in the absence of known risk factors, i.e. in a setting in which genetic factors are more likely than usual to play a role. Evidence that a rare non-synonymous variant in a logical candidate gene identified in this fashion is causative for AF can include segregation between the variant and other family members with AF and in vitro studies showing that the variant generates a dysfunctional protein.75, 76 While the use of functional studies to help classify the pathogenicity of rare genetic variants seems intuitively appealing, systematic and critical re-examination of gene-disease or variant-disease associations have called into question many of these associations. For example, among 21 genes associated with Brugada syndrome, systemic curation validated only one association (with SCN5A), and questioned the remaining 20 associaitons.77 Similar analyses found limited supportive evidence for 25/33 genes associated with hypertrophic cardiomyopathy (HCM)78 and more than half of 17 genes associated with long QT79. Hence, the validity of many associations between genetic variants and AF remain to be re-examined using contemporary validation criteria.
Very recently, exome or genome sequencing across populations has been used to identify rare variants associating with disease. Herman et al.80 sequenced the sarcomeric gene TTN, encoding titin, in patients with dilated cardiomyopathy (DCM) or HCM, or controls; TTN is the largest gene in the genome and a candidate gene for DCM.81 Because TTN is so big, most patients harbor at least one missense variant, so this analysis, and others that have followed, focused on nonsense variants, like early stop codons, that definitely produce a change in the encoded protein. They found nonsense mutations in 54/203 patients with DCM compared to 3/231 with HCM and 7/249 controls, and reported (as have others subsequently) that disease associated variants concentrated in the region of the gene encoding the A-band.80 Subsequent studies have reported that nonsense TTN variants are risk factors for cardiomyopathies provoked by pregnancy82 or cancer chemotherapy.83 The National Heart, Lung, and Blood Institute’s Trans-Omics for Precision Medicine (TOPMed) initiative examined whole exome or genome sequences in 2781 European ancestry cases of eoAF, and 4959 controls without AF, and found that 2.1% of the cases carried rare nonsense TTN variants, compared to 1.1% of the controls. Interestingly, the prevalence of TTN nonsense variants in the case group varied by age, and was as high as 6.5% in those presenting with AF at age under 30 (Figure 4).84 In a group of 25 patients referred to a tertiary care arrhythmia clinic for early onset AF (eoAF), defined as age of onset ≤ 45, TTN nonsense variants were found in 4/25, and a pathogenic rare variant in RBM20 (a DCM-associated gene known to modulate titin splicing85) in 1/25.86 While loss of function variants in TTN are the most prevalent abnormalities identified in young participants with AF, they only account for a relatively small proportion of cases. Further studies are necessary to more completely explain the genetic architecture of eoAF.
Figure 4. The association of titin (TTN) LOF in patients with AF.
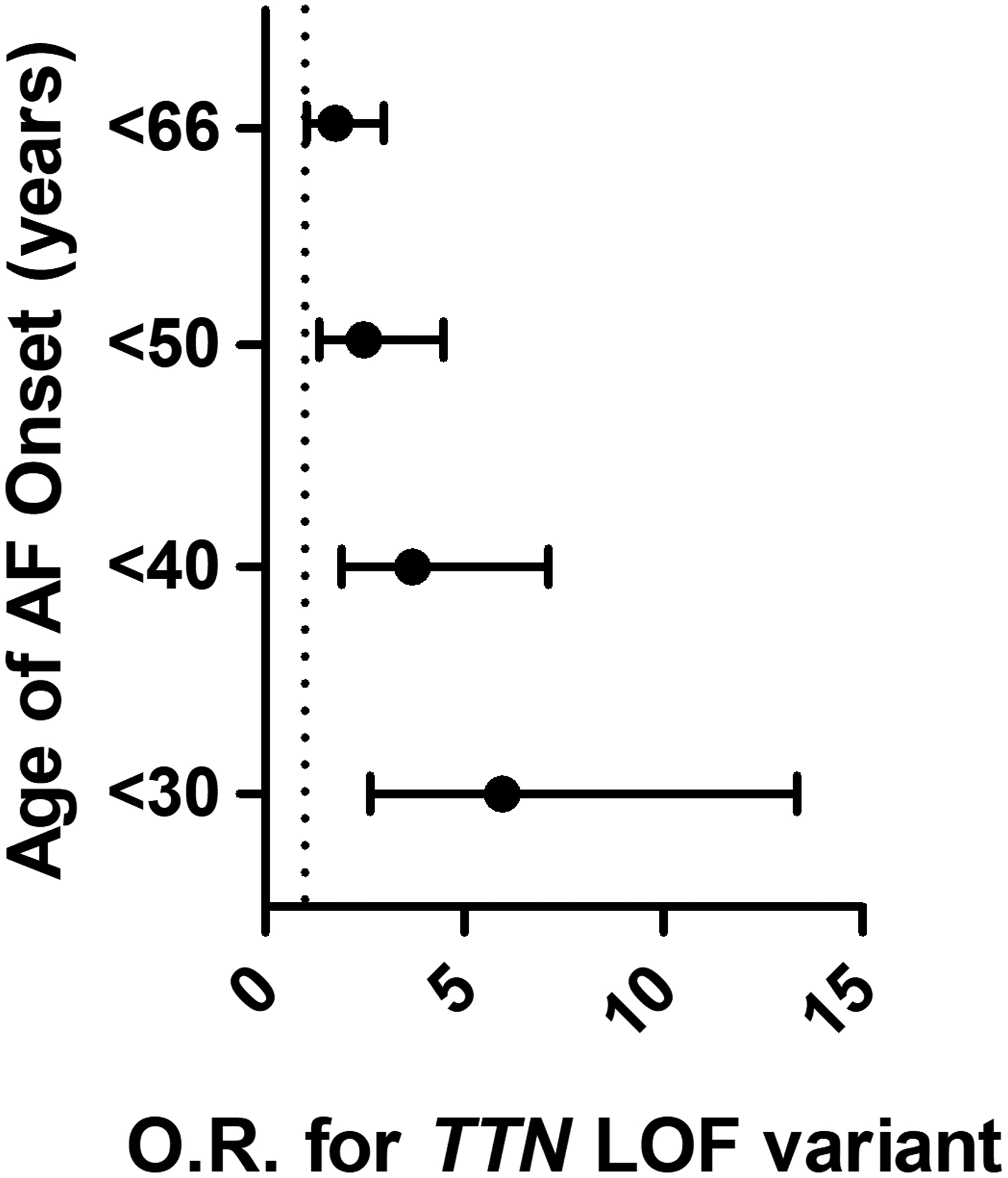
The odds of having TTN LOF compared to control participants increases with younger age at AF diagnosis and was found to be highest in individuals diagnosed before the age of 30 years. Reproduced and modified with permission from JAMA. 2019. 320 (22): 2354–2364. Copyright © 2019 American Medical Association. All rights reserved.84
Clinical Implications of Rare Variants and Early Onset AF
Though AF is the most common cardiac arrhythmia and confers significant risk of morbidity and mortality with increasing age,87 approximately 0.5% of AF is identified before the age of 45 and the clinical significance of detecting AF at such an early age is not well understood.88 The descriptor “lone” AF was previously used to designate AF without known risk factors or underlying heart disease and the term connoted a benign clinical course in younger patients. However, the identification of AF in the young, in the absence of conventional qualifiers, is not necessarily free of risk for stroke, syncope, and early heart failure and mortality. Very few studies have been published that investigate the association between rare variants and AF outcomes.89 This reflects the need for large study cohorts to establish a clinical role for any rare variant.
Rare inherited disorders associated with AF
Many of the genes associated with inherited ion channelopathies, cardiomyopathies and other rare familial disorders have independently been linked to AF (Figure 5). This suggests a shared mechanism and has implications on the clinical genetic testing that could be recommended.
Figure 5. Rare Inherited Disorders Associated with AF.
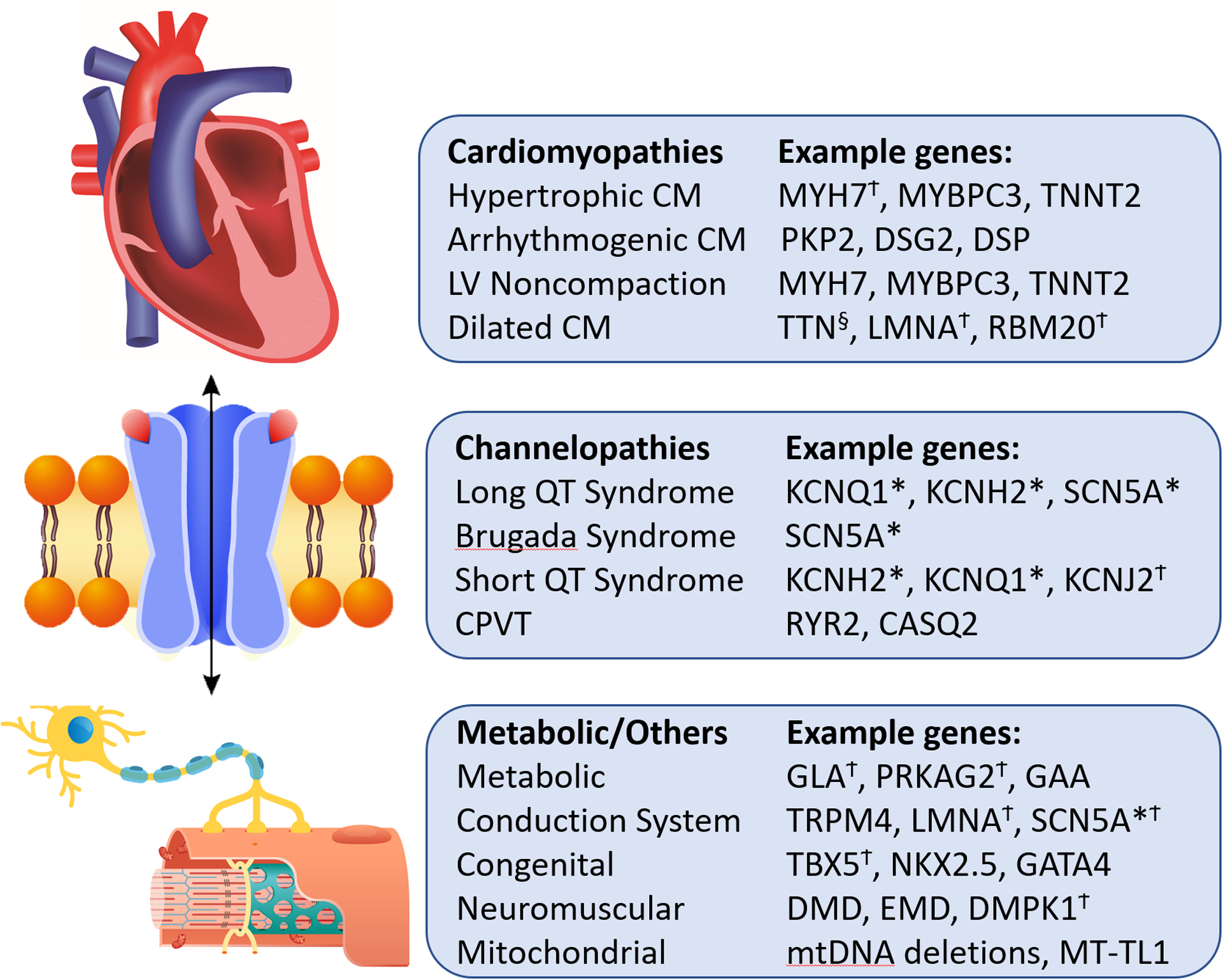
Displayed are examples of up to 3 genes associated with each inherited disorder, many of which have independently been linked with AF. * Genes associated with AF in linkage analysis. § Gene association with AF in rare variant association study. Ϯ Genes associated with high frequency of AF in corresponding disease. CM- cardiomyopathy. LV- left ventricular. CPVT- catecholaminergic polymorphic VT.
While the incidence of AF in the young is low in the overall population, the prevalence of AF is higher among patients with primary arrhythmia syndromes, ranging from 2% in long QT syndrome (LQTS) to 30% in short QT syndrome (SQTS).90 Individuals with LQTS have a greater than 15-fold higher risk of AF before the age of 50 compared with population-based matched controls (RR 17.5).91 In Brugada Syndrome, concomitant AF is associated with increased rates of syncope and ventricular arrhythmias.92, 93
AF is the most common documented arrhythmia in patients with inherited cardiomyopathies and portends a worse prognosis. In those with hypertrophic cardiomyopathy (HCM), cross-sectional studies reveal that AF is found in 15–25% of patients.94, 95 In patients with HCM, rare pathogenic variants in MYH7 are associated with a greater frequency of AF compared to other pathogenic variants.96 The presence of AF in patients with HCM has been linked to decreased exercise tolerance,97 clinical deterioration, and increased risk of death98, 99 compared to those diagnosed with HCM without AF. Similarly, AF occurs in 14–20% of patients with arrhythmogenic cardiomyopathy (ACM) and is present at a significantly younger age than in the general population.100 AF in ACM has been associated with worse RV function and increased mortality.101 MRIs in patients with AF and ACM show increased atrial size but – in contrast to AF in the general population – no increased fibrosis. Similarly, AF in patients with left ventricular non-compaction cardiomyopathy (LVNC) has been associated with worse heart failure symptoms, decreased ejection fraction and increased mortality.102 In addition, those with rare, non-sarcomeric variants demonstrated higher frequency of AF, lower LVEF, and increased rates of composite heart transplantation and death.103 Poor outcomes in the presence of AF are not only observed in cardiac muscle disorders, but also neuromuscular disorders. Atrial arrhythmias occur in up to 25–30% of patients with myotonic dystrophy type 1 and are associated with increased mortality.104, 105
Combinations of rare and common variants
TTN variants in exons highly expressed in the heart were found in 0.44% of subjects in the UK biobank, and 14% had AF, while in the 0.44% of subjects with the highest PRS for AF, 9.3% had AF. Interestingly, the PRS explained 4.7% of the variance in AF susceptibility, while the TTN variants accounted for only 0.2%,106 indicating that both common and rare variants will likely be used in any attempt to identify individuals at genetically-driven increased risk for AF. There is also precedent for thinking that common variant-based PRS may modulate the penetrance of rare variants. In one study in 12 families each with multiple individuals with AF and a rare culprit variant implicated, common variants at 4q25 were very strong determinants of penetrance, particularly at age <50.107 In the UK Biobank study, AF was seen in 21.5% of those with the highest PRS tertile compared to 6.7% in the lowest tertile in the presence of a TTN loss of function variant.
CLINICAL INTEGRATION
Since the publication of the first human genome sequence in 2001,108, 109 there has been a public expectation that genetic testing would quickly play a central role in allowing us to tailor specific treatments based on individual characteristics. While understanding of the genetic architecture of rare and common diseases has advanced at a rapid pace, the implementation of this knowledge in the clinical setting has been slower.
Clinical testing for very early onset AF
In the most recent guideline statements, clinical genetic testing for AF was not recommended1, 110. Emerging data, however, suggests that clinical testing with targeted gene sequencing may play a role in evaluating patients with early onset AF. Whether there is sufficient evidence to recommend the clinical implementation of genetic testing depends heavily on the clinical setting. The role of clinical genetic testing, using targeted sequencing of a handful of genes, in the diagnosis of rare cardiovascular disease has been well established and is now routinely recommended for several inherited arrhythmic conditions110. Genetic testing can also be cost-effective in the screening of family members at risk for inherited conditions like long QT syndrome111.
A contemporary view of AF etiology posits a role for both genetic as well as environmental factors, and their interactions. Thus, until we understand better the genetic basis of AF, widespread genetic testing runs the risk of being low yield and identifying many false positives or variants of uncertain significance. On the other hand, there now seems little doubt that identifiable genetic variants play a prominent role in mediating AF susceptibility in some patient populations. One obvious group are those with eoAF, particularly in those who have a family history of eoAF and no conventional risk factors.
Early studies focused on the idea that AF was an ion channel disease, but screening for variants in channelopathy genes was very low yield.112, 113 However, in a study in patients with AF onset before age 45 described above, rare pathogenic variants were found in 6/25 subjects, including variants in TTN and RBM20 in patients with previously undiagnosed cardiomyopathy86. Although young patients may be at low risk for stroke, early reports show a high incidence of cardiomyopathy among eoAF patients. In one study, 9/11 had an abnormal cardiac MRI despite a normal echocardiogram86, and eoAF patients also had a higher than expected family history of cardiomyopathy and/or sudden cardiac death. Thus, patients with eoAF may well have an underlying susceptibility to cardiomyopathy and be at increased risk of developing more overt structural abnormalities. Further, detection of early cardiomyopathy should lead to more careful surveillance in family members. Long term follow-up of a larger number of eoAF patients will be needed to help better define these risks.
Practical Recommendations for Evaluating Early Onset AF in clinic
Figure 6 shows one model for the workup of patients who are candidates for a genetics-focused evaluation of AF. The emphasis is on those with eoAF (AF onset before age 45) and without identifiable risk factors, or those with a family history of eoAF. The description of an established inherited arrhythmia clinic model follows and may serve as a platform for other growing centers.
Figure 6. Proposed evaluation of individuals with AF onset before age 45 or with a family history of early onset AF.
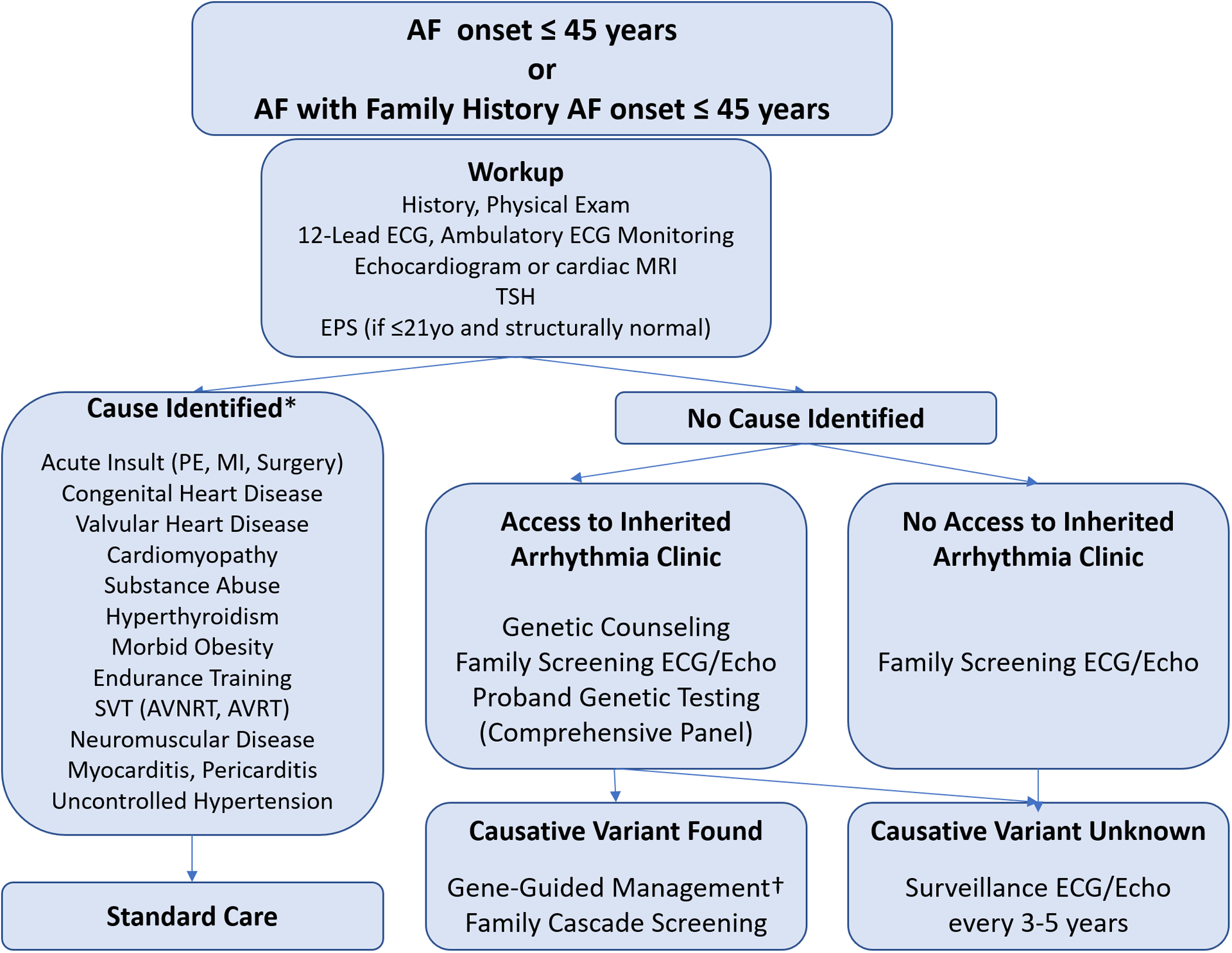
*The presence of a causative factor does not preclude evaluating the individual for a genetic etiology if the clinical features or family history suggest an underlying genetic susceptibility exits. † Gene-guided management includes additional diagnostic testing (e.g. sodium-channel blocker challenge, signal averaged ECG) or treatments (e.g. physical activity restrictions, implantable cardioverter defibrillators) for specific inherited syndromes. EPS- electrophysiology study. PE- pulmonary embolism. MI- myocardial infarction. SVT- supraventricular tachycardia. AVNRT- atrioventricular nodal reentrant tachycardia. AVRT- atrioventricular reentrant tachycardia.
Given the complexity of potential screening and need for diverse expertise, an inherited arrhythmia clinic consists of multiple team members ideally including adult and pediatric electrophysiologists, clinicians with expertise in cardiomyopathy, nurses specializing in inherited disease, and genetic counselors with experience in counseling, educating patients and their families on genetic testing and variant interpretation. Genetic counseling services in cardiovascular medicine have been growing at a rapid pace and are becoming more widely available.114
Patients are often referred to an inherited arrhythmia clinic based on identification of an abnormal arrhythmia at an early age or a suspicious family history. A pre-visit telephone interview between patient and genetic counselor or specialist nurse is valuable in obtaining a brief history of present illness and an initial inquiry of family history. Given the substantial importance of family history, this allows an opportunity for the patient to further explore any background uncertainties prior to the visit. Following the initial call, it is essential for the clinic to assist the patient in pursuing any record of prior cardiac testing results such as electrocardiography, Holter, exercise treadmill test, echocardiogram, etc. Occasionally, a referral to complete such studies is recommended prior to the in-person visit on a case by case basis. During the visit, ample time is allotted for several layers of the interview which focus on personal clinical history, as well as an extensive family history taken in concert by the specialist nurse, electrophysiologist, and genetic counselor (Supplementary Table II). The use of open-ended questions115 as well as focused inquiries116 are helpful in creating a multi-generation pedigree for every patient which is then reviewed in a “working case,” roundtable format, the purpose being to identify any suspicious trends and the extent of possibly affected members. Additional testing, such as dedicated cardiac imaging or wearable patch recorders, may be recommended to guide clinical decision making. When indicated and felt to be high yield, either following the initial visit or following resulted additional testing, patients receive extensive genetic counseling and may elect to undergo genetic testing after informed consent.
During the evaluation of eoAF, it is important that the clinic maintains resources to provide ongoing counseling, expert advice, and support for the proband. Based on inheritance patterns, a systemic process for cascade screening should be instituted for patients to receive written recommendations to communicate with any biologic relatives who may be at-risk. Individualized recommendations are made to each family member as carriers of pathogenic variants are identified; generated letters can be effective. It is equally critical to recognize several limitations that may arise during the evaluation process. Cultural differences, individuals no longer in communication, adopted family members, and inability to obtain records of elderly or geographically distant family members are all barriers to establishing a comprehensive family pedigree. Clinically, it is important for the program to furthermore be prepared to interpret genetic test results that may be of uncertain significance, pathogenic but incidental to the observed phenotype, or discrepant involving a mismatch between an individual’s phenotypic evidence of early onset AF and that of other family members.
Identification of a pathogenic rare variant associated not just with a familial cardiomyopathy but also with other arrhythmic conditions such as neuromuscular or mitochondrial disease can have significant implications for the patient and the family members. These findings will likely change the clinical care offered to the patient and will permit cascade genetic screening of the family, which is indicated in most of these conditions. Patients should be informed ahead of time that these discoveries are possible. Clinicians should be ready to provide guidance for addressing pathogenic variants linked to the broad set of disorders tested, particularly as genomic panels continue to expand.
Regardless, in all scenarios it is imperative that if clinical suspicion of a latent arrhythmic syndrome or cardiomyopathy remains high, despite genetic testing, surveillance with periodic interview, physical exam, and occasionally cardiac testing – is established to follow course of individuals long-term. As in other areas, the generation of large national and international databases linking genotypes and phenotypes is a useful tool for continuously updating the role of specific genes (and specific variants) in AF pathogenesis,117 especially as the cost of sequencing continues to fall and thus more variants of uncertain significance are identified.
Figure 6 also emphasizes that genetic susceptibility is not the only potential explanation for eoAF. Congenital heart disease118, Wolf Parkinson White Syndrome119, substance abuse120 and endurance athleticism121, 122 are well-described causes of early manifestations of AF. While there may be a genetic contribution to AF in these scenarios, these entities should be independently considered and potentially ruled out with clinical history and testing including ECG and echocardiogram. For now, the data summarized suggests that a thorough family history which includes assessment of cardiomyopathy and sudden death should be an integral part of the evaluation of eoAF. Further studies are needed to better define whether channelopathy or cardiomyopathy clinical testing should become routine practice in some patients.
Similarly, while there are emerging data that PRS can identify patients at risk for AF, or modulate penetrance of rare pathogenic variants, the clinical role of AF PRS will require further study. When direct-to-consumer genetic testing was first introduced, results of predisposition to common diseases such as AF using common genomic variants was included. However, the Food and Drug Administration (FDA) recognized many of these challenges and has since begun to regulate this testing and has given clearance for marketing of genetic testing for some conditions,123 although this has not included AF.
Bringing Clinical Genetics of early onset AF to the inherited arrhythmia clinic
The studies discussed above are pointing to a shift in the value of genetic testing with broad cardiac panels in patients with eoAF. This area is also ripe for further investigation as clinical genetic testing in these patients begins to move into the inherited arrhythmia clinic. One salient question is at what age should genetic sequencing be recommended. The prevalence of AF begins to rise at approximately 55 years of age,124 suggesting that at approximately this age, the genetic and environmental contributions become more complex. However, the yield of rare variant genetic testing will need to be described at different ages. It is also possible that only certain families, such as those with multiple members with AF or established cardiomyopathy, will benefit from genetic testing.
FUTURE STUDY DIRECTIONS
Predicting AF risk – an integrated view
There remain several major research questions to be addressed in the field of clinical integration of genetics of AF (Table). While increasingly sophisticated genomic approaches can identify patients at increased risk for AF, other factors – such as environmental influences or yet unidentified genomic variation – might enhance the value of predictive algorithms for AF. It is, for example, not yet established to what extent a strong family history predicts the development of AF independent of known genetic risk predictors; indeed, methods to capture and quantitate a “strong” family history have not been standardized.
Table.
Major questions to integrate AF genetics into clinical practice.
| Genetic Testing for AF |
| 1A. Who should be tested? 1B. How should age at AF diagnosis, clinical risk factors, and family history be factored? |
| 2. What genes should be sequenced on a panel for AF? |
| 3A. Should polygenic risk for AF be measured? 3B. What is the optimal polygenic risk score for AF (number and selection of SNPs)? |
| Genetic Subtypes of AF |
| 4A. What is the cardiac phenotype and natural history of individuals found to have a rare variant associated with AF? 4B. What is the penetrance and natural history of heart failure in patients who present with AF and are found to have a cardiomyopathy-associated rare variant? 4C. What is the penetrance and natural history of ventricular arrhythmias in those who present with AF and are found to have an arrhythmia-associated rare variant? |
| 5A. Which other AF susceptibility genes (e.g. those that are near AF GWAS loci but are not cardiomyopathy or arrhythmia genes), are monogenic causes of AF? 5B. Do unique cardiac and non-cardiac phenotypes exist for rare variant carriers in the other AF susceptibility genes? |
| 6A. What is the cardiac phenotype and natural history of individuals with high polygenic risk for AF? |
| Personalized AF Care using Genetics |
| 7. Should enhanced AF surveillance be used for individuals at high clinical/genetic risk for AF? |
| 8. Should empiric anticoagulation be used for stroke prophylaxis in individuals with a high CHADS-VASC score who are at high clinical/genetic risk for AF? |
| 9. Are there genetic subtypes of AF associated with arrhythmia recurrence after catheter ablation for AF? |
| 10. Are there genetic subtypes of AF associated with arrhythmia recurrence or adverse drug reactions with antiarrhythmic drug therapy? |
Clinical variables have also been used to predict the development of AF. The Cohorts for Heart and Aging Research in Genomic Epidemiology-Atrial Fibrillation (CHARGE-AF) consortium reported a risk prediction instrument using readily obtained variables from a series of epidemiologic studies.125 One appeal of this approach is that the variables could also be extracted from electronic health records (EHRs), and an EHR-based study in 33,494 subjects did show nearly identical performance characteristics compared to the CHARGE-AF epidemiologic cohort-derived performance (C-statistic 0.708 [CHARGE-AF] vs 0.709).126 A more recent study used 412,085 EHRs to derive a model that included sex, age, race, smoking, height, weight, diastolic blood pressure, hypertension, hyperlipidemia, heart failure, coronary heart disease, valvular disease, prior stroke, peripheral arterial disease, chronic kidney disease, hypothyroidism, and quadratic terms for height, weight, and age. Its performance (C-statistic: 0.777; 95% confidence interval [CI:] 0.771 to 0.783) was equivalent or superior to other models such as CHA2DS2-VASc or that proposed by CHARGE-AF,127 although some these variables may not be coded in all EHRs. Further, a UK biobank study found that a polygenic risk predictor and “poor health behaviors” (e.g. smoking, physical inactivity, diet) generated additive risk for incident AF, CAD, stroke, hypertension, and diabetes with no interaction between genomics and behaviors.128 A challenge in this area is that “poor health behaviors”, and other increasingly well-recognized predictors of AF such as sleep apnea, may not be readily captured in most current EHRs.
Thus, one opportunity that seems ripe for study is the optimization of tools like PRS, clinical risk factors, and family history to capture AF, develop methods to integrate these into validated risk predictors (Figure 7), and then to show that deploying them can identify patients with previously-undetected AF and prevent adverse AF-related outcomes. A key component of this vision is the development of robust methods to monitor for AF in high-risk subjects, discussed further below.129 Genomic tools including common variant-based PRS and rare variants would be a component of such risk prediction. Others might include family history, individual conventional risk factors and their combinations, newer clinical risk factors such as sleep apnea or inflammatory stimuli, and biomarkers. Validation would require studies in epidemiologic cohorts like FHS, Atherosclerosis Risk in Communities (ARIC), Women’s Health Initiative (WHI), the Multi-Ethnic Study of Atherosclerosis (MESA) and others, and very large epidemiologic or EHR-based cohorts, such as the UK biobank,130 the Million Veterans Program (MVP),131 the Electronic Medical Records and Genomics (eMERGE) network,132, 133 and the All of Us (AoU) Program.134 A major limitation of most genomic and other studies in this area to date is that they have been conducted primarily in European ancestry populations. The extent to which any predictive tool works well only in one ancestral population is a potential source of increasing healthcare disparities,135 so an early priority in future studies should be extending the knowledge base to all ancestries. Datasets like MVP, WHI and AoU have extensive and large non-EA populations. Further, population stratification even within major ancestral groups may influence the utility of PRS.136 It will be especially important to demonstrate and quantify added value of any algorithm that requires new resources (including genomics or new biomarkers), and to identify populations in which risk prediction is especially useful or not useful.
Figure 7. Proposed evaluation for early-onset AF that integrates family history and genetic risk assessment with traditional clinical risk assessment.
HF- heart failure. SCD- sudden cardiac death. DCM- dilated cardiomyopathy. RCTs- randomized controlled trials.
Several questions remain prior to considering implementation of PRS for the purpose of preventing AF-related outcomes such as strokes. First, what is the optimal genetic risk score? As already seen, this is likely an iterative process as newer and larger datasets continue to be introduced, but will PRS for stroke continue to be derived only from AF GWAS or can more specific scores be developed to identify subgroups of patients with AF at higher risk for stroke? For example, would a GWAS of AF patients with and without stroke further refine AF-related stroke risk? Second, what clinical interventions are recommended for individuals with a high genetic risk for AF? Risk of stroke in AF can be defined by integrating multiple clinical risk factors such as the CHADS2-VASC score.137 However, individuals with higher PRS for AF are known to be younger and have fewer cardiovascular comorbidities. Will PRS for AF primarily identify individuals who are at low overall risk for stroke (CHADS2-VASC=0 or 1) and for whom anticoagulation is not recommended even following detection of AF? If so, should secondary prevention of stroke be a major focus of future trials? Patients with cryptogenic stroke without a prior diagnosis of AF but a high genetic risk for AF may be started on anticoagulation due to a presumed cardioembolic source. Recent data to support this idea comes from a study that demonstrated polygenic risk for AF is associated with strokes determined to be from cardioembolic, but not other causes of stroke such as large artery atherosclerosis or small artery occlusion.138
There should also be a focus on genome-wide discovery efforts to identify individual common variants specifically associated with recurrence after ablation, and to explore genetic associations with intermediate phenotypes of AF mechanisms139–141 that can be targeted by specific ablation techniques (e.g. LA voltage, non-PV triggers, ganglionic plexi) ultimately moving the field towards a personalized approach to AF ablation and away from a one-size fits all approach relying solely on PV isolation with empiric adjunctive ablation. These are among some of the many questions to be addressed over the next 5 years, along with the logistical challenges of deploying polygenic risk models in the clinic and hospital setting.
Device-based prolonged monitoring of cardiac rhythm
A paradigm shift in AF is that direct-to-consumer technologies, such as smartwatches, are now capable of monitoring for AF. In the Apple Heart Study, 0.16% and 0.37% of participants in the 22–39 and the 40–54 year-old age groups were found to have an irregular pulse consistent with AF129. These younger consumers have thus far been the early adopters of these technologies and are therefore going to be increasingly exposed to AF monitoring with wearable devices and future technologies. The proportions of young participants found to have AF may remain small, but with screening at a population scale, there may be large numbers of young patients with newly diagnosed AF. While these patients may not be at high risk of stroke, they may be at risk for underlying cardiovascular disease. Future studies will need to examine the role of rare variant testing in young patients who present after detection with new digital health technologies and continue to study whether there is a threshold level of atrial arrhythmia that increases the risk of stroke, heart failure or other outcomes.
Another area ripe for investigation is the role of rare genetic variation or polygenic risk in early onset AF due to environmental stimuli such as alcohol consumption or endurance athleticism. As discussed above, truncating variants in TTN have now been associated with increased risk of peri-partum and post-chemotherapy cardiomyopathy.82, 83 Thus, future studies should explore the interaction between rare variation and environmental exposures in AF.
Role of Stem Cells and Gene Therapy in AF
Stem cell therapy, with delivery of stem cells into the myocardium, has demonstrated a modest clinical effect in the treatment of heart failure and has not yet been studied in AF. In 2012, the first FDA-approved gene therapy became available142 and several gene therapies, including those that use small interfering RNA, are signaling a new wave of therapeutics. Although animal studies of gene therapies for AF have shown early promise,143 none are yet available for the treatment of AF. These technologies need to overcome several hurdles, including improved gene delivery to myocardial tissue, high cost and minimization of unintended mutagenesis.
The use of human-derived stem cells to model “disease in a dish” along with genome editing has opened several possibilities for the future role of clinical genetics in AF. In LQTS, patient-derived induced pluripotent stem cells, in combination with gene editing tools, have been used to evaluate the pathogenicity of variants of unclear significance.144 Stem cell models have also been used to model response to antiarrhythmic drug therapy. More recently, human induced pluripotent stem cell-derived atrial myocytes have been used to model AF.145 While these models will need to be developed further, there are early examples of how patient-specific genetic information could be used to recreate an individual patient’s disease in a cellular model and use it to optimize therapy. Over the next few years, as stem cell models are improved and gene editing tools are further optimized, we will use these to better understand the role of rare variants in AF and perhaps someday use them to truly provide precision medical care.
SUMMARY
AF is common, and confers important risks including stroke, heart failure, and increased mortality. Thus, the major challenge to the field is to identify patients at risk before they present, with the goal of instating preventive therapies. Predictors of AF include a positive family history, specific comorbidities, and rare and common genetic variants. Within the next decade, the role of genetic testing will be further clarified and the increasingly widespread use of robust wearable technologies will enable large trials to identify patients with early AF. Preventive therapies may include currently available or new drugs targeting specific mechanisms such as inflammation, drugs to prevent stroke, or entirely new drug classes whose development will come from increased understanding of AF genetic, molecular and cellular mechanisms.
Supplementary Material
Sources of Funding
Grant Support
American Heart Association (18SFRN34110369; MBS, DMR), National Institute of Health (R01HL149826, DMR; 1R01HL136390, MVP; K23HL127704, MBS).
Disclosures
Grant Support from and Consultant for Apple Inc. (MVP); Consultant for Boehringer-Ingelheim (MVP); No disclosures reported for DMR, MBS and RLS.
References
- 1.Kirchhof P, Benussi S, Kotecha D, et al. 2016 ESC Guidelines for the Management of Atrial Fibrillation Developed in Collaboration With EACTS. Rev Esp Cardiol (Engl Ed). 2017;70:50. [DOI] [PubMed] [Google Scholar]
- 2.Manolio TA. Genomewide Association Studies and Assessment of the Risk of Disease. The New England Journal of Medicine. 2010;363:166–176. [DOI] [PubMed] [Google Scholar]
- 3.Gudbjartsson DF, Arnar DO, Helgadottir A, et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature. 2007;448:353–7. [DOI] [PubMed] [Google Scholar]
- 4.Helgadottir A, Thorleifsson G, Manolescu A, et al. A Common Variant on Chromosome 9p21 Affects the Risk of Myocardial Infarction. Science. 2007;316:1491–1493. [DOI] [PubMed] [Google Scholar]
- 5.Samani NJ, Erdmann J, Hall AS, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357:443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McPherson R, Pertsemlidis A, Kavaslar N, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arking DE, Pfeufer A, Post W, et al. A common genetic variant in the NOS1 regulator NOS1AP modulates cardiac repolarization. Nature genetics. 2006;38:644–51. [DOI] [PubMed] [Google Scholar]
- 8.Offit K Genomic profiles for disease risk: predictive or premature? Jama. 2008;299:1353–5. [DOI] [PubMed] [Google Scholar]
- 9.Crotti L, Monti MC, Insolia R, Peljto A, Goosen A, Brink PA, Greenberg DA, Schwartz PJ and George AL Jr. NOS1AP Is a Genetic Modifier of the Long-QT Syndrome. Circulation. 2009;120:1657–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tomas M, Napolitano C, De GL, et al. Polymorphisms in the NOS1AP gene modulate QT interval duration and risk of arrhythmias in the long QT syndrome. J Am Coll Cardiol. 2010;55:2745–2752. [DOI] [PubMed] [Google Scholar]
- 11.Kolder IC, Tanck MW, Postema PG, et al. Analysis for Genetic Modifiers of Disease Severity in Patients With Long-QT Syndrome Type 2. Circulation Cardiovascular genetics. 2015;8:447–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jamshidi Y, Nolte IM, Dalageorgou C, et al. Common variation in the NOS1AP gene is associated with drug-induced QT prolongation and ventricular arrhythmia. Journal of the American College of Cardiology. 2012;accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kao WH, Arking DE, Post W, et al. Genetic Variations in Nitric Oxide Synthase 1 Adaptor Protein Are Associated With Sudden Cardiac Death in US White Community-Based Populations. Circulation. 2009;119:940–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roselli C, Chaffin MD, Weng LC, et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nature genetics. 2018;50:1225–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huizar JF, Ellenbogen KA, Tan AY and Kaszala K. Arrhythmia-Induced Cardiomyopathy. J Am Coll Cardiol. 2019;73:2328–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orgain ES, Wolff L and White PD. Uncomplicated auricular fibrillation and auricular flutter: Frequent occurrence and good prognosis in patients without other evidence of cardiac disease. Archives of Internal Medicine. 1936;57:493–513. [Google Scholar]
- 17.Wolff L Familial Auricular Fibrillation. New England Journal of Medicine. 1943;229:396–398. [Google Scholar]
- 18.Fox CS, Parise H, D’Agostino RB Sr., Lloyd-Jones DM, Vasan RS, Wang TJ, Levy D, Wolf PA and Benjamin EJ. Parental Atrial Fibrillation as a Risk Factor for Atrial Fibrillation in Offspring. JAMA: The Journal of the American Medical Association. 2004;291:2851–2855. [DOI] [PubMed] [Google Scholar]
- 19.Ellinor PT, Yoerger DM, Ruskin JN and Macrae CA. Familial aggregation in lone atrial fibrillation. HumGenet. 2005;118:179–184. [DOI] [PubMed] [Google Scholar]
- 20.Darbar D, Herron KJ, Ballew JD, Jahangir A, Gersh BJ, Shen WK, Hammill SC, Packer DL and Olson TM. Familial atrial fibrillation is a genetically heterogeneous disorder. Journal of the American College of Cardiology. 2003;41:2185–2192. [DOI] [PubMed] [Google Scholar]
- 21.Weng LC, Choi SH, Klarin D, et al. Heritability of Atrial Fibrillation. Circulation Cardiovascular genetics. 2017;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lubitz SA, Yin X, Fontes JD, et al. Association between familial atrial fibrillation and risk of new-onset atrial fibrillation. Jama. 2010;304:2263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lubitz SA, Sinner MF, Lunetta KL, et al. Independent susceptibility markers for atrial fibrillation on chromosome 4q25. Circulation. 2010;122:976–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lubitz SA, Lunetta KL, Lin H, et al. Novel genetic markers associate with atrial fibrillation risk in europeans and Japanese. J Am Coll Cardiol. 2014;63:1200–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lubitz SA, Yin X, Lin HJ, et al. Genetic Risk Prediction of Atrial Fibrillation. Circulation. 2017;135:1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khera AV, Chaffin M, Aragam KG, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nature genetics. 2018;50:1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ueberham L, Bollmann A, Shoemaker MB, Arya A, Adams V, Hindricks G and Husser D. Genetic ACE I/D polymorphism and recurrence of atrial fibrillation after catheter ablation. Circulation Arrhythmia and electrophysiology. 2013;6:732–7. [DOI] [PubMed] [Google Scholar]
- 28.Wu G, Cheng M, Huang H, Yang B, Jiang H and Huang C. A variant of IL6R is associated with the recurrence of atrial fibrillation after catheter ablation in a Chinese Han population. PLoS One. 2014;9:e99623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Husser D, Adams V, Piorkowski C, Hindricks G and Bollmann A. Chromosome 4q25 variants and atrial fibrillation recurrence after catheter ablation. J Am Coll Cardiol. 2010;55:747–753. [DOI] [PubMed] [Google Scholar]
- 30.Shoemaker MB, Muhammad R, Parvez B, et al. Common atrial fibrillation risk alleles at 4q25 predict recurrence after catheter-based atrial fibrillation ablation. Heart rhythm : the official journal of the Heart Rhythm Society. 2013;10:394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shoemaker MB, Bollmann A, Lubitz SA, et al. Common Genetic Variants and Response to Atrial Fibrillation Ablation. Circulation: Arrhythmia and Electrophysiology. 2015;8:296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi EK, Park JH, Lee JY, et al. Korean Atrial Fibrillation (AF) Network: Genetic Variants for AF Do Not Predict Ablation Success. J Am Heart Assoc. 2015;4:e002046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gretarsdottir S, Thorleifsson G, Manolescu A, et al. Risk variants for atrial fibrillation on chromosome 4q25 associate with ischemic stroke. Ann Neurol. 2008;64:402–9. [DOI] [PubMed] [Google Scholar]
- 34.Gudbjartsson DF, Holm H, Gretarsdottir S, et al. A sequence variant in ZFHX3 on 16q22 associates with atrial fibrillation and ischemic stroke. Nat Genet. 2009;41:876–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tada H, Shiffman D, Smith JG, et al. Twelve-single nucleotide polymorphism genetic risk score identifies individuals at increased risk for future atrial fibrillation and stroke. Stroke. 2014;45:2856–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weng LC, Preis SR, Hulme OL, et al. Genetic Predisposition, Clinical Risk Factor Burden, and Lifetime Risk of Atrial Fibrillation. Circulation. 2018;137:1027–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calkins H, Hindricks G, Cappato R, et al. 2017 HRS/EHRA/ECAS/APHRS/SOLAECE expert consensus statement on catheter and surgical ablation of atrial fibrillation: Executive summary. Heart Rhythm. 2017;14:e445–e494. [DOI] [PubMed] [Google Scholar]
- 38.Al-Hijji MA, Deshmukh AJ, Yao X, et al. Trends and predictors of repeat catheter ablation for atrial fibrillation. Am Heart J. 2016;171:48–55. [DOI] [PubMed] [Google Scholar]
- 39.Husser D, Adams V, Piorkowski C, Hindricks G and Bollmann A. Chromosome 4q25 variants and atrial fibrillation recurrence after catheter ablation. J Am Coll Cardiol. 2010;55:747–53. [DOI] [PubMed] [Google Scholar]
- 40.Shoemaker MB, Bollmann A, Lubitz SA, et al. Common genetic variants and response to atrial fibrillation ablation. Circ Arrhythm Electrophysiol. 2015;8:296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benjamin Shoemaker M, Muhammad R, Parvez B, et al. Common atrial fibrillation risk alleles at 4q25 predict recurrence after catheter-based atrial fibrillation ablation. Heart Rhythm. 2013;10:394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen F, Yang Y, Zhang R, et al. Polymorphism rs2200733 at chromosome 4q25 is associated with atrial fibrillation recurrence after radiofrequency catheter ablation in the Chinese Han population. Am J Transl Res. 2016;8:688–97. [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao LQ, Zhang GB, Wen ZJ, et al. Common variants predict recurrence after nonfamilial atrial fibrillation ablation in Chinese Han population. Int J Cardiol. 2017;227:360–366. [DOI] [PubMed] [Google Scholar]
- 44.Miyazaki S, Ebana Y, Liu L, et al. Chromosome 4q25 variants and recurrence after second-generation cryoballoon ablation in patients with paroxysmal atrial fibrillation. Int J Cardiol. 2017;244:151–157. [DOI] [PubMed] [Google Scholar]
- 45.Rattanawong P, Chenbhanich J, Vutthikraivit W and Chongsathidkiet P. A Chromosome 4q25 Variant is Associated with Atrial Fibrillation Recurrence After Catheter Ablation: A Systematic Review and Meta-Analysis. J Atr Fibrillation. 2018;10:1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mommersteeg MT, Brown NA, Prall OW, de Gier-de Vries C, Harvey RP, Moorman AF and Christoffels VM. Pitx2c and Nkx2–5 are required for the formation and identity of the pulmonary myocardium. Circ Res. 2007;101:902–9. [DOI] [PubMed] [Google Scholar]
- 47.Parvez B, Chopra N, Rowan S, Vaglio JC, Muhammad R, Roden DM and Darbar D. A common beta1-adrenergic receptor polymorphism predicts favorable response to rate-control therapy in atrial fibrillation. J Am Coll Cardiol. 2012;59:49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liggett SB, Mialet-Perez J, Thaneemit-Chen S, et al. A polymorphism within a conserved {beta}1-adrenergic receptor motif alters cardiac function and {beta}-blocker response in human heart failure. Proceedings of the National Academy of Sciences. 2006;103:11288–11293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aleong RG, Sauer WH, Murphy GA, et al. Prevention of atrial fibrillation by bucindolol is dependent on the beta(1)389 Arg/Gly adrenergic receptor polymorphism. JACC Heart failure. 2013;1:338–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Piccini JP, Connolly SJ, Abraham WT, et al. A genotype-directed comparative effectiveness trial of Bucindolol and metoprolol succinate for prevention of symptomatic atrial fibrillation/atrial flutter in patients with heart failure: Rationale and design of the GENETIC-AF trial. Am Heart J. 2018;199:51–58. [DOI] [PubMed] [Google Scholar]
- 51.Piccini JP, Abraham WT, Dufton C, et al. Bucindolol for the Maintenance of Sinus Rhythm in a Genotype-Defined HF Population: The GENETIC-AF Trial. JACC Heart Fail. 2019;7:586–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parvez B, Vaglio J, Rowan S, Muhammad R, Kucera G, Stubblefield T, Carter S, Roden D and Darbar D. Symptomatic Response to Antiarrhythmic Drug Therapy Is Modulated by a Common Single Nucleotide Polymorphism in Atrial Fibrillation. J Am Coll Cardiol. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parvez B, Shoemaker MB, Muhammad R, Richardson R, Jiang L, Blair MA, Roden DM and Darbar D. Common genetic polymorphism at 4q25 locus predicts atrial fibrillation recurrence after successful cardioversion. Heart Rhythm. 2013;10:849–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lahtinen AM, Noseworthy PA, Havulinna AS, et al. Common genetic variants associated with sudden cardiac death: the FinSCDgen study. PloS one. 2012;7:e41675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Briasoulis A, Sharma S, Telila T, Mallikethi-Reddy S, Papageorgiou N, Oikonomou E and Tousoulis D. MicroRNAs in Atrial Fibrillation. Current medicinal chemistry. 2019;26:855–863. [DOI] [PubMed] [Google Scholar]
- 56.Ko D, Benson MD, Ngo D, et al. Proteomics Profiling and Risk of New-Onset Atrial Fibrillation: Framingham Heart Study. J Am Heart Assoc. 2019;8:e010976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang TJ, Larson MG, Levy D, Benjamin EJ, Leip EP, Omland T, Wolf PA and Vasan RS. Plasma natriuretic peptide levels and the risk of cardiovascular events and death. N Engl J Med. 2004;350:655–63. [DOI] [PubMed] [Google Scholar]
- 58.Patton KK, Ellinor PT, Heckbert SR, Christenson RH, DeFilippi C, Gottdiener JS and Kronmal RA. N-terminal pro-B-type natriuretic peptide is a major predictor of the development of atrial fibrillation: the Cardiovascular Health Study. Circulation. 2009;120:1768–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kurl S, Ala-Kopsala M, Ruskoaho H, Makikallio T, Nyyssonen K, Vuolteenaho O, Sivenius J, Salonen JT and Laukkanen JA. Plasma N-terminal fragments of natriuretic peptides predict the risk of stroke and atrial fibrillation in men. Heart. 2009;95:1067–71. [DOI] [PubMed] [Google Scholar]
- 60.Sinner MF, Stepas KA, Moser CB, et al. B-type natriuretic peptide and C-reactive protein in the prediction of atrial fibrillation risk: the CHARGE-AF Consortium of community-based cohort studies. Europace. 2014;16:1426–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wells QS, Gupta DK, Smith JG, et al. Accelerating Biomarker Discovery Through Electronic Health Records, Automated Biobanking, and Proteomics. J Am Coll Cardiol. 2019;73:2195–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mosley JD, Benson MD, Smith JG, et al. Probing the Virtual Proteome to Identify Novel Disease Biomarkers. Circulation. 2018;138:2469–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Salem JE, Shoemaker MB, Bastarache L, et al. Association of Thyroid Function Genetic Predictors With Atrial Fibrillation: A Phenome-Wide Association Study and Inverse-Variance Weighted Average Meta-analysis. JAMA cardiology. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ellervik C, Roselli C, Christophersen IE, et al. Assessment of the Relationship Between Genetic Determinants of Thyroid Function and Atrial Fibrillation: A Mendelian Randomization Study. JAMA Cardiol. 2019;4:144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen YH, Xu SJ, Bendahhou S, et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. 2003;299:251–254. [DOI] [PubMed] [Google Scholar]
- 66.Hodgson-Zingman DM, Karst ML, Zingman LV, et al. Atrial Natriuretic Peptide Frameshift Mutation in Familial Atrial Fibrillation. The New England Journal of Medicine. 2008;359:158–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Orr N, Arnaout R, Gula LJ, et al. A mutation in the atrial-specific myosin light chain gene (MYL4) causes familial atrial fibrillation. Nat Commun. 2016;7:11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhao J, Yao H, Li Z, Wang L, Liu G, Wang DW, Wang DW and Liang Z. A novel nonsense mutation in LMNA gene identified by Exome Sequencing in an atrial fibrillation family. European journal of medical genetics. 2016;59:396–400. [DOI] [PubMed] [Google Scholar]
- 69.Fatkin D, MacRae C, Sasaki T, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715–24. [DOI] [PubMed] [Google Scholar]
- 70.Lieve KV, Verkerk AO, Podliesna S, et al. Gain-of-function mutation in SCN5A causes ventricular arrhythmias and early onset atrial fibrillation. International journal of cardiology. 2017;236:187–193. [DOI] [PubMed] [Google Scholar]
- 71.Olson TM, Michels VV, Ballew JD, Reyna SP, Karst ML, Herron KJ, Horton SC, Rodeheffer RJ and Anderson JL. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. Journal of the American Medical Association. 2005;293:447–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tucker NR, Mahida S, Ye J, et al. Gain-of-function mutations in GATA6 lead to atrial fibrillation. Heart rhythm : the official journal of the Heart Rhythm Society. 2017;14:284–291. [DOI] [PubMed] [Google Scholar]
- 73.Zhang X, Chen S, Yoo S, et al. Mutation in nuclear pore component NUP155 leads to atrial fibrillation and early sudden cardiac death. Cell. 2008;135:1017–1027. [DOI] [PubMed] [Google Scholar]
- 74.Fatkin D, MacRae C, Sasaki T, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. New England Journal of Medicine. 1999;341:1715–1724. [DOI] [PubMed] [Google Scholar]
- 75.Hoffmann S, Paone C, Sumer SA, et al. Functional Characterization of Rare Variants in the SHOX2 Gene Identified in Sinus Node Dysfunction and Atrial Fibrillation. Frontiers in genetics. 2019;10:648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang Y, Xia M, Jin Q, et al. Identification of a KCNE2 Gain-of-Function Mutation in Patients with Familial Atrial Fibrillation. AmJ HumGenet. 2004;75:899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hosseini SM, Kim R, Udupa S, et al. Reappraisal of Reported Genes for Sudden Arrhythmic Death. Circulation. 2018;138:1195–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ingles J, Goldstein J, Thaxton C, et al. Evaluating the Clinical Validity of Hypertrophic Cardiomyopathy Genes. Circ Genom Precis Med. 2019;12:e002460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Adler A, Novelli V, Amin AS, et al. An International, Multicentered, Evidence-Based Reappraisal of Genes Reported to Cause Congenital Long QT Syndrome. Circulation. 2020;141:418–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Herman DS, Lam L, Taylor MRG, et al. Truncations of Titin Causing Dilated Cardiomyopathy. New England Journal of Medicine. 2012;366:619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Norton N, Li D, Rampersaud E, et al. Exome sequencing and genome-wide linkage analysis in 17 families illustrate the complex contribution of TTN truncating variants to dilated cardiomyopathy. Circulation Cardiovascular genetics. 2013;6:144–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ware JS, Li J, Mazaika E, et al. Shared Genetic Predisposition in Peripartum and Dilated Cardiomyopathies. N Engl J Med. 2016;374:233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Garcia-Pavia P, Kim Y, Restrepo-Cordoba MA, et al. Genetic Variants Associated With Cancer Therapy-Induced Cardiomyopathy. Circulation. 2019;140:31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Choi SH, Weng LC, Roselli C, et al. Association Between Titin Loss-of-Function Variants and Early-Onset Atrial Fibrillation. JAMA. 2018;320:2354–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Refaat MM, Lubitz SA, Makino S, et al. Genetic variation in the alternative splicing regulator RBM20 is associated with dilated cardiomyopathy. Heart rhythm : the official journal of the Heart Rhythm Society. 2012;9:390–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Goodyer WR, Dunn K, Caleshu C, et al. Broad Genetic Testing in a Clinical Setting Uncovers a High Prevalence of Titin Loss-of-Function Variants in Very Early Onset Atrial Fibrillation. Circ Genom Precis Med. 2019;12:e002713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wolf PA, Mitchell JB, Baker CS, Kannel WB and D’Agostino RB. Impact of atrial fibrillation on mortality, stroke, and medical costs. Arch Intern Med. 1998;158:229–34. [DOI] [PubMed] [Google Scholar]
- 88.Sankaranarayanan R, Kirkwood G, Dibb K and Garratt CJ. Comparison of Atrial Fibrillation in the Young versus That in the Elderly: A Review. Cardiology research and practice. 2013;2013:976976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Husser D, Ueberham L, Hindricks G, et al. Rare variants in genes encoding the cardiac sodium channel and associated compounds and their impact on outcome of catheter ablation of atrial fibrillation. PLoS One. 2017;12:e0183690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Giustetto C, Di Monte F, Wolpert C, et al. Short QT syndrome: clinical findings and diagnostic-therapeutic implications. Eur Heart J. 2006;27:2440–7. [DOI] [PubMed] [Google Scholar]
- 91.Johnson JN, Tester DJ, Perry J, Salisbury BA, Reed CR and Ackerman MJ. Prevalence of early-onset atrial fibrillation in congenital long QT syndrome. Heart Rhythm. 2008;5:704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bordachar P, Reuter S, Garrigue S, Cai X, Hocini M, Jais P, Haissaguerre M and Clementy J. Incidence, clinical implications and prognosis of atrial arrhythmias in Brugada syndrome. Eur Heart J. 2004;25:879–84. [DOI] [PubMed] [Google Scholar]
- 93.Bigi MA, Aslani A and Shahrzad S. Clinical predictors of atrial fibrillation in Brugada syndrome. Europace. 2007;9:947–50. [DOI] [PubMed] [Google Scholar]
- 94.Rowin EJ, Hausvater A, Link MS, et al. Clinical Profile and Consequences of Atrial Fibrillation in Hypertrophic Cardiomyopathy. Circulation. 2017;136:2420–2436. [DOI] [PubMed] [Google Scholar]
- 95.Guttmann OP, Rahman MS, O’Mahony C, Anastasakis A and Elliott PM. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart (British Cardiac Society). 2014;100:465–72. [DOI] [PubMed] [Google Scholar]
- 96.Lee SP, Ashley EA, Homburger J, et al. Incident Atrial Fibrillation Is Associated With MYH7 Sarcomeric Gene Variation in Hypertrophic Cardiomyopathy. Circ Heart Fail. 2018;11:e005191. [DOI] [PubMed] [Google Scholar]
- 97.Azarbal F, Singh M, Finocchiaro G, Le VV, Schnittger I, Wang P, Myers J, Ashley E and Perez M. Exercise capacity and paroxysmal atrial fibrillation in patients with hypertrophic cardiomyopathy. Heart. 2014;100:624–30. [DOI] [PubMed] [Google Scholar]
- 98.Maron BJ, Casey SA, Poliac LC, Gohman TE, Almquist AK and Aeppli DM. Clinical course of hypertrophic cardiomyopathy in a regional United States cohort. JAMA : the journal of the American Medical Association. 1999;281:650–5. [DOI] [PubMed] [Google Scholar]
- 99.Siontis KC, Geske JB, Ong K, Nishimura RA, Ommen SR and Gersh BJ. Atrial fibrillation in hypertrophic cardiomyopathy: prevalence, clinical correlations, and mortality in a large high-risk population. Journal of the American Heart Association. 2014;3:e001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Camm CF, James CA, Tichnell C, Murray B, Bhonsale A, te Riele AS, Judge DP, Tandri H and Calkins H. Prevalence of atrial arrhythmias in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Heart Rhythm. 2013;10:1661–8. [DOI] [PubMed] [Google Scholar]
- 101.Saguner AM, Ganahl S, Kraus A, et al. Clinical role of atrial arrhythmias in patients with arrhythmogenic right ventricular dysplasia. Circulation journal : official journal of the Japanese Circulation Society. 2014;78:2854–61. [DOI] [PubMed] [Google Scholar]
- 102.Stollberger C, Blazek G, Winkler-Dworak M and Finsterer J. Atrial fibrillation in left ventricular noncompaction with and without neuromuscular disorders is associated with a poor prognosis. International journal of cardiology. 2009;133:41–5. [DOI] [PubMed] [Google Scholar]
- 103.Li S, Zhang C, Liu N, Bai H, Hou C and Pu J. Clinical implications of sarcomere and nonsarcomere gene variants in patients with left ventricular noncompaction cardiomyopathy. Mol Genet Genomic Med. 2019;7:e874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Brembilla-Perrot B, Schwartz J, Huttin O, et al. Atrial flutter or fibrillation is the most frequent and life-threatening arrhythmia in myotonic dystrophy. Pacing and clinical electrophysiology : PACE. 2014;37:329–35. [DOI] [PubMed] [Google Scholar]
- 105.Russo V, Rago A, Ciardiello C, Russo MG, Calabro P, Politano L and Nigro G. The Role of the Atrial Electromechanical Delay in Predicting Atrial Fibrillation in Myotonic Dystrophy Type 1 Patients. J Cardiovasc Electrophysiol. 2016;27:65–72. [DOI] [PubMed] [Google Scholar]
- 106.Choi SH, Jurgens SJ, Weng LC, et al. Monogenic and Polygenic Contributions to Atrial Fibrillation Risk: Results from a National Biobank. Circ Res. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ritchie MD, Rowan S, Kucera G, Stubblefield T, Blair M, Carter S, Roden DM and Darbar D. Chromosome 4q25 variants are genetic modifiers of rare ion channel mutations associated with familial atrial fibrillation. J Am Coll Cardiol. 2012;60:1173–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. [DOI] [PubMed] [Google Scholar]
- 109.Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science. 2001;291:1304–51. [DOI] [PubMed] [Google Scholar]
- 110.Priori SG, Wilde AA, Horie M, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart rhythm : the official journal of the Heart Rhythm Society. 2013;10:1932–63. [DOI] [PubMed] [Google Scholar]
- 111.Perez MV, Kumarasamy NA, Owens DK, Wang PJ and Hlatky MA. Cost-effectiveness of genetic testing in family members of patients with long-QT syndrome. Circ Cardiovasc Qual Outcomes. 2011;4:76–84. [DOI] [PubMed] [Google Scholar]
- 112.Olesen MS, Andreasen L, Jabbari J, Refsgaard L, Haunso S, Olesen SP, Nielsen JB, Schmitt N and Svendsen JH. Very early-onset lone atrial fibrillation patients have a high prevalence of rare variants in genes previously associated with atrial fibrillation. Heart rhythm : the official journal of the Heart Rhythm Society. 2014;11:246–51. [DOI] [PubMed] [Google Scholar]
- 113.Hayashi K, Konno T, Tada H, et al. Functional Characterization of Rare Variants Implicated in Susceptibility to Lone Atrial Fibrillation. Circ Arrhythm Electrophysiol. 2015;8:1095–104. [DOI] [PubMed] [Google Scholar]
- 114.Moscarello T and Caleshu C. A review of the Emergence and Expansion of Cardiovascular Genetic Counseling. C Curr Cardiovasc Risk Rep. 2019;13. [Google Scholar]
- 115.Takemura Y, Sakurai Y, Yokoya S, Otaki J, Matsuoka T, Ban N, Hirata I, Miki T and Tsuda T. Open-ended questions: are they really beneficial for gathering medical information from patients? Tohoku J Exp Med. 2005;206:151–4. [DOI] [PubMed] [Google Scholar]
- 116.Vetto JT, Richert-Boe K, Desler M, DuFrain L and Hagen H. Tumor board formats: “fascinating case” versus “working conference”. J Cancer Educ. 1996;11:84–8. [DOI] [PubMed] [Google Scholar]
- 117.Manolio TA, Rowley R, Williams MS, et al. Opportunities, resources, and techniques for implementing genomics in clinical care. Lancet. 2019;394:511–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bouchardy J, Therrien J, Pilote L, Ionescu-Ittu R, Martucci G, Bottega N and Marelli AJ. Atrial arrhythmias in adults with congenital heart disease. Circulation. 2009;120:1679–86. [DOI] [PubMed] [Google Scholar]
- 119.Schwieler JH, Zlochiver S, Pandit SV, Berenfeld O, Jalife J and Bergfeldt L. Reentry in an accessory atrioventricular pathway as a trigger for atrial fibrillation initiation in manifest Wolff-Parkinson-White syndrome: a matter of reflection? Heart rhythm : the official journal of the Heart Rhythm Society. 2008;5:1238–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lowenstein SR, Gabow PA, Cramer J, Oliva PB and Ratner K. The role of alcohol in new-onset atrial fibrillation. Arch Intern Med. 1983;143:1882–5. [PubMed] [Google Scholar]
- 121.Elosua R, Arquer A, Mont L, Sambola A, Molina L, Garcia-Moran E, Brugada J and Marrugat J. Sport practice and the risk of lone atrial fibrillation: a case-control study. Int J Cardiol. 2006;108:332–7. [DOI] [PubMed] [Google Scholar]
- 122.Mont L, Sambola A, Brugada J, Vacca M, Marrugat J, Elosua R, Pare C, Azqueta M and Sanz G. Long-lasting sport practice and lone atrial fibrillation. Eur Heart J. 2002;23:477–82. [DOI] [PubMed] [Google Scholar]
- 123.Gill J, Obley AJ and Prasad V. Direct-to-Consumer Genetic Testing: The Implications of the US FDA’s First Marketing Authorization for BRCA Mutation Testing. Jama. 2018;319:2377–2378. [DOI] [PubMed] [Google Scholar]
- 124.Go AS, Hylek EM, Phillips KA, Chang Y, Henault LE, Selby JV and Singer DE. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. Jama. 2001;285:2370–5. [DOI] [PubMed] [Google Scholar]
- 125.Alonso A, Krijthe BP, Aspelund T, et al. Simple risk model predicts incidence of atrial fibrillation in a racially and geographically diverse population: the CHARGE-AF consortium. J Am Heart Assoc. 2013;2:e000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kolek MJ, Graves AJ, Xu M, et al. Evaluation of a Prediction Model for the Development of Atrial Fibrillation in a Repository of Electronic Medical Records. JAMA cardiology. 2016;1:1007–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hulme OL, Khurshid S, Weng LC, et al. Development and Validation of a Prediction Model for Atrial Fibrillation Using Electronic Health Records. JACC Clin Electrophysiol. 2019;5:1331–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Said MA, Verweij N and van der Harst P. Associations of Combined Genetic and Lifestyle Risks With Incident Cardiovascular Disease and Diabetes in the UK Biobank Study. JAMA cardiology. 2018;3:693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Perez MV, Mahaffey KW, Hedlin H, et al. Large-Scale Assessment of a Smartwatch to Identify Atrial Fibrillation. N Engl J Med. 2019;381:1909–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Allen NE, Sudlow C, Peakman T, Collins R and Biobank oboU. UK Biobank Data: Come and Get It. Science Translational Medicine. 2014;6:224ed4. [DOI] [PubMed] [Google Scholar]
- 131.Gaziano JM, Concato J, Brophy M, et al. Million Veteran Program: A mega-biobank to study genetic influences on health and disease. Journal of clinical epidemiology. 2016;70:214–23. [DOI] [PubMed] [Google Scholar]
- 132.Gottesman O, Kuivaniemi H, Tromp G, et al. The Electronic Medical Records and Genomics (eMERGE) Network: past, present, and future. Genetics in medicine : official journal of the American College of Medical Genetics. 2013;15:761–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.eMERGE. Harmonizing clinical sequencing and interpretation for the EMERGE III Network. American Journal of Human Genetics. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.The “All of Us” Research Program. New England Journal of Medicine. 2019;381:668–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM and Daly MJ. Clinical use of current polygenic risk scores may exacerbate health disparities. Nature genetics. 2019;51:584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mostafavi H, Harpak A, Conley D, Pritchard JK and Przeworski M. Variable prediction accuracy of polygenic scores within an ancestry group. bioRxiv. 2019:629949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lip GY, Nieuwlaat R, Pisters R, Lane DA and Crijns HJ. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach: the euro heart survey on atrial fibrillation. Chest. 2010;137:263–72. [DOI] [PubMed] [Google Scholar]
- 138.Pulit SL, Weng LC, McArdle PF, et al. Atrial fibrillation genetic risk differentiates cardioembolic stroke from other stroke subtypes. Neurol Genet. 2018;4:e293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ebana Y, Nitta J, Takahashi Y, et al. Association of the Clinical and Genetic Factors With Superior Vena Cava Arrhythmogenicity in Atrial Fibrillation. Circ J. 2017;82:71–77. [DOI] [PubMed] [Google Scholar]
- 140.Husser D, Ueberham L, Dinov B, et al. Genomic contributors to atrial electroanatomical remodeling and atrial fibrillation progression: Pathway enrichment analysis of GWAS data. Sci Rep. 2016;6:36630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mohanty S, Hall AW, Mohanty P, et al. Novel association of polymorphic genetic variants with predictors of outcome of catheter ablation in atrial fibrillation: new directions from a prospective study (DECAF). J Interv Card Electrophysiol. 2016;45:7–17. [DOI] [PubMed] [Google Scholar]
- 142.Gaudet D, Methot J, Dery S, et al. Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPLS447X) gene therapy for lipoprotein lipase deficiency: an open-label trial. Gene Ther. 2013;20:361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Amit G, Kikuchi K, Greener ID, Yang L, Novack V and Donahue JK. Selective molecular potassium channel blockade prevents atrial fibrillation. Circulation. 2010;121:2263–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Garg P, Oikonomopoulos A, Chen H, et al. Genome Editing of Induced Pluripotent Stem Cells to Decipher Cardiac Channelopathy Variant. J Am Coll Cardiol. 2018;72:62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Argenziano M, Lambers E, Hong L, et al. Electrophysiologic Characterization of Calcium Handling in Human Induced Pluripotent Stem Cell-Derived Atrial Cardiomyocytes. Stem Cell Reports. 2018;10:1867–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chen YH, Xu SJ, Bendahhou S, et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. 2003;299:251–4. [DOI] [PubMed] [Google Scholar]
- 147.Zhang X, Chen S, Yoo S, et al. Mutation in nuclear pore component NUP155 leads to atrial fibrillation and early sudden cardiac death. Cell. 2008;135:1017–27. [DOI] [PubMed] [Google Scholar]
- 148.Olson TM, Michels VV, Ballew JD, Reyna SP, Karst ML, Herron KJ, Horton SC, Rodeheffer RJ and Anderson JL. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. Jama. 2005;293:447–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hodgson-Zingman DM, Karst ML, Zingman LV, et al. Atrial natriuretic peptide frameshift mutation in familial atrial fibrillation. N Engl J Med. 2008;359:158–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.