

Abstract
The analysis of proteins expressed on circulating extracellular vesicles (EVs) could facilitate the diagnosis of different types of cancers. EV assays however have lengthy sample workups and limited throughput and sensitivity, making them unsuitable for routine clinical use. Here, we report a high-throughput assay that integrates EV enrichment, via antibody-coated magnetic beads, with the detection of EV-bound antibodies, via an electrochemical reaction. The assay requires less than one hour, is performed on plasma samples, and its 96-well plate format enables measurements in parallel via a prototype reader. Using samples from patients with colorectal cancer or healthy volunteers, we identified a panel of biomarkers (EGFR, EpCAM, CD24, GPA33) in circulating EVs that, when combined, showed higher diagnostic accuracy (>96%) than conventional assays. In a prospective cohort, the combined biomarker profile enabled assigning patients to a high- or a low-risk 5-year disease-free survival group, and the serial monitoring of EVs during therapy showed values declined after surgery yet increased upon relapse. Biomarker panels from plasma EVs may be suitable for the non-invasive monitoring of disease trajectory.
Assessing circulating biomarkers, also known as liquid biopsy, is an emerging approach to obtaining molecular information about patients’ tumours through repeated yet minimally invasive sampling1–4. One appealing target for such assessment is extracellular vesicles (EVs) — membrane particles secreted by cells5,6. In addition to being generally abundant and stable, EVs have been reported to carry biomolecules (e.g., proteins7,8, nucleic acids9–11, lipids12) of parent cells. Analysing tumour-derived EVs in particular has considerable potential to reveal tumours’ dynamic status13–15 and thereby improve current cancer diagnostics10,11,14–20. Establishing clinical EV tests, however, faces multiple technical challenges, namely i) laborious manual sample preparation, ii) existing tools’ limited sensitivity and throughput; and iii) high cost of test equipment or assays (e.g., sequencing). In short, for EV diagnostics to become clinically useful, new integrative methods for EV isolation and molecular analyses are needed, ideally ones that are amenable to high-throughput operations21. Equally important is to analyse large clinical samples to establish robust EV biomarker baselines for disease status.
In the current study, we aimed technical advances towards clinical EV tests. First, we developed a clinically adoptable high-throughput EV analysis technology termed HiMEX (high-throughput integrated magneto-electrochemical extracellular vesicle). This technology streamlined EV analyses by combining EV enrichment and electrochemical detection in a single assay, enabling EV protein profiling directly from clinical samples; the total assay time was <1 hour (in a 96-well format) and the analytical signal was read out in parallel from all (96) detection probes. We next applied HiMEX to analyse clinical samples, demonstrating HiMEX’s practical advantages. Specifically, we analysed blood samples from 102 colorectal cancer (CRC) patients as they underwent surgery and chemotherapy plus 40 non-CRC controls (n = 142 total). HiMEX analyses revealed EVs’ diverse potentials for CRC management. i) EVs that reflected parental tumours’ key protein signatures were present in circulation, and detecting such CRC-derived EVs led to highly accurate cancer diagnoses (overall accuracy >96%). ii) Serial changes in CRC-EVs could be related to patients’ treatment responses. Importantly, CRC-EV levels decreased in all patients after curative surgery but rebounded from each patient’s baseline values with tumour recurrence. iii) Preoperative CRC-EV levels showed significant correlation with patients’ 5-year disease-free survival (n = 90), effectively categorizing patients into high- and low-risk groups. These outcomes have implications for timely, better-informed cancer care, broadening EVs’ clinical utility for CRC diagnosis, recurrence monitoring, and prognosis.
Results
HiMEX approach for clinical EV tests.
Our study aimed to evaluate HiMEX for applications in CRC diagnosis, treatment monitoring, and prognosis (Fig. 1a and Supplementary Fig. 1). Overall, we analysed plasma sample of 102 CRC patients and 40 non-CRC controls (n = 142 total). For CRC diagnostics, we first defined a CRC-EV signature based on published results, in-vitro studies, and tissue immunohistochemistry. We next measured these markers in plasma EVs. A total of 131 patient samples were used: a training cohort consisting of 25 non-CRC controls and 58 CRC patients; and a testing cohort of 15 non-CRC controls and 33 CRC patients. For patient monitoring, we followed additional 11 CRC patients as they underwent standard clinical care. Serial blood samples were collected at defined time points (i.e., before and after surgery and during chemotherapy) and analysed for CRC EV markers. The EV-profiling results were then compared against clinical information, including levels of conventional tumour markers (carcinoembryonic antigen, CEA; carbohydrate antigen, CA19-9)22,23, radiologic assessment, and treatment outcomes. Finally, for the prognostic use of EV profiling, we correlated 5-year disease-free survival with pre-surgery EV CRC levels among 90 CRC patients.
Fig. 1 |. HiMEX approach for clinical EV analyses.
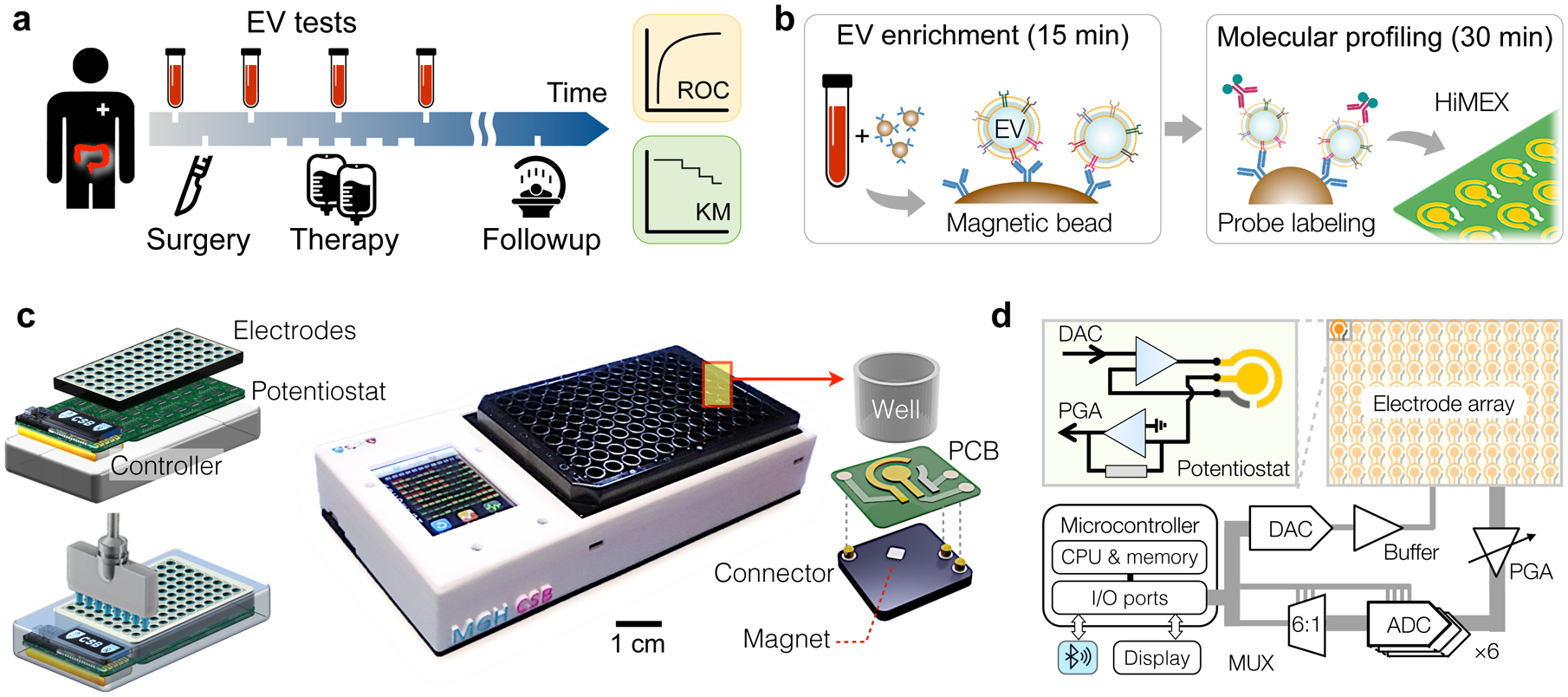
a, Study design. We collected blood samples from colorectal cancer (CRC) patients (n = 91) during their standard clinical care. Extracellular vesicles (EVs) as well as conventional markers (i.e., CEA, CA19-9) were analysed from blood samples and cross-compared with clinical outcomes, including immunohistology, radiologic reports, and survival. b, Two-step HiMEX assay protocol. EVs are enriched via immunomagnetic capture. Bead-bound EVs are then labelled with probing antibodies for signal generation through electrochemical reaction. The assay is simple and fast (<1 hour), directly analysing plasma samples without requiring purification steps. c, We developed a compact HiMEX reader with a touchscreen interface. The reader accommodated an array of 96 electrodes in a conventional 96-well plate. Under each electrode were push-pin connectors to make electrical contacts and a magnet to concentrate bead-bound EVs. PCB, printed circuit board. d, The HiMEX reader was designed to carry out 96 parallel measurements within 2 minutes. Each electrode was connected its own potentiostat. A microcontroller applied electrical potential through a digital-to-analogue converter (DAC), initiating electrochemical reaction. Resulting currents from the electrode were read out by an analogue-to-digital converter (ADC) via rapid multiplexing (MUX). The microcontroller processed and displayed data on a touchscreen. The reader also communicated with an external device (e.g., smartphones) for data logging. PGA, programmable gain amplifier.
To streamline EV analyses, we optimized the HiMEX technology, integrating sample preparation and measurement within a single assay (Fig. 1b). We employed immunomagnetic pulldown to enrich target-specific EVs, which allowed for rapid (≈15 min) EV isolation directly from native samples without additional manipulation. We then harnessed magnetic beads as substrates for subsequent signal generation; bead-bound EVs were labelled with probing antibodies functionalized with catalysing enzymes for electrochemical reaction16,24. The HiMEX strategy had the following merits: i) sample handling was simple (i.e., magnetic pulling), with no extensive EV purification required; ii) the assay benefitted from fast binding kinetics and high efficiency in EV capture and labelling (~30 min in total); iii) analytical signal was amplified via enzymatic reaction; and iv) the detection modality (i.e., electrical measurements) was amenable to scale-up for high-throughput measurements.
HiMEX detection system for parallel measurements.
Exploiting HiMEX’s unique technical advantages, we developed a compact system capable of parallel measurements (Fig. 1c). The system was compatible with a 96-well plate to enable batch processing. EV capture and labelling were performed in a conventional 96-well plate; a custom-designed magnet array (Supplementary Fig. 2a) was used for bead handling (e.g., washing, buffer exchange). EV-bound beads were then spotted on a 12 × 8 probe array (Supplementary Fig. 2b) that was loaded to the detection device. Each probe in the array, which contained three electrodes (i.e., working, counter, reference), was connected to a potentiostat through quick push-pin connectors and was placed over a small embedded magnet to concentrate magnetic beads (Fig. 1c and Supplementary Fig. 2c). The detection system also featured a touchscreen interface for device control and data display, and wirelessly communicated (via Bluetooth) with external servers for data storage.
As an analytical signal, electrical currents between working and counter electrodes were monitored while a constant voltage was applied between working and reference electrodes. To enable fast readouts, we designed the system electronics to rapidly (100 Hz) poll each probe during measurements (Fig. 1d): an embedded microcontroller sequentially accessed individual probes through six analogue-to-digital converters and a 6-to-1 multiplexer. This scheme effectively executed real time, parallel measurements (Supplementary Fig. 2d). The total readout time for all 96 probes was < 2 min.
We benchmarked the system performance using samples with varying K4Fe(CN)6 concentrations. We particularly focused on assessing the reproducibility among 96 probes. For a given K4Fe(CN)6 concentration, different probes in the array reported consistent signal (Supplementary Fig. 3a). Four standard curves, generated through parallel measurement of K4Fe(CN)6 dilution series, showed an excellent linear relationship between current levels and K4Fe(CN)6 concentrations (Supplementary Fig. 3b). Importantly, these curves were statistically identical, demonstrating high fidelity among probes (i.e., low intra-probe variations) for the parallel detection.
HiMEX assay characterization.
We next optimized key assay steps, namely EV capture and electrochemical detection, to maximize analytical signal. For EV capture, we prepared three types of magnetic beads, each specific to one of tetraspanins (i.e., CD63, CD9, CD81) enriched in EVs. When mixed with EVs, these beads were effectively covered with vesicles (Fig. 2a); control beads conjugated with IgG antibodies showed minimal non-specific binding (Supplementary Fig. 4). We further used either single types or a cocktail of capture beads for EV pull-down and compared EV tetraspanin levels (Fig. 2b). Using a mixture of capture beads (CD63, CD9, CD81) led to higher signal across all tetraspanin levels than using any single-marker bead alone (Fig. 2b and Supplementary Fig. 5a), presumably reflecting EV heterogeneity25–27. Single EV imaging (Supplementary Fig. 6) supported the observation; the number of EVs visible under microscopy was the highest when the cocktail of CD63, CD9, and CD81 antibodies were used for fluorescent staining. The immunomagnetic capture was efficient in plasma as well (Supplementary Fig. 5b); based on CD63 levels before and after immunocapture, the pull-down yield was estimated to be 82%.
Fig. 2 |. HiMEX assay optimization and characterization.
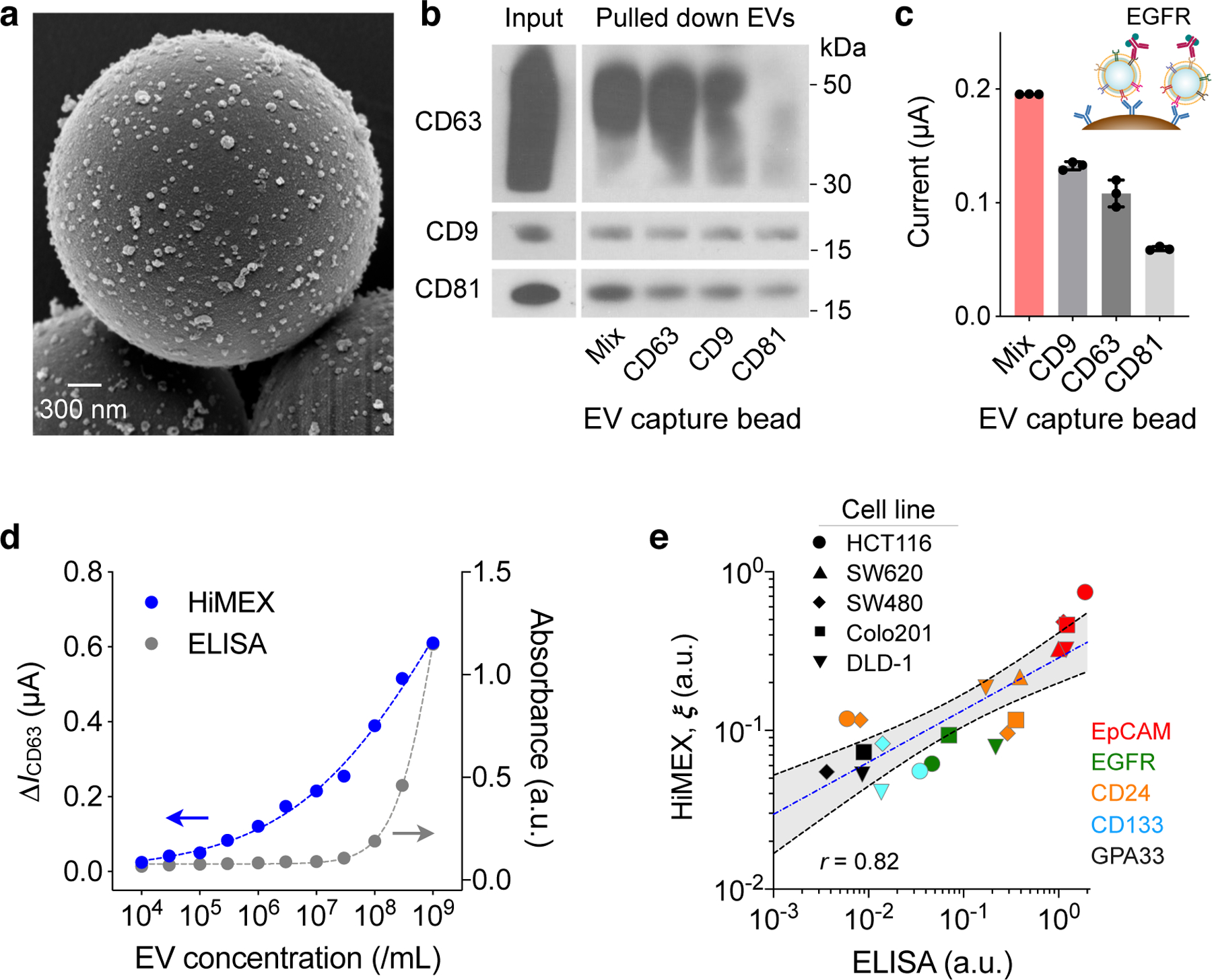
a, Scanning electron micrograph of microbeads after incubation with EV samples. The beads (diameter, 3 μm), functionalized with antibodies against CD63, captured EVs isolated from a cell culture media (HCT116 cell line). A representative image is selected from technical duplicate samples. b, Three types of magnetic beads, with each type specific to a different tetraspanin, and their mixture (Mix) were used to capture EVs. Key tetraspanin expression was then measured on captured EVs. The bead cocktail most efficiently overcame EV heterogeneity. A representative image is selected from duplicate measurements. Full western blot images are shown in Supplementary Information. c, EVs (107/mL) from HT29 cell lines were captured and further labelled for EGFR. Mixed bead types led to the highest HiMEX signal. The data are displayed as mean ± SD from technical triplicates. d, The HiMEX assay had superior sensitivity and wider dynamic range than conventional ELISA. The limit of detection was ~104 EVs/mL for HiMEX and ~107 EVs/mL for ELISA. a.u., arbitrary unit. EV numbers were estimated from nanoparticle tracking analysis. The data points represent mean from technical duplicates. e, EVs from different CRC cell lines were profiled to a set of protein markers. The HiMEX results showed a good match (Pearson correlation coefficient, r = 0.82) with those from ELISA. To obtain the HiMEX expression (ξ) of a target protein marker (M), the marker-associated current level was normalized against the loading control, the current level of CD63. For ELISA, the same amount of EVs (109 EVs/mL) was used for each marker. Data were plotted in log scales to better display small values. The blue-dashed line indicates the best linear fit in the log-log scale, and the shaded grey area 95% confidence band. HiMEX data represent mean values from technical duplicates.
To generate detection signal, we used a probing antibody to label captured EVs with an oxidizing enzyme (horseradish peroxidase, HRP) and mixed the conjugates with chromogenic electron mediators (3,3′,5,5′-tetramethylbenzidine, TMB). The redox reaction quickly reached an equilibrium, with electrical currents plateauing within 1 min (Supplementary Fig. 7). We recorded the current between 50 and 55 sec after initiating the reaction and used the average value (I) as an analytical metric. Because it consistently produced maximum HiMEX signal (Fig. 2c), we used the three-bead cocktail for EV capture throughout the rest of the study.
Our next step was to characterize HiMEX’s analytical capacities. To begin, we prepared samples with varying EV concentrations. Using the CD63, CD9, and CD81 bead mixture, we captured EVs and labelled them for CD63; we generated control samples by labelling captured EVs with IgG antibodies. These samples were then measured using the HiMEX detector (Supplementary Fig. 7). We used the net current ΔICD63 = ICD63 – IIgG as an analytical metric to minimize the effect of common-mode errors (e.g., temperature fluctuation, non-specific bindings). From the titration results (Fig. 2d), the limit of detection (LOD) of the HiMEX technology was estimated at ~104 EVs, >103-fold lower than conventional ELISA. HiMEX also displayed much wider dynamic ranges (~105 EVs) than ELISA (<103 EVs). We next assessed assay reproducibility. On three different days, we prepared EV samples and probed them for CD63, CD9, and CD81 using the HiMEX array. For a given marker, the observed signals were statistically identical among different days (Supplementary Fig. 8). We further compared signal levels with EVs spiked either in plasma or serum (Supplementary Fig. 9) and observed similar signal changes in both sample types. We opted for plasma as it is considered the physiological medium for EVs, containing less number of platelet-derived vesicles28.
To test whether EVs, captured by the bead mixture, carry CRC-relevant markers, we performed a competitive assay (Supplementary Fig. 10a). Samples were prepared by spiking EVs from a CRC cell line (SW480; EpCAM and CD24 positive) into human plasma. EV immunomagnetic capture was carried out in the presence of excess, free-floating capture antibodies (a mixture of CD63, CD9, CD81). We then probed bead-bound EVs for CD63, EpCAM, and CD24 expression. The measured HiMEX signal decreased as the concentration of free antibodies increased (Supplementary Fig. 10b), indicating the presence of CRC markers on bead-captured EVs.
For a quantitative HiMEX assay, we established the following protocol. For a given marker (M) of interest, we measured HiMEX signal (ΔIM = IM – IIgG) after EV capture and labelling. As an EV-loading control, we also measured ΔICD63 using a same-sample aliquot. The expression level (ξM) of the target marker was then estimated by scaling IM against the loading control (ξM = ΔIM / ΔICD63). We applied the method to profile EVs for different protein markers. The results showed a good linear correlation (Pearson r = 0.82) with ELISA (Fig. 2e).
High-throughput screening of cell-line derived EVs.
To choose the initial EV markers relevant for CRC detection, we applied a custom-designed bioinformatic pipeline to public databases (see Methods; Supplementary Fig. 11). From Human Protein Atlas, we retrieved protein expression profiles for CRC and normal tissues and chose markers that are over-expressed in CRC. This collection was then cross-referenced with UniProt annotations to select proteins containing an extracellular domain. We further narrowed down the list by applying two criteria: i) reported marker presence in EVs (database: Vesiclepedia); and ii) availability of ELISA-compatible antibodies (Supplementary Table 1). This algorithm selected nine markers (CD44, B7-H3, EGFR, ABCG2, GPA33, EpCAM, HER2, MUC1, MET). We augmented the list by including markers that were observed to be over-expressed in CRC29–34(CD44v6, CD24, CD166, STEAP1, ALDH1) and those used for drug-resistance monitoring35–37 (MRP1, MDR1, TS, CD133).
We then measured the candidate markers across CRC cell-derived EVs, utilizing HiMEX’s high-throughput capacity. This preclinical study focused on i) investigating the correlation in marker expression between EVs and their originating cells; and ii) checking the presence of candidate markers in EVs. The cell-line panel included cells representing different CRC stages38 (SW480, Dukes’ classification Type B; DLD-1 and SW620, Type C; Colo201, Type D) as well as drug-resistant (irinotecan) phenotypes (HT29, HCT116). As a negative control, we profiled EVs from a normal colon cell line (CCD-18Co) as well as plasma from healthy donors; inclusion of healthy plasma samples was justifiable as the analytical goal was to identify markers over-expressed in CRC EVs. After collecting EVs, we profiled them by HiMEX (Supplementary Fig. 12a); cellular expression was measured by flow cytometry. Overall, the selected markers’ expression profiles closely matched between EVs and their parent cells (r = 0.89; Supplementary Fig. 12b), supporting the use of EVs as cellular surrogates7,10,14,17,39. For further testing with clinical EV samples, we chose EpCAM, EGFR, CD24, and CD133, based on their relatively higher expression in CRC cell lines (Supplementary Fig. 12c). We also included GPA33 for its clinical relevance in CRC diagnostics40.
Defining EV protein signature for CRC diagnostics in clinical sample.
We next applied HiMEX to detect CRC EVs in clinical samples. We first examined whether circulating EVs from CRC patients carry key protein markers found in CRC tissue. From a pilot cohort of 12 CRC patients, we obtained both preoperative blood samples and tissue specimens during surgery. We assessed the expression of top three markers (EpCAM, EGFR, CD24) identified from the cell-line study. Tissue specimens were stained (immunohistochemistry, IHC) for these markers, and the expression was graded by a pathologist as a fraction of marker-positive cells among total cancer cells (from 0 to 1 with the increment of 0.1; see Methods for details). EVs were screened for the same markers via HiMEX (Fig. 3a). Overall, marker expression between tissue and EV samples displayed good concordance (Fig. 3b and Supplementary Fig. 13) and a positive correlation (Spearman’s rank coefficient ρs = 0.65, p < 0.0001). These results indicated the presence of CRC-derived EVs in circulation, thereby supporting the rationale of assessing tumour molecular status through EV profiling.
Fig. 3 |. EV profiling for CRC detection.
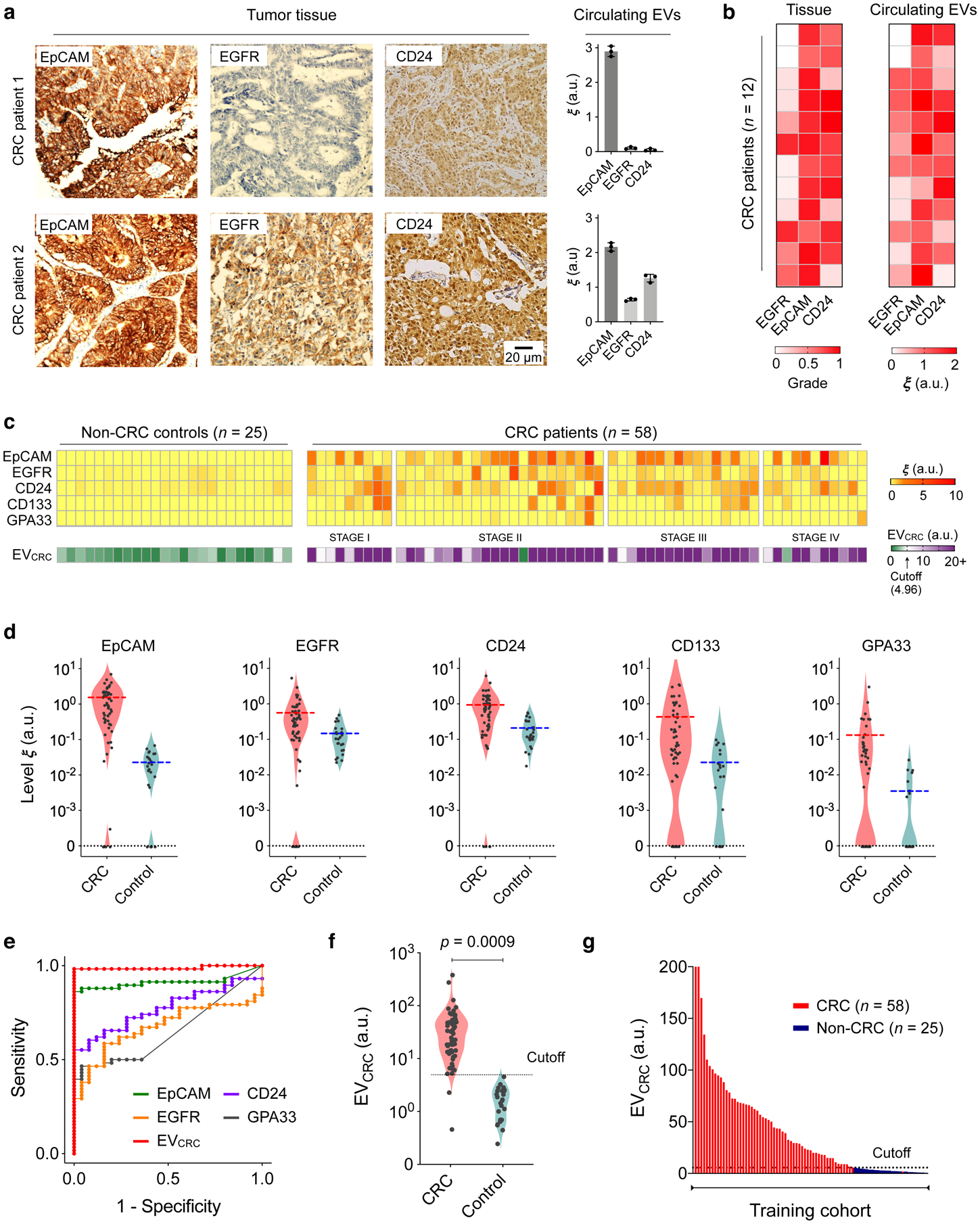
a, Tumour tissue and plasma EV analyses. Three key CRC protein markers (EpCAM, EGFR, CD24) were assessed in tumour tissues (immunohistochemistry) and blood samples (HiMEX) from each patient. Expression profiles showed a qualitative match. Data from two representative patients are shown. The bar graphs show mean ± SEM from technical triplicate measurements. Full images of these samples are shown in Supplementary Information. b, Tumour tissue and plasma EV samples from 12 CRC patients were analysed for EpCAM, EGFR, and CD24. Tissue staining was graded as the fraction of marker-positive cells among total cancer cells. The pathological score and the EV expression profile showed significant correlation (Spearman rank coefficient ρs = 0.65, p < 0.0001; two-sided test). c, HiMEX analyses for CRC diagnosis. As a training set, plasma samples from 58 CRC patients before surgery and 25 non-CRC controls were analysed. The expression of five CRC markers (EpCAM, EGFR, CD24, CD133, and GPA33) was measured by HiMEX. The diagnostic metric, EVCRC, was determined as a weighted sum of four marker levels (EpCAM, EGFR, CD24, GPA33) through logistic regression. d, The average level of each marker was higher in CRC patients than in non-CRC controls. The marker distribution, however, overlapped between patients and controls, reducing the classification power of single markers. e, For each CRC marker and EVCRC, receiver operating characteristic (ROC) curves were constructed. The area under curve (AUC) of EVCRC was 0.98, significantly larger than those of single markers (all p < 0.001). Cutoff levels that maximized the sum of sensitivity and specificity were determined from the ROC curves. f–g, EVCRC effectively differentiated CRC patients from controls (p = 0.0009; unpaired two-sided t-test). In the training cohort, the diagnostic accuracy was 98%. The cutoff value from the EVCRC ROC curve was 4.96.
To establish EV-CRC diagnostics, we expanded the cohort and used the full marker set in EV profiling. Blood was collected from CRC patients before surgery. Control samples were obtained from healthy donors as well as non-CRC patients. All blood samples were collected and consistently processed per our established protocol (see Methods). Circulating EVs were assessed by HiMEX for EpCAM, EGFR, CD133, GPA33, CD24, and CD63 levels. Aliquots of samples were also assayed for the clinically relevant serum markers, CEA and CA19-9.
Figure 3c summarizes EV marker expression profiles from the initial training cohort (58 CRC patients, 25 non-CRC controls). The average levels of all five markers were higher in CRC patients than in controls (Fig. 3d). However, the two distributions overlapped, making classification based on a single marker difficult. We therefore considered multi-marker combinations. Specifically, we used weighted sums of EV markers, wherein optimal weights were determined by logistic regression (see Methods for details). For both individual markers and combinations thereof, we constructed receiver operating characteristic (ROC) curves (Fig. 3e and Supplementary Fig. 14). In each ROC curve, we determined a cutoff value that maximized the sum of sensitivity and specificity41. The combination of EGFR, EpCAM, CD24, and GPA33 was found to be most optimal; this metric was thus defined as the EVCRC score. The area under the curve (AUC) of EVCRC was significantly higher (all p < 0.001, DeLong’s test) than those of the individual EV markers (Fig. 3e) and two- or three-marker combinations. Compared to the five-marker combination, EVCRC showed statistically identical (p = 0.17, DeLong’s test) classification performance. Overall, EVCRC with the cutoff value of 4.96 showed a high diagnostic power (Figs. 3f, g) with the training cohort, achieving detection sensitivity of 97%, specificity of 100%, and accuracy of 98% (see Methods for details; summary in Table 2).
Table 2 |.
Summary of CRC diagnostic statistics for conventional and EV markers.
| Markers | Cutoff | Training cohort (n = 83) | Testing cohort (n = 48) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| AUC | Sensitivity (%) | Specificity (%) | Accuracy (%) | Sensitivity (%) | Specificity (%) | Accuracy (%) | |||
| Serum | CEA | 1.65 | 0.85 | 79 | 88 | 82 | 76 (58–89) |
60 (32–84) |
71 (56–83) |
| CA199 | 12.9 | 0.74 | 64 | 84 | 70 | 52 (34–69) |
87 (61–98) |
63 (48–77) |
|
| EV single markers | EGFR | 0.23 | 0.67 | 59 | 84 | 66 | 52 (34–69) |
87 (60–98) |
63 (48–77) |
| EpCAM | 0.08 | 0.91 | 86 | 100 | 90 | 85 (68–95) |
93 (68–100) |
88 (75–95) |
|
| CD24 | 0.60 | 0.77 | 55 | 100 | 69 | 39 (23–58) |
100 (78–100) |
58 (43–72) |
|
| GPA33 | 0.02 | 0.65 | 47 | 96 | 61 | 42 (25–61) |
73 (45–92) |
52 (37–67) |
|
| CD133 | 0.03 | 0.70 | 66 | 76 | 69 | 58 (39–75) |
53 (27–79) |
56 (41–71) |
|
| EGFR + EpCAM | 2.69 | 0.94 | 90 | 100 | 93 | 88 (72–97) |
100 (78–100) |
92 (80–98) |
|
| EGFR + CD24 | 3.15 | 0.86 | 60 | 90 | 71 | 45 (28–64) |
100 (78–100) |
63 (47–76) |
|
| EpCAM + CD24 | 4.23 | 0.97 | 91 | 100 | 94 | 88 (72–97) |
100 (78–100) |
92 (80–98) |
|
| EV weighted combination | EGFR + EpCAM + CD24 | 4.23 | 0.97 | 91 | 100 | 94 | 88 (72–97) |
100 (78–100) |
92 (80–98) |
| EGFR + EpCAM + CD24 + CD133 | 6.15 | 0.99 | 95 | 100 | 96 | 88 (72–97) |
93 (68–100) |
90 (77–97) |
|
| EGFR + EpCAM + CD24 + GPA33 | 4.96 | 0.98 | 97 | 100 | 98 | 94 (80–99) |
100 (78–100) |
96 (85–99) |
|
| EGFR + EpCAM + CD24 + GPA33 + CD133 | 5.63 | 0.99 | 98 | 100 | 99 | 100 (89–100) |
93 (68–100) |
96 (89–100) |
|
We further tested the statistical model by analysing samples from a prospective testing cohort (33 preoperative CRC patients and 15 non-CRC controls; Fig. 4a). With the same cutoffs from the training sets applied (Figs. 4b, c), EVCRC maintained excellent diagnostic statistics (sensitivity, 94%; specificity, 100%; accuracy, 96%). EVCRC’s diagnostic accuracy was superior to that of conventional serum markers (Supplementary Fig. 15a), presumably due to the CRC-specific nature of EV analyses. EVCRC showed no significant correlation (Supplementary Fig. 15b) with CEA (ρs = −0.12, p = 0.24) or CA19-9 (ρs = −0.11, p = 0.29); combining either of these serum markers with EVCRC (via logistic regression) did not significantly improve the AUC (p = 0.20 for the addition of CEA; p = 0.18 for the addition of CA19-9, DeLong’s test).
Fig. 4 |. Analyses of prospective cohorts for CRC diagnosis.
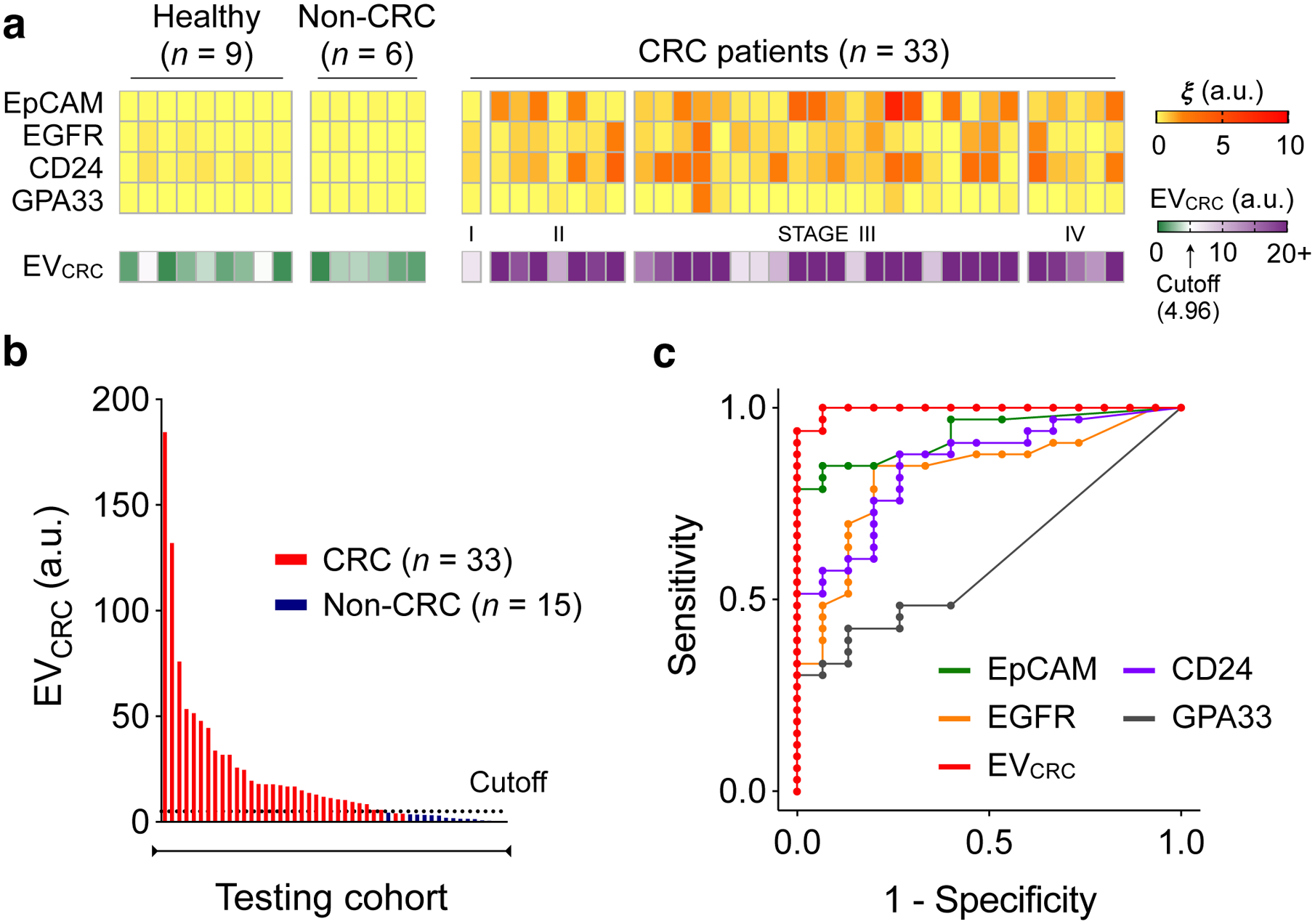
a, Plasma EVs from 33 CRC patients, 9 healthy donors, and 6 non-CRC patients (gastrointestinal stromal tumour, n = 2; small bowel internal herniation, n = 2; appendicitis, n = 2) were analysed for the expression EpCAM, EGFR, CD24, and GPA33. EVCRC was calculated according to the same formula as with the training cohorts. b, EVCRC levels were significantly higher in CRC patients than in healthy controls, validating the EV-based diagnostic algorithm. The same cutoff value (4.96) from the training set was applied. c, EVCRC remained superior in CRC detection, with its AUC significantly larger than those of single markers. Detailed statistics are in Table 2.
Longitudinal CRC EV monitoring during clinical care.
Next, we tracked longitudinal EV profile changes as patients underwent standard clinical care. We first compared EV levels before and after surgery (Fig. 5a). Blood samples (n = 13) were collected 24 hours before surgery and within one week thereafter. Overall EV loads showed no significant changes (p = 0.634; paired t-test) or directionality. In contrast, EVCRC decreased in all patients (p < 0.0001; paired t-test), presumably due to the presence of fewer CRC-derived EVs after curative tumour resection. The reasoning was further supported when we analysed blood samples of patients (n = 4) who underwent abdominal surgery for non-CRC diseases (e.g., appendicitis, small bowel internal herniation). For these patients, EVCRC values remained statistically identical (p = 0.77, paired t-test) before and after surgery (Supplementary Fig. 16). On the patient-wide average, CEA and CA19-9 levels decreased after surgery, but in pairwise comparison (pre- and post-surgery) the downward trend was not statistically significant (paired t-test, p = 0.19 for CEA and p = 0.190 for CA19-9).
Fig. 5 |. HiMEX analyses of longitudinal CRC patient samples.
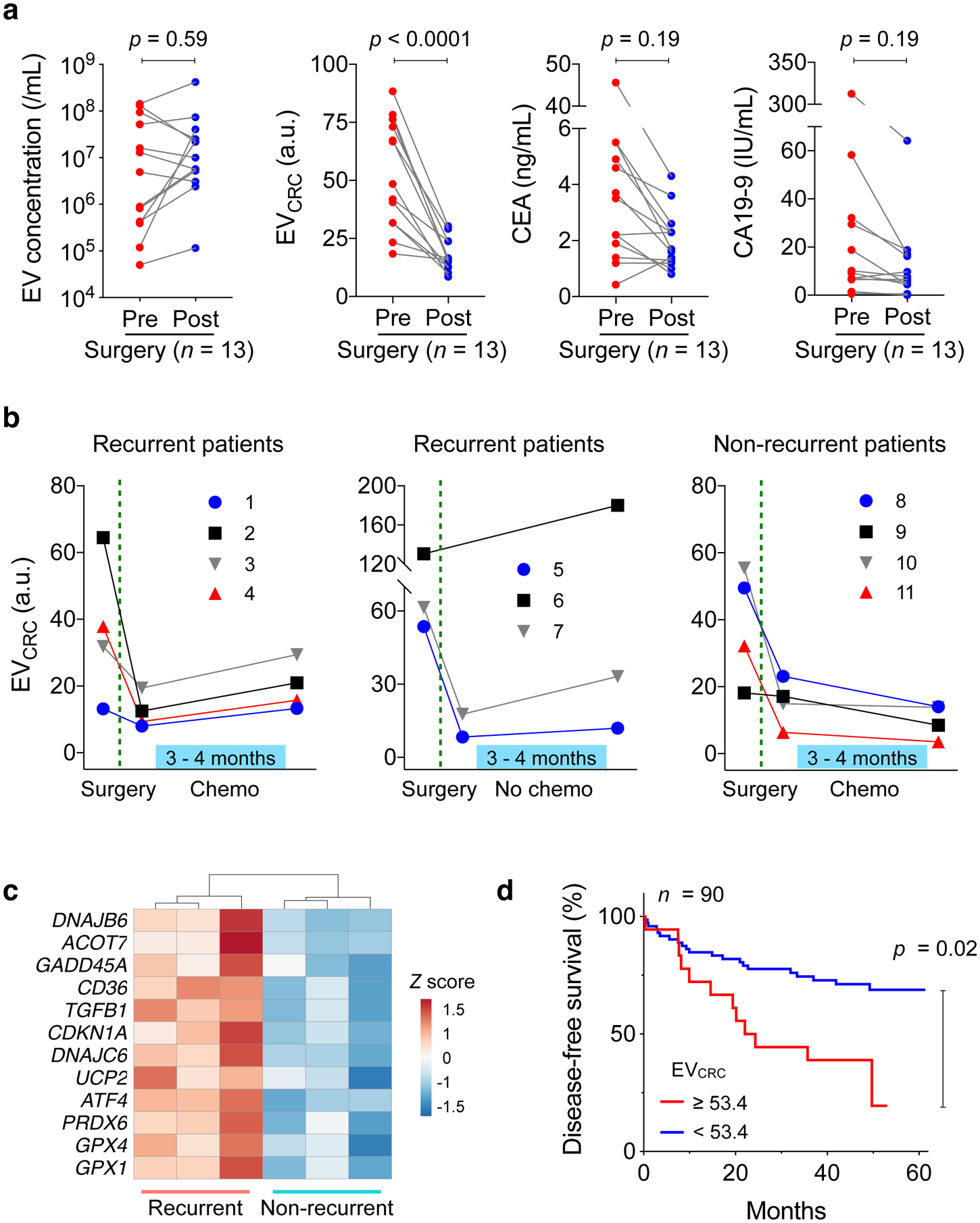
a, CRC patient blood samples were analysed before and after surgery (n = 13). Molecular EV profiling revealed that EVCRC values decreased in all patients (p < 0.0001; paired two-sided t-test), whereas total EV concentrations (p = 0.59; paired two-sided t-test) and the levels of conventional serum markers, CEA (p = 0.19; paired two-sided t-test) and CA19-9 (p = 0.19; paired two-sided t-test), showed no significant changes. Each data point in the graph represents the mean value from technical duplicates. b, Plasma EVs were further monitored after surgery, as some patients underwent chemotherapy. EVCRC values rebounded in all patients (n = 7) with recurrent tumours. By contrast, in patients without recurrent tumour (n = 4), EVCRC continued to decrease or stabilized from the post-surgery level. Each data point in the graph represents mean ± SEM from technical duplicates. c, EVs from recurrent (n = 3) and non-recurrent (n = 3) CRC patients were analysed for mRNA expression. Highly variable genes were plotted. Genes involved in DNA repair, protection against oxidative pressure, and chemoresistance (5-FU) showed higher levels in recurrent patients’ EVs. d, CRC patients (n = 90) were monitored up to five years for disease-free survival (DFS). Kaplan-Meier estimator for DFS was plotted, stratified by preoperative EVCRC levels. High EVCRC values (≥ 53.4) were associated with poor prognostics (p = 0.02, two-sided log-rank test).
To better ascertain temporality, we monitored additional CRC patients (n = 11) for six months after surgery. Eight patients, either at Stage II with risk factors (inadequate lymph node sampling, perforation, or lymphovascular invasion) or at Stage III, received adjuvant treatment (FOLFOX) with 5-FU/Leucovorin and oxaliplatin. The remaining three patients, all Stage II, were excluded from therapy (Supplementary Table 2 for patient details). For treated patients, blood samples were collected before and after therapy; for non-treated patients, samples were collected around 6 months after surgery. Tumour recurrence status was confirmed later either via surgical resection or radiological detection of lesions that increased in size over time. Among treated patients (n = 8), four patients were classified as non-recurrent and the rest as recurrent; tumours recurred in all non-treated patients (n = 3). Blind to these classifications, we performed HiMEX EV profiling. In all patients, EVCRC values decreased after surgery (Supplementary Fig. 17). From the post-surgery EVCRC baseline, EVCRC increased in all recurrent patients regardless of treatment status. In contrast, EVCRC decreased or became stable in non-recurrent patients (Fig. 5b). Serial changes in EVCRC (ΔEVCRC) thus showed potential as an indicator of short-term tumour relapse (Supplementary Fig. 18; p = 0.0004, unpaired two-sided t-test). Changes in CEA (p = 0.13, t-test) and CA19-9 (p = 0.76, t-test) were similar regardless of recurrence status.
EV capture and RNA analyses.
The immunomagnetic capture employed in HiMEX enabled us to enrich EVs not only for CRC protein detection (HiMEX) but also for other molecular assays. We applied this technique to aid in EV-RNA analyses in blood samples collected after adjuvant chemotherapy. As in the HiMEX assay, we used a cocktail of tetraspanin magnetic beads to capture EVs. Plasma samples of recurrent (n = 3) and non-recurrent (n = 3) patients were processed. Captured EVs were then lysed and their RNA contents were sequenced for quantitative gene expression profiling (see Materials and Methods for details). Principal component analysis, based on mRNA reads, separated the two cohorts (i.e., recurrent vs. non-recurrent), and more genes were up-regulated in the recurrent patient group (Supplementary Fig. 19a). We conducted differential analysis to identify highly variable genes across samples (Supplementary Fig. 19b). Interestingly, selected genes all had higher expression in the recurrent patients (Fig. 5c). These genes are involved in DNA damage repair (GADD45A, CDKN1A)42,43, protection against oxidative stress (GPX1, GPX4, PRDX6, UCP2)44–46, fatty acid oxidation (ACOT7, CD36)44,47, and resistance to 5-FU (TGFB1, DNAJB6, DNAJC6, ATF4)48–51.
Assessing CRC EV signature for prognosis of five-year disease-free survival.
Finally, we evaluated EV analyses’ capacity to predict disease-free survival. High CRC EV burden may indicate large primary lesions or malignant phenotypes; it is thus conceivable that high EVCRC levels before surgery could correlate with the risk of disease recurrence, likely caused by either residual tumour presence after surgery or metastasis52. We prospectively followed 90 CRC patients (Supplementary Table 3), who all had preoperative blood tests and underwent tumour resection, up to five years. From EV analyses, we indeed found that initial EVCRC scores were higher in the patient group whose tumours eventually progressed into metastasis (Supplementary Fig. 20; p = 0.041, unpaired two-sided t-test). Survival analyses further confirmed the association between high EVCRC values and poor disease-free survival (DFS). We stratified patients into two groups according to their EVCRC scores, with a score threshold of 53.4 selected to maximize the absolute log-rank statistic between the two strata53. Thirteen out of the 18 patients with high EVCRC scores reported tumour metastasis over the five years of follow-up, compared to 21 out of the 72 patients with EVCRC scores below the threshold; comparing the two survival curves suggested significant prognostic differences between the two groups (Fig. 5d; p = 0.02, two-sided log-rank test; see Methods for details). In contrast, preoperative CEA concentrations (cutoff, 7 ng/mL), often used for CRC prognostics22,54, displayed borderline performance with respect to DFS in this cohort (Supplementary Fig. 21; p = 0.07, two-sided log-rank test).
Discussion
Analysing tumour-derived EVs can provide real-time snapshots of tumours’ molecular makeup, potentially offering timely, better-informed opportunities for clinical intervention. We developed HiMEX to position such EV-based testings closer to clinical reality. The HiMEX approach integrates EV isolation and protein detection in a continuous workflow to enable batch-processing of clinical samples. In particular, HiMEX offers the following technical advantages: i) direct use of plasma samples for target specific EV protein analyses; ii) superior detection sensitivity (about 1000-fold higher than that of ELISA), through magnetic enrichment and enzymatic signal amplifications; and iii) a compact device executing parallel measurements without using mechanical scanners. To prove the concept, we implemented a compact HiMEX device that uses a 96-electrode array. We also showed that the entire HiMEX assay, from EV isolation to analysis, was complete within 1 hr.
HiMEX can make EV profiling simple in standard laboratory settings; the technology is cost-effective and capable of fast, parallel EV protein detection. Immunomagnetic pulling, the initial assay step in HiMEX, can also facilitate EV collection for other molecular assays. For example, in surveying drug resistance status, we immunomagnetically capture EVs directly from plasma; a portion of bead-captured EVs were then used for CRC protein detection by HiMEX, and the rest for targeted mRNA sequencing. To further improve the system, one could engineer detection electrodes to enhance assay sensitivity and throughput. Miniaturizing electrodes into the micrometre scale will improve the mass-detection limit (by reducing sample volumes) and facilitate the construction of dense arrays. Detection electrode surfaces can also be textured with nano-structures to achieve exquisite sensitivity55. These improvements may empower HiMEX to detect even scarce EV targets (e.g., mutated proteins inside vesicles, protein modification, nucleic acids), thereby enabling comprehensive molecular profiling in a single assay system.
To use HiMEX in CRC detection, we initially performed a bioinformatic survey to determine over-expressed protein biomarkers for CRC diagnostics. While many proteins have been associated with CRC, we narrowed them down to a panel consisting of EGFR, EpCAM, CD24, and GPA33. Interestingly, the CRC EV panel achieved high accuracy in our discovery (98%) and testing (96%) cohorts, which could be attributed to avid tumour EV release into circulation. Moreover, serial EV profiling revealed a correlation between EVs’ molecular signature and temporal changes in tumour burden. CRC EV levels dropped in all patients immediately after surgery; for those patients without near-term recurrence, CRC EV levels continued to decrease over time, while for those patients with recurrence, CRC EV levels rebounded with tumour progression. We further observed that initial EV burden can be a predictive risk factor of short term (5-year) tumour relapse. Combined, these outcomes would widen EVs’ potential as CRC biomarkers, not only for diagnostics but also for treatment monitoring and prognosis.
Several limitations of the current work, however, need to be addressed in future studies. Our comparison between tissue and circulating EVs was carried out for three markers (EpCAM, EGFR, CD24), restricted by insufficient tissue amount for IHC optimization. Expanding both markers (e.g., CD133, GPA33) and sample numbers will firmly establish the presence of CRC-derived EVs as representative of tumour cells. Serial EV tracking, particularly for treatment monitoring, also requires more frequent sampling and larger prospective cohorts than the current study, to obtain robust statistics. As for the patient class, we enrolled resectable patients with established disease, because we were interested in monitoring EV changes during treatment. Invariably, many of our patients were at Stages 2 and 3, with only a few at Stage I. In future research, it may be interesting to adapt a panel for a CRC screening tool. Additional molecular markers (e.g., KRASG12D, KRASG13D, KRASG12V, BRAFV600E, MLH1, MSH3, MSH6, IGF2)56–58 could be tested for this purpose.
Going forward, we envision expanding the current study to broaden its impact. First, we could enlarge the study cohort, both CRC patients and controls, to further validate EV-based CRC diagnostics. Acquiring CRC patient samples from multi-institutions would enable us to account for ethnic and geographical diversities; including non-CRC patients but with potentially confounding bowel symptoms (e.g., irritable bowel syndrome, Crohn’s disease) would serve to refine EV biomarkers and their cutoffs. With its throughput and low equipment cost, HiMEX will expedite processing such sample volumes even in standard laboratory settings. Second, we could design new EV tests to discriminate among different CRC stages to complement conventional imaging. Improved non-invasive CRC staging and molecular profiling would be a powerful tool to i) select patients for neoadjuvant rather than adjuvant chemotherapies, ii) identify groups at high risk for relapse, and iii) select patients for targeted clinical trials. Achieving such classification power will require larger numbers of patients at each CRC stage than in the current study. Third, we could expand EV screening to obtain diverse clinically relevant information. For example, tumour-derived EVs have been shown to present distinct integrin patterns; these EVs can be taken up by resident cells in an organ-specific manner, preparing pre-metastatic niches59,60. Others have posited that chemotherapy can further trigger a primary tumour to release EVs with pro-metastatic potential61,62. HiMEX’s capacity to enrich tumour-specific EVs will facilitate detecting these EV subpopulations to help understand, monitor, and even predict metastasis.
Methods
Collecting clinical samples.
The study was approved by the Institutional Review Board (IRB) of Kyungpook National University Medical Centre (PI: Jun Seok Park) where all clinical samples were collected. The procedures followed were in accordance with institutional guidelines. Informed consent was obtained from all subjects. Peripheral blood (~15 mL) was withdrawn from patients and centrifuged at 400 g for 15 min to separate plasma from red blood cells and buffy coat. We used 20 μL of plasma to analyse each marker.
Constructing the HiMEX system.
The HiMEX device consisted of a micro-controller (ATSAMD21G18, Atmel Corporation), a digital-to-analogue converter (DAC8552, Texas Instruments), an analogue-to-digital converter (AD7490, Analog Devices), a multiplexer (ADG708, Analog Devices), and 96 potentiostats. Each potentiostat had two operational amplifiers (AD8606, Analog Devices): one amplifier maintained the potential difference between a working and a reference electrode, and the other one functioned as a transimpedance amplifier to convert current to a voltage signal. The current-measuring range of the transimpedance amplifier was ±7.5 μA. A 12 × 8 electrode array (96×220, DropSens, Spain) was used.
Preparing assay reagents.
(i) Immunomagnetic beads. 5 mg of magnetic beads with epoxy groups (e.g., 14302D, Dynabeads M-270 Epoxy, Invitrogen) were suspended in 1 mL of 0.1 M sodium phosphate solution at room temperature (RT) for 10 min. Magnetic beads were separated from the solution with a permanent magnet and re-suspended in 100 μL of the same solution. 100 μg of antibodies against CD63, CD9, or CD81 (see Supplementary Table 1 for details) were added and mixed thoroughly. Next, 100 μL ammonium sulphate solution (3 M) was added, and the whole mixture was incubated for 2 h at RT and then for overnight at 4 °C with slow tilt rotation. Beads were washed twice with phosphate buffer saline (PBS) solution and finally re-suspended in 2 mL of PBS with 1% bovine serum albumin (BSA). (ii) Labelling antibodies. 10 mM Sulfo-NHS-Biotin (A39257, Pierce) solution in PBS was incubated with antibodies for 2 h at RT. Unreacted Sulfo-NHS-Biotin was removed using a Zeba spin desalting column, 7K MWCO (89882, Thermo Scientific). Biotinylated antibodies were kept at 4 °C until use.
HiMEX assay.
We mixed EV samples (10 μL of EV-spiked PBS or 20 μL of plasma) with 50 μL of the immunomagnetic bead solution for 15 min. We then washed the beads by collecting them with a permanent magnet, discarding supernatant, and adding a fresh buffer (50 μL, PBS with 1% BSA). 10 μL of biotinylated antibodies of interest (20 μg/mL in PBS) were added and the mixture was incubated for 15 min. We separated and washed the beads as described above and then added 5 μL of streptavidin-conjugated HRP enzymes (21130, Pierce, 1:100 diluted in PBS; 15 min at 21 °C). Beads were separated, washed, and finally suspended in 7 μL of PBS. To generate signal, the prepared bead solution and 20 μL of UltraTMB solution (34028, ThermoFisher Scientific) were loaded on top of the screen-printed electrode. After 3 minutes, chronoamperometry measurement was started. The current levels between 50 and 55 sec were averaged. Total assay time was about 1 hour. All assay steps were carried out at 21 °C.
EV capture.
Antibodies for EV capture (anti-CD63, anti-CD9, or anti-CD81) were coupled to magnetic beads, at a ratio of 10 μg of total antibody per 100 μL of beads, by overnight incubation at 4 °C with rotation. Beads were washed three times with 500 μL of PBST buffer (PBS pulse 0.001% Tween 20) and resuspended in 100 μL of the same buffer. EVs were isolated by OptiPrep density gradient ultracentrifugation as described previously63. The final pellet was resuspended in 100 μL of PBS. Collected EVs (5 μL, 5 × 109 vesicles/mL) were then mixed with 25 μL of antibody-coated beads, followed by addition of 170 μL of PBST and incubation at 4 °C with rotation. Beads with pulled-down EVs were collected and washed three times with 500 μL PBST. Flow-through was concentrated to 50 μL using Amicon Ultra 10kDa filters (UFC501096, Millipore Sigma). For western blotting, samples were lysed with non-reducing LDS sample buffer, boiled for 5 min at 95 °C, and loaded on gel. Proteins were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and immunostained with the following antibodies: anti-CD63 (clone H5C6, BD Biosciences, 1:200 dilution); anti-CD9 (clone D8O1A, Cell Signaling, 1:1000 dilution); and anti-CD81 (clone 1.3.3.22, Thermo Fisher, 1:500 dilution). HRP-conjugated secondary antibodies were added, then chemiluminescence substrate (WesternBright Sirius, Advansta), and blots were developed using autoradiographic films (Fig. 2b and Supplementary Fig. 22). Films were digitized and quantification of signal intensity was performed using ImageJ.
Enzyme-linked immunosorbent assay (ELISA).
CD63 antibody (Ancell) and IgG1 antibody (Ancell) were diluted in PBS (5 μg/mL) and transferred to the Maxisorp 96 well plate (Nunc) for overnight incubation at 4 °C. After washing with PBS, 2% BSA in PBS blocking solution was added to the plate (1 hr incubation at RT). Subsequently, EV samples (in 100 μL PBS) were added to each well for 1 hour incubation at RT. After discarding the blocking solution, antibodies (1 μg/mL) against various markers were inserted in each well and incubated another 1 hr at RT. Unbounded antibodies were triple-washed with PBS. Streptavidin-HRP molecules then were added to each well, and the mixture was incubated for 1 hr at RT. After washout with PBS, chemiluminescence signal was measured by a plate reader (Tecan).
Flow cytometry.
About 106 cells per marker were used for flow cytometry experiments. Cells were fixed with 4% paraformaldehyde for 10 min at RT and then washed with PBS (0.5% BSA). Next, cells were blocked with BSA (0.5% in PBS) and incubated with primary antibodies (4 μg/mL). Labelled cells were washed, incubated with fluorophore-conjugated secondary antibodies (2 μg/mL; Abcam), and washed again. Control samples were similarly labelled using isotype-matched IgG and secondary antibodies. As a background estimator, an aliquot of cells was incubated with secondary antibodies only. Blocking and incubation with antibodies (primary and secondary) were performed for 30 min each at RT. Every washing step comprised three 5-min washes at 300 g with PBS (0.5% BSA). Fluorescence signals from the labelled cells were measured using BD LSRII Flow Cytometer (BD Biosciences). Mean fluorescent intensities (MFIs) were obtained from three types of samples (i.e., targeted, IgG, secondary-only). The expression level of a target marker was obtained as (MFItarget – MFIIgG)/MFIsecondary. Gating information is shown in Supplementary Fig. 23.
Cell culture.
A panel of CRC cell lines was purchased (ATCC) and grown in the vendor-recommended media: DLD-1 (RPMI-1640, Cellgro); HT29 (MacCoy’s 5a modified with 2% NaHCO3, Cellgro); HCT116 (MacCoy’s 5a, Cellgro); Colo201 (RPMI-1640 modified with 1% sodium pyruvate, Cellgro); SW480 and SW620 (Dulbecco’s modified essential medium, Cellgro); CCD-18Co (Eagle’s minimum essential medium, Cellgro); CCD-112CoN (Eagle’s minimum essential medium, Cellgro); CCD-33Co (Eagle’s minimum essential medium, Cellgro). All media were supplemented with 10% FBS and penicillin-streptomycin (Cellgro). All cell lines were tested and determined to be free of mycoplasma contamination (MycoAlert Mycoplasma Detection Kit, Lonza, LT07-418).
Isolating EVs from cell culture.
For spike-in experiments, we prepared pure EVs from cell culture. Cells at passages 1–15 were cultured in vesicle-depleted medium (with 5% depleted FBS) for 48 hours. Conditioned medium from ~107 cells was collected and centrifuged at 300 g (5 min). Supernatant was filtered through a 0.2-μm membrane filter (Millipore) and concentrated by 100,000 g centrifugation (1 hr). After the supernatant was removed, the EV pellet was washed with PBS and centrifuged at 100,000 g (EV). Collected EVs were resuspended in PBS and stored at 4 °C. For the stock solution samples, EV concentrations were estimated through nanoparticle tracking analysis (NTA).
Immunohistochemistry.
Tumoural and non-neoplastic tissue sections from CRC patients were subjected to immunohistochemical staining (Fig. 3a and Supplementary Fig. 24). Primary monoclonal antibodies against EGFR (EGFR1, Abcam, 1:50 dilution), EpCAM (VU-1D9, Abcam, 1:100 dilution), and CD24 (eBioSN3, ebioscience, 1:100 dilution) were used and the staining was conducted on a Ventana BenchMark XT automated slide stainer (Ventana Medical Systems, Inc.), according to the manufacturer’s recommendations. Stained images were scored, based on the fraction of positive cells among total cancer cells, by a pathologist (G.Y.) who was blind to clinicopathological variables and EV profiling results. Positive cells were defined by positively stained cytoplasmic or membranous pattern within cancer cells in the face of concurrent negative labelling in non-neoplastic tissues. The marker expression was ranked from 0 (negative) to 1 (positive) with the increment of 0.1; this level was used as an ordinal variable in Spearman’s rank test with EV profiling results.
Tumour recurrence status determination.
Recurrence was diagnosed pathologically by either surgical resection or radiological detection of lesions that increased in size over time. Radiologists and pathologists independently assessed the radiological imaging and pathological specimens. Local recurrence was defined as any recurrence within the pelvic cavity or the perineum. Systemic recurrence was defined as any recurrence outside the pelvic cavity.
EV mRNA analyses.
We used about 3 mL of patient plasma samples as recommended by the vendor. EVs from patient plasma were captured on magnetic beads and RNA was isolated using exoRNeasy Serum/Plasma Starter kit (77023, Qiagen). Libraries were prepared with QIAseq Targeted RNA panel according to the manufacturer’s instructions (Human Molecular Toxicology Transcriptome, Qiagen). Libraries were quantified using Qubit dsDNA HS Assay kit (Q32850, ThermoFisher Scientific) and QIAseq library Quant Assay kit, and the library size was determined using Agilent 2100 Bioanalyzer (High Sensitivity DNA Analysis Kit, Agilent Technologies). Prepared libraries were pooled (4 nM for each library), and the pooled mixture (6.5 pM) was run on an Illumina MiSeq (MiSeq Reagent Kit v2, Illumina). Data were analysed using the DESeq2 package in R version 3.6.1.
Single EV imaging.
EVs from CRC cell lines (SW480, HT29; 108 vesicles/ml) were incubated at RT for 30 min on a glass slide (63429–04, Electron Microscopy Science). The solution was drained out with paper towel followed by 15 min 4% paraformaldehyde solution (AAJ19943K2, Thermoscientific) incubation at RT for EV fixation. The slide was washed two times with PBS. Perm/Wash buffer (554723, BD Science) was added for EV permeabilization, and the slide was incubated for 5 min at RT. After five times PBS washing step, the slide was blocked with 2% BSA in PBS for 30 min. 10 μl of AF647 (1434, Click Chemistry Tools) labelled anti-CD63 (215–020, Ancell), CD9 (555675, BD science), CD81 (555370, BD Science) antibodies (10 μg/ml in PBS with 1% BSA) were added for EV labelling overnight at 4 °C. After three times PBS washing, the slide covered with cover slide glass and EVs were imaged with BX63 Fluorescent microscope (Olympus).
Bioinformatic algorithm for selecting initial markers.
From the Human Protein Atlas (HPA) portal, we downloaded immunohistochemistry data on CRC and normal tissues. Numerical values were assigned to staining levels (high, 3; medium, 2; low, 1; undetected, 0) and a mean staining value was obtained for each protein. For each tissue type (i.e., CRC and normal), these values were normalized z-scores. Markers were then ranked according to their differential z-score (CRC - normal). This list was narrowed down by cross-referencing UniProt to select proteins at specific cellular domains (e.g., transmembrane). We further filtered the list using EV databases to collect markers reportedly found in EVs. Analyses were performed in R using custom-written scripts.
Statistical analyses.
Statistical analysis was performed using Stata version 12.1 (StataCorp LP), GraphPad Prism version 8.0 (GraphPad Software Inc.), or R version 3.6.1. For all statistical tests, p values <0.05 were considered significant. (i) Marker selection for CRC detection. We selected candidate predictive markers from the EV profiling data (Fig. Sa) by applying the least absolute shrinkage and selection operator (Lasso) regression. Note that we augmented non-CRC controls with EV data from healthy plasma samples. We calculated the cross-validation error (CVE) and determined the tuning parameter (λ) that minimized CVE. We used the glmnet package in R. (ii) CRC diagnosis. To assess the ability of the selected protein markers, either individually or in combination, to predict CRC cases, we conducted an ROC analysis using the training cohort (n = 83). The specific individual biomarkers under consideration were EGFR, EpCAM, CD24, GPA33, and CD133. We also considered the the linear combination of two markers (EGFR + CD24, EGFR + EpCAM, and EpCAM + CD24), three markers (EGFR + EpCAM + CD24), four-marker combinations (EGFR + EpCAM + CD24 + GPA33 and EGFR + EpCAM + CD24 + CD133), and five-marker combinations (EGFR + EpCAM + CD24 + GPA33 + CD133). For each biomarker combination, optimal weights were determined via logistic regression. We constructed ROC curves and determined level cutoffs that maximized the sum of sensitivity and specificity. Standard formulas were used to define sensitivity (true positive rate), specificity (true negative rate), and accuracy [(true positive + true negative)/(positive + negative)]. AUCs were compared following Delong’s method64. We next analysed the prospective cohort (n = 48) using the selected cutoff values, determining the classification performance of each biomarker and biomarker combination. Exact 95% CIs for the sensitivity, specificity, and accuracy were also estimated. Analyses were performed using the pROC package in R. (iii) Survival analyses. We monitored 90 CRC patients, who underwent curative surgery, up to 61 months. DFS was defined as the interval between surgery and either the first radiologic recurrence or death as a result of CRC. We used the Kaplan-Meier method to calculate the survival functions stratified by EVCRC or CEA levels. The chosen EVCRC cutoff for patient classification was the value that maximized the absolute log-rank score statistic between the two classification groups: the p-value for comparing the corresponding survival curves for the two groups above vs. below the selected EVCRC cutoff was obtained using conditional Monte Carlo simulation with 10000 replications to approximate the null distribution of the maximally selected rank statistic53. The log-rank test was used to compare survival curves under the conventional CEA cutpoint. Analyses were performed using the maxstat and the survival packages in R.
Reporting summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
Table 1 |.
Clinical information of cohorts involved in CRC diagnostics.
| Training cohort | Testing cohort | Total | |||
|---|---|---|---|---|---|
| Case | Non-CRC | CRC | Non-CRC | CRC | |
| 25 | 58 | 15 | 33 | 131 | |
| Age | |||||
| Median | 40 | 66 | 59 | 63 | 61 |
| Range | 18–80 | 38–80 | 25–76 | 34–82 | 18–82 |
| Sex | |||||
| Male | 14 (56%) | 35 (60%) | 10 (67%) | 19 (58%) | 78 (60%) |
| Female | 11 (44%) | 23 (40%) | 5 (33%) | 14 (42%) | 53 (40%) |
| Stage | |||||
| I | - | 9 (16%) | - | 1 (3%) | 10 (11%) |
| II | - | 22 (38%) | - | 7 (21%) | 29 (32%) |
| III | - | 16 (28%) | - | 21 (64%) | 37 (41%) |
| IV | - | 11 (18%) | - | 4 (12%) | 15 (16%) |
| Serum marker (median / range) | |||||
| CA19-9 (IU/mL) | 6.9 (0.3–24.9) |
17.8 (0.5–2167) |
4.2 (0.11–18.8) |
14.7 (0.8–188.7) |
|
| CEA (ng/mL) | 1.4 (0.4–5.2) |
3.5 (0.4–7671) |
1.5 (0.2–4.8) |
5.1 (0.4–37.8) |
|
Acknowledgements
The authors thank Dr. Breakefield (Massachusetts General Hospital) for helpful discussion. This work was supported in part by grants from the V-Foundation for Cancer Research (R.Weissleder, C.M.C.); the American Cancer Society (C.M.C.); the Robert Wood Johnson Foundation / Amos Medical Faculty Development Program (C.M.C.); U.S. NIH Grants R01CA229777 (H.Lee), R21DA049577 (H.Lee), R01CA204019 (R.Weissleder), U01CA233360 (H.Lee, C.M.C.), TR000931 (B.S.C.), U01CA230697 (B.S.C., L.B.), CA069246 (B.S.C.); US DOD-W81XWH1910199 (H.Lee), DOD-W81XWH1910194 (H.Lee); MGH Scholar Fund (H.Lee), MGH Fund for Medical Discovery Fellowship (H.-Y.L.); Basic Science Research Program NRF-2019R1C1C1008792 (J.P.), NRF-2020R1A4A1016093 (J.P.), NRF-2017M3A9G8083382 (J.S.P.) from the Ministry of Education, South Korea.
Footnotes
Data availability
The main data supporting the results in this study are available within the paper and its supplementary information. The raw patient datasets generated and analysed during the study are available from the corresponding authors on reasonable request, subject to approval from the Institutional Review Board of the Kyungpook National University Hospital. For initial marker selection, we used public databases: Human Protein Atlas (https://www.proteinatlas.org/) and UniProt (https://www.uniprot.org/).
Code availability
Source codes for the marker selection are available at https://csb.mgh.harvard.edu/bme_software.
Competing interests
R.W. declares that he has received consultancy payments from Accure Health, and that he is a shareholder of Lumicell. H.L. declares that he has received consultancy payments from Accure Health. Patents: all patents associated with R.W. and H.L. have been assigned to and handled by Massachusetts General Hospital.
References
- 1.Heitzer E, Haque IS, Roberts CES & Speicher MR Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet 20, 77–88 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Siravegna G, Marsoni S, Siena S & Bardelli A Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol 14, 531–548 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Chi KR The tumour trail left in blood. Nature 532, 269–271 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Pantel K & Alix-Panabieres C Real-time liquid biopsy in cancer patients: fact or fiction? Cancer Res. 73, 6384–6388 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Théry C, Ostrowski M & Segura E Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol 9, 581–593 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Xu R et al. Extracellular vesicles in cancer—implications for future improvements in cancer care. Nat. Rev. Clin. Oncol 15, 617–638 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Shao H et al. Protein typing of circulating microvesicles allows real-time monitoring of glioblastoma therapy. Nat. Med 18, 1835–1840 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi D, Spinelli C, Montermini L & Rak J Oncogenic Regulation of Extracellular Vesicle Proteome and Heterogeneity. Proteomics 19, 1800169 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Skog J et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol 10, 1470–1476 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shao H et al. Chip-based analysis of exosomal mRNA mediating drug resistance in glioblastoma. Nat. Commun 6, 6999 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoshioka Y et al. Ultra-sensitive liquid biopsy of circulating extracellular vesicles using ExoScreen. Nat. Commun 5, 3591 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skotland T, Sandvig K & Llorente A Lipids in exosomes: current knowledge and the way forward. Prog. Lipid Res 66, 30–41 (2017). [DOI] [PubMed] [Google Scholar]
- 13.EL Andaloussi S, Mäger I, Breakefield XO & Wood MJ Extracellular vesicles: biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov 12, 347–357 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Im H et al. Label-free detection and molecular profiling of exosomes with a nano-plasmonic sensor. Nat. Biotechnol 32, 490–495 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu C et al. Low-cost thermophoretic profiling of extracellular-vesicle surface proteins for the early detection and classification of cancers. Nat. Biomed. Eng 3, 183–193 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Jeong S et al. Integrated magneto–electrochemical sensor for exosome analysis. ACS Nano 10, 1802–1809 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang KS et al. Multiparametric plasma EV profiling facilitates diagnosis of pancreatic malignancy. Sci. Transl. Med 9, eaal3226 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang P et al. Ultrasensitive detection of circulating exosomes with a 3D-nanopatterned microfluidic chip. Nat. Biomed. Eng 3, 438–451 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang K et al. Nanoplasmonic quantification of tumour-derived extracellular vesicles in plasma microsamples for diagnosis and treatment monitoring. Nat. Biomed. Eng 1, 0021 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis JM et al. Integrated Analysis of Exosomal Protein Biomarkers on Alternating Current Electrokinetic Chips Enables Rapid Detection of Pancreatic Cancer in Patient Blood. ACS Nano 12, 3311–3320 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Shao H et al. New technologies for analysis of extracellular vesicles. Chem. Rev 118, 1917–1950 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Locker GY et al. ASCO 2006 update of recommendations for the use of tumour markers in gastrointestinal cancer. J. Clin. Oncol 24, 5313–5327 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Tao S, Hundt S, Haug U & Brenner H Sensitivity estimates of blood-based tests for colorectal cancer detection: impact of overrepresentation of advanced stage disease. Am. J. Gastroenterol 106, 242–253 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Park J et al. Integrated Kidney Exosome Analysis for the Detection of Kidney Transplant Rejection. ACS Nano 11, 11041–11046 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fraser K et al. Characterization of single microvesicles in plasma from glioblastoma patients. Neuro. Oncol 21, 606–615 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jeppesen DK et al. Reassessment of Exosome Composition. Cell 177, 428–445.e18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee K et al. Multiplexed Profiling of Single Extracellular Vesicles. ACS Nano 12, 494–503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramirez MI et al. Technical challenges of working with extracellular vesicles. Nanoscale 10, 881–906 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Zhao LH et al. CD44v6 expression in patients with stage II or stage III sporadic colorectal cancer is superior to CD44 expression for predicting progression. Int. J. Clin. Exp. Pathol 8, 692–701 (2015). [PMC free article] [PubMed] [Google Scholar]
- 30.Weichert W et al. Cytoplasmic CD24 expression in colorectal cancer independently correlates with shortened patient survival. Clin. Cancer Res 11, 6574–6581 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Weichert W, Knösel T, Bellach J, Dietel M & Kristiansen G ALCAM/CD166 is overexpressed in colorectal carcinoma and correlates with shortened patient survival. J. Clin. Pathol 57, 1160–1164 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee CH et al. The Prognostic Role of STEAP1 Expression Determined via Immunohistochemistry Staining in Predicting Prognosis of Primary Colorectal Cancer: A Survival Analysis. Int. J. Mol. Sci 17, 592 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ingebrigtsen VA et al. B7-H3 expression in colorectal cancer: nuclear localization strongly predicts poor outcome in colon cancer. Int. J. Cancer 131, 2528–2536 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Deng Y et al. ALDH1 is an independent prognostic factor for patients with stages II-III rectal cancer after receiving radiochemotherapy. Br. J. Cancer 110, 430–434 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ong CW et al. CD133 expression predicts for non-response to chemotherapy in colorectal cancer. Mod. Pathol 23, 450–457 (2010). [DOI] [PubMed] [Google Scholar]
- 36.Peters GJ et al. Induction of thymidylate synthase as a 5-fluorouracil resistance mechanism. Biochim. Biophys. Acta 1587, 194–205 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Ekblad L, Kjellström J & Johnsson A Reduced drug accumulation is more important in acquired resistance against oxaliplatin than against cisplatin in isogenic colon cancer cells. Anticancer Drugs 21, 523–531 (2010). [DOI] [PubMed] [Google Scholar]
- 38.Mouradov D et al. Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer. Cancer Res. 74, 3238–3247 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Skog J et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol 10, 1470 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garinchesa P et al. Organ-specific expression of the colon cancer antigen A33, a cell surface target for antibody-based therapy. Int. J. Oncol 9, 465–471 (1996). [DOI] [PubMed] [Google Scholar]
- 41.Zou KH, O’Malley AJ & Mauri L Receiver-operating characteristic analysis for evaluating diagnostic tests and predictive models. Circulation 115, 654–657 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Liu J et al. Down-regulation of GADD45A enhances chemosensitivity in melanoma. Sci. Rep 8, 4111 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang P et al. Chemotherapy-driven increases in the CDKN1A/PTN/PTPRZ1 axis promote chemoresistance by activating the NF-κB pathway in breast cancer cells. Cell Commun. Signal 16, 92 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cuadrado A et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov 18, 295–317 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Derdak Z et al. The mitochondrial uncoupling protein-2 promotes chemoresistance in cancer cells. Cancer Res. 68, 2813–2819 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nicolussi A, D’Inzeo S, Capalbo C, Giannini G & Coppa A The role of peroxiredoxins in cancer. Mol. Clin. Oncol 6, 139–153 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang J & Li Y CD36 tango in cancer: signaling pathways and functions. Theranostics 9, 4893–4908 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Romano G et al. The TGF-β pathway is activated by 5-fluorouracil treatment in drug resistant colorectal carcinoma cells. Oncotarget 7, 22077–22091 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu J et al. Heat Shock Proteins and Cancer. Trends Pharmacol. Sci 38, 226–256 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Sharma A, Upadhyay AK & Bhat MK Inhibition of Hsp27 and Hsp40 potentiates 5-fluorouracil and carboplatin mediated cell killing in hepatoma cells. Cancer Biol. Ther 8, 2106–2113 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Shi Z et al. Activation of the PERK-ATF4 pathway promotes chemo-resistance in colon cancer cells. Sci. Rep 9, 3210 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Phallen J et al. Direct detection of early-stage cancers using circulating tumour DNA. Sci. Transl. Med 9, eaan2415 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hothorn T & Lausen B On the exact distribution of maximally selected rank statistics. Comput. Stat. Data Anal 43, 121–137 (2003). [Google Scholar]
- 54.Duffy MJ et al. Clinical utility of biochemical markers in colorectal cancer. Eur. J. Cancer 39, 718–727 (2003). [DOI] [PubMed] [Google Scholar]
- 55.Das J et al. An electrochemical clamp assay for direct, rapid analysis of circulating nucleic acids in serum. Nat. Chem 7, 569–575 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Thierry AR et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumour DNA. Nat. Med 20, 430–435 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Network CGA Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Siravegna G et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med 21, 795–801 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hoshino A et al. Tumour exosome integrins determine organotropic metastasis. Nature 527, 329–335 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Peinado H et al. Pre-metastatic niches: organ-specific homes for metastases. Nat. Rev. Cancer 17, 302–317 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Keklikoglou I et al. Chemotherapy elicits pro-metastatic extracellular vesicles in breast cancer models. Nat. Cell Biol 21, 190–202 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Syn N, Wang L, Sethi G, Thiery JP & Goh BC Exosome-Mediated Metastasis: From Epithelial-Mesenchymal Transition to Escape from Immunosurveillance. Trends Pharmacol. Sci 37, 606–617 (2016). [DOI] [PubMed] [Google Scholar]
- 63.Van Deun J et al. The impact of disparate isolation methods for extracellular vesicles on downstream RNA profiling. J. Extracell. Vesicles 3, 24858 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.DeLong ER, DeLong DM & Clarke-Pearson DL Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics 44, 837–845 (1988). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.