

Abstract
The deep and durable antitumor effects of antibody-based immunotherapies such as immune checkpoint inhibitors (ICIs) have revolutionized oncology and transformed the therapeutic landscape for many cancers. Several anti–programmed death receptor 1 and anti–programmed death receptor ligand 1 antibodies have been approved for use in advanced solid tumors, including melanoma, non–small cell lung cancer (NSCLC), bladder cancer, and other cancers. ICIs are under development across many tumor types and preliminary results are compelling. However, ICIs have been associated with severe immune-related adverse events (irAEs), including rash, diarrhea, colitis, hypophysitis, hepatotoxicity, and hypothyroidism, which in some cases lead to high morbidity, are potentially life-threatening, and limit the duration of treatment. The incidence of severe irAEs increases further when programmed cell death-1 and programmed cell death ligand-1 inhibitors are combined with anti-CTLA-4 and/or other multi-drug regimens. Probody™ therapeutics, a new class of recombinant, proteolytically activated antibody prodrugs are in early development and are designed to exploit the hallmark of dysregulation of tumor protease activity to deliver their therapeutic effects within the tumor microenvironment (TME) rather than peripheral tissue. TME targeting, rather than systemic targeting, may reduce irAEs in tissues distant from the tumor. Probody therapeutic technology has been applied to multiple antibody formats, including immunotherapies, Probody drug conjugates, and T-cell–redirecting bispecific Probody therapeutics. In preclinical models, Probody therapeutics have consistently maintained anti-cancer activity with improved safety in animals compared with the non-Probody parent antibody. In the clinical setting, Probody therapeutics may expand or create therapeutic windows for anti-cancer therapies.
Keywords: immunotherapy, PD-1 pathway
Introduction
Evasion of antitumor immunity is a hallmark of cancer. Therefore, immunotherapies were developed to activate, expand, and/or redirect tumor-reactive T cells to enhance cell-based antitumor immune responses, including antibody-based therapies such as immune checkpoint inhibitor (ICIs) and T-cell–redirecting bispecifics (TCBs) (1–4). Although immunotherapies prolong survival in patients with various tumor types, they can result in toxicity because the desired systemic immunostimulatory effects on the tumor also occur in healthy tissue. Immune-related adverse events (irAEs) are the result of treatment-induced inflammation. Although irAEs can present anywhere in the body, common sites include skin, liver, and the endocrine system (1–4). Such toxicities can be life-threatening and lead to treatment discontinuation. Therefore, the National Comprehensive Cancer Network recently published guidelines on the management of irAEs with ICIs (5).
Despite the often-durable clinical benefits of ICIs, many patients do not respond, respond only transiently, or develop resistance; therefore, immunotherapy combinations are under investigation to improve response rates and durability of response. However, the proportion of patients with toxicities increases with immunotherapy combination, and irAEs are often more difficult to manage versus those expected with individual therapies (6–8). Toxicities can be so severe that the development of otherwise promising immunotherapy regimens is discontinued because therapeutic doses are not safe.
Given the important link between immunotherapy efficacy and toxicity, identifying strategies to uncouple the two is important in cancer drug development. One potential solution is to preferentially activate drugs in tumors and spare healthy tissue through an antibody prodrug or “pro-antibody” approach. Similar to non-biologic prodrug medicines that have been proven safe and effective in a variety of therapeutic areas including cancer (9,10), antibody prodrugs may enable administration of the antibody at otherwise intolerable doses or in combination with a chemotherapeutic agent that would otherwise have a high toxicity rate, thereby allowing longer durations of therapy than achievable by the parent antibody alone.
In this review, we discuss the strengths and weaknesses of current immunotherapeutic strategies, focusing on ICIs, and describe potential advantages of antibody prodrugs, using the novel Probody therapeutic platform as a model.
Immune Checkpoint Inhibitors: Efficacy, Safety, and Considerations for Combination Therapy
Antibodies blocking the inhibitory checkpoints cytotoxic T-lymphocyte–associated antigen-4 (CTLA-4) and programmed death 1 (PD-1), or its ligand PD-L1, restore T-cell–mediated antitumor immune responses and have emerged as effective immune-based cancer treatments (11). One CTLA-4 inhibitor (ipilimumab) and six PD-1/PD-L1 inhibitors (pembrolizumab, nivolumab, atezolizumab, durvalumab, cemiplimab, and avelumab) are approved for the treatment of specific cancers (11–13). Although ICIs demonstrate anticancer efficacy with variable response rates across tumor types and patient populations, most patients are nonresponsive to monotherapy (12); thus, combination strategies are being explored.
Although ICI monotherapy is generally well tolerated compared with traditional chemotherapy, potentially life-threatening irAEs can occur during and up to 1 year after treatment (2,14–16). irAEs result from an immune response against self-antigens, with subsequent target organ inflammation, and commonly include thyroiditis, colitis, and pneumonitis (16). In a recent meta-analysis of 13 studies, rates of hypothyroidism, pneumonitis, colitis, and hypophysitis were higher with anti–PD-1/PD-L1 antibodies compared with control treatments (14). These events are generally managed with high-dose corticosteroids and other immunosuppressants, and ICI therapy can usually continue after mild irAEs, with close monitoring. However, moderate to severe irAEs may result in severe declines in organ function and quality of life, and, in some cases, death. Furthermore, corticosteroids could reduce therapy effectiveness (17). New strategies to maintain efficacy and reduce toxicity are needed.
Because ICIs activate a broad-based immune response, irAEs represent an on-target, off-tumor toxicity for which incidence correlates with efficacy in some cases (eg, the PD-1 inhibitor nivolumab in melanoma and non–small cell lung cancer [NSCLC]) (18–20). A retrospective analysis of nivolumab-treated melanoma (N=148), demonstrated statistically significant improvements in overall survival in patients with rash (hazard ratio [HR], 0.423; 95% confidence interval [CI], 0.243–0.735; P=0.001) and vitiligo (HR, 0.184; 95% CI, 0.036–0.94; P=0.012) (18). In an observational cohort study of nivolumab-treated NSCLC (N=38), patients with irAEs had significantly higher objective response rates (ORR) than patients without irAEs (63.6% vs 7.4%; P<0.01) (20). Similarly, irAEs positively correlated with progression-free survival (HR, 0.525; 95% CI, 0.287–0.937; P=0 .03) and overall survival (HR, 0.282; 95% CI, 0.101–0.667; P=0 .003) in patients with advanced or recurrent NSCLC treated with second-line nivolumab (N=134) (18). The association between toxicity and response is not predictive for individual patients because some patients with irAEs do not achieve clinical efficacy with ICI therapy (21).
PD-1/PD-L1 inhibitors may have greater antitumor efficacy with fewer irAEs than CTLA-4 inhibitors (22). A study comparing adjuvant nivolumab (n=453) and ipilimumab (n=453) in patients with resected stage III/IV melanoma demonstrated a significantly greater rate of 12-month recurrence-free survival (70.5% vs 60.8%, respectively) and a lower rate of grade 3/4 treatment-related AEs (TRAEs; 14.4% vs 45.9%, respectively) with nivolumab (21). In a meta-analysis and systematic review of 73 studies of ICIs (N=3418), the incidence of irAEs was highest with CTLA-4 inhibitors (53.8%), followed by PD-1 inhibitors (26.5%), and was lowest with PD-L1 inhibitors (17.1%) (22). Conversely, overall response rates were lower with CTLA-4 inhibitors (11.2%) versus PD-1 inhibitors (27%) or PD-L1 inhibitors (22.2%) (22). Combination of PD-1/PD-L1 inhibitors with chemotherapy, CTLA-4 inhibitors, BRAF and/or MEK inhibitors, or vascular endothelial growth factor inhibitors improves response rates, but increases overall and grade ≥3 AEs.
Anti–CTLA-4 and anti–PD-1/PD-L1 antibody combinations have demonstrated superiority over anti–PD-1/PD-L1 antibody monotherapy in metastatic melanoma and renal cell carcinoma (RCC), but cause increased toxicity (24–26). In melanoma, 57.6% of patients treated with ipilimumab and nivolumab (n=314) had a Response Evaluation Criteria in Solid Tumors (RECIST) objective response, and 55% incurred a treatment-related grade ≥3 irAE; approximately one-third of patients discontinued therapy because of TRAEs (24). By comparison, of those receiving nivolumab monotherapy (n=316), ORR was 43.7%, and 16.3% of patients experienced grade ≥3 irAEs (7.7% discontinued therapy because of TRAEs). Results of a phase 3 trial of ipilimumab-nivolumab in intermediate- and high-risk advanced RCC are similar (ORR was 42%, grade 3 irAEs reported in 46% of patients [n=425], and discontinuation because of TRAEs was 22%) (25). Additionally, a single-center cohort of 64 patients with melanoma in an expanded-access program of nivolumab plus ipilimumab found that nearly three-fourths of patients required steroids, and over one-third were hospitalized for an irAE, some of which occurred months after treatment discontinuation (26). These toxicities have quality-of-life implications for patients and management of irAEs often requires high-dose steroids. These findings underscore the need for more tolerable combination therapies. Although multiple combination ICI studies are underway, only anti–PD-1/PD-L1 in combination with anti–CTLA-4 antibodies are currently approved in a limited number of indications.
Patients with pre-existing autoimmune disease or history of organ transplantation could be at high risk for AEs and are often excluded from clinical trials. Therefore, therapy that avoids off-tumor toxicities would be beneficial. Concerns about irAEs also limit the use of ICIs in patients with advanced thymic carcinoma, who are at higher risk of autoimmune disorders. In patients with thymic cancer (N=40), pembrolizumab was active, with an ORR of 22.5%; however, 15% had severe irAEs, including 5% with myocarditis (27).
T-cell–Engaging Bispecific Antibodies (TCBs)
TCBs are potent therapeutics designed to direct the activity of cytotoxic T cells to tumors. TCBs are dual-targeted and can bind to two different targets (ie, cell-surface receptors) on the same or different cells. Such dual binding potentially enhances therapeutic antitumor efficacy by simultaneously blocking multiple targets involved in pathogenesis, activating cell signaling, inducing antibody-dependent cell-mediated cytotoxicity, avoiding resistance and increasing antiproliferative effects, and temporarily engaging a patient’s own cytotoxic T cells to lyse cancer cells (3,4). Two TCBs are approved for cancer immunotherapy (catumaxomab, which targets CD3 and EpCAM to treat malignant ascites and blinatumomab which targets CD19 and CD3 to treat Philadelphia chromosome positive acute lymphoblastic leukemia) and more than 50 are in clinical development (3). These highly potent TCBs target healthy tissue even with low antigen expression, resulting in significant on-target, off-tumor toxicity (eg, cytokine release autoimmunity) that can limit dosing (3,4). Therefore, TCB levels necessary for therapeutic efficacy have been difficult to reach without excessive toxicity and novel methods are necessary to engage the potent antitumor activity of TCBs while limiting off-target toxicity.
Overcoming the Challenge of Immunotherapy-Associated AEs
New approaches are needed to optimize antitumor activity of antibody-based immunotherapeutics without compromising control of systemic immunity. One approach is local administration of low-dose immunotherapies via intratumoral or peritumoral injection (28–30). In preclinical mouse models, injection of low-dose, slow-release anti–CTLA-4 antibody formulation near the tumor resulted in effective antitumor CD8+ T-cell responses and tumor eradication, whereas serum levels of systemic antibodies remained low (28). Similarly, intratumoral co-injection of low-dose anti–CTLA-4 and anti-OX40 antibodies in tumor-bearing mice resulted in a systemic antitumor immune response (29). Intratumoral injection is under clinical investigation, though largely limited to individuals with palpable tumors, which challenges the potential scalability of this strategy (30). Furthermore, because not all metastatic tumors can be injected, either using image guidance (interventional radiology) or local subcutaneous intratumoral injection, these approaches must be demonstrated to yield systemic abscopal (ie, distant) anti-cancer, clinically meaningful effects.
Probody™ Therapeutics
A recent approach to overcoming AEs associated with immunotherapy is a new class of recombinant, proteolytically activated antibody prodrugs called Probody therapeutics, which exploit the hallmark dysregulation of protease activity in tumors and are designed to largely restrict drug activity to the tumor microenvironment (TME) (31,32). A Probody therapeutic consists of three modular components—an active anticancer monoclonal IgG antibody or fragment of a variable region, a masking peptide linked to the N-terminus of the light chain, and a protease-cleavable substrate linker peptide—produced as a single protein using recombinant antibody production methodology (Fig. 1) (31,32). In healthy tissue, the Probody therapy remains largely intact and blocked from target binding and retains the long circulatory half-life expected for monoclonal antibody therapies. When a Probody therapeutic reaches the TME, tumor-associated proteases cleave the substrate linker, which releases the masking peptide, enabling the antibody to bind target antigen (Fig. 1) (31,32). Measurement of tumor-associated proteases from human tissue across many tumors showed that >90% of tumors had sufficient protease activity in the TME for Probody therapeutic activation ex vivo (31).
Figure 1.
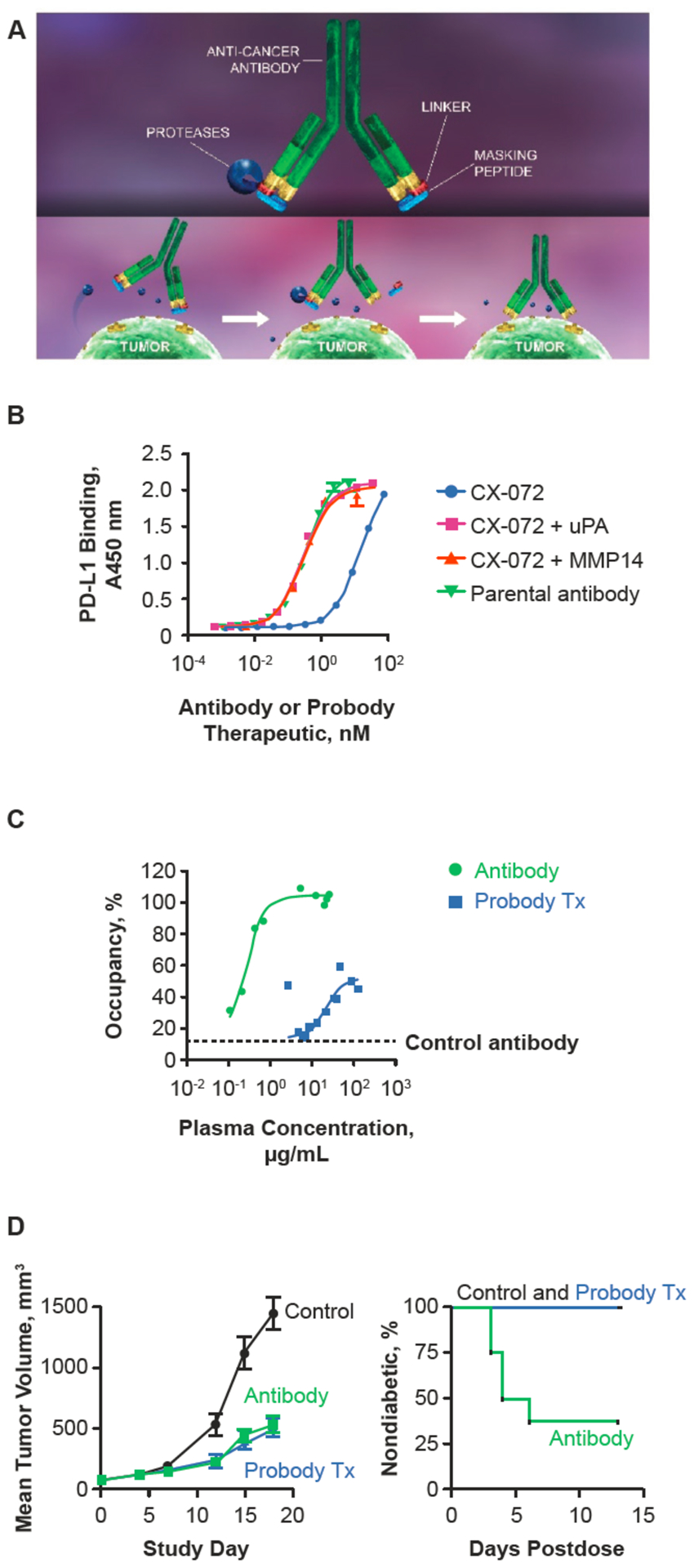
Schematic representation of Probody™ therapeutic activation in the tumor microenvironment. Probody therapeutics are fully recombinant antibody prodrugs designed to remain relatively inactive systemically and to be activated specifically in the tumor microenvironment by tumor-associated proteases. Figure redrawn with permission from CytomX.
In principle, a distinct advantage of Probody technology is its potential application to any therapeutic antibody. Preclinically, the technology has been successfully applied to several antibody-based therapies, including immune modulators/ICIs (eg, anti–PD-L1 [33]), antibody-drug conjugates (eg, anti-CD71 [34], anti-CD166 [35,36]), and TCBs (eg, targeting epidermal growth factor receptor (EGFR)-CD3 [37]). Although the Probody TCB targeting EGFR and CD3 (Pb-TCB) has not yet advanced into clinical development, preclinical results appear promising. In vitro studies demonstrated that an unmasked Pb-TCB exhibits potent dose-dependent tumor killing, while the masked molecule reduces cytotoxicity by more than 100,000-fold (37). In established HT29 xenograft tumor models in mice reconstituted with human PBMCs, the masked Pb-TCB demonstrated significant antitumor activity at 0.5 mg/kg and complete tumor regression at 1.5 mg/kg. EGFR-CD3 Pb-TCB has a significantly higher maximum tolerated dose than the unmasked TCB in nonhuman primates. Cynomolgus monkeys tolerated a dose of 4000 μg/kg of the Pb-TCB, whereas the maximum tolerated dose of the unmasked TCB was 60 μg/kg (37). The results of these studies suggest that the Pb-TCB might enable the development of T-cell–engaging bispecific therapeutics against broadly expressed targets such as EGFR.
Clinical trials evaluating Probody therapeutics are summarized in Table 1. Farthest along in development is CX-072, a Probody immunotherapy targeting PD-L1. Preclinical and preliminary clinical studies suggest that CX-072 has the potential to optimize anticancer efficacy without increasing toxicity. Like other Probody therapies, CX-072 is activated by tumor-associated proteases. In preclinical studies, occupancy of CX-072 on peripheral blood and splenic T cells was markedly reduced compared with that of the unmasked parental antibody at the same dose (33). CX-072 radiolabeled with zirconium-89 (89Zr) was used to study biodistribution into tumor versus lymphoid tissue; 89Zr-CX-072 accumulated in PD-L1-expressing tumors, with only minor uptake in murine peripheral lymphoid tissue (38). In mice bearing MC38 syngeneic tumors, CX-072 induced an antitumor response that was comparable to an unmasked parental antibody at the same dose (33). In addition, CX-072 provided protection from induction of autoimmune diabetes in a mouse model at doses that the parental antibody induced diabetes. Taken together, these preclinical findings suggest that CX-072 could induce an antitumor response similar to the parent antibody while remaining relatively inactive in peripheral tissue and potentially reduce the occurrence of systemic irAEs associated with other PD-1/PD-L1 inhibitors.(33) These data provided the rationale for further clinical development of an antibody-based Probody therapeutic targeting the T-cell checkpoint.
Table 1.
Summary of Ongoing Clinical Trials Evaluating Probody Therapeutics
| Compound Name | Target | Study /NCT Number (Sponsor) | Trial Phase | Patient Population(s) | Target N | Estimated Completion Date |
|---|---|---|---|---|---|---|
| CX-072 | Programmed death ligand-1 | PROCLAIM-CX-072 NCT03013491 (CytomX) |
1/2 | Advanced or recurrent solid tumors or lymphoma | 300 | December 2021 |
| CX-2009 | CD166 | PROCLAIM-CX-2009 NCT03149549 (CytomX) |
1/2 | Metastatic or locally advanced unresectable solid tumors (breast, non-small cell lung cancer, prostate, ovarian, endometrial, head and neck, cholangiocarcinoma) | 150 | December 2021 |
| BMS-986249 | Cytotoxic T-lymphocyte-associated protein-4 | CA030–001 NCT03369223 (Bristol-Myers Squibb) |
1/2 | Advanced solid tumors | 375 | October 2022 (primary) |
| CX-2029 | CD71 | PROCLAIM-CX-2029 NCT03543813 (CytomX) |
1/2 | Metastatic or locally advanced unresectable solid tumors (head and neck, non-small cell lung cancer, pancreatic) or diffuse large B-cell lymphoma | 150 | December 2022 |
| CX-072 | Programmed death ligand-1 | PROCLAIM-CX-072–002 NCT03993379 (CytomX) |
2 | Previously untreated solid tumors, relapsed solid tumors following checkpoint inhibitor therapy, solid tumors with progression during or after platinum therapy, or in neoadjuvant setting | 160 | January 2023 |
CX-072: From Proof-of-Concept to Clinical Trials
Launched in 2017, PROCLAIM-CX-072 (PRObody Clinical Assessment In Man; NCT03013491), is a proof-of-concept phase 1/2a, open-label, multicenter, dose-escalation, study to evaluate tolerability and antitumor activity of CX-072 as monotherapy or in selected combinations in patients with advanced, unresectable solid tumors or lymphoma for which a PD-1 or PD-L1 inhibitor was not approved by the US Food and Drug Administration (FDA) or other regulatory body (38,40). Patients were required to be naïve to ICI therapies. PROCLAIM-CX-072 includes dose-escalation groups (monotherapy and combinations), a stage testing biomarkers and efficacy in PD-L1+ tumors, and an indication expansion phase. Preliminary results are available for the monotherapy and combination dose-escalation phases.
In the monotherapy escalation phase, CX-072 is being evaluated for efficacy and safety (maximum tolerated dose) in dose-escalation patient cohorts, and preliminary results have been presented (39). As of April 2018, 37 patients who had a median of 3 prior therapies (range: 1–13) received CX-072 at increasing doses from 0.03 to 30.0 mg/kg. The median time on treatment was 2.1 months (range: 1–10 months). In 23 evaluable patients across all dose levels, investigator-assessed best tumor response included 2 patients with partial response (one each in patients with thymoma and triple-negative breast cancer) and 10 patients with stable disease. At the time of data cut-off, a maximum tolerated dose had not been reached; one dose-limiting toxicity (grade 3 febrile neutropenia) was observed in a thymoma patient receiving CX-072 at 3 mg/kg. Grade 3 or 4 treatment-related AEs were observed in 4 patients (10.8%), and irAEs with reversible grade 3 events were observed in 3 patients (8.1%), including thrombocytopenia, elevated aminotransferases, and dyspnea. Two patients (5.4%) discontinued because of AEs. Preliminary results from the monotherapy dose expansion at CX-072 10 mg/kg in cohorts with anal squamous cell carcinoma, cutaneous squamous cell carcinoma (cSCC), small bowel adenocarcinoma (SBA), triple-negative breast cancer with skin lesions (TNBC), or undifferentiated pleomorphic sarcoma (UPS) also have been presented (41). A total of 51 patients, with a median age of 63 years (range: 32–80) and median of 3 prior regimens (range: 1–12), were evaluated as of the November 2018 cut-off, at a median treatment duration of 1.8 months (range: 0.3–14.7 months). Partial responses (confirmed and unconfirmed) were observed in patients with cSCC (n = 1 of 3 total patients), TNBC (n = 2 of 2 total patients), and UPS (n = 1 of 16 total patients). One grade 3/4 treatment-related AE was observed (grade 3 rash), and 2 patients discontinued treatment because of AEs (nausea and sepsis; n = 1 each). Although direct comparison to FDA-approved anti–PD-1/PD-L1 antibodies is limited based on sample size and trial design, these preliminary results with CX-072 are very encouraging when compared with historic control data for PD-1/PD-L1 inhibitors.
Pharmacodynamic and pharmacokinetic studies performed on patients receiving CX-072 as monotherapy mirror preclinical research for this agent. As part of translational efforts, a cohort of 13 patients underwent paired baseline and on-treatment biopsies (42). Most patients (75%) in this paired biopsy cohort had protease activity that could be measured in their pre-treatment tumor sample (42). The proportion of patients with detectable intratumoral activation of CX-072 increased with increasing dose. Consistent with clinical activity observed with CX-072, this research supports the intended mechanism of action of Probody therapeutics. Moreover, this integrated clinical and translational data led to the selection of the CX-072 10-mg/kg dose for the expansion cohorts.
The CHECKMATE 067 trial provided evidence of enhanced efficacy with anti-PD-1 (nivolumab) and anti-CTLA-4 (ipilimumab) combination therapy in patients with melanoma (43). However, the improved efficacy of the ICI combination was at the expense of higher toxicity, with a markedly higher rate of immune-related toxicities observed with the combination compared with each agent alone (22,43). To evaluate the efficacy of combination treatment while potentially lowering the safety risk of traditional combination regimens, the PROCLAIM-CX-072 trial includes two combination treatment arms, one with ipilimumab and one with a BRAF inhibitor (vemurafenib), In the ipilimumab combination evaluation in the PROCLAIM-CX-072 study (44), patients (n=16) with advanced solid tumors who received a median of 3 prior cancer treatments (range: 1–12) were treated with CX-072 (0.3, 1.0, 3.0, and 10.0 mg/kg) plus ipilimumab (3.0 mg/kg or 6.0 mg/kg for the highest CX-072 dose level). The median number of ipilimumab doses received was 3. Best tumor response in 10 evaluable patients was one patient with confirmed complete response (anal squamous cell carcinoma), two with confirmed partial responses (testicular cancer [n=1] and small bowel [n=1]), and one with stable disease. The maximum tolerated dose was not reached as of the data cut-off date; however, preliminary data suggest that concomitant dosing of CX-072 and full-dose ipilimumab compares favorably with historical data for non-Probody-therapeutic–based PD-1 pathway inhibitors combined with ipilimumab (22,41). Grade 3 treatment-related irAEs occurred in two patients (colitis and dyspnea/pneumonitis), but no patients discontinued combination therapy because of treatment-related irAEs.
Summary
Antibodies targeting PD-L1 demonstrate antitumor activity against a variety of cancers and are being evaluated in combination with other immunotherapies and targeted agents to improve response rate and durability. However, combinations may be accompanied by increases in overall grade ≥3 AEs, particularly irAEs from immune system overactivation. Because anti–PD-L1/PD-L1 agent use is limited by on-target and off-tumor toxicities, novel strategies are necessary that allow antigen binding in tumors with limited healthy tissue binding. Probody technology was developed to limit off-tumor toxicity. Preliminary results of the first-in-human PROCLAIM-CX-072 study suggest an encouraging safety profile and antitumor activity for the PD-L1–directed Probody therapeutic CX-072. These preliminary findings support further exploration of CX-072 as monotherapy and in combination with other ICIs or targeted therapies.
Probody therapeutics are a new approach to overcome the AE challenges of immunotherapy because their activation is designed to be restricted to the TME. Therefore, systemic toxicity should be limited, risk-benefit improved, and more potent combination therapies may be exploited. A robust pipeline of Probody therapeutics in oncology is advancing through preclinical and clinical trials with the potential to broaden the range of effective doses and targets and enable new treatment combinations.
Acknowledgments
The authors would like to thank Bryan Irving and Chihunt Wong for their contributions to the preclinical development of CX-072. Writing and editorial support was provided by Chris Ontiveros, PhD (ApotheCom, New York, NY), Cathy Winter, PhD (ApotheCom, Yardley, PA), Julia Burke, PhD (ApotheCom, Auckland, New Zealand), and Amy Zannikos, PharmD (Echelon Brand Communications, Parsippany, NJ).
MSK author and trial conduct of PROCLAIM-CX-072 is supported in part by the NIH/NCI Cancer Center Support Grant P30 CA008748.
Financial Support:
Support for this review article was provided by CytomX Therapeutics, Inc.
Conflicts of Interest:
K. Autio has received research funding (to institution) from Merck, ARMO Sciences, CytomX Therapeutics, Inc., Eli Lilly, GlaxoSmithKline, and Pfizer.
A. Naing has received research funding from NCI, EMD Serono, MedImmune, Healios Onc. Nutrition, Atterocor, Amplimmune, ARMO BioSciences, Eli Lilly, Karyopharm Therapeutics, Incyte, Novartis, Regeneron, Merck, BMS, Pfizer, CytomX Therapeutics, Neon Therapeutics, Calithera Biosciences, TopAlliance Biosciences, Kymab, PsiOxus, and the Immune Deficiency Foundation (spouse); has received funding for advisory board participation for CytomX Therapeutics, Inc. and Novartis; and has received reimbursement for travel and accommodations from ARMO BioSciences.
Valentina Boni has received research funding from CytomX Therapeutics, Inc.
Rachel W. Humphrey is employed and has stock ownership from CytomX Therapeutics, Inc.
Footnotes
Ethics approval and consent to participate
PROCLAIM studies discussed herein are conducted in accordance with the current IRB/IEC approved clinical protocol, International Council for Harmonisation (ICH) Good Clinical Practice (GCP) Guidelines, and relevant policies and requirements of the national regulations and laws, including the Health Insurance Portability and Accountability Act of 1996 (HIPAA). Written informed consent/assent is required from each patient prior to any testing in PROCLAIM, including screening tests and evaluations.
Consent for publication
All authors consent to publish this review.
PROBODY is a trademark of CytomX Therapeutics, Inc. All other brands and trademarks referenced herein are the property of their respective owners.
References
- 1.King GT, Sharma P, Davis SL, Jimeno A. Immune and autoimmune-related adverse events associated with immune checkpoint inhibitors in cancer therapy. Drugs Today (Barc) 2018;54(2):103–22. [DOI] [PubMed] [Google Scholar]
- 2.Yoest JM. Clinical features, predictive correlates, and pathophysiology of immune-related adverse events in immune checkpoint inhibitor treatments in cancer: a short review. ImmunoTargets Ther 2017;6:73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trivedi A, Stienen S, Zhu M, Li H, Yuraszeck T, Gibbs J, et al. Clinical pharmacology and translational aspects of bispecific antibodies. Clin Trans Sci 2017;10(3):147–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yuraszeck T, Kasichayanula S, Benjamin J. Translation and clinical development of bispecific T-cell engaging antibodies for cancer treatment. Clin Pharmacol Ther 2017;101(5):634–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.National Comprehensive Cancer Network. Management of Immunotherapy-Related Toxicities (Version 1.2019). https://www.nccn.org/professionals/physician_gls/pdf/immunotherapy.pdf.Accessed: February 28, 2019. [DOI] [PubMed]
- 6.Kyi C, Postow MA. Immune checkpoint inhibitor combinations in solid tumors: opportunities and challenges. Immunotherapy 2016;8(7):821–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haanen J, Carbonnel F, Robert C, Kerr KM, Peters S, Larkin J, et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2017;28(suppl_4):iv119–iv42. [DOI] [PubMed] [Google Scholar]
- 8.Brahmer JR, Lacchetti C, Schneider BJ, Atkins MB, Brassil KJ, Caterino JM, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol 2018;36(17):1714–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waller DG, George CF. Prodrugs. Br J Clin Pharmacol 1989;28:497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giang I, Boland EL, Poon GMK. Prodrug applications for targeted cancer therapy. AAPS J 2014;16(5):899–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Reviews Clin Oncol 2016;13(5):273–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gong J, Chehrazi-Raffle A, Reddi S, Salgia R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer 2018;6(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.U.S. Food & Drug Administration. FDA approves cemiplimab-rwlc for metastatic or locally advanced cutaneous squamous cell carcinoma [press release]. September 28, 2018. Available at: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm622251.htm [Accessed October 30, 2018].
- 14.Baxi S, Yang A, Gennarelli RL, Khan N, Wang Z, Boyce L, et al. Immune-related adverse events for anti-PD-1 and anti-PD-L1 drugs: systematic review and meta-analysis. BMJ 2018;360:k793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fecher LA, Agarwala SS, Hodi FS, Weber JS. Ipilimumab and its toxicities: a multidisciplinary approach. Oncologist 2013;18(6):733–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Winer A, Bodor JN, Borghaei H. Identifying and managing the adverse effects of immune checkpoint blockade. J Thorac Dis 2018;10(Suppl 3):S480–s9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Margolin K, Ernstoff MS, Hamid O, Lawrence D, McDermott D, Puzanov I, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol 2012;13(5):459–65. [DOI] [PubMed] [Google Scholar]
- 18.Freeman-Keller M, Kim Y, Cronin H, Richards A, Gibney G, Weber JS. Nivolumab in resected and unresectable metastatic melanoma: characteristics of immune-related adverse events and association with outcomes. Clin Cancer Res 2016;22(4):886–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haratani K, Hayashi H, Chiba Y, Kudo K, Yonesaka K, Kato R, et al. Association of immune-related adverse events with nivolumab efficacy in non-small-cell lung cancer. JAMA Oncol 2018;4(3):374–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sato K, Akamatsu H, Murakami E, Sasaki S, Kanai K, Hayata A, et al. Correlation between immune-related adverse events and efficacy in non-small cell lung cancer treated with nivolumab. Lung Cancer 2018;115:71–4. [DOI] [PubMed] [Google Scholar]
- 21.Palmieri DJ, Carlino MS. Immune checkpoint inhibitor toxicity. Current Oncol Rep 2018;20(9):72. [DOI] [PubMed] [Google Scholar]
- 22.El Osta B, Hu F, Sadek R, Chintalapally R, Tang SC. Not all immune-checkpoint inhibitors are created equal: Meta-analysis and systematic review of immune-related adverse events in cancer trials. Critical Rev Oncology Hematol 2017;119:1–12. [DOI] [PubMed] [Google Scholar]
- 23.Weber J, Mandala M, Del Vecchio M, Gogas HJ, Arance AM, Cowey CL, et al. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N Engl J Med 2017;377(19):1824–35. [DOI] [PubMed] [Google Scholar]
- 24.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373(1):23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK, et al. Nivolumab plus Ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med 2018;378(14):1277–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shoushtari AN, Friedman CF, Navid-Azarbaijani P, Postow MA, Callahan MK, Momtaz P, et al. Measuring toxic effects and time to treatment failure for nivolumab plus ipilimumab in melanoma. JAMA Oncol 2018;4(1):98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giaccone G, Kim C, Thompson J, McGuire C, Kallakury B, Chahine JJ, et al. Pembrolizumab in patients with thymic carcinoma: a single-arm, single-centre, phase 2 study. Lancet Oncol 2018;19(3):347–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fransen MF, van der Sluis TC, Ossendorp F, Arens R, Melief CJ. Controlled local delivery of CTLA-4 blocking antibody induces CD8+ T-cell-dependent tumor eradication and decreases risk of toxic side effects. Clin Cancer Res 2013;19(19):5381–9. [DOI] [PubMed] [Google Scholar]
- 29.Marabelle A, Kohrt H, Sagiv-Barfi I, Ajami B, Axtell RC, Zhou G, et al. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J Clin Invest 2013;123(6):2447–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ray A, Williams MA, Meek SM, Bowen RC, Grossmann KF, Andtbacka RH, et al. A phase I study of intratumoral ipilimumab and interleukin-2 in patients with advanced melanoma. Oncotarget 2016;7(39):64390–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Desnoyers LR, Vasiljeva O, Richardson JH, Yang A, Menendez EE, Liang TW, et al. Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Sci Transl Med 2013;5(207):207ra144. [DOI] [PubMed] [Google Scholar]
- 32.Polu KR, Lowman HB. Probody therapeutics for targeting antibodies to diseased tissue. Expert Opin Biol Ther 2014;14(8):1049–53. [DOI] [PubMed] [Google Scholar]
- 33.Wong C, Mei L, Wong KR, Menendez EEM, Vasiljeva O, Richardson JH, et al. A PD-L1-targeted Probody provides antitumor efficacy while minimizing induction of systemic autoimmunity. Cancer Immunol Res 2016;4(1 suppl); abstract A081. [Google Scholar]
- 34.Singh S, Serwer L, Chauhan N, DuPage A, Krimm M, Wong K, et al. Optimizing a CD71-targeting Probody drug conjugate (PDC) for activity in multiple solid tumor and lymphoma models and for tolerability in nonhuman primates. Mol Cancer Ther 2018;17(1 suppl); abstract B116. [Google Scholar]
- 35.Garcia-Corbacho J, Spira A, Boni V, Feliu J, Middleton M, Burris H, et al. PROCLAIM-CX-2009: A first-in-human trial to evaluate CX-2009 in adults with metastatic or locally advanced unresectable solid tumors. Ann Oncol 2017;28(suppl 5); abstract 422TiP. [Google Scholar]
- 36.Weaver AY, Singh S, DuPage A, Sagert J, Flandez J, Menendez E, et al. Development of a probody drug conjugate (PDC) against CD166 for the treatment of multiple cancers. Mol Cancer Ther 2015;14(12 suppl 2); abstract C165. [Google Scholar]
- 37.Boustany LM, Wong L, White CW, Diep L, Huang Y, Liu S, et al. EGFR-CD3 bispecific Probody™ therapeutic induces tumor regressions and increases maximum tolerated dose >60-fold in preclinical studies. Mol Cancer Ther 2018;17(1 suppl); abstract A164. [Google Scholar]
- 38.Giesen D, Broer LN, Lub-De Hooge MN, Popova I, Howng B, Vasiljeva O3, et al. 89Zr-labeled anti-PD-L1 CX-072 PET imaging in human xenograft and syngeneic tumors. Ann Oncol. 2019February1;30(Supplement_1). pii: mdz029.001. [Google Scholar]
- 39.Boni V, Garcia-Corbacho J, Ott PA, Cho DC, Autio KA, Uboha N, et al. Preliminary results of PROCLAIM-CX-072: The first-in-human, dose-finding trial of PD-L1 Probody therapeutic CX-072 as monotherapy in patients (pts) with advanced solid tumors. Ann Oncol 2018;29(suppl 8); abstract 435P. [Google Scholar]
- 40.Autio KA, Arkenau H-T, O’Neil BH, Bendell JC, El-Khoueiry A, Strauss J, et al. Preliminary results of the first-in-human, dose-finding PROCLAIM-CX-072 trial of the PD-L1 Probody therapeutic CX-072 as monotherapy in patients (pts) with advanced solid tumors. J Clin Oncol 2018;36(15 suppl); abstract 3071. [Google Scholar]
- 41.Naing A, Thistlethwaite FC, Spira AI, Garcia-Corbacho J, Randhawa M, Eskens F, et al. CX-072, a PD-L1 Probody therapeutic, as monotherapy in patients with advanced solid tumors: Preliminary results of PROCLAIM-CX-072. J Clin Oncol. 2019; 37 (suppl); abstract 2513. [Google Scholar]
- 42.Lyman SK, Gordon J, DuPage A, Pramanik P, Howng B, Zein IA, et al. Preliminary evidence of intratumoral activation and immunomodulatory effect of CX-072, a Probody therapeutic antibody prodrug targeting PD-L1, in a phase 1/2 trial. J Immunother Cancer. 2018, 6(Suppl 1):114; abstract P87.30400835 [Google Scholar]
- 43.Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 2017;377(14):1345–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Plummer R, Sanborn RE, de Vries EGE, Lorusso P, Arkenau HT, Uboha N, et al. 436P - Preliminary results of the first-in-human, dose-finding PROCLAIM-CX-072 trial evaluating the PD-L1 probody therapeutic CX-072 in combination with ipilimumab (ipi) in patients (pts) with advanced solid tumors. Ann Oncol 2018;29(suppl 8); abstract 436P. [Google Scholar]