

To the Editor:
In a recent article in the American Journal of Human Genetics, biallelic pathogenic KDELR2 variants were described as a novel cause of autosomal recessive (AR) osteogenesis imperfecta (OI) (MIM: #166200) in four families with six affected individuals (van Dijk et al., 2020). The KDELR family of proteins is important in inter‐organelle communication by regulating protein trafficking between the Golgi apparatus and the endoplasmic reticulum (Capitani & Sallese, 2009). KDELR2‐related OI results from the inability of HSP47 (heat shock protein 47) to bind KDELR2, leading to failure of HSP47 to dissociate from collagen type 1. HSP47‐bound extracellular collagen cannot form collagen fibers in individuals with pathogenic biallelic KDELR2 variants (Figure 1; van Dijk et al., 2020). We read the authors' work with great enthusiasm and would like to share clinical and genetic information from two additional unrelated consanguineous families with three affected children with OI with additional phenotypic features, therefore expanding the phenotypic spectrum of KDELR2‐related OI.
FIGURE 1.
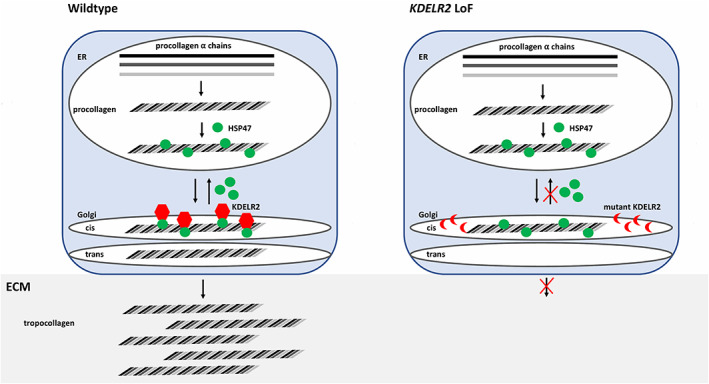
KDELR2 loss of function (LoF) leads to inability of heat shock protein 47 (HSP47) to dissociate from procollagen. In wildtype cells, alpha collagen fibers assemble to form procollagen. Procollagen binds HSP47 and is transferred to the Golgi apparatus where KDELR2 binds HSP47 and leads to dissociation of HSP47 from procollagen. HSP47 is recycled back to the ER. Procollagen is further processed in the Golgi and secreted into the extracellular matrix (ECM) as tropocollagen. In mutant KDELR2 cells, KDELR2 is unable to bind HSP47. HSP47 cannot dissociate from procollagen and is retained in the Golgi and not secreted into the extracellular matrix [Color figure can be viewed at wileyonlinelibrary.com]
OI is a clinically and genetically heterogeneous connective tissue disorder hallmarked by increased susceptibility to bone fractures and is most commonly caused by monoallelic de novo pathogenic variants in COL1A1 (MIM: 120150) or COL1A2 (MIM: 120160). However, biallelic variants in genes involved in collagen type I biosynthesis have been frequently reported in consanguineous populations (Essawi et al., 2018; van Dijk et al., 2020; Van Dijk & Sillence, 2014). Currently, 20 different types of OI are identified in Online Mendelian Inheritance in Man (OMIM) (Amberger et al., 2015) with variable severity and phenotypic spectrum affecting primarily the skeletal system, although neurodevelopmental and other systemic complications have been observed in some autosomal recessive forms (e.g., MESD, MIM: 618644) (Moosa et al., 2019).
Here, we describe three affected children from two unrelated consanguineous families in order to expand the phenotype and further support the role of KDELR2 in AR OI. Informed consent, including consent to publish photographs, was obtained from the childrens' parents and institutional review board approval was obtained. All three children were clinically diagnosed with progressively deforming OI and neurodevelopmental delay. Three children had motor delay and two of three children had speech delay. The detailed clinical features of each patient are described in Table 1. Pedigrees, radiographs, and brain magnetic resonance images (MRIs) are shown in Figure 2. Common features observed in the affected patients include musculoskeletal abnormalities, including short stature and failure to thrive, Wormian bones, bowed limbs, chest deformity, hypotonia, joint hypermobility, and dysmorphic facies (Figure 2). Family 1 consists of two affected children, a boy and a girl (P1, P2), born to consanguineous (first cousins) parents of Pakistani origin. Both patients have marked motor delay with inability to walk independently at 6 years and 2 years 8 months of age, respectively. The older child crawls as a means of ambulation and has never walked. He has had four fractures in his lifetime, the last at 4 years of age. The younger sister has not had any documented fracture to date at 2 years and 8 months of age. She is not independently ambulatory but can take few steps with great support. In addition, she has speech delay with the first word spoken recently at 2 years of age. Common dysmorphic features in both siblings include epicanthus inversus, deep, sunken eyes, short neck, and thin, sparse hair. Brain MRI obtained from P1 at 6 years of age shows brachycephaly but is otherwise unremarkable (Figure 2(e)). P3 was born to consanguineous first cousin Turkish parents with two prior miscarriages of unknown etiology. He was prenatally suspected to have OI due to ultrasounds showing abnormal bone structure. The patient has one unaffected sibling who does not carry the variant (Figure 2). The patient's first fracture occurred at 21 days of age (Figure 2(d)). Additional features observed include dentinogenesis imperfecta, blue sclera, scoliosis, and neurodevelopmental delay involving both motor and speech. Independent ambulation and speech emerged at 2 years of age; currently at age 4 years he is comparable to his neurotypical peers. Therefore, although he may have had early childhood developmental delay with speech and motor affected, he has caught up to his peers and it is therefore difficult to dissect if the KDELR2 variant identified contributes to the speech delay observed or if it is due to lack of exposure or other unidentified genetic etiologies. Additionally, at 4 years of age, he is currently independently ambulatory. Neurodevelopmental cognition (developmental quotient/intelligence quotient) of all three patients is unknown nor has formal testing been performed in any of the patients.
TABLE 1.
Comparison of clinical features in patients with KDELR2‐related osteogenesis imperfecta
| This study | Published in van Dijk et al., 2020 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Individual | P1 | P2 | P3 | P1 | P2‐1 | P2‐2 | P3 | P4‐1 | P4‐2 |
| Ethinicity | Pakistani | Pakistani | Turkish | Pakistani | Dutch | Dutch | Spanish | Dutch | Dutch |
| GeneVariant (NM_006854) | c.13C > T (p.Arg5Trp) hmz | c.13C > T (p.Arg5Trp) hmz | c.485A > G (p.Tyr162Cys) hmz | c.448dupC (p.His150fs*24), hmz | c.34C > G (p.His12Asp), hmz | N/A | c.398C > T (p.Pro133Leu), hmz | c.34C > G (p.His12Asp), c.360G > A (p.Trp120*) | c.34C > G (p.His12Asp), c.360G > A (p.Trp120*) |
| Age, first assessement | 4 years 5 months | 15 months | 24 days | 5 years | 29 years | N/A | 1.5 mo | 24 weeks of gestation | N/A |
| Age, last assessment | 6 years | 2 years 8 months | 4 years 3 months | 14 years | 39 years | N/A | 43 years | N/A | N/A |
| OFC, first assessment (cm, Z‐score) | 47 cm (−2.5) | 43 cm (−2.3) | N/A | ||||||
| Height, last assessment (cm, Z‐score) | 77 cm (−3.1) | 66.5 cm (−5.2) | 83.5 cm (−3) | 130 (−4.0) | 121 (N/A) | 115 (N/A) | 138 (N/A) | N/A | N/A |
| Weight, last assessment (kg, Z‐score) | 10 kg (−3.9) | 7 kg (−4.1) | 10.2 kg (−3.5) | N/A | N/A | N/A | N/A | N/A | N/A |
| OFC, last assessment (cm, Z‐score) | N/A | N/A | 50.5 cm (1.1) | N/A | N/A | N/A | N/A | N/A | N/A |
| Prenatal fractures | U | U | + | − | − | − | − | + | + |
| Wormian bones | + | + | + | − | U | U | + | N/A | N/A |
| Age at first fracture | 1 year | N/A | 21 days | 40 | 32 | U | 24 | In utero | In utero |
| Estimated number of sustained fractures | 4 | 0 | >2 | N = 12 | N = 26 | N = 15 aged 25 years | N > 30 | N/A | N/A |
| Last sustained fracture | 4 years 5 months | N/A | 4 years | right femur age 10 years | right femur age 28 and right femoral neck age 29 | U | right femur, age 37 | N/A | N/A |
| Color of sclera | White | Blue | Blue | White | White | White | White | U | U |
| Dentinogenesis imperfecta | − | + | + | − | − | − | − | N/A | N/A |
| Hypermobility of joints | + | + | + | + | + | U | + | N/A | N/A |
| Hearing impairment | − | − | − | − | − | − | − | N/A | N/A |
| Chest deformity | Barrel shaped with pectus excavatum | Bell shaped | Barrel shaped, asymmetrical mild carinatum, increased A‐P diameter | Barrel shaped with pectus excavatum | Barrel shaped with pectus excavatum | + | Bell shaped | − | − |
| Cardiac abnormalities | − | − | mild mitral and tricuspid regurgitation | − | − | + | U | − | − |
| Vertebral fractures | + | − | + | + | + | U | + | N/A | N/A |
| Scoliosis | − | + | + | − | + | + | + | − | − |
| Bowing of upper extremities | + | − | − | − | + | + | + | − | − |
| Bowing of lower extremities | + | − | + | − | + | + | + | + | + |
| Shortening of upper extremities | − | − | − | − | + | + | + | + | + |
| Shortening of lower extremities | + | − | − | − | + | + | + | + | + |
| Surgical correction for bone deformation | − | − | − | + | + | + | + | N/A | N/A |
| Age at BP treatment (start/end) | 4 years 8 months | N/A | 2‐month‐old /still every 6 months | 5/9 years | 29/37 years | N/A | 39/42 years | N/A | N/A |
| BP type and dosage | Pamidronate 0.5 mg/kg monthly for 8 months | N/A | Pamidronate 0.5 mg/kg every 6 months | Neridronate 2 mg/kg body weight, IV, every 3 months | Alendronic acid 70 mg, weekly | N/A | Zoledronate 5 mg, IV, yearly | N/A | N/A |
| DEXA scores before BP treatment | N/A | N/A | N/A | Z score: *L2–L4, −3.7; *TBLH, −1.9 | Z score: *L2–L4, −3.09; *femoral neck (R), −2.05; *trochanter, −2.50 | N/A | U: severe osteoporosis on X‐rays | N/A | N/A |
| DEXA scores after BP treatment | N/A | N/A | N/A | Z score: *L2–L4, −2.4 | Z‐score: *L1–L4, −3.4 | N/A | U | N/A | N/A |
| Calcium—level (mmol/L) | 9.9 | 10.5 | 9.1 | 2.36 | 2.55 | U | 2.49 | N/A | N/A |
| Alkaline phosphatase at first visit (U/L) | N/A | 592 | 261 (normal for age) | 201 | 69 | U | U | N/A | N/A |
| Alkaline phosphatase at last visit (U/L) | 183 | 368 | 159 (normal for age) | 198 | 56 | U | U | N/A | N/A |
| Vascular abnormalities | − | − | − | N/A | N/A | N/A | N/A | N/A | N/A |
| Skin/nail | − | − | − | N/A | N/A | N/A | N/A | N/A | N/A |
| MRI brain | Brachycephaly, otherwise normal | N/A | N/A, CT head was normal | N/A | N/A | N/A | N/A | N/A | N/A |
| Mobility | Crawls | Walks with much support | Walks independently | mobile | Wheelchair since age of 4.5 years | Wheelchair | Wheelchair since age of 18 years | N/A | N/A |
| Intelligence | U | U | U | Normal | Normal | Normal | Normal | N/A | N/A |
| Hypotonia | + | + | + | N/A | N/A | N/A | N/A | N/A | N/A |
| Muscle weakness | Mild | Mild | − | N/A | N/A | N/A | N/A | N/A | N/A |
| Speech delay | − | + | + | N/A | N/A | N/A | N/A | N/A | N/A |
| Motor delay | + | + | + | N/A | N/A | N/A | N/A | N/A | N/A |
| Family miscarriages | − | − | 2 | N/A | N/A | N/A | N/A | N/A | N/A |
Abbreviations: hmz, homozygous; U, unknown; N/A, not applicable; BP, bisphosphonate; TBLH, total body less head.
FIGURE 2.
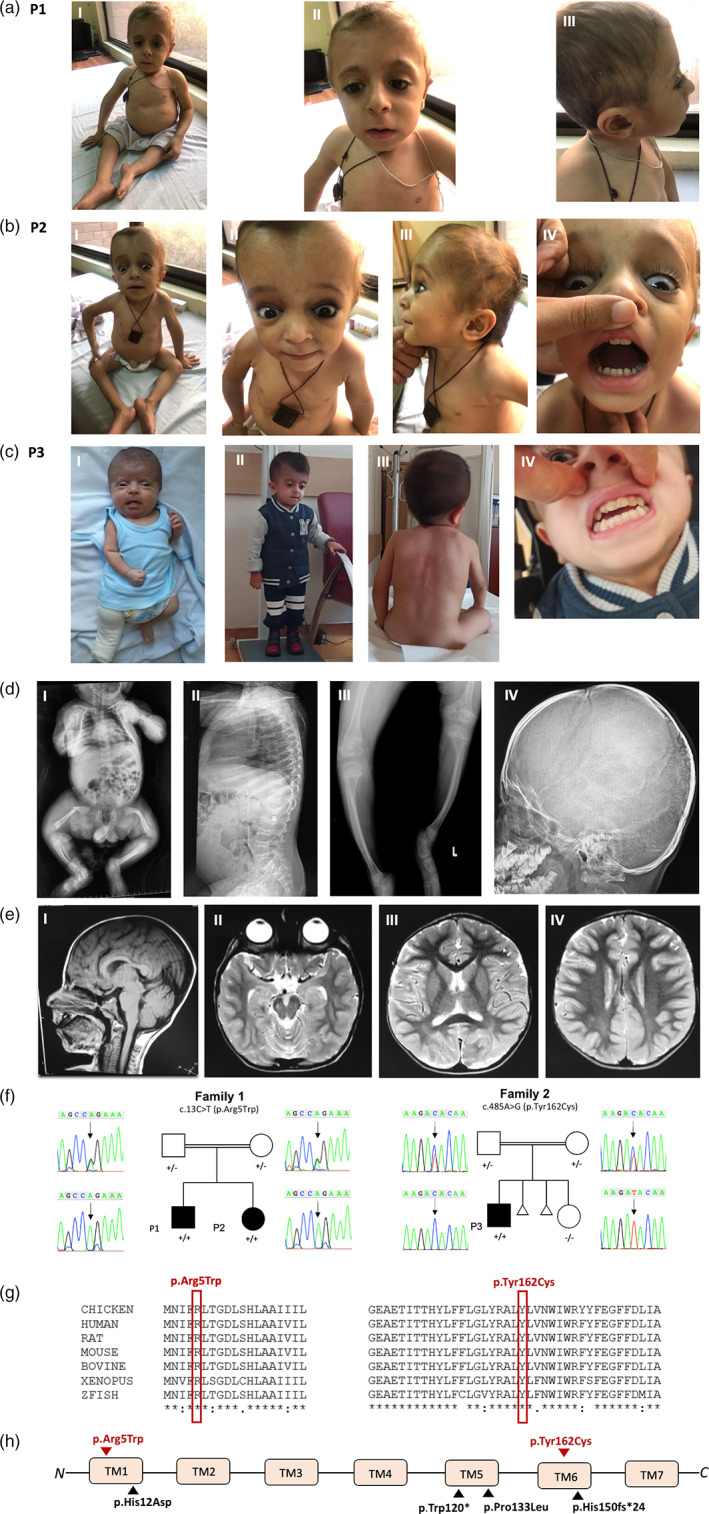
Three affected patients with KDELR2‐related osteogenesis imperfecta from two consanguineous families. (a) Photographs of patient P1 showing short stature, barrel shaped chest (I), sunken eyes, epicanthus inversus (II), and sparse thin hair (III). (b) Photographs of P2 showing short stature, barrel shaped chest (I), blue sclera (II), sunken eyes secondary to molding of the soft cranium (II), thin sparse hair (III), and dentinogenesis imperfecta (IV). (c) Photographs of P3 showing infantile short stature a right leg cast following a pathological femoral fracture (I), current short stature at age 4 years (II), scoliosis (III), and dentinogenesis imperfecta (IV). (d) Radiographs of affected subjects depicting infantile femoral fracture from P3 (I), vertebral compression fractures and platyspondyly from patient P1 (II), short bowed limbs from P1 (III), and Wormian bones from P1 (IV). (e) Brain MRI sections from P1 obtained at 6 years of age. (I) Sagittal T1 showing normal brain appearance. (II) Axial T2 showing brachycephaly. (III and IV) Axial T2 images showing age‐appropriate myelination. (f) Sanger segregation of KDELR2 variants in family 1 and 2. (g) Conservation of amino acid residues across species for both variants. (h) Location of current (red) and previously reported (black) KDELR2 pathogenic variants. All identified variants to date affect transmembrane domains (TMs) 1, 5, and 6 of the KDELR2 protein product [Color figure can be viewed at wileyonlinelibrary.com]
Family‐based exome sequencing (ES) with rare variant analysis was performed in both families followed by Sanger segregation for the identified variants as described before (Efthymiou et al., 2019; Manole et al., 2020). All three affected subjects were found to have homozygous variants in KDELR2 (GenBank: NM_006854.3). P1 and P2 have a c.13C > T (p.Arg5Trp) missense variant and P3 has a c.485 A>G (p.Tyr162Cys) missense variant (Table 2). Neither variant is present in gnomAD and both variants are predicted to be pathogenic via in silico prediction analysis (CADD v1.4, MutationTaster, PolyPhen, SIFT). All current and previously reported variants affect highly conserved amino acids located in the KDELR2 transmembrane domains (Figure 1(h)).
TABLE 2.
Summary of pathogenic KDELR2 variant alleles
| Family | Individual | Ethnicity | Position (hg19) | Nucleotide change | Protein change | Zygosity | gnomAD allele count | REVEL score | CADD score | ACMG classification |
|---|---|---|---|---|---|---|---|---|---|---|
| This study | ||||||||||
| 1 | P1 | Pakistani | Chr7:6523676 G > A | c.13C > T | p.Arg5Trp | hmz | 0 htz, 0 hmz | 0.64 | 35 | PP1, PM2 |
| 1 | P2 | Pakistani | Chr7:6523676 G > A | c.13C > T | p.Arg5Trp | hmz | 0 htz, 0 hmz | 0.64 | 35 | PP1, PM2 |
| 2 | P3 | Turkish | Chr7:6505821 T > C | c.485A > G | p.Tyr162Cys | hmz | 0 htz, 0 hmz | 0.576 | 32 | PM2 |
| van Dijk et al., 2020 | ||||||||||
| 1 | P1 | Pakistani | Chr7:6505858 G > GG | c.448dupC | p.His150fs*24 | hmz | 0 htz, 0 hmz | — | — | PM2 |
| 2 | P2‐1 | Dutch | Chr7:6523655 G > C | c.34C > G | p.His12Asp | hmz | 0 htz, 0 hmz | 0.776 | 28 | PP1, PM2 |
| 2 | P2‐2 | Dutch | Chr7:6523655 G > C | c.34C > G | p.His12Asp | hmz | 0 htz, 0 hmz | 0.776 | 28 | PP1, PM2 |
| 3 | P3 | Spanish | Chr7:6505908 G > A | c.398C > T | p.Pro133Leu | hmz | 0 htz, 0 hmz | 0.863 | 30 | PM2 |
| 4 | P4‐1 | Dutch | Chr7:6523655 G > CChr7:6505946 C > T | c.34C > G c.360G > A | p.His12Asp p.Trp120* | cmp htz | 0 htz, 0 hmz 0 htz, 0 hmz | 0.776; — | 2841 | PP1, PM2 |
| 4 | P4‐2 | Dutch | Chr7:6523655 G > CChr7:6505946 C > T | c.34C > G c.360G > A | p.His12Asp p.Trp120* | cmp htz | 0 htz, 0 hmz 0 htz, 0 hmz | 0.776; — | 2841 | PP1, PM2 |
Abbreviations: CADD, Combined Annotation‐Dependent Depletion; cmp htz, compound heterozygous; hmz, homozygous; htz, heterozygous; REVEL, rare exome variant ensemble learner.
The role of KDELR2 in human development has not been well established until this point. However, animal studies of KDELR2 loss of function (LoF) demonstrate an essential role in embryonic development. The characterization of Kdelr2‐LoF mice by the International Mouse Phenotypic Consortium (IMPC)(Dickinson et al., 2016) scored several statistically significant phenotypes, including preweaning lethality, decreased animal size, bone structural abnormalities, abnormalities in head shape and size, facial dysmorphology, and abnormal body wall structure (Table 3), features which overlap with human biallelic pathogenic KDELR2 variants.
TABLE 3.
International mouse phenotyping consortium Kdelr2 LOF phenotypes
| Phenotype | Zygosity | Life stage | p‐value |
| Abnormal embryo size | htz, hmz | E9.5, E18.5 | 0.00 |
| Abnormal head size | hmz | E18.5 | 0.00 |
| Abnormal heart looping | htz | E.9.5 | 0.00 |
| Increased exploratory behavior | htz | early adult | 1.17 × 10−7 |
| Abnormal bone mineralization | htz | early adult | 1.39 × 10−6 |
| Abnormal facial morphology | hmz | E18.5 | 0.00 |
| Preweaning lethality, incomplete penetrance | hmz | early adult | 0.00 |
| Abnormal head shape | hmz | E18.5 | 0.00 |
| Abnormal bone structure | htz | early adult | 1.75 × 10−7 |
| Abnormal body wall morphology | hmz | E18.5 | 0.00 |
Abbreviations: hmz, homozygous; htz, heterozygous.
In conclusion, the data presented here support the role of KDELR2 in AR OI and expand the phenotypic spectrum of recessive KDELR2‐related AR OI first described by van Dijk et al. (2020) to include neurodevelopmental disorders such as motor and speech delay, as well as blue sclerae, dentinogenesis imperfecta, and hypotonia. However, motor delay and hypotonia are common features of OI and one reason they have not previously been reported may have been due to the small sample size of patients with this newly identified genetic etiology of OI. Additionally, it is unclear if the speech delay seen in early development is related to KDELR2, lack of exposure, or some other unidentified etiology. Noteworthy, the phenotypic spectrum of IMPC‐generated Kdelr2‐LoF mice overlaps with human KDELR2‐OI patients and provides a model system in which to better characterize this type of AR OI. Combined data from humans and mouse models could lead to further studies investigating the pathologic mechanism of KDELR2‐related OI and to the development of novel disease treatments. With the current rate of novel disease gene discovery and pathogenic disease mechanisms, it is expected that more as of yet undiscovered molecular causes of OI exist. Therefore, it becomes important to perform family‐based genetic analysis in these molecular undiagnosed patients in order to work toward a diagnosis with implications for prognosis, family planning, and potential treatments to mitigate the clinical consequences of this deforming disorder.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest.
AUTHORS' CONTRIBUTIONS
Stephanie Efthymiou and Isabella Herman performed data collection, analysis, manuscript drafting, and designed the study. Fatima Rahman, Najwa Anwar, Shazia Maqbool, Reza Maroofian, Janice Yip, Tadahiro Mitani, Daniel G. Calame, Jill V. Hunter, V. Reid Sutton, Elif Yilmaz Gulec, Ruizhi Duan, Jawid M. Fatih, Dana Marafi, Davut Pehlivan, Shalini N. Jhangiani, Richard A. Gibbs and Jennifer E. Posey organized participant recruitment and performed data collection. James R. Lupski and Henry Houlden sponsored the research, assisted in study design, and supervised the laboratory studies and clinical integration. All coauthors assisted with manuscript preparation and writing and all coauthors approved of the final manuscript.
ACKNOWLEDGMENTS
This study was supported in part by the US National Human Genome Research Institute (NHGRI) and National Heart Lung and Blood Institute (NHBLI) to the Baylor‐Hopkins Center for Mendelian Genomics (BHCMG, UM1 HG006542, J.R.L); NHGRI grant to Baylor College of Medicine Human Genome Sequencing Center (U54HG003273 to R.A.G.), US National Institute of Neurological Disorders and Stroke (NINDS) (R35NS105078 to J.R.L.) and Muscular Dystrophy Association (MDA) (512848 to J.R.L.). D.M. is supported by a Medical Genetics Research Fellowship Program through the United States National Institute of Health (T32 GM007526‐42). T.M. is supported by the Uehara Memorial Foundation. D.P. is supported by a Clinical Research Training Scholarship in Neuromuscular Disease partnered by the American Academy of Neurology (AAN), American Brain Foundation (ABF) and Muscle Study Group (MSG), and NIH—Brain Disorders and Development Training Grant (T32 NS043124‐17). J.E.P. was supported by NHGRI K08 HG008986. Family 1 was collected as part of the SYNaPS Study Group collaboration funded by the Wellcome Trust and strategic award (Synaptopathies) funding (WT093205 MA and WT104033AIA) and research was conducted as part of the Queen Square Genomics group at University College London, supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre. HH is funded by the MRC (MR/S01165X/1, MR/S005021/1, G0601943), the National Institute for Health Research University College London Hospitals Biomedical Research Centre, Rosetree Trust, Ataxia UK, MSA Trust, Brain Research UK, Sparks GOSH Charity, Muscular Dystrophy UK (MDUK), Muscular Dystrophy Association (MDA USA).
Efthymiou, S., Herman, I., Rahman, F., Anwar, N., Maroofian, R., Yip, J., Mitani, T., Calame, D. G., Hunter, J. V., Sutton, V. R., Yilmaz Gulec, E., Duan, R., Fatih, J. M., Marafi, D., Pehlivan, D., Jhangiani, S. N., Gibbs, R. A., Posey, J. E., SYNAPS Study Group, Maqbool, S., Lupski, J. R., & Houlden, H. (2021). Two novel bi‐allelic KDELR2 missense variants cause osteogenesis imperfecta with neurodevelopmental features. American Journal of Medical Genetics Part A Part A, 185A:2241–2249. 10.1002/ajmg.a.62221
Stephanie Efthymiou and Isabella Herman contributed equally to this study.
Funding information Medical Research Council, Grant/Award Numbers: G0601943, MR/S005021/1, MR/S01165X/1; Muscular Dystrophy Association, Grant/Award Number: 512848; National Human Genome Research Institute, Grant/Award Numbers: BHCMG, UM1 HG006542, K08 HG008986, U54HG00327; National Institute of Neurological Disorders and Stroke, Grant/Award Number: R35NS105078; NIH Clinical Center, Grant/Award Numbers: T32 GM007526‐42, T32 NS043124‐17; Uehara Memorial Foundation; Wellcome Trust, Grant/Award Numbers: WT093205 MA, WT104033AIA
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Amberger, J. S., Bocchini, C. A., Schiettecatte, F., Scott, A. F., & Hamosh, A. (2015). OMIM.Org: Online Mendelian inheritance in man (OMIM[R]), an online catalog of human genes and genetic disorders. Nucleic Acids Research, 43, D789–D798. 10.1093/nar/gku1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capitani, M., & Sallese, M. (2009). The KDEL receptor: New functions for an old protein. FEBS Letters, 583(23), 3863–3871. 10.1016/j.febslet.2009.10.053 [DOI] [PubMed] [Google Scholar]
- Dickinson, M. E., Flenniken, A. M., Ji, X., Teboul, L., Wong, M. D., White, J. K., Meehan, T. F., Weninger, W. J., Westerberg, H., Adissu, H., Baker, C. N., Bower, L., Brown, J. M., Caddle, L. B., Chiani, F., Clary, D., Cleak, J., Daly, M. J., Denegre, J. M., Doe, B., … Murray, S. A. (2016). High‐throughput discovery of novel developmental phenotypes. Nature, 537(7621), 508–514. 10.1038/nature19356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efthymiou, S., Salpietro, V., Malintan, N., Poncelet, M., Kriouile, Y., Fortuna, S., De Zorzi, R., Payne, K., Henderson, L. B., Cortese, A., Maddirevula, S., Alhashmi, N., Wiethoff, S., Ryten, M., Botia, J. A., Provitera, V., Schuelke, M., Vandrovcova, J., SYNAPS Study Group, Walsh, L., … Houlden, H. (2019). Biallelic mutations in neurofascin cause neurodevelopmental impairment and peripheral demyelination. Brain, 142(10), 2948–2964. 10.1093/brain/awz248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essawi, O., Symoens, S., Fannana, M., Darwish, M., Farraj, M., Willaert, A., Essawi, T., Callewaert, B., De Paepe, A., Malfait, F., & Coucke, P. J. (2018). Genetic analysis of osteogenesis imperfecta in the Palestinian population: Molecular screening of 49 affected families. Molecular Genetics & Genomic Medicine, 6(1), 15–26. 10.1002/mgg3.331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manole, A., Efthymiou, S., O'Connor, E., Mendes, M. I., Jennings, M., Maroofian, R., Davagnanam, I., Mankad, K., Lopez, M. R., Salpietro, V., Harripaul, R., Badalato, L., Walia, J., Francklyn, C. S., Athanasiou‐Fragkouli, A., Sullivan, R., Desai, S., Baranano, K., Zafar, F., Rana, N., … Houlden, H. (2020). De novo and bi‐allelic pathogenic variants in NARS1 cause neurodevelopmental delay due to toxic gain‐of‐function and partial loss‐of‐function effects. American Journal of Human Genetics, 107(2), 311–324. 10.1016/j.ajhg.2020.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moosa, S., Yamamoto, G. L., Garbes, L., Keupp, K., Beleza‐Meireles, A., Moreno, C. A., Valadares, E. R., de Sousa, S. B., Maia, S., Saraiva, J., Honjo, R. S., Kim, C. A., Cabral de Menezes, H., Lausch, E., Lorini, P. V., Lamounier, A., Jr, Carniero, T., Giunta, C., Rohrbach, M., Janner, M., … Netzer, C. (2019). Autosomal‐recessive mutations in MESD cause osteogenesis Imperfecta. American Journal of Human Genetics, 105(4), 836–843. 10.1016/j.ajhg.2019.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk, F. S., Semler, O., Etich, J., Köhler, A., Jimenez‐Estrada, J. A., Bravenboer, N., Claeys, L., Riesebos, E., Gegic, S., Piersma, S. R., Jimenez, C. R., Waisfisz, Q., Flores, C. L., Nevado, J., Harsevoort, A. J., Janus, G., Franken, A., van der Sar, A. M., Meijers‐Heijboer, H., Heath, K. E., … Micha, D. (2020). Interaction between KDELR2 and HSP47 as a key determinant in osteogenesis imperfecta caused by bi‐allelic variants in KDELR2 . American Journal of Human Genetics, 107(5), 989–999. 10.1016/j.ajhg.2020.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dijk, F. S., & Sillence, D. O. (2014). Osteogenesis imperfecta: Clinical diagnosis, nomenclature and severity assessment. American Journal of Medical Genetics. Part A, 164A(6), 1470–1481. 10.1002/ajmg.a.36545 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.