

Abstract
Sepsis and septic shock driven by microbial infections are still among the most challenging health problems, causing 11 million deaths worldwide every year. How does the host’s response to pathogen infections effectively restore homeostasis instead of precipitating pathogenic and potentially fatal feedforward reactions? Recently there have been significant new advances in our understanding of the interface between mammalian immunity and coagulation (“immunocoagulation”) and its impact on sepsis. In particular, the release and activation of F3 (the main initiator of coagulation) from and on myeloid or epithelial cells is facilitated by activating inflammasomes and consequent GSDMD-mediated pyroptosis, coupled to signaling via HMGB1, STING1 or SQSTM1. Pharmacological modulation of the immunocoagulation pathways emerges as novel and potential therapeutic strategies for sepsis.
Medical challenge: sepsis and septic shock
Historically, the term sepsis is derived from the Greek “sepo”, meaning “I rot”. Two thousand years ago, Hippocrates used the word ‘sepidon’ to denote “a potentially dangerous biological decay that may occur in the body”. Since 1991, the definition of sepsis has been revised three times by the international consensus conference, providing clinicians with a useful diagnostic framework and emphasizing the need for early identification of underlying causes. According to the last definition termed ‘Sepsis 3.0’ (BOX1) [1], sepsis is a life-threatening organ dysfunction stemming from an impaired host response to infection that can be bacterial, fungal, protozoal, or viral in origin, including the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) responsible for coronavirus disease 2019 (COVID-19) [2]. Sepsis is still a serious global health problem and the most common cause of death in hospitals [3]. In 2017, there were 48.9 million cases and 11 million deaths from sepsis worldwide, accounting for 19.7% of all global deaths [3]. The World Health Organization recently urged member states to improve the prevention, diagnosis, and clinical management of sepsis [4]. However, the prerequisite for achieving these goals is to better understand the underlying mechanisms leading to sepsis. Sepsis is a biphasic disease characterized by overwhelming inflammation in its initial stage, followed by immunosuppression. Although the pathophysiology of sepsis is complex, disseminated intravascular coagulation (DIC) caused by the host immune response is associated with disease severity and mortality [5]. Normally, this so-called immunocoagulation-- describing the crosstalk between inflammation and coagulation-- is part of innate immunity and can serve as a first line of defense against infection. In this review, we delineate recently described molecular mechanisms of immunocoagulation underlying pathogen infections, but also highlight novel candidate therapeutic targets and agents that might help treat sepsis.
BOX1. New definitions and diagnosis: Sepsis 3.0.
The International Consensus Definition of Sepsis and Septic Shock (Sepsis 3.0) released in 2016 (https://jamanetwork.com/journals/jama/fullarticle/2492881) represents a fundamental difference from the previous definition of sepsis in 1991 (Sepsis 1.0) and 2001 (Sepsis 2.0). The Sepsis 3.0 consensus abandons the concept of sepsis called systemic inflammatory response syndrome (SIRS) due to infection, which has become the diagnostic standard in Sepsis 2.0. Instead, it states: “Sepsis should be defined as life-threatening organ dysfunction caused by a dysregulated host response to infection (suspected or confirmed)” [1]. The updated clinical criteria for sepsis in Sepsis 3.0 that was derived and retrospectively validated in a cohort of 148,907 patients emphasizes organ dysfunction in the presence of infection, which can be quantified using the sequential organ failure assessment (SOFA) score. SOFA uses objective measurement results to classify the functions of 6 organ systems (respiratory, cardiovascular, liver, renal, coagulation, and neurologic variables) from 0 to 4 based on the degree of dysfunction. The organ dysfunction of sepsis is defined as an increase in SOFA score of 2 points or more. The increase in SOFA score is associated with an increase in septic death. The Sepsis 3.0 consensus also abandoned the use of “severe sepsis”, that is, sepsis with acute organ dysfunction. It is recommended to use only “sepsis” and “septic shock” to describe the severity of infection affecting organ function. Septic shock is defined as “hypotension that does not respond to fluid resuscitation” with the added requirement for vasopressors to maintain mean arterial pressure ≥65 mm Hg and lactate > 2 mmol/L. The new definition of septic shock highlights hyperlactatemia, suggesting that glucose metabolism disorders may be an important pathogenesis of sepsis. The purpose of revising these new definitions and standards in Sepsis 3.0 is to promote earlier recognition and more timely treatment of patients with sepsis or at risk of developing sepsis, although they may not be able to identify severely infected patients before organ dysfunction occurs.
Gemini twins: inflammation and coagulation
Inflammation is triggered when innate immune cells (e.g., macrophages, monocytes, and neutrophils) recognize pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs) through germline-encoded pattern recognition receptors (PRRs) during infection or tissue damage (BOX2). A fine-tuned inflammatory response helps to eradicate microorganisms and repair tissue damages, while unrestricted inflammatory activation often leads to diseases, such as inflammatory bowel disease [6]. During infection in mammals, sepsis is characterized by an aggravated initial inflammatory response or cytokine storm, which is integrally linked to excessive pro-coagulant and impaired fibrinolytic responses [7]. These cytokines trigger not only the activation of coagulation factors and platelets, but also the downregulation of anticoagulant mechanisms (e.g., the antithrombin system, the activated protein C system, and tissue factor pathway inhibitor), thereby causing vascular leakage, DIC, and extensive deposition of fibrin in blood vessels or tissues [7]. In the later stages of sepsis, apoptosis of lymphocytes and dendritic cells (DCs) can lead to immune paralysis by reducing the host’s ability to clear invading microorganisms and increasing the susceptibility to secondary infections [8, 9]. As a driver of multiple organ failure in sepsis, DIC is characterized by extensive microvascular thrombosis, followed by consumption of platelets and coagulation factors, and eventually, a bleeding diathesis [7]. Clinical observations and animal (mice or rats) studies to simulate sepsis (especially using lipopolysaccharides [LPS, a constituent of the outer membrane of the cell wall of gram-negative bacteria] to induce endotoxemia, or the cecal ligation and puncture [CLP]-mediated polymicrobial sepsis model) have demonstrated that pathogen infections lead to an extensive feedback loop between inflammation and coagulation pathways in immune and endothelial cells by activating cytokine storm or causing cell death [10–13].
BOX2. Danger triangle: PAMPs, DAMPs, and PRRs in mammals.
There are two types of danger signals (also known as alarms or the signal 0s), which can be recognized by the pattern recognition receptor (PRR) to trigger the innate immune response in the body [101]. The type I is pathogen-associated molecular patterns (PAMPs), which are structural components or metabolites produced by microorganisms. A well-known PAMP is lipopolysaccharide (LPS), which is located on the outer cell wall of Gram-negative bacteria. Gram-negative bacterial infections account for 30% of blood infections in the hospital Intensive Care Unit and are the main pathogenic bacteria leading to sepsis globally [102]. Extracellular and intracellular LPS are specifically recognized by toll-like receptor (TLR)-4 and CASP11/4/5, respectively. Other PAMPs include bacterial flagellin (recognized by TLR5), lipoprotein acid from Gram-positive bacteria (recognized by TLR2), peptidoglycan (recognized by TLR2), and viruses double-stranded RNA (recognized by TLR3), or unmethylated CpG motifs (recognized by TLR3). The type II is damage-associated molecular patterns (DAMPs), which are derived from endogenous components of cells, including protein and non-protein subtypes. The release of DAMPs not only occurs in dead or dying cells, but also in stressed cells or activated immune cells after PAMP stimulation. Functionally, DAMPs are mediators of sterile inflammation during tissue damage. The most studied protein type DAMP is the high mobility group box 1 (HMGB1), which is released in various types of cell death (e.g., apoptosis, necroptosis, pyroptosis, and ferroptosis) and is an important target in human health and diseases [52]. The activity of extracellular HMGB1 depends on many factors, including its receptors (e.g., TLRs and advanced glycosylation end-product specific receptor [AGER]) and modification (e.g., oxidation). Other protein types of DAMP include proteins from the nucleus (e.g., histone), cytoplasm (e.g., sequestosome 1 [SQSTM1]), endoplasmic reticulum (e.g., heat shock protein family A (Hsp70) member 5 [HSPA5, also known as BIP]) and mitochondria (e.g., transcription factor A, mitochondrial [TFAM]). The non-protein types of DAMP that have been studied in depth are adenosine triphosphate (ATP) and host DNA from the nucleus or mitochondria. PRRs generally divided into membrane-bound PRRs (e.g., TLR2, TLR4, TLR5, AGER, insulin receptor [INSR], and C-type lectin receptors [CLRs]) and cytoplasmic PRRs (e.g., TLR3, TLR7, TLR9, NOD-like receptors [NLRs], RIG-I–like receptors [RLRs], CASP11, and cyclic GMP-AMP synthase [CGAS]) [97]. The activation of PRR by PAMP and/or DAMP can not only trigger the immune response to eliminate pathogens or promote tissue repair, but also accelerate infection and tissue damage [97]. For this reason, PAMPs, DAMPs and PRRs should be considered as druggable intervention points for the treatment of excessive inflammation including sepsis.
Tissue factor F3
Coagulation can be activated by extrinsic and intrinsic pathways that converge to trigger fibrin formation (Fig. 1). The initiator of the extrinsic pathway is coagulation factor III (F3, best known as tissue factor), a transmembrane glycoprotein receptor for coagulation factor VII/VIIa. The pathological role of F3 in sepsis is confirmed by preclinical or clinical studies of endotoxemia or cytokinemia. Global deletion of F3 (F3−/−) or intraperitoneal injection of anti-F3 neutralizing antibodies similarly prevents endotoxemia and DIC in mice [14, 15]. While exogenous LPS stimulates transcriptional upregulation of F3 expression in myeloid/endothelial cells [16], recent studies have revealed that LPS transported to the cytoplasm can activate the non-canonical inflammasome-mediated pyroptosis to induce F3 release, or cell surface expression of F3 leading to septic death in mice (see below) [14, 17, 18]. Pro-inflammatory cytokines, such as tumor necrosis factor (TNF), interleukin (IL)-6, and IL33, also have the ability to activate an F3-related coagulation response in mouse models of sepsis [19, 20]. In turn, several coagulation proteases (e.g., F3 [21], thrombin [22], or coagulation factor XII [23]) and products (e.g., fibrin [24] or fibrinogen [25]) bind receptors (e.g., protease activated receptors [26]) in immune and non-immune cells to trigger the release of pro-inflammatory cytokines (e.g., IL1A [22]), that can aggravate sepsis. In contrast, anticoagulation proteins, such as antithrombin [27] and activated protein C [28], block inflammatory responses in macrophages/monocytes and mouse endotoxemia models. These studies extend our understanding of the mechanisms leading to the activation of immunocoagulation.
Figure 1. The coagulation cascade in mammals.
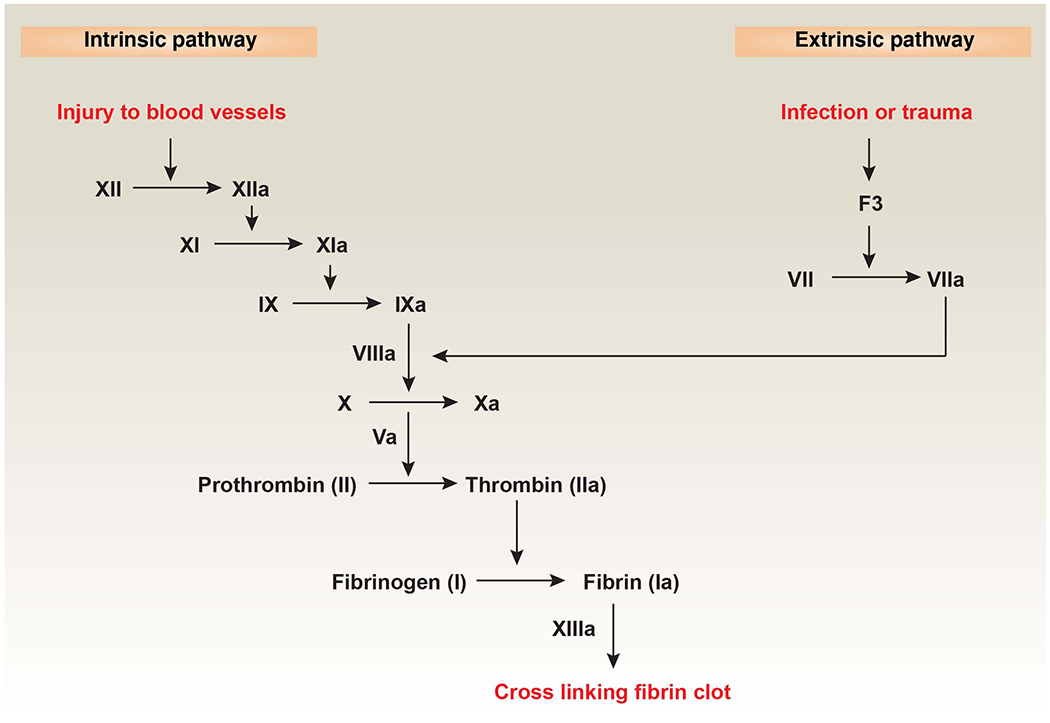
Coagulation is the process of forming a blood clot to reduce blood loss or pathogen spread after blood vessel damage or microbial infections. The intrinsic pathway is also known as the contact pathway, which is activated by damage inside the vasculature. Platelets, exposed/damaged endothelium, collagen, and other chemicals can activate this arm of the pathway [7]. The extrinsic pathway is also called the F3 pathway, which is activated by trauma or pathogen infection. When coagulation factor X is activated through the intrinsic or extrinsic pathways, it activates prothrombin (also known as coagulation factor II) and uses coagulation factor V to convert it into thrombin. Thrombin then cleaves fibrinogen into fibrin, forming a mesh that binds and strengthens platelet embolism, completes coagulation, and stops bleeding [7]. Abbreviations: F3, coagulation factor III.
Platelets
Platelets are small circulating anucleate cells that express many receptors (e.g., integrins) involved in the regulation of coagulation and inflammation in sepsis [29]. Inappropriate aggregation and activation of platelets by pro-inflammatory mediators (e.g., platelet activating factor), PAMPs (e.g., LPS), or coagulation products (e.g., thrombin) seem to be an early indicator of sepsis [30]. In contrast, patients with DIC exhibit a low or rapidly declining platelet count, correlating with the severity of sepsis [30]. For example, severe COVID-19 patients are usually in a hypercoagulable state (with elevated fibrinogen and D-dimer/fibrinogen) with associated thrombocytopenia [31]. Platelets exert immunomodulatory or procoagulant effects directly through cell-to-cell contact, or indirectly through the release of endogenous proteins, such as platelet factor 4 (PF4) [32]) or high mobility group box 1 (HMGB1) [33]. Platelets not only trigger endothelial cell adhesion and leukocyte extravasation at inflammatory sites, but also promote the activation of neutrophils (including their phagocytotic activity and the formation of neutrophil extracellular traps, NETs) and subsequent fibrin deposition [34]. Additionally, platelets regulate the function of T cells by releasing granular proteins, thereby affecting adaptive immunity in sepsis [35]. Although platelets have potential anti-inflammatory functions, they can activate both innate and adaptive immune functions and trigger excessive inflammatory and coagulation responses during sepsis in humans or mice [36].
New players in immunocoagulation
In recent years, new studies have provided increasing support for the concept that immunocoagulation represents a pathologic response in sepsis. Here, we review emerging molecular regulators of this host response in the context of lethal infection.
GSDMD
Gasdermin D (GSDMD) is one of the six members of the gasdermin family, and is expressed in immune and non-immune cells. GSDMD is the key downstream effector of pyroptosis (Fig. 2) [37, 38], which is a type of regulated cell death initially documented in activated macrophages during bacterial infection. The induction of pyroptosis is mainly triggered by inflammasomes, which are multiprotein complexes assembled in response to PAMPs, DAMPs, or environmental stress [39] (BOX 2).
Figure 2. Mechanism and regulation of GSDMD-mediated F3 release and activation in mouse sepsis.
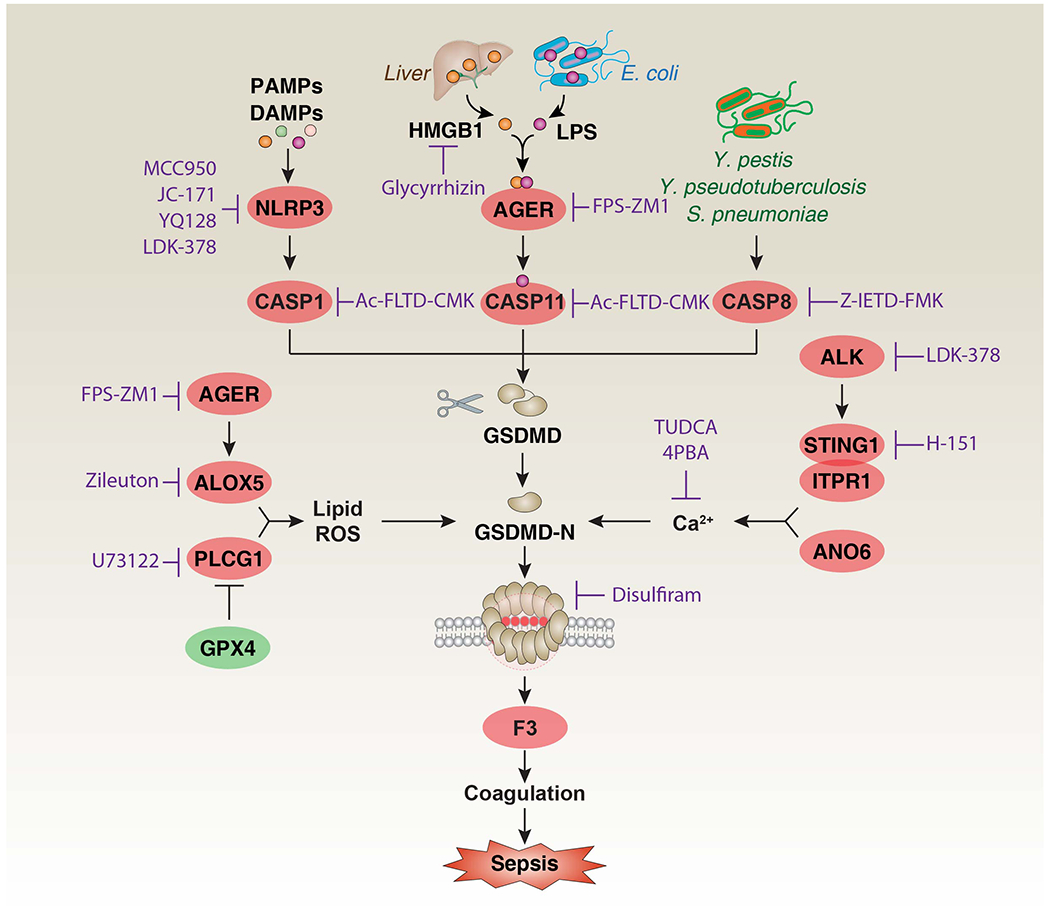
GSDMD is the substrate of CASP1, CASP11, and CASP8, producing two fragments: GSDMD-N and GSDMD-C. GSDMD-N triggers pyroptosis and subsequent F3 release and activation by forming pores in the plasma membrane [14, 17, 18]. The activation of GSDMD-N is positively regulated by lipid ROS and Ca2+ [121, 122]. The generation of lipid ROS is related to AGER-dependent ALOX5 activation and GPX4 depletion-induced PLCG1 activation [59, 80]. The increase in Ca2+ influx is related to the STING1-ITPR1 complex-mediated ER calcium release and ANO6 signal in the plasma membrane [17, 18]. In addition, HMGB1 released from the liver can deliver extracellular LPS to the inside to activate CASP11 through its receptor AGER [55]. These inflammatory immune pathways can ultimately lead to the F3-mediated coagulation cascade in myeloid or epithelial cells, and have been associated with sepsis [17, 18, 55, 67]. Abbreviations: AGER, advanced glycosylation end-product specific receptor; ALK, ALK receptor tyrosine kinase; ANO6, anoctamin 6; ALOX5, arachidonate 5-lipoxygenase; CASP1, caspase 1; CASP8, caspase 8; CASP11, caspase 11; F3, coagulation factor III; GSDMD, gasdermin D; GPX4, glutathione peroxidase 4; ITPR1, inositol 1,4,5-trisphosphate receptor type 1; HMGB1, high mobility group box 1; LPS, lipopolysaccharides; NLRP3, NLR family pyrin domain containing 3; PLCG1, phospholipase C gamma 1; ROS, reactive oxygen species; STING1, stimulator of interferon response CGAMP interactor 1.
There is a context-dependent GSDMD regulatory network that plays a prominent role in inflammatory diseases and conditions (BOX 3). In particular, global or conditional knockout of inflammasome, caspases or GSDMD genes in mice (Nlrp3−/−, Casp1−/−, Casp11−/−, or Gsdmd−/−), has been shown to protect the animals against septic shock [40–47] or lethal endotoxemia [48], rendering these molecules promising candidate targets for the treatment of sepsis.
Box 3. Inflammasomes and Gasdermin D in mammals.
Canonical inflammasome assembly usually involves a sensor (a cytoplasmic PRR), an adaptor (apoptosis-associated speck-like protein containing a caspase recruitment domain [ASC]), and a zymogen (pro-CASP1), leading to the activation of CASP1 and subsequent cleavage of pro-inflammatory IL1 family cytokines, such as IL1B and IL18. Inflammasome-associated PRRs have different types, such as nod-like receptors (NLRs, e.g., NLR family pyrin domain containing 3 [NLRP3]), absent in melanoma 2 (AIM2), and pyrin [39]. Of note, ASC is not required for activation of CASP1 via certain PRRs (e.g., NLR family CARD domain containing 4 [NLRC4] and NLR family pyrin domain containing 1B [NLRP1B]). The activation of CASP1 leads to the cleavage of GSDMD to produce N-terminal fragments (GSDMD-N) [37, 103], which oligomerize to form pores in the plasma membrane, resulting in its permeabilization. Hence, GSDMD-N triggers pyroptotic cell death and the subsequent release of IL1 family cytokines as well as DAMPs (e.g., HMGB1) [104, 105]. Whether IL1B release always requires N-GSDMD-mediated plasma membrane pores is still controversial [61–63].
The non-canonical inflammasome relies on CASP11 (in mice) or CASP4/5 (in humans), which are receptors for cytoplasmic LPS, leading to GSDMD-N-mediated pyroptosis [48, 106–108]. Analysis of crystal structures revealed that CASP1 and CASP4/11 have unique recognition domains in GSDMD at its linker region of N-terminal domain (NTD) and C-terminal domain (CTD) as well as the CTD [109, 110]. The basal expression of CASP11 in macrophages is usually low, and a priming signal (e.g., via toll like receptor [TLR] ligands or interferons [IFNs]) is required to induce its expression for subsequent pyroptosis [48, 111]. As a feedforward mechanism, the production of GSDMD-N during the activation of non-canonical inflammasomes can cause the activation of NLRP3 inflammasomes, and lead to the maturation and release of IL1B and IL18 [48, 111]. Recently, CASP8 (an initiator caspase in the death receptor-mediated apoptotic pathway) was reported to also produce GSDMD-N, thereby inducing pyroptosis in macrophages during bacterial infection in mice (especially with Yersinia pestis and Yersinia pseudotuberculosis) [112–114] or in intestinal epithelial cells [115]. CASP1 also mediates BH3 interacting domain death agonist (BID)-dependent apoptosis in mouse cortical neurons and mast cells in the absence of GSDMD [116]. Moreover, GSDMD- or GSDME-mediated mitochondrial damage has enhanced the sensitivity of cells to classical CASP3-mediated apoptosis in human lymphoid cell line CEM-C7 [117], while GSDMD-dependent pyroptosis has prevented CASP8 activation in mice [118]. These findings reveal a complex relationship between apoptosis and pyroptosis, both of which are co-regulated by an overlapping set of caspases.
In addition to the direct regulation by caspases, the cleavage and activity of GSDMD are controlled by many factors, including but not limited to transcriptional factors (e.g., interferon regulatory factor 2 [IRF2] [119]), post-translational modifications (e.g., succination [120]), ion transport (e.g., K+ efflux [65]), lipid peroxidation (e.g., phospholipase C gamma 1 [PLCG1] [80] and the advanced glycosylation end-product specific receptor (AGER, also known as RAGE) [59]), second messengers (e.g., reactive oxygen species [ROS] [121], calcium [122], and cyclic adenosine monophosphate [cAMP] [123]), protein-protein interactions (e.g., NIMA related kinase 7 [NEK7] [124]), metabolic pathways (e.g., glycolysis [125]), the membrane repair machinery (e.g., endosomal sorting complexes required for transport [ESCRT]-III [126]), autophagy regulators (e.g., GABA type A receptor associated protein like 2 [GABARAPL2/Gate16] [127]), and the apoptotic peptidase activating factor 1 (APAF1)-dependent pyroptosome [128].
In 2019, a report provided the first evidence that GSDMD could mediates the coagulation response in mice by promoting the release of F3 into serum [14]. The authors mainly studied a specific PAMP from Escherichia coli, the type III secretion system (T3SS) inner rod protein EprJ, to understand the mechanism of CASP1 activation in the regulation of F3 release and coagulation in mouse primary bone-marrow derived macrophages (BMDMs) or C57BL/6J mice [14]. Systemic coagulation and animal death caused by intravenous injection of EprJ were limited in Casp1−/−, Casp1/11−/− , Gsdmd−/− or F3−/− mice (but not in Tlr4−/−, Casp11−/− mice or mice lacking the receptors for IL1B [Il1r−/−] and IL18 [Il18r−/−]) [14]. This provided evidence that CASP1/GSDMD could mediate F3 release (but not IL1B and IL18 production) and subsequent systemic coagulation during sepsis [14]. Moreover, the authors used western blot analysis to show that bacterial Rod proteins (including E. coli EprJ, Burkholderia BsaK, Salmonella PrgJ, and EPEC EscI) as well as a needle protein (E. coli EprI) caused the release of F3 from mouse BMDMs and human myeloid THP1 cells through a pathway that required GSDMD-mediated membrane rupture, but not pore formation [14]. Subsequent blood assays revealed that LPS-induced F3 release and lethal coagulation in mice that were primed with TLR3 agonist poly(I:C), required CASP11 (but not CASP1)-mediated GSDMD activation [14]. Moreover, the depletion of macrophages and monocytes using clodronate-containing liposomes protected mice that were treated with LPS from CASP1- or CASP11-mediated F3 release and systemic coagulation, indicating that myeloid cells were an important source of circulating F3 in sepsis [14]. Although GSDMD-independent pyroptosis required the activation of the purinergic receptor P2X7 (P2RX7) [49], depletion of P2rx7 (P2rx7−/−) failed to affect the coagulation cascade through blood assays in endotoxemic mice [14, 18], highlighting a unique role for GSDMD in promoting coagulation. We propose that human polymorphisms in GSDMD might alter F3 release and coagulation responses [50], but this hypothesis requires further confirmation. An additional point of interest is that intraperitoneal injection of necrosulfonamide (an inhibitor of the necroptosis effector mixed lineage kinase domain like pseudokinase [MLKL]) also acts as a direct inhibitor of GSDMD, thus protecting mice from sepsis relative to controls [51]. These findings thus highlight the GSDMD system as an attractive putative therapeutic target for treating sepsis, and certainly merit further attention.
HMGB1
The human or mouse HMGB1 protein is mostly located in the nucleus and acts as a DNA chaperone to sustain chromatin homeostasis [52]. Once actively secreted by immune or epithelial cells, or is passively released by damaged cells, extracellular HMGB1 can act as a DAMP and mediator of lethality during critical illness, including mouse or human sepsis [53], as well as COVID-19 [54]. Recent findings report that HMGB1 plays a role in mediating cell interaction-induced inflammasome activation in mouse models of sepsis [55]. Specifically, exogenous HMGB1 can directly interact with LPS to form molecular complexes that trigger CASP11-dependent pyroptosis and subsequent NLRP3 inflammasome activation in mouse macrophages and lung endothelial cells [55]. Studies using mice with a conditional Hmgb1 deletion in either myeloid cells (LysM-Cre;Hmgb1f/f) or hepatocytes (Alb-Cre;Hmgb1f/f) revealed that the circulating HMGB1 (using enzyme-linked immunosorbent assay [ELISA]) during endotoxemia and CLP-induced sepsis stemmed mainly from liver, but not myeloid cells [55]. The study also determined that extracellular HMGB1-LPS complexes were transported via the plasma membrane advanced glycosylation end-product specific receptor (AGER, best known as RAGE) into the lysosomes of mouse macrophages and endothelial cells, where HMGB1 destabilized lysosomal membranes and facilitated the release of LPS into the cytosol to activate CASP11 for pyroptosis, thus promoting lethal coagulation (Fig. 2) [55]. These findings established a DAMP-dependent pathway for the delivery of LPS into the cytosol, activating CASP11 and coagulation, independently from previously reported non-DAMP pathways through bacteria-released outer membrane vesicles (OMV) [56] or human guanylate binding protein 1 (GBP1) [57, 58]. Thus, transport of LPS into cells is likely to involve multiple pathways, and collectively, these studies highlight the importance of cell-surface as well as cytosolic LPS sensing in promoting cellular pathways that lead to coagulation activation in sepsis.
There is also evidence for additional HMGB1-independent pathways for activating coagulation in sepsis. AGER-mediated lipid peroxidation by arachidonate 5-lipoxygenase (ALOX5) drives CASP11 inflammasome activation in mouse macrophages [59]. Moreover, global disruption (Ager−/−) or conditional depletion of Ager in myeloid cells (LysM-Cre;Agerf/f) protects poly(I:C)-primed mice against LPS-induced death through survival assays [59]. Of note, TLR4 is essential for LPS-induced gene expression of Casp11, TLR4 is not required for the coagulation cascade and septic death in poly(I:C)-primed and LPS-challenged mice through coagulation and survival assays, respectively [18]. These findings, in combination with previous work showing that TLR4 could drive other essential cell-specific functions (such as bacterial clearance) [60], might help explain the disappointing outcome of clinical trials that attempted to treat sepsis with TLR4 inhibitors. In mice, HMGB1-mediated GSDMD activation and pore formation is required to activate F3 through phosphatidylserine (PS) exposure, rather than GSDMD-mediated pyroptosis [55]. Accordingly, PS binding proteins, such as lactadherin and MFG-E8, limit cytoplasmic LPS-induced F3 activation in mouse macrophages [18]. An unexpected finding was that glycine (an osmotic protectant) inhibited the release of F3 in EprJ-infected BMDMs [14], whereas it had no influence on cytoplasmic LPS-induced F3 activation in mouse macrophages [18]. These seemingly contradictory results from different laboratories extend to the controversial effects of GSDMD on promoting or not affecting IL1B release [61–63]. Indeed, at this point it is difficult to distinguish whether proteins such as IL1B are actively secreted from stressed and dying cells or they are passively released from dead cells. One possibility is that GSDMD activation and pore formation kinetics may have rapid changes relative to the inflammatory stimulus gradients, respectively, thereby promoting or not affecting IL1B release. Alternatively, ninjurin 1-mediated plasma membrane rupture during lytic cell death (including pyroptosis) might also participate in HMGB1 release and F3 activation during lethal infection in mice based on ELISA assay [64].
In vitro studies conducted in mouse macrophages show that GSDMD-mediated Ca2+ influx (rather than K+ efflux [65]) promotes F3 activation, at least in part through anoctamin 6 (ANO6, also known as TMEM16F), which is a Ca2+-dependent phospholipid scramblase [18]. As expected, calcium chelators (e.g., BAPTA-AM and EDTA) reduced F3 release/activity in human THP1 cells or murine macrophages based on ELISA or western blot assays [17, 18]. It will be interesting to see whether the depletion of Ano6 can prevent systemic coagulation in a mouse model of sepsis. Different from GSDMD-mediated pyroptosis that mediates the release of HMGB1 in macrophages, GSDMD-deficient (Gsdmd−/−) mice normally secrete HMGB1 during endotoxemia [66]. Thus, these findings increase the possibility of overlapping and diversified functions of GSDMD and inflammasomes in the regulation of HMGB1 release, which may engage in feedback mechanism to control inflammation and coagulation.
STING1
Stimulator of interferon response cGAMP interactor 1 (STING1, also known as STING or TMEM173) is a transmembrane adaptor protein normally expressed in the endoplasmic reticulum (ER) in human or mouse cells [67]. As a component of the cellular innate immune response to infections, especially cytoplasmic DNA from pathogens or hosts, the robust activation of STING1 in myeloid cells (macrophages and monocytes) has recently been associated with inflammation, coagulation and tissue damage in mouse models of sepsis, including CLP-induced polymicrobial infection or bacteremia with E. coli or Streptococcus pneumoniae [17, 68, 69]. Consequently, global depletion (Sting1−/−) or the conditional ablation of Sting1 in myeloid cells (LysM-Cre;Sting1f/f) or mutation of the pore formation site in GSDMD (GsdmdI105N/I105N) [37] protected against lethal coagulation in mouse models of sepsis [17]. A classical function of STING1 is to induce type I IFN signaling by activating upstream cyclic GMP-AMP (cGAMP) synthase (CGAS) and downstream TANK binding kinase 1 (TBK1) and interferon regulatory factor 3 (IRF3). The global depletion of Cgas (Cgas−/−), type I IFN receptor (Ifnar1−/−) and Irf3 (Irf3−/−) failed to affect CLP-induced coagulation activation in mice [17]; however, type I IFN (including IFNA and IFNB) could induce HMGB1 release, leading to CASP11-dependent GSDMD activation in macrophages based on western blot and ELISA assays [70]. Thus, these findings suggest an intricate but minor role for type 1 IFNs, downstream of STING1, in the pathogenesis of sepsis in mice.
In another study, STING1-mediated F3 release from THP1 cells was found to be independent of the transcriptional induction of F3 expression, as well as of STING1 trafficking to the endoplasmic-reticulum–Golgi intermediate compartment (ERGIC) during infection with E. coli- or S. pneumoniae based on mRNA or western blot assays [17]. In contrast, STING1 bound to inositol 1,4,5-trisphosphate receptor type 1 (ITPR1) to trigger Ca2+ release from the ER to cytosol, which led to GSDMD-dependent pyroptosis and F3 release from membranes (Fig. 2) [17]. This procoagulant activity was enhanced by the V155M or N154S STING1 variants, which are gain-of-function mutations leading to the constitutive activation of STING1 in patients with STING1-associated vasculopathy with onset in infancy (SAVI) [71]. Moreover, this procoagulation activity was inhibited by blocking cytosolic Ca2+ influx by means of Itpr1 knockdown or overexpression of ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2 (ATP2A2) in mouse BMDMs [17]. In addition, STING1-mediated Ca2+ release appeared to have a broad role in promoting GSDMD-N production mediated by the activation of CASP1, CASP11 (during E. coli infection) or CASP8 (during S. pneumoniae infection), which triggered F3 release in human or mouse macrophages/monocytes [17]. Of note, STING1-mediated apoptosis in mouse CD4+ T cells in vitro may promote inflammation-induced immunosuppression during endotoxemia in mice [72], indicating that STING1 is a therapeutic target for T cell-mediated diseases. Moreover, after CGAS recognizes cytosolic DNA, the activated STING1 can be transported to the lysosome, leading to lysosomal rupture and subsequent K+ efflux, ultimately causing the activation of the NLRP3 inflammasome in human myeloid cells in vitro [73]. In turn, by measuring type I IFN gene production, GSDMD may restrict or induce CGAS-mediated activation of STING1 in mouse macrophages and endothelial cells, respectively [74, 75], suggesting the dual role of GSDMD in the feedback control of STING1 activation. Overall, the activity of STING1 is regulated by various binding proteins or posttranslational modifications linked to immunity, autophagy, and cell death, highlighting the complex contribution of STING1 to the pathogenesis of disease, especially sepsis and coagulation-related diseases [76].
SQSTM1
Sequestosome 1 (SQSTM1) is not only a key autophagy receptor, but also a multifunctional protein involved in signal transduction pathways during inflammation and oxidative stress [77]. Recent studies have emphasized that extracellular SQSTM1 is a mediator of septic death in vitro and in vivo [78]. Specifically, LPS induces human or mouse macrophages/monocytes to secrete SQSTM1 by first activating TLR4 -mediated transactivation of the SQSTM1 gene, coupled to STING1-mediated phosphorylation of the SQSTM1 protein on Ser403 [78]. Cytoplasmic LPS also causes GSDMD-dependent pyroptosis to promote the passive release of SQSTM1 from macrophages and monocytes in vitro [78]. Extracellular SQSTM1 also acts as a new pro-inflammatory mediator through its binding to the insulin receptor (INSR), leading to a nuclear factor kappa B (NFKB)-dependent metabolic switch towards aerobic glycolysis, followed by pro-inflammatory M1 polarization of macrophages (Fig. 3) [78]. Unlike the insulin-INSR axis [79], the SQSTM1-INSR axis-mediated NFKB activation and inflammatory response occurs in macrophages through a pathway that requires PLCG1-dependent lipid peroxidation [78] and subsequent elevated pyroptosis [80]. Conditional depletion of glutathione peroxidase 4 (GPX4, which reduces hydrogen peroxide, organic hydroperoxides and lipid peroxides) in myeloid cells (LysM-Cre;Gpx4f/f) increases the activity of PLCG1 to induce pyroptosis based on western blot assays [80]. Therefore, in the course of infection, unlike insulin-mediated INSR activation that limits the inflammatory response, the SQSTM1-mediated INSR pathway may lead to aerobic glycolysis in macrophages, thereby accelerating the inflammatory response.
Figure 3. Release and activity of SQSTM1 in mouse sepsis.
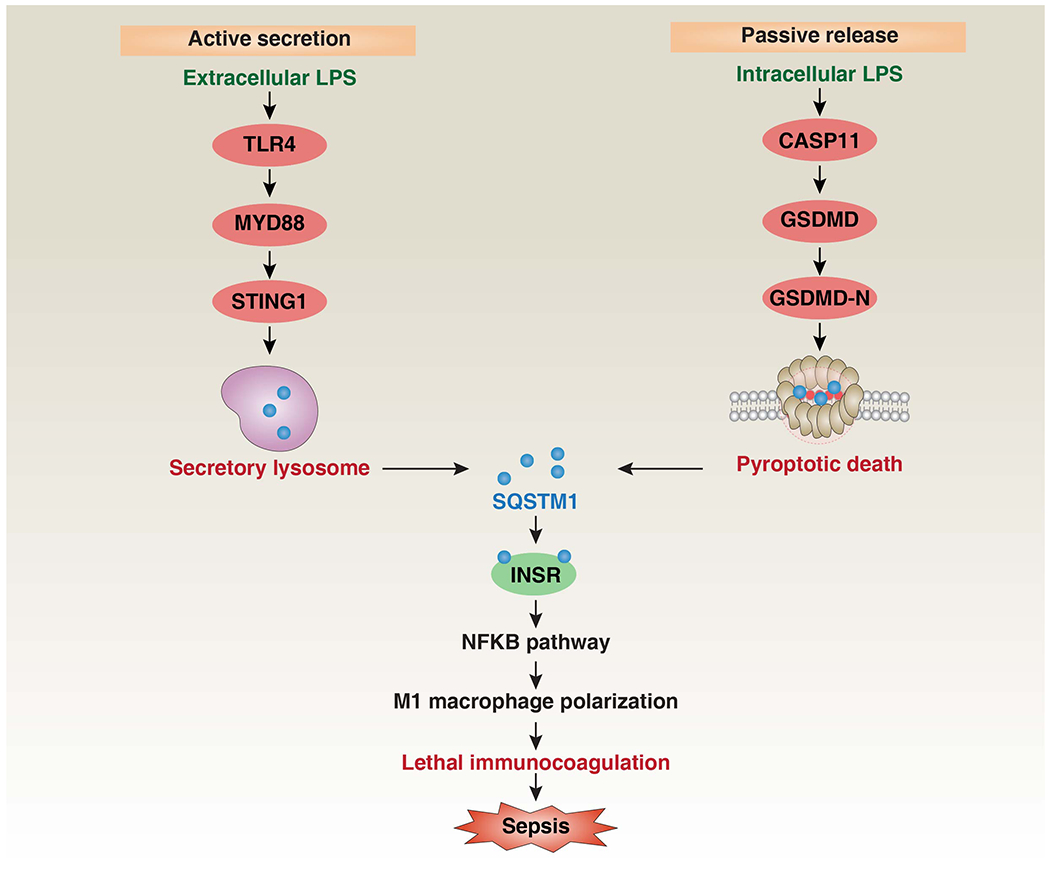
There are two mechanisms by which macrophages release SQSTM1 into the extracellular environment (active secretion or passive release). Extracellular LPS induces SQSTM1 lysosomal secretion by activating the TLR4-MYD88-STING1 pathway, while intracellular LPS triggers the pyroptotic release of SQSTM1 by activating the CASP11-GSDMD-GSDMD-N pathway [78]. After its release, SQSTM1 binds to the receptor INSR to activate the NFκB pathway, causing the polarization of pro-inflammatory macrophages (aka ‘M1’), and finally mediating septic death in mice through excessive inflammation and coagulation [78]. Abbreviations: CASP11, caspase 11; GSDMD, gasdermin D; INSR, insulin receptor; LPS, lipopolysaccharides; MYD88, MYD88 innate immune signal transduction adaptor; NFKB, nuclear factor-κB; PLCG1, phospholipase C gamma 1; ROS, reactive oxygen species; STING1, stimulator of interferon response CGAMP interactor 1; SQSTM1, sequestosome 1; TLR4, toll like receptor 4.
Furthermore, intraperitoneal administration of anti-SQSTM1–neutralizing monoclonal antibodies or conditional depletion of Insr (LysM-Cre;Insrf/f) or Sqstm1 (LysM-Cre;Sqstm1f/f) in myeloid cells efficiently protects mice against lethal sepsis (induced by CLP or infection with E. coli or S. pneumoniae) and endotoxemia relative to IgG or wild-type controls, and it is also accompanied by a decreased occurrence of systemic coagulation response and multiple organ failure [78]. In patients with bacterial sepsis, SQSTM1 and INSR mRNA increase in peripheral blood mononuclear cells together with serum SQSTM1 concentrations, further supporting for the hypothesis that the SQSTM1-INSR pathway might play a pathological role during sepsis in humans [78]. Moreover, an elevated serum SQSTM1 concentration has also been identified as an independent risk factor for patients with steatosis and liver inflammation [81]. Thus, it is foreseeable that extracellular SQSTM1 might be incriminated in the pathogenesis of other inflammatory diseases, once its pro-inflammatory potential is further revealed, and pending future investigations.
Potential inhibitors of immunocoagulation
Over the past few decades, our understanding of the processes that trigger the production of pro-inflammatory cytokines (such as IL1, IL6, and TNF) in sepsis has made significant progress. Unfortunately, these findings have not yet been translated into effective treatments of bacterial sepsis. Monoclonal antibodies that block IL6 signaling have also failed in COVID-19 patients with sepsis [82]. While the reasons for the failure of such clinical trials are multifactorial, the need for novel targets and new approaches in sepsis is obvious. Based on the emerging immunocoagulation mechanism described above (Fig. 2), we argue that inhibitors targeting NLRP3 (e.g., MCC950, JC-171, and YQ128), CASP1/4/5/11 (e.g., Ac-FLTD-CMK), CASP8 (e.g., Z-IETD-FMK), GSDMD (e.g., disulfiram), HMGB1 (e.g., glycyrrhizin), AGER (e.g., FPS-ZM1), PLCG1 (e.g., U73122), ALOX5 (e.g., zileuton), STING1 (e.g., LDK-378 and H-151) or ER stress (e.g., tauroursodeoxycholic acid [TUDCA] and 4-phenyl butyric acid [4PBA]) have the potential for further research in translational and clinical studies (Table 1). As these inhibitors have been shown to inhibit inflammation and/or coagulation in vitro or in rodent models of sepsis (endotoxemia and CLP) [54, 55, 59, 83–91], their evaluation in human trials is urgently awaited.
Table 1.
Selected therapeutic drugs for putatively targeting immunocoagulation pathways in sepsis
| Drug | Year | Target | Mechanism | Usage | Model | Ref |
|---|---|---|---|---|---|---|
| Glycyrrhizin | 2007 | HMGB1 | Bind HMGB1 and inhibit its release and activity |
In vitro: 5-50 μM In vivo: 10-50 mg/kg |
In vitro: BMDM, THP1; In vivo: endotoxemia, CLP (mouse or rat) |
[54, 83, 90, 91] |
| MCC950 | 2015 | NLRP3 | Interact with the Walker B motif within the NLRP3 NACHT domain and block NLRP3-induced ASC oligomerization instead of K+ efflux, Ca2+ flux or NLRP3-ASC interaction |
In vitro: 0.1-10 μM In vivo: 10-50 mg/kg |
In vitro: BMDM, HMDM; In vivo: CLP (mouse or rat) |
[55, 83–86] |
| JC-171 | 2017 | NLRP3 | Block NLRP3 inflammasome activation |
In vitro: 1-100 μM In vivo: 100 mg/kg |
In vitro: BMDM, J774A.1; In vivo: endotoxemia (mouse) |
[129] |
| LDK-378 | 2017 | ALK | Inhibit NLRP3 inflammasome and STING1 activation |
In vitro: 1-10 μM In vivo: 20 mg/kg |
In vitro: BMDM;THP1 In vivo: endotoxemia, CLP (mouse) |
[130, 131] |
| Ac-FLTD-CMK | 2018 | CASP1/4/5/11 | Block CASP1/4/5/11-mediated GSDMD-N production |
In vitro: 10 μM In vivo: N/A |
In vitro: BMDM; In vivo: N/A |
[132] |
| Z-IETD-FMK | 2018 | CASP8 | Block CASP8 activity mediated GSDMD-N production |
In vitro: 10 μM In vivo: N/A |
In vitro: BMDM; In vivo: N/A |
[17] |
| U73122 | 2018 | PLCG1 | Inhibit GSDMD-N-mediated pyroptosis |
In vitro: 10 μM In vivo: 30 mg/kg |
In vitro: BMDM; In vivo: CLP (mouse) |
[17, 80] |
| H-151 | 2018 | STING1 | Inhibits STING1 by covalently binding to STING at the transmembrane cysteine residue at position 91 |
In vitro: 2 μM In vivo: 750 nM/mice |
In vitro: primary human or mouse macrophages In vivo: Trex1−/− mice |
[78, 133] |
| YQ128 | 2019 | NLRP3 | A blood-brain barrier permeable and selective NLRP3 inflammasome inhibitor |
In vitro: 10-100 μM In vivo: 10-20 mg/kg |
In vitro: BMDM, J774A.1; In vivo: endotoxemia (mouse) |
[134] |
| FPS-ZM1 | 2019 | AGER | A high-affinity blocker of AGER V domain-mediated ligand binding |
In vitro: 0.1-1 μM In vivo: 10 mg/kg, 75 μg/day |
In vitro: BMDM; In vivo: endotoxemia, A. baumannii infection (mouse) |
[59, 87, 88] |
| Zileuton | 2019 | ALOX5 | Block ALOX5-mediated lipid peroxidation |
In vitro: 5 μM In vivo: 30 mg/kg |
In vitro: BMDM; In vivo: endotoxemia (mouse) |
[59, 89] |
| Disulfiram | 2020 | GSDMD | Prevent the formation of GSDMD pores by covalently modifying human/mouse GSDMD Cys191/Cys192 |
In vitro: 1-30 μM In vivo: 15-50 mg/kg |
In vitro: BMDM, THP1; In vivo: endotoxemia (mouse) |
[135] |
| TUDCA | 2020 | ER stress and Ca2+ | Inhibit ER stress and calcium release |
In vitro: 50 μM In vivo: 200 mg/kg |
In vitro: THP1 In vivo: CLP (mouse) |
[17] |
| 4PBA | 2020 | ER stress and Ca2+ | Inhibit ER stress and calcium release |
In vitro: 1 mM In vivo: 20 mg/kg |
In vitro: THP1 In vivo: CLP (mouse) |
[17] |
Concluding Remarks
Characterized by excessive immunocoagulation, multiple organ failure, and limited treatment options, sepsis remains a major challenge for basic and clinical research. Targeting a single mediator (e.g., activated protein C) has so far failed to reduce the mortality of patients with sepsis [92]. The use of mouse models has revealed new mechanisms of sepsis, but many factors (e.g., crucial genetic differences between humans and mice) limit the translation to the human applications. Nevertheless, in recent years, many molecular connections between inflammation and coagulation have been discovered [14, 17, 18], thus increasing our understanding of the nature, characteristics, and consequences of the host response to systemic microbial infections. However, these exciting advances have raised additional scientific questions that need to be answered (see Outstanding Questions). A keystone discovery may be the new function of inflammasomes in regulating the release and activation of F3, driving an explosion of research into the recognition of exogenous or endogenous danger signals, as well as defining the structural basis of inflammatory caspase activation and the molecular machinery of regulated cell death. Through integrated metabolic pathways, the activation of inflammasome-dependent coagulation in sepsis is further enhanced by STING1, HMGB1, and SQSTM1. However, the biggest challenge concerns the fact that almost all immune mediators or coagulation factors play diverse and inter-connected roles in the pathophysiology of sepsis, meaning that their interception may simultaneously affect homeostatic and disease-amplifying circuitries. It is our hope that, despite this complexity, further in-depth elucidation of the initial signals, intermediate connections, final effector mechanisms and feedback loops of immunocoagulation might reveal novel candidate therapeutic approaches that can benefit patients with lethal infection. Given that 50% of bacterial infections in sepsis are Gram-positive [3], it will also be interesting to compare the differences in immunocoagulation caused by Gram-negative or positive bacteria, as they may trigger different host responses. In addition to NLRP3, NLRC4 might be a promising candidate target for anti-coagulation treatment in sepsis in individuals infected with Salmonella, Legionella, and Pseudomonas aeruginosa, because NLRC4 mediates host response to these pathogens [93].
Outstanding Questions.
How does GSDMD play different roles in different immune cells? In addition to caspases, other enzymes can also cleave GSDMD. Recent studies have shown that the cleavage and activation of GSDMD in human or mouse neutrophils is mediated by neutrophil elastase (ELANE) in a caspase-independent manner [94], which helps to form neutrophil extracellular traps (NETs) for bacterial cleavage [95]. Thus, different cleavage sites in GSDMD may result in different activities in immunocoagulation. Additionally, the complementarity of the functions of GSDMD family members remains a mystery.
Do distinct cell death modalities have different effects on immunocoagulation? Depending on their cell types or the initiating stimuli, cells can succumb to an ever-expanding variety of cell death subroutines [96]. The release of F3, HMGB1 and SQSTM1, the type and function of DAMPs and their receptors may affect immunocoagulation during infection and tissue damage in mouse models of sepsis [97].
Does immunocoagulation affect the course of immunosuppression in septic shock? After excessive inflammation, most patients with sepsis quickly show obvious signs of immunosuppression, which favors secondary infections [98]. It would be interesting to further understand the dynamic relationship and modulators (e.g., STING1 [17, 72]) that link immunocoagulation and immunosuppression during sepsis.
What are the physiological and pathological differences between mice and humans? In sepsis research, mice are most often used due to the ease of experimentation, the availability of genetically engineered species, and the relatively low cost. However, in the past few decades, clinical translation based on mouse experimental results has been disappointing [99].
How to establish precision medicine for the treatment of sepsis? Precision therapy is a treatment tailored to individual genetic status and has been first practiced in cancer patients. Sepsis is a very heterogeneous syndrome [100], including different genetic states and functional changes of the immunocoagulation pathways. It is foreseeable that certain new technologies (such as single-cell whole-genome sequencing) will assist individualized medical applications in patients with sepsis.
When might experimental drugs to treat sepsis be ready? Drug development is a lengthy, complex and costly process. Whether the drug can be successfully transformed from subclinical stage to clinical application is still uncertain. Obviously, this challenge requires the joint efforts and cooperation of industry, academia, and relevant government agencies.
Acknowledgments:
We thank the numerous colleagues in the field of immunocoagulation and sepsis. We also apologize to the researchers who were not referenced due to space limitations.
Glossary
- Advanced glycosylation end-product specific receptor (AGER)
a transmembrane receptor of the immunoglobulin superfamily, which mediates inflammation by recognizing various ligands.
- Arachidonate 5-lipoxygenase (ALOX5)
a non-heme iron-containing enzyme of the lipoxygenase family, which mediated lipid peroxidation.
- Anoctamin 6 (ANO6)
a Ca2+-dependent phospholipid scramblase, which mediates phosphatidylserine exposure during cell death.
- Bleeding diathesis
an unusual susceptibility to bleed mostly due to a defect in the system of coagulation.
- Cytokine storm
a severe immune response in which the body releases too many cytokines into the circulatory system.
- Damage-associated molecular patterns (DAMPs)
host or environmental molecules that can trigger an immune response when released.
- Disseminated intravascular coagulation (DIC)
pathological condition that can cause abnormal blood clotting throughout the body’s blood vessels.
- Ninjurin 1
a cell surface adhesion protein, which mediates plasma membrane rupture during lytic cell death.
- Pathogen-associated molecular patterns (PAMPs)
structural components or products of pathogens that can trigger innate immune activation.
- Pattern recognition receptors (PRRs)
receptors that can recognize PAMPs, DAMPs or stressors.
- Pyroptosis
a lytic pro-inflammatory type of cell death that is mainly initiated through caspases (e.g., CASP1, CASP4/5/11, CASP8, or CASP3) and mediated through gasdermins (e.g., GSDMD or GSDME).
- Steatosis
an abnormal retention of fat or lipids within cells or organs.
Footnotes
Disclaimer Statement: No conflicts of interest to declare.
References
- 1.Singer M et al. (2016) The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315 (8), 801–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou P et al. (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579 (7798), 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rudd KE et al. (2020) Global, regional, and national sepsis incidence and mortality, 1990-2017: analysis for the Global Burden of Disease Study. Lancet 395 (10219), 200–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reinhart K et al. (2017) Recognizing Sepsis as a Global Health Priority - A WHO Resolution. N Engl J Med 377 (5), 414–417. [DOI] [PubMed] [Google Scholar]
- 5.Gando S et al. (2019) Role of disseminated intravascular coagulation in severe sepsis. Thromb Res 178, 182–188. [DOI] [PubMed] [Google Scholar]
- 6.Lopez-Otin C and Kroemer G (2021) Hallmarks of Health. Cell 184 (1), 33–63. [DOI] [PubMed] [Google Scholar]
- 7.Levi M and van der Poll T (2017) Coagulation and sepsis. Thromb Res 149, 38–44. [DOI] [PubMed] [Google Scholar]
- 8.Qi AL et al. (2020) Recombinant human ulinastatin improves immune dysfunction of dendritic cells in septic mice by inhibiting endoplasmic reticulum stress-related apoptosis. Int Immunopharmacol 85, 106643. [DOI] [PubMed] [Google Scholar]
- 9.Deng W et al. (2018) The Circadian Clock Controls Immune Checkpoint Pathway in Sepsis. Cell Rep 24 (2), 366–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schneck E et al. (2020) Blood Levels of Free-Circulating Mitochondrial DNA in Septic Shock and Postsurgical Systemic Inflammation and Its Influence on Coagulation: A Secondary Analysis of a Prospective Observational Study. J Clin Med 9 (7), 2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Higgins SJ et al. (2018) Tie2 protects the vasculature against thrombus formation in systemic inflammation. J Clin Invest 128 (4), 1471–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pan B et al. (2017) The fifth epidermal growth factor like region of thrombomodulin alleviates LPS-induced sepsis through interacting with GPR15. Thromb Haemost 117 (3), 570–579. [DOI] [PubMed] [Google Scholar]
- 13.Xu S et al. (2018) BMSCs ameliorate septic coagulopathy by suppressing inflammation in cecal ligation and puncture-induced sepsis. J Cell Sci 131 (3), jcs211151. [DOI] [PubMed] [Google Scholar]
- 14.Wu C et al. (2019) Inflammasome Activation Triggers Blood Clotting and Host Death through Pyroptosis. Immunity 50 (6), 1401–1411 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pawlinski R et al. (2004) Role of tissue factor and protease-activated receptors in a mouse model of endotoxemia. Blood 103 (4), 1342–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oeth P et al. (1998) Retinoic acid selectively inhibits lipopolysaccharide induction of tissue factor gene expression in human monocytes. Blood 91 (8), 2857–65. [PubMed] [Google Scholar]
- 17.Zhang H et al. (2020) TMEM173 Drives Lethal Coagulation in Sepsis. Cell Host Microbe 27 (4), 556–570 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang X et al. (2019) Bacterial Endotoxin Activates the Coagulation Cascade through Gasdermin D-Dependent Phosphatidylserine Exposure. Immunity 51 (6), 983–996 e6. [DOI] [PubMed] [Google Scholar]
- 19.Kamegashira A et al. (2020) Histamine- or vascular endothelial growth factor-induced tissue factor expression and gap formation between vascular endothelial cells are synergistically enhanced by lipopolysaccharide, tumor necrosis factor-alpha, interleukin (IL)-33 or IL-1beta. J Dermatol 47 (11), 1293–1300. [DOI] [PubMed] [Google Scholar]
- 20.Gao H et al. (2018) Porcine IL-6, IL-1beta, and TNF-alpha regulate the expression of pro-inflammatory-related genes and tissue factor in human umbilical vein endothelial cells. Xenotransplantation 25 (5), e12408. [DOI] [PubMed] [Google Scholar]
- 21.Queiroz KC et al. (2011) Tissue factor-dependent chemokine production aggravates experimental colitis. Mol Med 17 (9-10), 1119–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burzynski LC et al. (2019) The Coagulation and Immune Systems Are Directly Linked through the Activation of Interleukin-1alpha by Thrombin. Immunity 50 (4), 1033–1042 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vorlova S et al. (2017) Coagulation factor XII induces pro-inflammatory cytokine responses in macrophages and promotes atherosclerosis in mice. Thromb Haemost 117 (1), 176–187. [DOI] [PubMed] [Google Scholar]
- 24.Cole HA et al. (2014) Fibrin accumulation secondary to loss of plasmin-mediated fibrinolysis drives inflammatory osteoporosis in mice. Arthritis Rheumatol 66 (8), 2222–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hsieh JY et al. (2017) Differential regulation of macrophage inflammatory activation by fibrin and fibrinogen. Acta Biomater 47, 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niessen F et al. (2008) Dendritic cell PAR1-S1P3 signalling couples coagulation and inflammation. Nature 452 (7187), 654–8. [DOI] [PubMed] [Google Scholar]
- 27.Minnema MC et al. (2000) Recombinant human antithrombin III improves survival and attenuates inflammatory responses in baboons lethally challenged with Escherichia coli. Blood 95 (4), 1117–23. [PubMed] [Google Scholar]
- 28.Schouten M et al. (2015) The cytoprotective effects of endogenous activated protein C reduce activation of coagulation during murine pneumococcal pneumonia and sepsis. Thromb Res 135 (3), 537–43. [DOI] [PubMed] [Google Scholar]
- 29.Middleton EA et al. (2019) Sepsis alters the transcriptional and translational landscape of human and murine platelets. Blood 134 (12), 911–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao L et al. (2020) Platelets as a prognostic marker for sepsis: A cohort study from the MIMIC-III database. Medicine (Baltimore) 99 (45), e23151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang D et al. (2020) The hallmarks of COVID-19 disease. PLoS Pathog 16 (5), e1008536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brousseau-Nault M et al. (2017) Chronic periodontitis is associated with platelet factor 4 (PF4) secretion: A pilot study. J Clin Periodontol 44 (11), 1101–1111. [DOI] [PubMed] [Google Scholar]
- 33.Vogel S et al. (2015) Platelet-derived HMGB1 is a critical mediator of thrombosis. J Clin Invest 125 (12), 4638–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDonald B et al. (2017) Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 129 (10), 1357–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Albayati S et al. (2020) P2Y12 antagonism results in altered interactions between platelets and regulatory T cells during sepsis. J Leukoc Biol doi: 10.1002/JLB.3A0220-097R. Online ahead of print. [DOI] [PubMed] [Google Scholar]
- 36.Assinger A et al. (2019) Platelets in Sepsis: An Update on Experimental Models and Clinical Data. Front Immunol 10, 1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kayagaki N et al. (2015) Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526 (7575), 666–71. [DOI] [PubMed] [Google Scholar]
- 38.Shi J et al. (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526 (7575), 660–5. [DOI] [PubMed] [Google Scholar]
- 39.Xue Y et al. (2019) Emerging Activators and Regulators of Inflammasomes and Pyroptosis. Trends Immunol 40 (11), 1035–1052. [DOI] [PubMed] [Google Scholar]
- 40.Mao K et al. (2013) Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res 23 (2), 201–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gong Z et al. (2015) Curcumin suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Mol Nutr Food Res 59 (11), 2132–42. [DOI] [PubMed] [Google Scholar]
- 42.Wang P et al. (2015) Exogenous Carbon Monoxide Decreases Sepsis-Induced Acute Kidney Injury and Inhibits NLRP3 Inflammasome Activation in Rats. Int J Mol Sci 16 (9), 20595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moon JS et al. (2015) UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J Clin Invest 125 (2), 665–80. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Luo YP et al. (2014) Hemin inhibits NLRP3 inflammasome activation in sepsis-induced acute lung injury, involving heme oxygenase-1. Int Immunopharmacol 20 (1), 24–32. [DOI] [PubMed] [Google Scholar]
- 45.Long H et al. (2016) Artemisinin protects mice against burn sepsis through inhibiting NLRP3 inflammasome activation. Am J Emerg Med 34 (5), 772–7. [DOI] [PubMed] [Google Scholar]
- 46.Kalbitz M et al. (2016) Complement-induced activation of the cardiac NLRP3 inflammasome in sepsis. FASEB J 30 (12), 3997–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mandal P et al. (2018) Caspase-8 Collaborates with Caspase-11 to Drive Tissue Damage and Execution of Endotoxic Shock. Immunity 49 (1), 42–55 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kayagaki N et al. (2011) Non-canonical inflammasome activation targets caspase-11. Nature 479 (7371), 117–21. [DOI] [PubMed] [Google Scholar]
- 49.Yang D et al. (2015) Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity 43 (5), 923–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rathkey JK et al. (2020) Human polymorphisms in GSDMD alter the inflammatory response. J Biol Chem 295 (10), 3228–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rathkey JK et al. (2018) Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol 3 (26), eaat2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kang R et al. (2014) HMGB1 in health and disease. Mol Aspects Med 40, 1–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang H et al. (1999) HMG-1 as a late mediator of endotoxin lethality in mice. Science 285 (5425), 248–51. [DOI] [PubMed] [Google Scholar]
- 54.Chen R et al. (2020) HMGB1 as a potential biomarker and therapeutic target for severe COVID-19. Heliyon 6 (12), e05672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deng M et al. (2018) The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immunity 49 (4), 740–753.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Peng Y et al. (2020) Bacterial outer membrane vesicles induce disseminated intravascular coagulation through the caspase-11-gasdermin D pathway. Thromb Res 196, 159–166. [DOI] [PubMed] [Google Scholar]
- 57.Santos JC et al. (2020) Human GBP1 binds LPS to initiate assembly of a caspase-4 activating platform on cytosolic bacteria. Nat Commun 11 (1), 3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fisch D et al. (2019) Human GBP1 is a microbe-specific gatekeeper of macrophage apoptosis and pyroptosis. EMBO J 38 (13), e100926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen R et al. (2019) AGER-Mediated Lipid Peroxidation Drives Caspase-11 Inflammasome Activation in Sepsis. Front Immunol 10, 1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deng M et al. (2013) Lipopolysaccharide clearance, bacterial clearance, and systemic inflammatory responses are regulated by cell type-specific functions of TLR4 during sepsis. J Immunol 190 (10), 5152–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Karmakar M et al. (2020) N-GSDMD trafficking to neutrophil organelles facilitates IL-1beta release independently of plasma membrane pores and pyroptosis. Nat Commun 11 (1), 2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Monteleone M et al. (2018) Interleukin-1beta Maturation Triggers Its Relocation to the Plasma Membrane for Gasdermin-D-Dependent and -Independent Secretion. Cell Rep 24 (6), 1425–1433. [DOI] [PubMed] [Google Scholar]
- 63.Evavold CL et al. (2018) The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 48 (1), 35–44 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kayagaki N et al. (2021) NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 591 (7848), 131–136. [DOI] [PubMed] [Google Scholar]
- 65.Banerjee I et al. (2018) Gasdermin D Restrains Type I Interferon Response to Cytosolic DNA by Disrupting Ionic Homeostasis. Immunity 49 (3), 413–426 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Volchuk A et al. (2020) Indirect regulation of HMGB1 release by gasdermin D. Nat Commun 11 (1), 4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barber GN (2015) STING: infection, inflammation and cancer. Nat Rev Immunol 15 (12), 760–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zeng L et al. (2017) ALK is a therapeutic target for lethal sepsis. Sci Transl Med 9 (412), eaan5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Heipertz EL et al. (2017) STING and TRIF Contribute to Mouse Sepsis, Depending on Severity of the Disease Model. Shock 47 (5), 621–631. [DOI] [PubMed] [Google Scholar]
- 70.Yang X et al. (2020) The role of type 1 interferons in coagulation induced by gram-negative bacteria. Blood 135 (14), 1087–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu Y et al. (2014) Activated STING in a vascular and pulmonary syndrome. N Engl J Med 371 (6), 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Long J et al. (2020) Notch signaling protects CD4 T cells from STING-mediated apoptosis during acute systemic inflammation. Sci Adv 6 (39), eabc5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gaidt MM et al. (2017) The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell 171 (5), 1110–1124 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ma C et al. (2020) Gasdermin D in macrophages restrains colitis by controlling cGAS-mediated inflammation. Sci Adv 6 (21), eaaz6717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Huang LS et al. (2020) mtDNA Activates cGAS Signaling and Suppresses the YAP-Mediated Endothelial Cell Proliferation Program to Promote Inflammatory Injury. Immunity 52 (3), 475–486 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hopfner KP and Hornung V (2020) Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol 21 (9), 501–521. [DOI] [PubMed] [Google Scholar]
- 77.Sanchez-Martin P et al. (2019) p62/SQSTM1: ‘Jack of all trades’ in health and cancer. FEBS J 286 (1), 8–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhou B et al. (2020) Extracellular SQSTM1 mediates bacterial septic death in mice through insulin receptor signalling. Nat Microbiol 5 (12), 1576–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Caldwell A et al. (2018) Impact of insulin on the intestinal microcirculation in a model of sepsis-related hyperglycemia. Microvasc Res 119, 117–128. [DOI] [PubMed] [Google Scholar]
- 80.Kang R et al. (2018) Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host Microbe 24 (1), 97–108.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang X et al. (2018) Defective lysosomal clearance of autophagosomes and its clinical implications in nonalcoholic steatohepatitis. FASEB J 32 (1), 37–51. [DOI] [PubMed] [Google Scholar]
- 82.Stone JH et al. (2020) Efficacy of Tocilizumab in Patients Hospitalized with Covid-19. N Engl J Med 383 (24), 2333–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kang R et al. (2016) A novel PINK1- and PARK2-dependent protective neuroimmune pathway in lethal sepsis. Autophagy 12 (12), 2374–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Coll RC et al. (2015) A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21 (3), 248–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cornelius DC et al. (2019) NLRP3 inflammasome activation in platelets in response to sepsis. Physiol Rep 7 (9), e14073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cornelius DC et al. (2020) NLRP3 inflammasome inhibition attenuates sepsis-induced platelet activation and prevents multi-organ injury in cecal-ligation puncture. PLoS One 15 (6), e0234039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nielsen TB et al. (2017) Diabetes Exacerbates Infection via Hyperinflammation by Signaling through TLR4 and RAGE. mBio 8 (4), e00818–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Deane R et al. (2012) A multimodal RAGE-specific inhibitor reduces amyloid beta-mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest 122 (4), 1377–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Collin M et al. (2004) Reduction of the multiple organ injury and dysfunction caused by endotoxemia in 5-lipoxygenase knockout mice and by the 5-lipoxygenase inhibitor zileuton. J Leukoc Biol 76 (5), 961–70. [DOI] [PubMed] [Google Scholar]
- 90.Zhao F et al. (2017) Glycyrrhizin Protects Rats from Sepsis by Blocking HMGB1 Signaling. Biomed Res Int 2017, 9719647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mollica L et al. (2007) Glycyrrhizin binds to high-mobility group box 1 protein and inhibits its cytokine activities. Chem Biol 14 (4), 431–41. [DOI] [PubMed] [Google Scholar]
- 92.Cavaillon JM et al. (2020) Sepsis therapies: learning from 30 years of failure of translational research to propose new leads. EMBO Mol Med 12 (4), e10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wu R et al. (2021) Inflammasome-Dependent Coagulation Activation in Sepsis. Front Immunol 12, 641750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kambara H et al. (2018) Gasdermin D Exerts Anti-inflammatory Effects by Promoting Neutrophil Death. Cell Rep 22 (11), 2924–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sollberger G et al. (2018) Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol 3 (26), eaar6689. [DOI] [PubMed] [Google Scholar]
- 96.Tang D et al. (2019) The molecular machinery of regulated cell death. Cell Res 29 (5), 347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gong T et al. (2020) DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol 20 (2), 95–112. [DOI] [PubMed] [Google Scholar]
- 98.van Vught LA et al. (2016) Incidence, Risk Factors, and Attributable Mortality of Secondary Infections in the Intensive Care Unit After Admission for Sepsis. JAMA 315 (14), 1469–79. [DOI] [PubMed] [Google Scholar]
- 99.Guillon A et al. (2019) Preclinical septic shock research: why we need an animal ICU. Ann Intensive Care 9 (1), 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Davenport EE et al. (2016) Genomic landscape of the individual host response and outcomes in sepsis: a prospective cohort study. Lancet Respir Med 4 (4), 259–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tang D et al. (2012) PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev 249 (1), 158–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Peleg AY and Hooper DC (2010) Hospital-acquired infections due to gram-negative bacteria. N Engl J Med 362 (19), 1804–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.He WT et al. (2015) Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res 25 (12), 1285–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ding J et al. (2016) Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535 (7610), 111–6. [DOI] [PubMed] [Google Scholar]
- 105.Liu X et al. (2016) Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535 (7610), 153–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shi J et al. (2014) Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514 (7521), 187–92. [DOI] [PubMed] [Google Scholar]
- 107.Hagar JA et al. (2013) Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341 (6151), 1250–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kayagaki N et al. (2013) Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341 (6151), 1246–9. [DOI] [PubMed] [Google Scholar]
- 109.Wang K et al. (2020) Structural Mechanism for GSDMD Targeting by Autoprocessed Caspases in Pyroptosis. Cell 180 (5), 941–955 e20. [DOI] [PubMed] [Google Scholar]
- 110.Liu Z et al. (2020) Caspase-1 Engages Full-Length Gasdermin D through Two Distinct Interfaces That Mediate Caspase Recruitment and Substrate Cleavage. Immunity 53 (1), 106–114 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rathinam VA et al. (2012) TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150 (3), 606–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Orning P et al. (2018) Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362 (6418), 1064–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sarhan J et al. (2018) Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci U S A 115 (46), E10888–E10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Demarco B et al. (2020) Caspase-8-dependent gasdermin D cleavage promotes antimicrobial defense but confers susceptibility to TNF-induced lethality. Sci Adv 6 (47), eabc3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schwarzer R et al. (2020) FADD and Caspase-8 Regulate Gut Homeostasis and Inflammation by Controlling MLKL- and GSDMD-Mediated Death of Intestinal Epithelial Cells. Immunity 52 (6), 978–993 e6. [DOI] [PubMed] [Google Scholar]
- 116.Tsuchiya K et al. (2019) Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat Commun 10 (1), 2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rogers C et al. (2019) Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun 10 (1), 1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Schneider KS et al. (2017) The Inflammasome Drives GSDMD-Independent Secondary Pyroptosis and IL-1 Release in the Absence of Caspase-1 Protease Activity. Cell Rep 21 (13), 3846–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Benaoudia S et al. (2019) A genome-wide screen identifies IRF2 as a key regulator of caspase-4 in human cells. EMBO Rep 20 (9), e48235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Humphries F et al. (2020) Succination inactivates gasdermin D and blocks pyroptosis. Science 369 (6511), 1633–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang Y et al. (2019) Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J Mol Cell Biol 11 (12), 1069–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Aglietti RA et al. (2016) GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A 113 (28), 7858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chen R et al. (2019) cAMP metabolism controls caspase-11 inflammasome activation and pyroptosis in sepsis. Sci Adv 5 (5), eaav5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chen X et al. (2019) NEK7 interacts with NLRP3 to modulate the pyroptosis in inflammatory bowel disease via NF-kappaB signaling. Cell Death Dis 10 (12), 906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Aki T et al. (2020) Extracellular glucose is crucially involved in the fate decision of LPS-stimulated RAW264.7 murine macrophage cells. Sci Rep 10 (1), 10581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ruhl S et al. (2018) ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362 (6417), 956–960. [DOI] [PubMed] [Google Scholar]
- 127.Eren E et al. (2020) Irgm2 and Gate-16 cooperatively dampen Gram-negative bacteria-induced caspase-11 response. EMBO Rep 21 (11), e50829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Xu W et al. (2021) Apaf-1 Pyroptosome Senses Mitochondrial Permeability Transition. Cell Metab 33 (2), 424–436. [DOI] [PubMed] [Google Scholar]
- 129.Guo C et al. (2017) Development and Characterization of a Hydroxyl-Sulfonamide Analogue, 5-Chloro-N-[2-(4-hydroxysulfamoyl-phenyl)-ethyl]-2-methoxy-benzamide, as a Novel NLRP3 Inflammasome Inhibitor for Potential Treatment of Multiple Sclerosis. ACS Chem Neurosci 8 (10), 2194–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zeng L et al. (2017) ALK is a therapeutic target for lethal sepsis. Sci Transl Med 9 (412). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhang B et al. (2018) ALK is required for NLRP3 inflammasome activation in macrophages. Biochem Biophys Res Commun 501 (1), 246–252. [DOI] [PubMed] [Google Scholar]
- 132.Yang J et al. (2018) Mechanism of gasdermin D recognition by inflammatory caspases and their inhibition by a gasdermin D-derived peptide inhibitor. Proc Natl Acad Sci U S A 115 (26), 6792–6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Haag SM et al. (2018) Targeting STING with covalent small-molecule inhibitors. Nature 559 (7713), 269–273. [DOI] [PubMed] [Google Scholar]
- 134.Jiang Y et al. (2019) Discovery of Second-Generation NLRP3 Inflammasome Inhibitors: Design, Synthesis, and Biological Characterization. J Med Chem 62 (21), 9718–9731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hu JJ et al. (2020) FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat Immunol 21 (7), 736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]