

Abstract
Rationale: There is an urgent need for simple, cost-effective prognostic biomarkers for idiopathic pulmonary fibrosis (IPF); biomarkers that show potential include monocyte count.
Objectives: We used pooled data from pirfenidone and IFNγ-1b trials to explore the association between monocyte count and prognosis in patients with IPF.
Methods: This retrospective pooled analysis included patients (active and placebo arms) from the following four phase III, randomized, placebo-controlled trials: ASCEND (NCT01366209), CAPACITY (NCT00287729 and NCT00287716), and INSPIRE (NCT00075998). Outcomes included IPF progression (≥10% absolute decline in FVC% predicted, ≥50 m decline in 6-minute-walk distance, or death), all-cause hospitalization, and all-cause mortality over 1 year. The relationship between monocyte count (defined as time-dependent) and outcomes was assessed using bivariate and multivariable models.
Measurements and Main Results: This analysis included 2,067 patients stratified by monocyte count (at baseline: <0.60 × 109 cells/L [n = 1,609], 0.60 to <0.95 × 109 cells/L [n = 408], and ≥0.95 × 109 cells/L [n = 50]). In adjusted analyses, a higher proportion of patients with monocyte counts of 0.60 to <0.95 × 109 cells/L or ≥0.95 × 109 cells/L versus <0.60 × 109 cells/L experienced IPF progression (P = 0.016 and P = 0.002, respectively), all-cause hospitalization (P = 0.030 and P = 0.003, respectively), and all-cause mortality (P = 0.005 and P < 0.001, respectively) over 1 year. Change in monocyte count from baseline was not associated with any of the outcomes over 1 year and did not appear to be affected by study treatment.
Conclusions: In patients with IPF, elevated monocyte count was associated with increased risks of IPF progression, hospitalization, and mortality. Monocyte count may provide a simple and inexpensive prognostic biomarker in IPF.
Keywords: prognosis, pulmonary fibrosis, biomarkers
At a Glance Commentary
Scientific Knowledge on the Subject
The disease course of idiopathic pulmonary fibrosis (IPF) is highly variable, and prognosis can be difficult to predict, creating an urgent need for simple, cost-effective prognostic biomarkers to identify patients at risk of more rapid disease progression. Several potential prognostic biomarkers have been identified, but measurement of these biomarkers has been relatively complex, labor intensive, and costly. A recent retrospective analysis identified monocyte count as a potential prognostic biomarker for IPF, finding that high monocyte counts (≥0.95 × 109 cells/L) were associated with an increased risk of mortality (vs. <0.95 × 109 cells/L).
What This Study Adds to the Field
In this retrospective pooled analysis of the ASCEND, CAPACITY, and INSPIRE trials of patients with IPF, elevated monocyte count was associated with significantly increased risks of IPF progression, hospitalization, and mortality over 1 year. In addition to a monocyte count of ≥0.95 × 109 cells/L, which was investigated in the previous study, our analysis also demonstrated that a monocyte count of 0.60 to <0.95 × 109 cells/L was associated with worse 1-year outcomes (vs. <0.60 × 109 cells/L). Our findings provide a rationale for prospective clinical studies investigating monocyte count as a simple and inexpensive prognostic biomarker for IPF.
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, fibrosing form of interstitial lung disease (ILD) (1, 2) with an estimated median survival time of 2−5 years after diagnosis in the absence of antifibrotic treatment (3, 4). Two antifibrotics, pirfenidone and nintedanib, are approved for the treatment of IPF and are associated with reduced lung-function decline (5–7). The disease course of IPF is highly variable, and prognosis can be difficult to predict (1, 3), creating an urgent need for the identification of simple, clinician-friendly, cost-effective prognostic biomarkers that can be used to stratify patients with similar clinical presentations at diagnosis who are at risk of more rapid disease progression (8). Identification of such biomarkers may aid in the development of more personalized treatment plans and help guide more intensive disease management strategies, such as lung transplant allocation (8).
Several studies have reported on the potential of circulating biomarkers, gene expression signatures, and telomere length as prognostic biomarkers in patients with IPF (9–19). Measurement of such biomarkers can be relatively complex, labor intensive, and costly, therefore presenting a challenge for their implementation in clinical practice (8, 20).
In other respiratory diseases, including asthma, chronic obstructive pulmonary disease, and lung cancer, there has been a focus on whole blood and peripheral blood cells as potential sources of prognostic and predictive biomarkers (21–23). A retrospective pooled analysis of data from three large health record databases and a cohort of patients with systemic sclerosis recently identified monocyte count elevation as a potential prognostic biomarker for mortality in IPF and other fibrotic disorders, including systemic sclerosis, hypertrophic cardiomyopathy, and myelofibrosis (8). A recent analysis of the Australian IPF registry data also found an association between monocyte concentration and mortality (24). Given that monocyte count is a component of a standard blood test, this may represent a simple and inexpensive biomarker, warranting its further investigation.
We used data pooled from trials of pirfenidone (ASCEND, NCT01366209; CAPACITY Study 004, NCT00287716; and CAPACITY Study 006, NCT00287729) and IFNγ-1b (INSPIRE; NCT00075998) to further explore the association between monocyte count elevation and prognosis in patients with a confirmed diagnosis of IPF. Some of the results of this analysis have been previously reported in the form of an abstract (25).
Methods
Patient Population
This retrospective, pooled analysis included data from four phase III, randomized, placebo-controlled trials in IPF, as follows: ASCEND, CAPACITY (two studies), and INSPIRE. The eligibility criteria have been described previously (5, 6, 26) (see Appendix E1 in the online supplement). Patients who received 2,403 mg/d pirfenidone, 200 μg IFNγ-1b three times a week, or placebo were included in this analysis (patients who received 1,197 mg/d pirfenidone in CAPACITY Study 004 were excluded) (5, 6, 26).
The trials were conducted in accordance with International Conference on Harmonization Guidelines and the Declaration of Helsinki. All patients provided written, informed consent before participation, and study protocols were approved by the institutional review board/ethics committee at each center.
Sample Collection and Measurements
Monocyte count was recorded at baseline and scheduled trial visits (see Appendix E1) as part of complete blood counts and was defined as a time-dependent variable in analyses described herein (i.e., monocyte count reflects the most recently available measurement on or before the time at which an event occurred). The thresholds used to stratify patients by monocyte count were defined initially in increments of 0.20 × 109 cells/L for values <0.80 × 109 cells/L (i.e., <0.20 × 109 cells/L, 0.20 to <0.40 × 109 cells/L, 0.40 to <0.60 × 109 cells/L, and 0.60 to <0.80 × 109 cells/L), whereas for values ≥0.80 × 109 cells/L, strata were defined as 0.80 to <0.95 × 109 cells/L and ≥0.95 × 109 cells/L on the basis of the analysis by Scott and colleagues that reported higher all-cause mortality among patients with monocyte count ≥0.95 × 109 cells/L (8). Strata for values <0.95 × 109 cells/L were subsequently modified on the basis of findings from bivariate analyses, suggesting that risks of study outcomes were comparable among patients within monocyte count strata defined as <0.60 × 109 cells/L (<0.20 × 109 cells/L, 0.20 to <0.40 × 109 cells/L, and 0.40 to <0.60 × 109 cells/L) and those defined as 0.60 to <0.95 × 109 cells/L (0.60 to <0.80 × 109 cells/L and 0.80 to <0.95 × 109 cells/L). Results from a goodness-of-fit analysis corroborated the use of the modified thresholds for the analyses described herein. In supplemental analyses, other components of white blood cells (e.g., lymphocytes and neutrophils), which were recorded at baseline and scheduled trial visits (see Appendix E1), were also considered and were defined as continuous time-dependent variables.
Outcomes
Outcomes over 1 year were IPF progression (≥10% absolute decline in FVC% predicted, ≥50 m decline in 6-min-walk distance [6MWD], or death), all-cause hospitalization, and all-cause mortality. Mean monocyte count by chronic immunosuppressant use, mean change from baseline in monocyte count by treatment (placebo, pirfenidone, or IFNγ-1b), and mean change from baseline in FVC% predicted by monocyte count and treatment, all over 1 year, were also assessed.
Statistical Analysis
Patients in the intent-to-treat populations with baseline monocyte count data were included in analyses; there was no imputation for missing values.
Shared frailty models (an extension of the Cox proportional hazards model that adjusts for intracluster correlation [i.e., clustering by trial]) were employed to examine the relationship between monocyte count (defined as time-dependent), longitudinal change from baseline in monocyte count, and study outcomes over 1 year. Bivariate models were used without adjustment, and multivariable models were used with adjustment for patient demographics, physiologic function (time-dependent), comorbidity profile, and chronic immunosuppressant use (time-dependent). The monocyte stratification variable, change from baseline in monocyte count variable, and dummy variables for pirfenidone and IFNγ-1b were forced into models as regressors; other predictors of outcomes were selected for inclusion in the multivariable models via a backward selection method (P < 0.10). Supplemental analyses of monocyte count and counts of other components of white blood cells were similarly conducted.
Survival analyses were performed using Kaplan-Meier techniques, and monocyte count comparisons were undertaken using the log-rank test. For time-to-event analyses, patients were censored at the time of loss to follow-up or lung transplantation (or mortality for all-cause hospitalization only) or at the end of 1-year follow-up, whichever occurred first.
Results
Patient Baseline Characteristics
Overall, 2,067 patients (ASCEND, n = 555; CAPACITY, n = 692; INSPIRE, n = 820) were included in this analysis. The patient demographics and disease characteristics have been previously described for the individual study populations (5, 6, 26) and are summarized for the pooled population in Table 1. The mean age (SD) for the pooled population was 66.7 (7.6) years, and 73.0% of patients were male (Table 1).
Table 1.
Summary of Patient Baseline Characteristics in the Pooled ASCEND, CAPACITY, and INSPIRE Population by Monocyte Count
| All Patients (n = 2,067) | Patients, by Monocyte Count |
|||
|---|---|---|---|---|
| <0.60 × 109 cells/L (n = 1,609) | 0.60 to <0.95 × 109 cells/L (n = 408) | ≥0.95 × 109 cells/L (n = 50) | ||
| Age, mean (SD), yr | 66.7 (7.6) | 66.4 (7.7) | 67.8 (7.3) | 67.7 (8.1) |
| Sex, n (%) | ||||
| M | 1,508 (73.0) | 1,136 (70.6) | 328 (80.4) | 44 (88.0) |
| Monocytes, × 109 cells/L | ||||
| Mean* (SD) | 0.49 (0.18) | 0.41 (0.10) | 0.71 (0.09) | 1.14 (0.29) |
| Quintile, minimum–maximum | ||||
| First | 0.10−0.34 | 0.10−0.32 | 0.60−0.62 | 0.95−0.96 |
| Second | 0.35−0.42 | 0.33−0.38 | 0.63−0.66 | 0.97−1.01 |
| Third | 0.43−0.50 | 0.39−0.44 | 0.67−0.71 | 1.02−1.09 |
| Fourth | 0.51−0.61 | 0.45−0.51 | 0.72−0.77 | 1.11−1.21 |
| Fifth | 0.62−2.81 | 0.52−0.59 | 0.78−0.94 | 1.24−2.81 |
| FVC% predicted, mean (SD) | 69.0 (15.0) | 69.7 (15.1) | 67.4 (14.3) | 63.1 (14.3) |
| DlCO% predicted, mean (SD) | 43.6 (11.4) | 44.1 (11.4) | 41.9 (11.2) | 41.4 (9.4) |
| 6MWD, mean (SD), m | 382.0 (120.6) | 385.3 (120.5) | 374.7 (119.6) | 336.3 (122.8) |
| UCSD-SOBQ total score, mean (SD) | 39.1 (24.6) | 38.6 (24.4) | 39.7 (24.9) | 47.0 (26.8) |
| Comorbidities,†n (%) | ||||
| GERD | 645 (31.2) | 473 (29.4) | 154 (37.7) | 18 (36.0) |
| CAD | 444 (21.5) | 310 (19.3) | 111 (27.2) | 23 (46.0) |
| COPD | 114 (5.5) | 83 (5.2) | 30 (7.4) | 1 (2.0) |
| MI | 111 (5.4) | 73 (4.5) | 34 (8.3) | 4 (8.0) |
| Pulmonary hypertension | 50 (2.4) | 27 (1.7) | 22 (5.4) | 1 (2.0) |
| DVT | 48 (2.3) | 39 (2.4) | 8 (2.0) | 1 (2.0) |
| CV risk factors, n (%) | ||||
| Smoker‡ | 1,372 (66.4) | 1,064 (66.1) | 276 (67.6) | 32 (64.0) |
| Hypertension | 1,068 (51.7) | 815 (50.7) | 230 (56.4) | 23 (46.0) |
| Obesity§ | 897 (43.4) | 686 (42.6) | 187 (45.8) | 24 (48.0) |
| Hypercholesterolemia | 864 (41.8) | 650 (40.4) | 190 (46.6) | 24 (48.0) |
| Diabetes | 454 (22.0) | 348 (21.6) | 95 (23.3) | 11 (22.0) |
| Chronic immunosuppressant use,ǁn (%) | ||||
| Steroid (systemic) | 135 (6.5) | 102 (6.3) | 24 (5.9) | 9 (18.0) |
| Nonsteroid | 33 (1.6) | 25 (1.6) | 7 (1.7) | 1 (2.0) |
| Neither | 1,918 (92.8) | 1,496 (93.0) | 382 (93.6) | 40 (80.0) |
Definitionof abbreviations: 6MWD = 6-minute-walk distance; CAD = coronary artery disease; COPD = chronic obstructive pulmonary disease; CV = cardiovascular; DVT = deep vein thrombosis; GERD = gastroesophageal reflux disease; MI = myocardial infarction; UCSD-SOBQ = University of California, San Diego, Shortness of Breath Questionnaire.
At time of event (idiopathic pulmonary fibrosis progression, defined as ≥10% absolute decline in FVC% predicted, ≥50 m decline in 6MWD, or death) or censor.
Includes comorbidities reported in >2% of the total patient population.
Current/former.
Body mass index >30 kg/m2.
At any time from baseline through to end of 1-year follow-up period; values may sum more than 100%.
The mean (SD) monocyte count (at time of event [IPF progression] or censor) for the pooled population was 0.49 (0.18) × 109 cells/L (ASCEND, 0.52 [0.21] × 109 cells/L; CAPACITY, 0.48 [0.16] × 109 cells/L; INSPIRE, 0.47 [0.18] × 109 cells/L; Table E1). Other mean white blood cell counts at time of event or censor were also found to be relatively consistent across the included trials (Table E1). A total of 77.8% (n = 1,609), 19.7% (n = 408), and 2.4% (n = 50) of patients had baseline monocyte counts of <0.60 × 109 cells/L, 0.60 to <0.95 × 109 cells/L, and ≥0.95 × 109 cells/L, respectively. Physiologic characteristics at baseline were generally balanced between monocyte count groups, although patients in the ≥0.95 × 109 cells/L group had a numerically lower 6MWD and a numerically higher University of California, San Diego, Shortness of Breath Questionnaire score versus the <0.60 × 109 cells/L and 0.60 to <0.95 × 109 cells/L groups (Table 1).
The most frequently reported comorbidities in the overall population were gastroesophageal reflux disease (31.2%) and coronary artery disease (CAD; 21.5%; Table 1). CAD was more common in patients in the ≥0.95 × 109 cells/L monocyte group (46.0%) versus the <0.60 × 109 cells/L and 0.60 to <0.95 × 109 cells/L groups (19.3% and 27.2%, respectively). A higher percentage of patients in the ≥0.95 × 109 cells/L monocyte group (18.0%) had reported chronic systemic steroid immunosuppressant use versus the <0.60 × 109 cells/L and 0.60 to <0.95 × 109 cells/L groups (6.3% and 5.9%, respectively; Table 1).
One-Year Risk of Study Outcomes by Monocyte Count
Findings based on the initial stratification scheme for monocyte count are presented in Table E2. In bivariate analyses, after modification of the stratification scheme, times to first evidence of IPF progression, all-cause hospitalization, and all-cause mortality were shorter in patients with a monocyte count of ≥0.95 × 109 cells/L and 0.60 to <0.95 × 109 cells/L versus those with a count of <0.60 × 109 cells/L (Figure 1).
Figure 1.

Kaplan-Meier curves for time to first (A) IPF progression (≥10% absolute decline in FVC% predicted, ≥50 m decline in 6-minute-walk distance, or death), (B) all-cause hospitalization, and (C) all-cause mortality over 1 year by monocyte count, in which monocyte count was defined as a time-dependent variable. IPF = idiopathic pulmonary fibrosis.
In adjusted analyses, a significantly higher percentage of patients with a monocyte count of either 0.60 to <0.95 × 109 cells/L or ≥0.95 × 109 cells/L versus those with a count of <0.60 × 109 cells/L experienced IPF progression (hazard ratio [HR], 1.25; 95% confidence interval [CI], 1.04–1.50; P = 0.016 and HR, 1.80; 95% CI, 1.23–2.63; P = 0.002, respectively), all-cause hospitalization (HR, 1.29; 95% CI, 1.03–1.63; P = 0.030 and HR, 2.14; 95% CI, 1.30–3.51; P = 0.003, respectively), and all-cause mortality (HR, 1.78; 95% CI, 1.19–2.66; P = 0.005 and HR, 4.05; 95% CI, 2.00–8.19; P < 0.001, respectively) over 1 year (Figure 2 and Table E3). This pattern was maintained when the models were adjusted only for age, sex, FVC% predicted, DlCO% predicted, and 6MWD (Table E4) and when nonchronic immunosuppressant use was considered (Table E5).
Figure 2.
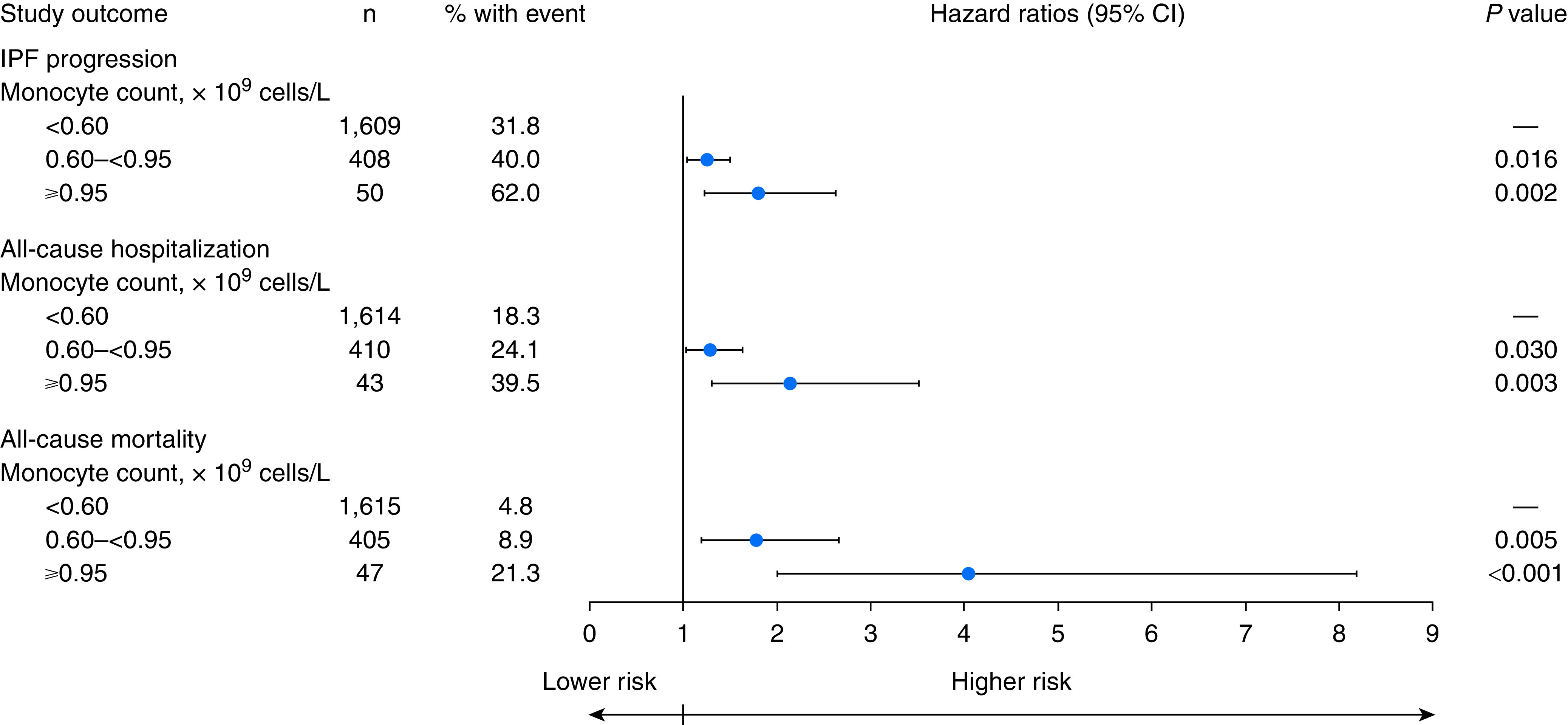
Adjusted hazard ratios for IPF progression, all-cause hospitalization, and all-cause mortality by monocyte count. Monocyte count and other model covariates were defined as time-dependent variables, as appropriate. CI = confidence interval; IPF = idiopathic pulmonary fibrosis.
When other white blood cell counts were added to the adjusted multivariable analyses that included all model covariates, significant associations were observed between neutrophil count (positive association) and lymphocyte count (negative association) and study outcomes (Table E6). However, associations between elevated monocyte counts and increased risk of study outcomes were still observed when other white blood cell counts were accounted for in the adjusted model, which were significant for increased risk of IPF progression (for monocyte counts of 0.60 to <0.95 × 109 cells/L and ≥0.95 × 109 cells/L vs. <0.60 × 109 cells/L: HR, 1.22; 95% CI, 1.01–1.48; P = 0.039 and HR, 1.61; 95% CI, 1.08–2.39; P = 0.019, respectively) and all-cause hospitalization (for monocyte counts of ≥0.95 × 109 cells/L vs. <0.60 × 109 cells/L: HR, 1.75; 95% CI, 1.05–2.93; P = 0.031) over 1 year (Table E6).
One-Year Risk of Study Outcomes by Change in Monocyte Count from Baseline
The mean monocyte count ranged from 0.47 to 0.53 × 109 cells/L over the 1-year follow-up period (Figure E1), and large changes in monocyte count from baseline were rare (n = 18 or 19 for change of ≥0.50 × 109 cells/L). In the unadjusted model, changes in monocyte count of 0.10 to <0.50 × 109 cells/L (HR, 1.26; 95% CI, 1.01–1.58; P = 0.042) or ≥0.50 × 109 cells/L (HR, 2.46; 95% CI, 1.36–4.43; P = 0.003) from baseline were significantly associated with IPF progression versus a change of <−0.10 × 109 cells/L over 1 year (Table E7). Change in monocyte count of ≥0.50 × 109 cells/L from baseline was significantly associated with all-cause hospitalization versus a change of <−0.10 × 109 cells/L (HR, 2.31; 95% CI, 1.07–5.00; P = 0.033) over 1 year. No significant association was found between amounts of change in monocyte count from baseline and all-cause mortality (Table E7).
In adjusted analyses, no significant association was found between change in monocyte count from baseline and any of the study outcomes over 1 year (Table E3).
Effect of Treatments on Monocyte Count
In descriptive analyses, mean monocyte count over 1 year was similar for patients receiving or not receiving chronic (systemic steroid or nonsteroid) immunosuppressants (Figure E2). Although some small differences in mean changes in monocyte count from baseline over 1 year were observed across treatment groups (placebo, pirfenidone, or IFNγ-1b [presented for all patients, those receiving chronic steroid treatment, those receiving nonchronic steroid treatment, and those who did not receive steroid treatment]), these were not considered clinically meaningful (Figure E3). Moreover, mean change in FVC% predicted did not differ considerably between patients with monocyte counts of <0.60 × 109 cells/L, 0.60 to <0.95 × 109 cells/L, and ≥0.95 × 109 cells/L throughout the 1-year treatment period; this was generally the case regardless of treatment group, although conclusions regarding the ≥0.95 × 109 cells/L subgroup are limited because of low patient numbers (Figure E4).
Discussion
This retrospective, pooled analysis found that patients with a confirmed diagnosis of IPF who had a monocyte count of 0.60 to <0.95 × 109 cells/L or ≥0.95 × 109 cells/L had a higher 1-year risk of IPF progression, all-cause hospitalization, and all-cause mortality versus patients with a monocyte count of <0.60 × 109 cells/L after adjustment for demographics, physiologic function, comorbidity profile, and chronic immunosuppressant use.
These findings are consistent with those of a previous study evaluating a possible link between monocyte count and prognosis in patients with IPF (8). In the previous retrospective, pooled analysis, a monocyte count of ≥0.95 × 109 cells/L was significantly associated with all-cause mortality versus a monocyte count of <0.95 × 109 cells/L after adjustment for FVC (HR, 2.47; P = 0.0063) and gender, age, and physiology index (HR, 2.06; P = 0.0068) in a subset of patients with confirmed IPF. In the same study, a higher monocyte count was also associated with shortened survival in patients with other fibrotic diseases, including systemic sclerosis, myelofibrosis, and hypertrophic cardiomyopathy (8). Further data supporting these findings come from an analysis of 231 patients from three Australian states in the Australian IPF registry (24). Analysis of these registry data found an association between monocyte counts of ≥0.95 × 109 cells/L and increased mortality after adjustment for age, sex, and baseline FVC% predicted (HR, 2.36; P = 0.02) (24).
Although the previous analyses only looked at ≥0.95 × 109 cells/L versus <0.95 × 109 cells/L (8, 24), we also demonstrated that a monocyte count of 0.60 to <0.95 × 109 cells/L was significantly associated with a higher 1-year risk of IPF progression, hospitalization, and mortality versus patients with a monocyte count of <0.60 × 109 cells/L. Moreover, this association was found after adjustment for age, sex, FVC% predicted, DlCO% predicted, and 6MWD, suggesting that monocyte count may provide added predictive value to these clinical variables. In contrast, we found that change from baseline in monocyte count over 1 year was not associated with study outcomes. This suggests that monocyte count has potential as a prognostic biomarker rather than as a predictive biomarker for treatment response, although this has not been confirmed yet in prospective cohorts. Looking at real-world applications of our findings, a monocyte count of ≥0.60 × 109 cells/L could potentially be used to enrich the population of clinical trials for patients at greater risk of IPF progression. Furthermore, in clinical practice, a monocyte count of ≥0.60 × 109 cells/L could be used to alert healthcare professionals to a patient’s risk of IPF progression and worse prognosis, which may help to guide the decision to initiate treatment and the choice of therapeutic interventions. In addition, a monocyte count of ≥0.60 × 109 cells/L could be used to identify which patients to assess for their suitability for a lung transplant.
Preliminary analyses assessing the relationship between other white blood cell counts and study outcomes found significant associations for neutrophil count (positive association) and lymphocyte count (negative association) in multivariable analyses. However, elevated monocyte counts were still associated with a higher risk of study outcomes when the multivariable model was adjusted for other white blood cell counts, indicating that the relationship between monocyte counts and study outcomes is distinct from that of the other white blood cell counts. Additional analyses would be needed to further assess whether other white blood cell counts, apart from monocytes, have a relationship with the outcomes observed.
These results indicate an association between monocyte (and potentially other white blood cell) concentrations and IPF prognosis, which is in line with what is known about the possible roles of monocytes and other immune cells in the development of IPF and may help shed some light on the pathogenesis of IPF. Although the pathophysiology of IPF has not yet been fully elucidated, various immune cells have previously been linked with pathogenesis, including monocytes, neutrophils, and lymphocytes (27–33). The model of IPF pathogenesis currently favored is based on repeated epithelial injury leading, in genetically susceptible individuals, to aberrant repair and the formation of fibrotic tissue (34, 35). Fitting with this model, immune cells (including monocytes) migrate to the site of injury to aid repair, where they differentiate. Recruitment and differentiation of monocytes is driven by the surrounding microenvironment, with circulating monocytes having the potential to become interstitial or airway macrophages or dendritic cells (35–38). Disease progression in IPF has been linked to changes in the phenotype and function of alveolar macrophages. It follows that altered alveolar macrophage phenotype may be driven, in part, by changes in the populations of differentiated monocyte subsets, especially as monocyte-driven changes in airway macrophages are observed in healthy human aging (32, 39, 40). Single-cell RNA sequencing of lung collected from individuals with IPF has confirmed marked alveolar macrophage heterogeneity, something that has been linked to the evolution of fibrosis in murine models (41, 42). Accumulation of distinct populations of alveolar macrophages has been linked to disease progression and shortened survival in patients with IPF (43, 44).
The fibrocyte, a specialized cell derived from the monocyte cell lineage, has been postulated to be a precursor of the myofibroblast and has been implicated in the pathogenesis of IPF (45, 46). In patients with IPF, higher concentrations of circulating fibrocytes may be predictive of a worse prognosis in terms of disease progression and have shown to be markedly elevated in acute exacerbations of IPF (46, 47). C-C motif chemokine ligand 18, produced by fibrocytes and, to a greater extent, by alveolar macrophages, has shown potential as a serum biomarker of disease progression and mortality in IPF (45, 48, 49).
The emergence of readily measurable serum biomarkers that are reflective of IPF-related pathophysiology may help to better inform treatment approaches in IPF. In addition to the prediction of poorer prognosis, monocyte count has been linked with the occurrence of acute exacerbations of fibrosing ILDs, and regular monitoring may help to guide clinical decision-making with respect to the initiation of antifibrotic medications (8, 24, 50). The ease and speed with which biomarkers can be measured will govern their applicability within a clinical setting. Monitoring of circulating monocyte concentrations is easily incorporated into the routine assessment of patients with IPF as part of a standard blood count in clinical practice, and it represents a technically reproducible biomarker that is simpler, less labor intensive, and more cost-effective than measurement of other prognostic biomarkers identified for IPF, such as gene signatures (8, 15). Further prospective cohort studies are required to fully explore the relationships between monocyte count and change in monocytes from baseline on outcomes in IPF and response to treatment. These studies could also be designed to evaluate whether the findings in patients with IPF can be more broadly applied to patients with other forms of ILD.
There are a number of limitations that should be considered when interpreting the results of this analysis, including the relatively small number of patients included in the ≥0.95 × 109 cells/L monocyte count group versus the 0.60 to <0.95 × 109 cells/L and <0.60 × 109 cells/L groups. The low mortality rate across subgroups defined by change in monocyte count (and corresponding low statistical power) is another limitation. Furthermore, the effect of comorbidities (such as CAD, which may also result in high monocyte concentrations) (51) on the observed association between monocyte count and study outcomes is not known. In addition, steroid use can lead to reduced monocyte counts and possible shifts in monocyte phenotype (52), and although chronic steroid use was not found to considerably affect monocyte counts in this analysis, no data were available on the duration or timing of nonchronic steroid use, and, as such, their effect on monocyte counts could not be fully determined. We also considered that treatment group (placebo, pirfenidone, or IFNγ-1b) may also have affected monocyte count; this possible effect could not be adequately assessed because classification and identification of patients who were responders to therapy was not possible in a robust manner with the current data set. In a descriptive analysis, although some differences in mean change from baseline in monocyte count over 1 year were observed for pirfenidone and IFNγ-1b compared with placebo, these changes were not clinically meaningful. This was a retrospective, pooled analysis restricted to patients with IPF from clinical trials; therefore, the analysis population included more patients with greater short-term survival than real-world cohorts because patients with severe disease were excluded. This analysis may also underestimate the longer-term prognostic value of monocyte counts because relatively limited outcome data were available beyond the end of the trials. It should also be noted that the relationship between monocyte count and study outcomes did not appear to be linear, and thus a categorical (rather than continuous) measure for monocyte count was employed in most of these analyses.
Conclusions
In this retrospective analysis of pooled data from ASCEND, CAPACITY, and INSPIRE, elevated monocyte count was associated with increased risks of IPF progression, hospitalization, and mortality. Monocyte count may provide a novel, simple, and inexpensive prognostic biomarker in patients with IPF and should be investigated further in future prospective clinical studies.
Acknowledgments
Acknowledgment
Medical writing support was provided by Rebekah Waters, Ph.D., of CMC AFFINITY, McCann Health Medical Communications, funded by F. Hoffmann-La Roche, Ltd.
Footnotes
Supported by F. Hoffmann-La Roche, Ltd., which designed and performed this analysis and was involved in data interpretation and the writing of the manuscript in collaboration with the academic authors. The authors had full access to all of the data in this analysis and had final responsibility for the decision to submit for publication.
Author Contributions: All authors were involved in the conception and/or design of the work and interpretation of study results, contributed to the manuscript from the outset, and read and approved the final draft. All authors vouch for the accuracy of the content included in the final manuscript.
Data sharing statement: Qualified researchers may request access to individual patient-level data through the clinical study data request platform (https://vivli.org). Further details on Roche’s criteria for eligible studies are available at https://vivli.org/members/ourmembers. For further details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202003-0669OC on January 12, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Ley B, Collard HR, King TE., Jr Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183:431–440. doi: 10.1164/rccm.201006-0894CI. [DOI] [PubMed] [Google Scholar]
- 2. Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society. Diagnosis of idiopathic pulmonary fibrosis: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198:e44–e68. doi: 10.1164/rccm.201807-1255ST. [DOI] [PubMed] [Google Scholar]
- 3. Nathan SD, Shlobin OA, Weir N, Ahmad S, Kaldjob JM, Battle E, et al. Long-term course and prognosis of idiopathic pulmonary fibrosis in the new millennium. Chest. 2011;140:221–229. doi: 10.1378/chest.10-2572. [DOI] [PubMed] [Google Scholar]
- 4. Vancheri C, Failla M, Crimi N, Raghu G. Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. Eur Respir J. 2010;35:496–504. doi: 10.1183/09031936.00077309. [DOI] [PubMed] [Google Scholar]
- 5. King TE, Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, et al. ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–2092. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 6. Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, et al. CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377:1760–1769. doi: 10.1016/S0140-6736(11)60405-4. [DOI] [PubMed] [Google Scholar]
- 7. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–2082. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- 8. Scott MKD, Quinn K, Li Q, Carroll R, Warsinske H, Vallania F, et al. Increased monocyte count as a cellular biomarker for poor outcomes in fibrotic diseases: a retrospective, multicentre cohort study. Lancet Respir Med. 2019;7:497–508. doi: 10.1016/S2213-2600(18)30508-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Neighbors M, Cabanski CR, Ramalingam TR, Sheng XR, Tew GW, Gu C, et al. Prognostic and predictive biomarkers for patients with idiopathic pulmonary fibrosis treated with pirfenidone: post-hoc assessment of the CAPACITY and ASCEND trials. Lancet Respir Med. 2018;6:615–626. doi: 10.1016/S2213-2600(18)30185-1. [DOI] [PubMed] [Google Scholar]
- 10. Kahn N, Rossler A-K, Hornemann K, Muley T, Grünig E, Schmidt W, et al. C-proSP-B: a possible biomarker for pulmonary diseases? Respiration. 2018;96:117–126. doi: 10.1159/000488245. [DOI] [PubMed] [Google Scholar]
- 11. Tzouvelekis A, Herazo-Maya JD, Slade M, Chu JH, Deiuliis G, Ryu C, et al. Validation of the prognostic value of MMP-7 in idiopathic pulmonary fibrosis. Respirology. 2017;22:486–493. doi: 10.1111/resp.12920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maher TM, Oballa E, Simpson JK, Porte J, Habgood A, Fahy WA, et al. An epithelial biomarker signature for idiopathic pulmonary fibrosis: an analysis from the multicentre PROFILE cohort study. Lancet Respir Med. 2017;5:946–955. doi: 10.1016/S2213-2600(17)30430-7. [DOI] [PubMed] [Google Scholar]
- 13. Jenkins RG, Simpson JK, Saini G, Bentley JH, Russell AM, Braybrooke R, et al. Longitudinal change in collagen degradation biomarkers in idiopathic pulmonary fibrosis: an analysis from the prospective, multicentre PROFILE study. Lancet Respir Med. 2015;3:462–472. doi: 10.1016/S2213-2600(15)00048-X. [DOI] [PubMed] [Google Scholar]
- 14. Raghu G, Richeldi L, Jagerschmidt A, Martin V, Subramaniam A, Ozoux ML, et al. Idiopathic pulmonary fibrosis: prospective, case-controlled study of natural history and circulating biomarkers. Chest. 2018;154:1359–1370. doi: 10.1016/j.chest.2018.08.1083. [DOI] [PubMed] [Google Scholar]
- 15. Herazo-Maya JD, Sun J, Molyneaux PL, Li Q, Villalba JA, Tzouvelekis A, et al. Validation of a 52-gene risk profile for outcome prediction in patients with idiopathic pulmonary fibrosis: an international, multicentre, cohort study. Lancet Respir Med. 2017;5:857–868. doi: 10.1016/S2213-2600(17)30349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Korthagen NM, van Moorsel CH, Barlo NP, Ruven HJ, Kruit A, Heron M, et al. Serum and BALF YKL-40 levels are predictors of survival in idiopathic pulmonary fibrosis. Respir Med. 2011;105:106–113. doi: 10.1016/j.rmed.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 17. Stuart BD, Lee JS, Kozlitina J, Noth I, Devine MS, Glazer CS, et al. Effect of telomere length on survival in patients with idiopathic pulmonary fibrosis: an observational cohort study with independent validation. Lancet Respir Med. 2014;2:557–565. doi: 10.1016/S2213-2600(14)70124-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Richards TJ, Kaminski N, Baribaud F, Flavin S, Brodmerkel C, Horowitz D, et al. Peripheral blood proteins predict mortality in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;185:67–76. doi: 10.1164/rccm.201101-0058OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Organ LA, Duggan AR, Oballa E, Taggart SC, Simpson JK, Kang’ombe AR, et al. Biomarkers of collagen synthesis predict progression in the PROFILE idiopathic pulmonary fibrosis cohort. Respir Res. 2019;20:148. doi: 10.1186/s12931-019-1118-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kreuter M, Maher TM. Can monocytes predict prognosis of idiopathic pulmonary fibrosis? Lancet Respir Med. 2019;7:467–469. doi: 10.1016/S2213-2600(19)30050-5. [DOI] [PubMed] [Google Scholar]
- 21. Suissa S, Dell’Aniello S, Ernst P. Comparative effectiveness of LABA-ICS versus LAMA as initial treatment in COPD targeted by blood eosinophils: a population-based cohort study. Lancet Respir Med. 2018;6:855–862. doi: 10.1016/S2213-2600(18)30368-0. [DOI] [PubMed] [Google Scholar]
- 22. Xu-Welliver M, Carbone DP. Blood-based biomarkers in lung cancer: prognosis and treatment decisions. Transl Lung Cancer Res. 2017;6:708–712. doi: 10.21037/tlcr.2017.09.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Håkansson KEJ, Rasmussen LJH, Godtfredsen NS, Tupper OD, Eugen-Olsen J, Kallemose T, et al. The biomarkers suPAR and blood eosinophils are associated with hospital readmissions and mortality in asthma - a retrospective cohort study. Respir Res. 2019;20:258. doi: 10.1186/s12931-019-1234-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Teoh AKY, Jo HE, Chambers DC, Symons K, Walters EH, Goh NS, et al. Blood monocyte counts as a potential prognostic marker for idiopathic pulmonary fibrosis: analysis from the Australian IPF registry. Eur Respir J. 2020;55:1901855. doi: 10.1183/13993003.01855-2019. [DOI] [PubMed] [Google Scholar]
- 25. Kreuter M, Lee JS, Tzouvelekis AE, Oldham JM, Molyneaux PL, Weycker D, et al. Monocyte count as a prognostic biomarker in patients with idiopathic pulmonary fibrosis (IPF): a retrospective, pooled analysis from Ascend, Capacity, and Inspire [abstract] Am J Respir Crit Care Med. 2020;201:A6207. doi: 10.1164/rccm.202003-0669OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. King TE, Jr, Albera C, Bradford WZ, Costabel U, Hormel P, Lancaster L, et al. INSPIRE Study Group. Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): a multicentre, randomised, placebo-controlled trial. Lancet. 2009;374:222–228. doi: 10.1016/S0140-6736(09)60551-1. [DOI] [PubMed] [Google Scholar]
- 27. Gregory AD, Kliment CR, Metz HE, Kim K-H, Kargl J, Agostini BA, et al. Neutrophil elastase promotes myofibroblast differentiation in lung fibrosis. J Leukoc Biol. 2015;98:143–152. doi: 10.1189/jlb.3HI1014-493R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hou Z, Ye Q, Qiu M, Hao Y, Han J, Zeng H. Increased activated regulatory T cells proportion correlate with the severity of idiopathic pulmonary fibrosis. Respir Res. 2017;18:170. doi: 10.1186/s12931-017-0653-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moore BB, Fry C, Zhou Y, Murray S, Han MK, Martinez FJ, et al. The COMET Investigators. Inflammatory leukocyte phenotypes correlate with disease progression in idiopathic pulmonary fibrosis. Front Med. 2014;1:56. doi: 10.3389/fmed.2014.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Reilkoff RA, Peng H, Murray LA, Peng X, Russell T, Montgomery R, et al. Semaphorin 7a+ regulatory T cells are associated with progressive idiopathic pulmonary fibrosis and are implicated in transforming growth factor-β1-induced pulmonary fibrosis. Am J Respir Crit Care Med. 2013;187:180–188. doi: 10.1164/rccm.201206-1109OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xue J, Kass DJ, Bon J, Vuga L, Tan J, Csizmadia E, et al. Plasma B lymphocyte stimulator and B cell differentiation in idiopathic pulmonary fibrosis patients. J Immunol. 2013;191:2089–2095. doi: 10.4049/jimmunol.1203476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med. 2017;214:2387–2404. doi: 10.1084/jem.20162152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ziegenhagen MW, Zabel P, Zissel G, Schlaak M, Müller-Quernheim J. Serum level of interleukin 8 is elevated in idiopathic pulmonary fibrosis and indicates disease activity. Am J Respir Crit Care Med. 1998;157:762–768. doi: 10.1164/ajrccm.157.3.9705014. [DOI] [PubMed] [Google Scholar]
- 34. Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med. 2018;378:1811–1823. doi: 10.1056/NEJMra1705751. [DOI] [PubMed] [Google Scholar]
- 35. Betensley A, Sharif R, Karamichos D. A systematic review of the role of dysfunctional wound healing in the pathogenesis and treatment of idiopathic pulmonary fibrosis. J Clin Med. 2016;6:E2. doi: 10.3390/jcm6010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Boyette LB, Macedo C, Hadi K, Elinoff BD, Walters JT, Ramaswami B, et al. Phenotype, function, and differentiation potential of human monocyte subsets. PLoS One. 2017;12:e0176460. doi: 10.1371/journal.pone.0176460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Duffield JS, Lupher M, Thannickal VJ, Wynn TA. Host responses in tissue repair and fibrosis. Annu Rev Pathol. 2013;8:241–276. doi: 10.1146/annurev-pathol-020712-163930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Byrne AJ, Maher TM, Lloyd CM. Pulmonary macrophages: a new therapeutic pathway in fibrosing lung disease? Trends Mol Med. 2016;22:303–316. doi: 10.1016/j.molmed.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 39. Greiffo FR, Viteri-Alvarez V, Frankenberger M, Dietel D, Ortega-Gomez A, Lee JS, et al. CX3CR1-fractalkine axis drives kinetic changes of monocytes in fibrotic interstitial lung diseases. Eur Respir J. 2020;55:pii1900460. doi: 10.1183/13993003.00460-2019. [DOI] [PubMed] [Google Scholar]
- 40. Byrne AJ, Powell JE, O’Sullivan BJ, Ogger PP, Hoffland A, Cook J, et al. Dynamics of human monocytes and airway macrophages during healthy aging and after transplant. J Exp Med. 2020;217:e20191236. doi: 10.1084/jem.20191236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 2019;20:163–172. doi: 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, et al. Single Cell RNA-seq reveals ectopic and aberrant lung resident cell populations in Idiopathic Pulmonary Fibrosis [preprint] bioRxiv 2019. Available from: https://www.biorxiv.org/content/10.1101/759902v1 [DOI] [PMC free article] [PubMed]
- 43. Nouno T, Okamoto M, Ohnishi K, Kaieda S, Tominaga M, Zaizen Y, et al. Elevation of pulmonary CD163+ and CD204+ macrophages is associated with the clinical course of idiopathic pulmonary fibrosis patients. J Thorac Dis. 2019;11:4005–4017. doi: 10.21037/jtd.2019.09.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Allden SJ, Ogger PP, Ghai P, McErlean P, Hewitt R, Toshner R, et al. The transferrin receptor CD71 delineates functionally distinct airway macrophage subsets during idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2019;200:209–219. doi: 10.1164/rccm.201809-1775OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Walsh SM, Worrell JC, Fabre A, Hinz B, Kane R, Keane MP. Novel differences in gene expression and functional capabilities of myofibroblast populations in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2018;315:L697–L710. doi: 10.1152/ajplung.00543.2017. [DOI] [PubMed] [Google Scholar]
- 46. Heukels P, van Hulst JAC, van Nimwegen M, Boorsma CE, Melgert BN, van den Toorn LM, et al. Fibrocytes are increased in lung and peripheral blood of patients with idiopathic pulmonary fibrosis. Respir Res. 2018;19:90. doi: 10.1186/s12931-018-0798-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moeller A, Gilpin SE, Ask K, Cox G, Cook D, Gauldie J, et al. Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179:588–594. doi: 10.1164/rccm.200810-1534OC. [DOI] [PubMed] [Google Scholar]
- 48. Prasse A, Probst C, Bargagli E, Zissel G, Toews GB, Flaherty KR, et al. Serum CC-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179:717–723. doi: 10.1164/rccm.200808-1201OC. [DOI] [PubMed] [Google Scholar]
- 49. Cai M, Bonella F, He X, Sixt SU, Sarria R, Guzman J, et al. CCL18 in serum, BAL fluid and alveolar macrophage culture supernatant in interstitial lung diseases. Respir Med. 2013;107:1444–1452. doi: 10.1016/j.rmed.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 50. Kawamura K, Ichikado K, Anan K, Yasuda Y, Sekido Y, Suga M, et al. Monocyte count and the risk for acute exacerbation of fibrosing interstitial lung disease: a retrospective cohort study. Chron Respir Dis. 2020;17:1479973120909840. doi: 10.1177/1479973120909840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ji H, Li Y, Fan Z, Zuo B, Jian X, Li L, et al. Monocyte/lymphocyte ratio predicts the severity of coronary artery disease: a syntax score assessment. BMC Cardiovasc Disord. 2017;17:90. doi: 10.1186/s12872-017-0507-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yeager MP, Pioli PA, Collins J, Barr F, Metzler S, Sites BD, et al. Glucocorticoids enhance the in vivo migratory response of human monocytes. Brain Behav Immun. 2016;54:86–94. doi: 10.1016/j.bbi.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]