

Abstract
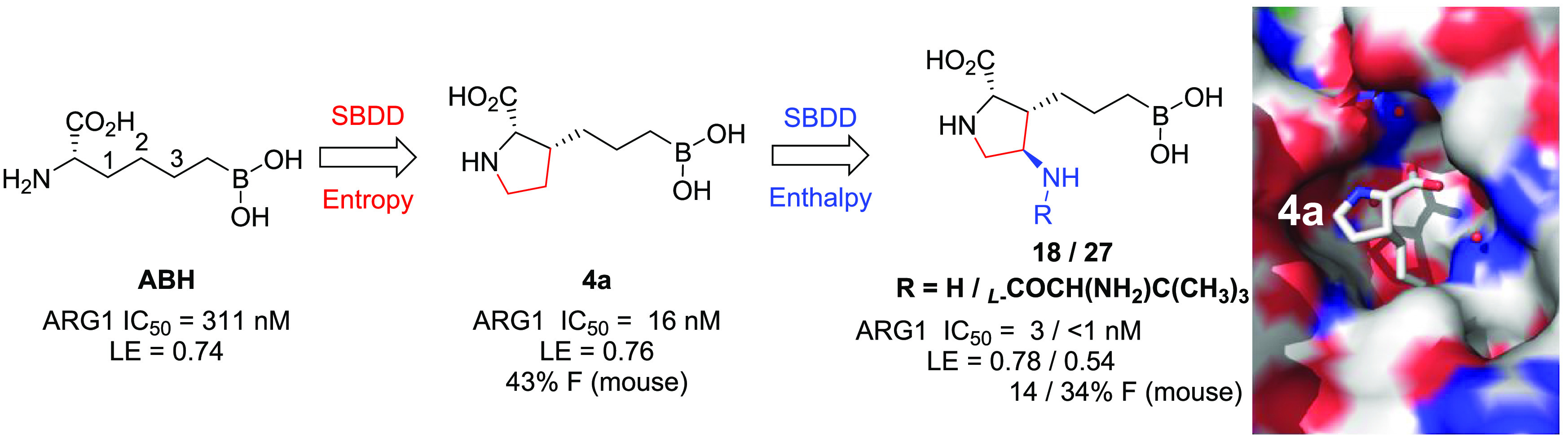
Recent data suggest that the inhibition of arginase (ARG) has therapeutic potential for the treatment of a number of indications ranging from pulmonary and vascular disease to cancer. Thus, high demand exists for selective small molecule ARG inhibitors with favorable druglike properties and good oral bioavailability. In light of the significant challenges associated with the unique physicochemical properties of previously disclosed ARG inhibitors, we use structure-based drug design combined with a focused optimization strategy to discover a class of boronic acids featuring a privileged proline scaffold with superior potency and oral bioavailability. These compounds, exemplified by inhibitors 4a, 18, and 27, demonstrated a favorable overall profile, and 4a was well tolerated following multiple days of dosing at concentrations that exceed those required for serum arginase inhibition and concomitant arginine elevation in a syngeneic mouse carcinoma model.
Keywords: Arginase inhibitor, structure-based drug design, oral bioavailability, cancer immunotherapy, proline
Harnessing the innate immune system to cure diseases has revived and witnessed great progress in the past few years, in particular, in the field of cancer immunotherapy.1 Some immune checkpoint inhibitors, such as mAbs for programmed cell death protein 1 (PD-1), programmed death ligand 1 (PD-L1), or cytotoxic T-lymphocyte antigen 4 (CTLA-4), have demonstrated unprecedented and durable efficacy in a variety of cancers, including some tough-to-treat subtypes.2,3 Despite these breakthroughs, the objective response rate of these novel antitumor therapies remains low in certain tumor types, thus limiting their potential to benefit a much broader patient population.4 To address this issue, different combination strategies are being intensively explored, including chemotherapy, radiotherapy, as well as those mechanisms capable of overcoming tumor-induced local immunosuppression.5
Arginase (ARG) is an enzyme that metabolizes arginine to ornithine and urea.6 The role of ARG1, secreted by myeloid derived suppressor cells (MDSCs)7 in the tumor microenvironment, has become increasingly recognized as an important mechanism of tumor immune evasion by (1) depriving T cells of arginine and (2) increasing the concentration of immunosuppressive metabolites.8,9 Elevated levels of ARG1 have been detected in tumors of patients with various types of cancer,10,11 and elevated plasma ARG1 and reduced l-arginine levels are reported to correlate with suppressed T-cell function and proliferation in patients with different histologies.12,13 Therefore, pharmacological inhibition of ARG1 by small molecules and its combination with anti-PD-1 or anti-PD-L1 agents have attracted great attention for cancer immunotherapy.14,15 Here we describe the discovery and optimization of a novel class of boronic acid ARG1 inhibitors16 guided by structural-based drug design (SBDD), which features a privileged natural proline moiety. Extensive studies were also carried out to evaluate and improve the oral bioavailability of this class of inhibitors, which occupies a unique property space that was proven to be challenging for oral absorption and bioavailability.17,18
As part of a comprehensive effort to discover novel small-molecule inhibitors of ARG to enhance cancer immunotherapy,21 we took inspiration from the reported human ARG1 (hARG1) cocrystal structures of (S)-2-amino-6-boronohexanoic acid (ABH, 1, Figure 1A).22 Our starting point, as exemplified by 1 (hARG1 IC50 = 311 nM), has a moderate enzymatic activity.23 Based on careful analysis of the binding site, in particular, the underexplored space not effectively occupied by 1, we envisioned that (1) a judicious cyclization of the α-amino group to the carbon backbone might be tolerated and fit better into the narrow pocket; and (2) the resulting N-heterocyclic core could be leveraged as a template to better engage in direct or water-mediated H-bonds with residues Asp183 and/or Asp181, as observed by Van Zandt and co-workers (2, Figure 1B)20 and ourselves previously.21 In an attempt to identify the most appropriate cyclization approach, we employed a modular synthetic strategy by investigating a broad scope of N-heterocyclic cores with a variety of linker sizes (Scheme 1). The strategy was enabled by readily available building blocks and robust cross-coupling methods, while subsequent hydroboration, hydrogenation, and global deprotection readily yielded a diverse set of core and linker combinations.24
Figure 1.
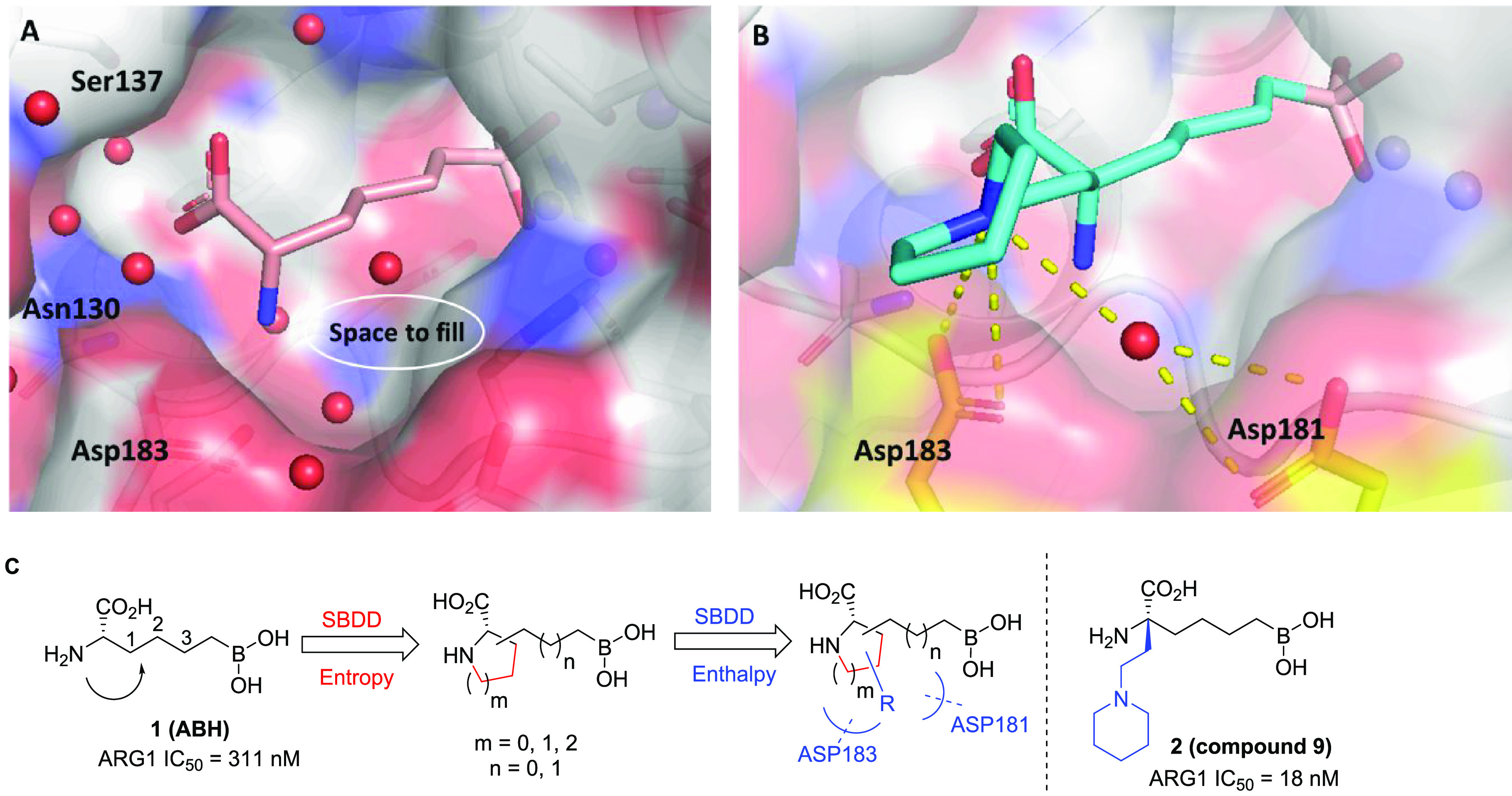
SBDD strategy: (A) Structure of 1 (pink) bound to hARG1 (PDB 6Q92)19 indicates underexplored space (white circle) between the ligand and active site. (B) Structure of 2 (cyan) bound to hARG1 (PDB 4HWW)20 reveals proximity between the piperidine ring and Asp 183 (gold) and Asp181 (through water, gold) respectively, which contributes to >15× improvement in biochemical potency. (C) Two-stage approach of introducing N-containing heterocyclic cores as well as H-bond interactions for entropic and enthalpic improvements in binding energies.
Scheme 1. Modular Strategy for Diverse Core SAR Exploration.

As summarized in Table 1, while each core–linker combination had comparable modeling overlay with ABH,40 significant differences of activities were observed in the hARG1 enzymatic assay23 between 1,2-cyclization [(±)-3 and (±)-4] and 1,3-cyclization approaches [(±)-5 and (±)-6], presumably due to the relatively small volume of the binding site to accommodate nonideal conformations, as further evidenced by the complete loss of activity in other combinations (data not shown). Nevertheless, we were encouraged by the identification of pipecolic acid [(±)-3] and proline [(±)-4] cores with comparable potencies as the starting point ABH (1), as mixtures of isomers from the hydrogenation step. In addition, as the individual isomers were resolved by chiral SFC separation, the absolute stereochemistry was also found to be crucial and (2S,3R)-4a was favored over (2S,3S)-4b by >25-fold in hARG1 IC50, while the unnatural (2R) isomers were completely inactive (data not shown). Interestingly, despite almost identical crystal structure binding modes between 3a and 4a (see overlay: Figure 2A), there was a remarkable 23-fold improvement as the ring size was reduced. As the ring size was further reduced to an l-azetidine-carboxylate, no further improvement could be achieved by either a cis- (7a) or trans-isomer (7b), presumably due to the most favorable conformational bias presented by the l-proline core. Introducing an additional stereoelectronic effect, represented by the morpholine core in 8, slightly improved the biochemical IC50. Overall, l-proline analogue 4a presented an optimal profile, in particular with a remarkable ligand efficiency (LE = 0.76).26 Recognizing the significant challenges associated with the nontraditional property space of known boronic acid ARG inhibitors (ALogP98 < −2, HBD ≥ 5, PSA > 120),16 we further evaluated these representative single isomers in rodent pharmacokinetic studies to assess in particular their oral bioavailability.21,25 Gratifyingly, these novel core modifications turned out to be beneficial for bioavailabilities when dosed orally (p.o.) at 10 mg/kg (mpk) in both C57BL/6 mice and Wister Han rats (Table 1).
Table 1. Selected SAR of N-Heterocyclic Core Exploration: General Design Strategy and Structuresa.

NA = data not available.
Figure 2.
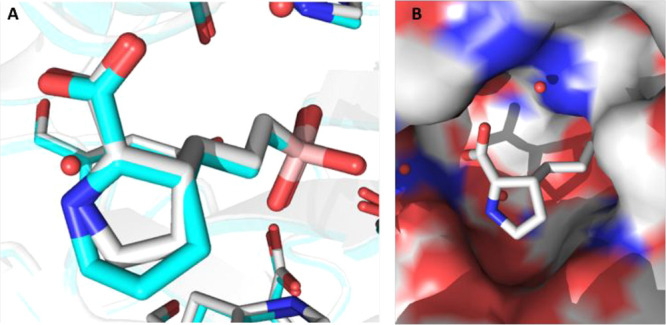
(A) Comparison of crystal structure binding modes for compounds 3a (cyan, PDB 7KLK) and 4a (white, PDB 7K4G) to hARG1. (B) Surface view of cocrystal structure of 4a (white) bound to hARG1 (PDB 7K4G).
Next, our attention was shifted to explore the feasibility of introducing additional substituents around the l-proline core. To enable broad assessment of diverse substitution patterns, we first envisioned the use of a key intermediate I on the route to 4a(24) for both stereoselective α-alkylation and δ-formylation, with the latter enabled by a novel photoexcited hydrogen atom abstraction (HAA) process (Scheme 2A).27 Alternatively, a previously reported building block II(28) was carefully selected as the linchpin for regio- and stereoselective β-enolate chemistry as well as sequential γ-reduction and displacement with a variety of nucleophiles. (Scheme 2B).40
Scheme 2. Novel Synthetic Routes for Enabling Facile Sidechain Exploration.

Reagents and conditions: (α) NaHMDS, THF, −40 °C; then allyl iodide or alkyl bromide, −40 °C to rt; (β) DBU (cat.), HCHO (37 w t% in H2O), THF, 0 °C to rt; (γ) KHMDS, THF, −78 °C; then (+)-CSA, THF, −78 °C; then MeOH, NaBH4, −78 °C to rt; (δ) benzophenone, phenylsulfonylmethanal O-benzyl oxime, CH3CN, 365 nM LED, rt.
Significant SAR exploration centered around various α-substitutions of ABH has been disclosed previously,29−32 with 2-piperidinyl-ethyl being one of the most privileged side chains (2, Figure 1);20 incorporation of the same moiety in our scaffold, however, resulted in a 4-fold drop in enzymatic IC50 (9, Table 2). While a preliminary survey of α-side chains did not further improve potency, modeling based on the cocrystal structure of 4a with hARG1 indicated that small polar substituents growing off β, γ, and δ-positions of the l-proline core might be not only tolerated but also beneficial for potency, presumably via polar interactions with residues Asp181 and Asp183 (Figure 2B).
Table 2. Selected SAR for Side Chain Optimization: General Design Strategy and Structuresa.


NA = data not available.
We started with the β-substitutions to expand SAR toward the solvent region. Similar to the SAR at the α-substitutions, although simple methylation resulted in a slight drop in potency (10, Table 2), a hydroxymethyl group was found to be neutral in terms of both activity and bioavailabilities (11). A dramatic improvement in potency was observed when a basic amine was introduced (12), albeit with a slightly diminished LE when compared with the baseline 4a (0.76 → 0.71). Encouraged by these results, we then shifted our attention toward the δ-substitutions, since the HAA-formylation chemistry turned out to be highly stereoselective, resulting in the desired trans-configuration as a single isomer (Scheme 2A). As summarized in Table 2, the methyl analogue 13 turned out to be one of the most potent, with a comparable LE to 4a and only a slight decrease in oral bioavailability (43% → 31%). Surprisingly, the SAR trend was somewhat reversed from β- to δ-substitutions, as additional hydroxy- (14) or amino-extension (15) did not further improve potency as designed to engage Asp181, but rather led to lower LEs (0.69 and 0.65, respectively). Further evaluation of the surface residues around the γ-carbon led us to introduce the same hydroxy and amino groups to directly engage the Asp181 residue and increase H-bond interactions with the backbone of the protein. Satisfyingly, both hydroxy and amino analogues (16 and 18) proved to be beneficial to the potency, with good to moderate bioavailabilities and achieving the best LEs (0.77 and 0.78) so far. As expected, methylation of the γ-hydroxy led to >10-fold loss of activity (17), presumably due to the disruption of a key H-bond interaction between the hydroxy group and Asp181.
The highly polar nature of this structural class of Arginase inhibitors led to lower than usual ALogP98 (−4.0 to −1.0) values than the ranges generally preferred for good oral absorption (1.0 ∼ 3.0).33 At first glance, it seemed surprising for compound 4a to still show good oral bioavailabilities (43% and 25%, mouse and rat). In addition to its low ALogP98 value,34 several other parameters including H-bond donor (HBD) counts and PSA also fall outside of typically preferred value ranges,33 which, unsurprisingly, led to significantly lower activities against intracellular ARG in the HEK293 cell line (Table 2), likely due to poor permeability.35 Since the elevated ARG1 and reduced l-arginine levels detected in cancer patient plasma are extracellular (vide supra), achieving high cellular potency was not key to our optimization strategy; in fact, we hypothesized it might ultimately lead to a widening of the therapeutic index. Nevertheless, understanding key parameters for obtaining good oral absorption would be key for further optimization. As shown in Figure 3, we plotted binned distributions of oral bioavailability in C57BL/6 mice against a variety of molecular descriptors, and good oral bioavailability (i.e., ≥20% F shown in green bands) tends to enrich in compounds with MW lower than 230, which seems to indicate the possibility of a paracellular transport mechanism.35 Furthermore, HBD counts also seem to have a significant impact on achieving good oral bioavailability, with less than five HBDs being seemingly optimal. These observations prompted us to focus on the most ligand efficient compound 18 to allow further exploration of several strategies including paracellular transport as well as groups that may allow for active uptake mechanisms.15,36
Figure 3.
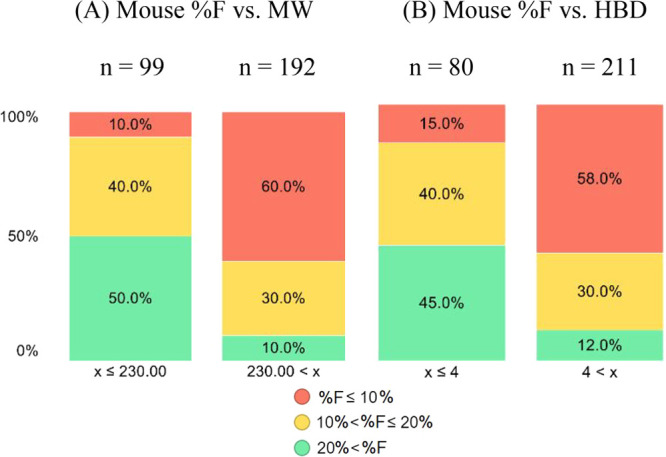
Percent distributions of mouse oral bioavailability (p.o., 10 mpk) vs (A) MW, and (B) HBD.
As shown in Scheme 3, leveraging our access to a key intermediate III, our strategy aimed at a general and robust displacement strategy of a superior leaving group, chloromethylsulfonate,37 that allowed the expeditious investigation of a variety of substitution patterns of the γ-amino group. From the outset, we looked to maintain or improve potency by further extending toward surface residues (i.e., Asp181 and Asp183) while focusing on (1) reducing HBD counts and (2) introducing strategically placed amino acid groups to explore whether the engagement of uptake transporters could potentially improve bioavailability.38 Although the former approach failed to yield any improvement in potency or bioavailability (Table 3, 18–22), we were very encouraged by the significant potency gain (>20-fold) when an l-alanine moiety was installed (23), compared to the corresponding acyl analogue (22). Interestingly, the peptide motif turned out to have relatively poor in vitro and in vivo stability in mouse hepatocytes and pharmacokinetic experiments, where rapid clearance of 23 was observed and hydrolysis product 18 was detected as the predominant species. Interestingly, the combined exposures of 18 and 23 when 23 was dosed orally (10 mpk) surpassed that of dosing 18 (27% vs 14% F) itself, indicating the potential contribution of an active uptake mechanism via recognition of the l-Ala moiety. Modeling indicated that the side chain of the amino acid motif would likely extend toward the solvent region without detrimental effects on binding affinity; therefore, a focused set of natural as well as unnatural amino acids were evaluated. Growing from l-Ala to l-Val dramatically improved potency (24a, IC50 = 2 nM), but good metabolic stability was not achieved. Furthermore, the natural S-configuration was found to be important for good potency, as evidenced by the >15-fold loss of activity when d-Val was introduced (24b, IC50 = 34 nM). Finally, a variety of unnatural amino acids as a follow-up of the l-Val result were evaluated to further improve metabolic stability. Interestingly, while moving the additional methyl group to the amine (25) or α′-carbon (26) resulted in suboptimal bioavailability and potency, respectively; the l-α-tert-butyl glycine analogue 27 proved to be hitting the sweet spot of potency, stability, and bioavailability. The superior profile presented by 27 supported the potential of this novel class of molecules.
Scheme 3. Modular Strategy for Diverse Core SAR Exploration and Modular Synthesis for γ-Side Chain Optimization.

Reagents and conditions: (1) 2,6-lutidine, ClCH2SO2Cl, CH2Cl2, 0 °C to rt; (2) CsOAc, 18-crown-6, toluene, 80 °C; (3) NaN3, DMSO, 80 °C; (4) Pd/C, H2, EtOAc, rt; (5) R2CHO, AcOH, NaBH3CN, MeOH, 0 °C to rt; (6) R2CO2H, HATU, Et3N, DMF, rt; (7a) HBpin, [Ir(COD)Cl]2, dppe, CH2Cl2, rt; (7b) (+)-pinanediolborane used instead of HBpin; (8a) 6N HCl, 80 °C; (8b) KOTMS, THF, rt; then 6N HCl, 35 to 60 °C; (9) K2CO3, MeOH, rt; (10) MeI, NaH, DMF, rt.
Table 3. Optimizing for Potency and Oral Bioavailabilitya.

Asterisk (*): Discernible exposures of 18 detected in plasma samples. NA = data not available.
To gain a detailed understanding of the binding mode of our inhibitors, cocrystal structures of 18 and 24a with hARG1 protein were obtained (Figure 4). As the structural overlays revealed, both 18 and 24a adopt a similar binding mode as that of the starting point 4a and the boronate, alkyl chain, and pyrrolidine overlay extremely well. As expected, the boronate, pyrrolidine, and carboxylate groups make numerous interactions within the active site. Interestingly, the additional amine moiety on the γ carbon of the pyrrolidine ring of 18 is hydrogen bonding to nearby Asp181, consistent with the original design, while the valine side chain of 24a is oriented to interact with the pocket entrance through favorable electrostatic and van der Waals interactions, as well as the water molecules of the first solvation shell. These fruitful interactions, therefore, allow the ligand to explore the area directly outside of the active site, though it does not make any direct interactions with Asp181 or Asp183.
Figure 4.
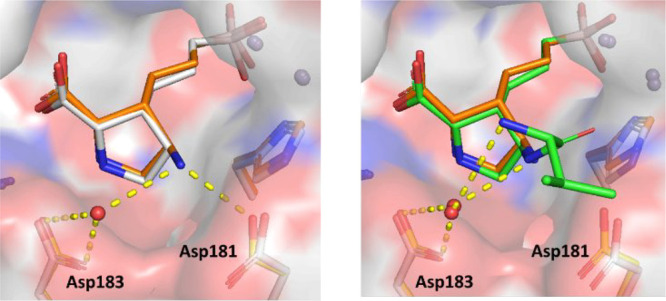
Overlay of cocrystal structures of (left) 4a (white, PDB 7K4G) and 18 (orange, PDB 7KLL); (right) 18 and 24a (green, PDB 7KLM)
Contemporaneous to the optimization efforts described in the preceding paragraphs, 4a was selected as a fit-for-purpose tool for further profiling in mouse pharmacokinetic–pharmacodynamic experiments (Figure 5). In our first study, 4a was administered to EMT6 tumor-implanted BALB/c mice at escalating doses of 30, 100, and 300 mpk twice daily (BID) by oral gavage (n = 5 for each dosing group). Gratifyingly, at 1 h time points after the fourth dose, serum and tumor levels of 4a showed comparable dose-dependent increases in compound exposures, all of which surpassed the enzymatic arginase IC50 of 16 nM by >300-fold (Figure 5 top, blue and red lines, respectively); accordingly, both serum and tumor arginine levels were also substantially elevated in a dose/exposure dependent manner, as compared to the control group (saline, n = 5).38 Encouraged by these results, 4a was then dosed alone (p.o., 250 mpk, BID) and in combination with a mouse anti-PD-1 mAb (mDX400, i.p., 5 mpk, Q5D) in a 14 day repeated dosing experiment (n = 10 for each dosing group).39 To our delight, 4a was well tolerated with no discernible body weight changes and demonstrated comparable pharmacodynamic effects on serum and tumor arginine levels with or without codosing of mDX400.
Figure 5.
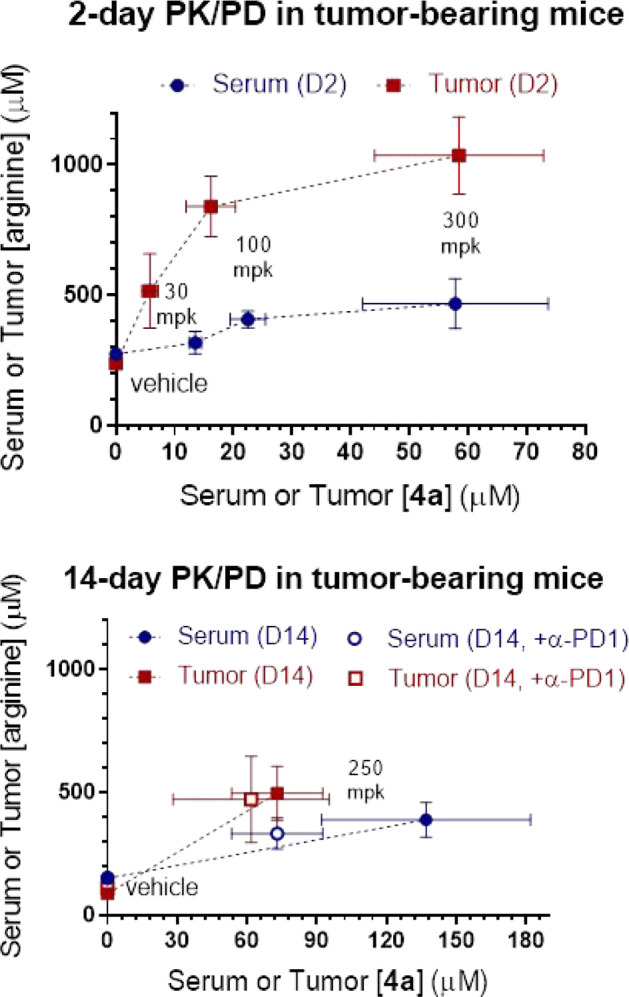
Pharmacokinetic–pharmacodynamic assessment of 4a (p.o.) in EMT6 tumor-bearing BALB/c mice in (top) 2 day (10, 30, and 300 mpk, BID) and (bottom) 14 day (250 mpk, BID) ± mDX400 (5 mpk, Q5D) experiments.
In conclusion, guided by SBDD, a novel class of ARG1 inhibitors featuring a proline core was identified. Further optimization of the side chain to improve potency while maintaining favorable pharmacokinetics was greatly accelerated by harnessing a variety of stereoselective late-stage functionalizations of late-stage building blocks. Compounds 4a and 27 were discovered and exhibited favorable overall profiles, including excellent potency, selectivity, and oral bioavailability. The binding model for this class of molecules was also established unambiguously by the X-ray cocrystal structure of compounds 4a, 18, and 24a with the hARG1 protein. Finally, 4a demonstrated promising pharmacodynamic effects in a mouse EMT6 tumor syngeneic model and was well tolerated. These findings provide rationale for further investigations of this novel series of hARG1 inhibitors as anticancer agents in order to add to the armamentarium of agents that activate the immune system to fight cancer, and future development will be communicated in due course.
Acknowledgments
The authors gratefully acknowledge Abdelghani Achab, Matthew Mitcheltree, and Theodore Martinot for contributions to the synthesis and biological profiling of compounds described here. We also recognize Jialiang Wang, Qingxin Li, Xiong Xiao, Peng Zhang, Wenqin Hu, Jiuyin Yi, Minfang Zhang, Zhenxiang Xie, Yaheng Wang, and Yang Wang from WuXi Apptec for their synthetic efforts. We thank Donovon Adpressa for NMR structure-elucidation work, Foster Tenkorang and Janet Diratsouian for high resolution mass spectrometry support, and Brian Hall and Angie Sun for providing purified protein for crystallography studies.
Biography
Min Lu received his Ph.D. in Organic Chemistry from Nanyang Technological University, Singapore, under the supervision of Prof. Guofu Zhong. After his Post Doctoral Fellowships at The Scripps Research Institute, CA, and Rice University, TX, in the laboratories of Prof. K. C. Nicolaou, he joined the Discovery Chemistry team at Merck & Co., Inc., Boston, MA, in 2015. In 2018 he was promoted to Associate Principal Scientist working on projects in various drug discovery phases. Recent publications include orally available non-nucleotide STING agonists with antitumor activity.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00195.
Experimental procedures for protein preparation; X-ray crystallography; synthesis of all new compounds (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
This work was funded entirely by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
The authors declare no competing financial interest.
Author Status
○ Peter Spacciapoli died April 15, 2019.
Dedication
This paper is dedicated to the memory of our colleague, Peter Spacciapoli.
Supplementary Material
References
- Whiteside T. L.; Demaria S.; Rodriguez-Ruiz M. E.; Zarour H. M.; Melero I. Emerging Opportunities and Challenges in Cancer Immunotherapy. Clin. Cancer Res. 2016, 22 (8), 1845–55. 10.1158/1078-0432.CCR-16-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S. C.; Duffy C. R.; Allison J. P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discovery 2018, 8 (9), 1069–1086. 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- Tang J.; Yu J. X.; Hubbard-Lucey V. M.; Neftelinov S. T.; Hodge J. P.; Lin Y. The clinical trial landscape for PD1/PDL1 immune checkpoint inhibitors. Nat. Rev. Drug Discovery 2018, 17 (12), 854. 10.1038/nrd.2018.210. [DOI] [PubMed] [Google Scholar]
- Tang J.; Shalabi A.; Hubbard-Lucey V. M. Comprehensive analysis of the clinical immuno-oncology landscape. Ann Oncol 2018, 29 (1), 84–91. 10.1093/annonc/mdx755. [DOI] [PubMed] [Google Scholar]
- Patel S. A.; Minn A. J. Combination Cancer Therapy with Immune Checkpoint Blockade: Mechanisms and Strategies. Immunity 2018, 48 (3), 417–433. 10.1016/j.immuni.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris S. M. Jr. Regulation of enzymes of the urea cycle and arginine metabolism. Annu. Rev. Nutr. 2002, 22, 87–105. 10.1146/annurev.nutr.22.110801.140547. [DOI] [PubMed] [Google Scholar]
- Pan W.; Sun Q.; Wang Y.; Wang J.; Cao S.; Ren X. Highlights on mechanisms of drugs targeting MDSCs: providing a novel perspective on cancer treatment. Tumor Biol. 2015, 36 (5), 3159–69. 10.1007/s13277-015-3363-9. [DOI] [PubMed] [Google Scholar]
- Munder M. Arginase: an emerging key player in the mammalian immune system. Br. J. Pharmacol. 2009, 158 (3), 638–51. 10.1111/j.1476-5381.2009.00291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez P. C.; Quiceno D. G.; Zabaleta J.; Ortiz B.; Zea A. H.; Piazuelo M. B.; Delgado A.; Correa P.; Brayer J.; Sotomayor E. M.; Antonia S.; Ochoa J. B.; Ochoa A. C. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004, 64 (16), 5839–49. 10.1158/0008-5472.CAN-04-0465. [DOI] [PubMed] [Google Scholar]
- Porembska Z.; Luboinski G.; Chrzanowska A.; Mielczarek M.; Magnuska J.; Baranczyk-Kuzma A. Arginase in patients with breast cancer. Clin. Chim. Acta 2003, 328 (1–2), 105–11. 10.1016/S0009-8981(02)00391-1. [DOI] [PubMed] [Google Scholar]
- Zea A. H.; Rodriguez P. C.; Atkins M. B.; Hernandez C.; Signoretti S.; Zabaleta J.; McDermott D.; Quiceno D.; Youmans A.; O’Neill A.; Mier J.; Ochoa A. C. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. 2005, 65 (8), 3044–8. 10.1158/0008-5472.CAN-04-4505. [DOI] [PubMed] [Google Scholar]
- Polat M. F.; Taysi S.; Polat S.; Boyuk A.; Bakan E. Elevated serum arginase activity levels in patients with breast cancer. Surg. Today 2003, 33 (9), 655–61. 10.1007/s00595-002-2563-2. [DOI] [PubMed] [Google Scholar]
- Wu C.-W.; Chi C.-W.; Lin E.-C.; Lui W.-Y.; Pʼeng F.-K.; Wang S.-R. Serum arginase level in patients with gastric cancer. J. Clin. Gastroenterol. 1994, 18 (1), 84–5. 10.1097/00004836-199401000-00019. [DOI] [PubMed] [Google Scholar]
- Pham T. N.; Liagre B.; Girard-Thernier C.; Demougeot C. Research of novel anticancer agents targeting arginase inhibition. Drug Discovery Today 2018, 23 (4), 871–878. 10.1016/j.drudis.2018.01.046. [DOI] [PubMed] [Google Scholar]
- Steggerda S. M.; Bennett M. K.; Chen J.; Emberley E.; Huang T.; Janes J. R.; Li W.; MacKinnon A. L.; Makkouk A.; Marguier G.; Murray P. J.; Neou S.; Pan A.; Parlati F.; Rodriguez M. L. M.; Van de Velde L. A.; Wang T.; Works M.; Zhang J.; Zhang W.; Gross M. I. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J. Immunother. Cancer 2017, 5 (1), 101. 10.1186/s40425-017-0308-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borek B.; Gajda T.; Golebiowski A.; Blaszczyk R. Boronic acid-based arginase inhibitors in cancer immunotherapy. Bioorg. Med. Chem. 2020, 28 (18), 115658. 10.1016/j.bmc.2020.115658. [DOI] [PubMed] [Google Scholar]
- Pudlo M.; Demougeot C.; Girard-Thernier C. Arginase Inhibitors: A Rational Approach Over One Century. Med. Res. Rev. 2017, 37 (3), 475–513. 10.1002/med.21419. [DOI] [PubMed] [Google Scholar]
- Abdelkawy K. S.; Lack K.; Elbarbry F. Pharmacokinetics and Pharmacodynamics of Promising Arginase Inhibitors. Eur. J. Drug Metab. Pharmacokinet. 2017, 42 (3), 355–370. 10.1007/s13318-016-0381-y. [DOI] [PubMed] [Google Scholar]
- Grobben Y.; Uitdehaag J. C. M.; Willemsen-Seegers N.; Tabak W. W. A.; de Man J.; Buijsman R. C.; Zaman G. J. R. Structural insights into human Arginase-1 pH dependence and its inhibition by the small molecule inhibitor CB-1158. J Struct Biol X 2020, 4, 100014. 10.1016/j.yjsbx.2019.100014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Zandt M. C.; Whitehouse D. L.; Golebiowski A.; Ji M. K.; Zhang M.; Beckett R. P.; Jagdmann G. E.; Ryder T. R.; Sheeler R.; Andreoli M.; Conway B.; Mahboubi K.; D’Angelo G.; Mitschler A.; Cousido-Siah A.; Ruiz F. X.; Howard E. I.; Podjarny A. D.; Schroeter H. Discovery of (R)-2-amino-6-borono-2-(2-(piperidin-1-yl)ethyl)hexanoic acid and congeners as highly potent inhibitors of human arginases I and II for treatment of myocardial reperfusion injury. J. Med. Chem. 2013, 56 (6), 2568–80. 10.1021/jm400014c. [DOI] [PubMed] [Google Scholar]
- Mitcheltree M. J.; Li D.; Achab A.; Beard A.; Chakravarthy K.; Cheng M.; Cho H.; Eangoor P.; Fan P.; Gathiaka S.; Kim H. Y.; Lesburg C. A.; Lyons T. W.; Martinot T. A.; Miller J. R.; McMinn S.; O’Neil J.; Palani A.; Palte R. L.; Sauri J.; Sloman D. L.; Zhang H.; Cumming J. N.; Fischer C. Discovery and Optimization of Rationally Designed Bicyclic Inhibitors of Human Arginase to Enhance Cancer Immunotherapy. ACS Med. Chem. Lett. 2020, 11 (4), 582–588. 10.1021/acsmedchemlett.0c00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Costanzo L.; Sabio G.; Mora A.; Rodriguez P. C.; Ochoa A. C.; Centeno F.; Christianson D. W. Crystal structure of human arginase I at 1.29-A resolution and exploration of inhibition in the immune response. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (37), 13058–63. 10.1073/pnas.0504027102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S.; Viola R. E. A spectrophotometric assay of arginase. Anal. Biochem. 2001, 295 (1), 117–9. 10.1006/abio.2001.5189. [DOI] [PubMed] [Google Scholar]
- Lyons T. W.; Martinot T. A.; He C. Q. X.; Qi J.; Shao G. X. Development of a Zinc-Mediated Approach to a 2,3-cis-Pyrrolidine Arginase Inhibitor. Org. Process Res. Dev. 2020, 24 (8), 1457–1466. 10.1021/acs.oprd.0c00171. [DOI] [Google Scholar]
- Blaszczyk R.; Brzezinska J.; Dymek B.; Stanczak P. S.; Mazurkiewicz M.; Olczak J.; Nowicka J.; Dzwonek K.; Zagozdzon A.; Golab J.; Golebiowski A. Discovery and Pharmacokinetics of Sulfamides and Guanidines as Potent Human Arginase 1 Inhibitors. ACS Med. Chem. Lett. 2020, 11 (4), 433–438. 10.1021/acsmedchemlett.9b00508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins A. L.; Keseru G. M.; Leeson P. D.; Rees D. C.; Reynolds C. H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discovery 2014, 13 (2), 105–21. 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- Kamijo S.; Takao G.; Kamijo K.; Hirota M.; Tao K.; Murafuji T. Photo-induced Substitutive Introduction of the Aldoxime Functional Group to Carbon Chains: A Formal Formylation of Non-Acidic C(sp(3))-H Bonds. Angew. Chem., Int. Ed. 2016, 55 (33), 9695–9699. 10.1002/anie.201603810. [DOI] [PubMed] [Google Scholar]
- Holladay M. W.; Lin C. W.; May C. S.; Garvey D. S.; Witte D. G.; Miller T. R.; Wolfram C. A.; Nadzan A. M. trans-3-n-propyl-L-proline is a highly favorable, conformationally restricted replacement for methionine in the C-terminal tetrapeptide of cholecystokinin. Stereoselective synthesis of 3-allyl- and 3-n-propyl-L-proline derivatives from 4-hydroxy-L-proline. J. Med. Chem. 1991, 34 (1), 455–7. 10.1021/jm00105a068. [DOI] [PubMed] [Google Scholar]
- Busnel O.; Carreaux F.; Carboni B.; Pethe S.; Goff S. V.; Mansuy D.; Boucher J. L. Synthesis and evaluation of new omega-borono-alpha-amino acids as rat liver arginase inhibitors. Bioorg. Med. Chem. 2005, 13 (7), 2373–9. 10.1016/j.bmc.2005.01.053. [DOI] [PubMed] [Google Scholar]
- Ilies M.; Di Costanzo L.; Dowling D. P.; Thorn K. J.; Christianson D. W. Binding of alpha,alpha-disubstituted amino acids to arginase suggests new avenues for inhibitor design. J. Med. Chem. 2011, 54 (15), 5432–43. 10.1021/jm200443b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golebiowski A.; Paul Beckett R.; Van Zandt M.; Ji M. K.; Whitehouse D.; Ryder T. R.; Jagdmann E.; Andreoli M.; Mazur A.; Padmanilayam M.; Cousido-Siah A.; Mitschler A.; Ruiz F. X.; Podjarny A.; Schroeter H. 2-Substituted-2-amino-6-boronohexanoic acids as arginase inhibitors. Bioorg. Med. Chem. Lett. 2013, 23 (7), 2027–30. 10.1016/j.bmcl.2013.02.024. [DOI] [PubMed] [Google Scholar]
- Golebiowski A.; Whitehouse D.; Beckett R. P.; Van Zandt M.; Ji M. K.; Ryder T. R.; Jagdmann E.; Andreoli M.; Lee Y.; Sheeler R.; Conway B.; Olczak J.; Mazur M.; Czestkowski W.; Piotrowska W.; Cousido-Siah A.; Ruiz F. X.; Mitschler A.; Podjarny A.; Schroeter H. Synthesis of quaternary alpha-amino acid-based arginase inhibitors via the Ugi reaction. Bioorg. Med. Chem. Lett. 2013, 23 (17), 4837–41. 10.1016/j.bmcl.2013.06.092. [DOI] [PubMed] [Google Scholar]
- General References. In Drug-like Properties: Concepts, Structure Design and Methods; Kerns E. H., Di L., Eds.; Academic Press: San Diego, 2008; p 492. [Google Scholar]
- Ghose A. K.; Viswanadhan V. N.; Wendoloski J. J. Prediction of hydrophobic (lipophilic) properties of small organic molecules using fragmental methods: An analysis of ALOGP and CLOGP methods. J. Phys. Chem. A 1998, 102 (21), 3762–3772. 10.1021/jp980230o. [DOI] [Google Scholar]
- Di L.; Artursson P.; Avdeef A.; Benet L. Z.; Houston J. B.; Kansy M.; Kerns E. H.; Lennernas H.; Smith D. A.; Sugano K. The Critical Role of Passive Permeability in Designing Successful Drugs. ChemMedChem 2020, 15, 1862. 10.1002/cmdc.202000419. [DOI] [PubMed] [Google Scholar]
- Varma M. V.; Ambler C. M.; Ullah M.; Rotter C. J.; Sun H.; Litchfield J.; Fenner K. S.; El-Kattan A. F. Targeting intestinal transporters for optimizing oral drug absorption. Curr. Drug Metab. 2010, 11 (9), 730–42. 10.2174/138920010794328850. [DOI] [PubMed] [Google Scholar]
- Shimizu T.; Hiranuma S.; Nakata T. Efficient method for inversion of secondary alcohols by reaction of chloromethanesulfonates with cesium acetate. Tetrahedron Lett. 1996, 37 (34), 6145–6148. 10.1016/0040-4039(96)01333-0. [DOI] [Google Scholar]
- Kumar S.; Jaipuri F. A.; Waldo J. P.; Potturi H.; Marcinowicz A.; Adams J.; Van Allen C.; Zhuang H.; Vahanian N.; Link C. Jr.; Brincks E. L.; Mautino M. R. Discovery of indoximod prodrugs and characterization of clinical candidate NLG802. Eur. J. Med. Chem. 2020, 198, 112373. 10.1016/j.ejmech.2020.112373. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Liu K.; Pu Q.; Achab A.; Ardolino M. J.; Cheng M.; Deng Y.; Doty A. C.; Ferguson H.; Fradera X.; Knemeyer I.; Kurukulasuriya R.; Lam Y. H.; Lesburg C. A.; Martinot T. A.; McGowan M. A.; Miller J. R.; Otte K.; Biju P. J.; Sciammetta N.; Solban N.; Yu W.; Zhou H.; Wang X.; Bennett D. J.; Han Y. Discovery of Amino-cyclobutarene-derived Indoleamine-2,3-dioxygenase 1 (IDO1) Inhibitors for Cancer Immunotherapy. ACS Med. Chem. Lett. 2019, 10 (11), 1530–1536. 10.1021/acsmedchemlett.9b00344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See the Supporting Information for more details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.