

Abstract
Salt sensitivity of blood pressure (SSBP) is an independent risk factor for cardiovascular mortality not only in hypertensive, but also in normotensive adults. The diagnosis of SSBP is not feasible in the clinic due to lack of a simple diagnostic test, making it difficult to investigate therapeutic strategies. Most research efforts to understand the mechanisms of SSBP have focused on renal regulation of sodium (Na+). However, salt retention or plasma volume expansion are not different between salt sensitive (SS) and salt resistant (SR) individuals. In addition, over 70% of extracellular fluid is interstitial and therefore not directly controlled by renal salt and water excretion. We discuss in this review how the seminal work by Harry Goldblatt paved the way for our attempts at understanding the mechanisms that underlie immune activation by salt in hypertension. We describe our findings that Na+, entering antigen presenting cells (APCs) via an epithelial Na+ channel (ENaC), triggers a PKC- and SGK1-stimulated activation of NADPH oxidase, which in turn enhances lipid oxidation with generation of highly reactive isolevuglandins (IsoLGs). IsoLGs adduct to proteins, with the potential to generate degraded peptide neo-antigens. Activated APCs increase production of the TH17 polarizing cytokines, IL6, IL1β, and IL23, which leads to differentiation and proliferation of IL-17A producing T-cells. Our laboratory and others have shown that this cytokine contributes to hypertension. We also discuss where this Na+-activation of APCs may occur in vivo and describe the multiple experiments, with pharmacological antagonists and knockout mice that we employed to unravel this sequence of events in rodents. Finally, we describe experiments in mononuclear cells obtained from normotensive or hypertensive volunteers, which confirm that analogous processes of salt-induced immunity take place in humans.
Introduction
Hypertension research continues being of the utmost importance, since hypertension is the worldwide leading cause of mortality and disability, accounting for 10.8 million or 19.2% of all attributable deaths in 2019.1 Also, control rates in the US had reached 53.8% in 2013–14, but declined to 43.7% in 2017–18, for several reasons.2 Among them, 6% of all hypertensive patients have truly resistant hypertension, and 10% of the latter have the most severe pattern of refractory hypertension.3 Finally, even controlled hypertension is associated with increased residual cardiovascular risk of unclear cause.4
Harry Goldblatt, a forefather of hypertension research, was stimulated by observations as a clinician (lack of hypertension in a patient dying of uremia after accidental removal of a horseshoe kidney) and as a pathologist (presence of renal arteriolar abnormalities in the autopsies of hypertensive patients) to hypothesize that hypertension required the presence of the kidneys. In order to explore the controversy on whether the renal arteriolar abnormalities were the cause or consequence of hypertension, he decided to mimic the situation of renal ischemia by constricting the main renal arteries of dogs. He was very cognizant of the difference between such a model and the intrarenal vascular abnormalities of essential hypertension but proceeded with it nonetheless, as the most feasible method to reproduce the suspected mechanism of renal ischemia.5
The revolutionary consequences of the results of his experiments were two-fold. First, the recognition that renal artery stenosis was sufficient to produce sustained blood pressure (BP) elevation, later leading to the recognition of renovascular hypertension in humans. Most importantly, after preliminary investigations on possible mechanisms, Goldblatt postulated that one or more humoral factors produced by the kidneys needed to be involved. A few years later, Page and Braun Menendez simultaneously identified angiotensin II (Ang II)6,7 which began a century of additional research on the role of this peptide in BP regulation.
Gavras et al showed that dependency of BP on the renin-angiotensin system (RAS) in rat models of Goldblatt hypertension was linked to the status of salt balance (natriuresis by the unclipped kidney with persistent hyper-reninemia in the unilateral model (2K-1C), as opposed to Na+ retention and renin suppression in the bilateral one, 1K-1C).8,9 Laragh et al proposed that Goldblatt’s findings were therefore applicable to essential hypertension because the heterogeneous arteriolar lesions of this disorder resulted in some ischemic glomeruli with impaired Na+ excretion, and other non-ischemic, hyperfiltering ones with enhanced natriuresis.10
The interplay between Ang II and salt in BP regulation acquired a new dimension once it was demonstrated that tissue-generated Ang II played ubiquitous roles in growth, proliferation and organ damage, central nervous system regulation of sympathetic tone, and most importantly, in renal Na+ transport by direct actions on transporters and by stimulation of aldosterone release.
More recently, a series of seemingly unrelated observations suggested participation of immunity and inflammation in the pathogenesis of hypertension. For example, hypertension was produced in normal rats by transfer of lymphocytes from rats with renal infarction or from splenocytes of rats with DOCA-salt hypertension.11,12 Conversely, immunosuppressants, thymectomy, antithymocyte serum or transplant of a normal thymus prevented or reversed hypertension in several hypertensive rodent models.11,13–15 A direct link between these observations and the previously recognized interplay between Ang II and Na+ was provided by the group of Harrison et al, who showed that Ang II and DOCA-salt hypertension were attenuated in mice genetically lacking lymphocytes.16
Other investigators showed that Na+ is involved in immune disorders unrelated to hypertension, e.g., experimental encephalomyelitis.17 We embarked on the investigation of how Na+ may trigger immune changes that underlie the pathophysiology of hypertension.
High Salt and Cardiovascular Disease
There is evidence that excess dietary salt in the Western diet is associated with inflammation, autoimmunity and cardiometabolic disease (CVD).18 Less than 10% of the U.S. population observes the current recommendation to limit Na+ intake to 2,300 mg per day.19,20 An additional problem with excess salt consumption is that 50% of the hypertensive and 25% of the normotensive population exhibit SSBP. This phenotype is characterized by changes in BP that parallel changes in salt intake and is an independent risk factor for CV morbidity and mortality. Consequently, strong evidence indicates that reducing Na+ intake decreases BP and cardiovascular events.21–24 Owing to the poor adherence of populations to a low salt diet, it is of the utmost importance to understand the mechanisms for salt-induced CVD, to identify targets that may reduce Na+-induced morbidity independent of salt intake.
High Salt, Inflammation and Hypertension
In classical physiology, the speculation about how Na+ increased BP was that expansion of intravascular volume would increase cardiac output acutely, whereas autoregulation of organ blood flow would lead to vasoconstriction with normalization of cardiac output, the hemodynamic pattern of most essential hypertension. The problem with this view is that changes in total peripheral resistance occur as rapidly as 24 hours in response to an acute Na+ load,25 inconsistent with the idea of long-term autoregulation of blood flow.
In view of the evidence above, we asked the question of whether Na+ could have rapid effects on immune cells and whether such effects could account for the underlying mechanisms of hypertension on a non-hemodynamic basis.
We first found that modestly increasing NaCl in excess of the physiological plasma concentrations leads to entry of Na+ into myeloid antigen presenting cells (APCs), including dendritic cells (DCs), which are the most potent APCs.26,27 Na+ entry into DCs is mediated through an amiloride-inhibitable epithelial Na+ channel (ENaC) and starts a chain of events leading to a state of oxidative stress in these cells (Figure 1). First, the increased intracellular Na+ alters the Na+/Ca2+ exchanger, resulting in an increase in intracellular Ca2+, confirmed by fluorescence photometry, and activation of protein kinase C (PKC). Exposure of DCs to NaCl for 15 minutes leads to PKC-induced, i.e., calphostin C-inhibitable, serine phosphorylation of the p47phox subunit of the NADPH oxidase enzyme, leading to its assembly with the gp91phox subunit with consequent enzyme activation and increased production of superoxide.
Figure 1: Sodium (Na+) sensing and activation of NADPH oxidase by antigen presenting cells.

Na+ enters antigen presenting cells through the epithelial Na+ channel (ENaC) leading to intracellular Ca2+ influx via the Na+/Ca2+ exchanger (NCX) and activation of PKC. PKC phosphorylates p47phox leading to assembly of NADPH oxidase and increased production of reactive oxygen species (ROS).
We also showed that Na+ entry into DCs increases expression of the salt sensing kinase serum/glucocorticoid kinase 1 (SGK1).27 This was associated with increased expression, activation and assembly of various NADPH oxidase subunits. That is, entry of Na+ into these cells via an ENaC channel leads to activation of NAPDH oxidase by a dual, PKC and SGK1 stimulation.
Activation of NADPH oxidase leads to increased production of reactive oxygen species (ROS), lipid peroxidation and accumulation of isolevuglandins (IsoLGs), also referred to as isoketals (Figure 2). These are highly reactive gamma ketoaldehydes formed by lipid peroxidation of arachidonic acid, which our group previously identified in immune cells during angiotensin II and deoxycorticosterone acetate (DOCA)-salt-induced hypertension.28 IsoLGs rapidly adduct to lysines on proteins, the denaturation of which produces neo-antigenic peptides. During this process, DCs acquire a pro-inflammatory and pro-hypertensive profile, with increased expression of the B7 ligand CD86. Presentation of neoantigens in the MHC complex plus the cytokines of activated DCs promote proliferation among memory T cells, which produce IL-17A. We found that proteins modified by other lipids including malondialdehyde (MDA), hydroxynonenal (HNE) or methylglyoxal (MGO) are much less potent or have no effect on DC immunogenicity, and that DCs pulsed with these lipid-modified proteins had little to no effect on T cell proliferation or activation.28 In contrast, DCs activated by IsoLGs produce cytokines that are known to polarize T cells to an IL-17 producing phenotype. Hypertension was associated with a 2-fold increase in IL-6 and IL1-β, and a 3-fold increase in IL-23 production by DCs. These cytokines are known to polarize T cells to IL-17 producing T cells, and these responses were completely normalized by co-treatment with 2-hydroxybenzylamine, a potent scavenger of IsoLGs.28 These T cells have the potential to migrate to target organs and induce vascular inflammation and enhanced renal Na+ reabsorption, leading to hypertension. When adoptively transferred into naïve mice, these DCs can prime hypertension in response to an otherwise sub-pressor dose of angiotensin II.26
Figure 2: Immune cell activation by isolevuglandins (IsoLGs).

Increased reactive oxygen species (ROS) in hypertension lead to oxidation of fatty acids such as arachidonic acid leading to formation of IsoLGs. IsoLGs adduct to and cross-link proteins leading to post-translational modifications of self-proteins. The IsoLG-adducted self-proteins lead to an auto-immune-like state in hypertension. Antigen presenting cells including dendritic cells (DCs) accumulate IsoLGs and are activated to produce pro-inflammatory cytokines including IL-1β, IL-6 and IL-23. They also activate T cell to proliferate and produce inflammatory cytokines IL-17A, TNF-α and IFN-γ which lead to hypertension. Excess dietary salt is a potent hypertensive stimulus leading to formation of IsoLGs.
The sequence of events above was confirmed with experiments in mice lacking the NADPH oxidase, DCs lacking the p22phox subunit, mice treated with IsoLG scavengers, or DCs treated gp91ds-tat peptide, an inhibitor of the p47phox-gp91phox interaction. These interventions inhibited superoxide generation, DC activation, salt-induced production of IsoLGs and its immunogenic adducts, hypertension and end organ damage.26,28 Analogously, the role of SGK1 was confirmed in mice lacking this enzyme in CD11c+ cells or in mice treated with a pharmacological inhibitor. These mice had reduced NADPH subunit expression and isoLG adducts, blunted pressor response to the salt feeding phase of the N-Nitro-L-arginine methyl ester hydrochloride (L-NAME/high salt) model of salt-sensitive hypertension, and protection from renal inflammation and endothelial dysfunction. Also, the ability of salt-treated DCs to prime a hypertensive response to subpressor Ang II in normal recipient mice was lost if inhibition of SGK1 was enacted in the donor during salt treatment of their DCs.
In addition to the murine studies of DCs, we also studied monocytes from humans, cultured in either normal (150 mM/L NaCl), or high salt media (190 mM NaCl) for 48 hours.29 Employing RNA sequencing and principal component analysis we found that genes clustered differently in normal salt- versus high salt-treated monocytes. The latter exhibited a gene expression pattern consistent with an activated proinflammatory phenotype. For example, when exposed to high Na+ concentration, but not to equiosmolar mannitol, monocytes showed increased expression of the DC activation markers CD209, CD80 and CD86, and increased production of the proinflammatory cytokines IL-1β, IL-6, IL-23 and TNF-α, the colony stimulating factors CSF 1 and 3, the migration-promoting chemokine receptors CCR2 and CCR5 and the highly reactive IsoLGs. The increase in IsoLGs produced by salt correlated with traditional cardiovascular risk factors (pulse pressure, BMI, cholesterol and glucose) suggesting that the inflammatory response of monocytes to salt stimulation is enhanced in subjects with a high CV risk profile. There was large interindividual variability in the IsoLG responses to salt. Whether this variability in salt-activation of immune cells reflects interindividual variability in SSBP is unknown to date.
In summary, we have unequivocally demonstrated that an elevated Na+ concentration, analogous to observations with Ang II and DOCA salt, is a sufficient and potent stimulus for APC activation and generation of IsoLG-adducted neoantigens in murine DCs through a mechanism involving Na+ entry via ENaC and subsequent activation of the NADPH oxidase.26
The Significance of ENaC in Immune Cells
Renal ENaC fine tunes Na+ excretion at the distal nephron. Its hyperactivity contributes to hypertension in inherited Liddle syndrome, in every form of hypertension with excess or inappropriate aldosterone secretion30 and in an amiloride-sensitive, spironolactone-resistant hypertensive phenotype observed in Blacks, which is possible related to abnormal inhibition of the channel by eicosanoids 31,32 or to SNPs that diminish channel activity.33
It is now known that ENaC is ubiquitously expressed. Endothelial ENaC overexpression results in endothelial dysfunction, increases endothelial stiffness and produces amiloride-sensitive hypertension in mice.34–37 Dahl-S rats do not suppress endothelial ENaC in response to a high salt diet, which is the normal response in Sprague Dawley rats, indicating a participation of vascular ENaC in salt-sensitive vasoconstriction.38,39 In rat brain, ENaC is involved in modulation of sympathetic tone and release of vasopressin.40,41 Brain ENaC hyperactivity in mice enhances pressor responses to CSF injection of Na+.42 ENaC channels in the renal pelvis signal via renal afferents to the forebrain and brainstem when stimulated by hyperosmolality, participating in resulting BP regulation.43 Finally, ENaC channels in tastebuds and in intestinal cells regulate salt appetite and intestinal absorption of Na+, respectively.44,45
We now add to this body of knowledge by documenting the expression of ENaC in APCs.27 As stated above, entry of Na+ into these cells is blocked by amiloride. Furthermore, we demonstrated in direct fashion that Na+-stimulated DCs exhibit increased expression of γENaC which co-immunoprecipitated with αENaC. Genetic deletion or pharmacological inhibition of SGK1 in DCs prevented the high salt induced expression of αγENaC, indicating that this kinase mediates the effect of Na+ on activation of the channel. Some have reported that increased intracellular Ca2+ concentration stimulates expression of the monomeric ENaC subunits in human embryonic kidney cells,46 while others noted that increases in intracellular Ca2+ inhibit ENaC by reducing its interaction with phosphatidylinositol 4, 5-bisphosphate (PIP2, an ENaC activator) and the scaffold protein MARCKS (myristoylated alanine-rich C-kinase substrate).33 Whether the increased Ca2+ concentration and PKC activation produced by Na+/Ca2+ exchange in our salt-loaded DCs alters ENaC activity remains to be determined. Therefore, in DCs, SGK1 creates a positive feedforward loop, by which Na+ entry into DCs via ENaC leads to further assembly and activity of this channel. Whether Ca2+ enhances or downregulates this effect of SGK1 is not known. In the kidney, an inactivating phosphorylation of the ubiquitin-ligase Nedd4–2 by SGK1, with impairment of removal of the channel from the membrane, is the predominant mechanism by which SGK1 stimulates ENaC activity. It is not known whether the effect we observed of SKG1 in the DC channel involves such mechanism.
Major regulation of ENaC function includes stimulation by aldosterone, which increases expression of αENaC and by other proteins that impact ENaC expression and activity, including SGK1, GILZ and CNK3,47 resulting in increased channel assembly and open probability. Inhibitors include the EETs, which are lipids generated from arachidonic acid by CYP-450 epoxygenases. They produce a specific threonine phosphorylation of γENaC48 that reduces channel open probability and also inhibit PKA, with consequent dephosphorylation of Nedd4–2, which activates ubiquitination of ENaC and its removal from the membrane. We have preliminary evidence (unpublished observations) suggesting that aldosterone and renally-generated EETs may regulate human monocyte ENaC activity, as assessed by relationships of these compounds with the generation of IsoLGs by high salt stimulation of these cells.
Several other processes regulate ENaC function in other tissues. For example, proteases such as furin, prostasin, matriptase, cathepsin B, elastase, kallikrein, urokinase and plasmin cleave and release inhibitory tracts of αENaC in vitro, transitioning the channels from a low or moderate to a high activity state.33 This has been confirmed in some experiments in vivo. For example, the non-selective serine protease inhibitor aprotinin induced natriuresis in mice with nephrotic syndrome.49 However, others have shown discrepancies between ENaC subunit cleavage and increases in channel activity.50 Finally, signaling lipids other than the EETs, such as PIP2 and PIP3, and palmitoylation of Cys residues also increase channel open probability and activate ENaC.33 It is not known whether any of these mechanisms participate in regulation of immune cell ENaC.
In summary, we discovered a role for ENaC that is upstream in the salt-induced hypertensive process, i.e., activation of autoimmunity in hypertension. Because this ultimately leads to T-cell proliferation and migration to target organs, where T-cell-induced inflammation certainly stimulates ENaC in renal epithelia,51 and perhaps ENaCs in other cell types, activation of this channel in immune cells may have a central role in salt-induced hypertension.
Where do immune cells encounter high Na+ concentration in vivo?
Several tissues have been implicated in the pathogenesis of Na+-induced immune cell activation, hypertension and end-organ damage including the kidneys, skin and the gut (Figure 3).
Figure 3: Role of the gut, kidney and skin interstitium in sodium (Na+)-induced immune cell activation.
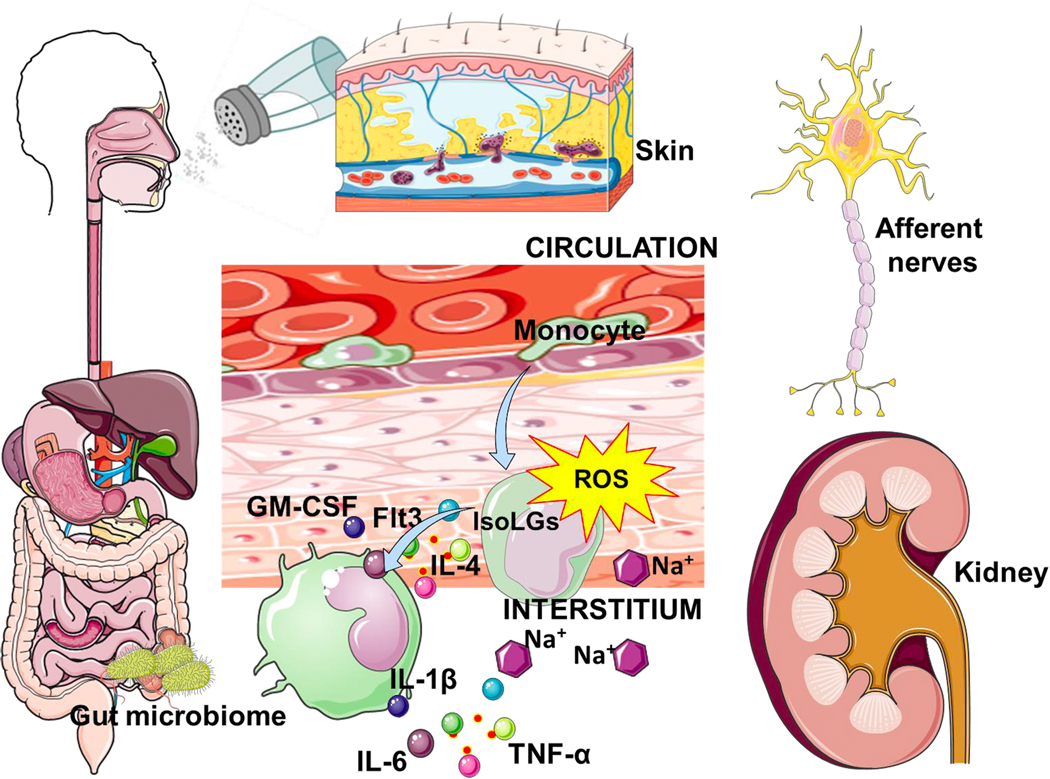
Monocytes encounter a high tissue Na+ environment and are activated to a pro-inflammatory phenotype which can activate T cells leading to increased inflammation. The gut microbiome has also been implicated and Na+-induced immune cell activation leading to hypertension.
The Kidneys:
Glomeruli filter about 22,000 to 25,000 mmols of Na+ per day, but only 1% or less of this amount is excreted in the urine. The countercurrent mechanism that facilitates Na+ reabsorption in the loop of Henle generates medullary hyperosmolality, which increases progressively from the corticomedullary junction to reach values as high as 1200 mOsm/L at the papilla.
Previous observations of our laboratory had shown T cell infiltration of the kidney in hypertensive mice, an observation that could be prevented by IsoLG scavenging.28 More recently, we examined sections of human kidneys and found that in hypertensive subjects, there was accumulation of APCs (macrophages, DCs and monocytes) starting at the cortico-medullary junction and extending into deeper regions of the medulla.29 Most importantly, the kidneys of patients with hypertensive arteriolar nephrosclerosis, the typical renal vascular lesion of essential hypertension, had a greater proportion of moderate to severe infiltration of CD11c+ cells compared to control samples. Taken together, these observations confirm that in hypertension, myeloid antigen presenting cells localize in renal areas where they can be activated by high Na+ concentration to attract proinflammatory T cells to the kidney, via an IsoLG-dependent mechanism.
Finally, in preliminary observations in humans, we found that urine levels of EETs, which reflect a renally synthesized pool,52 are inversely correlated with high-salt induced IsoLGs in the circulating monocytes of these subjects, also supporting the possibility that regulation of ENaC-dependent activation of immune cells may take place in the kidney.53 It is important to note that inflammatory cytokines have been found to play a regulatory role in salt-sensitivity. Studies by Ferreri et al have found that salt-resistant C56BL6 mice develop salt-sensitive hypertension when renal specific TNF-α is deleted or inhibited, and that TNF-α regulates NKCC2B in response to salt restriction.54,55 Nevertheless, several studies have found that cytokines including IL17A and IL-1beta modulate sodium transporters in the kidney and contribute to hypertension.56–59
The Skin:
Seventy percent of extracellular fluid is interstitial and hence not controlled directly by renal salt and water excretion. Excess dietary salt increases Na+ in the skin interstitium without significant changes in plasma Na+ concentration.60 Therefore, the traditional dogma that interstitial Na+ was in equilibrium with that of the plasma compartment has been challenged. Recent observations demonstrated that Na+ accumulates in microdomains of the interstitium, associated with glycosaminoglycans and without commensurate water retention. Efflux of Na+ from this dynamic compartment is mediated by VEGF-C-induced lymphangiogenesis. Several issues require further investigation to ascertain the link between skin Na+ and hypertension. Whether interstitial Na+ is iso- or hyperosmolar remains controversial.61 Also, in rodents, inhibition of Na+ efflux from the skin with antagonists of the lymphangiogenic VEGF-C pathway produces hypertension,60 which is consistent with the observation that Na+ accumulation in the skin and skeletal muscle, measured with 23Na MRI, is increased in human aging and human hypertension.62 However, in a study of normal humans, exaggerated pressor responses to salt in women, compared to men, were related to inability of women to increase Na+ influx into the skin, which opposes the previous interpretation.63
Regardless of the above, high Na+ concentrations in the interstitium have the potential to polarize immune cells toward an inflammatory phenotype. We recruited 67 subjects and non-invasively quantified their skin Na+ using 23Na MRI. We found that increased skin accumulation of Na+ was associated with a parallel increased expression of the activation marker CD83 and increased generation of IsoLG-adducts in their freshly isolated circulating monocytes obtained by flow cytometry.29 Although these data could be interpreted as evidence that immune cell activation by Na+ occurs in the skin, they are correlative, not necessarily causal. Furthermore, it is not known how interstitial Na+ is mobilized or whether it becomes osmotically available to activate immune cells. Obtaining these answers will require validated assays for precise measurement of tissue Na+ concentration and osmolality.
The Gut:
The gut is another potential site for immune cell activation by elevated Na+ since it is the first and largest location for Na+ absorption in the body. DCs survey the intestinal mucosa and regulate its immune homeostasis by inducing tolerance to harmless antigens and by initiating protective immunity against intestinal pathogens. Others had shown that excess salt induces pro-inflammatory and hypertensive effects by acting on the gut microbiome.64 We most recently found that a dietary salt intake that exceeds AHA recommendation alters the gut microbiome, and that this is associated with immune cell activation via IsoLG formation in DCs.65 We also showed that adoptive transfer of fecal material from conventionally housed high salt-fed mice to germ-free mice predisposed them to increased inflammation and hypertension.65 In additional studies, we demonstrated that intestinal tissue from humans with hypertension had a marked increase in arterial wall thickness, fibrosis, immune cell infiltration (T cells, monocytes and macrophages) and concomitant accumulation of IsoLGs.
Although it is conceivable that the high Na+ environment of the gut is responsible for triggering activation of immune cells, we have no direct measure of the actual Na+ concentrations at sites adjacent to the gut epithelium. We also do not know whether intestinal immune cells are resident or recruited and whether they leave the intestinal mucosa to activate T cell inflammation in systemic tissues or organs.
In conclusion, we have summarized the findings obtained by our laboratory, which provide evidence that immune activation may be an early and central pathogenic mechanism for salt-induced hypertension. Major gaps in our understanding remain but our studies will hopefully stimulate research in this area. Such gaps include: a) establishing with certainty the tissues that accumulate relevant in vivo concentrations of Na+ for immune cell activation, b) determining whether immune cells can exit original sites of activation and migrate to other cardiovascular tissues to mediate inflammatory damage, c) discovering the nature of the neoantigens generated by salt-induced lipid oxidation, d) discriminating the tissues in which immune activation occurs locally from those to which migrating, already activated cells are recruited, e) investigating the reasons for the large interindividual variability in the magnitude of salt-induced immune cell activation in humans, and f) determining whether such “salt sensitivity” of immune cells is a correlate, i.e., a marker, for SSBP.
Unraveling this knowledge will hopefully provide therapeutic targets for SSBP, a cardiovascular risk factor devoid of treatment to date. As an example, having established a possible central role for immune cell ENaC in the mechanisms of salt-induced hypertension, our laboratory is in the process of investigating the role of its blockade by amiloride on CV outcomes in a large electronic patient database available in our institution.
Acknowledgments
Sources of Funding
This study was supported by the National Institutes of Health grants K01HL130497, R03HL155041, R01HL144941 (AK), and R01HL147818 (TRK and AK).
Footnotes
Disclosures
The authors have nothing to disclose.
References
- 1.GBD 2019 Risk Factors Collaborators. Global burden of 87 risk factors in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. 2020;396:1223–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muntner P, Hardy ST, Fine LJ, Jaeger BC, Wozniak G, Levitan EB, Colantonio LD. Trends in blood pressure control among US adults with hypertension, 1999–2000 to 2017–2018. JAMA. 2020;324:1190–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grigoryan L, Pavlik VN, Hyman DJ. Characteristics, drug combinations and dosages of primary care patients with uncontrolled ambulatory blood pressure and high medication adherence. J Am Soc Hypertens. 2013;7:471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blacher J, Evans A, Arveiler D, Amouyel P, Ferrières J, Bingham A, Yarnell J, Haas B, Montaye M, Ruidavets JB, Ducimetière P, on behalf of the PRIME Study Group. Residual cardiovascular risk in treated hypertension and hyperlipidaemia: the PRIME study. J Hum Hypertens. 2010;24:19–26. [DOI] [PubMed] [Google Scholar]
- 5.Goldblatt H, Lynch J, Hanzal RF, Summerville WW. Studies on experimental hypertension. 1. The production of persistent elevation of systolic blood pressure by means of renal ischemia. J Exp Med. 1934;59:347–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Braun-Menendéz E, Fasciolo JC, Leloir LF, Muñoz JM. La sustancia hipertensora de la sangre del riñón isquemiado. Rev Soc Arg Biol. 1939;15:420–425. [Google Scholar]
- 7.Page IH, Helmer OM. A crystalline pressor substance (angiotonin) resulting from the reaction between renin and renin-activator. J Exp Med. 1940;71:29–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gavras H, Brunner HR, Vaughan D, Laragh JH. Angiotensin-sodium interaction in blood pressure maintenance of renal hypertensive and normotensive rats. Science. 1973;180:1369–1372. [DOI] [PubMed] [Google Scholar]
- 9.Gavras H, Brunner HR, Thurston H, Laragh JH. Reciprocation of renin dependency and sodium volume dependency in renal hypertension. Science. 1975;188:1316–1317. [DOI] [PubMed] [Google Scholar]
- 10.Sealey JE, Blumenfeld JD, Bell GM, Pecker MS, Sommers SC, Laragh JH. On the renal basis for essential hypertension: nephron heterogeneity with discordant renin secretion and sodium excretion causing a hypertensive vasoconstriction - volume relationship. J Hypertens. 1988;6:763–778. [DOI] [PubMed] [Google Scholar]
- 11.Okuda T, Grollman A. Passive transfer of autoimmune induced hypertension in the rat by lymph node cells. Tex Rep Biol Med. 1967;25:257–264. [PubMed] [Google Scholar]
- 12.Olsen F. Transfer of arterial hypertension by splenic cells from DOCA-salt hypertensive and renal hypertensive rats to normotensive recipients. Acta Pathol Microbiol Scand C. 1980;88:1–5. [DOI] [PubMed] [Google Scholar]
- 13.Dzielak DJ. Immune mechanisms in experimental and essential hypertension. Am J Physiol. 1991;260:R459–R467. [DOI] [PubMed] [Google Scholar]
- 14.Bendich A, Belisle EH, Strausser HR. Immune system modulation and its effect on the blood pressure of the spontaneously hypertensive male and female rat. Biochem Biophys Res Commun. 1981;99:600–607. [DOI] [PubMed] [Google Scholar]
- 15.Ba D, Takeichi N, Kodama T, Kobayashi H. Restoration of T cell depression and suppression of blood pressure in spontaneously hypertensive rats (SHR) by thymus grafts or thymus extracts. J Immunol. 1982;128:1211–1216. [PubMed] [Google Scholar]
- 16.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, Muller DN, Hafler DA. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. 2013;496:518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilck N, Balogh A, Marko L, Bartolomaeus H, Muller DN. The role of sodium in modulating immune cell function. Nature Rev Nephrol. 2019;15:546–558. [DOI] [PubMed] [Google Scholar]
- 19.Lev-Ran A, Porta M. Salt and hypertension: a phylogenetic perspective. Diabetes/metabolism Res Rev. 2005;21:118–131. [DOI] [PubMed] [Google Scholar]
- 20.Frisoli TM, Schmieder RE, Grodzicki T, Messerli FH. Salt and hypertension: is salt dietary reduction worth the effort? Am J Med. 2012;125:433–439. [DOI] [PubMed] [Google Scholar]
- 21.He FJ, MacGregor GA. Reducing population salt intake worldwide: from evidence to implementation. Prog Cardiovascr Dis. 2010;52:363–382. [DOI] [PubMed] [Google Scholar]
- 22.He FJ, Li J, Macgregor GA. Effect of longer term modest salt reduction on blood pressure: Cochrane systematic review and meta-analysis of randomised trials. BMJ. 2013;346:f1325. [DOI] [PubMed] [Google Scholar]
- 23.Aburto NJ, Ziolkovska A, Hooper L, Elliott P, Cappuccio FP, Meerpohl JJ. Effect of lower sodium intake on health: systematic review and meta-analyses. BMJ. 2013;346:f1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He FJ, MacGregor GA. Salt reduction lowers cardiovascular risk: meta-analysis of outcome trials. Lancet. 2011;378:380–382. [DOI] [PubMed] [Google Scholar]
- 25.Laffer CL, Scott RC III, Titze JM, Luft FC, Elijovich F. Hemodynamics and salt-and-water balance link sodium storage and vascular dysfunction in salt-sensitive subjects. Hypertension. 2016;68:195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barbaro NR, Foss JD, Kryshtal DO, et al. Dendritic cell amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Rep. 2017;21:1009–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Beusecum JP, Barbaro NR, McDowell Z, Aden LA, Xiao L, Pandey AK, Itani HA, Himmel LE, Harrison DG, Kirabo A. High salt activates CD11c(+) antigen-presenting cells via SGK (serum glucocorticoid kinase) 1 to promote renal inflammation and salt-sensitive hypertension. Hypertension. 2019;74:555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kirabo A, Fontana V, de Faria AP, et al. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest. 2014;124:4642–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barbaro NR, Van Beusecum J, Xiao L, et al. Sodium activates human monocytes via the NADPH oxidase and isolevuglandin formation. Cardiovasc Res. 2021;117:1358–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dudenbostel T, Calhoun DA, Use of aldosterone antagonists for treatment of uncontrolled resistant hypertension. Am J Hypertens. 2017;30:103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saha C, Eckert GJ, Ambrosius WT, Chun TY, Wagner MA, Zhao Q, Pratt JH. Improvement in blood pressure with inhibition of the epithelial sodium channel in blacks with hypertension. Hypertension. 2005;46:481–487. [DOI] [PubMed] [Google Scholar]
- 32.Laffer CL, Elijovich F, Eckert GJ, Tu W, Pratt JH, Brown NJ. Genetic variation in CYP4A11 and blood pressure response to mineralocorticoid receptor antagonism or ENaC inhibition: an exploratory pilot study in African Americans. J Am Soc Hypertens. 2014;8:475–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kleyman TR, Eaton DC. Regulating ENaC’s gate. Am J Physiol Cell Physiol. 2020;318:C150–C162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Knoepp F, Ashley Z, Barth D, et al. Shear force sensing of epithelial Na+ channel (ENaC) relies on N-glycosylated asparagines in the palm and knuckle domains of alphaENaC. Proc Natl Acad Sci USA. 2020;117:717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jeggle P; Callies C; Tarjus A; Fassot C; Fels J; Oberleithner; Jaisser F; Kusche-Vihrog K. Epithelial sodium channel stiffens the vascular endothelium in vitro and in Liddle mice. Hypertension. 2013;61:1053–1059. [DOI] [PubMed] [Google Scholar]
- 36.Wang ZR, Liu HB, Sun YY, et al. Dietary salt blunts vasodilation by stimulating epithelial sodium channels in endothelial cells from salt-sensitive Dahl rats. Br J Pharmacol. 2018;175:1305–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Faulkner JL; Belin de Chantemele EJ. Mineralocorticoid receptor and endothelial dysfunction in hypertension. Curr Hypertens Rep. 2019;21:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu HB, Zhang J, Sun YY, et al. Dietary salt regulates epithelial sodium channels in rat endothelial cells: adaptation of vasculature to salt. Br J Pharmacol. 2015;172:5634–5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kusche-Vihrog K, Tarjus A, Fels J, Jaisser F. The epithelial Na+ channel: a new player in the vasculature. Curr Opin Nephrol Hypertens. 2014;23:143–148. [DOI] [PubMed] [Google Scholar]
- 40.Amin MS, Wang HW, Reza E, Whitman SC, Tuana BS, Leenen FH. Distribution of epithelial sodium channels and mineralocorticoid receptors in cardiovascular regulatory centers in rat brain. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1787–R1797. [DOI] [PubMed] [Google Scholar]
- 41.Mills NJ, Sharma K, Haque M, Moore M, Teruyama R. Aldosterone mediated regulation of epithelial sodium channel (ENaC) subunits in the rat hypothalamus. Neuroscience. 2018;390:278–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leenen FH, Hou X, Wang HW, Ahmad M. Enhanced expression of epithelial sodium channels causes salt-induced hypertension in mice through inhibition of the alpha2-isoform of Na+, K+-ATPase. Physiol Rep. 2015;3:e12383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goodwill VS, Terrill C, Hopewood I, Loewy AD, Knuepfer MM. CNS sites activated by renal pelvic epithelial sodium channels (ENaCs) in response to hypertonic saline in awake rats. Auton Neurosci. 2017;204:35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sakamoto T, Fujii A, Saito N, Kondo H, Ohuchi A. Alteration of amiloride-sensitive salt taste nerve responses in aldosterone/NaCl-induced hypertensive rats. Neurosci Res. 2016;108:60–66. [DOI] [PubMed] [Google Scholar]
- 45.Nakamura T, Kurihara I, Kobayashi S, et al. Intestinal mineralocorticoid receptor contributes to epithelial sodium channel-mediated intestinal sodium absorption and blood pressure regulation. J Am Heart Assoc. 2018;7:e008259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rao US, Baker JM, Pluznick JL, Balachandran P. Role of intracellular Ca2+ in the expression of the amiloride-sensitive epithelial sodium channel. Cell Calcium. 2004;35:21–28. [DOI] [PubMed] [Google Scholar]
- 47.Soundararajan R, Pearce D, Ziera T. The role of the ENaC-regulatory complex in aldosterone-mediated sodium transport. Mol Cell Endocrinol. 2012;350:242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Capdevila JH, Pidkovka N, Mei S, Gong Y, Falck JR, Imig JD, Harris RC, Wang W. The Cyp2c44 epoxygenase regulates epithelial sodium channel activity and the blood pressure responses to increased dietary salt. J Biol Chem. 2014;289:4377–4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bohnert BN, Menacher M, Janessa A, et al. Aprotinin prevents proteolytic epithelial sodium channel (ENaC) activation and volume retention in nephrotic syndrome. Kidney Int. 2018;93:159–172. [DOI] [PubMed] [Google Scholar]
- 50.Frindt G, Yang L, Bamberg K, Palmer LG. Na restriction activates epithelial Na channels in rat kidney through two mechanisms and decreases distal Na+ delivery. J Physiol. 2018;596:3585–3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nguyen MTX, Lee DH, Delpire E, McDonough AA. Differential regulation of Na+ transporters along nephron during ANG II-dependent hypertension: distal stimulation counteracted by proximal inhibition. Am J Physiol Ren Physiol. 2013;305:F510–F519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Elijovich F, Milne GL, Brown NJ, Laniado-Schwartzman M, Laffer CL. Two pools of eicosatrienoic acids in humans: alterations in salt sensitive normotensive subjects. Hypertension. 2018;71:346–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sahinoz M, Elijovich F, Laffer CL, Pitzer A, Stewart TG, Ikizler TA, Kirabo A. The relationship between tissue sodium storage, immune cell activation and salt-sensitive hypertension. Abstract. Council on Hypertension. Hypertension. 2020;76:AP139. [Google Scholar]
- 54.Ferreri NR, Escalante BA, Zhao Y, An SJ, McGiff JC. Angiotensin II induces TNF production by the thick ascending limb: functional implications. Am J Physiol. 1998;274:F148–155. [DOI] [PubMed] [Google Scholar]
- 55.Hao S, Salzo J, Hao M, Ferreri NR. Regulation of NKCC2B by TNF-alpha in response to salt restriction. Am J Physiol Renal Physiol. 2020;318:F273–F282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS, Delpire E, Harrison DG, McDonough AA. Renal transporter activation during angiotensin-II hypertension is blunted in interferon-gamma−/− and interleukin-17A−/− mice. Hypertension. 2015;65:569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pitzer AL, Kirabo A. Dendritic Cell A20: Targeting hypertension in autoimmunity. Circ Res. 2019;125:1067–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu X, Rudemiller NP, Wen Y, Ren J, Hammer GE, Griffiths R, Privratsky JR, Yang B, Sparks MA, Crowley SD. A20 in myeloid cells protects against hypertension by inhibiting dendritic cell-mediated T-cell activation. Circ Res. 2019;125:1055–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang J, Rudemiller NP, Patel MB, Karlovich NS, Wu M, McDonough AA, Griffiths R, Sparks MA, Jeffs AD, Crowley SD. Interleukin-1 receptor activation potentiates salt reabsorption in angiotensin II-induced hypertension via the NKCC2 co-transporter in the nephron. Cell Metab. 2016;23:360–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Machnik A, Neuhofer W, Jantsch J, et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med. 2009;15:545–552. [DOI] [PubMed] [Google Scholar]
- 61.Rossitto G, Mary S, Chen JY, et al. Tissue sodium excess is not hypertonic and reflects extracellular volume expansion. Nat Comm. 2020;11:4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kopp C, Linz P, Dahlmann A, et al. 23Na magnetic resonance imaging-determined tissue sodium in healthy subjects and hypertensive patients. Hypertension. 2013;61:635–640. [DOI] [PubMed] [Google Scholar]
- 63.Selvarajah V, Maki-Petaja KM, Pedro L, Bruggraber SFA, Burling K, Goodhart AK, Brown MJ, McEniery CM, Wilkinson IB. Novel mechanism for buffering dietary salt in humans: effects of salt loading on skin sodium, vascular endothelial growth factor C and blood pressure. Hypertension. 2017;70:930–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wilck N, Matus MG, Kearney SM, et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature. 2017;551:585–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ferguson JF, Aden LA, Barbaro NR, et al. High dietary salt-induced dendritic cell activation underlies microbial dysbiosis-associated hypertension. JCI insight. 2019;5. [DOI] [PMC free article] [PubMed] [Google Scholar]