

Abstract
Molecular-level features of tumours can be tracked via single-cell analyses of circulating tumour cells (CTCs). Yet single-cell measurements of protein expression for rare CTCs are hampered by the presence of a large number of non-target cells. Here, we show that the antibody-mediated labelling of intracellular proteins in the nucleus, mitochondria, and cytoplasm of human cells with magnetic nanoparticles enables the analysis of target proteins at the single-cell level by sorting the cells according to their nanoparticle content in a microfluidic device with cell-capture zones sandwiched between arrays of magnets. We used the magnetic labelling and cell-sorting approach to track the expression of therapeutic protein targets in CTCs isolated from blood samples of mice with orthotopic prostate xenografts and from patients with metastatic castration-resistant prostate cancer. We also show that mutated proteins that are drug targets or markers of therapeutic response can be directly identified in CTCs and analysed at the single-cell level, and used to predict how mice with drug-susceptible and drug-resistant pancreatic tumour xenografts respond to therapy.
The liquid biopsy – a replacement for invasive tissue biopsies where tumour-derived material is collected from the blood - is an emerging approach with great promise for the non-invasive management of cancer. Circulating tumour cells (CTCs) are important targets for liquid biopsy as they permit the biology and heterogeneity of a tumour to be interrogated1. CTCs are rare cells shed from primary and metastatic tumour sites into the circulation and are often present at levels as low as one cell per milliliter of blood. Substantial effort has focused on the isolation and counting of CTCs based on their distinctive characteristics, such as surface protein expression2–5, size6–8, dielectric properties9, and invasiveness10, 11. However, to make liquid biopsy-based measurements focused on CTCs clinically actionable, it is imperative to move beyond enumeration of these cells and towards the collection of information that informs patient treatment. Evaluating expression levels and altered proteoforms for proteins that are targets of cancer therapeutics within CTCs would offer an important new capability for liquid biopsy testing. A key advantage of this type of noninvasive testing is that it can be applied in situations where tissue biopsy is not applicable (e.g. metastatic tumours). Measuring the expression of altered proteoforms in CTCs is a challenging goal, however, given the heterogeneity and rarity of these cells in blood samples. Ensemble measurements of intracellular proteins have been performed using conventional methods such as flow cytometry, immunoblotting and immunohistochemistry12. CTCs have been isolated with FACS and analysed for their protein content with multi-parameter imaging cytometry13 and microwestern blot arrays can be used for multiplexed protein analysis14, but neither approach has been used to evaluate the molecular level properties of tumours related to therapeutic response. Immunohistochemical assays are used in the clinic but are limited to few proteins owing to the spectral imaging limitations imposed by conventional filter sets. In addition, the low abundance of many intracellular proteins makes their detection using this approach problematic15.
The majority of protein assays that have been applied to single cells are single-analyte immunoassays; however, newer formats have improved multiplexing using fluorescent proteins16, DNA-barcoded antibodies/barcode sequencing17–20 and metal-isotope labeled antibodies for mass cytometry applications21. In addition, single-molecule enzyme-linked immunosorbent assays (SiMOA)22 and plasmonic ELISA23 have permitted the analysis of proteins with remarkable sensitivities. However, these methods are primarily limited to the analysis of free18–20, 23 and cell-surface proteins17, and require on-chip cell lysis24 or complex sequencing-based analysis17. Recently, imaging mass cytometry have shown a great potential for multiplexed analysis of proteins in liquid biopsies from metastatic prostate cancer patients25. Microfluidic western blotting has enabled highly specific single-cell analysis of proteins from low starting cell numbers, particularly when integrated with FACS26, and was recently employed to assay small numbers of patient-derived CTCs27. However, this type of approach requires a complex workflow and separate capture and analysis technologies that may cause phenotypic drift.
Recently, we have developed a magnetic ranking cytometry approach to analyse the expression of cell-surface proteins in rare bloodborne CTCs28. However, the method is not capable of analysing intracellular proteins due to the low magnetic susceptibility of cells achieved after targeting the intracellular proteins with antibodies labeled with magnetic nanoparticles. Herein, we developed magnetic cell labelling reagents that can target intracellular proteins and facilitate magnetic ranking of rare cells according to the expression levels of intracellular proteins. This approach allows the measurement of protein levels within the CTCs of cancer patients that are important indicators of metastatic potential and permits a single-cell level detection of protein markers that are important therapeutic targets.
Results and discussion
Our magnetic cell labelling approach exploits nanoparticle-mediated profiling of cancer cells at the single-cell level according to the expression level of a specific intracellular protein. An antibody specific to an intracellular protein target is chemically modified with streptavidin and then labeled with DNA strands via biotin–streptavidin coupling (Fig. 1a). The cells expressing the intracellular protein are fixed and permeabilized to facilitate the internalization of the labelled antibody. After incubation of the cells with the antibody, each DNA strand is hybridized with a pair of complementary DNA probes appended to iron oxide magnetic nanoparticles (MNPs), which triggers the formation of large aggregates of MNPs (Fig. 1b). Due to their large size, the aggregates become trapped inside the cells and consequently enhance their magnetic susceptibility, thus providing a generic and highly efficient way to translate the presence of an intracellular protein into a magnetic content. We have recently developed a similar strategy for the analysis of intracellular mRNAs in rare cells in which the mRNAs were directly targeted with a pair of MNPs-labeled complementary DNA probes to create larger clusters of MNPs29. Herein, we designed a family of magnetic cell labelling reagents to analyse intracellular proteins. Dynamic light scattering (DLS) measurements demonstrated that combining an antibody labeled with a DNA strand and a pair of MNPs-labeled complementary DNA probes produced larger aggregates of MNPs (Supplementary Fig. 1), a key feature enabling the detection of low abundance proteins.
Fig. 1 |. The single-cell intracellular protein analysis approach.
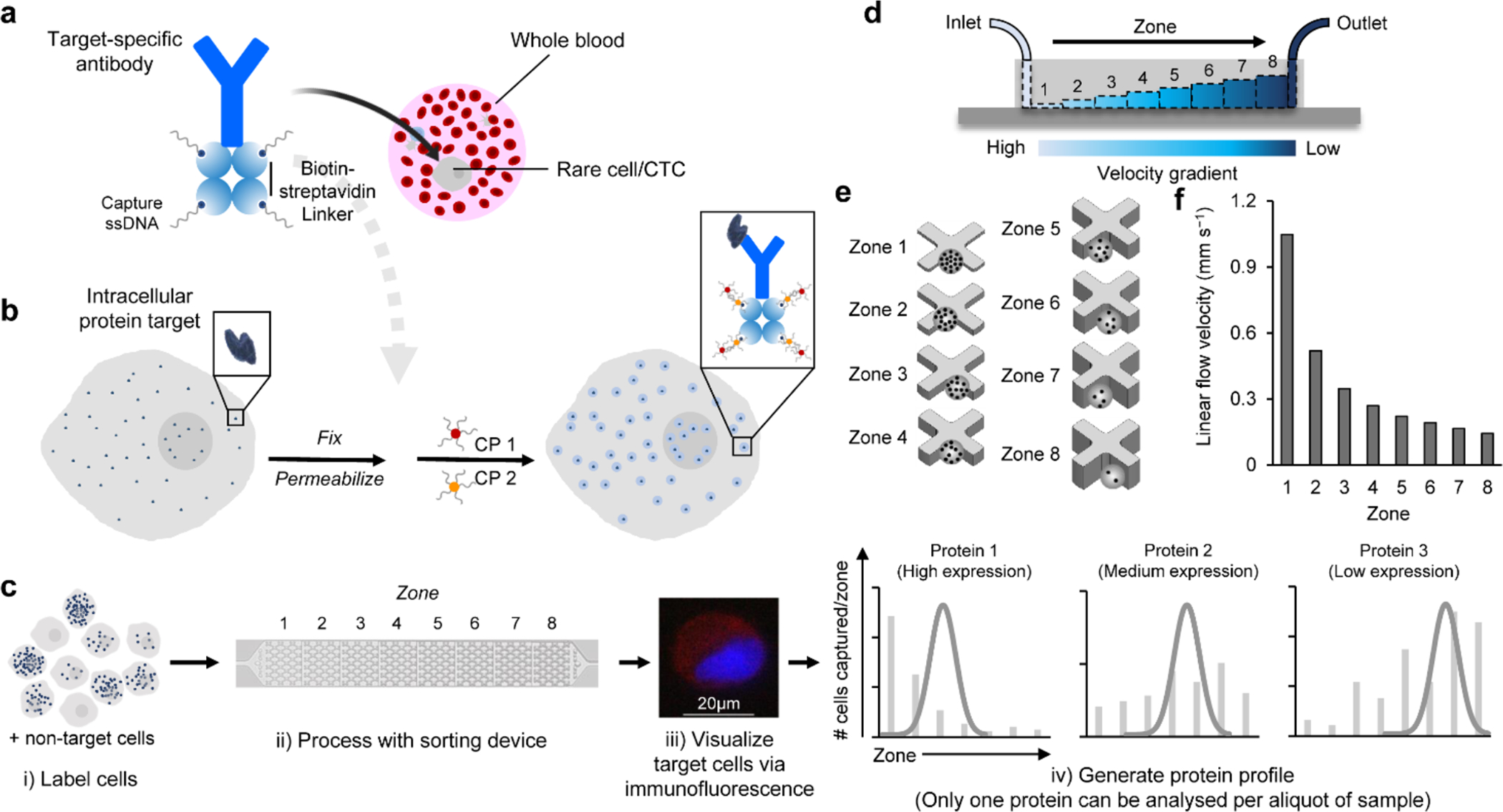
a, An antibody specific to the target intracellular protein is labeled with streptavidin then modified with single stranded DNAs (ssDNAs) via biotin-streptavidin coupling. b, The cells expressing the target intracellular protein are fixed and permeabilized. The cells are incubated with a protein specific antibody modified with ssDNAs. The ssDNAs are then hybridized with two capture probes (CP1 and CP2), which are composed of complementary DNA sequences modified at one end with magnetic nanoparticles (MNPs). Aggregates of MNPs are thus formed and trapped within the cells that express the intracellular protein. c, The cells are sorted using a microfluidic device, immunostained and counted to generate a profile characteristic for the target protein. Only one protein can be analysed per aliquot of sample. d, The microfluidic device features eight sequential zones with increasing heights to facilitate capturing cells with different magnetic content. e, Each zone features X-shaped microstructures to create regions of low flow velocity, thus facilitating cell capture. f, The sequential zones feature different average linear velocities of 1x, 0.52x, 0.35x, 0.27x, 0.22x, 0.19x, 0.17x, 0.14x from zone 1 to 8, respectively at a flow rate of 2 mL h−1.
The cells treated with the magnetic cell labelling reagents are sorted within a microfluidic device sandwiched between two arrays of magnets. The cells are immunostained and visualized within the device. The number of cells and their distribution are used to generate a protein expression profile, allowing for only one protein to be analysed per aliquot of sample (Fig. 1c). The device features eight capture zones with differing linear velocities to capture cells with varying levels of internalized magnetic nanoparticles (Fig. 1d, Supplementary Fig. 2). Owing to the low magnetic susceptibility of magnetic nanoparticles, each capture zone contains microfabricated structures to create localized regions of low flow velocity and enhanced capture dynamics (Fig. 1e). The first zone exhibits the highest linear velocity and thus retains cells with high magnetic content because the retaining magnetic force overcomes the drag force exerted by the locally high flow velocity. The ensuing seven zones exhibit gradually reduced linear velocities (Fig. 1f). This design allows cells with high levels of the intracellular protein target to be captured in the first zone of the device, whereas cells with lower expression levels become sorted in later zones according to protein levels. It is noteworthy that the production of the device masters, a major step in the fabrication of microfluidic devices, is carried out using a stereolithographic 3D printer to facilitate a large-scale production of devices for future commercialization. In addition, we optimized the device design and flow rate and found that using X-shaped microfabricated structures (H: 50 μm, W: 200 μm) and a flow rate of 2 mL h−1 resulted in the highest capture efficiency (Supplementary Fig. 3). Simulations for cell capture within the device are provided in the SI and the minimum number of beads required for cell capture was calculated for each capture zone (Supplementary Table 1). In addition, the probe sequences utilized in this study are provided in Supplementary Table 2.
In a first suite of experiments, we assessed the efficiency of a device designed to facilitate the analysis of an intracellular protein, c-Myc, and its ability to sort cells exhibiting different levels of the protein. The c-Myc protein is a nuclear transcription factor that regulates several cellular processes, including cell growth and proliferation, differentiation, apoptosis and motility30. In prostate cancer, c-Myc (nuclear and cytoplasmic) is upregulated in both androgen receptor-positive and androgen receptor-negative castration-resistant prostate cancers. In addition, c-Myc is involved in prostate cancer progression and its upregulation either at the mRNA or protein level in approximately 30% of patients is correlated with metastasis and biochemical recurrence, which can also affect the outcome of cancer radiotherapy and chemotherapy31. While c-Myc has been regarded as a challenging therapeutic target, recent progress is being made with new protein therapeutics advancing to the clinic32.
Three prostate cancer cell lines with varying phenotypic properties and c-Myc levels were utilized in this proof-of concept study: 22Rv1, PC3, and PC3M33,34. To test the efficiency of our capture and analysis approach, we spiked one millilitre of healthy blood with 100 cells from each cell line and incubated the samples with ssDNA-labeled c-Myc antibody then hybridized the ssDNAs with MNPs-conjugated CP1 and CP2. The cells were sorted within the microfluidic device and their sorting profiles were determined after immunostaining the cells for two epithelial markers, EpCAM and CK, and confirmation of the presence of cell nuclei using the nuclear stain (DAPI). Additionally, the lymphocyte antigen, CD45, was immunostained to enable the identification of white blood cells. In a parallel experiment, cells suspended in blood were captured with EpCAM antibody conjugated with MNPs to gauge the overall capture efficiency and provide an overall cell or CTC count. For each of the cell lines tested, the c-Myc-mediated capture efficiencies were high (22Rv1 86±6%, PC3 85±3%, PC3M 78±5%) (Fig. 2a and Supplementary Fig. 4) and comparable to EpCAM-mediated capture efficiencies (22Rv1 77±2%, PC3 83±1%, PC3M 83±4%) (Fig. 2a and Supplementary Fig. 5).
Fig. 2 |. Intracellular protein analysis and the sensitivity of the approach.
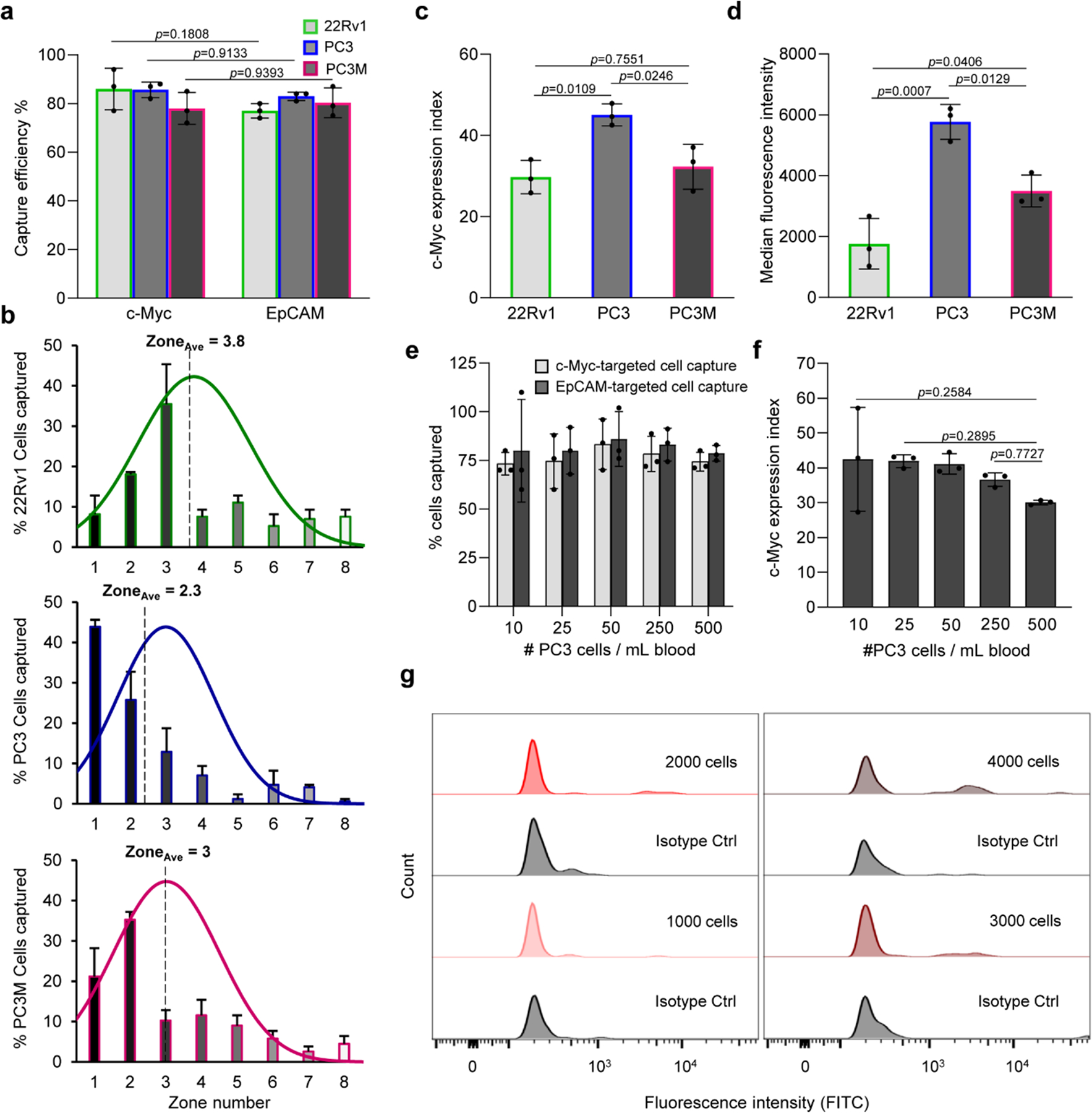
a, Capture efficiency of 22Rv1, PC3, and PC3M cell lines after fixation, permeabilization, and incubation with a c-Myc specific antibody modified with ssDNAs that were subsequently hybridized with CP1 and CP2. A parallel experiment was carried out in which the cells were captured using magnetic nanoparticles tagged anti-EpCAM antibody. One hundred cells were used in these trials. Error bars represent the s.d. of biological replicates (N=3). Two-way analysis of variance (ANOVA) tests were used to analyse the data and Sidak’s corrections with 95% confidence intervals and significance were used. b, Determination of the average capture zone (ZoneAve) of the three cell lines. The curves represent the normal distribution fit to the data. c, Cellular analysis of c-Myc protein in the three cell lines. The c-Myc expression index reflects the capture of cells using the ssDNA-tagged antibody followed with hybridization with CP1 and CP2 relative to cell capture with MNPs-labeled anti-EpCAM antibody. The c-Myc expression index is calculated from the formula (NIP * 100 / NEpCAM * ZoneAve), where NIP is the number of cells captured by targeting the intracellular protein (c-Myc), NEpCAM is the number of cells captured by targeting EpCAM, and ZoneAve is the median capture zone of the cells. d, Flow cytometric analysis of c-Myc protein in the three cell lines. e, Sensitivity of the approach tested by spiking different numbers of PC3 cells into 1 mL of blood. f, Dynamic range of the approach determined by measuring the variation of c-Myc expression index with the number of cells spiked in a blood sample. Error bars represent the s.d. of biological replicates (N=3). Ordinary one-way ANOVA tests were used to analyse the data and Turkey’s corrections with 95% confidence intervals and significance were used. f, Sensitivity of the flow cytometric analysis of c-Myc in PC3 cells spiked in whole blood. A high cell count (> 1000 cells) was needed for c-Myc detection in blood after RBCs removal using the Ficoll method.
For each cell line, the median zone of cell capture was calculated to consider the effect of intracellular protein levels on the overall cell distribution within the device and to provide a parameter that can be used to refine the calculation of protein expression. PC3 cells were mainly captured in the early zones of the device and had an average zone value of 2.3. The distribution of PC3M cells was shifted into later zones and an average zone value of 3 was calculated. 22Rv1 cells were binned in later zones and had an average zone value of 3.8 (Fig. 2b). An expression index (EI) for the c-Myc protein was then calculated for each cell line by dividing the number of cells captured by targeting the intracellular protein by the total number of cells captured by EpCAM antibody and the average zone parameter (Fig. 2c). For example, the average number of PC3 cells captured by c-Myc and EpCAM targeting are 85 and 83, respectively. The average zone value is 2.3 and thus an EI value of 45 is calculated.
Flow cytometry was used to measure the expression of c-Myc protein in the same cell lines (Fig. 2d and Supplementary Fig. 6). Our results show that the levels of c-Myc expression measured using the magnetic cell labelling approach and flow cytometry are comparable within measurement errors. The concordance of c-Myc expression index measurements and flow cytometry-based quantitation supports the notion that the magnetic cell labelling approach is quantitative. Also, our results revealed that the c-Myc expression levels in all cell lines are low compared to the levels of the standard cell-surface protein, EpCAM (Supplementary Fig. 7). However, comparable cell capture efficiencies were achieved by targeting c-Myc using the magnetic cell labelling reagents and EpCAM with anti-EpCAM conjugated with MNPs (Fig. 2a). This provides another support to the ability of the method to amplify the magnetic susceptibility of cells bearing low abundance of intracellular proteins.
We next sought to determine the sensitivity and dynamic range of the magnetic cell labelling approach. Analysis of c-Myc expression in as few as 10 cells in 1 millilitre of blood could be reproducibly achieved; lower cell counts could also be reliably analysed, but sampling error exists at concentrations below 10 cells/ml. The protein EI values were constant between 10 and 250 cells. Analysing larger number of target cells leads to the saturation of the initial zones, resulting in lower EI values (Fig. 2e, f and Supplementary Fig. 8). However, such high cell count would not be typically encountered in most clinical specimens. The performance of magnetic cell labelling approach was benchmarked against flow cytometry in order to assess the sensitivity of the method. PC3 cells immunostained for c-Myc and analysed with flow cytometry could be visualized at low cell counts when suspended in buffered solution. However, over 1,000 cells spiked in blood were required for c-Myc analysis even after the removal of red blood cells using the Ficoll method prior to analysis (Fig. 2g and Supplementary Fig. 9). This can be ascribed to the background signal of residual blood cells which can obscure the signal emitted from the fluorescently labeled c-Myc antibody.
We used this approach to analyse a series of other clinically-relevant intracellular proteins in the three prostate cancer cell lines. We analysed a panel of four intracellular proteins that exist in different subcellular locations, including cytoplasm, nucleus, and mitochondria. These proteins include vimentin, poly [ADP-ribose] polymerase 1 (PARP1), octamer-binding transcription factor 4 (Oct4), and DNA-directed RNA polymerase (POLRMT). Vimentin, a cytoplasmic intermediate filament protein, is a canonical marker of epithelial-mesenchymal transition (EMT)35. Upregulation of vimentin contributes to androgen-independent prostate cancer invasion and metastasis via Src regulation and is associated with poor survival outcomes in patients harboring vimentin-positive CTCs36. PARP1 is an abundant nuclear enzyme functionally involved in DNA damage repair and transcriptional regulation37. PARP1 interacts with the predominant TMPRSS2:ETS gene fusion product, ERG, to elicit protumorigenic effects in androgen-receptor-positive prostate cancer by transcriptionally regulating functional genes contributing to tumour growth, metastasis and resistance to therapy38. Oct4 is a transcription factor (nuclear and cytoplasmic) that regulates self-renewal and differentiation in embryonic stem cells and is overexpressed in various tumours, including prostate cancer39. POLRMT is a single-subunit mitochondrial RNA polymerase enzyme that controls the transcription of mitochondrial genome. POLRMT contributes functionally to tumour growth and is overexpressed in most malignancies40.
The expression pattern of each protein was analysed using the magnetic cell labelling approach (Supplementary Fig. 10,11) and the EI values were calculated for each protein in the three cell lines (Fig. 3). Flow cytometry was utilized to analyse the protein levels in the same cell lines (Fig. 3 and Supplementary Fig. 12). Both methods generated comparable results, again indicating that the approach can be used to analyse intracellular proteins in cancer cells. It also highlights the ability of the magnetic cell labelling reagents to reach subcellular organelles, such as nucleus and mitochondria, to facilitate the analysis of different classes of intracellular proteins. Also, we attempted to analyse the same proteins using antibodies directly labeled with magnetic nanoparticles. In these trials, the intracellular proteins were targeted with specific antibodies directly labeled with MNPs via biotin-streptavidin coupling and the cells were sorted using the same device (Supplementary Fig. 13). Lower capture efficiencies of cells (14±3%−57±4%) were achieved, which did not allow for an accurate determination of the protein levels (Supplementary Fig. 14). The results indicate that the magnetic cell labelling reagents are crucial for the analysis of low abundance proteins.
Fig. 3 |. Analysis of clinically-relevant intracellular proteins.
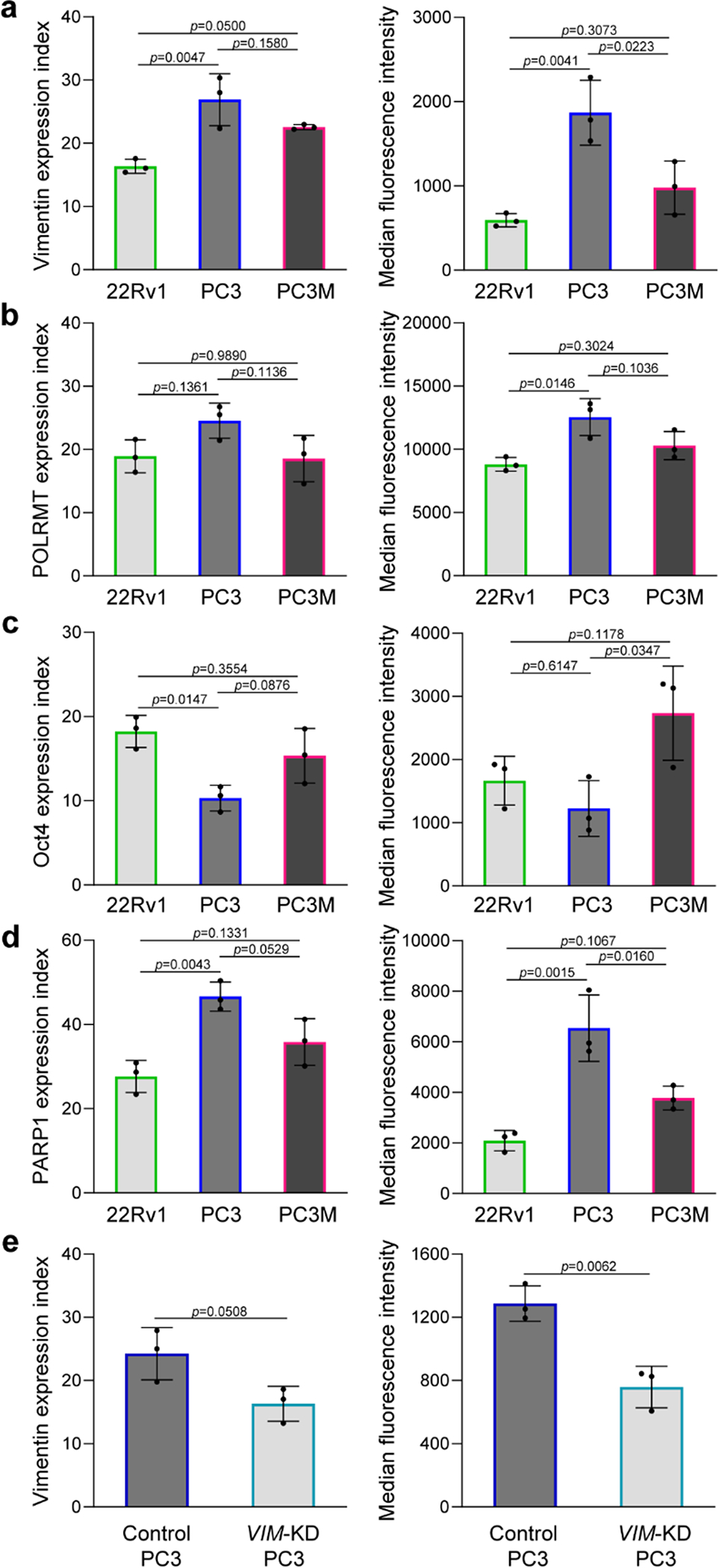
a–d, Cellular analysis of four intracellular proteins, including (a), vimentin, (b), POLRMT, (c), Oct4, and (d), PARP1 in three prostate cancer cell lines using the single-cell approach (left graphs) and flow cytometry (right graphs). A total of 100 cells and 100,000 cells were used for the single-cell analysis and flow cytometry trials, respectively. The agreement between the intracellular protein expression indices determined using the single-cell approach and flow cytometry indicates that the method is quantitative. Error bars represent the s.d. of biological replicates (N=3). Ordinary one-way ANOVA tests were used to analyse the data and Turkey’s corrections with 95% confidence intervals and significance were used. e, Analysis of vimentin in PC3 cells after knocking down the VIM gene using the single-cell approach (left graph) and flow cytometry (right graph). Error bars represent the s.d. of biological replicates (N=3). Unpaired two-tailed Student’s t-test was used to the analyse the data.
To demonstrate the selectivity of the approach, we analysed vimentin in PC3 cells before and after knocking down the VIM gene with small interfering RNAs (siRNAs). We found that the transfected PC3 cells exhibited lower EIvimentin, indicating 33±6.6% decrease in vimentin expression (Fig. 3e and Supplementary Fig. 15). Flow cytometric analysis of vimentin revealed that the protein level decreased by 41±5.9% (Fig. 3e and Supplementary Fig. 16). The results corroborated the protein expression data obtained using the magnetic cell labelling approach. In addition, fluorescence in situ hybridization (FISH) studies were carried out to determine the rate of entry of MNPs-labeled capture probes into PC3 and LNCaP cells after targeting vimentin with the DNA-labeled vimentin antibody. A similar rate of entry was observed in two different cell lines bearing different vimentin expression levels (Supplementary Fig. 17). The results demonstrate the efficient delivery of probes enabled by this approach. Additionally, the PC3 cells captured in each zone has a distinct level of vimentin protein that increases from zone 1 to 8 (Supplementary Fig. 18). The increase in protein content, measured with flow cytometry, correlated with the number of magnetic beads required for cell capture in each zone. The data show that the cell capture in a particular zone is only dependant on the intracellular protein content.
We then proceeded to assess the application of this approach in samples collected from tumour-bearing animals and cancer patients. First, we analysed the levels of c-Myc and vimentin in CTCs captured from the blood of mice bearing PC3 and PC3M orthotopic xenografts at 1 week and 3 weeks post-implantation. In each sample, the c-Myc and vimentin targeted capture of CTCs was conducted along with a total cell count determined by targeting EpCAM with anti-EpCAM conjugated with MNPs. For all of the xenografted mice, substantial numbers of CTCs were detected. The CTCs collected from mice xenografted with the less metastatic PC3 cells did not show a marked upregulation in the c-Myc and vimentin levels after 3 weeks post-implantation (Fig. 4a and 4b). Conversely, CTCs collected from animals bearing the highly metastatic PC3M-derived tumours demonstrated a marked increase in the c-Myc and vimentin levels after 3 weeks (Fig. 4c and d). Also, we studied the distribution of CTCs captured by targeting c-Myc among the eight capture zones of the microfluidic device. We observed that the CTCs obtained from the PC3M xenograft were mainly captured in earlier zones, whereas CTCs collected from the PC3 xenograft were sorted in later zones. This indicates a higher level of c-Myc in CTCs collected from the PC3M xenograft compared to those collected from the PC3 xenograft (Fig. 4e). A similar cell distribution pattern was demonstrated in vimentin-targeted capture of CTCs obtained from both xenografts, indicating a higher vimentin level in CTCs obtained from the PC3M xenograft (Fig. 4f). We found a strong correlation between the mRNA expression level (Supplementary Fig. 19−21) and the protein abundance previously demonstrated in Fig. 2C and 3A, with r=−0.94 and −0.99 for c-Myc and vimentin, respectively. The results agree with previous reports that demonstrated a similar correlation between the expression levels of c-Myc41 and vimentin mRNAs42 and the abundance of their proteins in prostate cancer.
Fig. 4 |. Analysis of c-Myc and vimentin in xenografts and clinical samples.
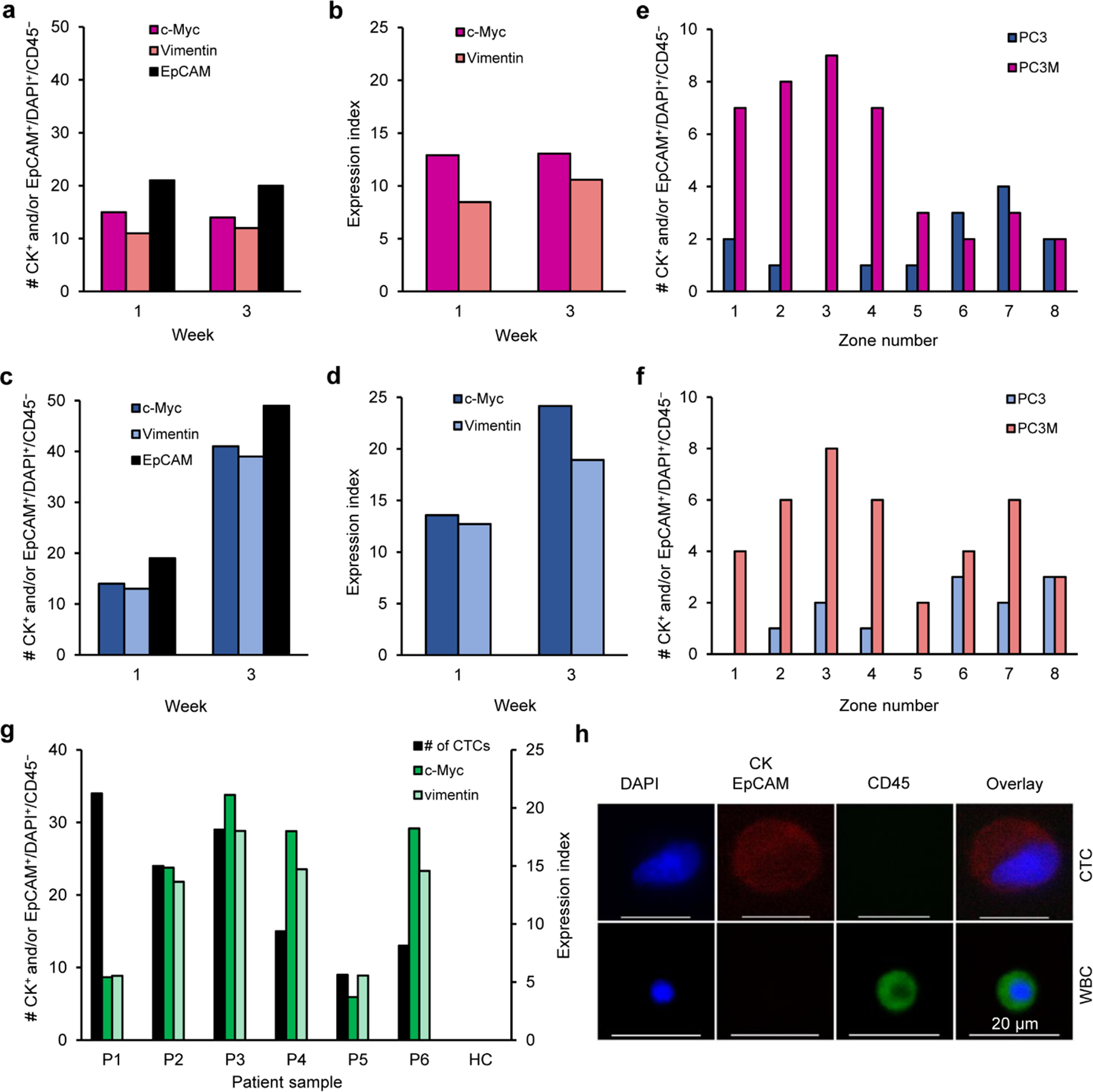
a, Number of CTCs captured from the blood of mice bearing PC3 xenografts after 1 and 3 weeks. CTCs were captured by targeting c-Myc, vimentin, and EpCAM subsequent to RBCs removal using the Ficoll method. b, Single-cell analysis of c-Myc and vimentin expression at the corresponding time points. c, Number of CTCs captured from the blood of mice bearing PC3M xenografts after 1 and 3 weeks. d, Single-cell analysis of c-Myc and vimentin expression at the corresponding time points. e–f, Number of CTC captured from the blood of mice bearing PC3M and PC3 xenografts after 3 weeks by targeting (e), c-Myc and (f), vimentin. g, Single-cell analysis of c-Myc and Vimentin expression in the CTCs collectively isolated from the blood samples of a cohort of six prostate cancer patients and one healthy control (HC). h, Representative image of a CTC captured from a prostate cancer patient blood sample versus a white blood cell (WBC). A single image was acquired for each cell type. The cells were stained with APC-labeled anti-CK, APC-labeled anti-EpCAM, AF488-labeled anti-CD45, and DAPI. Only CK+ and/or EpCAM+/CD45−/DAPI+ cells are counted as CTCs.
To showcase the clinical utility of the approach, we conducted a study in which we measured the levels of c-Myc and vimentin in CTCs captured from the blood samples collected from a small cohort of patients undergoing treatment for metastatic castration-resistant prostate cancer. An average of 10 millilitres of blood were analysed per patient and CTCs were identified using immunofluorescence. In 4 of the 6 patients tested, high levels of c-Myc and vimentin were detected in the CTCs isolated from their blood (Fig. 4g). Representative images of a CTC captured from patient samples versus a white blood cell are shown in Fig. 4h. The qPCR data revealed that the same 4 patients have exhibited high levels of c-Myc mRNA expression (r=−0.81). However, a discrepancy was noticed between the vimentin mRNA expression and the protein abundance (r=−0.69) (Supplementary Fig. 22).
Unlike conventional tumour markers that are simply associated with tumours, genetically-altered oncoproteins are produced only by tumour cells and are responsible for the initiation or progression of tumours43. Increasingly, proteins harbouring tumour-specific mutations or fusions are being targeted with tumour therapeutics43. Before these therapeutics can be administered, it is critical that patients are assessed to determine whether their tumours possess perturbations in specific molecular targets. While immunohistochemistry performed on biopsy specimens is a key tool for this type of analysis, disease found in metastatic cancer patients may not be accurately represented in a biopsy of a primary tumour, and an initial biopsy taken during diagnosis may not reflect the molecular changes that occur over the course of a patient’s treatment. Assessing drug targets via liquid biopsy, however, provides a means to profile the molecular markers present in primary and metastatic tumours at any time point required for therapeutic selection. We therefore sought to apply our magnetic cell labelling approach to a panel of intracellular protein markers that are validated targets of anticancer therapy. These proteins represent classes of oncogenic proteins that can arise from either gene splicing, gene fusion, or gene mutation.
Androgen-deprivation drugs are commonly used to treat advanced prostate cancer; these drugs, which include abiraterone and enzalutamide44, either suppress the synthesis of extragonadal androgens or target the androgen receptor directly. Approximately 20 to 40% of patients show no response to androgen-deprivation therapy (ADT), whereas those who initially have a response to ADT acquire a secondary resistance and thus require a second-line chemotherapy (e.g. docetaxel)45. One plausible explanation for IDT resistance may involve the presence of androgen receptors splice variants that encode for a truncated androgen receptor protein. The androgen receptor splice variant 7 (ARV7) is the most abundantly expressed variant and has been recently identified as a predictive biomarker for ADT46. Using our magnetic cell labelling approach, we were able to detect the splice variant in blood containing ARV7-positive cells. A negligible EI was measured for a blood sample containing ARV7-negative cells (Fig. 5a, Supplementary Fig. 23).
Fig. 5 |. Figure 5. Analysis of mutated proteins relevant for therapeutic selection.
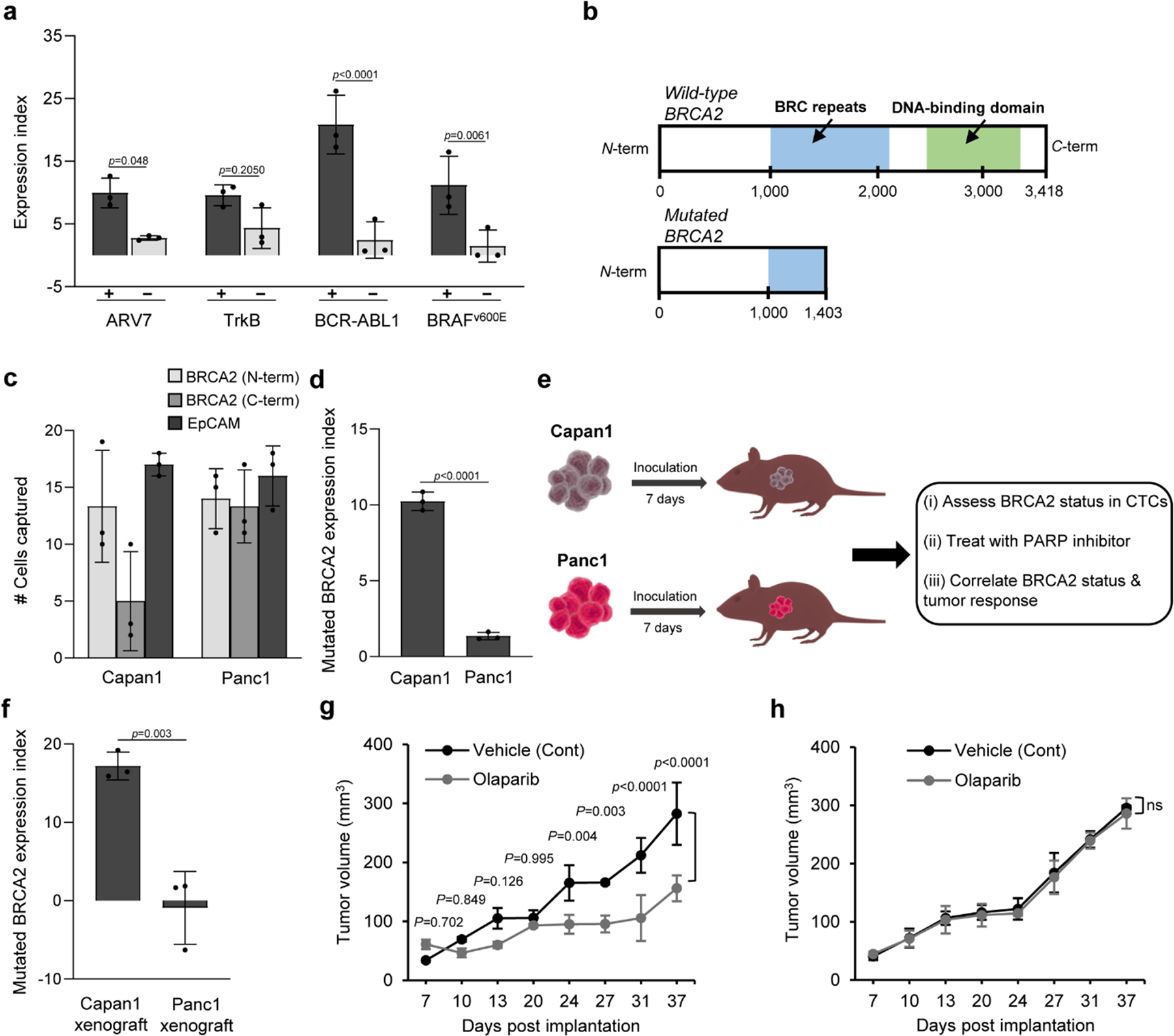
a, Analysis of ARV7 in 22Rv1 (+) and LNCaP (−) cells (30 cells/1mL blood), TrkB in H460 (+) and K562 (−) cells (30 cells/1mL PBS), BCR-ABL1 in K562 (+) and Jurkat cells (−) (30 cells/1mL PBS), and BRAFv600E in HT29 (+) and DU145 (−) cells (30 cells/1mL blood). After RBCs removal and cell fixation and permeabilization, cells were captured by targeting the intracellular protein. A parallel experiment was carried out in which cells were captured by targeting either EpCAM or CD45 (for K562 & Jurkat cells). Samples tested positive for the proteins with flow cytometry exhibited higher expression indices than those tested negative. b, Graphical representation of the structures of wild-type and mutated (truncated) BRCA2 proteins. c, Analysis of mutated BRCA2 protein in 1-mL blood samples spiked with Capan1 and Panc1 cells (30 cells). Cells were captured by targeting mutated BRCA2 protein with N-terminus specific antibody and C-terminus specific antibody with reference to the total number of cells captured with anti-EpCAM. d, Samples tested positive for mutated BRCA2 protein with flow cytometry exhibited higher expression indices than those tested negative. e, Schematic illustration for therapeutic protein analysis in xenografted mice. f, Analysis of mutated BRCA2 protein in CTCs captured from the blood of mice bearing either Capan1 or Panc1 xenograft. The analysis was carried out at day 7 after tumour formation. An extended analysis is provided in Supplementary Fig. 30. After tumour formation, the mice were randomly divided into control and treated groups (n=3). Mice in the treated group received 50 mg/kg olaparib every other day. Mice in the control group received only the vehicle. g–h, Tumour volume is plotted against length of treatment for (g), Capan1 and (h), Panc1 xenografts. Error bars represent the s.d. of biological replicates (N=3). Two-way ANOVA tests were used to analyse the data in (a), (g), and (h), and Holm–Sidak test was used for multiple comparisons with 95% confidence intervals. Unpaired two-tailed Student’s t-test was used to the analyse the data in (d), and (f).
We then proceeded to analyse other abnormal oncoproteins resulting from gene fusions. NTRK gene fusions cause overexpression of the transmembrane tropomyosin receptor kinase receptors referred to as TrkA, TrkB, and TrkC. Treatment of NTRK fusion-positive patients with Trk inhibitors, such as larotrectinib or entrectinib, is associated with high response rate (>75%), regardless of the tumour histology47. This finding has led to so-called “basket” clinical trials where patients are enrolled on the basis of NTRK fusion status rather than tumour type. The detection of NTRK fusions is challenging using sequencing-based analysis, however, because of the presence of large introns48. Identifying fusions as expressed proteins is therefore advantageous. The results shown in Figure 5A indicate that we can detect TrkB in blood samples spiked with lung cancer cells positive for the protein fusion. A lower EI was measured for TrkB in samples containing myelogenous leukemia cells (Supplementary Fig. 23). While the EI values measured for non-target cells were consistently low, it is noteworthy that statistical tests indicated that the variability in these measurements diminished the statistical significance attained with this target.
In chronic myeloid leukemia (CML), pathogenesis involves the fusion of the breakpoint cluster region (BCR) on chromosome 22 with the Abelson murine leukemia (ABL1) gene on chromosome 9, resulting in the formation of a fusion hybrid gene that encodes a cytoplasmic BCR-ABL1 oncoprotein with tyrosine kinase activity. FDA-approved tyrosine kinase inhibitors for CML treatment, such as nilotinib, imatinib, bosutinib, and dasatinib, have improved the overall survival rate of CML patients49. The magnetic cell labelling approach allowed for the detection of the mutant protein in samples containing BCR-ABL1 positive leukemia cells (Fig. 5a). A negligible EI was measured for BCR-ABL1 in control samples containing T-lymphocyte cells (Supplementary Fig. 23).
We also analysed abnormal oncoproteins resulting from gene mutations. Most BRAF mutations commonly found in melanoma and colorectal cancer patients result in an amino acid change of valine to glutamate in the nuclear and cytoplasmic protein, thus termed BRAFv600E. The BRAFv600E mutation causes constitutive activation of MAPK pathway, leading to drug- and immune-resistance, apoptosis evasion and cancer metastasis50. BRAFv600E-positive tumours can be controlled with FDA-approved BRAF inhibitors, such as vemurafenib and dabrafenib51. Our approach permitted the detection of the mutated protein in blood samples containing BRAFv600E-positive colon cancer cells. A negligible EI was measured for samples containing BRAFv600E-negative prostate cancer cells (Fig. 5a, Supplementary Fig. 23).
Another mutated protein that is an important marker in breast and prostate cancer is BRCA2. Breast cancer patients with BRCA2 gene mutations are more susceptible to breast cancer recurrence or development of ovarian cancer52. Most cancer-associated BRCA2 protein mutations are C-terminal truncating mutations that cause the protein to accumulate in the cytoplasm53. In 2018, the FDA approved a PARP inhibitor (olaparib) for the treatment of advanced breast and ovarian cancer with BRCA mutations54, and clinical evidence is mounting that the drug is also effective in prostate cancer patients harbouring BRCA2 truncations55. It is therefore critical to develop liquid biopsy tests for this mutated protein that can be used for therapeutic selection. The structures of wild-type and mutated BRCA2 proteins are schematically depicted in Fig. 5b. Blood samples were spiked with Capan1 cells and analysed for the truncated BRCA2 protein, and the cell capture results were compared those obtained using Panc1 cells expressing the wild-type BRCA2. Unlike BRAFv600E protein, there is no antibody specific for truncated BRCA2 protein. Thus, cells were immunostained and enumerated subsequent to targeting the N-terminus of BRCA2, C-terminus of BRCA2, and EpCAM (Fig. 5c, Supplementary Fig. 23). The results demonstrate the ability of the approach to detect mutated BRCA2 protein in blood samples spiked with low number of cells, with minimal interference from wild-type BRCA2 protein (Fig. 5d). Furthermore, all protein analysis results obtained using the magnetic cell labelling approach corroborated the flow cytometry data within error (Supplementary Fig. 24, 25). We also utilized this approach for parallelized analysis of 8 intracellular proteins, including c-Myc, vimentin, oct4, ARV7, BRCA2, TrkA, KRAS 2B, and CK in healthy blood samples spiked with MDA-MB-231 cells (Supplementary Fig. 26, 27) and the results corroborated the flow cytometry data (r=0.83) (Supplementary Fig. 28).
Finally, we set out to assess the utility of the approach for predicting the response of tumours to a molecularly-targeted therapy. BRCA2 mutations are common in tumours and lead to impaired homology-directed DNA repair (HDR) that makes these tumours sensitive to therapeutic agents that target HDR, such as PARP inhibitors56. We conducted a study in which we measured the level of mutated BRCA2 protein in CTCs captured from the blood of mice bearing either Capan1 or Panc1 xenograft, as representatives for BRCA2-deficient and BRCA2-proficient tumours, respectively. At day 7 post-implantation, the tumour-bearing mice were divided into control and treated groups. Mice in the treated groups received olaparib, whereas the control group only received the vehicle (Fig. 5e). In each sample, the C-terminus and N-terminus of BRCA2-targeted capture of CTCs were conducted along with a total cell count determined by targeting EpCAM with anti-EpCAM conjugated with MNPs. For all of the xenografted mice, substantial numbers of CTCs were detected. The CTCs collected from mice xenografted with Capan1 cells exhibited detectable levels of mutated BRCA2 protein at day 7, 22, and 37 after implantations, whereas CTCs from Panc1-xenografted mice did not exhibit similar levels of the mutated BRCA2 protein (Fig. 5f, Supplementary Fig. 29, 30). Monitoring the tumour growth in mice demonstrated that treatment with olaparib has resulted in a marked tumour growth inhibition in Capan1-xenograted mice, whereas Panc1-xenografted mice were not affected (Fig. 5g, h). The results validate the utility of the approach for predicting the therapeutic response of susceptible and resistant tumours in vivo and present a use case for how this technology could be used in the clinic.
Outlook
The method reported here allows direct isolation of rare CTCs from blood and intracellular protein analysis in a single experiment. This approach is quantitative when benchmarked against flow cytometry, and is amenable to the analysis of low (~10) numbers of cells. The analytical performance of the approach is benchmarked against FACS-based microfluidic Western blotting in Supplementary Table 3. The precision and detection sensitivity of such low cell counts obviates the need for post-isolation cell culture. The approach relies on immunostaining for identification of target cells and is thus not susceptible to interference from residual blood cells. Also, it does not require any enzymatic amplification steps. It is noteworthy that EpCAM-based CTC capture is used to generate an overall count of CTCs relative to those captured using specific markers targeted for quantitation, but given the ability to use any antibody with the method, a cocktail of antibodies can be utilized to generate the overall CTC count.
Furthermore, the method can be utilized for parallelized analysis of a panel of protein markers in CTCs − a capability particularly beneficial for studying tumour metastasis pathways that usually involve cascades of several proteins. Combining CTC isolation with protein profiling would make CTC taxonomy more substantial, as some CTCs do not resemble the primary tumour and would likely diverge at the proteomic level. Given the inherent long-term solution-phase storage stability of antibody-DNA conjugates, we foresee that the magnetic cell labelling reagents will continue to provide a useful tool for evaluation of subcellular proteins. More broadly, we envision the role of the magnetic cell labelling approach as a useful tool with relevance spanning from understanding CTC biology, early cancer detection and monitoring patient’s response to particular therapeutic regimens.
Methods
Device fabrication.
Devices were fabricated using a standard soft-lithography procedure. Masters were fabricated by a stereolithographic 3D printer (μMicrofluidics Edition 3D Printer, Creative CADworks, Canada), according to manufacturer’s protocol. Polydimethylsiloxane (PDMS, Dow Chemical, US) replicas were poured on masters and baked at 70°C for 2 hours. The cured replicas were then peeled off, punched, and plasma bonded to thickness no. 1 glass coverslips (Ted Pella, US). The bonded chips were kept at 100°C overnight to secure a robust bonding. Afterward, the silicon tubing was attached to the inlet and outlet of the device. Prior to use, the devices were conditioned with 1% Pluronic F68 (Sigma-Aldrich, US) in phosphate buffered saline (PBS) for at least 4 hours, to reduce the nonspecific adsorption. Each device was sandwiched between two arrays of N52 Nd FeB magnets (K&J Magnetics, US, 1.5 mm by 8 mm) with alternating polarity. A syringe pump (Chemyx, US) was used during the cell capture process.
Cell culture.
22Rv1, PC3M, LNCaP, H460, and Jurkat cell lines were cultured in RPMI-1640 medium (ATCC 30-2001). PC3 cell line was cultured in F-12K Medium (ATCC 30-2004). DU145 cell line was cultured in EMEM medium (ATCC 30-2003). MDA-MB-231 cell line was cultured in L-15 medium (ATCC 30-2008). HT29 cell line was cultured in McCoy’s 5A medium (ATCC 30-2007). Panc1 cell line was cultured in Dulbecco’s modified Eagle’s medium (ATCC 30-2002). Capan1 and K562 cell lines were cultured in IMDM medium (ATCC 30-2005). All media were supplemented with 10% FBS (20% FBS for Capan1 cell culture in IMDM) and 1% penicillin-streptomycin and the cells were cultured at 37°C and 5% CO2 in T75 flasks. Cells were harvested when they reached more than 70–80% confluence. With the exception of K562 and Jurkat cell lines, cell detachment from the culture dishes was performed using 1 mL of 0.25% (w/v) trypsin-0.53 mM EDTA solution for 3 min at 37°C. The cells were then filtered using a 40μm BD falcon cell strainer (Becton, Dickinson and Company, US).
Preparation of DNA-conjugated antibodies.
Fourteen antibodies, including antibodies specific to c-Myc (bs-4963R, Bioss Inc., US), vimentin (bs-0756R, Bioss Inc., US), Oct4 (bs-1111R, Bioss Inc., US), POLRMT (489004 Pab, USBiological, US), PARP1 (LS-C745005, LifeSpan Biosciences Inc., US), ARV7 splice variant antibody (31-1109-00, RevMAb Biosciences, US), BCR-ABL1 (ab187831, Abcam, US), BRAFv600E (31-1042-00, RevMAb Biosciences, US), TrkA (ab76291, Abcam, US), TrkB (ab134155, Abcam, US), BRCA2 (N-terminus specific, ab75335, Abcam, US), and BRCA2 (C-terminus specific, ARG10523, Arigo Biolaboratories Corp., US), pan cytokeratin (bs-1712R, Bioss Inc., US), and KRAS 2B (16155-1-AP, Proteintech, US) were first modified with streptavidin using a streptavidin conjugation kit (Abcam, US), according to the manufacturer’s protocol. Briefly, 100 μL of the antibody solution (1 mg mL−1) were gently shaken with 10 μL of the activator. Afterward, the activated antibodies were incubated with 33 μg streptavidin overnight at 4°C. Next, 10 μL of the quencher were added to stop the reaction. Then, 80 μL of the biotin-labeled DNA (1 mg mL−1) were added and the mixture was incubated at room temperature for 30 min. Finally, the solution was stored at 4°C until use.
Preparation of capture probes.
Briefly, 100 μL of 20 μM of the antisense oligonucleotide solution in Dulbecco’s phosphate-buffered saline (DPBS, Sigma-Aldrich, US) containing 1 mM dithiothreitol (DTT, Sigma-Aldrich, US), were heated for 5 min at 60°C for deaggregation. Afterward, the solution was transferred to a microtiter plate and incubated with 1.5 μL of 10 mg mL−1 streptavidin-coated magnetic nanoparticles (100nm, Chemicell, US) for 30 min at room temperature. Subsequently, the magnetic nanoparticles-labeled capture probe (CP1 or CP2) was pelleted using a magnetic-ring stand (Thermofisher Scientific, US) and washed three times with DPBS/DTT solution.
Intracellular protein analysis.
Prostate cancer cell lines (100 cells in 100 μL DPBS containing 0.2 mg mL−1 dextran sulfate) were fixed with 100 μL of 8% paraformaldehyde (PFA, Sigma-Aldrich, US) solution in DPBS/DTT for 15 min at 37°C. After centrifugation, the cells were incubated with 100 μL of 0.3% Triton X-100 (TX-100, Sigma-Aldrich, US) in DPBS/DTT for 10 min at room temperature. Afterward, the cells were gently shaken with 5 μL of the DNA-conjugated antibody for 30 min at room temperature. After centrifugation, the cells were gently shaken with a mixture of CP1 and CP2 (100 μL each in DPBS/DTT) for 3 h at room temperature. In parallel, 100 cells in 100 μL DPBS were gently shaken with 20 μL of MNPs-labeled EpCAM antibody (130-061-101, Miltenyi Biotec, US) for 30 min at room temperature. Finally, the cells were loaded into the microfluidic device at a flow rate of 2 mL h−1.
Cell staining and imaging.
Captured cells were counted using a fluorescence microscope. Prior to staining, captured cells were fixed inside the device using 100 μL of 4% PFA in DPBS/DTT then permeabilized with 100 μL of 0.2% TX-100 in DPBS/DTT. Captured cells were immunostained with a mixture of 3% APC-labeled pan cytokeratin antibody (APC-CK, GTX80205, Genetex, US), 3% APC-labeled EpCAM antibody (APC-EpCAM, 130-111-000, Miltenyi Biotec, US), and 3% AF488-labeled CD45 antibody (AF488-CD45, MHCD4520, Invitrogen, US) in 200 μL PBS containing 1% bovine serum albumin (BSA, Sigma-Aldrich, US) and 0.1% Tween-20 (Sigma-Aldrich, US). Immunostaining was carried out for 60 min at a flow rate of 200 μL h−1. After washing with 0.1% Tween-20 in PBS, the cells were stained with 1 drop of 4’,6-diamidino-2-phenylindole (DAPI Prolong Gold nuclear stain, Invitrogen, US) in 200 μL PBS for 10 min at a flow rate of 1200 μL h−1. After staining, the cells were washed with 0.1% Tween-20 in PBS, and stored at 4°C. Finally, chips were scanned using Nikon Ti-E Eclipse microscope with an automated stage controller and CMOS Camera (Andor Neo). The blue channel was used for DAPI staining, with a typical exposure time of 10–20 ms. The green channel was used for the AF488-CD45 staining, with a typical exposure time of 40–60 ms. The red channel was used for the APC-CK and APC-EpCAM staining, with a typical exposure time of 200–300 ms. The exposure time was set individually for each chip and kept constant in the course of scanning. Cells were counted by overlaying the bright field, red, blue, and green fluorescent images.
Calculation of expression index.
The intracellular protein expression index is calculated from formula 1:
| (1) |
NIP denotes the number of cancer cells captured by targeting the intracellular protein, NAb is the total number of cells in the sample captured by targeting EpCAM, and ZoneAve is the median capture zone determined from a normal distribution fit to the distribution of cell populations bearing varying expression levels of the target intracellular protein.
The BRCA2 truncated (mutated) protein expression index is calculated from formula 2:
| (2) |
NITP(Nterminus) denotes the number of cancer cells captured by targeting the N-terminus of the intracellular truncated protein, NITP(Cterminus) represents the number of cancer cells captured by targeting the C-terminus of the truncated protein, NAb is the total number of cells in the sample captured by targeting EpCAM, and ZoneAve(Nterminus) and ZoneAve(Cterminus) represent the median capture zones determined from a normal distribution fit to the distribution of cell populations calculated after targeting the N terminus and C terminus of intracellular truncated protein, respectively.
Dynamic light scattering (DLS).
DLS experiments were carried out using Zeta sizer Nano series (Malvern Instruments, UK) to confirm the formation of MNP aggregates upon hybridization between CP1, CP2, and the ssDNA conjugate of c-Myc antibody. Prior to analysis, CP1 and CP2, each composed of 15 μg MNPs modified with antisense oligonucleotide, were incubated with 5 μL of the DNA-conjugated c-Myc antibody for 3 h at room temperature. A control experiment was carried out in which 30 μg of MNPs were incubated directly with 10 μL of 100 μg mL−1 of biotin-labeled c-Myc antibody (bs-0842R-Biotin, Bioss, US) for 3 h at room temperature.
Flow cytometric analysis of proteins.
Flow cytometry was used to analyse the levels of c-Myc, vimentin, POLRMT, Oct4, and PARP1 in 22Rv1, PC3M, and PC3 cell lines. Briefly, PC3 cells (100,000 cells) were incubated with the blocking buffer (2% BSA in PBS) for 30 min on ice. Afterward, the cells were fixed with 4% PFA and permeabilized with 0.2% TX-100. The cells were incubated with 10 μL of 100 μg mL−1 of c-Myc antibody (bs-4963R, Bioss Inc., US), vimentin antibody (bs-0756R, Bioss Inc., US), Oct4 antibody (bs-1111R, Bioss Inc., US), POLRMT antibody (489004 Pab, USBiological, US), PARP1 antibody (LS-C745005, LifeSpan Biosciences Inc., US) for 30 min at room temperature. Control experiments were carried out in which the cells were incubated with 10 μL of 100 μg mL−1 of rabbit IgG isotype control (02–6102, Invitrogen, US) for 30 min at room temperature. After washing, the cells were incubated with 10 μL of FITC-labeled mouse anti-rabbit antibody (31584, Invitrogen, US) for 30 min at room temperature. In addition, flow cytometry was used to analyse the levels of ARV7 in 22Rv1 and LNCaP cell lines, BCR-ABL1 in K562 and Jurkat cell lines, BRAFv600E in HT29 and DU145 cell lines, TrkB in H460 and K562 cell lines, and truncated BRCA2 in Capan1 and Panc1 cell lines. In these trials, the fixed and permeabilized cells were incubated with 10 μL of 100 μg mL−1 of ARV7 splice variant antibody (31-1109-00, RevMAb Biosciences, US), BCR-ABL1 antibody (ab187831, Abcam, US), BRAFv600E (31-1042-00, RevMAb Biosciences, US), TrkA antibody (ab76291, Abcam, US), TrkB antibody (ab134155, Abcam, US), BRCA2 (N-terminus specific, ab75335, Abcam, US), BRCA2 (C-terminus specific, ARG10523, Arigo Biolaboratories Corp., US), pan cytokeratin antibody (bs-1712R, Bioss Inc., US), and KRAS 2B antibody (16155-1-AP, Proteintech, US) for 30 min at room temperature. Control experiments were carried out in which the cells were incubated with 10 μL of 100 μg mL−1 of rabbit IgG isotype control (02-6102, Invitrogen, US) for 30 min at room temperature. After washing, the cells were incubated with 10 μL of APC-labeled anti-rabbit IgG (F0111, R&D Biosystems, US) for 30 min at room temperature. For BCR-ABL1 analysis, the cells were incubated with 10 μL of APC-labeled anti-mouse IgG (F0101B, R&D Biosystems, US) for 30 min at room temperature. Control experiments were carried out in which the cells were incubated with 10 μL of 100 μg mL−1 of mouse IgG isotype control (10400C, Invitrogen, US) for 30 min at room temperature. After washing, the cells were incubated with 10 μL of APC-labeled anti-mouse IgG (F0101B, R&D Biosystems, US) for 30 min at room temperature. Prior to analysis, the cells were washed three times with 2% BSA in PBS. Finally, samples were injected into FACSCanto flow cytometer (BD Biosciences, US) and measurements were plotted as histograms. Absorbance values were normalized to unstained control. A total of 10,000 cells were analysed per cell line.
Knockdown of vimentin.
We used VIM specific Accel smart pool siRNA to knock down vimentin in PC3 cells, according to the manufacturer’s protocol. Briefly, PC3 (100,000 cells) were cultured at 37°C and 5% CO2 in 6-well plate overnight. A stock solution of 100 μM VIM specific Accel smart pool siRNA (E-0003551-00-0020, Dharmacon, US) in 1x siRNA buffer (B-002000-UB-100) was prepared. The siRNA was suspended in 300 μL Accel delivery medium (B-005000-500, Dharmacon, US) to a final concentration of 1 μM and the suspension was shaken for 90 min at room temperature. The suspension was centrifuged briefly. The culture medium was aspired from each well and the cells were incubated with 300 μL siRNA suspension in the Accel delivery medium for 96 h at 37°C and 5% CO2. A control experiment was carried out in which the cells were incubated with 1 μM non-targeting siRNA (D-001206-13-20, Dharmacon, US) in the Accel Delivery medium (300 μL) under the same conditions. Finally, the cells were trypsinized and the vimentin protein level was analysed using the microfluidic approach and flow cytometry.
RT-qPCR analysis.
Total RNA was isolated from the cells using a single-cell RNA purification kit (Cat. 51800, Norgen Biotek Corp., US), according to the manufacturer’s protocol. The isolated RNA was used for cDNA synthesis using SuperScript III first-strand synthesis system (Invitrogen, US), which contains random hexamer primers and Superscript III reverse transcriptase, according to the manufacturer’s protocol. A comparative Ct experiment was performed on ABI 7500 real-time PCR system (Applied Biosystems, US). The assay was carried out in triplicates using 10 ng cDNA for each sample in a 96-well plate. The 20μL reaction mix consisted of 10 μL of SsoFast EvaGreen master mix (Bio-Rad, US), 0.5 μL of forward primer (10 μM), 0.5 μL of reverse primer (10 μM), 8 μL of water and 1 μL of 10 ng μL−1 cDNA. Cycling conditions for the qPCR were 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min.
Prostate cancer xenografts.
All animal experiments were carried out in accordance with the protocol approved by the University of Toronto Animal Care Committee. Male athymic nude mice (6–8 weeks old) were purchased from Envigo, US, and maintained at the University of Toronto animal facility. Tumour xenografts were generated by injecting a suspension of 1×106 PC3M or PC3 cells in 25 μL Hank’s buffered salt solution (HPSS, Life Technologies, US), orthotopically into the right dorsolateral lobe of the prostate. The whole blood was collected every week from each mouse by cardiac puncture under anesthesia. All blood samples were collected in BD vacutainer blood collection tubes containing EDTA (BD, US). A total of 5 mL whole blood was collected from 5 mice after 1 and 3 weeks. The mononuclear cells were isolated using the Ficoll method and subsequently suspended in 1 mL of DPBS/DTT. Briefly, the blood sample was diluted with an equal volume of PBS containing 2% FBS. The density gradient medium was added to the SepMate tube (StemCell Technologies, Canada) by pipetting through the central hole of the insert. The sample was centrifuged at 1200 × g for 10 min at room temperature. The top layer containing the enriched mononuclear cells was poured into a new tube and washed 2 times with PBS containing 2% FBS. Each time, the sample was centrifuged at 300 × g for 8 min. For intracellular protein analysis, 200 μL of the supernatant were incubated with 200 μL of 8% PFA in DPBS/DTT for 15 min at 37°C. After centrifugation, the cells were incubated with 100 μL of 0.3% TX-100 in DPBS/DTT for 10 min at room temperature. Afterward, the cells were gently shaken with 5 μL of the DNA-conjugated antibody (specific for c-Myc or vimentin) for 30 min at room temperature. After centrifugation, the cells were gently shaken with a mixture of CP1 and CP2 in DPBS/DTT (100 μL each) for 3 h at room temperature. To determine the total number of cells in the sample, 200 μL of the supernatant were incubated with 20 μL of MNPs-labeled EpCAM antibody for 30 min at room temperature. The cells were loaded into the microfluidic device at a flow rate of 2 mL h−1 and subsequently stained with APC-labeled anti-EpCAM, APC-labeled anti-CK, AF488-labeled anti-CD45 antibody, and DAPI. To measure the expression of c-Myc and vimentin mRNAs, the remaining supernatant solution was incubated with 20 μL of MNPs-labeled EpCAM antibody for 30 min at room temperature. The cells were loaded into a cell-extraction microfluidic device57 at a flow rate of 8 mL h−1. After washing, the Tygon tubing connecting the zones were cut and the cells were gently pipetted out the device and stored at −80°C before RT-qPCR analysis.
Analysis of intracellular proteins in clinical specimens.
Metastatic castration-resistant prostate cancer (CRPC) patients were recruited from the Princess Margaret Hospital according to the University of Toronto Research Ethics Board approval protocol. All patients were enrolled subsequent to informed consent. Ten milliliters of peripheral blood samples were collected from CRPC patients in CellSearch tubes containing EDTA. All the samples were analysed within 24 h after collection. A set of patient blood samples (n=6) and 1 healthy control were analysed to determine the eligibility of the approach for the analysis of c-Myc and vimentin in CTCs. The mononuclear cells were isolated using the Ficoll method and subsequently suspended in 2 mL of DPBS/DTT. For intracellular protein analysis, 200 μL of the supernatant were incubated with 200 μL of 8% PFA in DPBS/DTT for 15 min at 37°C. After centrifugation, the cells were incubated with 100 μL of 0.3% TX-100 in DPBS/DTT for 10 min at room temperature. Afterward, the cells were gently shaken with 5 μL of the DNA-conjugated antibody (specific for c-Myc or vimentin) for 30 min at room temperature. After centrifugation, the cells were gently shaken with a mixture of CP1 and CP2 in DPBS/DTT (100 μL each) for 3 h at room temperature. To determine the total number of cells in the sample, 200 μL of the supernatant were incubated with 20 μL of MNPs-labeled EpCAM antibody for 30 min at room temperature. The cells were loaded into the microfluidic device at a flow rate of 2 mL h−1 and subsequently stained with APC-labeled anti-EpCAM, APC-labeled anti-CK, AF488-labeled anti-CD45 antibody, and DAPI. To measure the expression of c-Myc and vimentin mRNAs, the remaining supernatant solution was incubated with 20 μL of MNPs-labeled EpCAM antibody for 30 min at room temperature. The cells were loaded into a cell-extraction microfluidic device57 at a flow rate of 8 mL h−1. After washing, the Tygon tubing connecting the zones were cut and the cells were gently pipetted out the device and stored at −80°C before RT-qPCR analysis.
Pancreatic cancer xenografts.
All animal experiments were carried out in accordance with the protocol approved by the University of Toronto Animal Care Committee. Female Athymic Nude-Foxn1nu mice at 6–8 weeks of age were purchased from Envigo, US, and maintained at the University of Toronto animal facility. Tumour xenografts were generated by injecting 5×106 Capan1 or Panc1 cells in 0.1 ml of PBS subcutaneously into the flanks of each mouse. Tumour volumes were measured twice per week using the formula: Tumour volume = (length / 2) × (width2). After 1 week, the tumour-bearing mice were randomly divided into control and treated groups (n=3). Mice in the treated group received 50 mg/kg olaparib every other day for 4 weeks. Mice in the control group received only the vehicle. At day 7, 22 and 37, blood samples were collected from both treated and control mice by cardiac puncture under anesthesia. All blood samples were collected in BD vacutainer blood collection tubes containing EDTA (BD, US). A total of 1 mL whole blood was collected from each mouse. The mononuclear cells were isolated using the Ficoll method and subsequently suspended in 600 μL of DPBS/DTT. For intracellular protein analysis, 400 μL of the supernatant were incubated with 400 μL of 8% PFA in DPBS/DTT for 15 min at 37°C. After centrifugation, the cells were incubated with 100 μL of 0.3% TX-100 in DPBS/DTT for 10 min at room temperature. After adding 100 μL of DPBS/DTT, the cells were gently shaken with 5 μL of the DNA-conjugated antibody (specific for N-terminus or C-terminus of BRCA2 protein) for 30 min at room temperature. After centrifugation, the cells were gently shaken with a mixture of CP1 and CP2 in DPBS/DTT (100 μL each) for 3 h at room temperature. To determine the total number of cells in the sample, the remaining 200 μL of the supernatant were incubated with 20 μL of MNPs-labeled EpCAM antibody for 30 min at room temperature. The cells were loaded into the microfluidic device at a flow rate of 2 mL h−1 and subsequently stained with APC-labeled anti-EpCAM, APC-labeled anti-CK, AF488-labeled anti-CD45 antibody, and DAPI.
Statistical analyses.
All statistical analyses were performed using the GraphPad Prism software. The specifics of the statistical tests and number of replicates are stated in the figure legends. The threshold for significance in all tests was p < 0.05.
Reporting summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
Research reported in this publication was supported by the Canadian Institutes of Health Research (Grant #FDN-148415), the Natural Sciences and Engineering Research Council of Canada (Grant #2016-06090), the Province of Ontario though the Ministry of Research, Innovation and Science (Grant #RE05-009), and the National Cancer Institute of the National Institutes of Health (Grant # 1R33CA204574). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the other funding agencies. We also thank Dr. Anthony Joshua at the Princess Margaret Hospital for supplying the clinical specimens.
Footnotes
Competing interests
The authors declare no competing interests.
Supplementary information is available for this paper at: https://doi.org/10.1038/s41551-020-0590-1
Data availability
The main data supporting the results in this study are available within the paper and its Supplementary Information. The raw and analysed datasets generated during the study are too large to be publicly shared, yet they are available for research purposes from the corresponding authors on reasonable request.
References
- 1.Alix-Panabieres C & Pantel K Challenges in circulating tumour cell research. Nat. Rev. Cancer 14, 623–631 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Nagrath S et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 450, 1235–1239 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoon HJ et al. Sensitive capture of circulating tumour cells by functionalized graphene oxide nanosheets. Nat. Nanotechnol 8, 735–741 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao W et al. Bioinspired multivalent DNA network for capture and release of cells. Proc. Natl. Acad. Sci. U. S. A 109, 19626–19631 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stott SL et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc. Natl. Acad. Sci. U. S. A 107, 18392–18397 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ozkumur E et al. Inertial focusing for tumor antigen-dependent and -independent sorting of rare circulating tumor cells. Sci. Transl. Med 5, 179ra147 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee A et al. All-in-one centrifugal microfluidic device for size-selective circulating tumor cell isolation with high purity. Anal. Chem 86, 11349–11356 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Kim TH et al. FAST: size-selective, clog-free isolation of rare cancer cells from whole blood at a liquid-liquid interface. Anal. Chem 89, 1155–1162 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Adams AA et al. Highly efficient circulating tumor cell isolation from whole blood and label-free enumeration using polymer-based microfluidics with an integrated conductivity sensor. J. Am. Chem. Soc 130, 8633–8641 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Zhou L & Qin L High-throughput 3D cell invasion chip enables accurate cancer metastatic assays. J. Am. Chem. Soc 136, 15257–15262 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y, Zhang W & Qin L Mesenchymal-mode migration assay and antimetastatic drug screening with high-throughput microfluidic channel networks. Angew. Chem. Int. Ed. Engl 53, 2344–2348 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toriello NM et al. Integrated microfluidic bioprocessor for single-cell gene expression analysis. Proc. Natl. Acad. Sci. U. S. A 105, 20173–20178 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reyes EE et al. Quantitative characterization of androgen receptor protein expression and cellular localization in circulating tumor cells from patients with metastatic castration-resistant prostate cancer. J. Transl. Med 12, 313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ciaccio MF, Wagner JP, Chuu CP, Lauffenburger DA & Jones RB Systems analysis of EGF receptor signaling dynamics with microwestern arrays. Nat. Methods 7, 148–155 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rimm DL What brown cannot do for you. Nat. Biotechnol 24, 914–916 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Liu Y et al. Modulation of fluorescent protein chromophores to detect protein aggregation with turn-on fluorescence. J. Am. Chem. Soc 140, 7381–7384 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pollock SB et al. Highly multiplexed and quantitative cell-surface protein profiling using genetically barcoded antibodies. Proc. Natl. Acad. Sci. U. S. A 115, 2836–2841 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engelen W, Meijer LH, Somers B, de Greef TF & Merkx M Antibody-controlled actuation of DNA-based molecular circuits. Nat. Commun 8, 14473 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan R et al. Integrated barcode chips for rapid, multiplexed analysis of proteins in microliter quantities of blood. Nat. Biotechnol 26, 1373–1378 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nam JM, Thaxton CS & Mirkin CA Nanoparticle-based bio-bar codes for the ultrasensitive detection of proteins. Science 301, 1884–1886 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Angelo M et al. Multiplexed ion beam imaging of human breast tumors. Nat. Med 20, 436–442 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rissin DM et al. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat. Biotechnol 28, 595–599 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de la Rica R & Stevens MM Plasmonic ELISA for the ultrasensitive detection of disease biomarkers with the naked eye. Nat. Nanotechnol 7, 821–824 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Xue M et al. Chemical methods for the simultaneous quantitation of metabolites and proteins from single cells. J. Am. Chem. Soc 137, 4066–4069 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerdtsson E et al. Multiplex protein detection on circulating tumor cells from liquid biopsies using imaging mass cytometry. Converg. Sci. Phys. Oncol 4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hughes AJ et al. Single-cell western blotting. Nat. Methods 11, 749–755 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sinkala E et al. Profiling protein expression in circulating tumour cells using microfluidic western blotting. Nat. Commun 8, 14622 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poudineh M et al. Tracking the dynamics of circulating tumour cell phenotypes using nanoparticle-mediated magnetic ranking. Nat. Nanotechnol 12, 274–281 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Labib M et al. Single-cell mRNA cytometry via sequence-specific nanoparticle clustering and trapping. Nat. Chem 10, 489–495 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herold S, Herkert B & Eilers M Facilitating replication under stress: an oncogenic function of MYC? Nat. Rev. Cancer 9, 441–444 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Dang CV MYC on the path to cancer. Cell 149, 22–35 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beaulieu ME et al. Intrinsic cell-penetrating activity propels Omomyc from proof of concept to viable anti-MYC therapy. Sci. Transl. Med 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burke AJ, Ali H, O’Connell E, Sullivan FJ & Glynn SA Sensitivity profiles of human prostate cancer cell lines to an 80 kinase inhibitor panel. Anticancer Res. 36, 633–641 (2016). [PubMed] [Google Scholar]
- 34.Lakshman M et al. Dietary genistein inhibits metastasis of human prostate cancer in mice. Cancer Res. 68, 2024–2032 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Thiery JP Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2, 442–454 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Lindsay CR et al. Vimentin and Ki67 expression in circulating tumour cells derived from castrate-resistant prostate cancer. BMC Cancer 16, 168 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ray Chaudhuri A & Nussenzweig A The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol 18, 610–621 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schiewer MJ et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2, 1134–1149 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kosaka T et al. The prognostic significance of OCT4 expression in patients with prostate cancer. Hum. Pathol 51, 1–8 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Salem AF, Whitaker-Menezes D, Howell A, Sotgia F & Lisanti MP Mitochondrial biogenesis in epithelial cancer cells promotes breast cancer tumor growth and confers autophagy resistance. Cell Cycle 11, 4174–4180 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao L et al. Androgen receptor promotes ligand-independent prostate cancer progression through c-Myc upregulation. PLoS One 8, e63563 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu M et al. Proteome analysis of human androgen-independent prostate cancer cell lines: variable metastatic potentials correlated with vimentin expression. Proteomics 7, 1973–1983 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Stratton MR, Campbell PJ & Futreal PA The cancer genome. Nature 458, 719–724 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tran C et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 324, 787–790 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scher HI et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1–2 study. Lancet 375, 1437–1446 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Watson PA, Arora VK & Sawyers CL Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 15, 701–711 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sinkevicius KW et al. Neurotrophin receptor TrkB promotes lung adenocarcinoma metastasis. Proc. Natl. Acad. Sci. U. S. A 111, 10299–10304 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen Y & Chi P Basket trial of TRK inhibitors demonstrates efficacy in TRK fusion-positive cancers. J. Hematol. Oncol 11, 78–78 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wylie AA et al. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature 543, 733–737 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Holderfield M, Deuker MM, McCormick F & McMahon M Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat. Rev. Cancer 14, 455–467 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hauschild A et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 380, 358–365 (2012). [DOI] [PubMed] [Google Scholar]
- 52.Rebbeck TR et al. Prophylactic oophorectomy in carriers of BRCA1 or BRCA2 mutations. N. Engl. J. Med 346, 1616–1622 (2002). [DOI] [PubMed] [Google Scholar]
- 53.Jeyasekharan AD et al. A cancer-associated BRCA2 mutation reveals masked nuclear export signals controlling localization. Nat. Struct. Mol. Biol 20, 1191–1198 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eriksson I, Wettermark B & Bergfeldt K Real-world use and outcomes of olaparib: a population-based cohort study. Target. Oncol 13, 725–733 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mateo J et al. DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med 373, 1697–1708 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rowe BP & Glazer PM Emergence of rationally designed therapeutic strategies for breast cancer targeting DNA repair mechanisms. Breast Cancer Res. 12, 203 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Green BJ et al. Isolation of phenotypically distinct cancer cells using nanoparticle-mediated sorting. ACS Appl. Mater. Interfaces 9, 20435–20443 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The main data supporting the results in this study are available within the paper and its Supplementary Information. The raw and analysed datasets generated during the study are too large to be publicly shared, yet they are available for research purposes from the corresponding authors on reasonable request.