

ABSTRACT
Background
GBA mutations are a common risk factor for Parkinson's disease (PD). A recent study has suggested that GBA haplotypes, identified by intronic variants, can affect age at diagnosis of PD.
Objectives
In this study, we assess this hypothesis using long reads across a large cohort and the publicly available Accelerating Medicines Partnership–Parkinson's Disease (AMP‐PD) cohort.
Methods
We recruited a PD cohort through the Remote Assessment of Parkinsonism Supporting Ongoing Development of Interventions in Gaucher Disease study (RAPSODI) and sequenced GBA using Oxford Nanopore technology. Genetic and clinical data on the full AMP‐PD cohort were obtained from the online portal of the consortium.
Results
A total of 1417 participants were analyzed. There was no significant difference in age at PD diagnosis between the two main haplotypes of the GBA gene.
Conclusions
GBA haplotypes do not affect age at diagnosis of PD in the two independent cohorts studied. © 2021 The Authors. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson and Movement Disorder Society
Keywords: Parkinson's, GBA, haplotypes, intronic variants, genetics
Mutations in the GBA gene are an important risk factor for the development of Parkinson's disease (PD).1 The prevalence of GBA mutations in PD varies according to the population studied and whether the analysis includes all coding variants or only the most common; in general it ranges between 5% and 10% of sporadic PD cases.2 The Ashkenazi Jewish population is an exception, with a prevalence of GBA mutations as high as 30% in sporadic PD cases.3 The lifetime risk of development of PD in exonic GBA mutation carriers is estimated at 5% to 30%,4 and it is not clear what factors contribute to this incomplete penetrance. Moreover, reduced activity of the enzyme encoded by GBA, glucocerebrosidase, is observed in PD brains without coding GBA mutations.5
A recent study explored the hypothesis that intronic variants in the GBA gene might contribute to the risk of PD.6 Deep intronic variants are not commonly regarded as pathogenic as they do not result in amino acid changes in proteins. Nonetheless, some deep intronic variants have been linked directly to genetic disorders, such as Gaucher disease.7, 8 Two common haplotypes were identified, differentiated by three intronic single nucleotide polymorphisms in GBA. These correspond to the previously reported 1.1+ and 1.1− haplotypes.9, 10 These haplotypes had a significant effect on age at onset and age at diagnosis of PD.
In this article, we analyzed our cohort of patients with PD and the publicly available Accelerating Medicines Partnership–Parkinson's Disease (AMP‐PD) cohort to try to replicate these findings.
Patients and Methods
Recruitment of Participants
Patients with PD were recruited through the Remote Assessment of Parkinsonism Supporting Ongoing Development of Interventions in Gaucher Disease study (RAPSODI) (http://rapsodistudy.com) an online cohort study that recruits and genotypes people with PD through a dedicated portal (http://pdfrontline.com). After signing an online consent form, participants were assessed remotely, and demographic and clinical information was recorded. Participants provided saliva DNA for analysis. The London Queen Square Research Ethics Committee approved the project.
DNA Extraction and Sequencing
Saliva was collected using the Oragene DNA OG‐500 kit (DNA Genotek), and DNA was extracted according to the manufacturer's protocol. Sequencing of long GBA amplicons was carried out using Oxford Nanopore Technologies as described previously.11 Sequencing data were generated using MinKNOW, basecalled with Guppy (both available via the Nanopore community site; https://community.nanoporetech.com), and aligned to hg38 with NGMLR.12 Variants were called using Clair13 and phased with Whatshap.14 Haplotypes were identified using the R package Haplotypes (R Foundation for Statistical Computing). A detailed pipeline is reported in Figure S1.
AMP‐PD Cohort Data
Clinical and sequencing data were downloaded from the AMP‐PD initiative website (https://amp-pd.org). Full details on data collection can be found on the website. Age at diagnosis, sex, and the GBA gene mutation calls for cases diagnosed as “idiopathic PD” or “Parkinson's disease” in the AMP‐PD cohort were downloaded on July 20, 2020 (version 2019_v1release_1015).
Haplotype Definition
The full GBA gene sequence was analyzed for all participants with PD from both the RAPSODI and AMP‐PD cohorts. Carriers of coding GBA variants were excluded from the analysis. Each GBA allele was assigned to one of two haplotypes.6 Haplotype A was identified by alternate genotype at three intronic variants (rs9628662, rs762488, and rs2009578), whereas haplotype B was identified by the reference genotype at these variants. The minor allele population frequencies in non‐Finnish Europeans in the Genome Aggregation Database (GnomAD) data (https://gnomad.broadinstitute.org) are 0.295, 0.294, and 0.287, respectively. Participants who carried at least one allele that did not fall into this classification or for which quality of the alignment at any of the three positions was not good enough for confident calling were excluded from the analysis.
Statistical Analysis
R version 4.0.2 was used for all statistical analyses. To assess the effect of haplotypes on age at diagnosis of PD, two models were investigated: an additive model and a dominant model of haplotype B. For the additive model, linear regression was used, with the number of alleles carrying haplotype B as the dependent variable. For the dominant model of haplotype B, participants were divided into two groups according to whether they carried at least one allele with haplotype B, and ANOVA was used for the analysis.
Results
Participants and Genotypes
We analyzed 1417 patients, of whom 100 were recruited through RAPSODI, and the remainder were from AMP‐PD. More than 100 unique haplotypes were identified (Figure S2), and the overall allelic frequency of each haplotype was 0.302 for haplotype A and 0.691 for haplotype B. The number of participants carrying each haplotype, genotypes, and mean age at diagnosis of PD are reported in Table 1. Of note, five (0.5%) participants in the AMP‐PD cohort carried at least one allele that was not classifiable in either of the two haplotypes, and were excluded from the analysis. Moreover, five samples in the RAPSODI cohort (5.0%) were not classifiable into one of the two haplotypes. Upon visual inspection of the sequencing data, the quality was not adequate for unequivocal haplotype assignment, and they were thus also excluded. Ethnic backgrounds in the two cohorts are reported in Tables S1 and S2.
TABLE 1.
GBA haplotypes and age at diagnosis of PD
| All ages | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| RAPSODI cohort | AMP‐PD cohort | Merged cohorts (RAPSODI + AMP‐PD) | |||||||
| Haplotype | Number of participants | Mean age at diagnosis | Median age at diagnosis | Number of participants | Mean age at diagnosis | Median age at diagnosis | Number of participants | Mean age at diagnosis | Median age at diagnosis |
| Homozygous haplotype A | 10 | 61.5 | 59.5 | 131 | 60.1 | 61.0 | 141 | 60.2 | 61.0 |
| Heterozygous | 33 | 59.9 | 60.0 | 540 | 59.5 | 61.0 | 573 | 59.5 | 61.0 |
| Homozygous haplotype B | 52 | 61.3 | 61.0 | 641 | 60.1 | 61.0 | 693 | 60.2 | 61.0 |
| Other | 5 | 5 | 10 | ||||||
| Total | 100 | 1317 | 1417 | ||||||
| P value additive model | 0.7292 | 0.5819 | 0.528 | ||||||
| P value dominant haplotype B | 0.7892383 | 0.7981861 | 0.7552578 | ||||||
| Age at diagnosis > 50 years | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| RAPSODI cohort | AMP‐PD cohort | Merged cohorts (RAPSODI + AMP‐PD) | |||||||
| Haplotype | Number of participants | Mean age at diagnosis | Median age at diagnosis | Number of participants | Mean age at diagnosis | Median age at diagnosis | Number of participants | Mean age at diagnosis | Median age at diagnosis |
| Homozygous haplotype A | 10 | 61.5 | 59.5 | 104 | 64.0 | 64.0 | 114 | 63.8 | 64.0 |
| Heterozygous | 29 | 61.8 | 61.0 | 445 | 63.0 | 63.0 | 474 | 62.9 | 63.0 |
| Homozygous haplotype B | 48 | 62.7 | 61.0 | 526 | 63.6 | 63.0 | 574 | 63.5 | 63.0 |
| Other | 4 | 5 | 9 | ||||||
| Total | 91 | 1080 | 1171 | ||||||
| P value additive model | 0.5504 | 0.7416 | 0.6431 | ||||||
| P value dominant haplotype B | 0.7378356 | 0.3714943 | 0.4654556 | ||||||
PD, Parkinson's disease; AMP‐PD, Accelerating Medicines Partnership–Parkinson's Disease.
Haplotypes and Age at Diagnosis of PD
Mean and median age at diagnosis of PD are reported in Table 1 and shown in Figure 1. After considering both an additive model and a dominant effect of haplotype B model, age at diagnosis of PD was not significantly different in the RAPSODI and AMP‐PD cohorts separately. There was also no significant difference after merging the two cohorts together (P value > 0.3 for both the additive model and dominant haplotype B model).
FIG. 1.
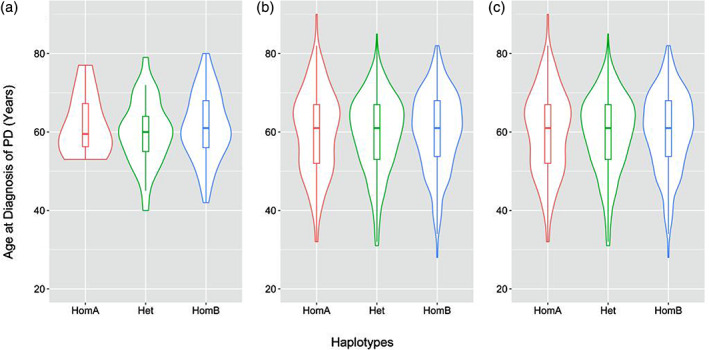
Distribution of age at diagnosis of PD symptoms by GBA haplotypes. The boxes show the medians, and the hinges are the first and third quartiles (25th and 75th percentiles): (a) RAPSODI cohort, (b) AMP‐PD cohort, and (c) RAPSODI and AMP‐PD cohorts together. RAPSODI, Remote Assessment of Parkinsonism Supporting Ongoing Development of Interventions in Gaucher Disease study;AMP‐PD, Accelerating Medicines Partnership–Parkinson's Disease; Het, heterozygous; HomA, homozygous haplotype A; HomB, homozygous haplotype B; PD, Parkinson's disease.
Because a significant number of participants with early‐onset PD (EOPD) can carry mutations in other PD‐causing genes,15 we repeated the analysis after removing all participants with an age at diagnosis younger than 50. Following this adjustment, there were 91 participants in the RAPSODI cohort and 883 participants in the AMP‐PD cohort. Still, no significant effect of the two GBA haplotypes on age at diagnosis of PD was observed. Data on this additional analysis are reported in Table S1.
Discussion
In this article, we attempted to validate the recent report that common haplotypes, identified by deep intronic variants in the GBA gene, could affect age at diagnosis of PD.6 This hypothesis is intriguing as it could help explain the reduced penetrance of GBA mutations and the role of intronic variants in the pathogenesis of PD.
To this end, we analyzed our original cohort, generated through the RAPSODI portal, and the publicly available AMP‐PD cohort. We investigated both an additive effect of the haplotypes and a dominant effect of haplotype B, but did not observe any effect of haplotypes on age at diagnosis of PD.
Both the RAPSODI and AMP‐PD cohorts included some participants who received a diagnosis of PD earlier than age 50 and would thus be classified as EOPD. Because a significant number of patients with EOPD can carry variants in other PD‐causing genes, we repeated the analysis after excluding all patients with EOPD. We still did not see any significant differences in age of onset between the different haplotypes.
The RAPSODI and AMP‐PD cohorts are similar in their ethnic profiles. In RAPSODI, 96% of participants identified themselves as “White UK,” and in the AMP‐PD cohort 93% of participants identified as “White.” The remarkably similar minor allele frequencies in our cohort to the European GNOMAD samples support this ethnic classification. It is possible that the cohort studied by Schierding et al.14 has a different balance of ethnic backgrounds, which might explain in part the discrepancy of results, although this information was not provided. Moreover, the inclusion of EOPD in the article by Schierding et al. might have influenced the results.
One limitation of our study is that we could not assess age at onset of PD symptoms, which had also been reported as variable by haplotype, as this was not captured in the RAPSODI and AMP‐PD cohorts.
Our study does not exclude a possible role for intronic variants. Although it is true that the majority of alleles could be grouped into one of the two main haplotypes according to their genotypes in three deep intronic variants, more than 50 unique intronic haplotypes were identified (Figure S2), and the role of each single intronic variant in PD might extend beyond that of these haplotypes and merits further study.
Conclusions
In this study, we were not able to confirm a role for common GBA haplotypes in determining age at diagnosis of PD.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript: A. Writing of the First Draft, B. Review and Critique.
M.T.: 1A, 2B, 2C, 3A
C.P.: 1A, 3B
X.C.: 1C, 2C, 3B
A.H.: 1C, 3B
C.L.: 1C, 3B
S.M.: 3B
S.K.: 1C, 3B
M.E.: 3B
F.J.S.: 3B
A.H.V.S.: 1A, 3B
Financial Disclosures
M.T. is a recipient of a student bourse from University College London. C.P., A.H., C.L., and S.K. are employees of University College London. S.M. works for the NHS. X.C. and M.E. are employees of Illumina Inc. F.J.S. is an employee of Baylor College of Medicine. A.H.V.S. is an employee of University College London and a consultant to Kyowa.
Supporting information
Appendix S1: Supporting information
Acknowledgments
Data used in the preparation of this article were obtained from the Accelerating Medicines Partnership–Parkinson's Disease (AMP PD) Knowledge Platform. For up‐to‐date information on the study, visit https://www.amp-pd.org. AMP PD, a public‐private partnership, is managed by the Foundation for the National Institutes oh Health (FNIH) and funded by Celgene, GlaxoSmithKline (GSK), The Michael J. Fox Foundation for Parkinson's Research, the National Institute of Neurological Disorders and Stroke, Pfizer, Sanofi, and Verily. Clinical data and biosamples used in preparation of this article were obtained from the Fox Investigation for New Discovery of Biomarkers (BioFIND), the Harvard Biomarker Study (HBS), the Parkinson's Progression Markers Initiative (PPMI), and the Parkinson's Disease Biomarkers Program (PDBP). BioFIND is sponsored by The Michael J. Fox Foundation for Parkinson's Research with support from the National Institute for Neurological Disorders and Stroke. The BioFIND investigators have not participated in reviewing the data analysis or content of the article. For up‐to‐date information on the study, visit michaeljfox.org/biofind. HBS and is a collaboration of HBS investigators (full list of HBS investigator found at https://www.bwhparkinsoncenter.org/biobank) and funded through philanthropy and National Institutes of Health and non–National Institutes of Health funding sources. The HBS Investigators have not participated in reviewing the data analysis or content of the article. PPMI, a public‐private partnership, is funded by the Michael J. Fox Foundation for Parkinson's Research and funding partners (the full names of all of the PPMI funding partners can be found at www.ppmi-info.org/fundingpartners). The PPMI investigators have not participated in reviewing the data analysis or content of the article. For up‐to‐date information on the study, visit www.ppmi-info.org. The PDBP consortium is supported by the National Institute of Neurological Disorders and Stroke at the National Institutes of Health. A full list of PDBP investigators can be found at https://pdbp.ninds.nih.gov/policy. The PDBP Investigators have not participated in reviewing the data analysis or content of the article.
Funding agencies: This study was funded by the Medical Research Council (MRC, MR/M006646/1), Cure Parkinson Trust, Kattan Trust, and an investigator‐initiated research grant (IIR‐GBR‐001110) provided by Shire International GmbH, a member of the Takeda group of companies.
Relevant conflicts of interest/financial disclosures: X.C. and M.E. are employees of Illumina Inc.
References
- 1.Blauwendraat C, Nalls MA, Singleton AB. The genetic architecture of Parkinson's disease. Lancet Neurol 2020;19(2):170–178. 10.1016/S1474-4422(19)30287-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schapira AHV. Glucocerebrosidase and Parkinson disease: recent advances. Mol Cell Neurosci 2015;66:37–42. https://www.sciencedirect.com/science/article/pii/S1044743115000421#bb0030. Accessed November 13, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neumann J, Bras J, Deas E, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson's disease. Brain 2009;132(7):1783–1794. 10.1093/brain/awp044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O'Regan G, DeSouza R‐M, Balestrino R, Schapira AH. Glucocerebrosidase mutations in Parkinson disease. J Parkinsons Dis 2017;7(3):411–422. 10.3233/JPD-171092 [DOI] [PubMed] [Google Scholar]
- 5.Gegg ME, Schapira AHVV. The role of glucocerebrosidase in Parkinson disease pathogenesis. FEBS J 2018;285(19):3591–3603. 10.1111/febs.14393 [DOI] [PubMed] [Google Scholar]
- 6.Schierding W, Farrow S, Fadason T, et al. Common variants co‐regulate expression of GBA and modifier genes to delay Parkinson's disease onset. Mov Disord 2020;35(8):1346–1356. 10.1002/mds.28144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vaz‐Drago R, Custódio N, Carmo‐Fonseca M. Deep intronic mutations and human disease. Hum Genet 2017;136(9):1093–1111. 10.1007/s00439-017-1809-4. [DOI] [PubMed] [Google Scholar]
- 8.Malekkou A, Sevastou I, Mavrikiou G, et al. A novel mutation deep within intron 7 of the GBA gene causes Gaucher disease. Mol Genet Genomic Med 2020;8(3):e1090. 10.1002/mgg3.1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zimran A, Gelbart T, Beutler E. Linkage of the PvuII polymorphism with the common Jewish mutation for Gaucher disease. Am J Hum Genet 1990;46(5):902–905. [PMC free article] [PubMed] [Google Scholar]
- 10.Beutler E, West C, Gelbart T. Polymorphisms in the human glucocerebrosidase gene. Genomics 1992;12(4):795–800. 10.1016/0888-7543(92)90311-F [DOI] [PubMed] [Google Scholar]
- 11.Leija‐Salazar M, Sedlazeck FJ, Toffoli M, et al. Evaluation of the detection of GBA missense mutations and other variants using the Oxford Nanopore MinION. Mol Genet Genomic Med 2019;7(3):e564. 10.1002/mgg3.564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sedlazeck FJ, Rescheneder P, Smolka M, et al. Accurate detection of complex structural variations using single‐molecule sequencing. Nat Methods 2018;15(6):461–468. 10.1038/s41592-018-0001-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo R, Wong C‐L, Wong Y‐S, et al. Exploring the limit of using a deep neural network on pileup data for germline variant calling. Nat Mach Intell 2020;2(4):220–227. 10.1038/s42256-020-0167-4 [DOI] [Google Scholar]
- 14.Martin M, Patterson M, Garg S, et al. WhatsHap: fast and accurate read‐based phasing. bioRxiv 2016;085050. 10.1101/085050 [DOI] [Google Scholar]
- 15.Alcalay RN, Caccappolo E, Mejia‐Santana H, et al. Frequency of known mutations in early‐onset Parkinson disease: implication for genetic counseling: the consortium on risk for early onset Parkinson disease study. Arch Neurol 2010;67(9):1116–1122. 10.1001/archneurol.2010.194 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting information