

Abstract
The current pharmaceutical market lacks therapeutic agents designed to modulate behavioral disturbances associated with dementia. To address this unmet medical need, we designed multifunctional ligands characterized by a nanomolar affinity for clinically relevant targets that are associated with the disease pathology, namely, the 5-HT2A/6/7 and D2 receptors. Compounds that exhibited favorable functional efficacy, water solubility, and metabolic stability were selected for more detailed study. Pharmacological profiling revealed that compound 11 exerted pronounced antidepressant activity (MED 0.1 mg/kg), outperforming commonly available antidepressant drugs, while compound 16 elicited a robust anxiolytic activity (MED 1 mg/kg), exceeding comparator anxiolytics. In contrast to the existing psychotropic agents tested, the novel chemotypes did not negatively impact cognition. At a chronic dose regimen (25 days), 11 did not induce significant metabolic or adverse blood pressure disturbances. These promising therapeutic-like activities and benign safety profiles make the novel chemotypes potential treatment options for dementia patients.
Introduction
The very first portrait of a dementia patient, described by Alois Alzheimer in 1907, reported an elderly woman who, along with severe cognitive decline, manifested disturbing neuropsychiatric symptoms.1 Nowadays, the symptoms of abnormal behavior have been termed as behavioral and psychological symptoms of dementia (BPSD) and are recognized as an inherent element of Alzheimer’s disease (AD) and other types of dementia.2 The spectrum of behavioral abnormalities includes agitation, aggression, irritability, apathy, depressive mood, anxiety, psychosis, and reduced sociability. BPSD affects approximately 90% of dementia individuals and is considered to be an even more disturbing symptom in everyday care than memory impairment.2,3
The pharmacological management of BPSD poses an enormous challenge to psychiatrists, since specifically designed and approved pharmacotherapies remain inaccessible.4 Consequently, the most severe BPSD such as depressive mood, aggression, agitation, and psychosis have had to be addressed with available psychotropic drugs.5 Antidepressants have been used in an attempt to treat the depressive mood commonly exhibited by dementia individuals.6 However, the effectiveness of these medications remains dubious, as shown by the recent systematic review by Cochrane.7 Moreover, treatment with antidepressants poses a risk of side effects that geriatric patients are particularly sensitive to, such as cognitive slowing or cardiac arrhythmias.8−11 Clinical practice showed that the use of atypical antipsychotics in the treatment of dementia-related psychosis or agitation/aggression gives modest efficacy at the expense of a high risk of side reactions.12,13 Elderly individuals appear to be particularly responsive to antipsychotic-induced metabolic and cardiovascular side reactions as well as cognitive deterioration, which results in significantly higher mortality.14,15 In addition, clinicians do not support the routine use of benzodiazepines in dementia patients because these drugs induce excessive daytime sleepiness with resulting cognitive slowing and increased risk of abnormal falls and resulting injuries.16
Given the unfavorable status related to the current pharmacotherapy of BPSD, more emphasis should be placed on the development of specifically designed and well-tolerated medications for BPSD. Clinicians accentuate that, to achieve optimal clinical efficacy, BPSD patients should be treated with therapeutic agents that interact with pharmacologically relevant targets, matching the pathogenesis of the disease.17,18 BPSD are a result of complex pathological processes induced by neurodegeneration and aging, running in parallel for several years. These processes cause shifts in neurotransmitter abundance, which, in turn, trigger changes in the expression and function of certain receptors.19
Genetic studies have made important strides in uncovering the specific receptor changes that contribute to the onset of BPSD. For instance, a robust association between the polymorphism of the serotonin 5-HT2A receptor and the onset of psychosis in AD patients was found.20,21 This evidence supports the relevance of targeting the 5-HT2A receptor to mitigate psychotic symptoms in dementia patients. The pertinence of this strategy was confirmed in clinical use, and a 5-HT2A inverse agonist pimavanserin is pending approval for the treatment of dementia-related psychosis.22 Another relevant molecular target that closely correlates with the BPSD pathology includes the serotonin 5-HT6 receptor. Postmortem studies have revealed a reduced density of 5-HT6 receptors in the prefrontal cortex of AD patients who manifested neuropsychiatric symptoms, compared to age-matched healthy controls.23−25 The results from animal models showed that pharmacological modulation of 5-HT6 receptor activity accounts for anxiolytic, antidepressant, and memory-enhancing properties.26 These shreds of evidence validate the 5-HT6 receptor as a therapeutic target in managing neuropsychiatric symptoms in dementia patients.
Similar observations were found after pharmacological modulation of another serotonin receptor subtype—5-HT7.27 In experimental studies, 5-HT7 receptor antagonists elicited pronounced antidepressant and anxiolytic-like activity. Reduced levels of serotonin 5-HT7 receptors were observed in the brain of aging rodents,28 and decreased expression of 5-HT7 receptors in the hippocampus was linked to age-related memory impairment.29 The dopamine D2 receptor acquired considerable importance as a potential target for BPSD after clinical studies revealed changes in the abundance of dopamine D2 receptors in AD patients that manifest behavioral abnormalities.30−32 Other studies suggested that dysfunction of dopaminergic signaling may result in the onset of apathy and loss of enjoyment in AD patients. Therefore, targeting D2 receptors to mitigate behavioral symptoms in dementia patients constitutes a rational intervention.
Given the multifactorial origin of BPSD, acting on an individual target might be insufficient to deliver the desired therapeutic efficacy. In fact, a recent belief that “single drugs do not cure complex diseases” has shifted drug discovery paradigms toward the development of multifunctional drugs, which can hit simultaneously multiple targets.33,34 Such a strategy offers a prospect of tackling several biological targets relevant to the disease and, thus, achieve a superior therapeutic window, compared to target-selective therapies. The attractiveness of such an approach inspired us to design multifunctional ligands dedicated to the sensitive population of dementia patients. In the pursuit of a novel anti-BPSD agent, we sought to obtain multifunctional molecules that will modulate the activity of the target combination that underlines BPSD pathology. We identified 5-HT2A, 5-HT6, 5-HT7, and D2 receptors as major druggable targets in the development of potential anti-BPSD agents because their dysregulation underlies many pathophysiological conditions associated with BPSD. Herein we report the design, synthesis and comprehensive in vitro characterization of a series of novel promising chemotypes based on the arylsulfone and methylopyrrolidine modifications of 6-fluoro-3-[3-(pyrrolidin-1-yl)propyl]benzo[d]isoxazole scaffolds that elicit a multifunctional profile in relation to laboratory-based models representing BPSD. To confirm the therapeutic potential of the presented chemotypes, the most promising molecules were comprehensively characterized in extended pharmacological studies. Finally, considering the specific sensitivity of dementia patients to drug-induced adverse reactions, in the final stage, we determined whether these novel compounds exhibit favorable safety effects that would be compatible with the further development of these dual chemotypes as successful anti-BPSD agents.
Results and Discussion
Design
One of the strategies practiced in pursuit of multifunctional agents is “the framework combination approach”, which takes two distinct pharmacophores and combines them into a single chemical entity that possesses the activity of both original fragments. In this approach, the degree of framework overlap is systematically increased until the frameworks overlap maximally to give the simplest single molecule, with druglike properties. The framework combination strategy has been already successfully applied in drug discovery to develop multifunctional drugs that have been approved for human clinical use.35 On the basis of this strategy, we have previously successfully identified several series of druglike multifunctional ligands acting on a defined set of serotonin and dopamine receptors, with a representative molecule 1 portrayed in Figure 1. We found that the superimposition of the selective 5-HT7 receptor antagonist, SB 258719, with the potent 5-HT6 receptor antagonist, SB214111, showed a remarkable overlap of the pharmacophore features to yield a common N-(3-(piperidin-1-yl)propyl)benzenesulfonamide fragment, with molecular recognition for both 5-HT6 and 5-HT7 receptors. Structure–activity relationship (SAR) studies around this scaffold revealed that the replacement of the piperidine ring with pyrrolidine markedly altered the geometry of the molecule and conferred a balanced 5-HT2A/D2 receptor functionality to the 6-fluoro-3-(3-(pyrrolidin-1-yl)propyl)benzo[d]isoxazole core. We took advantage of the basic nitrogen atom, which is present in both frameworks, and used it to merge both fragments to construct the smallest possible molecule with acceptable physicochemical properties, namely, compound 1.36 Although 1 displayed a well-balanced affinity for the defined biological targets (pKi > 7), which encouraged us to further explore this chemotype, it was characterized by high microsomal clearance, requiring the need for possible improvements. We found that all of the molecules in this series, including 1, underwent metabolic degradation with the predominant transformation routes involving sulfonamide bond cleavage37 and pyrrolidine N-dealkylation (Supporting Information Figure S1). Therefore, we reasoned that the metabolic stability of this compound might be rationally tuned by the replacement of the labile sulfonamide function with a bioisosteric sulfone (series I) and the pyrrolidine ring substituted by methylpyrrolidine (series II). The latter modification was to introduce steric hindrance against CYP450-mediated oxidation, thus reducing N-dealkylation.38 In the present study, we undertook structural modifications of the core 6-fluoro-3-[3-(pyrrolidin-1-yl)propyl]benzo[d]isoxazole moiety, in pursuit of more metabolically stable chemotypes, and verification of how these systemic modifications would affect the receptor profile of the resulting novel compounds (Figure 1).
Figure 1.

Design strategy based on “the framework combination approach” to deliver multifunctional ligand 1, containing a central basic nitrogen atom within the overlapping boxes representing the combined scaffolds. Further structural modifications resulted in the novel series based on arylsulfone and methylpyrrolidine fragments.
The compounds complied with Lipinski and Veber criteria (except for compounds 19 and 22, due to exceeded molecular weight, and compound 20, with exceeded MW and log P). The pKa values of the most basic nitrogen atom of the compounds did not exceed 8, which limits the risk of poor cell membrane permeability due to ionization issues, or P-gp efflux susceptibility. The series was characterized by promising values of Central Nervous System Multiparameter Optimization (CNS MPO) reaching up to 5.1 (median 4.1). Moreover, none of the compounds contained substructural features recognized as pan assay interference compounds (PAINS, reported by SwissADME). The predicted molecular properties suggested a druglike profile, possibly favorable bioavailability and low risk of attrition of the designed compounds (see Supporting Information Table S1). In addition, we expected that, by changing the substitution pattern around the aryl ring, we could enhance the biological activity profile of the new chemotypes, as arylsulfone moieties have been previously reported to demonstrate an affinity for 5-HT6 and 5-HT7.39,40 The structural changes were monitored using a structure-based molecular modeling approach to predict the binding capabilities of the modified core structures. Docking into the biological targets of interest proved that the binding modes of the designed compounds resembled those of the prototype representatives (Figure 2).36 Such an observation suggested that the affinity should remain high, despite the modifications that were primarily made to increase metabolic stability. The chemical structures of the novel series are presented in Tables 1 and 2. In series II, featuring a methylpyrrolidine ring, we decided to investigate the role of the 3S,5S enantiomer, due to the superior docking score for all of the targeted receptors.
Figure 2.

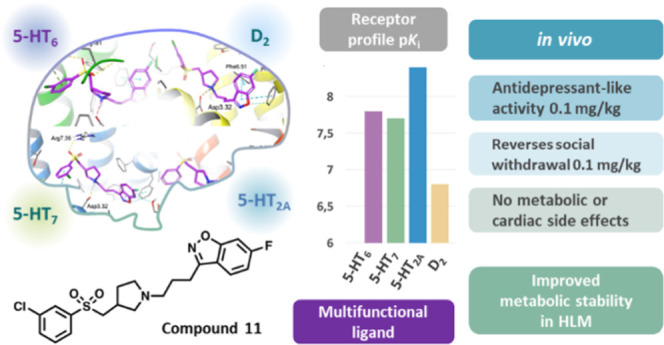
Proposed binding mode of representative compound 11 with the targeted receptors. The arylsulfone fragment substituted with an alkylarylamine moiety satisfied the required interactions for both the 5-HT7 and 5-HT6 receptor binding sites (homology models based on 2RH1 and 4IAR, respectively), mimicking the interactions of their reference ligands.41−43 6-Fluoro-benzo[d]isoxazole linked to the propylamine moiety constitutes a pharmacophore with blocking properties for the 5-HT2A and D2 receptors (homology models based on 4IB4 and 3PBL, respectively).44,45 The design stage resulted in a series of 21 ligands potentially characterized by high affinity for both the desirable biological targets. Key amino acid residues engaged in ligand binding (within 4 Å from the ligand atoms) are displayed as thick sticks together with their interactions: salt bridges (dotted magenta lines), H-bonds (dotted yellow lines), halogen bonds (dotted violet lines), π–π stacking (dotted cyan lines), or cation−π (dotted green lines). The detailed complexes of lead compounds representing series I (compound 7) and series II (compound 16) are shown in Supporting Information Figures S2–S5. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.).
Table 1. Structure and Binding Affinities for 5-HT2A, 5-HT6, 5-HT7, and D2 Receptors of Compounds 2–12 (Series I) and Reference Drugsa.


Binding affinity values are represented as pKi (i.e., −log Ki) and expressed as mean ± standard error of the mean (SEM) from at least three experiments performed in duplicate.
Table 2. Structure and Binding Affinities for 5-HT2A, 5-HT6, 5-HT7, and D2 Receptors of Compounds 13–22 (Series II) and Reference Drugsa.


Binding affinity values are represented as pKi (i.e., −log Ki) and expressed as mean ± SEM from at least three experiments performed in duplicate.
Synthesis
The series I compounds 2–12 were prepared in a four-step synthesis, as portrayed in Scheme 1. The synthesis commenced with the preparation of key building blocks III a–k. The starting thiol derivatives were S-alkylated with N-Boc-3-chloromethyl pyrrolidine and then subjected to an oxidation reaction in the presence of m-chloroperbenzoic acid (mCPBA), to deliver the corresponding sulfone derivatives II a–k. The latter were first deprotected with HCl/EtOAc to give key amines III a–k, which then reacted with 3-(3-chloropropyl)-6-fluorobenzo[d]isoxazole IV to yield the final products, 2–12.
Scheme 1. Reagents and Conditions: (a) K2CO3, EtOH, 80 °C, 2–6 h; (b) mCPBA, Dichloromethane (DCM), rt, 1 h then 35 °C, 12 h (c) 1 M HCl in EtOAC, rt, 12 h; (d) K2CO3, MeCN, KI, 60 °C, 24 h.

We next addressed the synthesis of series II, namely, Compounds 13–22 (Scheme 2). The key intermediate VI was constructued from commercially available 5-methylpyrrolidin-3-amine and 3-(3-chloropropyl)-6-fluorobenzo[d]isoxazole IV, which were first reacted in the presence of K2CO3 and KI to give an intermediate, tert-butyl (1-(3-(6-fluorobenzo[d]isoxazol-3-yl)propyl)-5-methylpyrrolidin-3-yl)carbamate, V. Next, the removal of Boc protecting moiety under acidic conditions yielded VI, which was further alkylated with corresponding sulfonyl chlorides to produce the desired molecules, Compounds 13–22.
Scheme 2. (a) K2CO3, CH3CN, 60 °C, 48 h; (b) HCl/EtOAc 12 h, rt; and (c) Cs2CO3, 4-Dimethylaminopyridine (DMAP), DCM, rt, 12 h.

Structure–Activity Relationships
Series I
All of the synthesized compounds were characterized in radioligand binding assays to establish their affinity for target serotonin 5-HT2A, 5-HT6, 5-HT7, and dopamine D2 receptors (Tables 1 and 2). The vast majority of compounds performed well in the in vitro assays and displayed marked affinities for all of the desired receptors, with pKi values ranging from 6.19 to 9.61. It is noteworthy that we observed a favorable difference between the pKi for the 5-HT2A receptor and the pKi for the D2 receptor for all compounds, within the range of 1–1.5, which translates to a 10-fold to 100-fold preference for the 5-HT2A receptor. This testifies its promising safety, particularly in terms of the appearance of potential extrapyramidal side effects. The potent 5-HT2A binding capabilities were proven for a majority of the sulfone derivatives, with pKi values ranging between 7.40-the 5-HT6, 5-HT7, and dopamine D2 receptors varied depending on the structure of the aryl fragment. Our first attempts focused on assessing the impact of various substituents incorporated into the meta position of the phenyl ring on the resulting interactions with 5-HT6/5-HT7 and D2 receptors. The affinity values were compared to the derivative containing a 3-Cl benzyl function, which was well tolerated by all targets (pKi5-HT6 = 7.78, pKi5-HT7 = 7.67, pKiD2 = 6.79, pKi5-HT2A = 8.36), and, based on these studies, 11 was chosen as the lead structure for this series.
In series I, we observed that the replacement of the chlorine atom at the meta position with a methyl group (compound 12) resulted in a negligible change in affinity for all tested receptors. On the other hand, introducing a fluorine atom at the meta position (compound 10) led to a decrease in activity for all targets, which was the most prominent for the 5-HT6 and 5-HT2A receptors (pKi5-HT6 = 6.82 and pKi5-HT2A = 7.40 vs pKi5-HT6 = 7.78 and pKi5-HT2A = 8.36 for 11).
Incorporation of a CF3 substituent at the meta position of the phenyl ring (compound 8) resulted in reduced affinity for 5-HT7 (pKi = 7.17) and D2 (pKi = 6.52) receptors, with no substantial changes toward the 5-HT6 and 5-HT2A receptors. Shifting the CF3 group from the meta to para position (compound 7) slightly improved affinity for the D2, 5-HT2A, and 5-HT7 receptors (pKi = 7.02, 8.71, 7.33, respectively), with no influence on the 5-HT6 receptor (pKi = 7.81). Compared to lead structure 11, the para CF3 derivative 7 maintained a high affinity for the 5-HT6 receptor, a slightly improved affinity for D2 (pKi = 7.02) and 5-HT2A (pKi = 8.71) receptors, but showed a small decrease in affinity for the 5-HT7 receptor (pKi = 7.33). Introducing a fluorine atom at the para position (9) led to a slight decrease in affinity for all targets, in comparison to 11.
Further studies focused on the exploration of different types of 3,4-disubstituted derivatives. The introduction of a second chlorine atom at the para position was well tolerated and compound 5, featuring a 3,4-dichlorophenyl group, showed high affinity for all receptor targets (pKi = 7.30–8.77), comparable with the mono-substituted compound, 11. In the case of the 3-chloro-4-fluorophenyl derivative, 6, no substantial changes in affinity were observed. Compound 4, featuring a 3,4-difluorophenyl group, maintains almost the same level of activity as its mono-substituted para analogue, 9. In comparison with the 3-methylphenyl derivative, 12, incorporation of a second methyl group at the 4-position of the phenyl ring (compound 3, 3,4-dimethylphenyl derivative) generally improved affinity for all targets, with exception of the 5-HT7 receptor, where a marked drop in affinity was observed (pKi = 6.69). Replacing the phenyl ring with a naphthyl ring (compound 2) maintained a high affinity for all targets (pKi = 7.81–9.61), and was particularly favorable for the 5-HT2A and 5-HT6 receptors (Table 1).
Series II
Further SAR studies revealed that the methylpyrrolidine series exhibited a high affinity for the 5-HT2A receptor with pKi values in the range of 7.89–9.19, while the affinity for the remaining receptors varied depending on the aryl fragment (Table 2). The affinities of the methylpyrrolidine series were compared to the 3-Cl benzyl derivative, 13, which was established as a starting point. Introduction of the 3-chloro-4-fluorophenyl ring, resulting in compound 14, maintained a high affinity for all targets, and pKi values were comparable to the mono-substituted analogue, 13. Replacement of the phenyl ring with 3-benzothiophene (15) or 2-benzothiophene (16) improved affinity for the 5-HT6 receptor (pKi = 7.58 and 7.79 respectively) and the D2 receptor (pKi = 7.27 and 7.26). In the case of 2-benzothiophene (16), we noticed also an improvement in binding affinity for the 5-HT2A receptor, with a pKi value of 9.16. Incorporation of the 2-benzofuran ring (17) slightly reduced affinities for the 5-HT6 (pKi = 6.93) and 5-HT7 (pKi = 6.75) receptors, while the activity against the D2 receptor increased (pKi = 7.52), compared with 13. Introduction of a 6-fluoro-2-benzothiophene ring (compound 18), improved interaction with 5-HT2A (pKi = 9.19), 5-HT7 (pKi = 7.31) and D2 (pKi = 7.27) receptors, while a slight reduction was observed in the affinity for the 5-HT6 receptor (pKi = 7.05). Swapping the fluorine atom (compound 18) for chlorine at the 6-position of the 2-benzothiophene ring in compound 19 resulted in a substantial decrease in affinity for the 5-HT2A receptor (pKi = 8.38), but a slight decrease in affinity for 5-HT7 (pKi = 6.90), D2 (pKi = 6.95), and 5-HT6 (pKi = 6.99) receptors. Similar activities were observed for compound 20 (5-chloro-3-methyl-1-benzothiophene derivative). When the phenyl ring was replaced with a 2-naphthyl ring, compound 21, affinities for all receptors increased. However, placing an additional chlorine atom in the naphthyl ring (22) resulted in a considerable decrease in affinity for all targets. Based on the above results, we observed that unsubstituted heterocycles make the compounds more favorable for achieving proportionate interactions with all four receptors (Table 2).
In Vitro Functional Activity
The most promising compounds, 2, 3, 5, 7, 11, 15, 16, 18, and 21 were subsequently submitted to functional studies. The majority of compounds exerted a prominent antagonist effect against all targeted receptors with nanomolar KB values (pKB 6.60–8.30) (Table 3) The sulfone derivatives (series I) performed slightly better than the methylpyrrolidine derivatives (series II) and a majority of compounds from series I demonstrated KB values < 100 nM (pKB > 7). Among the methylpyrrolidine derivatives, compounds bearing 3-benzothiophene (15), 2-benzothiophene (16), and 2-naphthyl (21) demonstrated the most efficacious antagonistic responses, with pKB values > 6.34.
Table 3. Functional Data for the Selected Compoundsb.
| antagonist
effect pKB ± SEM |
||||
|---|---|---|---|---|
| compd | 5-HT6R | 5-HT7R | D2R | 5-HT2AR |
| 2 | 7.26 ± 0.01 | 7.06 ± 0.02 | 7.62 ± 0.01 | 8.22 ± 0.20 |
| 3 | 7.02 ± 0.01 | 6.85 ± 0.01 | 7.81 ± 0.01 | 8.40 ± 0.34 |
| 5 | 7.41 ± 0.02 | 7.26 ± 0.01 | 7.22 ± 0.02 | 7.96 ± 0.04 |
| 7 | 7.44 ± 0.03 | 7.42 ± 0.03 | 8.21 ± 0.01 | 7.99 ± 0.07 |
| 11 | 7.12 ± 0.01 | 7.36 ± 0.01 | 7.34 ± 0.03 | 7.81 ± 0.58 |
| 15 | 6.82 ± 0.02 | 7.12 ± 0.01 | 6.87 ± 0.01 | 6.82 ± 0.01 |
| 16 | 6.74 ± 0.05 | 6.40 ± 0.01 | 6.73 ± 0.02 | 7.79 ± 0.58 |
| 18 | 6.41 ± 0.02 | 5.62 ± 0.01 | 6.79 ± 0.01 | 8.30 ± 0.19 |
| 21 | 6.77 ± 0.07 | 6.34 ± 0.02 | 6.60 ± 0.02 | 7.63 ± 0.01 |
| SB-42457 | 9.47 ± 0.01 | |||
| SB-269970 | 9.7 ± 0.01 | |||
| Chlorpromazine | 8.80 ± 0.0 | |||
| Pimavanserin | 9.17 ± 0.01 | |||
All of the functional activity values were expressed as mean from at least three experiments performed in duplicate.
Blank spaces—compounds not tested in the assays.
Determination of Interaction with Off-Target Receptors
It is becoming increasingly evident that dementia patients are acutely sensitive to drug-induced adverse reactions and show a higher prevalence of metabolic syndrome, stroke, and related conditions caused by psychotropic medications (antidepressants and antipsychotics in particular).46,47 The results from population-based studies clearly demonstrate that these undesirable events result in higher mortality among the geriatric dementia population. Higher occurrence of drug-induced side effects has been closely related to psychotropic drugs that exert high affinity for off-targets, such as the histamine (H1), muscarinic (M1), and α1 adrenergic receptors (e.g., imipramine, amitriptyline, doxepin, or antipsychotics: quetiapine and olanzapine).48 Small molecules interacting with histamine H1 receptors have been linked with drug-induced metabolic disturbances, weight gain, and excessive daytime sleepiness.49,50 Moreover, clinical studies found an association between high-H1 affinity antidepressants (e.g., amitriptyline, doxepin) and dysregulation of glucose homeostasis, resulting in hyperglycemia.51 Blockade of muscarinic M1 receptors may induce tachycardia, which drives precarious hemodynamic conditions, changes blood flow in intraparenchymal microcirculation which, in turn, can increase the risk of stroke (olanzapine and quetiapine).52,53 Furthermore, blockade of M1 receptors escalates the cholinergic deficit and intensifies memory impairment in dementia subjects who suffer from cognitive deterioration. Anticholinergic activity generates additional unpleasant reactions, namely, severe constipation and urine retention, which adds another layer of undesirable effects. Lastly, treating dementia subjects with antidepressants or antipsychotics may trigger orthostatic hypotension, which has been closely related to the blockade of the adrenergic α1 receptor. Consequently, it has been suggested that potential anti-BPSD agents should be devoid of any interactions with adverse targets, in particular: histamine H1, muscarinic M1, and adrenergic α1 receptors.54
Considering the above, we conducted in vitro screening to verify potential interactions of the two series of compounds with the above-mentioned undesirable targets. The two series of compounds proved to elicit a less disruptive profile than the reference antidepressants and antipsychotics, exhibiting substantially lower values of % inhibition of control specific binding at a concentration of 1.0 × 10–6 M, compared to the off-targets (Table 4). The two series of compounds showed an almost negligible binding with muscarinic M1 receptors. This observation is in line with our previous studies which show that arylsulfonamide-based hybrids bind poorly to muscarinic M1 receptors, due to insufficient space within the receptor binding site.45 However, we observed a slight tendency toward binding with the H1 and α1 receptors, particularly among the sulfone series of compounds 2, 3, 5, 7, 11 (α1: 43-66% at 1.0 × 10–6 M, H1: 24–58% at 1.0 × 10–6 M), but this was still substantially lower than that of the reference drugs (α1: 75–82% at 1.0 × 10–6 M, H1: 75–99%). This could be attributed to the presence of an aryl fragment featured with a basic pyrrolidine in these compounds, a common pharmacophore shared with other monoaminergic receptors. In general, an increased tendency to interact with α1 and H1 receptors was observed for the 3,4-disubstituted analogues, 3 and 5. Interestingly, the methylpyrrolidine series (15, 16, 18, and 21) did not show a significant affinity for the H1 or α1 receptors, indicating that the methyl group around the pyrrolidine moiety impairs H1 and α1 receptor binding. In summary, the majority of compounds proved to have a relatively high safety window, showing negligible interaction with the M1 receptor. In the case of the methylpyrrolidine series, we observed no significant affinity for the H1 and α1 receptors, while the sulfone series showed a limited interaction with H1 and α1 receptors. These promising biological activities permitted us to consider all of the compounds as eligible for further in vitro profiling.
Table 4. Off-Target Activity Data for the Selected Molecules.
| % activity
at 1.0 × 10–6 Ma |
||||
|---|---|---|---|---|
| compd | H1R (%) | α1R (%) | M1R (%) | hERGb (%) |
| 2 | 39 | 48 | –15 | 23 |
| 3 | 58 | 66 | 5 | 34 |
| 5 | 54 | 39 | 4 | 31 |
| 7 | 29 | 56 | –7 | 28 |
| 11 | 24 | 43 | –5 | 29 |
| 15 | 10 | 13 | 11 | 29 |
| 16 | 2 | 14 | –1 | 40 |
| 18 | 19 | 19 | 7 | 38 |
| 21 | 4 | 15 | –6 | 47 |
| Phentolamine | 91 | 76 | ||
| Mepyramine | 95 | 55 | ||
| Amitriptyline | 99 | 82 | 78 | |
| Doxepin | 97 | 92 | ||
| Quetiapine | 76 | 75 | 88 | |
| Olanzapine | 89 | 82 | 89 | |
| Verapamil | 91 | |||
% inhibition of control specific binding at the concentration of 1.0 × 10–6 M. Assays carried out in duplicate (n = 2). Blank spaces—compounds not tested in the assay.
% inhibition of hERG-mediated potassium currents at a concentration of 1.0 × 10–6 M.
Additionally, we established the potency of selected compounds to block the human hERG potassium channel, which is the most common mechanism related to drug-induced cardiotoxicity, via prolongation of the QT interval. The compounds were tested in a whole-cell electrophysiological assay at a single concentration of 1.0 × 10–6 M, and induced inhibition of hERG in the range of 20–47%. This would suggest that these compounds would display a relatively low occurrence of cardiac side effects.
Developability Analysis—In Vitro Assays
In the present study, our goal was to develop metabolically stable compounds from within the series I and II compound syntheses. We also paid close attention to the physicochemical properties by measuring the thermodynamic solubility of selected compounds.55 In general, sulfone analogues were characterized by superior solubility, ranging from 62 to 306 μg/mL (Table 5). Two compounds, 11 and 7, displayed remarkable solubilities (306.29 and 204.75 μg/mL, respectively), exceeding the reference drug metoprolol (193 μg/mL). Benzothiophene derivatives, 16 and 18, showed acceptable water solubility, while the other two 15, 21 were markedly less soluble (<0.1 μg/mL), and with a high probability that they would require more complex formulations for in vivo use. Therefore, compounds X and Y were excluded from further development. At this point, we selected the most promising compounds from series I namely: 2, 7, 11, and two methylpyrrolidine derivatives from series II, namely, 16 and 18. The compound selection was guided by the overall in vitro properties of potency and efficacy, as well as aqueous solubility.
Table 5. Thermodynamic Solubility Data of Selected Compounds.
| compda | aqueous solubility μg/mL |
|---|---|
| 2 | 128.38 ± 1.61 |
| 3 | 132.7 ± 0.74 |
| 5 | 62.44 ± 1.05 |
| 7 | 204.75 ± 1.68 |
| 11 | 306.29 ± 0.80 |
| 15 | <0.1 |
| 16 | 1.62 ± 0.1 |
| 18 | 9.16 ± 0.42 |
| 21 | <0.1 |
| Metoprolol | 193 ± 1.02 |
Prior to the measurement, compounds were converted to the corresponding hydrochloride salts.
The selected compounds, 2, 7, 11, 16, and 18, were subsequently tested for metabolic stability using human liver microsomes. We determined the quality of these compounds by comparing them head-to-head with the previously developed sulfonamide counterparts.36 In general, the derivatives were less heavily metabolized than the parent sulfonamides (Figure 3). This was particularly true for the sulfone derivatives, which showed very promising biological results and were characterized by increased metabolic stability. The highest stability was observed for compound 7, bearing a 4-CF3-phenyl moiety, which displayed desirable values, including a half-life of over 60 min and a hepatic clearance value below 115 μL/(min mg). The remaining sulfone derivatives composed of a naphthyl ring (compound 2) and a 3-chlorophenyl substituent (compound 11), also displayed a substantial increase in stability over the parent compounds, A and C, and half-life increased almost 2-fold, along with improved clearance values (CLint = 304.3 μL/(min mg) for 2 and CLint = 217.3 μL/(min mg) for 11). Among the methylpyrrolidine series, compound 16, bearing the 2-benzothiophene motif, showed prominent improvement of stability, and its clearance value was 2-fold lower (CLint = 252.1 μL/(min mg)) in comparison to its sulfonamide analogue, D (CLint = 584.5 μL/(min mg)). The addition of one fluorine atom into the 2-benzothiophene ring gave a modest improvement of stability, as compound 18 showed a half-life of only 11 min, with a metabolic clearance CLint = 610.4 μL/(min mg). Although these values were improved in comparison to the corresponding sulfonamide analogue, E (CLint = 1204.3 μL/(min mg)), compound 18 remained heavily metabolized and, consequently, 18 was ruled out from further evaluation.
Figure 3.

Metabolic stability of selected compounds in human liver microsomes. The study was performed at a concentration of 1.0 × 10–7 M. Determination of % of compound recovered after selected periods of time (0, 5, 15, 30, 45, and 60 min) was measured according to previously published protocols.56 Internal references: verapamil: t0.5 = 26 min, CLint = 267 mL/min/mg, imipramine: t0.5 = >60 min, CLint = <115.5 mL/min/mg.
Altogether, the above findings indicate that chemical modifications of the compounds aimed at blocking the predominant metabolic pathways, namely, sulfonamide bond cleavage and pyrrolidine N-dealkylation, resulted in a reduction of microsomal turnover. At this point of the study, compounds 7 and 11 emerged as the most promising compounds for further evaluation, because of their markedly superior metabolic stability. We decided also to include a methylpyrrolidine representative, compound 16, and explore it further.
In Vivo Studies: Pharmacokinetics
The pharmacokinetic (PK) properties of key molecules, 7, 11, and 16, were assessed in vivo. Plasma concentration values versus time were examined by a noncompartmental approach. Figures 4A, 5A, and 6A show that all of the studied compounds administered at a dose of 2 mg/kg i.p. were rapidly absorbed from the peritoneal cavity (tmax 5.0–22.5 min). Peak plasma concentrations of 7 and 16 were relatively high (Cmax = 193.44 and 179.33 ng/mL, respectively), while Cmax of 11 was lower (Cmax = 69.68 ng/mL). All of the tested compounds were characterized by a slow terminal elimination, resulting in a favorable value for serum elimination half-time (t0.5λz = 318.59 min for 11, 96.09 min for 7, and 55.67 min for 16). The area under the concentration–time curve from the time of dosing to infinity (AUC0–∞) for serum was 36 269.95 ng·min/mL for 7, 17 368.37 ng·min/mL for 11 and 7060.67 ng·min/mL for 16. The apparent volume of distribution (Vz/F) during the terminal phase was 7.96 L/kg for 7, 52.99 L/kg for 11, and 23.04 L/kg for 16. Clearance (Cl/F) was 0.12 L/min for 11, 0.06 L/min for 7 and 0.29 L/min for 16 (Table 6, Figures 4A, 5A, and 6A). Notably, the concentrations of sulfone derivatives 7 and 11 in brain tissue were particularly high, reaching up to 333.65 and 178.32 ng/g after 30 min, respectively (Figures 4B and 5B). The concentrations determined in plasma at this point ranged between 32.23 and 116.43 ng/mL. In the case of the methylpyrrolidine derivative, 16, the plasma concentrations were in the mid-range of 51.35 ng/mL, whereas the brain concentrations were relatively low (36.62 ng/g), 30 min after administration (Figure 6B). Both the sulfone derivatives maintained high brain concentrations 180 min after administration (between 107.72 and 232.26 ng/mL), suggesting superior bioavailability and metabolic stability, compared to the corresponding parameter for the methylpyrrolidine derivative, which was noticeably lower (7.67 ng/g) (Figures 4B, 5B, and 6B). Collectively, these results suggest that all compounds displayed favorable druglike pharmacokinetic properties in the rat, which prompted their pharmacodynamics evaluation.
Figure 4.

A. Time course of plasma concentrations (mean ± SD) of compound 7 administered intraperitoneally at a dose of 2 mg/kg to male Wistar rats (n = 3 per time point). B. Serum and brain concentrations of 7 administered at a dose of 2 mg/kg i.p. (n = 3).
Figure 5.

A. Time course of plasma concentrations (mean ± SD) of compound 11 administered intraperitoneally at a dose of 2 mg/kg to male Wistar rats (n = 3–4 per time point). B. Serum and brain concentrations of 11 administered at a dose of 2 mg/kg i.p. (n = 3).
Figure 6.

A. Time course of plasma concentration (mean ± SD) of compound 16 following i.p. administration of a dose of 2 mg/kg to rats (n = 3 per time point). B. Serum and brain concentrations of 16 administered at a dose of 2 mg/kg i.p. (n = 3).
Table 6. Pharmacokinetic Parameters (Mean ± Standard Deviation (SD)) of the Studied Compounds Administered to Ratsa at a Dose of 2 mg/kg i.p. Using Noncompartment Analysis.
| parameter | 7 | 11 | 16 |
|---|---|---|---|
| tmax (min) | 22.5 ± 10.61 | 8.3 ± 5.77 | 5.0 ± 0.00 |
| Cmax (ng/mL) | 193.44 ± 49.76 | 69.68 ± 5.86 | 179.33 ± 34.11 |
| λz (min–1) | 0.007 ± 0.0003 | 0.002 ± 0.0002 | 0.013 ± 0.0009 |
| t0.5λz (min) | 96.09 ± 4.25 | 318.59 ± 26.46 | 55.67 ± 4.09 |
| AUCinf (ng·min/mL) | 36 269.95 ± 8836.54 | 17 368.37 ± 1680.24 | 7060.67 ± 1056.47 |
| Vz/F (L/kg) | 7.96 ± 2.34 | 52.99 ± 6.05 | 23.04 ± 3.21 |
| CL/F (L/(min kg)) | 0.06 ± 0.01 | 0.12 ± 0.01 | 0.29 ± 0.05 |
| MRT (min) | 146.10 ± 4.67 | 435.68 ± 39.99 | 64.98 ± 4.06 |
The PK studies and all of the in vivo studies were conducted using male Wistar rats (n = 3).
In Vivo Pharmacological Profiling
The main goal of the behavioral studies was to obtain proof-of-concept at the in vivo level for the novel ligand activities in a head-to-head comparison to a series of reference drugs commonly prescribed to manage BPSD. Therefore, we selected a group of behavioral tests to evaluate the pharmacological profiles of 11 and 16 against comparator psychotropic drugs (Table 7). Given the multireceptor profile of the selected compounds, characterized by high affinities for 5-HT2A, 5-HT7, 5-HT6, and D2 receptors, we expected antidepressant, anxiolytic or antipsychotic activities to be revealed in in vivo models. Therefore, key compounds were surveyed in the forced swim test (FST, or Porsolt test), one of the most widely used procedures to assess antidepressant-like activities, as well as the Vogel conflict drinking test, to reflect potential anxiolytic-like activity and MK-801-induced hyperlocomotion, to examine antipsychotic-like activity (Table 7). In light of the fact that social skills and cognitive safety are crucial issues regarding the fragile population of geriatric patients, these results provide an indication that it could be possible to develop specifically tailored multifunctional pharmacophores that could form the basis of more advanced, safe, and selective therapeutics to address the critical unmet medical need of this patient group.
Table 7. Characterization of Selected Compounds in Behavioral In Vivo Pharmacological Tests in Comparison to Reference Drugsa.
| compd | FST MED [mg/kg] | Vogel test MED [mg/kg] | EPM MED [mg/kg] | MK-801-induced hyperactivity MED [mg/kg] | spontaneous locomotor activity MSD [mg/kg] |
|---|---|---|---|---|---|
| 7 | >1 | >1 | >3 | ||
| 11 | 0.1 | >10 | >10 | >0.3 | |
| 16 | >3 | 1 | 1 | >10 | >3 |
| Imipramine | 10 | 10 | |||
| S-citalopram | 20 | ||||
| Quetiapine | 100 | 3 | 1 | >100 | 100 |
| Risperidone | 3 | 0.3 | 1 | ||
| Olanzapine | 3 | 1 | 3 | 1 | |
| Diazepam | 5 | 2.5 | 3 | ||
| Buspirone | 1.25 | 1.25 | |||
| Aripiprazole(64) | 3 | 1064 | >364 | >100 | 10 |
| Pimavanserin(68) | 0.1b | 0.1b |
Tests carried out on Wistar rats (n = 8) after intraperitoneal (i.p.) administration. MED: minimum effective dose; MSD: minimum sedative dose, blank spaces—compounds not tested in the assay.
Experiments performed in mice.68
Forced Swim Test (FST)
Among the tested compounds, only 11 elicited pronounced antidepressant-like activity, as determined in the FST, at relatively low doses: 0.1 and 0.3 mg/kg, 100- and 66-fold lower than those observed for the reference drugs imipramine or escitalopram (MED = 10 and 20 mg/kg, respectively). Of note, alterations of mood manifested by dementia subjects, such as depressive symptoms, apathy, and irritability have been attempted to be managed with a “pharmacological cocktail” composed of antidepressants and adjunctive antipsychotics. In fact, it has been widely accepted that the addition of antipsychotic drugs to SSRI treatment produces a synergic effect in the treatment of unresponsive depressive patients.57−59 Antipsychotics like olanzapine or clozapine exert 5-HT6 and 5-HT7 receptor antagonism in the cortex and mesolimbic areas.60,61 Moreover, atypical antipsychotics block D2 and 5-HT2A receptors, which may promote indirect enhanced dopamine release in medial prefrontal circuits.62 These characteristic features are believed to provide synergic antidepressant effects which help to reach therapeutically acceptable responses in depressed patients. Notably, 5-HT6, 5-HT7, 5-HT2A, and D2 receptors antagonism remain unreachable for most antidepressants.63 Compound 11 shares some similarities with antipsychotics in terms of visible affinity for the 5-HT6, 5-HT7, 5-HT2A, and D2 receptors. We decided to compare the performance of 11 in the FST against the selected antipsychotics: risperidone, quetiapine, and olanzapine. We observed that the reference drugs developed antidepressant-like activity at 10-fold to 100-fold higher doses, namely: 3 mg/kg (risperidone, olanzapine, and aripiprazole) and at 100 mg/kg (quetiapine), compared with 11.64 Moreover, while being active in the FST, compound 11 did not influence the spontaneous locomotor activity at active doses (0.1 and 0.3 mg/kg), clearly suggesting that the observed results arise from a specific antidepressant effect of 11. Furthermore, 11 is characterized by proportionate affinities and functional activities toward all four receptors. The observed results confirm the earlier assumption that the robust antidepressant-like effect of 11 might be attributed to the extensive blockade of 5-HT6 and 5-HT7 receptors, which is potentiated by synergic efficacy derived from its interaction with 5-HT2A and D2 receptors. The functional interaction between 5-HT6, 5-HT7, 5-HT2A, and D2 receptors may affect the activity of prefrontal–subcortical circuits, in a similar manner that antipsychotics exert antidepressant responses.62 Compounds 7 and 16 did not show statistically significant efficacy in the FST in the range of tested doses (Table 7).
Vogel Conflict Drinking Test
Next, we investigated the potential anxiolytic activity of the key compounds in the Vogel conflict drinking test. Among the tested compounds, 16 produced statistically significant anxiolytic effects at a minimum effective dose of 1 mg/kg. In this regard, 16 showed similar efficacy to quetiapine (MED = 3 mg/kg) and the anxiolytic medication buspirone (MED = 1.25 mg/kg), while being more active than the multimodal antipsychotic, aripiprazole (MED = 10 mg/kg) and the gold standard anxiolytic, diazepam (MED = 5 mg/kg). Highly noteworthy, in contrast to diazepam,65 and aripiprazole, 16 did not influence spontaneous locomotor activity at a tested dose of 3 mg/kg (Table 7). The anxiolytic-like activity of 16 in the Vogel conflict drinking test seems to be specific, since the compound given at a dose of 1 mg/kg did not affect the pain reaction and unpunished water consumption in water-deprived rats.
The compounds’ efficacy was further explored in the elevated plus maze (EPM) test, in which 16 significantly and dose-dependently enhanced the total open arm time. Together, 16 showed consistent anxiolytic-like efficacy at a low dose (1 mg/kg) in two separate tests of anxiolytic activity in rats. Compound 16 displayed a distinctive high affinity for the 5-HT2A receptor (1-fold pKi) compared to the other serotonin subtypes 5-HT6, 5-HT7, as well as D2 receptors. The 5-HT2A receptor modulates a diverse array of behavioral responses related to fear and anxiety. Genetic studies with 5-HT2A receptor knock-out mice showed anxiety-like symptoms with the absence of depression-related behaviors.66 Further studies suggested that pharmacological modulation of 5-HT2A receptor activity may restore cortical signaling and normalize anxiety-like behavior. We believe that the dominant blockade of the 5-HT2A receptor accounts for the pronounced anxiolytic effect displayed by 16. These results are in line with other authors’ observations verifying the pronounced blockade of 5-HT2A receptor activity mediates predominantly anxiolytic-like effect.67 Compounds 7 and 11 did not show statistically significant efficacy in the Vogel conflict drinking test in the range of tested doses (Table 7).
MK-801-Induced Hyperactivity
Key compounds were further probed in the MK-801-induced hyperactivity test. Compounds 7 at 1 mg/kg, 11 at 3 mg/kg, and 16 at 3 mg/kg tended to decrease the total distance animals traveled. However, the effects were not statistically significant.
Despite possessing an affinity for D2 and 5-HT2A receptors, the tested compounds did not show a clear antipsychotic-like efficacy in terms of significant suppression of MK-801-induced hyperlocomotion. We compared the functional responses of 7, 11, and 16 at D2 receptor with structurally related sulfonamide series previously developed by our group.36 The lead structure of the latter series was characterized by pronounced antipsychotic-like activity and relatively high antagonistic properties at D2 receptors (pKB = 9.03) compared to 5-HT6 (pKB = 7.41), 5-HT7 (pKB = 8.72), and 5-HT2A (pKB = 8.79) receptors. The structural modifications resulting in sulfone and methylpyrrolidine series were primarily introduced to improve metabolic stability and aqueous solubility compared to the original sulfonamides. The change in the in vivo pharmacological profile that was simultaneously encountered may be attributed to the comparatively lower antagonistic properties for D2 (pKB = 6.73–8.21) and 5-HT2A (pKB = 7.79–7.99) receptors versus 5-HT6 (pKB = 6.74–7.44) and 5-HT7 (pKB = 6.40–7.42) receptors. These seemingly subtle differences might have possibly influenced the in vivo efficacy and resulted in nonsignificant alteration of MK-801-induced hyperlocomotion. Similar features have been observed by other authors who reported multifunctional ligands with surmountable antagonism for D2R and the lack of significant antipsychotic-like activity.68
The antipsychotic activity of small molecules may also be attributed to interaction with the 5-HT2A receptor. A selective 5-HT2A inverse agonist, pimavanserin exerts an antipsychotic-like effect in rodents and has been recently approved for the treatment of Parkinson’s disease-related psychosis. However, one has to notice that pimavanserin possesses a unique mechanism of action and outstandingly high affinity for 5-HT2A receptor (pKi = 10) and the lack of significant affinity for 5-HT6 and 5-HT7 receptors, which accounts for its pronounced antipsychotic-like effect.69 In contrast, compounds 7, 11, and 16 exert lower affinity for 5-HT2A receptor compared to pimavanserin and high affinity for 5-HT6, 5-HT7 receptors, which may explain the diverse pharmacological profile of the novel chemotypes.
Social Interaction Test (SIT)
One of the symptoms commonly observed in dementia subjects is social withdrawal that poses a great challenge for caregivers and family members. An animal model of impaired social functioning relies on a single administration of MK-801 to rats, which induces significant deficiencies in their social activities.70 We have used MK-801 to induce social withdrawal in rats and next examined the influence of the most promising compounds, 11 and 16, on social skills. Remarkably, administration of 11 at 0.1 and 0.3 mg/kg significantly reversed the social deficit induced by MK-801. The group treated with compound 11 spent significantly more time exploring an unknown rat, compared to the MK-801-treated group. For comparison, quetiapine, the antipsychotic drug used in the therapy of BPSD, was active in the SIT at a dose of 1.0 mg/kg. Compound 16, at 3 mg/kg, reversed social withdraw induced by MK-801 (Figure 7). These results suggest that both compounds display a favorable effect on social interactions, which is of particular significance in the view of the disrupted social functioning in BPSD patients.
Figure 7.

Effects of 11 and 16 on MK-801-induced deficits in the social interaction test (SIT) in Wistar rats. Control—vehicle-treated group (white bar), MK-801 group—after administration of 0.1 mg/kg (light gray bar), tested compound 11 (lavender blue), compound 16 (navy bar), quetiapine (dark gray). N = 7–8 pairs of rats per group Statistical analysis: one-way analysis of variance (ANOVA) (Bonferroni’s post hoc); *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 vs control (saline, white); ∧p < 0.05, ∧∧p < 0.01, ∧∧∧p < 0.001 vs MK-801-treated group (light gray).
Assessment of Cognitive Safety
Potential cognitive impairment has been recognized as a critical adverse reaction, which may be triggered by CNS-acting drugs. BPSD patients seem to be particularly susceptible to cognitive slowing induced by psychotropic medications. Therefore, assessing cognitive safety remains a crucial issue for compounds designed for the fragile population of geriatric BPSD patients. To assess the cognitive safety of the lead molecules developed in this study, we used the novel object recognition task (NORT), which is a commonly applied behavioral paradigm to examine cognitive performance.71,72 Administration of both compounds, 11 and 16, at pharmacologically effective doses, namely: 11 at 0.1–0.3 mg/kg and 16 at 1–3 mg/kg, did not affect the NORT performance, suggesting their safety regarding cognitive performance (Figure 8). All of the groups explored the novel object for a similar amount of time. Under these conditions, the reference compound quetiapine, administered at a dose of 1 mg/kg, markedly decreased NORT performance, confirming its harmful effect on cognitive functions.
Figure 8.

Novel object recognition task (NORT) with MK-801-induced memory deficit. Control group—white bar, compound 11—lavender, 16—blue, quetiapine—gray. N = 7–8 rats per group. Statistical analysis: one-way ANOVA (Bonferroni’s post hoc); **p < 0.01 vs control (saline, white).
Altogether, these results suggest that compounds 11 and 16 appear to be useful for addressing depressive-like or anxiety-like symptoms, respectively, at relatively low doses (0.1 and 1 mg/kg, respectively). Both compounds do not impair social functioning. In contrast to the majority of psychotropic medications, both compounds do not influence spontaneous locomotor activity and do not induce cognitive deficits. These findings appear to be remarkably important regarding the targeted population of dementia patients and further validates our pharmacological strategy to treat BPSD.
Metabolic and Cardiac Safety
As stated in numerous clinical observations, dementia patients are highly sensitive to drug-induced metabolic syndrome and harmful cardiovascular events, particularly those that result from interaction with undesirable biological targets. The interesting pharmacological profile that emerged for 11 prompted us to choose it as a model anti-BPSD agent to investigate its effect on blood pressure, lipid profile, glucose levels, body fat, and weight as well as its effect on selected liver enzymes in plasma.
During the 25-day study, the animals were fed according to a “cafeteria diet”, a protocol developed previously in our labs, which was successfully implemented in the evaluation of metabolic safety of novel compounds.73−76 The cafeteria diet relies on the animals’ free access and preference for palatable products, which they tend to consume in excess, and this paradigm facilitates the development of the potential metabolic syndrome. We used olanzapine (at a dose of 2 × 2 mg/kg b.w./day, i.p.) as a comparator in this study, due to its well-known ability to induce metabolic syndrome. After the 25-day study, we observed that the animals exposed to a palatable diet and chronic dosing of 11 (2 × 2 mg/kg b.w., i.p.), had significantly lower weight compared to the control group (animals fed with palatable diet and treated with vehicle) (Figure S15 Supporting Information). Notably, compound 11, in contrast to olanzapine, did not significantly affect plasma glucose, triglyceride, or total cholesterol levels (Figure 9A–C), and favorably increased HDL-cholesterol levels (Figure 9D). Moreover, compound 11 did not affect the activity of alanine aminotransferase in rats that experienced chronic dosing, whereas olanzapine significantly increased its activity at the chronic dosing used (Figure 9E). At the end of the study (25th day), we observed a slight decrease in systemic blood pressure (systolic and diastolic) in the test group; however, the level of blood pressure was adjusted to the level of arterial pressure observed in lean rats, which clearly indicates the significant benefit of compound 11 at the dosage tested (Figure 10A,B). The effect of 11 was beneficially differentiated from olanzapine, which significantly reduced both systolic and diastolic blood pressure to a very large extent, when tested under the same experimental conditions.
Figure 9.
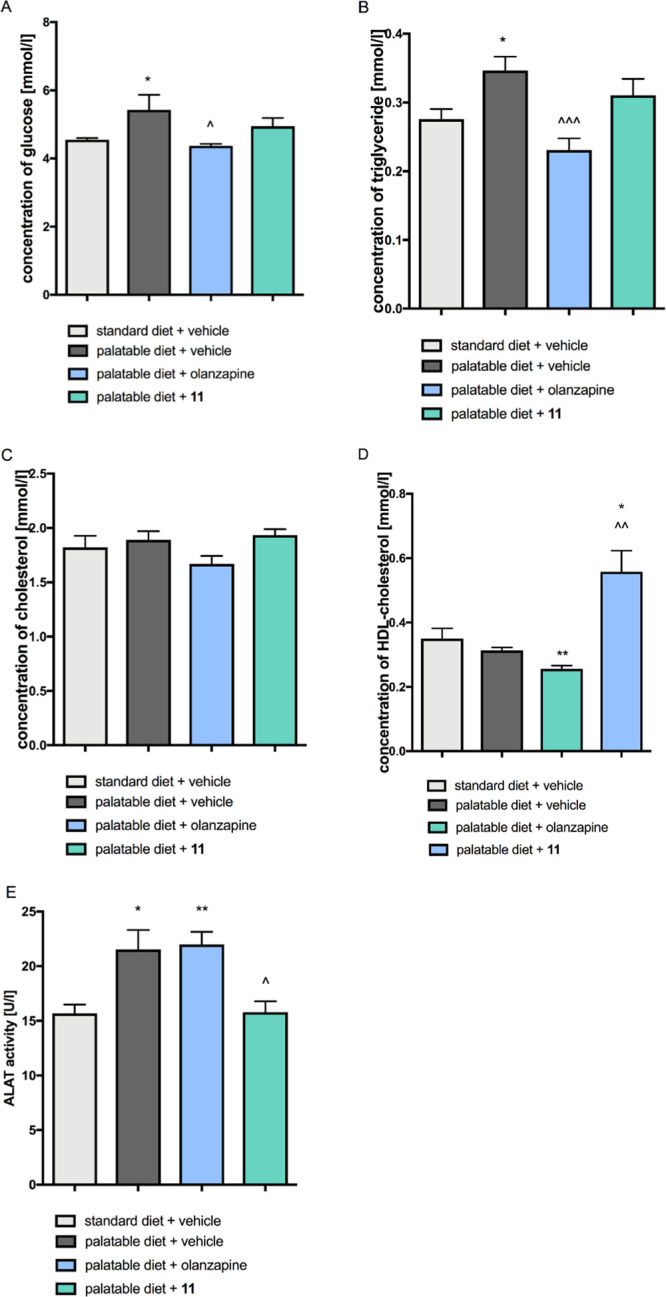
Effect of 11 and olanzapine on plasma biochemical parameters: A. glucose, B. triglyceride, C. total cholesterol, D. HDL-cholesterol, E. alanine aminotransferase. Statistical analysis: one-way ANOVA (Tukey post hoc); *p < 0.05, **p < 0.01 vs vehicle + standard diet; ∧p < 0.05, ∧∧p < 0.01 vs vehicle + palatable diet; mean ± SEM, n = 6.
Figure 10.

Time course of the effect of 11 or olanzapine on blood pressure: A. systolic, B. diastolic. Statistical analysis: two-way ANOVA with repeated measure (Bonferroni post hoc); *p < 0.05, **p < 0.01, ***p < 0.001 vehicle + standard diet vs vehicle + palatable diet; ∧∧∧p < 0.001 vehicle + palatable diet vs olanzapine + palatable diet; #p < 0.05 vehicle + palatable diet vs 11 + palatable diet; +p < 0.05 vehicle + standard diet vs 11 + palatable diet; $p < 0.05, $$p < 0.01, $$$p < 0.001 vehicle + standard diet vs olanzapine + palatable diet; mean ± SEM, n = 6.
In summary, these results indicate that, along with its promising therapeutic-like efficacy, compound 11 does not induce adverse reactions, such as significant metabolic disturbances nor fluctuations of blood pressure, in rats at the dosages tested. These results seem to be particularly important, considering that elderly patients are highly sensitive to drug-induced metabolic disturbances and orthostatic hypotension.
Conclusions
The hypothesis that BPSD patients might benefit from specifically designed agents that interact with clinically relevant targets led us to construct multifunctional ligands, 2–22, that interact simultaneously with a defined set of molecular targets: 5-HT2A, 5-HT6, 5-HT7, and D2 receptors, which are believed to provide therapeutically acceptable profiles. Using the framework combination strategy, we merged a 6-fluoro-3-propylbenzo[d]isoxazole core, as a privilege structure for achieving binding to D2 and 5-HT2A receptors, with 3-((arylsulfonyl)methyl)pyrrolidine or N-(5-methylpyrrolidin-3-yl)benzenesulfonamide, an essential scaffold for providing interaction with 5-HT6 and 5-HT7 receptors. These hybrid chemophores enabled us to design a series of multifunctional ligands that exert pronounced interactions in the nanomolar range with essentially matched biological targets and also ensured adequate functional responses. The novel chemotypes were surveyed within the cascade of in vitro studies, which led us to select the most promising molecules characterized by a low affinity for off-targets: α1, H1, and M1 receptors and hERG channels, as well as showing favorable aqueous solubility. Further, thorough profiling revealed that novel chemotypes are characterized by appreciable metabolic stability, compared to parent sulfonamide counterparts previously reported on.
The present study also led to the conclusion that arylsulfone-based chemotypes, such as compound 11, display pronounced antidepressant-like activity (MED = 0.1 mg/kg), outperforming marketed antidepressant medications in this regard. This feature is presumably attributed to the high affinity of 11 for 5-HT6, 5-HT7, and 5-HT2A receptors and synergic activity for these serotonin subtypes. Compound 16, featuring arylsulfonamide/methylpyrrolidine scaffolds, elicited robust anxiolytic activity, superior to reference anxiolytic drugs. In this regard, novel chemotypes differ substantially from the previously reported sulfonamide derivatives that exerted significant antipsychotic-like activity.36 The chemical modifications were introduced to solve two important issues—increase metabolic stability and obtain well-soluble druglike compounds of a given receptor profile. Therefore, the current chemical series constitutes a conscious extension of the previously reported one, which, in addition to achieving the above goals, resulted in a favorable and nonobvious pharmacological activity. In the previous series, the leading functional activity was D2R antagonism—it dominated 5-HT6 and 5-HT7 receptors function, and was even higher than the antagonism of the 5-HT2AR. The incremental structural alterations retained the general receptor profile, but introduced relative weakening of affinity and antagonist activity for D2R, and relatively stronger interactions with 5-HT6 and 5-HT7 receptors, making the 5-HT2AR antagonism the major mechanism of action. The receptor profile appears to be more balanced in the current series, even though the absolute values of the 5-HT2AR functional activity are lower. The above-mentioned in vitro activity differences caused an interesting change in the results of behavioral tests. The compounds lost antipsychotic-like activity (evidently mediated by antagonism of the D2 and 5-HT2A receptors) and expressed pure antidepressant- and anxiolytic-like activity.
Alongside their therapeutic-like activities, both compounds did not induce severe side reactions, such as impoverished cognitive functioning, characteristic of most psychotropic medications. The most promising compound, 11, did not impair glucose homeostasis and did not adversely affect weight and blood pressure. Given that dementia patients are highly vulnerable to severe drug-induced adverse reactions, the pharmacological approach encompassing the two key molecules described in this study, namely, 11 and 16, may provide a competitive and promising alternative to psychotropic medications used currently in BPSD patients.
Experimental Section
Molecular Modeling
Molecular property analysis and prediction of interactions in the binding sites of the target ligands aided in the design of the two series of molecules described in this study. Instant JChem was used for structure database management, search and property prediction, Instant JChem 20.19.0, 2020, ChemAxon (http://www.chemaxon.com). The biological chemistry of the molecules was evaluated in terms of Lipinski’s rule of five criteria: Log P o/w (octanol/water partition coefficient) < 5, molecular weight (MW) < 500, H-bond acceptors (HBA) < 10, H-bond donors (HBD) < 5, and the Veber rule criteria: rotatable bonds, preferably RB < 10 and topological polar surface area (TPSA) < 140 A2. Metabolic pathway and clearance prediction was conducted using the ADMET Predictor (Simulations Plus, Inc.). The designed structures were examined using the SwissADME tool (http://www.swissadme.ch) for known classes of reactive assay interference compounds (PAINS), and toxicophore structures (Brenk alert).
Docking studies were carried out using the previously developed homology models of the D2, 5-HT2A, 5-HT6, and 5-HT7 receptors. The procedure for obtaining ligand-optimized models of high predictive value, utilizing Induced-fit docking (IFD), was characterized in detail previously.36,77,78 The dopamine D2 receptor homology model was based on the 3PBL structure, the serotonin 5-HT2A receptor was built on 4IB4, the 5-HT6 receptor on 4IAR, and the 5-HT7 receptor—on the 2RH1 crystal structure.
Ligand structures were optimized using the LigPrep tool. The Glide SP flexible docking procedure was carried out using OPLS3 force field. H-bond constraint, as well as centroid of a grid box for docking to the receptor models were located on Asp3.32. The selection of the presented complexes was based on scoring function and interaction analysis. Molecular modeling studies were supported by the Small-Molecule Drug Discovery Suite (Schrödinger, Inc.).
General Chemistry Information
Unless otherwise indicated, all of the starting materials were purchased from commercial suppliers and were used without further purification. Analytical thin-layer chromatography (TLC) was performed on Merck Kieselgel 60 F254 (0.25 mm) precoated aluminum sheets (Merck, Darmstadt, Germany). Visualization was performed with a 254 nm UV lamp. Column chromatography was performed using silica gel (particle size 0.063–0.200 mm; 70–230 Mesh ATM) purchased from Merck. The ultraperformance liquid chromatography-mass spectrometry (UPLC-MS) or UPLC-MS/MS analyzes were run on an UPLC-MS/MS system comprising a Waters ACQUITY UPLC (Waters Corporation, Milford, MA) coupled with a Waters TQD mass spectrometer (electrospray ionization mode ESI with tandem quadrupole). Chromatographic separations were carried out using an ACQUITY UPLC BEH (bridged ethyl hybrid) C18 column: 2.1 mm × 100 mm and 1.7 μm particle size. The column was maintained at 40 °C and eluted under gradient conditions using 95–0% of eluent A over 10 min, at a flow rate of 0.3 mL/min. Eluent A: 0.1% solution of formic acid in water (v/v); eluent B: 0.1% solution of formic acid in acetonitrile (v/v). A total of 10 μL of each sample were injected, and chromatograms were recorded using a Waters eλ PDA detector. The spectra were analyzed in the range of 200–700 nm with 1.2 nm resolution and at a sampling rate of 20 points/s. MS detection settings of the Waters TQD mass spectrometer were as follows: source temperature 150 °C, desolvation temperature 350 °C, desolvation gas flow rate 600 l/h, cone gas flow 100 l/h, a capillary potential 3.00 kV, and cone potential 20 V. Nitrogen was used for both nebulizing and drying. The data were obtained in a scan mode ranging from 50 to 1000 m/z at 0.5 s intervals; 8 scans were summed up to obtain the final spectrum. Collision activated dissociation (CAD) analyzes were carried out using an energy of 20 eV, and all of the fragmentations were observed in the source. Consequently, the ion spectra were obtained in the range from 50 to 500 m/z. MassLynx V 4.1 software (Waters) was used for data acquisition. Standard solutions (1 mg/mL) of each compound were prepared in a mixture comprising analytical grade acetonitrile/water (1/1, v/v). The UPLC/MS purity of all of the test compounds and key intermediates was determined to be >95%.
1H NMR, 13C NMR, and 19F NMR spectra were obtained in a Varian Mercury spectrometer (Varian, Inc., Palo Alto, CA), in CDCl3, CD3OD, or DMSO-d6, operating at 300 MHz (1H NMR), 75 MHz (13C NMR), and 282 MHz (19F NMR). Chemical shifts are reported in terms of δ values (ppm) relative to TMS δ = 0 (1H) as an internal standard. The J values are expressed in Hertz (Hz). Signal multiplicities are represented by the following abbreviations: s (singlet), br s (broad singlet), d (doublet), dd (doublet of doublets), ddd (doublet of doublets of doublets), dt (doublet of triplets), t (triplet), td (triplet of doublets), tdd (triplet of doublet of doublets), q (quartet), dq (doublet of quartets), qd (quartet of doublets), quin (quintet), m (multiplet) and related.
Enantiomeric purity was determined using a chiral high-performance liquid chromatography (HPLC) technique on a Shimadzu Prominence LC-2030C SD Plus system (Shimadzu Corporation, Kyoto, Japan) equipped with an Amylose-C (250 mm × 4.6 mm) chiral column. The analysis was performed under the following conditions; column temperature: 45 °C, mixture of eluents: hexane/i-PrOH/TFA = 93/6.9/0.1 (v/v), flow rate: 1.4 mL/min, injection volume: 10 μL, analysis time: 70 min. (isocratic), detection at the wavelength λ = 200–800 nm. Enantiomeric purity is expressed in %.Detailed procedures for preparation of the synthesis intermediates I–VI are provided in the Supporting Information.
General Procedure for the Synthesis of Final Compounds 2–22
A mixture of appropriate amine IIIa–k (1 equiv), anhydrous potassium carbonate (3 equiv), 3-(3-chloropropyl)-6-fluorobenzo[d]isoxazole IV (1 equiv), and a catalytic amount of potassium iodide in acetonitrile (10 mL) was stirred at 60 °C for 24 h. After that time, the anhydrous potassium carbonate was filtered off and the solvent was evaporated under reduced pressure. The reaction mixture was purified by flash column chromatography using n-hexane/dichloromethane/methanol (60:35:5) (v/v/v) as eluent.
6-Fluoro-3-(3-(3-(((naphthalene-2-yl-phenyl)sulfonyl)methyl)pyrrolidin-1-yl)propyl)benzo[d]isoxazole (2)
The title compound was prepared starting from 3-{[(naphthalene-2-yl-phenyl)sulfonyl]methyl}pyrrolidine hydrochloride (0.3 mmol, 1 equiv, 0.094 g), 3-(3-chloropropyl)-6-fluorobenzo[d]isoxazole (0.3 mmol, 1 equiv, 0.064 g), potassium carbonate (0.9 mmol, 3 equiv, 0.124 g), and a catalytic amount of potassium iodide. Yield: 32%, brown oil. 1H NMR (300 MHz, CDCl3, δ): 8.48 (s, 1H), 8.04–7.96 (m, 2H), 7.93 (d, J = 7.6 Hz, 1H), 7.86 (dd, J = 1.8, 8.8 Hz, 1H), 7.72–7.55 (m, 3H), 7.21 (dd, J = 2.1, 8.5 Hz, 1H), 7.05 (dt, J = 2.3, 8.8 Hz, 1H), 3.33–3.17 (m, 2H), 2.98 (t, J = 7.3 Hz, 2H), 2.85–2.76 (m, 1H), 2.73–2.41 (m, 6H), 2.15–1.91 (m, 3H), 1.54 (qd, J = 6.6, 14.7 Hz, 1H). 13C NMR (75 MHz, CDCl3, δ): 163.6 (d, J = 13.8 Hz), 164.2 (d, J = 252.0 Hz), 158.1, 136.4, 135.3, 132.2, 129.8, 129.7, 129.4, 129.3, 128.0, 127.8, 122.6, 122.2 (d, J = 11.5 Hz), 118.2, 112.4 (d, J = 25.3 Hz), 97.3 (d, J = 27.7 Hz), 61.3, 59.2, 55.0, 53.1, 31.9, 30.5, 26.4, 22.9. LC-MS (ESI) calcd for C25H25FN2O3S 452.54 [M + H+], found 453 [M + H+].
6-Fluoro-3-(3-(3-(((3,4-dimethylphenyl)sulfonyl)methyl)-1-pyrrolidinyl)propyl)benzo[d]isoxazole (3)
The title compound was prepared starting from 3-{[(3,4-dimethylphenyl)sulfonyl]methyl}pyrrolidine hydrochloride (0.3 mmol, 1 equiv, 0.087 g), 3-(3-chloropropyl)-6-fluorobenzo[d]isoxazole (0.3 mmol, 1 equiv, 0.064 g), potassium carbonate (0.9 mmol, 3 equiv, 0.124 g) and catalytic amounts of potassium iodide. Yield: 27%, cream oil.1H NMR (300 MHz, CDCl3, δ): 7.69–7.55 (m, 3H), 7.33–7.27 (m, 1H), 7.22 (dd, J = 2.1, 8.5 Hz, 1H), 7.10–7.01 (m, 1H), 3.23–3.09 (m, 2H), 3.01 (t, J = 7.3 Hz, 2H), 2.88–2.79 (m, 1H), 2.73–2.48 (m, 5H), 2.33 (s, 6H), 2.27–2.18 (m, 1H), 2.10–1.99 (m, 3H), 1.65–1.54 (m, 1H). 13C NMR (75 MHz, CDCl3, δ): 163.8 (d, J = 18.4 Hz), 164.2 (d, J = 253.0 Hz), 158.0, 143.5, 138.2, 136.6, 130.4, 128.7, 125.5, 122.2 (d, J = 11.5 Hz), 118.2, 112.5 (d, J = 25.3 Hz), 97.3 (d, J = 27.7 Hz), 61.1, 59.1, 55.0, 53.2, 31.8, 30.5, 26.2, 22.9, 20.0, 19.8. LC-MS (ESI) calcd for C23H27FN2O3S 430.53 [M + H+], found 431 [M + H+].
6-Fluoro-3-(3-(3-(((3,4-difluorophenyl)sulfonyl)methyl)pyrrolidin-1-yl)propyl)benzo[d]isoxazole (4)
The title compound was prepared starting from 3-{[(3,4-difluorophenyl)sulfonyl]methyl} pyrrolidine hydrochloride (0.3 mmol, 1 equiv, 0.089 g), 3-(3-chloropropyl)-6-fluorobenzo[d]isoxazole (0.3 mmol, 1 equiv, 0.064 g), potassium carbonate (0.09 mmol, 3 equiv, 0.124 g), and a catalytic amount of potassium iodide. Yield: 25%, yellowish oil. 1H NMR (300 MHz, CDCl3, δ): 7.80–7.65 (m, 2H), 7.59 (dd, J = 5.0, 8.5 Hz, 1H), 7.43–7.31 (m, 1H), 7.22 (dd, J = 2.1, 8.5 Hz, 1H), 7.10–7.00 (m, 1H), 3.26–3.09 (m, 2H), 2.99 (t, J = 7.3 Hz, 2H), 2.80–2.70 (m, 1H), 2.66–2.36 (m, 6H), 2.15–1.92 (m, 3H), 1.52 (tdd, J = 6.4, 8.2, 12.9 Hz, 1H). 13C NMR (75 MHz, CDCl3, δ): 163.6 (d, J = 13.8 Hz), 164.2 (d, J = 251.0 Hz), 158.2, 150.5 (d, J = 268.3 Hz), 150.3 (d, J = 270.0 Hz), 136.7–136.1 (m), 125.4–125.2 (m), 122.2 (d, J = 11.5 Hz), 118.6 (d, J = 18.4 Hz), 118.3, 118.0 (d, J = 20.0 Hz), 112.4 (d, J = 25.3 Hz), 97.3 (d, J = 26.5 Hz), 61.4, 59.2, 54.9, 53.0, 31.8, 30.5, 26.5, 22.9. LC-MS (ESI) calcd for C21H21F3N2O3S 438.46 [M + H+], found 439 [M + H+].
6-Fluoro-3-(3-(3-(((3,4-dichlorophenyl)sulfonyl)methyl)-1-pyrrolidinyl)propyl)benzo[d]isoxazole (5)
The title compound was prepared starting from 3-{[(3,4-dichlorophenyl)sulfonyl]methyl}pyrrolidine hydrochloride (0.3 mmol, 1 equiv, 0.099 g), 3-(3-chloropropyl)-6-fluorobenzo[d]isoxazole (0.3 mmol, 1 equiv, 0.064 g), potassium carbonate (0.9 mmol, 3 equiv, 0.124 g), and a catalytic amount of potassium iodide. Yield: 57%, yellowish oil. 1H NMR (300 MHz, CDCl3, δ): 7.99 (d, J = 1.8 Hz, 1H), 7.77–7.69 (m, 1H), 7.67–7.63 (m, 1H), 7.60 (dd, J = 5.0, 8.5 Hz, 1H), 7.22 (dd, J = 2.1, 8.5 Hz, 1H), 7.06 (dt, J = 1.8, 8.8 Hz, 1H), 3.26–3.09 (m, 2H), 3.00 (t, J = 7.6 Hz, 2H), 2.82–2.72 (m, 1H), 2.68–2.39 (m, 6H), 2.18–1.93 (m, 3H), 1.53 (tdd, J = 6.5, 8.4, 13.1 Hz, 1H). 13C NMR (75 MHz, CDCl3, δ): 163.6 (d, J = 13.8 Hz), 164.2 (d, J = 251.0 Hz), 158.1, 139.3, 138.9, 134.2, 131.6, 130.0, 127.0, 122.2 (d, J = 11.5 Hz), 118.3, 112.5 (d, J = 25.3 Hz), 97.3 (d, J = 27.6 Hz), 61.4, 59.2, 54.9, 53.1, 31.7, 30.5, 26.5, 22.9. LC-MS (ESI) calcd for C21H21Cl2FN2O3S 471.37 [M + H+], found 471 [M + H+].
6-Fluoro-3-(3-(3-(((3-chloro-4-fluorophenyl)sulfonyl)methyl)pyrrolidin-1-yl)propyl)benzo[d]isoxazole (6)
The title compound was prepared starting from 3-{[(3-chloro-4-fluorophenyl)sulfonyl]methyl}pyrrolidine hydrochloride (0.3 mmol, 1 equiv, 0.094 g), 3-(3-chloropropyl)-6-fluorobenzo[d]isoxazole (0.3 mmol, 1 equiv, 0.064 g), potassium carbonate (0.9 mmol, 3 equiv, 0.124 mg), and a catalytic amount of potassium iodide. Yield: 8%, yellowish oil. 1H NMR (300 MHz, CDCl3, δ): 7.98 (dd, J = 2.1, 6.7 Hz, 1H), 7.81 (ddd, J = 2.3, 4.4, 8.5 Hz, 1H), 7.60 (dd, J = 5.0, 8.5 Hz, 1H), 7.33 (t, J = 8.5 Hz, 1H), 7.22 (dd, J = 2.1, 8.5 Hz, 1H), 7.06 (dt, J = 1.8, 8.8 Hz, 1H), 3.27–3.09 (m, 2H), 3.00 (t, J = 7.6 Hz, 2H), 2.82–2.71 (m, 1H), 2.69–2.37 (m, 6H), 2.16–1.94 (m, 3H), 1.53 (tdd, J = 6.4, 8.2, 12.9 Hz, 1H). 13C NMR (75 MHz, CDCl3, δ): 163.6 (d, J = 13.8 Hz), 164.2 (d, J = 251.0 Hz), 161.4 (d, J = 258.3 Hz), 158.1, 136.7 (d, J = 3.5 Hz), 131.1, 128.6 (d, J = 8.1 Hz), 123.0, 122.2 (d, J = 11.4 Hz), 118.2, 117.7 (d, J = 23.0 Hz), 112.5 (d, J = 25.3 Hz), 97.3 (d, J = 27.7 Hz), 61.5, 59.2, 54.9, 53.1, 31.7, 30.5, 26.5, 22.9. LC-MS (ESI) calcd for C21H21ClF2N2O3S 454.91 [M + H+], found 455 [M + H+].
6-Fluoro-3-(3-(3-(((4-(trifluoromethyl)phenyl)sulfonyl)methyl)pyrrolidin-1-yl)propyl)benzo[d]isoxazole (7)
The title compound was prepared starting from 3-{[(4-(trifluoromethyl)phenyl) sulfonyl] methyl}pyrrolidine hydrochloride (0.3 mmol, 1 equiv, 0.099 g), 3-(3-chloropropyl)-6-fluorobenzo[d]isoxazole (0.3 mmol, 1 equiv, 0.064 g), potassium carbonate (0.9 mmol, 3 equiv, 0.124 g), and a catalytic amount of potassium iodide. Yield: 67%, yellowish oil. 1H NMR (300 MHz, CDCl3, δ): 8.05 (d, J = 8.2 Hz, 2H), 7.84 (d, J = 8.2 Hz, 2H), 7.59 (dd, J = 5.0, 8.5 Hz, 1H), 7.22 (dd, J = 2.1, 8.5 Hz, 1H), 7.11–7.00 (m, 1H), 3.28–3.10 (m, 2H), 3.00 (t, J = 7.3 Hz, 2H), 2.82–2.71 (m, 1H), 2.69–2.38 (m, 6H), 2.17–1.93 (m, 3H), 1.53 (tdd, J = 6.7, 8.3, 13.0 Hz, 1H). 13C NMR (75 MHz, CDCl3, δ): 163.6 (d, J = 13.8 Hz), 164.2 (d, J = 249.9 Hz), 158.1, 143.1, 135.5 (q, J = 33.4 Hz), 128.6 (2C), 126.5 (q, J = 3.5 Hz, 2C), 122.2 (d, J = 10.4 Hz), 118.2, 123.1 (q, J = 275.0 Hz), 112.4 (d, J = 25.3 Hz), 97.3 (d, J = 27.0 Hz), 61.2, 59.2, 54.9, 53.0, 31.7, 30.5, 26.5, 22.9. LC-MS (ESI) calcd for C22H22F4N2O3S 470.48 [M + H+], found 471 [M + H+].
6-Fluoro-3-(3-(3-(((3-(trifluoromethyl)phenyl)sulfonyl)methyl)pyrrolidin-1-yl)propyl)benzo[d]isoxazole (8)
The title compound was prepared starting from 3-{[(3-(trifluoromethyl)phenyl) sulfonyl] methyl}pyrrolidine hydrochloride (0.3 mmol, 1 equiv, 0.099 g), 3-(3-chloropropyl)-6-fluorobenzo[d]isoxazole (0.3 mmol, 1 equiv, 0.064 g), potassium carbonate (0.9 mmol, 3 equiv, 0.124 g), and a catalytic amount of potassium iodide. Yield: 63%, yellowish oil. 1H NMR (300 MHz, CDCl3, δ): 8.18 (s, 1H), 8.11 (d, J = 7.6 Hz, 1H), 7.92 (br d, J = 8.2 Hz, 1H), 7.79–7.67 (m, 1H), 7.59 (dd, J = 4.7, 8.8 Hz, 1H), 7.22 (dd, J = 2.1, 8.5 Hz, 1H), 7.05 (dt, J = 1.8, 8.8 Hz, 1H), 3.29–3.10 (m, 2H), 3.00 (t, J = 7.6 Hz, 2H), 2.822.73 (m, 1H), 2.71–2.38 (m, 6H), 2.17–1.94 (m, 3H), 1.54 (tdd, J = 6.4, 8.2, 12.9 Hz, 1H). 13C NMR (75 MHz, CDCl3, δ): 163.6 (d, J = 14.0 Hz), 164.2 (d, J = 249.9 Hz), 158.1, 140.9, 132.1 (q, J = 33.4 Hz), 131.3, 130.5 (q, J = 3.5 Hz), 130.2, 125.1 (q, J = 3.5 Hz), 122.2 (d, J = 11.5 Hz), 118.2, 123.0 (q, J = 273.0 Hz), 112.4 (d, J = 25.3 Hz), 97.3 (d, J = 27.7 Hz, 1C), 61.3, 59.2, 54.9, 53.1, 31.6, 30.5, 26.5, 22.9. LC-MS (ESI) calcd for C22H22F4N2O3S 470.48 [M + H+], found 471 [M + H+].
6-Fluoro-3-(3-(3-(((4-fluorophenyl)sulfonyl)methyl)-1-pyrrolidinyl)propyl)benzo[d]isoxazole (9)
The title compound was prepared starting from the 3-{[(4-fluorophenyl)sulfonyl]methyl}pyrrolidine hydrochloride (0.3 mmol, 1 equiv, 0.084 g), 3-(3-chloropropyl)-6-fluorobenzo[d]isoxazole (0.3 mmol, 1 equiv, 0.064 g), potassium carbonate (0.9 mmol, 3 equiv, 0.124 g), and a catalytic amount of potassium iodide. Yield: 38%, yellowish oil. 1H NMR (300 MHz, CDCl3, δ): 7.92 (dd, J = 5.0, 9.1 Hz, 2H), 7.59 (dd, J = 5.3, 8.8 Hz, 1H), 7.29–7.19 (m, 3H), 7.10–7.00 (m, 1H), 3.24–3.09 (m, 2H), 2.99 (t, J = 7.3 Hz, 2H), 2.80–2.71 (m, 1H), 2.67–2.36 (m, 6H), 2.14–1.94 (m, 3H), 1.58–1.44 (m, 1H).13C NMR (75 MHz, CDCl3, δ): 166.3 (d, J = 13.8 Hz), 165.8 (d, J = 256.0 Hz), 164.7 (d, J = 180.8 Hz), 158.2, 135.6 (d, J = 3.5 Hz), 130.9 (d, J = 10.4 Hz, 2C), 122.2 (d, J = 10.4 Hz), 118.3, 116.7 (d, J = 21.9 Hz, 2C), 112.4 (d, J = 26.5 Hz), 97.3 (d, J = 26.5 Hz), 61.5, 59.3, 55.0 53.1, 31.8, 30.5, 26.6, 22.9. LC-MS (ESI) calcd for C21H22F2N2O3S 420.47 [M + H+], found 421 [M + H+].
6-Fluoro-3-(3-(3-(((3-fluorophenyl)sulfonyl)methyl)-1-pyrrolidinyl)propyl)benzo[d]isoxazole (10)
The title compound was prepared starting from the 3-{[(3-fluorophenyl)sulfonyl]methyl} pyrrolidine hydrochloride (0.3 mmol, 1 equiv, 0.084 g), 3-(3-chloropropyl)-6-fluorobenzo[d]isoxazole (0.3 mmol, 1 equiv, 0.064 g), potassium carbonate (0.9 mmol, 3 equiv, 0.124 g), and a catalytic amount of potassium iodide. Yield: 82%, yellowish oil. 1H NMR (300 MHz, CDCl3, δ): 7.74–7.66 (m, 1H), 7.63–7.50 (m, 3H), 7.40–7.30 (m, 1H), 7.22 (dd, J = 2.1, 8.5 Hz, 1H), 7.05 (dt, J = 2.1, 8.9 Hz, 1H), 3.26–3.10 (m, 2H), 2.99 (t, J = 7.6 Hz, 2H), 2.76 (dd, J = 7.0, 9.4 Hz, 1H), 2.66–2.37 (m, 6H), 2.16–2.02 (m, 2H), 1.98–1.92 (m, 1H), 1.60–1.46 (m, 1H).19F NMR (282 MHz, CDCl3, δ): −108.85 (s, 1F), -109.57 (s, 1F). 13C NMR (75 MHz, CDCl3, δ): 172.8, 163.6 (d, J = 13.8 Hz), 164.2 (d, J = 251.1 Hz), 162.5 (d, J = 252.0 Hz), 158.1, 141.6 (d, J = 5.8 Hz), 131.3 (d, J = 8.1 Hz), 123.8 (d, J = 3.5 Hz), 122.2 (d, J = 11.5 Hz), 121.1 (d, J = 21.0 Hz), 115.4 (d, J = 24.2 Hz), 112.5 (d, J = 25.3 Hz), 97.3 (d, J = 27.6 Hz), 61.2, 59.2, 55.0, 53.0, 31.7, 30.5, 26.4, 22.9. LC-MS (ESI) calcd for C21H22F2N2O3S 420.47 [M + H+], found 421 [M + H+].
6-Fluoro-3-(3-(3-(((3-chlorophenyl)sulfonyl)methyl)-1-pyrrolidinyl)propyl)benzo[d]isoxazole (11)
The title compound was prepared starting from the 3-{[(3-chlorophenyl)sulfonyl]methyl} pyrrolidine hydrochloride (0.3 mmol, 1 equiv, 0.089 g), 3-(3-chloropropyl)-6-fluorobenzo[d]isoxazole (0.3 mmol, 1 equiv, 0.064 g), potassium carbonate (0.9 mmol, 3 equiv, 0.124 g), and a catalytic amount of potassium iodide. Yield: 59%, yellowish oil. 1H NMR (300 MHz, CDCl3, δ): 7.89 (t, J = 1.8 Hz, 1H), 7.82–7.74 (m, 1H), 7.67–7.56 (m, 2H), 7.55–7.46 (m, 1H), 7.22 (dd, J = 2.1, 8.5 Hz, 1H), 7.06 (dt, J = 1.8, 8.8 Hz, 1H), 3.26–3.09 (m, 2H), 3.00 (t, J = 7.6 Hz, 2H), 2.77 (dd, J = 7.0, 9.4 Hz, 1H), 2.70–2.35 (m, 6H), 2.15–1.95 (m, 3H), 1.61–1.43 (m, 1H).13C NMR (75 MHz, CDCl3, δ): 163.6 (d, J = 13.8 Hz), 164.2 (d, J = 250.6 Hz), 158.2, 141.3, 135.6, 133.9, 130.7, 128.1, 126.1, 122.2 (d, J = 11.4 Hz), 118.2, 112.4 (d, J = 25.3 Hz), 97.3 (d, J = 27.6 Hz), 61.3, 59.3, 55.0, 53.1, 31.7, 30.5, 26.5, 22.9. LC-MS (ESI) calcd for C21H22FClN2O3S 436.93 [M + H+], found 437 [M + H+].
6-Fluoro-3-(3-(3-(((3-methylphenyl)sulfonyl)methyl)-1-pyrrolidinyl)propyl)benzo[d]isoxazole (12)
The title compound was prepared starting from the 3-{[(3-methylphenyl)sulfonyl]methyl}pyrrolidine hydrochloride (0.3 mmol, 1 equiv, 0.083 g), 3-(3-chloropropyl)-6-fluorobenzo[d]isoxazole (0.3 mmol, 1 equiv, 0.064 g), potassium carbonate (0.9 mmol, 1 equiv, 0.124 g), and a catalytic amount of potassium iodide. Yield: 45%, cream oil. 1H NMR (300 MHz, CDCl3, δ): 7.73–7.65 (m, 2H), 7.60 (dd, J = 5.0, 8.5 Hz, 1H), 7.48–7.39 (m, 2H), 7.22 (dd, J = 2.1, 8.5 Hz, 1H), 7.09–7.01 (m, 1H), 3.24–3.08 (m, 2H), 2.99 (t, J = 7.3 Hz, 2H), 2.77 (dd, J = 7.6, 9.4 Hz, 1H), 2.68–2.46 (m, 5H), 2.43 (s, 3H), 2.39 (dd, J = 6.4, 9.4 Hz, 1H), 2.07–1.94 (m, 3H), 1.52 (qd, J = 6.8, 13.5 Hz, 1H).13C NMR (75 MHz, CDCl3, δ): 163.6 (d, J = 13.8 Hz), 164.2 (d, J = 251.0 Hz), 158.2, 139.6, 139.4, 134.5, 129.2, 128.2, 125.1, 122.2 (d, J = 11.5 Hz), 118.3, 112.4 (d, J = 25.3 Hz), 97.3 (d, J = 26.5 Hz), 61.3, 59.3, 55.0, 53.2, 31.8, 30.5, 26.5, 22.9, 21.3. LC-MS (ESI) calcd for C22H25FN2O3S 416.51 [M + H+], found 417 [M + H+].
General procedure for the preparation of final molecules 13–22
Cesium carbonate (2.0 equiv), a catalytic amount of DMAP, and appropriate arylsulfonyl chloride (1.2 equiv) were added to a suspension of (3S,5S)-(1-(3-(6-fluorobenzo[d]isoxazol-3-yl)propyl)-5-methylpyrrolidin-3-amine hydrochloride (VI) (1 equiv) in dry dichloromethane at room temperature. The reaction mixture was stirred for 12 h, then the cesium carbonate was filtered, and the solvent was evaporated under reduced pressure. Crude reaction mixtures were purified by flash column chromatography over silica gel using dichloromethane/methanol 90:10 (v/v) as eluent.
3-Chloro-N-((3S,5S)-1-(3-(6-fluorobenzo[d]isoxazol-3-yl)propyl)-5-methylpyrrolidin-3-yl)benzenesulfonamide (13)
The title compound was prepared using (3S,5S)-tert-(1-(3-(6-fluorobenzo[d]isoxazol-3-yl)propyl)-5-methylpyrrolidin-3-amine hydrochloride VI (0.44 mmol, 1 equiv, 0.137 g), 3-chlorobenzene-1-sulfonyl chloride (0.53 mmol, 1.2 equiv, 0.112 g), DMAP (catalytic amount), and cesium carbonate (0.88 mmol, 2 equiv, 0.288 g) in DCM (5 mL). Yield 48%, yellowish oil. 1H NMR (300 MHz, CDCl3, δ): 7.86 (t, J = 2.1 Hz, 1H), 7.74 (td, J = 1.3, 7.9 Hz, 1H), 7.61–7.51 (m, 2H), 7.47–7.42 (m, 1H), 7.25 (dd, J = 2.1, 8.5 Hz, 1H), 7.07 (dt, J = 2.3, 8.8 Hz, 1H), 3.77 (quin, J = 7.03 Hz, 1 H), 3.35–3.25 (m, 1 H), 3.04–2.86 (m, 2 H), 2.78 (dt, J = 12.16 Hz, 2 H), 2.66–2.58 (m, 1 H), 2.30–2.19 (m, 1 H), 2.12–1.89 (m, 3 H), 1.75–1.62 (m, 2 H), 0.96 (d, J = 5.86 Hz, 3 H). 13C NMR (75 MHz, CDCl3, δ): 165.9 (d, J = 13.8 Hz), 163.5 (d, J = 250.2 Hz), 158.1, 142.6, 135.3, 132.7, 130.4, 127.1, 125.1, 122.1 (d, J = 10.4 Hz), 118.2 (d, J = 1.7 Hz), 112.5 (d, J = 26.0 Hz), 97.4 (d, J = 27.0 Hz), 60.5, 54.5, 52.9, 51.8, 32.4, 26.1, 22.8, 18.32. LC-MS (ESI) calcd for C21H23ClFN3O3S 451.94 [M + H+], found 452 [M + H+].
3-Chloro-4-fluoro-N-((3S,5S)-1-(3-(6-fluorobenzo[d]isoxazol-3-yl)propyl)-5-methylpyrrolidin-3-yl)benzenesulfonamide (14)
The title compound was prepared using (3S,5S)-tert-(1-(3-(6-fluorobenzo[d]isoxazol-3-yl)propyl)-5-methylpyrrolidin-3-amine hydrochloride VI (0.44 mmol, 1 equiv, 0.137 g), 3-chloro-4-fluorobenzene-1-sulfonyl chloride (0.528 mmol, 1.2 equiv, 0.120 g), DMAP (catalytic amount), and cesium carbonate (0.88 mmol, 2 equiv, 0.288 g) in DCM (5 mL). Yield 52%, yellowish oil. 1H NMR (300 MHz, CDCl3, δ): 7.98–7.90(m, 1H), 7.77 (ddd, J = 8.65, 4.25, 2.34 Hz, 1H), 7.58 (dd, J = 8.79, 5.27 Hz, 1H), 7.32–7.19 (m, 2H), 7.06 (dt, J = 2.3, 8.8 Hz, 1H), 3.83–3.59 (m, 2H), 3.56–3.40 (m, 1H), 3.35–3.23 (m, 1H), 3.21–3.12 (m, 1H), 2.98 (dq, J = 15.53, 7.62 Hz, 2H), 2.86–2.58 (m, 2H), 2.336–2.24 (m, 1H), 2.20–1.91 (m, 3H), 1.86–1.59 (m, 2H), 1.00–0.96 (m, 3H). 13C NMR (75 MHz, CDCl3, δ): 165.9 (d, J = 13.8 Hz), 163.5 (d, J = 250.2 Hz), 162.0, 158.1, 142.6, 135.3, 132.7, 130.4, 127.1, 125.1, 122.1 (d, J = 10.4 Hz), 118.2 (d, J = 1.7 Hz), 112.5 (d, J = 26.0 Hz), 97.4 (d, J = 27.0 Hz), 60.5, 54.5, 52.9, 51.8, 32.4, 26.1, 22.8, 18.32. LC-MS (ESI) calcd for C21H22ClF2N3O3S 469.93 [M + H+], found 469 [M + H+].
N-((3S,5S)-1-(3-(6-Fluorobenzo[d]isoxazol-3-yl)propyl)-5-methylpyrrolidin-3-yl)benzo[b]thiophene-3-sulfonamide (15)
The title compound was prepared using (3S,5S)-tert-(1-(3-(6-fluorobenzo[d]isoxazol-3-yl)propyl)-5-methylpyrrolidin-3-amine hydrochloride VI (0.44 mmol, 1 equiv, 0.137 g), benzo[b]thiophene-3-sulfonyl chloride (0.528 mmol, 1.2 equiv, 0.122 g), DMAP (catalytic amount), and cesium carbonate (0.88 mmol, 2 equiv, 0.288 g) in DCM (5 mL). Yield 79%, yellowish oil, chiral HPLC ee > 99%. 1H NMR (300 MHz, CDCl3, δ): 8.23 (s, 1H), 8.15 (d, J = 8.2 Hz, 1H), 7.87 (d, J = 8.8 Hz, 1H), 7.55 (dd, J = 5.3, 8.8 Hz, 1H), 7.47 – 7.35 (m, 2H), 7.25 7.22 (m, 1H), 7.06 (dt, J = 2.1, 8.9 Hz, 1H), 3.99–3.84 (m, 1H), 3.36 (dd, J = 9.67, 6.74 Hz, 2H), 3.03–2.50 (m, 4H), 2.25 (ddd, J = 12.16, 7.18, 5.27 Hz, 2H), 2.15–2.05 (m, 3H), 2.00–1.86 (m, 2H), 0.96 (d, J = 5.86 Hz, 2H). 19F NMR (282 MHz, CDCl3, δ): −109.6 (s, 1F); 13C NMR (75 MHz, CDCl3, δ): 165.9 (d, J = 250.2 Hz), 163.5 (d, J = 13.8 Hz), 157.8, 140.2, 134.3, 133.8, 125.7, 122.9, 122.1 (d, J = 11.5 Hz), 118.2, 112.5 (d, J = 26.0 Hz), 110.0, 97.5 (d, J = 26.5 Hz), 97.1, 60.2, 54.5, 52.8, 51.9, 32.3, 29.7, 25.9, 22.8, 18.22. LC-MS (ESI) calcd for C23H24FN3O3S2 473.58 [M + H+], found 474 [M + H+].
N-((3S,5S)-1-(3-(6-Fluorobenzo[d]isoxazol-3-yl)propyl)-5-methylpyrrolidin-3-yl)benzo[b]thiophene-2-sulfonamide (16)
The title compound was prepared using (3S,5S)-tert-(1-(3-(6-fluorobenzo[d]isoxazol-3-yl)propyl)-5-methylpyrrolidin-3-amine hydrochloride VI (0.44 mmol, 1 equiv, 0.137 g), benzo[b]thiophene-2-sulfonyl chloride (0.528 mmol, 1.2 equiv, 0.122 g), DMAP (catalytic amount), and cesium carbonate (0.67 mmol, 2 equiv, 0.288 g) in DCM (5 mL). Yield 87%, yellowish oil, chiral HPLC ee > 99%. 1H NMR (300 MHz, CDCl3, δ): 8.25 (s, 1H), 8.16 (d, J = 8.2 Hz, 1H), 7.90 (d, J = 8.8 Hz, 1H), 7.49–7.35 (m, 3H), 7.26–7.22 (m, 1H), 7.04 (dt, J = 2.1, 8.9 Hz, 1H), 3.99–3.82 (m, 1H), 3.21 (dd, J = 9.67, 6.74 Hz, 2H), 3.03–2.92 (m, 4H), 2.73 (ddd, J = 12.16, 7.18, 5.27 Hz, 2H), 2.19–2.05 (m, 3H), 2.00–1.91 (m, 2H), 0.89 (d, J = 5.86 Hz, 2H). 19F NMR (282 MHz, CDCl3, δ): −109.6 (s, 1F); 13C NMR (75 MHz, CDCl3, δ): 165.9 (d, J = 250.2 Hz), 163.5 (d, J = 13.8 Hz), 157.8, 140.2, 134.3, 133.8, 125.7, 122.9, 122.1 (d, J = 11.5 Hz), 118.2, 112.5 (d, J = 26.0 Hz), 110.0, 97.5 (d, J = 26.5 Hz), 97.1, 60.2, 54.5, 52.8, 51.9, 32.3, 29.7, 25.9, 22.8, 18.22. LC-MS (ESI) calcd for C23H24FN3O3S2 473.58 [M + H+], found 474 [M + H+].
N-((3S,5S)-1-(3-(6-Fluorobenzo[d]isoxazol-3-yl)propyl)-5-methylpyrrolidin-3-yl)benzofuran-2-sulfonamide (17)
The title compound was prepared using (3S,5S)-tert-(1-(3-(6-fluorobenzo[d]isoxazol-3-yl)propyl)-5-methylpyrrolidin-3-amine hydrochloride VI (0.44 mmol, 1 equiv, 0.137 g), benzofuran-2-sulfonyl chloride (0.528 mmol, 1.2 equiv, 0.113 g), DMAP (catalytic amount), and cesium carbonate (0.88 mmol, 2 equiv, 0.288 g) in DCM (5 mL). Yield 66%, yellowish oil. 1H NMR (300 MHz, CDCl3, δ): 8.62–8.53 (m, 1H), 7.69–7.63 (m, 1H), 7.61–7.03 (m, 6H), 4.18–3.90 (m, 1H), 3.68–3.37 (m, 2H), 3.34 (br s, 1H), 3.28–3.13 (m, 1H), 3.09–2.84 (m, 1H), 2.84–2.73 (m, 1H), 2.48–2.15 (m, 3H), 2.05 (dt, J = 14.51, 7.11 Hz, 2H), 1.43–1.13 (m, 1H), 1.08–1.06 (m, 3H); 13C NMR (75 MHz, CDCl3, δ): 165.9 (d, J = 250.2 Hz), 163.6 (d, J = 13.8 Hz), 162.6, 158.1, 155.6, 150.6, 127.6, 126.0, 124.2, 122.9, 122.2 (d, J = 11.5 Hz), 118.2, 112.5 (d, J = 25.4 Hz), 112.1, 97.5 (d, J = 26.5 Hz), 60.5, 54.6, 53.1, 52.0, 32.5, 26.1, 22.9, 18.9. LC-MS (ESI) calcd for C23H24FN3O4S 457.51 [M + H+], found 458 [M + H+].
6-Fluoro-N-((3S,5S)-1-(3-(6-fluorobenzo[d]isoxazol-3-yl)propyl)-5-methylpyrrolidin-3-yl)benzo[b]thiophene-2-sulfonamide (18)
The title compound was prepared using (3S,5S)-tert-(1-(3-(6-fluorobenzo[d]isoxazol-3-yl)propyl)-5-methylpyrrolidin-3-amine hydrochloride VI (0.44 mmol, 1 equiv, 0.137 g), 6-fluorobenzo[b]thiophene-2-sulfonyl chloride (0.528 mmol, 1.2 equiv, 0.131 g), DMAP (catalytic amount), and cesium carbonate (0.88 mmol, 2 equiv, 0.288 g) in DCM (5 mL). Yield 69%, yellowish oil. 1H NMR (300 MHz, CDCl3, δ): 7.85–7.79 (m, 2H), 7.58–7.49 (m, 2H), 7.26–7.16 (m, 2H), 7.07–7.00 (m, 1H), 3.94–3.85 (m, 1H), 3.38–3.32 (m, 1H), 3.04–2.85 (m, 2H), 2.78 (t, J = 7.6 Hz, 2H), 2.64–2.54 (m, 1H), 2.26 (ddd, J = 2.9, 10.0 Hz, 1H), 2.13 (dd, J = 9.96, 7.03 Hz, 1H), 2.00–1.87 (m, 2H), 1.84–1.68 (m, 2H), 0.98-0.96 (m, 3H); 13C NMR (75 MHz, CDCl3, δ): 171.8 (d, J = 250.2 Hz), 163.6 (d, J = 13.8 Hz), 162.6 (d, J = 249.0 Hz), 158.1, 154.0 (d, J = 2.2 Hz), 137.6, 128.7, 127.1, 126.9 (d, J = 9.3 Hz), 122.0 (d, J = 11.5 Hz), 118.2, 115.1 (d, J = 24.3 Hz), 112.5 (d, J = 26.0 Hz), 108.8 (d, J = 25.2 Hz), 97.4 (d, J = 26.5 Hz), 60.4, 54.5, 53.2, 51.9, 32.4, 26.2, 22.9, 18.4. LC-MS (ESI) calcd for C23H23F2N3O3S2 491.57 [M + H+], found 492 [M + H+].
6-Chloro-N-((3S,5S)-1-(3-(6-fluorobenzo[d]isoxazol-3-yl)propyl)-5-methylpyrrolidin-3-yl)benzo[b]thiophene-2-sulfonamide (19)
The title compound was prepared using (3S,5S)-tert-(1-(3-(6-fluorobenzo[d]isoxazol-3-yl)propyl)-5-methylpyrrolidin-3-amine hydrochloride VI (0.44 mmol, 1 equiv, 0.137 g), 6-chlorobenzo[b]thiophene-2-sulfonyl chloride (0.528 mmol, 1.2 equiv, 0.140 g), DMAP (catalytic amount), and cesium carbonate (0.88 mmol, 2 equiv, 0.288 g) in DCM (5 mL). Yield 71%, yellowish oil. 1H NMR (300 MHz, CDCl3, δ): 7.84–7.75 (m, 2H), 7.56 (dd, J = 8.50, 4.98 Hz, 1H), 7.41 (dd, J = 8.79, 1.76 Hz, 1H), 7.26–7.16 (m, 2H), 7.07–7.00 (m, 1H), 3.95–3.86 (m, 1H), 3.49–3.31 (m, 1H), 3.04–2.86 (m, 2H), 2.83–2.72 (m, 2H), 2.66–2.55 (m, 1H), 2.31–2.10 (m, 1H), 2.03–1.87 (m, 2H), 1.82–1.68 (m, 2H), 0.98-0.96 (m, 4H); 13C NMR (75 MHz, CDCl3, δ): 171.8 (d, J = 250.2 Hz), 163.6 (d, J = 13.8 Hz), 162.6 (d, J = 249.0 Hz), 158.1, 154.0 (d, J = 2.2 Hz), 137.6, 128.7, 127.1, 126.9 (d, J = 9.3 Hz), 122.0 (d, J = 11.5 Hz), 118.2, 115.1 (d, J = 24.3 Hz), 112.5 (d, J = 26.0 Hz), 108.8 (d, J = 25.2 Hz), 97.4 (d, J = 26.5 Hz), 60.4, 54.5, 53.2, 51.9, 32.4, 26.2, 22.9, 18.4. LC-MS (ESI) calcd for C23H23ClFN3O3S2 508.02 [M + H+], found 509 [M + H+].
5-Chloro-N-((3S,5S)-1-(3-(6-fluorobenzo[d]isoxazol-3-yl)propyl)-5-methylpyrrolidin-3-yl)-3-methylbenzo[b]thiophene-2-sulfonamide (20)
The title compound was prepared using (3S,5S)-tert-(1-(3-(6-fluorobenzo[d]isoxazol-3-yl)propyl)-5-methylpyrrolidin-3-amine hydrochloride VI (0.44 mmol, 1 equiv, 0.137 g), 5-chloro-3-methylbenzo[b]thiophene-2-sulfonyl chloride (0.528 mmol, 1.2 equiv, 0.148 g), DMAP (catalytic amount), and cesium carbonate (0.88 mmol, 2 equiv, 0.288 g) in DCM (5 mL). Yield 92%, yellowish oil. 1H NMR (300 MHz, CDCl3, δ): 7.78–7.72 (m, 2H), 7.55 (dd, J = 5.3, 8.8 Hz, 1H), 7.42 (dd, J = 2.1, 8.5 Hz, 1H), 7.19 (dd, J = 1.8, 8.8 Hz, 1H), 7.07–7.01 (m, 1H), 3.94 – 3.92 (m, 1H), 3.36–3.30 (m, 1H), 3.33 (dd, J = 9.67, 6.74 Hz, 2H), 3.03–2.90 (m, 2H), 2.73 (ddd, J = 12.16, 7.18, 5.27 Hz, 2H), 2.63 (s, 3H), 2.12–2.08 (m, 1H), 1.96–1.92 (m, 2H), 1.74–1.70 (m, 2H), 0.95 (d, J = 5.86 Hz, 3H). 19F NMR (282 MHz, CDCl3, δ): −109.6 (s, 1F); 13C NMR (75 MHz, CDCl3, δ): 165.9 (d, J = 250.2 Hz), 163.5 (d, J = 13.8 Hz), 158.0, 140.8, 137.5, 135.8, 131.5, 127.8, 123.7, 123.3 (d, J = 11.5 Hz), 118.2, 112.5 (d, J = 26.0 Hz), 122.2, 97.5 (d, J = 26.5 Hz), 97.1, 59.8, 57.8, 51.9, 51.04, 40.5, 26.0, 22.9;18.35, 12.4. LC-MS (ESI) calcd for C24H25ClFN3O3S2 521,10 [M + H+], found 522 [M + H+].
N-((3S,5S)-1-(3-(6-Fluorobenzo[d]isoxazol-3-yl)propyl)-5-methylpyrrolidin-3-yl)naphthalene-2-sulfonamide (21)
The title compound was prepared using (3S,5S)-tert-(1-(3-(6-fluorobenzo[d]isoxazol-3-yl)propyl)-5-methylpyrrolidin-3-amine hydrochloride VI (0.44 mmol, 1 equiv, 0.137 g), naphthalene-2-sulfonyl chloride (0.58 mmol, 1.2 equiv, 0.118 g), DMAP (catalytic amount), and cesium carbonate (0.88 mmol, 2 equiv, 0.288 g) in DCM (5 mL). Yield 95%, yellowish oil, chiral HPLC ee > 99%. 1H NMR (300 MHz, CDCl3, δ): 8.44 (s, 1H), 7.95–7.83 (m, 1H), 7.65–7.49 (m, 1H), 7.19 (d, J = 1.76 Hz, 2H), 7.16 (d, J = 2.34 Hz, 1H), 7.03 (d, J = 1.76 Hz, 1H), 7.00 (d, J = 1.76 Hz, 2H), 6.97 (d, J = 1.76 Hz, 1H), 3.85–3.79 (m, 1H), 3.33–3.29 (m, 1H), 2.97–2.86 (m, 2H), 2.84–2.72 (m, 1H), 2.09–2.06 (m, 2H), 2.14 (dd, J = 9.67, 7.33 Hz, 1H), 1.96–1.86 (m, 2H), 1.78–1.59 (m, 2H), 1.25–1.19 (m, 1H), 0.93 (d, J = 6.45 Hz, 3H); 13C NMR (75 MHz, CDCl3, δ): 165.8 (d, J = 250.2 Hz), 163.5 (d, J = 13.8 Hz), 158.0, 137.5, 135.3, 132.9, 130.7, 130.4, 128.6, 128.1, 126.7, 123.5, 122.1 (d, J = 11.5 Hz), 118.1, 112.5 (d, J = 26.0 Hz), 97.4 (d, J = 26.5 Hz), 59.8, 57.9, 52.0, 50.7, 40.46, 29.7, 26.0, 22.9, 18.4. LC-MS (ESI) calcd for C25H26FN3O3S 467.55 [M + H+], found 468 [M + H+].
6-Chloro-N-((3S,5S)-1-(3-(6-fluorobenzo[d]isoxazol-3-yl)propyl)-5-methylpyrrolidin-3-yl)naphthalene-2-sulfonamide (22)
The title compound was prepared using (3S,5S)-tert-(1-(3-(6-fluorobenzo[d]isoxazol-3-yl)propyl)-5-methylpyrrolidin-3-amine hydrochloride VI (0.44 mmol, 1 equiv, 0.137 g), 6-chloronaphthalene-2-sulfonyl chloride (0.528 mmol, 1.2 equiv, 0.137 g), DMAP (catalytic amount), and cesium carbonate (0.88 mmol, 2 equiv, 0.288 g) in DCM (5 mL). Yield 91%, yellowish oil. 1H NMR (300 MHz, CDCl3, δ): 8.41 (s, 1H), 7.90–7.80 (m, 4H), 7.54–7.50 (m, 2H), 7.20–7.17 (m, 1H), 7.02–6.98 (m, 1H), 3.81–3.79 (m, 1H), 3.29–3.23 (m, 1H), 2.94–2.86 (m, 2H), 2.75–2.71 (m, 1H), 2.56–2.54 (m, 1H), 2.09–2.06 (m, 2H), 1.96–1.92 (m, 2H), 1.74–1.70 (m, 2H), 1.27–1.17 (m, 1H), 0.95 (d, J = 5.86 Hz, 3H); 13C NMR (75 MHz, CDCl3, δ): 165.8 (d, J = 250.2 Hz), 163.5 (d, J = 13.8 Hz), 158.0, 137.5, 135.3, 132.9, 130.7, 130.4, 128.6, 128.1, 126.7, 123.5, 122.1 (d, J = 11.5 Hz), 118.1, 112.5 (d, J = 26.0 Hz), 97.4 (d, J = 26.5 Hz), 59.8, 57.9, 52.0, 50.7, 40.46, 29.7, 26.0, 22.9, 18.4. LC-MS (ESI) calcd for C25H25ClFN3O3S 502.0 [M + H+], found 503 [M + H+].
In Vitro Studies
The tested compounds were examined for known classes of assay interference compounds. None of the compounds contains substructural features recognized as pan assay interference compounds (PAINS), according to the SwissADME tool.79
Radioligand Binding Studies
Preparation of Solutions of Test and Reference Compounds
Stock solutions (10 mM) of tested compounds were prepared in 100% dimethyl sulfoxide (DMSO). Serial dilutions of tested compounds were prepared in 96-well microplate in assay buffers using an automated pipetting system epMotion 5070 (Eppendorf). Each compound was tested in six concentrations from 10–5 to 10–10 M (final concentration).
5-HT2A Receptor Binding Assay
Radioligand binding assay was performed using membranes from CHO-K1 cells stably transfected with the human 5-HT2A receptor provided by PerkinElmer. All tests were carried out in duplicate. First, 50 μL of working solution of the tested compounds, 50 μL of [3H]-ketanserin (final concentration 1 nM), and 150 μL of diluted membranes (7 μg protein per well) prepared in assay buffer (50 mM Tris, pH 7.4, 4 mM CaCl2, 0.1% ascorbic acid) were transferred to a polypropylene 96-well microplate using a Rainin Liquidator (Mettler Toledo) 96-well pipetting station. Mianserin (10 μM) was used to define nonspecific binding. The microplate was covered with sealing tape, mixed, and incubated for 60 min at 27 °C. The reaction was stopped by fast filtration through a GF/B filter mat presoaked with 0.5% polyethylenimine for 30 min. Ten rapid washes with 200 μL of 50 mM Tris buffer (4 °C, pH 7.4) were performed using a Harvester-96 MACH III FM automated harvester system (Tomtec). The filter mats were dried at 37 °C in a forced air fan incubator, and then solid scintillator MeltiLex was melted onto the filter mats at 90 °C for 5 min. Radioactivity was measured in a MicroBeta2 scintillation counter (PerkinElmer). Data were fitted to a one-site curve-fitting equation with Prism 6 (GraphPad Software), and Ki values were estimated from the Cheng–Prusoff equation.80
5-HT6 Receptor Binding Assay
Radioligand binding was performed using membranes from CHO-K1 cells stably transfected with the human 5-HT6 receptor (PerkinElmer). All assays were carried out in duplicate. First, 50 μL of working solution of the tested compounds, 50 μL of [3H]-LSD (final concentration 1 nM), and 150 μL of diluted membranes (8 μg protein per well) prepared in assay buffer (50 mM Tris, pH 7.4, 10 mM MgCl2, 0.1 mM ethylenediamine tetraacetic acid (EDTA)) were transferred to a polypropylene 96-well microplate using a Rainin Liquidator 96-well pipetting station (Mettler Toledo). Methiothepin (10 μM) was used to define nonspecific binding. The microplate was covered with sealing tape, mixed, and incubated for 60 min at 37 °C. The reaction was completed by rapid filtration through a GF/A filter mat presoaked with 0.5% polyethylenimine for 30 min. Ten rapid washes with 200 μL of 50 mM Tris buffer (4 °C, pH 7.4) were performed using a Harvester-96 MACH III FM automated harvester system (Tomtec). The filter mats were dried at 37 °C in a forced air fan incubator, and then solid scintillator MeltiLex was melted onto the filter mats at 90 °C for 5 min. Radioactivity was counted in a MicroBeta2 scintillation counter (PerkinElmer). Data were fitted to a one-site curve-fitting equation with Prism 6 (GraphPad Software), and Ki values were estimated from the Cheng–Prusoff equation.80
5-HT7 Receptor Binding Assay
Radioligand binding was performed using membranes from CHO-K1 cells stably transfected with the human 5-HT7 receptor (PerkinElmer). All assays were carried out in duplicate. First, 50 μL of working solution of the tested compounds, 50 μL of [3H]-LSD (final concentration 3 nM), and 150 μL of diluted membranes (17 μg protein per well) prepared in assay buffer (50 mM Tris, pH 7.4, 10 mM MgSO4, 0.5 mM EDTA) were transferred to a polypropylene 96-well microplate using a Rainin Liquidator 96-well pipetting station (Mettler Toledo). Methiothepin (10 μM) was used to define nonspecific binding. The microplate was covered with sealing tape, mixed, and incubated for 120 min at 27 °C. The reaction was terminated by rapid filtration through a GF/A filter mat presoaked with 0.3% polyethylenimine for 30 min. Ten rapid washes with 200 μL of 50 mM Tris buffer (4 °C, pH 7.4) were performed using a Harvester-96 MACH III FM an automated harvester system (Tomtec). The filter mats were dried at 37 °C in a forced air fan incubator, and then solid scintillator MeltiLex was melted onto the filter mats at 90 °C for 5 min. Radioactivity was counted in a MicroBeta2 scintillation counter (PerkinElmer). Data were fitted to a one-site curve-fitting equation with Prism 6 (GraphPad Software), and Ki values were estimated from the Cheng–Prusoff equation.80
D2 Receptor Binding Assay
Radioligand binding was performed using membranes from CHO-K1 cells stably transfected with the human D2 receptor (PerkinElmer). All assays were carried out in duplicate. First, 50 μL of working solution of the tested compounds, 50 μL of [3H]-methylspiperone (final concentration 0.4 nM), and 150 μL of diluted membranes (3 μg protein per well) prepared in assay buffer (50 mM Tris, pH 7.4, 50 mM HEPES, 50 mM NaCl, 5 mM MgCl2, 0.5 mM EDTA) were transferred to a polypropylene 96-well microplate using a Rainin Liquidator 96-well pipetting station (Mettler Toledo). Haloperidol (10 μM) was used to define nonspecific binding. The microplate was covered with sealing tape, mixed, and incubated for 60 min at 37 °C. The reaction was terminated by rapid filtration through a GF/B filter mat presoaked with 0.5% polyethylenimine for 30 min. Ten rapid washes with 200 μL of 50 mM Tris buffer (4 °C, pH 7.4) were performed using a Harvester-96 MACH III FM automated harvester system (Tomtec). The filter mats were dried at 37 °C in a forced air fan incubator, and then solid scintillator MeltiLex was melted onto the filter mats at 90 °C for 5 min. Radioactivity was counted in a MicroBeta2 scintillation counter (PerkinElmer). Data were fitted to a one-site curve-fitting equation with Prism 6 (GraphPad Software), and Ki values were estimated from the Cheng–Prusoff equation.80
M1 Receptor Binding Assay
Radioligand binding was performed using membranes from CHO-K1 cells stably transfected with the human M1 receptor (PerkinElmer). All assays were carried out in duplicate. The working solution (50 μL) of the tested compounds, [3H]-Scopolamine (50 μL) (final concentration 0.14 nM), and diluted membranes (150 μL) (35 μg protein per well) prepared in PBS (pH 7.4) were transferred to a polypropylene 96-well microplate using a Rainin Liquidator 96-well pipetting station (Mettler Toledo). Pirenzepine (10 μM) was used to define nonspecific binding. The microplate was covered with sealing tape, mixed, and incubated for 120 min at 27 °C. The reaction was terminated by rapid filtration through a GF/B filter mat presoaked with 0.3% polyethyleneimine for 30 min. Ten rapid washes with 200 μL of 50 mM Tris buffer, 154 mM NaCl (4 °C, pH 7.4) were performed using a 96-well FilterMate harvester (PerkinElmer). The filter mats were dried at 37 °C in a forced air fan incubator and then solid scintillator MeltiLex was melted onto the filter mats at 90 °C for 4 min. The radioactivity on the filters was measured in a MicroBeta TriLux 1450 scintillation counter (PerkinElmer). Data were fitted to a one-site curve-fitting equation with Prism 6 (GraphPad Software). Results from the assay are represented as a percentage of [3H]-scopolamine inhibition by tested compounds.
H1 Receptor Binding Assay
Radioligand binding was performed using membranes from CHO-K1 cells stably transfected with the human H1 receptor (PerkinElmer). All assays were carried out in duplicate. The working solution of the tested compounds (50 μL), [3H]-Pyrilamine (50 μL) (final concentration 1.5 nM), and diluted membranes (150 μL) (5 μg protein per well) prepared in assay buffer (50 mM Tris, pH 7.4, 5 mM MgCl2) were transferred to a polypropylene 96-well microplate using a Rainin Liquidator 96-well pipetting station (Mettler Toledo). Mepyramine (10 μM) was used to define nonspecific binding. The microplate was covered with sealing tape, mixed, and incubated for 60 min at 27 °C. The reaction was terminated by rapid filtration through a GF/B filter mat presoaked with 0.5% polyethyleneimine for 30 min. Ten rapid washes with 200 μL of 50 mM Tris buffer (4 °C, pH 7.4) were performed using a 96-well FilterMate harvester (PerkinElmer). The filter mats were dried at 37 °C in a forced air fan incubator and then solid scintillator MeltiLex was melted onto the filter mats at 90 °C for 4 min. The radioactivity on the filters was measured in a MicroBeta TriLux 1450 scintillation counter (PerkinElmer). Data were fitted to a one-site curve-fitting equation with Prism 6 (GraphPad Software). Results from the assay are represented as a percentage of [3H]-Pyrilamine inhibition by tested compounds.
α1-Adrenergic Receptor Binding Assay
Radioligand binding was performed using rat cortex tissue. All assays were carried out in duplicate. The working solution of the tested compounds (50 μL), [3H]-prazosin (50 μL) (final concentration 0.3 nM), and tissue suspension prepared in assay buffer (150 μL) (50 mM Tris-HCl, pH 7.6) were transferred to a polypropylene 96-well microplate using a Rainin Liquidator 96-well pipetting station (Mettler Toledo). Phentolamine (10 μM) was used to define nonspecific binding. The microplate was covered with sealing tape, mixed, and incubated for 30 min at 30 °C. The reaction was terminated by rapid filtration through a GF/B filter mat. Ten rapid washes with 200 μL of 50 mM Tris buffer (4 °C, pH 7.6) were performed using a 96-well FilterMate harvester (PerkinElmer). The filter mats were dried at 37 °C in a forced air fan incubator and then solid scintillator MeltiLex was melted onto the filter mats at 90 °C for 4 min. The radioactivity on the filter was measured in a MicroBeta TriLux 1450 scintillation counter (PerkinElmer). Data were fitted to a one-site curve-fitting equation with Prism 6 (GraphPad Software). Results from the assay are represented as a percentage of [3H]-prazosin inhibition by tested compounds.81
Functional Assay for the 5-HT2A Receptor
Test and reference compounds were dissolved in dimethyl sulfoxide (DMSO) at a concentration of 1 mM. Serial dilutions were prepared in a 96-well microplate in assay buffer and 8 to 10 different concentrations of each compound were tested. A cellular aequorin-based functional assay was performed with recombinant CHO-K1 cells expressing mitochondrially targeted aequorin, human GPCR, and the promiscuous G protein α16 for 5-HT2A. The assay was executed according to the previously described protocol. After thawing, the cells were transferred to assay buffer (DMEM/HAM’s F12 with 0.1% protease-free bovine serum albumin (BSA)) and centrifuged. The cell pellet was resuspended in assay buffer and coelenterazine h was added at a final concentration of 5 μM. The cell suspension was incubated at 16 °C, protected from light, with constant agitation for 16 h and then diluted with assay buffer to a concentration of 100 000 cells/mL. After 1 h of incubation, 50 μL of the cell suspension was dispensed using automatic injectors built into the MicroBeta2 LumiJET radiometric and luminescence plate counter (PerkinElmer) into white opaque 96-well microplates preloaded with test compounds. Immediate light emission generated following calcium mobilization was recorded for 30 s. In the antagonist mode, after 30 min of incubation, the reference agonist was added to the above assay mix and light emission was recorded again. The final concentration of the reference agonist was equal to EC80 (30 nM serotonin).
Functional Assay for the 5-HT6 Receptor
Test and reference compounds were dissolved in dimethyl sulfoxide (DMSO) at a concentration of 1 mM. Serial dilutions were prepared in a 96-well microplate in assay buffer and 8 to 10 different concentrations of each compound were tested. A cellular aequorin-based functional assay was performed with recombinant CHO-K1 cells expressing mitochondrially targeted aequorin, human GPCR, and the promiscuous G protein α16 for 5-HT6. The assay was executed according to the previously described protocol. After thawing, the cells were transferred to assay buffer (DMEM/HAM’s F12 with 0.1% protease-free BSA) and centrifuged. The cell pellet was resuspended in assay buffer and coelenterazine h was added at a final concentration of 5 μM. The cell suspension was incubated at 16 °C, protected from light, with constant agitation for 16 h and then diluted with assay buffer to a concentration of 100 000 cells/mL. After 1 h of incubation, 50 μL of the cell suspension was dispensed using the automatic injectors built into the MicroBeta2 LumiJET radiometric and luminescence plate counter (PerkinElmer) into white opaque 96-well microplates preloaded with test compounds. Immediate light emission generated following calcium mobilization was recorded for 60 s. In the antagonist mode, after 30 min of incubation, the reference agonist was added to the above assay mix and light emission was recorded again. The final concentration of the reference agonist was equal to EC80 (40 nM serotonin).
Functional Assay for the 5-HT7 Receptor
Test and reference compounds were dissolved in dimethyl sulfoxide (DMSO) at a concentration of 1 mM. Serial dilutions were prepared in a 96-well microplate in assay buffer and 8 to 10 different concentrations of each compound were tested. For the 5-HT7, adenylyl cyclase activity was monitored using cryopreserved CHO-K1 cells expressing the human serotonin 5-HT7 receptor. A functional assay based on cells expressing the human 5-hydroxytryptamine (serotonin) receptor 7 was performed. CHO-K1 cells were transfected with a β-lactamase reporter gene under the control of the cyclic AMP response element (CRE) (Life Technologies). Thawed cells were resuspended in stimulation buffer (HBSS, 5 mM HEPES, 0.5 IBMX, and 0.1% BSA at pH 7.4) at 2 × 105 cells/mL for the 5-HT7 receptor. The same volume (10 μL) of cell suspension was added to tested compounds for the 5-HT7 receptor. Samples were loaded onto a white opaque half-area 96-well microplate. The antagonist response experiment was performed with 10 nM serotonin as the reference agonist for the 5-HT7 receptor. The agonist and antagonist were added simultaneously. Cell stimulation was performed for 1 h at room temperature. After incubation, cAMP measurements were performed with the homogeneous time-resolved fluorescence resonance energy transfer (TR-FRET) immunoassay using the LANCE Ultra cAMP kit (PerkinElmer). 10 μL of EucAMP Tracer Working Solution and 10 μL of ULight-anti-cAMP Tracer Working Solution were added, mixed, and incubated for 1 h. The TR-FRET signal was read on an EnVision microplate reader (PerkinElmer). IC50 and EC50 were determined by nonlinear regression analysis using GraphPad Prism 6.0 software.
Functional Assay for the D2 Receptor
Test and reference compounds were dissolved in dimethyl sulfoxide (DMSO) at a concentration of 1 mM. Serial dilutions were prepared in 96-well microplate in assay buffer and 8 to 10 different concentrations of each compound were tested. A cellular aequorin-based functional assay was performed with recombinant CHO-K1 cells expressing mitochondrially targeted aequorin, human GPCR, and the promiscuous G protein Gαqi/5 for the D2 receptor. The assay was executed according to the previously described protocol.1 After thawing, the cells were transferred to assay buffer (DMEM/HAM’s F12 with 0.1% protease-free BSA) and centrifuged. The cell pellet was resuspended in assay buffer and coelenterazine h was added at a final concentration of 5 μM. The cell suspension was incubated at 16 °C, protected from light, with constant agitation for 16 h and then diluted with assay buffer to a concentration of 100 000 cells/mL. After 1 h of incubation, 50 μL of the cell suspension was dispensed using the automatic injectors built into the MicroBeta2 LumiJET radiometric and luminescence plate counter (PerkinElmer) into white opaque 96-well microplates preloaded with test compounds. Immediate light emission generated following calcium mobilization was recorded for 30 s. In the antagonist mode, after 30 min of incubation, the reference agonist was added to the above assay mix and light emission was recorded again. The final concentration of the reference agonist was equal to EC80 (30 nM apomorphine).
Thermodynamic Solubility Assay
LC/MS grade methanol, LC/MS grade acetonitrile, Dulbecco’s phosphate-buffered saline (DPBS), and formic acid (>98%) were from Sigma-Aldrich. HPLC grade water was obtained from the HLP 5 apparatus (HYDROLAB Poland) and was filtered through a 0.2 μm filter before use. Each of the analyzed compounds (1.0 mg) was weighed in a volumetric flask using an analytical balance. The volume was brought to 1 mL with methanol to obtain a 1.0 mg mL–1 solution. A series of dilutions of the standard solutions of the investigated compounds were prepared by diluting 500 μL of the stock solutions with methanol to make 1 mL and, afterward, diluting 500 μL of the obtained solutions again with water to make 1 mL. The procedure was repeated several times to obtain solutions with the concentration of the compounds in the range of 15.6–1000 μg mL–1. Each of the resulting solutions was analyzed in triplicate using 1 μL injections, and the results were used to obtain calibration curves. Each of the analyzed compounds (1.0 mg) was weighed in a SEPARA vial (GVS, Italy) using an analytical balance. The volume was brought to 0.5 mL with DPBS and the mixture was constantly agitated at 20 °C for 24 h in a thermoshaker. Afterward, the mixture was filtered directly in the vial and the filtrate was analyzed. Each sample was injected in triplicate in two series: using 1 and 10 μL. For the determination of the thermodynamic solubility of the investigated compounds, the quantitative UPLC method was developed. Chromatograms were recorded using a Waters eλ PDA detector. Spectra were analyzed in the 200–700 nm range with 1.2 nm resolution and a sampling rate of 20 points s–1. For calibration and quantification, areas under the peaks of the investigated compounds on DAD chromatograms were used. All analytical data were processed using MassLynx V4.1 software (Waters Corporation, Milford, MA). Calibration and quantitative methods’ validation were made using Statistica v. 13. The thermodynamic solubility of the investigated compounds was calculated using the formula
where Sol is the thermodynamic solubility in DPBS in 20 °C in μg mL–1, c is the concentration of the sample in μg mL–1 calculated using the obtained calibration curve, and Vinj is the volume of the injection in μL.
Metabolic Stability Studies
The metabolic stability studies were performed by Eurofins Pharma Discovery Services, according to well-established procedures. Further methodological details are available on the company website (www.eurofinsdiscoveryservices.com) and the appropriate publications.
Pharmacological Evaluation
Behavioral Studies
The experiments were performed on male Wistar rats (220–300 g) purchased from an accredited animal facility, the Jagiellonian University Medical College (Krakow, Poland). The animals were kept in a dark–light 12/12 h cycle (lights on at 8.00 a.m.), in groups of 4 per polycarbonate Makrolon type 3 cage (dimensions 26.5 × 15 × 42 cm3), at an ambient temperature of 21 ± 2 °C and relative humidity of 50–60%. Standard laboratory food (LSM-B) and filtered water was provided ad libitum. All of the experiments were carried out during the light phase, between 9.00 and 14.00. Each experimental group consisted of 6–8 rats/dose (with the exception of the social interaction test (SIT), where one group consisted of 7–8 pairs of rats). The animals were used only once and were killed immediately after experiments. All of the experimental procedures involving animals were carried out in full accordance with the ethical standards laid down by respective Polish and European (Directive no. 86/609/EEC) regulations and were approved by the I Local Ethics Commission at the Jagiellonian University Medical College in Krakow (Approval No 40/2018). The assays using reference drugs were performed at Jagiellonian University Medical College (Krakow, Poland) and the Institute of Psychiatry and Neurology, and the experimental procedures were approved by the IV Local Ethics Commission in Warsaw (Approval No 40/2008).
Drugs and Treatment
The following compounds were used: 7, 11, 16, imipramine (hydrochloride, Adamed, Pienkow, Poland), escitalopram (hydrochloride; Adamed, Pienkow, Poland), diazepam (Adamed, Pienkow, Poland), quetiapine (Adamed, Pienkow, Poland), dizocilpine ((+)-MK-801, hydrogen maleate, Sigma-Aldrich, U.K.). Compounds 7, 11, and 16 and quetiapine were suspended in a 1.5% solution of Tween 80 (Sigma-Aldrich, U.K.), while diazepam, imipramine, escitalopram, and MK-801 were dissolved in saline immediately before administration and injected intraperitoneally (i.p.) at a volume of 2 mL/kg body mass. All of the compounds, with the exception of imipramine and MK-801, which were injected 15 min before the test, were administered 60 min before the tests. Control animals received a vehicle (1.5% Tween 80 or saline) according to the same injection regime.
Forced Swim Test (FST) in Rats
The test was carried out according to the previously described method of Porsolt et al.82 On the first day of the experiment, rats were individually placed in Plexiglas cylinders of height 40 cm and diameter 18 cm, filled with water up to 15 cm maintained at 25 °C. After a 15 min period, rats were removed from the water and dried under a 60 W light bulb. On the following day, rats were placed again in the cylinders and the total time of immobility was recorded throughout the 5 min test period. The water was changed after each animal.
MK-801-Induced Hyperlocomotor Activity Test in Rats
The experiment was performed in a darkened room using a Motor Monitor System (Campden Instruments, Ltd., U.K.) consisting of two Smart Frame Open Field stations (40 × 40 × 38 cm3) with 16 × 16 beams, located in a sound-attenuating chamber and connected to a PC software via a control chassis; 60 min after vehicle or compound administration and 15 min after MK-801 (0.2 mg/kg), the treated rats were individually placed in the center of the station. An automated Motor Monitor System recorded ambulation (in both the X and Y axes), the number of rearing and the total distance covered by each rat for 30 min, with data registered every 5 min.65
Vogel Conflict Drinking Test in Rats
The testing procedure, based on a method described by Vogel et al.,83 was performed using the Anxiety Monitoring System “Vogel test” produced by TSE Systems. The apparatus consisted of a polycarbonate cage (dimensions 26.5 × 15 × 42 cm3), equipped with a grid floor made from stainless steel bars and a drinking bottle containing tap water. Experimental chambers (two) were connected to PC software by a control chassis and a device that generates electric shocks. In this “conditional” model an electric shock is applied as a noxious stimulus. The testing procedure consisted of a 2-day habituation/adaptation period and an actual test. On the first day of the experiment, the rats were adapted to the test chamber for a 10 min adaptation period, during which they had free access to the water drinking bottle, followed by a 24 h water deprivation period. Afterward, they were allowed a 30 min free-drinking session in their home cages. This protocol of 24 h deprivation and adaptation period was repeated on the second day. On the third day, the animals were placed again in the test chamber 60 min after administration of the vehicle or the test compound and were given free access to the water drinking bottle for 5 min. Recording data started immediately after the first lick and after every 20 licks, rats were punished with an electric shock (0.5 mA, lasting 1 s). The impulses were released via the spout of the drinking bottle. The number of shocks received throughout a 5 min experimental session was recorded automatically and was used as an indication of anticonflict activity.
Hot Plate and Free-Drinking Tests in Rats
To exclude possible drug-induced changes in shock sensitivity or an increasing influence on thirst drive, which can lead to false-positive results in the Vogel conflict drinking test, stimulus threshold and water consumption during a free-drinking session were determined in separate groups of rats. In both of these two studies, the rats were manipulated similarly to the Vogel conflict drinking test, including two 24 h water deprivation periods separated by a 10 min adaptation session in experimental cages and 30 min of water availability in their home cages. In the free-drinking test, each animal was allowed to freely drink from the drinking bottle and the amount of water (g) consumed during 5 min was recorded for each rat. The pain threshold was evaluated using a hot plate test (Commat Ltd, Turkey) in rats. The plate was enclosed with a transparent Plexiglass cylinder (35 cm high) to keep the animal on the heated surface of the plate. The latency to pain reaction (lick a hind paw or jump) when the rat was placed on a hot plate (52.5 ± 0.5 °C, 19 cm diameter) was measured. The rat was removed from the plate immediately upon visible pain reaction or if no response occurred within 30 s.
Elevated Plus-Maze (EPM) Test in Rats
The testing procedure was based on a method described by Pellow and File.84 The Plus Maze apparatus (an automated device produced by Campden Instruments Ltd., United Kingdom) made of durable, high density, nonporous black plastic, elevated to a height of 50 cm, consisted of two open arms (50 × 10 cm2) and two closed arms (50 cm × 10 cm, and 30 cm high walls), arranged so that the two arms of each type were opposite each other. The floor of the Plus Maze was made of infrared transparent material which means that there are no visible sensors. The Plus Maze apparatus was connected to PC software via a control chassis. The experiments were conducted in a darkened room, with only the center of the maze illuminated with low-intensity light (30 lux measured on the maze level). Each rat was gently placed in the center of the Plus Maze, facing one of the closed arms, immediately after a 5 min adaptation period in a plastic black box (60 × 60 × 35 cm3), to increase the overall activity in the EPM. During a 5 min test period, the automated Motor Monitor System recorded the number of entries into the closed and open arms and the time spent in either type of arm. The device counted an effective arm-entry when the four paws of a rat were into any arm. The maze was thoroughly cleaned after each trial. The number of open-arms entries, total time spent in open arms, and the percentages of these parameters were used as indications of anxiolytic-like activity.
Exploratory Activity Measured in the EPM in Rats
To assess the influence of the tested compounds on the general exploratory activity of rats and control possible changes within, total ambulation (the total distance covered by a rat, and ambulation along both X and Y axes) and the total number of entries (into open and closed arms) were taken during a 5 min test period (i.e., the time equal to the observation period in the EPM test). The experiment was performed using EPM apparatus (details see above).
Social Interaction Test
Five days before the experiment, male Wistar rats were placed individually in home cages (dimensions 26.5 × 15 × 42 cm3) and exposed daily with the touch and presence of the experimenter. The social interaction test was conducted in opaque black boxes with dimensions of 60 × 60 × 60 cm3 in an experimental room dimly illuminated with the diffused light of 30–40 Lx. On the fifth day of social isolation, 24 h before the test, rats were individually placed in experimental cages for 10 min and allowed to freely explore the arena. After this time, the rats were transferred to their home cages. Each social interaction experiment involving two rats was carried out during the light phase of the light/dark cycle. The rats were selected from separate housing cages to make a pair for the study. The paired rats were matched for body weight within 15 g. The social interaction was measured 30 min after i.p. administration of MK-801 at a dose of 0.1 mg/kg, and 60 min after administration of the tested compound. Rats in pair received the same treatment. Each pair of rats was diagonally placed in opposite corners of the box facing away from each other. The behavior of the animals was measured over a 10 min period. The test box was wiped clean between each trial. Social interaction between two rats was expressed as the total time spent in social behavior, such as sniffing, genital investigation, chasing, and fighting with each other.
Novel Object Recognition Test (NORT) in Rats
The protocol was adapted from the original work of Ennaceur and Delacour.85 The experiment was conducted in opaque black boxes with dimensions of 60 × 60 × 60 cm3. The 2-day procedure consists of habituation to the test arena (without any objects) for 5 min on the first day and a test session comprising two 3 min trials separated by a 1 h intertrial interval on the second day. During the first trial (familiarization, T1), two identical objects (A1 and A2) were presented in opposite corners of the arena, approximately 10 cm from the walls. During the second trial (recognition, T2), one of the A objects was replaced by a novel object B so that the animals were presented with A (familiar) and B (novel) objects. Both trials lasted for 3 min and the animals were returned to their home cages after T1. Metal Coca-Cola cans and glass jars filled with sand were used as the objects. The heights of the objects were comparable (approximately 12 cm) and the objects were heavy enough so that the animals could not displace them. The sequence of presentations and the location of the objects were randomly assigned to each rat. The animals explored the objects by looking, licking, sniffing, or touching them while sniffing, but not when leaning against, standing, or sitting on the objects. Any rat exploring the two objects for <5 s within the 3 min duration of T1 or T2 was eliminated from the study. An experimenter, blind to the drug treatment, measured the exploration time of the objects. Based on the exploration time (E) of the two objects during T2, the discrimination index (DI) was calculated according to the formula: DI = (EB – EA)/(EA + EB). Using this metric, scores approaching zero reflect no preference, while positive values reflect a preference for the novel object and negative numbers reflect a preference for the familiar object. In the test variant, which used MK-801 to induce memory impairment, MK-801 was administered at a dose of 0.1 mg/kg (i.p.) 30 min before the familiarization phase (T1).
Open Field Test (OFT) in Rats
The compounds active in FST, SIT, and/or MK-801-induced hyperlocomotor activity test were tested for their impact on spontaneous locomotor activity at doses that were active in these previous studies. The experiment was performed as described above. Individual vehicle- or drug-injected animals were gently placed in the center of the station. An automated Motor Monitor System recorded ambulation (in X and Y axes), the number of rearing, and total distance covered by a rat for 30 min, with data registered every 5 min.
Statistical Analysis
All of the data are presented as the mean ± SEM. The statistical significance of the results was evaluated by a one-way ANOVA, followed by a Bonferroni’s Comparison Test.
Metabolic Safety
The experiments were carried out on female Wistar rats, with an initial body weight of 185–195 g. The animals were housed in pairs in plastic cages in constant temperature facilities exposed to a light–dark cycle; water and food were available ad libitum. Both control and experimental groups consisted of six animals. All experiments were conducted according to the guidelines of the Animal Use and Care Committee of the Jagiellonian University and were approved for realization (2018, Poland; Permissions No 126/2018). Statistical calculations were performed using GraphPad Prism 6 software. Results are given as arithmetic means with a standard error of the mean. Statistical significance was calculated using a two-way variance analysis (ANOVA) with repeated measures, followed by a Bonferroni post hoc test (daily changes in body weight and spontaneous activity) or a one-way variance analysis (ANOVA) followed by a Tukey post hoc test (biochemical analysis, differences were considered statistically significant at: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001).
Palatable Western-Style Cafeteria Diet as a Reliable Method for Metabolic Syndrome Development
Female Wistar rats were housed in pairs. Four groups of 6 rats were fed on diets consisting of milk chocolate with nuts, cheese, salted peanuts, and 7% condensed milk with additional access to standard feed (Labofeed B, Morawski Manufacturer Feed, Poland) and water ad libitum, for 25 days. The palatable control group (palatable diet + vehicle) received vehicle (1% Tween 80, i.p.), while the palatable test groups (palatable diet + compound) received 11 or olanzapine injected intraperitoneally (i.p.) at a dose of 2 × 2 mg/kg b.w./day dissolved in 1% Tween 80. The body weights were measured daily immediately prior to administration of drugs.
On the 26th day, 20 min after i.p. administration of heparin (1000 units/rat) and thiopental (70 mg/kg b.w.), blood was collected from the animals.
The palatable diet contained: peanuts—100 g, 614 kcal; condensed milk—100 mL, 131 kcal; milk chocolate with hazelnuts—100 g, 195 kcal; cheese (Greek type)—100 g, 270 kcal. The standard diet contained 100 g of feed—280 kcal.
Biochemical Analysis
On the 25th day of the experiment, 20 min after intraperitoneal administration of heparin (5000 units/rat) and thiopental (70 mg/kg b.w.), blood was collected from the left carotid artery and then centrifuged at 600g (15 min, 4 °C) to obtain the plasma. The influence of the tested compounds on glucose, total cholesterol, HDL-cholesterol or triglyceride levels, and activity of alanine aminotransferase activity in plasma was analyzed. To determine the glucose, total cholesterol, HDL-cholesterol or triglyceride levels, and activity of alanine aminotransferase in plasma, standard enzymatic and spectrophotometric tests (Biomaxima S.A. Lublin, Poland) were used.
Determination of the Effect on Blood Pressure
On the 26th day, rats were anesthetized with thiopental (70 mg/kg) by i.p. injection. The left carotid artery was cannulated with tubing filled with heparin solution in saline to facilitate pressure measurements using Apparatus PowerLab 4/35 (ADInstruments, Australia). Blood pressure was measured: before intraperitoneal administration of AK37, olanzapine or 1% Tween 80—time 0 min (control pressure) and during the next 40 min.75,86,87
Pharmacokinetics Study in Rats
Extraction Procedure
Plasma samples (150 μL) containing compounds 7, 11, and 16 were transferred to Eppendorf tubes and 10 μL of internal standard solution ((3-chloro-4-fluorophenyl)(4-fluoro-4-((((5-methylpyridin-2-yl)methyl)amino)methyl)piperidin-1-yl)methanone—50 ng/mL for compounds 7 and 11, and 3-chloro-4-fluorophenyl)(4-fluoro-4-(((2-(2-(methylamino)phenoxy) ethyl)amino)methyl)piperidin-1-yl)methanone—500 ng/mL for 16 was added. The samples were alkalized with 20 μΛ of 4 M sodium hydroxide solution and after vortex-mixed were extracted with 1 mL of ethyl acetate/hexane (30:/, v/v) mixture for 20 min on a shaker (VXR Vibrax, IKA, Germany). Then, the samples were centrifuged (TDx Centrifuge, Abbott Laboratories), and the organic layers were transferred into new Eppendorf tubes containing 100 μL of methanol and 0.1 M sulfuric acid mixture (10:90, v/v). The samples were shaken and centrifuged again. Finally, 85–90 μL of each acidic layer was subjected to analysis via the HPLC system.
Frozen rat brains were thawed at room temperature, weighed, and then homogenized (4 mL/g) in distilled water with a tissue homogenizer TH220 (Omni International, Inc.). The tissue homogenates (2 mL) were transferred to glass tubes. Then, 10 μL of proper internal standard solution was added and samples were alkalized with 100 μL of 4 M sodium hydroxide solution. After vortex-mixing, samples were extracted with 6 mL of extraction mixture for 20 min on a shaker and centrifuged (Universal 32, Hettich, Germany). Subsequently, the organic layers were transferred into new glass tubes containing 150 μL of methanol and 0.1 M sulfuric acid mixture (10:90, v/v). Then, after shaking, the samples were centrifuged and 90 μL of each acidic layers was injected into the HPLC system.
Equipment and Chromatographic Conditions
The analysis of all tested compounds was performed in an HPLC system consisting of a P100 pump (Thermo Separation Products, San Jose, CA), an L-2200 autosampler, and an L-2420 UV/VIS detector (Merck-Hitachi). EZChrome Elite v. 3.2 software (Merck-Hitachi) was used for data acquisition. The chromatographic separation of all compounds and the internal standard was achieved at room temperature (22 ± 1 °C) on the Supelcosil PCN column 250 mm × 4.6 mm (Sigma-Aldrich, Germany) with 5 μm particles, protected with the Supelcosil LC-PCN guard column (Sigma-Aldrich, Germany) with the same packing material under isocratic conditions. The mobile phase consisted of 25 mM potassium dihydrogen phosphate buffer, pH 4.6: methanol: acetonitrile (51:40:9, v/v/v). The flow rate was 1.0 mL/min, and the detector wavelength was 203 nm. Validation of the chromatographic method was performed in accordance with the FDA guideline (Bioanalytical method validation, 2013). This included selectivity, linearity, accuracy and precision, limit of detection (LOD) and limit of quantitation (LOQ), recovery, and stability.
Pharmacokinetics Study
The compounds were administered via the intraperitoneal (i.p.) route to permit a direct comparison to the reference compounds, which have been previously studied following i.p. administration. Male Wistar rats weighing 250–300 g were housed in conditions of constant temperature with a 12:12 h light–dark cycle with access to food and water ad libitum. The animals were divided into two groups. In the first group (n = 3 for each compound), the animals were implanted with catheters (SAI Infusion Technologies) in the jugular vein under ketamine/xylazine anesthesia at least 2 days prior to the experiment. The animals (n = 3) received a single dose of 2 mg/kg (as a free base) of all tested compounds intraperitoneally and at 5, 15, 30, 60, 120, 180, and 300 min after dosing, blood samples were collected from the cannulated jugular vein into heparinized tubes. Plasma was separated by centrifugation at 3000 rpm for 10 min and stored at −80 °C until analysis. In the second group (n = 9 for each compound) animals (without catheters) were exsanguinated 30, 60, and 180 min post-dose (n = 3 per time point). Plasma and brains were harvested and stored at −80 °C until analysis. All animal procedures were approved by the 2nd Local Institutional Animal Care and Use Committee in Krakow.
Pharmacokinetics Analysis
Plasma concentration versus time data was analyzed by a noncompartmental approach (NCA) in Phoenix WinNonlin v. 7.0 (Pharsight Corporation, a Certara Company, Princeton, NJ). The maximum concentration (Cmax) and the time to reach peak concentration (tmax) in plasma following intraperitoneal dosing were obtained directly from the concentration versus time data. The terminal elimination rate constant (λz) was assessed by linear regression and terminal half-life (t0.5λz) was calculated as ln 2/λz. The area under the concentration–time curve from the time of dosing to infinity (AUC0–∞) was calculated by the linear trapezoidal rule. The extrapolated terminal area was defined as Cn/λz, where Cn is the last data point. Clearance (CL/F) was calculated as D/AUC0–∞, the volume of distribution based on the terminal phase (Vz/F) was estimated as D/(λz·AUC0–∞), where F is the fraction absorbed, and mean residence time (MRT) as calculated as AUC0–∞/AUMC0–∞, where AMUC0–∞ is the area under the first moment curve from the time of dosing to infinity. Data were presented as mean ± SD.
Acknowledgments
This study was financially supported by The National Science Centre, Poland (NCN), Grant No. 2014/15/D/NZ7/01789.
Glossary
Abbreviations Used
- BPSD
behavioral and psychological symptoms of dementia
- CNS MPO
Central Nervous System Multiparameter Optimization
- EPS
elevated plus maze test
- FST
forced swimming test (Porsolt test)
- SIT
social interaction test
- NORT
novel object recognition task
- MK-801
dizocilpine
- SB214111
4-bromo-N-[4-methoxy-3-(4-methylpiperazin-1-yl)phenyl]benzenesulfonamide
- SB258719
3-methyl-N-[(1R)-1-methyl-3-(4-methyl-1-piperidinyl)propyl]-N-methylbenzenesulfonamide hydrochloride
- PAINS
pan assay interference compounds
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00497.
In silico predictions of biotransformation routes, molecular druglike properties, and the detailed characteristics of binding modes in the target receptors; detailed procedures for the preparation of the intermediates I–VI; HPLC traces, 1H NMR, and 13C NMR spectra of the final products 7, 11, and 16; and the effect of 11 and olanzapine on body weight (PDF)
5-HT2AR homology model (PDB)
5-HT6R homology model (PDB)
5-HT7R homology model (PDB)
D2R homology model (PDB)
Molecular Formula Strings (CSV)
Author Contributions
All authors have contributed to the work and have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Paper | ALZFORUM. https://www.alzforum.org/papers/uber-eine-eigenartige-erkrankung-der-hirnrinde/en (accessed 2021-02-01).
- Cerejeira J.; Lagarto L.; Mukaetova-Ladinska E. B. Behavioral and Psychological Symptoms of Dementia. Front. Neurol. 2012, 3, 73. 10.3389/fneur.2012.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller-Spahn F. Behavioral Disturbances in Dementia. Dialogues Clin. Neurosci. 2003, 5, 49–59. 10.31887/DCNS.2003.5.1/fmuellerspahn. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinkowska M.; Śniecikowska J.; Fajkis N.; Paśko P.; Franczyk W.; Kołaczkowski M. Management of Dementia-Related Psychosis, Agitation and Aggression: A Review of the Pharmacology and Clinical Effects of Potential Drug Candidates. CNS Drugs 2020, 34, 243–268. 10.1007/s40263-020-00707-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach L. B.; Kales H. C. Managing Behavioral and Psychological Symptoms of Dementia. Psychiatr. Clin. North Am. 2018, 41, 127–139. 10.1016/j.psc.2017.10.010. [DOI] [PubMed] [Google Scholar]
- Lyketsos C. G.; Olin J. Depression in Alzheimer’s Disease: Overview and Treatment. Biol. Psychiatry 2002, 52, 243–252. 10.1016/S0006-3223(02)01348-3. [DOI] [PubMed] [Google Scholar]
- Orgeta V.; Tabet N.; Nilforooshan R.; Howard R. Efficacy of Antidepressants for Depression in Alzheimer’s Disease: Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2017, 58, 725–733. 10.3233/JAD-161247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Gao S.; Hendrie H. C.; Kesterson J.; Campbell N. L.; Shekhar A.; Callahan C. M. Antidepressant Use in the Elderly Is Associated with an Increased Risk of Dementia. Alzheimer Dis. Assoc. Disord. 2016, 30, 99–104. 10.1097/WAD.0000000000000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho A. F.; Sharma M. S.; Brunoni A. R.; Vieta E.; Fava G. A. The Safety, Tolerability and Risks Associated with the Use of Newer Generation Antidepressant Drugs: A Critical Review of the Literature. Psychother. Psychosom. 2016, 85, 270–288. 10.1159/000447034. [DOI] [PubMed] [Google Scholar]
- Coupland C.; Hill T.; Morriss R.; Moore M.; Arthur A.; Hippisley-Cox J. Antidepressant Use and Risk of Cardiovascular Outcomes in People Aged 20 to 64: Cohort Study Using Primary Care Database. BMJ 2016, 352, i1350. 10.1136/bmj.i1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxman T. E. Antidepressants and Cognitive Impairment in the Elderly. J. Clin. Psychiatry 1996, 57, 38–44. [PubMed] [Google Scholar]
- Farlow M. R.; Shamliyan T. A. Benefits and Harms of Atypical Antipsychotics for Agitation in Adults with Dementia. Eur. Neuropsychopharmacol. 2017, 27, 217–231. 10.1016/j.euroneuro.2017.01.002. [DOI] [PubMed] [Google Scholar]
- Parker C.; Coupland C.; Hippisley-Cox J. Antipsychotic Drugs and Risk of Venous Thromboembolism: Nested Case-Control Study. BMJ 2010, 341, c4245. 10.1136/bmj.c4245. [DOI] [PubMed] [Google Scholar]
- Tampi R. R.; Tampi D. J.; Balachandran S.; Srinivasan S. Antipsychotic Use in Dementia: A Systematic Review of Benefits and Risks from Meta-Analyses. Ther. Adv. Chronic Dis. 2016, 7, 229–245. 10.1177/2040622316658463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider L. S.; Dagerman K. S.; Insel P. Risk of Death with Atypical Antipsychotic Drug Treatment for Dementia: Meta-Analysis of Randomized Placebo-Controlled Trials. JAMA 2005, 294, 1934–1943. 10.1001/jama.294.15.1934. [DOI] [PubMed] [Google Scholar]
- Markota M.; Rummans T. A.; Bostwick J. M.; Lapid M. I. Benzodiazepine Use in Older Adults: Dangers, Management, and Alternative Therapies. Mayo Clin. Proc. 2016, 91, 1632–1639. 10.1016/j.mayocp.2016.07.024. [DOI] [PubMed] [Google Scholar]
- Kales H. C.; Lyketsos C. G.; Miller E. M.; Ballard C. Management of Behavioral and Psychological Symptoms in People with Alzheimer’s Disease: An International Delphi Consensus. Int Psychogeriatr 2019, 31, 83–90. 10.1017/S1041610218000534. [DOI] [PubMed] [Google Scholar]
- Garay R. P.; Citrome L.; Grossberg G. T.; Cavero I.; Llorca P.-M. Investigational Drugs for Treating Agitation in Persons with Dementia. Expert Opin. Invest. Drugs 2016, 25, 973–983. 10.1080/13543784.2016.1193155. [DOI] [PubMed] [Google Scholar]
- Vogt I.; Prinz J.; Campillos M. Molecularly and Clinically Related Drugs and Diseases Are Enriched in Phenotypically Similar Drug-Disease Pairs. Genome Med. 2014, 6, 52 10.1186/s13073-014-0052-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes C.; Arranz M. J.; Powell J. F.; Collier D. A.; Lovestone S. 5-HT2A and 5-HT2C Receptor Polymorphisms and Psychopathology in Late Onset Alzheimer’s Disease. Hum. Mol. Genet. 1998, 7, 1507–1509. 10.1093/hmg/7.9.1507. [DOI] [PubMed] [Google Scholar]
- Assal F.; Alarcón M.; Solomon E. C.; Masterman D.; Geschwind D. H.; Cummings J. L. Association of the Serotonin Transporter and Receptor Gene Polymorphisms in Neuropsychiatric Symptoms in Alzheimer Disease. Arch. Neurol. 2004, 61, 1249–1253. 10.1001/archneur.61.8.1249. [DOI] [PubMed] [Google Scholar]
- ACADIA Pharmaceuticals Announces U.S. FDA Accepted for Filing the Supplemental New Drug Application for NUPLAZID (pimavanserin) for the Treatment of Hallucinations and Delusions Associated with Dementia-Related Psychosis | Acadia Pharmaceuticals Inc. https://ir.acadia-pharm.com/news-releases/news-release-details/acadia-pharmaceuticals-announces-us-fda-accepted-filing (accessed 2021-02-01).
- Lorke D. E.; Lu G.; Cho E.; Yew D. T. Serotonin 5-HT2A and 5-HT6 Receptors in the Prefrontal Cortex of Alzheimer and Normal Aging Patients. BMC Neurosci. 2006, 7, 36. 10.1186/1471-2202-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan R.; Nabulsi N.; Gaiser E.; Gallezot J.-D.; Henry S.; Planeta B.; Lin S.; Ropchan J.; Williams W.; Morris E.; D’Souza D. C.; Huang Y.; Carson R. E.; Matuskey D. Age-Related Change in 5-HT6 Receptor Availability in Healthy Male Volunteers Measured with 11C-GSK215083 PET. J. Nucl. Med. 2018, 59, 1445–1450. 10.2967/jnumed.117.206516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Alloza M.; Hirst W. D.; Chen C. P. L.-H.; Lasheras B.; Francis P. T.; Ramírez M. J. Differential Involvement of 5-HT 1B/1D and 5-HT 6 Receptors in Cognitive and Non-Cognitive Symptoms in Alzheimer’s Disease. Neuropsychopharmacology 2004, 29, 410–416. 10.1038/sj.npp.1300330. [DOI] [PubMed] [Google Scholar]
- Dupuis D. S.; Mannoury la Cour C.; Chaput C.; Verrièle L.; Lavielle G.; Millan M. J. Actions of Novel Agonists, Antagonists and Antipsychotic Agents at Recombinant Rat 5-HT6 Receptors: A Comparative Study of Coupling to G Alpha s. Eur. J. Pharmacol. 2008, 588, 170–177. 10.1016/j.ejphar.2008.04.039. [DOI] [PubMed] [Google Scholar]
- Nikiforuk A.; Kos T.; Fijał K.; Hołuj M.; Rafa D.; Popik P. Effects of the Selective 5-HT7 Receptor Antagonist SB-269970 and Amisulpride on Ketamine-Induced Schizophrenia-like Deficits in Rats. PLoS One 2013, 8, e66695 10.1371/journal.pone.0066695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saroja S. R.; Kim E.-J.; Shanmugasundaram B.; Höger H.; Lubec G. Hippocampal Monoamine Receptor Complex Levels Linked to Spatial Memory Decline in the Aging C57BL/6J. Behav. Brain Res. 2014, 264, 1–8. 10.1016/j.bbr.2014.01.042. [DOI] [PubMed] [Google Scholar]
- Beaudet G.; Bouet V.; Jozet-Alves C.; Schumann-Bard P.; Dauphin F.; Paizanis E.; Boulouard M.; Freret T. Spatial Memory Deficit across Aging: Current Insights of the Role of 5-HT7 Receptors. Front. Behav. Neurosci. 2015, 8, 448. 10.3389/fnbeh.2014.00448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X.; Kaminga A. C.; Wen S. W.; Wu X.; Acheampong K.; Liu A. Dopamine and Dopamine Receptors in Alzheimer’s Disease: A Systematic Review and Network Meta-Analysis. Front. Aging Neurosci. 2019, 11, 175. 10.3389/fnagi.2019.00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell R. A.; Herrmann N.; Lanctôt K. L. The Role of Dopamine in Symptoms and Treatment of Apathy in Alzheimer’s Disease. CNS Neurosci. Ther. 2011, 17, 411–427. 10.1111/j.1755-5949.2010.00161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y.; Meguro K.; Yamaguchi S.; Ishii H.; Watanuki S.; Funaki Y.; Yamaguchi K.; Yamadori A.; Iwata R.; Itoh M. Decreased Striatal D2 Receptor Density Associated with Severe Behavioral Abnormality in Alzheimer’s Disease. Ann. Nucl. Med. 2003, 17, 567–573. 10.1007/BF03006670. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Jiang X.; He S.; Jiang H.; Feng F.; Liu W.; Qu W.; Sun H. Rational Design of Multitarget-Directed Ligands: Strategies and Emerging Paradigms. J. Med. Chem. 2019, 62, 8881–8914. 10.1021/acs.jmedchem.9b00017. [DOI] [PubMed] [Google Scholar]
- Morphy R.; Rankovic Z. Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem. 2005, 48, 6523–6543. 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- Morphy R.; Rankovic Z.. Medicinal Chemistry Approaches for Multitarget Drugs. In Burger’s Medicinal Chemistry and Drug Discovery; American Cancer Society, 2010; pp 249–274. [Google Scholar]
- Bucki A.; Marcinkowska M.; Śniecikowska J.; Zagórska A.; Jamrozik M.; Pawłowski M.; Głuch-Lutwin M.; Siwek A.; Jakubczyk M.; Pytka K.; Jastrzębska-Więsek M.; Partyka A.; Wesołowska A.; Mierzejewski P.; Kołaczkowski M. Multifunctional 6-Fluoro-3-[3-(Pyrrolidin-1-Yl)Propyl]-1,2-Benzoxazoles Targeting Behavioral and Psychological Symptoms of Dementia (BPSD). Eur. J. Med. Chem. 2020, 191, 112149 10.1016/j.ejmech.2020.112149. [DOI] [PubMed] [Google Scholar]
- Song D.; Liu H.; Zhang A.; Qu J. Fragmentation of Typical Sulfonamide Drugs via Heterolytic Bond Cleavage and Stepwise Rearrangement. RSC Adv. 2014, 4, 48426–48432. 10.1039/C4RA07737G. [DOI] [Google Scholar]
- Di L.; Kerns E. H.. Drug-like Properties: Concepts, Structure Design and Methods: From ADME to Toxicity Optimization, 1st ed.; Academic Press: Amsterdam; Boston, 2008. [Google Scholar]
- Raubo P.; Beer M. S.; Hunt P. A.; Huscroft I. T.; London C.; Stanton J. A.; Kulagowski J. J. Aminoalkyl Phenyl Sulfones-a Novel Series of 5-HT(7) Receptor Ligands. Bioorg. Med. Chem. Lett. 2006, 16, 1255–1258. 10.1016/j.bmcl.2005.11.067. [DOI] [PubMed] [Google Scholar]
- Bernotas R.; Lenicek S.; Antane S.; Zhang G. M.; Smith D.; Coupet J.; Harrison B.; Schechter L. E. 1-(2-Aminoethyl)-3-(Arylsulfonyl)-1H-Indoles as Novel 5-HT6 Receptor Ligands. Bioorg. Med. Chem. Lett. 2004, 14, 5499–5502. 10.1016/j.bmcl.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Hirst W. D.; Minton J. A. L.; Bromidge S. M.; Moss S. F.; Latter A. J.; Riley G.; Routledge C.; Middlemiss D. N.; Price G. W. Characterization of [125I]-SB-258585 Binding to Human Recombinant and Native 5-HT6 Receptors in Rat, Pig and Human Brain Tissue. Br. J. Pharmacol. 2000, 130, 1597–1605. 10.1038/sj.bjp.0703458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes I. T.; Dabbs S.; Duckworth D. M.; Jennings A. J.; King F. D.; Lovell P. J.; Brown A. M.; Collin L.; Hagan J. J.; Middlemiss D. N.; Riley G. J.; Thomas D. R.; Upton N. (R)-3,N-Dimethyl-N-[1-Methyl-3-(4-Methylpiperidin-1-Yl)Propyl]Benzenesulfonamide: The First Selective 5-HT7 Receptor Antagonist. J. Med. Chem. 1998, 41, 655–657. 10.1021/jm970519e. [DOI] [PubMed] [Google Scholar]
- Kołaczkowski M.; Nowak M.; Pawłowski M.; Bojarski A. J. Receptor-Based Pharmacophores for Serotonin 5-HT7R Antagonists-Implications to Selectivity. J. Med. Chem. 2006, 49, 6732–6741. 10.1021/jm060300c. [DOI] [PubMed] [Google Scholar]
- Fielding S.; Novick W. J.; Geyer H. M.; Petko W. W.; Wilker J. C.; Davis L.; Klein J. T.; Cornfeldt M. The Preclinical Antipsychotic Evaluation of HRP 913, a Novel Benzisoxazole Derivative. Drug Dev. Res. 1983, 3, 233–243. 10.1002/ddr.430030305. [DOI] [Google Scholar]
- Kołaczkowski M.; Marcinkowska M.; Bucki A.; Pawłowski M.; Mitka K.; Jaśkowska J.; Kowalski P.; Kazek G.; Siwek A.; Wasik A.; Wesołowska A.; Mierzejewski P.; Bienkowski P. Novel Arylsulfonamide Derivatives with 5-HT6/5-HT7 Receptor Antagonism Targeting Behavioral and Psychological Symptoms of Dementia. J. Med. Chem. 2014, 57, 4543–4557. 10.1021/jm401895u. [DOI] [PubMed] [Google Scholar]
- Tata L. J.; West J.; Smith C.; Farrington P.; Card T.; Smeeth L.; Hubbard R. General Population Based Study of the Impact of Tricyclic and Selective Serotonin Reuptake Inhibitor Antidepressants on the Risk of Acute Myocardial Infarction. Heart 2005, 91, 465–471. 10.1136/hrt.2004.037457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coupland C.; Dhiman P.; Morriss R.; Arthur A.; Barton G.; Hippisley-Cox J. Antidepressant Use and Risk of Adverse Outcomes in Older People: Population Based Cohort Study. BMJ 2011, 343, d4551. 10.1136/bmj.d4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olten B.; Bloch M. H. Meta Regression: Relationship between Antipsychotic Receptor Binding Profiles and Side-Effects. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 84, 272–281. 10.1016/j.pnpbp.2018.01.023. [DOI] [PubMed] [Google Scholar]
- Salvi V.; Barone-Adesi F.; D’Ambrosio V.; Albert U.; Maina G. High H1-Affinity Antidepressants and Risk of Metabolic Syndrome in Bipolar Disorder. Psychopharmacology 2016, 233, 49–56. 10.1007/s00213-015-4085-9. [DOI] [PubMed] [Google Scholar]
- Starrenburg F. C. J.; Bogers J. P. A. M. How Can Antipsychotics Cause Diabetes Mellitus? Insights Based on Receptor-Binding Profiles, Humoral Factors and Transporter Proteins. Eur. Psychiatry 2009, 24, 164–170. 10.1016/j.eurpsy.2009.01.001. [DOI] [PubMed] [Google Scholar]
- Derijks H. J.; Meyboom R. H. B.; Heerdink E. R.; De Koning F. H. P.; Janknegt R.; Lindquist M.; Egberts A. C. G. The Association between Antidepressant Use and Disturbances in Glucose Homeostasis: Evidence from Spontaneous Reports. Eur. J. Clin. Pharmacol. 2008, 64, 531–538. 10.1007/s00228-007-0441-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.-S.; Wang S.-C.; Gau S. S.-F.; Tsai H.-J.; Cheng Y.-C. Association of Stroke with the Receptor-Binding Profiles of Antipsychotics-a Case-Crossover Study. Biol. Psychiatry 2013, 73, 414–421. 10.1016/j.biopsych.2012.07.006. [DOI] [PubMed] [Google Scholar]
- Douglas I. J.; Smeeth L. Exposure to Antipsychotics and Risk of Stroke: Self Controlled Case Series Study. BMJ 2008, 337, a1227. 10.1136/bmj.a1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ÜÇOK A. L. P.; GAEBEL W. Side Effects of Atypical Antipsychotics: A Brief Overview. World Psychiatry 2008, 7, 58–62. 10.1002/j.2051-5545.2008.tb00154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Research, C. for D. E. and. Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System. Guidance for Industry. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/waiver-vivo-bioavailability-and-bioequivalence-studies-immediate-release-solid-oral-dosage-forms (accessed 2021-02-19).
- Obach R. S.; Baxter J. G.; Liston T. E.; Silber B. M.; Jones B. C.; MacIntyre F.; Rance D. J.; Wastall P. The Prediction of Human Pharmacokinetic Parameters from Preclinical and in Vitro Metabolism Data. J. Pharmacol. Exp. Ther. 1997, 283, 46–58. [PubMed] [Google Scholar]
- Komossa K.; Depping A. M.; Gaudchau A.; Kissling W.; Leucht S. Second-Generation Antipsychotics for Major Depressive Disorder and Dysthymia. Cochrane Database Syst. Rev. 2010, CD008121 10.1002/14651858.CD008121.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisler R. H.; Montgomery S. A.; Earley W. R.; Szamosi J.; Lazarus A. Efficacy of Extended Release Quetiapine Fumarate Monotherapy in Patients with Major Depressive Disorder: A Pooled Analysis of Two 6-Week, Double-Blind, Placebo-Controlled Studies. Int. Clin. Psychopharmacol. 2012, 27, 27–39. 10.1097/YIC.0b013e32834d6f91. [DOI] [PubMed] [Google Scholar]
- Nyberg S.; Widzowski D. PW01-27 - Translational Pharmacology of Quetiapine and Norquetiapine: Preclinical Findings Support Multifunctional Psychotropic Properties. Eur. Psychiatry 2010, 25, 1446. 10.1016/S0924-9338(10)71429-2. [DOI] [Google Scholar]
- Carr G. V.; Schechter L. E.; Lucki I. Antidepressant and Anxiolytic Effects of Selective 5-HT6 Receptor Agonists in Rats. Psychopharmacology 2011, 213, 499–507. 10.1007/s00213-010-1798-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Żmudzka E.; Sałaciak K.; Sapa J.; Pytka K. Serotonin Receptors in Depression and Anxiety: Insights from Animal Studies. Life Sci. 2018, 210, 106–124. 10.1016/j.lfs.2018.08.050. [DOI] [PubMed] [Google Scholar]
- Ichikawa J.; Ishii H.; Bonaccorso S.; Fowler W. L.; O’Laughlin I. A.; Meltzer H. Y. 5-HT(2A) and D(2) Receptor Blockade Increases Cortical DA Release via 5-HT(1A) Receptor Activation: A Possible Mechanism of Atypical Antipsychotic-Induced Cortical Dopamine Release. J. Neurochem. 2001, 76, 1521–1531. 10.1046/j.1471-4159.2001.00154.x. [DOI] [PubMed] [Google Scholar]
- Owens M. J.; Morgan W. N.; Plott S. J.; Nemeroff C. B. Neurotransmitter Receptor and Transporter Binding Profile of Antidepressants and Their Metabolites. J. Pharmacol. Exp. Ther. 1997, 283, 1305–1322. [PubMed] [Google Scholar]
- Kołaczkowski M.; Mierzejewski P.; Bienkowski P.; Wesołowska A.; Newman-Tancredi A. Antipsychotic, Antidepressant, and Cognitive-Impairment Properties of Antipsychotics: Rat Profile and Implications for Behavioral and Psychological Symptoms of Dementia. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2014, 387, 545–557. 10.1007/s00210-014-0966-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mierzejewski P.; Kolaczkowski M.; Marcinkowska M.; Wesolowska A.; Samochowiec J.; Pawlowski M.; Bienkowski P. Antipsychotic-like Effects of Zolpidem in Wistar Rats. Eur. J. Pharmacol. 2016, 773, 51–58. 10.1016/j.ejphar.2016.01.009. [DOI] [PubMed] [Google Scholar]
- Weisstaub N. V.; Zhou M.; Lira A.; Lambe E.; González-Maeso J.; Hornung J.-P.; Sibille E.; Underwood M.; Itohara S.; Dauer W. T.; Ansorge M. S.; Morelli E.; Mann J. J.; Toth M.; Aghajanian G.; Sealfon S. C.; Hen R.; Gingrich J. A. Cortical 5-HT2A Receptor Signaling Modulates Anxiety-like Behaviors in Mice. Science 2006, 313, 536–540. 10.1126/science.1123432. [DOI] [PubMed] [Google Scholar]
- Pytka K.; Głuch-Lutwin M.; Żmudzka E.; Sałaciak K.; Siwek A.; Niemczyk K.; Walczak M.; Smolik M.; Olczyk A.; Gałuszka A.; Śmieja J.; Filipek B.; Sapa J.; Kołaczkowski M.; Pańczyk K.; Waszkielewicz A.; Marona H. HBK-17, a 5-HT1A Receptor Ligand With Anxiolytic-Like Activity, Preferentially Activates ß-Arrestin Signaling. Front. Pharmacol. 2018, 9, 1146. 10.3389/fphar.2018.01146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pytka K.; Głuch-Lutwin M.; Kotańska M.; Żmudzka E.; Jakubczyk M.; Waszkielewicz A.; Janiszewska P.; Walczak M. HBK-15 Protects Mice from Stress-Induced Behavioral Disturbances and Changes in Corticosterone, BDNF, and NGF Levels. Behav. Brain Res. 2017, 333, 54–66. 10.1016/j.bbr.2017.06.032. [DOI] [PubMed] [Google Scholar]
- Hacksell U.; Burstein E. S.; McFarland K.; Mills R. G.; Williams H. On the Discovery and Development of Pimavanserin: A Novel Drug Candidate for Parkinson’s Psychosis. Neurochem. Res. 2014, 39, 2008–2017. 10.1007/s11064-014-1293-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanover K. E.; Weiner D. M.; Makhay M.; Veinbergs I.; Gardell L. R.; Lameh J.; Del Tredici A. L.; Piu F.; Schiffer H. H.; Ott T. R.; Burstein E. S.; Uldam A. K.; Thygesen M. B.; Schlienger N.; Andersson C. M.; Son T. Y.; Harvey S. C.; Powell S. B.; Geyer M. A.; Tolf B.-R.; Brann M. R.; Davis R. E. Pharmacological and Behavioral Profile of N-(4-Fluorophenylmethyl)-N-(1-Methylpiperidin-4-Yl)-N’-(4-(2-Methylpropyloxy)Phenylmethyl) Carbamide (2R,3R)-Dihydroxybutanedioate (2:1) (ACP-103), a Novel 5-Hydroxytryptamine(2A) Receptor Inverse Agonist. J. Pharmacol. Exp. Ther. 2006, 317, 910–918. 10.1124/jpet.105.097006. [DOI] [PubMed] [Google Scholar]
- Deiana S.; Watanabe A.; Yamasaki Y.; Amada N.; Kikuchi T.; Stott C.; Riedel G. MK-801-Induced Deficits in Social Recognition in Rats: Reversal by Aripiprazole, but Not Olanzapine, Risperidone, or Cannabidiol. Behav. Pharmacol. 2015, 26, 748–765. 10.1097/FBP.0000000000000178. [DOI] [PubMed] [Google Scholar]
- Rajagopal L.; Massey B. W.; Huang M.; Oyamada Y.; Meltzer H. Y. The Novel Object Recognition Test in Rodents in Relation to Cognitive Impairment in Schizophrenia. Curr. Pharm. Des. 2014, 20, 5104–5114. 10.2174/1381612819666131216114240. [DOI] [PubMed] [Google Scholar]
- Kotańska M.; Śniecikowska J.; Jastrzębska-Więsek M.; Kołaczkowski M.; Pytka K. Metabolic and Cardiovascular Benefits and Risks of EMD386088—A 5-HT6 Receptor Partial Agonist and Dopamine Transporter Inhibitor. Front Neurosci 2017, 11, 50. 10.3389/fnins.2017.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotańska M.; Lustyk K.; Bucki A.; Marcinkowska M.; Śniecikowska J.; Kołaczkowski M. Idalopirdine, a Selective 5-HT6 Receptor Antagonist, Reduces Food Intake and Body Weight in a Model of Excessive Eating. Metab. Brain Dis. 2018, 33, 733–740. 10.1007/s11011-017-0175-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek M.; Knutelska J.; Bednarski M.; Nowiński L.; Zygmunt M.; Kazek G.; Mordyl B.; Głuch-Lutwin M.; Zaręba P.; Kulig K.; Sapa J. Pyrrolidin-2-One Derivatives May Reduce Body Weight in Rats with Diet-Induced Obesity. Eur. J. Pharmacol. 2016, 776, 146–155. 10.1016/j.ejphar.2016.02.047. [DOI] [PubMed] [Google Scholar]
- Kurczab R.; Ali W.; Łażewska D.; Kotańska M.; Jastrzębska-Więsek M.; Satała G.; Więcek M.; Lubelska A.; Latacz G.; Partyka A.; Starek M.; Dąbrowska M.; Wesołowska A.; Jacob C.; Kieć-Kononowicz K.; Handzlik J. Computer-Aided Studies for Novel Arylhydantoin 1,3,5-Triazine Derivatives as 5-HT6 Serotonin Receptor Ligands with Antidepressive-Like, Anxiolytic and Antiobesity Action In Vivo. Molecules 2018, 23, 2529. 10.3390/molecules23102529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucki A.; Marcinkowska M.; Śniecikowska J.; Więckowski K.; Pawłowski M.; Głuch-Lutwin M.; Gryboś A.; Siwek A.; Pytka K.; Jastrzębska-Więsek M.; Partyka A.; Wesołowska A.; Mierzejewski P.; Kołaczkowski M. Novel 3-(1,2,3,6-Tetrahydropyridin-4-Yl)-1H-Indole-Based Multifunctional Ligands with Antipsychotic-Like, Mood-Modulating, and Procognitive Activity. J. Med. Chem. 2017, 60, 7483–7501. 10.1021/acs.jmedchem.7b00839. [DOI] [PubMed] [Google Scholar]
- Kołaczkowski M.; Bucki A.; Feder M.; Pawłowski M. Ligand-Optimized Homology Models of D1 and D2 Dopamine Receptors: Application for Virtual Screening. J. Chem. Inf. Model. 2013, 53, 638–648. 10.1021/ci300413h. [DOI] [PubMed] [Google Scholar]
- Daina A.; Michielin O.; Zoete V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y.-C.; Prusoff W. H. Relationship between the Inhibition Constant (KI) and the Concentration of Inhibitor Which Causes 50 per cent Inhibition (I50) of an Enzymatic Reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Marcinkowska M.; Kotańska M.; Zagórska A.; Śniecikowska J.; Kubacka M.; Siwek A.; Bucki A.; Pawłowski M.; Bednarski M.; Sapa J.; Starek M.; Dąbrowska M.; Kołaczkowski M. Synthesis and Biological Evaluation of N-Arylpiperazine Derivatives of 4,4-Dimethylisoquinoline-1,3(2H,4H)-Dione as Potential Antiplatelet Agents. J. Enzyme Inhib. Med. Chem. 2018, 33, 536–545. 10.1080/14756366.2018.1437155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porsolt R. D.; Anton G.; Blavet N.; Jalfre M. Behavioural Despair in Rats: A New Model Sensitive to Antidepressant Treatments. Eur. J. Pharmacol. 1978, 47, 379–391. 10.1016/0014-2999(78)90118-8. [DOI] [PubMed] [Google Scholar]
- Vogel J. R.; Beer B.; Clody D. E. A Simple and Reliable Conflict Procedure for Testing Anti-Anxiety Agents. Psychopharmacologia 1971, 21, 1–7. 10.1007/BF00403989. [DOI] [PubMed] [Google Scholar]
- Pellow S.; File S. E. Anxiolytic and Anxiogenic Drug Effects on Exploratory Activity in an Elevated Plus-Maze: A Novel Test of Anxiety in the Rat. Pharmacol., Biochem. Behav. 1986, 24, 525–529. 10.1016/0091-3057(86)90552-6. [DOI] [PubMed] [Google Scholar]
- Ennaceur A.; Delacour J. A New One-Trial Test for Neurobiological Studies of Memory in Rats. 1: Behavioral Data. Behav. Brain Res. 1988, 31, 47–59. 10.1016/0166-4328(88)90157-X. [DOI] [PubMed] [Google Scholar]
- Dudek M.; Knutelska J.; Bednarski M.; Nowiński L.; Zygmunt M.; Mordyl B.; Głuch-Lutwin M.; Kazek G.; Sapa J.; Pytka K. A Comparison of the Anorectic Effect and Safety of the Alpha2-Adrenoceptor Ligands Guanfacine and Yohimbine in Rats with Diet-Induced Obesity. PLoS One 2015, 10, e0141327 10.1371/journal.pone.0141327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek M.; Marcinkowska M.; Bucki A.; Olczyk A.; Kołaczkowski M. Idalopirdine - a Small Molecule Antagonist of 5-HT6 with Therapeutic Potential against Obesity. Metab. Brain Dis. 2015, 30, 1487–1494. 10.1007/s11011-015-9736-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.