

Abstract
The binding of adenosine 5’-triphosphate (ATP) and adenosine 5’-monophosphate (AMP) to adenylate kinase (AdK) drives closure of lids over the substrate adenosyl groups. We test the hypothesis that this conformational change activates AdK for catalysis. The rate constants for Homo sapiens adenylate kinase 1 (HsAdK1)-catalyzed phosphoryl group transfer to AMP, kcat/Km = 7.0 x 106 M−1 s−1, and phosphite dianion, (kHPi)obs ≤ 1 x 10−4 M−1 s−1, show that the binding energy of the adenosyl group effects a ≥7.0 x 1010-fold rate acceleration of phosphoryl transfer from ATP. The third-order rate constant of kcat/KHPiKEA = 260 M−2 s−1 for 1-(β-d-erythrofuranosyl)adenine (EA)-activated phosphoryl transfer to phosphite dianion was determined, and the isohypophosphate reaction product characterized by 31P NMR. The results demonstrate: (i) a ≥14.7 kcal/mol stabilization of the transition state for phosphoryl transfer by the adenosyl group of AMP and a ≥2.6 x 106-fold rate acceleration from the EA-driven conformational change, and (ii) the recovery of ≥8.7 kcal/mol of this transition state stabilization for EA-activated phosphoryl transfer from ATP to phosphite.
Graphical Abstract
The phosphodianion of phosphate monoester substrates for metabolic reactions provides ca. 12 kcal/mol of binding energy for stabilization of transition states for enzyme-catalyzed proton transfer, hydride transfer,1, 2 decarboxylation,2, 3 and phosphoryl transfer reactions.4 From 33–66% of this binding energy is utilized in phosphite dianion activation of these enzymes for catalysis of the reactions of phosphodianion truncated substrates.1–5 Our model to rationalize the reactions of substrate pieces can be generalized to any enzyme that undergoes a substrate-driven conformational change,6 but phosphite dianion is the only identified activating substrate piece.
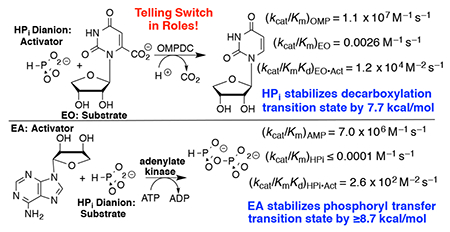
Adenylate kinase (AdK) catalyzes the transfer of a phosphoryl group from ATP to AMP to form two molecules of ADP (Scheme 1). The binding of nucleotide substrates to AdK drives the closure of ATP- and AMP-lids over the substrate adenosine groups.7 We predict that these protein-adenosine interactions activate AdK for catalysis of phosphoryl transfer, and that AdK is an effective catalyst of the reaction of the AMP pieces 1-(β-d-erythrofuranosyl)adenine (EA) and phosphite dianion. This is a reversal of the roles of phosphite dianion (substrate) and truncated nucleoside EA (activator) from that for phosphite dianion activation of orotidine monophosphate decarboxylase (OMPDC, Scheme 2).8
Scheme 1.
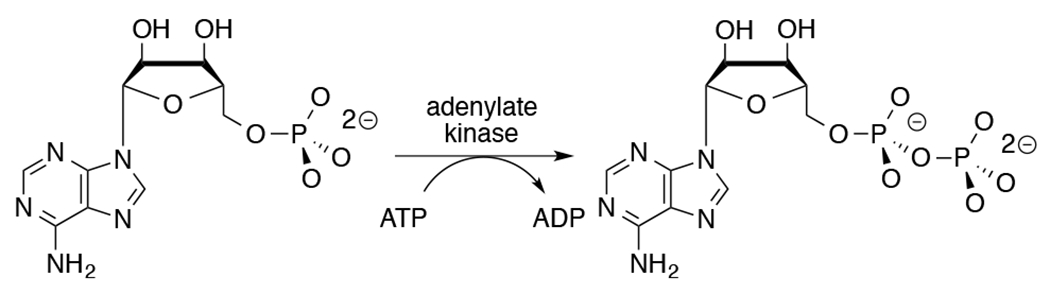
Adenylate Kinase-Catalyzed Phosphoryl Transfer from ATP to ADP.
Scheme 2.
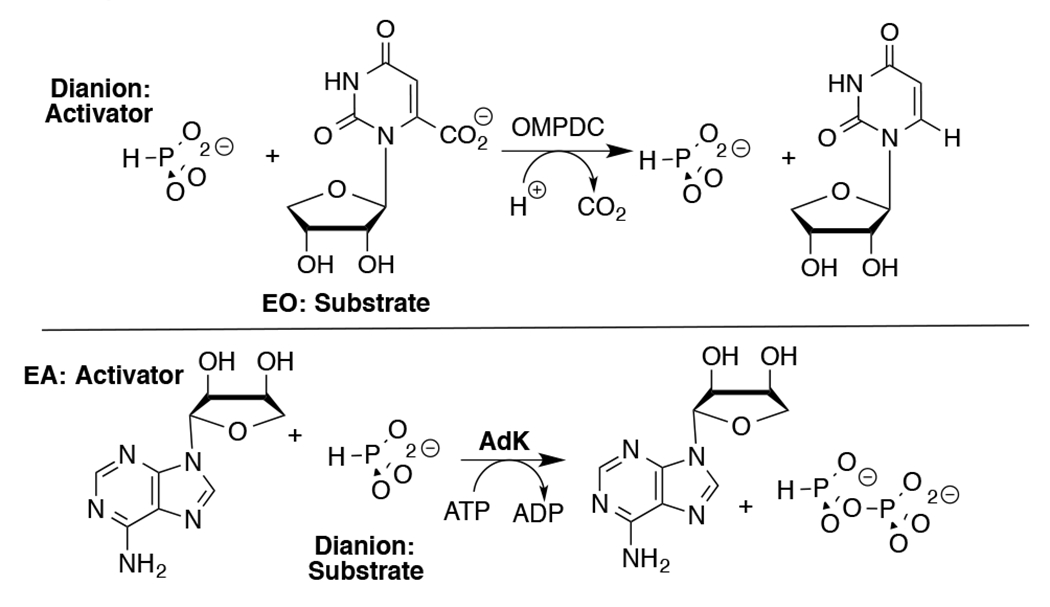
Roles of Dianion and Nucleoside (EO and EA) Substrate Pieces in Reactions Catalyzed by OMPDC and AdK.
The commercial sources for all materials, and the methods for the preparation of Homo sapiens adenylate kinase 1 (HsAdK1) and tobacco etch virus (TEV) protease are given in the Supporting Information (SI). The following protocols are described in the SI: (i) assays for AdK-catalyzed phosphoryl transfer from ATP to AMP, for AdK-catalyzed hydrolysis of ATP, and for unactivated and EA-activated AdK-catalyzed phosphoryl transfer from ATP to phosphite dianion and (ii) protocol for product determination using 31P NMR.
His6-tag labelled HsAdK1 was expressed in E. coli, purified over a Ni2+-column, and the His6-tag removed by tobacco etch virus (TEV)-protease. The purified HsAdK1 showed a single band by SDS-PAGE (Figure S1). The initial velocity, vo, at 25 °C for HsAdK1-catalyzed phosphoryl transfer from ATP to AMP was determined by a standard enzyme assay (SI).9 Figure S2 shows a plot of vo/[E] against AMP for HsAdK1-catalyzed phosphoryl transfer from 1 mM ATP (saturating)10 to AMP to form 2 ADPs at 25 °C and pH 7.5, which gave the kinetic parameters kcat/Km = (7.0 ± 0.4) x 106 M−1 s−1, kcatt = 475 ± 8 s−1 and Km = 68 ± 4 μM.
HsAdK1-catalyzed hydrolysis of ATP to ADP was monitored by coupling the formation of ADP to the oxidation of NADH by pyruvate, catalyzed by lactate dehydrogenase. Figure S3A shows the dependence of vo on [HsAdK1] for catalysis of hydrolysis of 1 mM ATP (saturating)10 to form ADP. The slope of this correlation, khyd = 5 x 10−6 s−1, is similar to khyd = 2 x 10−6 s−1 reported for adenylate kinase from E. coli.7
Figure S3B shows the increase in khyd = 5 x 10−6 s−1 for HsAdK1-catalyzed (72 μM) conversion of ATP (1 mM) to ADP in the presence of increasing concentrations of phosphite dianion. The slope of this correlation, (kHPi)obs = (9.9 ± 0.3) x 10−5 M−1 s−1, is the sum of the rate constants for HsAdK1-catalyzed phosphoryl transfer from ATP to phosphite to form isohypophosphate (HPPi), (kHPi)E, and for activation of the HsAdK1-catalyzed hydrolysis reaction by a specific salt effect, (kHPi)salt. The formation of ADP by the reaction of phosphite with ATP (Scheme 3) would result in a low yield of HPPi, undetectable by the 31P analytical methods described below. Therefore, the value of (kHPi)obs = (9.9 ± 0.3) x 10−5 M−1 s−1 (Figure S3B) sets an upper limit of (kHPi)E ≤ 1 x 10−4 M−1 s−1 for unactivated HsAdK1-catalyzed phosphoryl transfer from ATP to phosphite (Table 1).
Scheme 3.
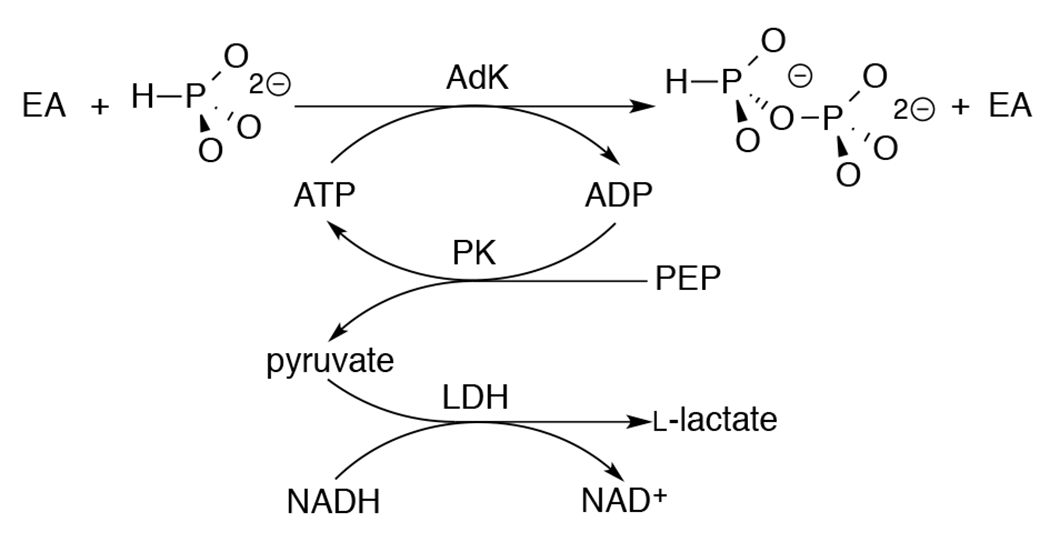
Assay to Monitor AdK-Catalyzed Phosphoryl Transfer from ATP to Phosphite Dianion.
Table 1.
Rate Constants and Transition-State Binding Energies ΔΔG‡ for HsAdK1-Catalyzed Phosphoryl Transfer from 1.0 mM ATP to AMP and to Phosphite Dianion.a
| Phosphoryl Acceptor | Kinetic Parameter | ΔΔG‡ (kcal/mol) b |
|---|---|---|
| AMP | kcat/Km (7.0 ± 0.4) x 106 M−1 s−1 | ≥ 14.7 c |
| HPO32− + EA | (kcat)HPi•EA/KHpiKEA (2.58 ± 0.05) x 102 M−2 s−1 d | ≥ 8.7 e |
| HPO32− | (kHpi)E ≤1.0 x 10−4 M−1 s−1 f |
At 25 ° C and pH 7.5.
Calculated from the ratio of rate constants for HsAdK1-catalyzed reactions of AMP and the substrate piece (HPO32−) or pieces (HPO32− + EA).
Assuming similar intrinsic nucleophilic reactivities for AMP and phosphite dianion.
Calculated from the ratio of rate constant for HsAdK1-catalyzed reactions of phosphite in the presence and absence of EA.18
The slope from Figure S3B (see text).
The HsAdK1-catalyzed conversion of ATP to ADP in the presence of phosphite dianion is strongly activated by EA. Figure 1A shows the effect of increasing [EA] on (vobs – vo) for HsAdK1-catalyzed (2 μM) reactions of saturating ATP (1 mM), where vobs is the total reaction velocity, and vo is a ≤1.1% correction for the velocity at [EA] = 0 M. The increase in (vobs – vo) is consistent with EA-activation of HsAdK1 for catalysis of phosphoryl transfer from ATP to HPO32− to form HPPi (Scheme 3). We confirm this by determining the reaction products using 31P NMR.
Figure 1.
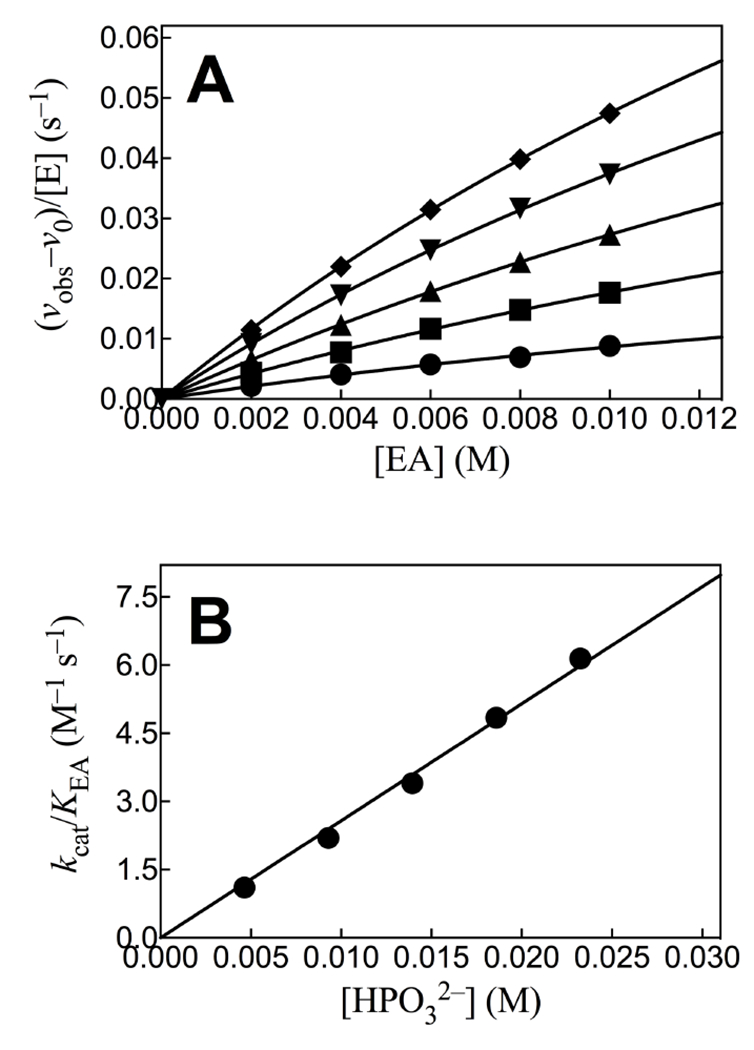
(A) The effect of increasing [EA] on the velocity for HsAdK1-catalyzed reactions of ATP (1 mM) at 25 °C. Key: ♦, 25 mM [HPi]T, (93% dianion); ▼, 20 mM [HPi]T; ▲, 15 mM [HPi]T; ■, 10 mM [HPi]T; ●, 5 mM [HPi]T. (B) The effect of increasing [HPO32−] on the values of (kcat/KEA)obs determined for Figure 1A.
The HsAdK1-catalyzed conversion of ATP to ADP in the presence of phosphite dianion was monitored in a 1.0 mL solution in D2O that contains 50 mM TEA (pD 7.5), 10 mM EA, 3 mM MgCl2, 30 mM KCl, 25 mM phosphite (93% dianion),1 1 mM ATP, 30 mM phosphoenolpyruvate (PEP), 10 mM phosphonoacetate (31P-standard), 1 U pyruvate kinase, and 67 μM HsAK1. This reaction was monitored continuously for 16 h at 25 °C by 31P NMR spectroscopy on a Varian Inova 500 MHz spectrometer, as described in the SI. Figure 2 shows the relevant changes in the 31P NMR spectra during this time. The integrated areas for the singlet for PEP (−0.62 ppm) and doublet for phosphite (3.05 ppm, d, 1JPH = 568 Hz) decrease, as peaks for HPPi appear (−4.50 ppm, dd, 1JPH = 646 Hz, 2JPP = 17 Hz; −5.44 ppm, d, 2JPP = 17 Hz).11 No inorganic phosphate from adenylate-kinase catalyzed hydrolysis of ATP, or from hydrolysis of HPPi was detected. This is consistent with the published 12 h halftime for hydrolysis of HPPi at 60 °C and pH 7.4.11
Figure 2.
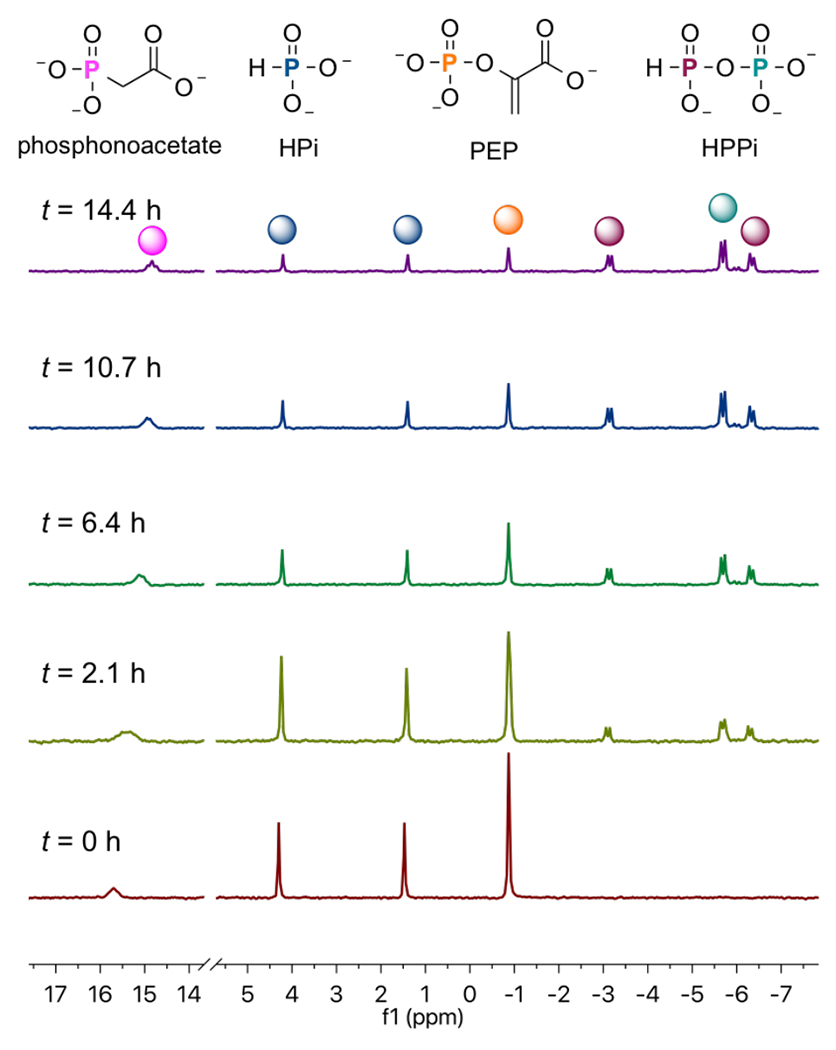
31P NMR spectra that show the change with time in the areas of the peaks AR at −0.62 and 3.05 ppm for PEP and phosphite reactants, and Ap at −4.50 and 5.44 ppm for HPPi product of EA-activated HsAdK1-catalyzed reactions (Scheme 3).
The sum of the normalized 31P peaks areas for the reactants PEP and phosphite (AR) and the HPPi product (AP) were determined using a constant peak area (AS) for the phosphonoacetate standard. There is no significant change in the sum of normalized areas of peaks for reactants and products during the reaction. Figure 3 compares the time courses for the decrease in the fraction of PEP and phosphite reactants remaining (Ai = AR) with the increase in the fraction of reactants converted to HPPi (Ai = AP), with normalization of (AR + AP). The solid lines in Figure 3 show the fit of these data to the rate equation for the apparent first-order conversion of (30 mM PEP + 25 mM phosphite) to HPPi, using a rate constant of kobs = 0.16 h−1 and reaction end-points of 22 mM HPPi and 11 mM (PEP + phosphite). These fits show that the reactants are converted to HPPi as essentially the exclusive product.
Figure 3.
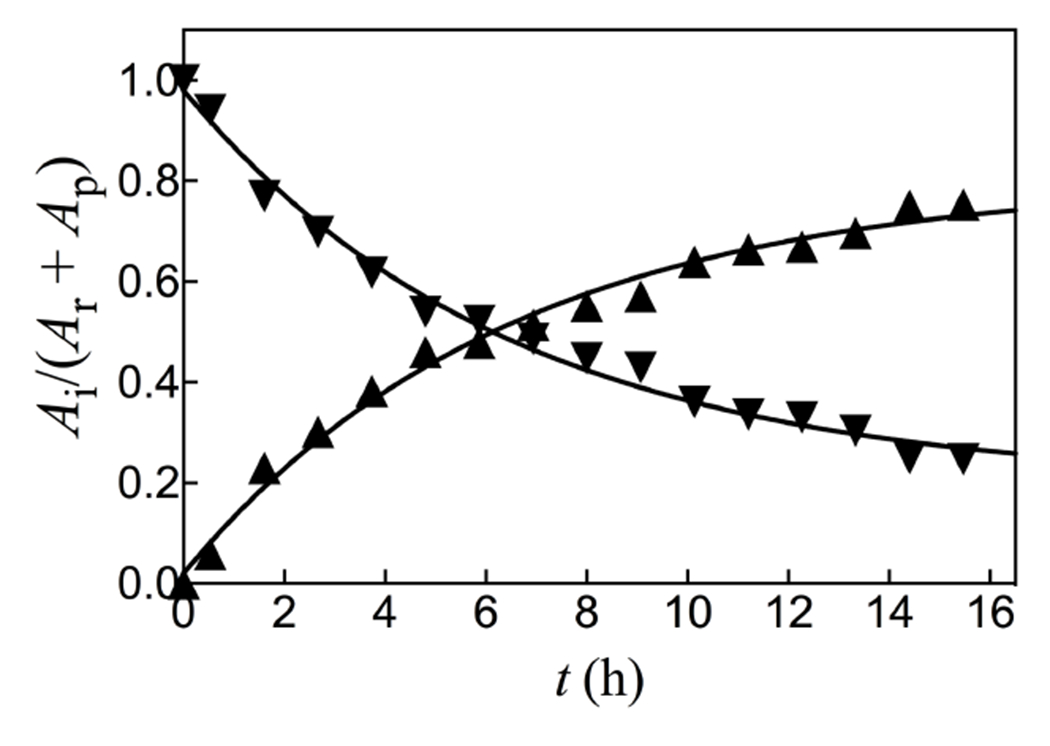
The fractions of reactants PEP and phosphite remaining (▼, Ai = AR) and product HPPi formed (▲, Ai = AP at time t, determined by integration of the peaks from Figure 2.
Figure 3 shows that HsAdK1-catalyzed phosphoryl transfer from ATP to phosphite dianion to form HPPi can account for essentially the entire increase in (vobs – vo) from Figure 1A. The solid lines for Figures 1A show the nonlinear least squares fits of the kinetic data to eq 1, derived for Scheme 4, assuming KHPi >> [HPi] and using the values of (kcat/KEA)obs determined for reactions at different [HPO32−]. Figure 1B shows the linear plot (eq 2) of values of (kcat/KEA)obs from Figure 1A, against [HPO32−]. The slope of this correlation is (kcat)HPi•EA/KHPiKEA = (2.58 ± 0.05) x 102 M−2 s−1 (Table 1).
Scheme 4.
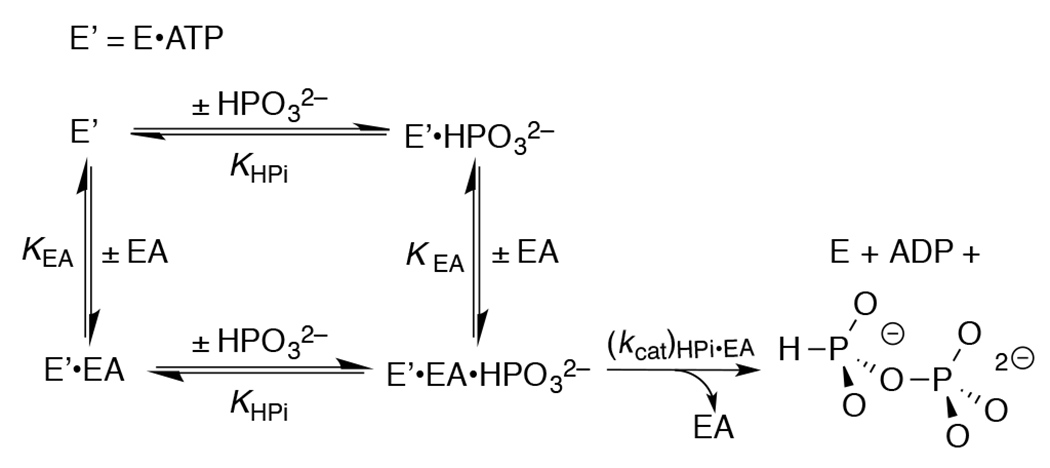
Kinetic Mechanism for Activation of HsAdK1-Catalyzed Phosphoryl Transfer from ATP to Phosphite Dianion by the Substrate Piece EA.
Table 1 summarizes data that defines the role of the adenosyl group of AMP in activating HsAdK1 for catalysis of phosphoryl transfer from enzyme saturated with ATP (1 mM). The values of kcat/Km = 7.0 x 106 M−1 s−1 and (kHPi)E ≤ 1 x 10−4 M−1 s−1, respectively for the catalyzed reactions of AMP and phosphite dianion show that the AMP adenosyl group is responsible for a ≥7.0 x 1010-fold rate acceleration for phosphoryl group transfer. This is a lower limit, because the full AMP binding interactions are probably not expressed at the transition state for the enzyme conformational change, which limits the rate of reaction of AMP.7 This corresponds to a ≥14.7 kcal/mol stabilization of the transition state for phosphoryl transfer by the AMP adenosyl group. The ratio of the rate constants for HsAdK1-catalyzed phosphoryl transfer from ATP to HPO32− in the presence and absence of the EA activator, ≥2.6 x 106 M, shows that ≥8.7 kcal/mol of the adenosyl binding energy is recovered as stabilization of the transition state by the activator. This binding energy is utilized to hold AdK in an active closed conformation.6, 12–15 By comparison, the transition state for OMPDC-catalyzed decarboxylation is stabilized 12 kcal/mol by interactions with the OMP phosphodianion and 19 kcal/mol by interactions with the OMP ribosyl and orotate moieties.8
Inorganic tripolyphosphate binds weakly to chicken muscle adenylate kinase (Kd = 1 mM) and undergoes phosphoryl transfer to AMP with kcat that is ca (104–105)-fold smaller than for the reaction of ATP.16 This shows that the adenosyl group of ATP is essential for optimal phosphoryl donor activity. HsAdK1 shows a higher specificity for the phosphoryl acceptor AMP compared to the donor ATP,17 which suggests a larger activating adenosyl binding energy for the acceptor compared with the donor nucleotide.
| (1) |
| (2) |
Adenylate kinase functions in muscle tissues to maintain an equilibrium concentration of ATP, ADP, and AMP in order to optimize [ATP] under conditions of energy stress, created by the rapid myosin-catalyzed hydrolysis of ATP. This important function creates pressure to optimize enzyme activity, similar to that for the glycolytic enzyme TIM.19, 20 In these and other cases, the pressure to optimize catalytic activity has driven evolution of protein motifs, where substrate binding energy is utilized to drive large, enzyme-activating, conformation changes.2, 6
X-ray crystal structures for AdK show that the adenosyl group of AMP sits distant from the reacting phosphates, and is held at the enzyme by hydrogen bonds with protein side chains and backbone amides.21, 22 The ligand phosphates interact with many cationic amino acid side chains and backbone amides, so that the activator-driven conformational changes move disordered polar groups into positions that provide for optimal stabilizing electrostatic interactions with the phosphate oxygens at the transition state for HsAdK1-catalyzed phosphoryl transfer.23 We propose that the ≥8.7 kcal/mol transition state stabilization from binding of the EA activator is due to the incremental tightening of many stabilizing polar interactions that result from the ligand-driven enzyme conformational change.
Supplementary Material
Funding Sources
The authors acknowledge the National Institutes of Health Grant GM134881 for support of this work.
ABBREVIATIONS
- AdK
adenylate kinase
- AMP
adenosine 5’-monophosphate
- ADP
adenosine 5’-diphosphate
- ATP
adenosine 5’-triphosphate
- HsAdK1
Homo sapiens adenylate kinase 1
- EA
1-(β-d-erythrofuranosyl)adenine
- HPPi
isohypophosphate
- OMP
orotidine 5’-monophosphate
- OMPDC
orotidine 5’-monophosphate decarboxylase
- SI
Supporting Information
- TEA
triethanolamine
- TEV
tobacco etch virus
- TIM
triosephosphate isomerase
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental Section with (i) the sources of the enzymes and chemicals used in this work, (ii) procedures for the preparation and purification of TEV protease and HsAdK1, and (iii) protocol for the determination of kinetic parameters for HsAdK1-catalyzed reactions of the whole substrate AMP and the substrate pieces phosphite and EA. Results Section with (i) SDS-PAGE gel for purified TEV protease and HsAdK1 (Figure S1), (ii) kinetic data for HsAdK1-catalyzed reactions of ATP with AMP (Figure S2), and (iii) kinetic data for HsAdK1-catalyzed hydrolysis of the ATP (Figure S3A) and for the effect of increasing [HPi] on the velocity HsAdK1-catalyzed conversion of ATP to ADP (Figure S3B). (PDF)
The authors declare no competing financial interests.
Contributor Information
Patrick L. Fernandez, Department of Chemistry, University at Buffalo, The State University of New York at Buffalo, Buffalo, New York 14260-3000, United States.
John P. Richard, Department of Chemistry, University at Buffalo, The State University of New York at Buffalo, Buffalo, New York 14260-3000, United States.
REFERENCES
- [1].Tsang W-Y, Amyes TL, and Richard JP (2008) A Substrate in Pieces: Allosteric Activation of Glycerol 3-Phosphate Dehydrogenase (NAD+) by Phosphite Dianion. Biochemistry 47, 4575–4582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Fernandez PL, Nagorski RW, Cristobal JR, Amyes TL, and Richard JP (2021) Phosphodianion Activation of Enzymes for Catalysis of Central Metabolic Reactions. J. Am. Chem. Soc 143, 2694–2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Amyes TL, Richard JP, and Tait JJ (2005) Activation of orotidine 5’-monophosphate decarboxylase by phosphite dianion: The whole substrate is the sum of two parts. J. Am. Chem. Soc 127, 15708–15709. [DOI] [PubMed] [Google Scholar]
- [4].Ray WJ Jr., Long JW, and Owens JD (1976) An analysis of the substrate-induced rate effect in the phosphoglucomutase system. Biochemistry 15, 4006–4017. [DOI] [PubMed] [Google Scholar]
- [5].Amyes TL, and Richard JP (2007) Enzymatic catalysis of proton transfer at carbon: activation of triosephosphate isomerase by phosphite dianion. Biochemistry 46, 5841–5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Richard JP (2019) Protein Flexibility and Stiffness Enable Efficient Enzymatic Catalysis. J. Am. Chem. Soc 141, 3320–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kerns SJ, Agafonov RV, Cho Y-J, Pontiggia F, Otten R, Pachov DV, Kutter S, Phung LA, Murphy PN, Thai V, Alber T, Hagan MF, and Kern D (2015) The energy landscape of adenylate kinase during catalysis. Nature 22, 124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Richard JP, Amyes TL, and Reyes AC (2018) Orotidine 5’-Monophosphate Decarboxylase: Probing the Limits of the Possible for Enzyme Catalysis. Acc. Chem. Res 51, 960–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rhoads DG, and Lowenstein JM (1968) Initial Velocity and Equilibrium Kinetics of Myokinase. J. Biol. Chem 243, 3963–3972. [PubMed] [Google Scholar]
- [10].Sheng XR, Li X, and Pan XM (1999) An Iso-random Bi Bi Mechanism for Adenylate Kinase. J. Biol. Chem 274, 22238–22242. [DOI] [PubMed] [Google Scholar]
- [11].Carroll RL, and Mesmer RE (1967) Isohypophosphate: Kinetics of Hydrolysis and Potentiometric and Nuclear Magnetic Resonance Studies on the Acidity and Complexing. Inorg. Chem 6, 1137–1142. [Google Scholar]
- [12].Amyes TL, Malabanan MM, Zhai X, Reyes AC, and Richard JP (2017) Enzyme activation through the utilization of intrinsic dianion binding energy. Prot. Eng. Des. & Sel 30, 157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Thomas JA, and Koshland DE Jr. (1960) Competitive inhibition by substrate during enzyme action. Evidence for the induced-fit theory. J. Am. Chem. Soc 82, 3329–3333. [Google Scholar]
- [14].Koshland DE Jr. (1958) Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc. Natl. Acad. Sci. U. S. A 44, 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Amyes TL, and Richard JP (2013) Specificity in transition state binding: The Pauling model revisited. Biochemistry 52, 2021–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sanders CR, Tian G, and Tsai MD (1989) Mechanism of adenylate kinase. Is there a relationship between local substrate dynamics, and local binding energy, and the catalytic mechanism? Biochemistry 28, 9028–9043. [DOI] [PubMed] [Google Scholar]
- [17].Panayiotou C, Solaroli N, and Karlsson A (2014) The many isoforms of human adenylate kinases. Int. J. Biochem. & Cell. Biol 49, 75–83. [DOI] [PubMed] [Google Scholar]
- [18].Reyes AC, Zhai X, Morgan KT, Reinhardt CJ, Amyes TL, and Richard JP (2015) The Activating Oxydianion Binding Domain for Enzyme-Catalyzed Proton Transfer, Hydride Transfer and Decarboxylation: Specificity and Enzyme Architecture. J. Am. Chem. Soc 137, 1372–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Knowles JR, and Albery WJ (1977) Perfection in enzyme catalysis: the energetics of triosephosphate isomerase. Acc. Chem. Res 10, 105–111. [Google Scholar]
- [20].Albery WJ, and Knowles JR (1976) Evolution of enzyme function and the development of catalytic efficiency. Biochemistry 15, 5631–5640. [DOI] [PubMed] [Google Scholar]
- [21].Bellinzoni M, Haouz A, Graña M, Munier-Lehmann H, Shepard W, and Alzari PM (2006) The crystal structure of Mycobacterium tuberculosisadenylate kinase in complex with two molecules of ADP and Mg2+ supports an associative mechanism for phosphoryl transfer. Prot. Sci 15, 1489–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Müller CW, and Schulz GE (1992) Structure of the complex between adenylate kinase from Escherichia coli and the inhibitor Ap5A refined at 1.9 Å resolution: A model for a catalytic transition state. J. Mol. Biol 224, 159–177. [DOI] [PubMed] [Google Scholar]
- [23].Warshel A, Sharma PK, Kato M, Xiang Y, Liu H, and Olsson MHM (2006) Electrostatic basis for enzyme catalysis. Chem. Rev 106, 3210–3235. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.