

PURPOSE
Poly (ADP-ribose) polymerase (PARP) inhibitors have emerged as promising therapy in cancers with homologous recombination repair deficiency. However, efficacy is limited by both intrinsic and acquired resistance. The Olaparib Combinations basket trial explored olaparib alone and in combination with other homologous recombination–directed targeted therapies. Here, we report the results of the arm in which olaparib was combined with the orally bioavailable ataxia telangiectasia and RAD3-related inhibitor ceralasertib in patients with relapsed or refractory cancers harboring DNA damage response and repair alterations, including patients with BRCA-mutated PARP inhibitor–resistant high-grade serous ovarian cancer (HGSOC).
PATIENTS AND METHODS
Germline and somatic mutations had to be deleterious by COSMIC or ClinVar for eligibility. Olaparib was administered at 300 mg twice daily and ceralasertib at 160 mg daily on days 1-7 in 28-day cycles until progression or unacceptable toxicities. Primary end points were confirmed complete response (CR) or partial response (PR) rates and clinical benefit rate (CBR; CR + PR + stable disease [SD] at 16 weeks).
RESULTS
Twenty-five patients were enrolled, with median four prior therapies. Five patients required dose reductions for myelosuppression. Overall response rate was 8.3% and CBR was 62.5% among the entire cohort. Two of five patients with tumor harboring ATM mutation achieved CR or SD ongoing at 24+ months, respectively (CBR 40%). Of seven patients with PARP inhibitor–resistant HGSOC, one achieved PR (–90%) and five had SD ranging 16-72 weeks (CBR 86%).
CONCLUSION
Olaparib with ceralasertib demonstrated preliminary activity in ATM-mutated tumors and in PARP inhibitor–resistant BRCA1/2–mutated HGSOC. These data warrant additional studies to further confirm activity in these settings.
INTRODUCTION
DNA damage response and repair (DDR) is an essential function to maintain viability in all cells. Homologous recombination (HR) repair is a high-fidelity process used to repair DNA double-strand breaks (DSB), during the S and G2 phases.1 Germline mutations in HR genes and other genes involved in DDR and the genes giving rise to Fanconi anemia significantly increase the lifetime risk of certain cancers. HR and DDR pathway genes mutations with accompanying loss of heterozygosity (LOH) have been observed in 17%-21% of patients in large, pancancer data sets.2,3
CONTEXT
Key Objective
Despite initial benefit from platinum and poly (ADP-ribose) polymerase (PARP) inhibitor–based therapy in high-grade serous ovarian cancer and in other cancers harboring BRCA alterations or other homologous recombination repair defects, most tumors develop resistance resulting in treatment failure. Reversing resistance and enhancing response to PARP inhibitors is an area of unmet need.
Knowledge Generated
Inhibition of the ATR-CHK1 pathway reverses restored homologous recombination and compromises replication fork stability in BRCA-mutated tumors with acquired PARP inhibitor resistance. Ceralasertib, an ataxia telangiectasia and RAD3-related inhibitor, combined with olaparib in heavily pretreated ovarian cancer patients with BRCA-mutated cancers resistant to PARP inhibitors produced an encouraging early clinical benefit signal.
Relevance
Ceralasertib combined with olaparib was well-tolerated and effective in a subset of BRCA-mutated patients with PARP inhibitor–resistant high-grade serous ovarian cancer in this exploratory trial and should be further studied in this population.
HR deficiency has been shown to be synthetically lethal with inhibition of poly (ADP-ribose) polymerase (PARP) leading to the advent of PARP inhibitors in HR-deficient tumors, particularly in BRCA-mutated high-grade serous ovarian cancer (HGSOC), as well as breast, pancreatic, and prostate cancers.4-6 TCGA data sets have shown that biallelic HR gene inactivation may be present in other cancer types as well and is associated with genomic features of HR deficiency.7 In HGSOC, approximately 50% are characterized by genetic and epigenetic alterations of the HR pathway genes, particularly BRCA genes.8-10 HR deficiency has been an important therapeutic target in ovarian cancer. In patients with recurrent disease, PARP inhibitors have been used in patients with tumors harboring BRCA alterations with response rates typically exceeding 30%.11-14 Additionally, these agents are now commonly considered irrespective of BRCA mutation in the second-line maintenance setting, after a response to a platinum-based chemotherapy.8-10 Recent work has also examined PARP inhibition in the first-line maintenance setting.15,16 More common PARP inhibitors use has highlighted the importance of both acquired and de novo resistance. The outlook for patients with HGSOC with acquired or de novo HR proficiency is poor so that reversal of resistance is a pressing clinical problem.
Ataxia telangiectasia and RAD3-related (ATR) is a member of the phosphoinositide 3-kinase-related kinase family. ATR governs checkpoints that serve to ensure cell survival after replication stress or DNA damage. ATR is recruited to stalled replication forks, where it mediates CHK1 activation resulting in cell cycle arrest in S phase. ATR and CHK1 phosphorylate PALB2 and RAD51, respectively, facilitating HR repair.17,18 ATR also initiates the cascade of events culminating in G2 arrest following DNA damage. Preclinical data have supported the synergism of ATR and PARP inhibition in BRCA-mutated PARP inhibitor–sensitive ovarian and breast cancer models.19 Mechanistically, PARP inhibition leads to G2 accumulation; the addition of ATR inhibition promotes release from G2 with premature mitotic entry with increased chromosomal aberrations and apoptosis.20
ATR inhibition is also a promising strategy to overcome PARP inhibitor resistance in BRCA-mutated cancers.20 PARP inhibitor–resistant BRCA-deficient cells are increasingly dependent on ATR for genomic stability and survival. Preclinical data suggest that ATR inhibition targets PARP inhibitor resistance through two potential mechanisms, including disruption of BRCA1-independent RAD51 loading to sites of DSB and reversal of BRCA1-independent replication fork protection.20 Combined ATR-CHK1 axis and PARP inhibition has been shown to be cooperative in the PARP inhibitor–resistant setting, with synergistic increases in replication fork stalling, DSB, and apoptosis, coupled with compromised HR repair, translating to improved survival in preclinical ovarian cancer models.20,21
Like ATR, ataxia telangiectasia-mutated (ATM) has both DNA damage–induced checkpoint and repair functions. ATM deficiency is expected to sensitize malignant cells to ATR inhibition, which has been demonstrated both in preclinical models and in clinical trials.22-25 Additionally, in preclinical pancreatic and lung cancer models, ATM deficiency also sensitizes to PARP inhibition, suggesting that combined ATR and PARP inhibition may be useful.26,27 In contrast, other studies showed low sensitivity of ATM-deficient prostate cancers to PARP inhibition.28,29 Although the role of PARP inhibitor monotherapy in ATM-deficient tumors is not fully clarified, combined ATR and PARP inhibition may still provide clinical benefit.
The Olaparib Combinations (OLAPCO) trial is a basket trial that has explored olaparib alone and in combination with other HR-directed targeted therapies. The objective of this arm of the OLAPCO trial was to assess the efficacy of the combined regimen of olaparib and the ATR inhibitor ceralasertib in previously treated patients with DDR-deficient solid tumors.
PATIENTS AND METHODS
Patient Selection
For the ceralasertib-olaparib arm of OLAPCO (NCT02576444), patients with tumor mutations in HR and other DDR genes were identified by tests performed in a Clinical Laboratory Improvement Amendments–certified laboratory, either locally at one of the participating sites or at a commercial testing facility, before participation in the trial. These platforms included standard local (including Oncomine30 or Oncopanel31) or commercial (including Myriad, FoundationOne, or Tempus) platforms. Patients with tumors harboring deleterious mutations in HR genes (including BRCA1, BRCA2, PALB2, and other genes) and other DNA repair pathway genes (including ATM and CHEK2), as well as mutations in TCA cycle genes implicated in HR defects,32 were enrolled. Patients with HGSOC harboring germline or somatic mutations in BRCA1/2 genes and who had prior progression on PARP inhibitors were also permitted to enroll. Germline and somatic mutations had to be deleterious by COSMIC or ClinVar for eligibility.
Eligibility
Eligible patients had to have received standard first-line therapy for metastatic cancer (except for tumors for which no first-line therapy exists) with progressive disease at the time of study entry. Other eligibility criteria include measurable disease by RECIST v1.1 and age ≥ 18 years with life expectancy ≥ 16 weeks. Enrolled patients had Eastern Cooperative Oncology Group performance score of 0-1, adequate hematologic function with no features suggestive of myelodysplastic syndrome/acute myelomonocytic leukemia, and adequate hepatic and renal function. Prior treatment with a PARP inhibitor was required in patients with BRCA-mutated HGSOC during the second stage of the study, but there was no specification regarding the duration or response to prior PARP inhibition. The number of prior lines of therapy was not restricted on this study.
Protocol Treatment
Patients received olaparib 300 mg orally twice a day continuously and ceralasertib 160 mg orally on days 1-7 in 28-day cycles until disease progression or unacceptable toxicities. This regimen was based on the recommended phase II dose of ceralasertib that could be combined with full-dose olaparib that was established during a prior phase I study. Toxicities were evaluated using the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0. Dose reductions for olaparib were 250 mg (dose level –1) and 200 mg (dose level –2) daily, whereas for ceralasertib, dose reductions were 160 mg for day 1-4 for hematologic toxicities or 120 mg oral daily for day 1-7 for nonhematologic toxicities (dose level –1) and 120 mg oral day 1-4 for hematologic or nonhematologic toxicities (dose level –2). Dose reescalation was not permitted.
Ceralasertib and olaparib could be reduced in stepwise fashion for anemia, where dose reduction for olaparib was recommended first, followed by a dose reduction for ceralasertib if the adverse event (AE) recurred. Simultaneous reduction was also allowed depending on the severity and duration of anemia. If a dose reduction was required for neutropenia, leukopenia, or thrombocytopenia, olaparib and ceralasertib were reduced simultaneously because of the greater frequency of these events associated with ceralasertib.
End Points
The primary objectives were to determine overall response rate (ORR) by RECIST v1.1 and clinical benefit rate (CBR), defined as ORR and stable disease (SD) after 16 weeks of treatment.33 Additional objectives were to determine progression-free survival, duration of ORR and SD, and AEs.
Statistical Design, Sample Size Justification, and Decision Rules
A two-stage accrual design was used.34,35 A 30% ORR was considered worthy of further study. Initially, 16 eligible patients were to be treated. If there were < 2 responses, accrual would be terminated on the basis of the likelihood of an ORR of ≤ 10%. If ≥ 2 responded in the first stage, the study would continue until 25 patients were treated. This design provided 90% power with a significance level of < .10 (type I error). The second stage was activated with the goal of evaluating more patients with BRCA-mutated, PARP inhibitor–resistant HGSOC on the basis of an initial signal observed among the first 16 patients.
Additionally, an early stopping rule for safety was incorporated. If among the first 16 patients, ≥ 4 experienced unacceptable toxicity, enrollment would be terminated early. Unacceptable toxicity was defined as grade 4 hematologic and grade 3 nonhematologic toxicities that failed to resolve to grade 1 despite appropriate supportive care, as defined by Common Terminology Criteria for Adverse Events version 4.0. With this design, the probability of terminating the arm early was .07 if the true but unknown unacceptable toxicity rate was 10% and 0.75 if the true toxicity rate was 30%.
RESULTS
Patient Characteristics
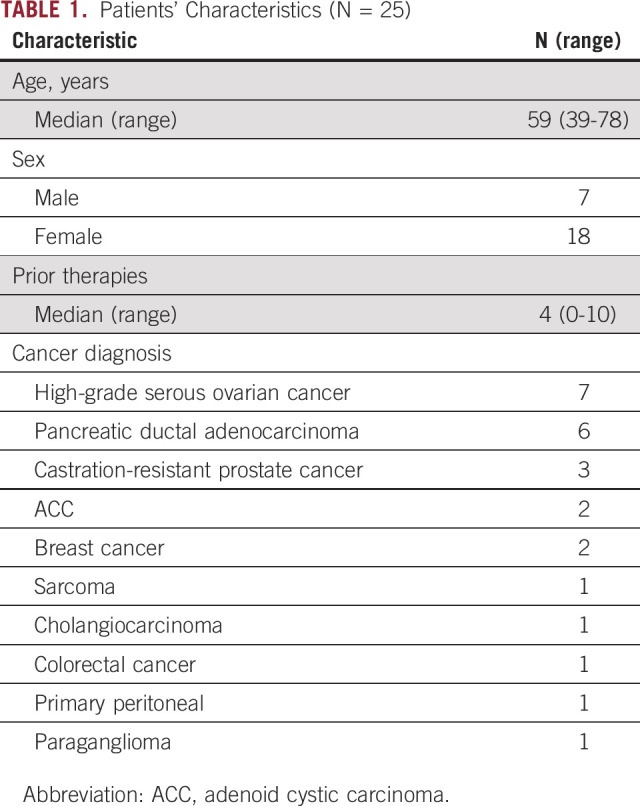
Twenty-five patients were enrolled over 14 months. The median age was 59 years (39-79), including 18 females and seven males. The median number of prior therapies was 4 (0-10). Patient characteristics are presented in Table 1.
TABLE 1.
Patients' Characteristics (N = 25)
Safety and Tolerability
The combined regimen of olaparib and ceralasertib was well-tolerated. Probable or definite treatment-related AEs as judged by the treating investigator are listed in Table 2. All patients had grade 1 AEs that were deemed to be mild and required no dose alterations. These events occurred after many cycles on treatment. Only the most severe grade for each individual patient toxicity is entered. Hematologic toxicity was the most common event as expected from previous clinical trials of each agent. One patient, a woman with germline BRCA1-mutated HGSOC, who had received eight prior regimens, experienced grade 3 anemia, grade 4 neutropenia, and thrombocytopenia and required two dose reductions for olaparib and one reduction for ceralasertib. A second patient with grade 4 neutropenia also required a dose reduction for olaparib alone. Three more patients were dose-reduced because of anemia. All adverse hematologic toxicities were reversible. Nonhematologic toxicities were rare and all were ≤ grade 2. No treatment-related deaths occurred. Among the two patients who achieved objective response, one patient with BRCA1-mutated PARP inhibitor–resistant HGSOC required dose reduction but continued to have response to the combination therapy.
TABLE 2.
Summary of Treatment-Related AEs

Efficacy
The individual germline and somatic mutations of enrolled patients are listed in Table 3, along with diagnosis and clinical outcome. One patient was not evaluable because she withdrew consent after being enrolled. Among the 24 evaluable patients, there was one complete response (CR) and one partial response (ORR 8.3%; Fig 1). These were durable, confirmed responses that occurred among the first 16 patients, allowing the study to progress to the second stage. Thirteen patients (54.2%) had SD for at least 16 weeks with a CBR of 62.5% (Fig 1). The median duration of response was 22 months (18-26+ months), and the median duration of clinical benefit was 5 months (4-26+ months).
TABLE 3.
Cancer Sites, Mutational Status, and Extent and Duration of Response of Patients Who Received Olaparib and Ceralasertib/AZD6738

FIG 1.

Efficacy of ceralasertib and olaparib in all patients enrolled in the study. A swimmer plot demonstrating time to response and duration of study treatment. CR, complete response; HGSOC, high-grade serous ovarian cancer; PR, partial response.
Five patients with ATM mutations were included. The ORR was 20% (1 of 5) and CBR was 40% (2 of 5, including one patient with durable CR and one patient with durable SD; Fig 2). The CR occurred in a patient with estrogen receptor–positive metastatic breast cancer and a germline ATM mutation with LOH. CR was initially achieved at 4 months and has been ongoing for 26+ months. A second patient with primary adenoid cystic carcinoma of minor salivary gland and germline ATM mutation with LOH has had an ongoing 22% reduction in target lesions and has also remained on treatment for 26+ months. This patient had previously been treated with surgery for a primary tumor of the sella turcica, proton beam radiation, and cisplatin and has multiple lytic bone metastases. Neither patient has had significant toxicity or required a dose reduction. Two patients with PDAC and ATM mutation (unknown LOH status) progressed rapidly. A patient with colon cancer had a 29% reduction in target lesions at 2 months but progressed at 4 months with new lesions.
FIG 2.
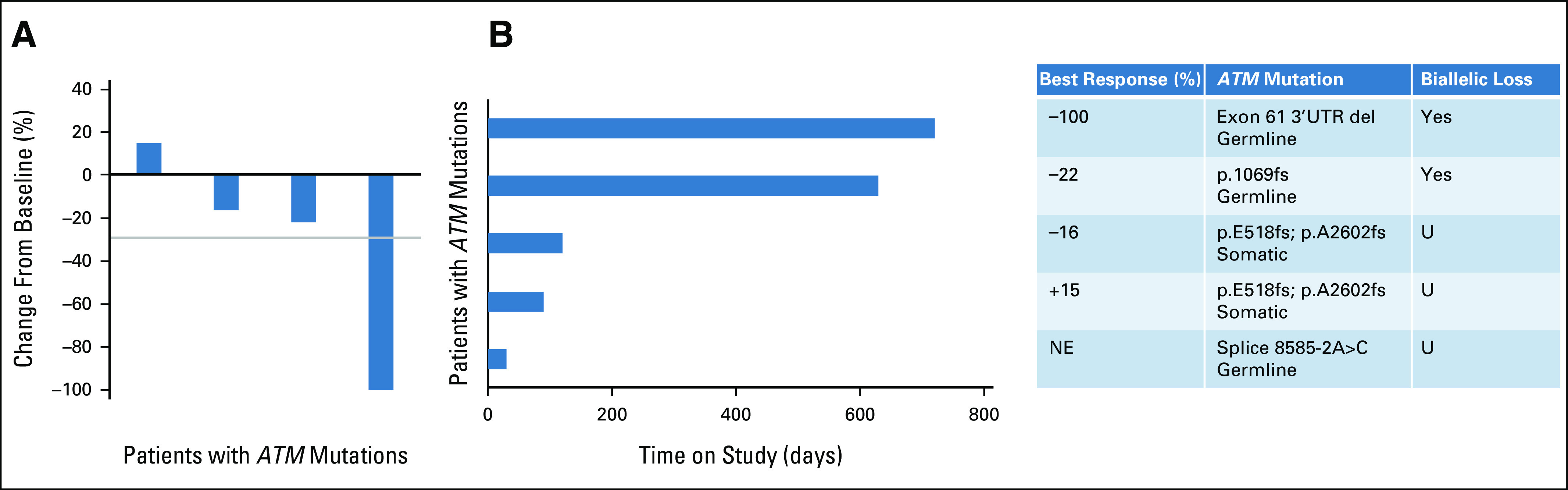
Efficacy of ceralasertib and olaparib in a subset of patients with ATM mutation. (A) Waterfall plot of the best objective response measured as the maximum change from baseline in the sum of the longest diameter of each target lesion. (B) Swimmer plot demonstrating time to response and duration of study treatment. The table on the right defines the cohort, extent of response, ATM mutation details, and biallelic status. NE, non-evaluable; U, unknown.
Seven patients with HGSOC with BRCA1/2 mutations represented the largest group (Table 4). All patients were heavily pretreated; the median number of prior regimens was 5 (range 2-11). Furthermore, all patients had received 1-3 prior PARP inhibitor–based regimens and had progressed during their most recent PARP inhibitor exposure. The ORR was 14% (1 of 7) and the SD rate was 71% (5 of 7), leading to a CBR of 85.7% (6 of 7; Fig 3). The median duration of clinical benefit among the seven patients was 8 months (2-18 months). The duration of benefit from olaparib and ceralasertib exceeded the initial duration of response to the PARP inhibitor in these patients (median 8 months, range 2-18 months v 4 months, range 2-12 months; Table 4). Among the five patients with germline BRCA1 mutations, one had a partial response (–90% tumor reduction for 18 months) and two others had minor responses (–13% for 11 months; –27% for 8 months).
TABLE 4.
Prior Therapy, Extent and Duration of Response of Patients With BRCA1/2–Mutated High-Grade Serous Ovarian Cancer Post-PARP Inhibition Received Olaparib and Ceralasertib/AZD6738

FIG 3.
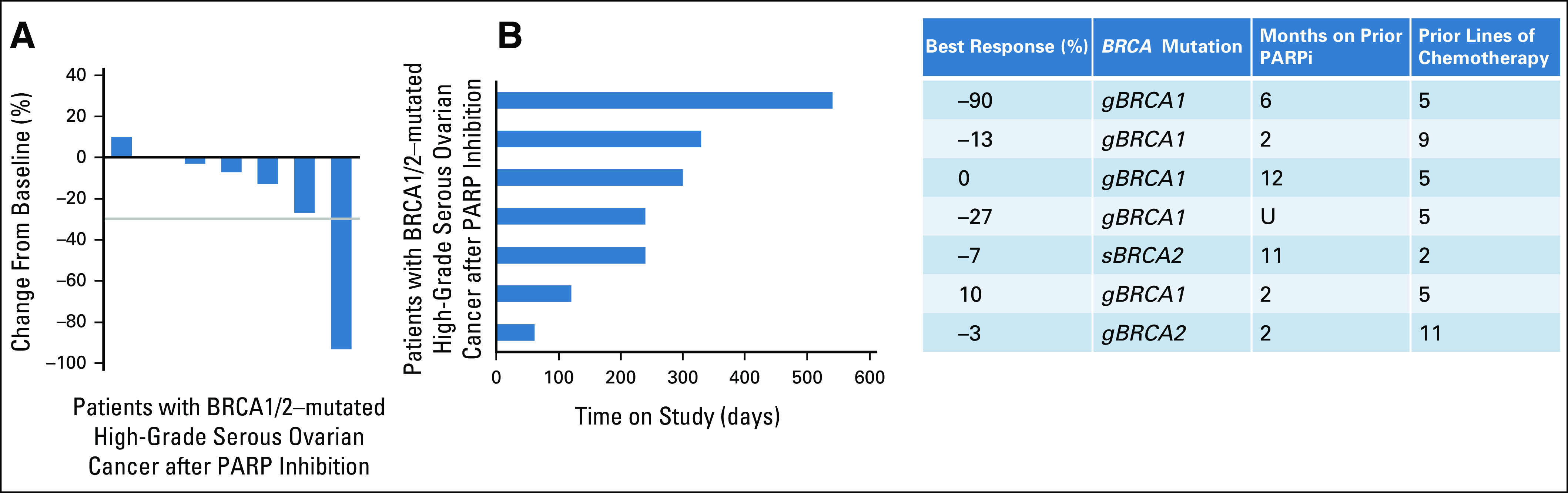
Efficacy of ceralasertib and olaparib in a subset of patients with BRCA1/2–mutated high-grade serous ovarian cancer after PARP inhibition. (A) Waterfall plot of the best objective response measured as the maximum change from baseline in the sum of the longest diameter of each target lesion. (B) Swimmer plot demonstrating time to response and duration of study treatment. The table on the right defines the cohort, BRCA mutation details, extent of response and number of prior lines of therapy as well as the duration of therapy on prior PARP inhibition. PARPi, poly (ADP-ribose) polymerase inhibitor. U, unknown.
In other solid tumors with pathogenic BRCA mutation, three patients had brief periods of SD. Among four patients with PALB2 mutations, one with metastatic adenoid cystic carcinoma of salivary gland has had ongoing disease stability at 14+ months. Notably, this patient had a 23% increase in pulmonary target lesions in the year preceding study initiation and < 1% increase in these target lesions while receiving olaparib and ceralasertib. Two patients with MUS81 mutation (presumed to be germline from the family history) and CHEK2 mutation, respectively, did not derive benefit.
Two patients who had received olaparib monotherapy as part of another cohort in OLAPCO received olaparib and ceralasertib on disease progression. A patient with PDAC harboring PALB2 mutation received olaparib alone for 11 months previously with SD (23% reduction by RECIST 1.1) but progressed without response on olaparib and ceralasertib. In contrast, a second patient with an IDH1-mutated chondrosarcoma had SD for 7 months on olaparib monotherapy and subsequently experienced SD for an additional 7 months on combined olaparib and ceralasertib.
DISCUSSION
The OLAPCO trial is an exploratory basket study that includes several olaparib combinations in genomically targeted patient subsets. In this arm, patients with tumors harboring DDR alterations were treated with combined ceralasertib and olaparib. The regimen used full-dose olaparib along with ceralasertib on the basis of prior phase I data. The ORR was 8.3%, making it a negative trial, although the CBR of 62.5% was promising in this heavily pretreated population. Responses and instances of clinical benefit were observed in subsets of patients with tumors harboring ATM mutation and in patients with BRCA-mutated HGSOC with acquired PARP inhibitor resistance. These findings warrant further investigation in larger groups of patients.
ATR inhibition has been shown to be synthetically lethal with ATM deficiency in preclinical models, translating to responses to monotherapies that have been reported with agents such as BAY1895344 and M6620.25,36 The complete loss of ATM protein is ideally confirmed by protein immunohistochemistry, but this is not yet a standard laboratory test. Genetic testing is not as certain, although LOH can sometimes be confirmed. In this trial, two patients with germline ATM mutation and evidence of biallelic loss in tumor had durable clinical benefit, with one patient achieving a CR. Loss of the wild-type ATM allele could be identified in these two patients as determined by variant allelic frequency in the tumor when compared with germline control. The activity of PARP inhibition in the ATM-deficient setting has primarily been studied in prostate cancer and is less clear, although responses have been reported.29 Ultimately, randomized trials in ATM-deficient cancers will be required to determine whether the activity is driven by ATR inhibition alone or whether the combination contributed to the benefit observed.
PARP inhibition is now part of the standard armamentarium for HGSOC so that it is critical to develop strategies addressing acquired resistance. This exploratory experience suggests promise for combined ATR and PARP inhibition in this setting. There are several postulated mechanisms of PARP inhibitor resistance including expression of drug efflux pumps or loss of PARP1 protein expression. Restoration of HR pathway function is also a major resistance mechanism that may occur by somatic reversion or restoration of an open reading frame, epigenetic reversion of BRCA1 promoter hypermethylation, express of a hypomorphic protein with residual BRCA function, or by loss of end resection regulation.37 Additionally, stabilization of replication forks represents another major mechanism of PARP inhibitor resistance.20,37
Preclinical evidence supports the importance of the ATR-CHK1 pathway in BRCA-mutated cancers, where it is used to maintain genomic stability. PARP inhibitor–resistant BRCA1-deficient cells become increasingly dependent on ATR for survival.20,21 Despite the lack of BRCA1, PARP inhibitor–resistant cells regain RAD51 loading to DNA double-stranded breaks and stalled replication forks, enabling both restored HR and replication fork stabilization as resistance mechanisms. ATR inhibition will compromise HR and destabilize replication forks to overcome both resistance mechanisms. ATR inhibition also overcomes PARP inhibitor resistance in BRCA2-mutated ovarian cancer models. In general, responses in PARP inhibitor–resistant preclinical models are superior with combined ATR and PARP inhibition compared with ATR inhibition alone.21 Our results suggest that the ceralasertib-olaparib combination has potential clinical activity with manageable toxicity in BRCA-mutated PARP inhibitor–resistant HGSOC, with a duration of benefit that exceeded the duration achieved on prior PARP inhibitor monotherapy.
The current schedule uses full-dose olaparib with attenuated ceralasertib, which may be most appropriate in less heavily treated, PARP inhibitor–naïve patients. In both the ATM-deficient and PARP inhibitor–resistant settings, a schedule maximizing ceralasertib may be preferable. Dose-finding efforts are underway in other clinical trials with olaparib at 100-150 mg twice daily, which may afford substantially higher doses of ceralasertib that may ultimately be critical for maximizing clinical activity.
Further work will be required for insights into nonresponding patients with tumors harboring DDR alterations. It is possible that BRCA, PALB2, and other mutations have different functional relevance in certain cancer types38 or that routine assessment of LOH will carry high importance. Additionally, assessment of HR function and replication fork stability at baseline will also be important in future studies to better understand clinical outcomes. Such assessments could include an IHC-based RAD51 assay or DNA fiber assays in organoid cultures derived from patient biopsies.39 Future studies should include planned translational analyses of paired tissue biopsies and serial circulating tumor DNA samples to assess mechanisms of PARP inhibitor resistance and to identify predictors of response and determinants of resistance to the combination regimen.
This study has several limitations. Although we were able to determine biallelic loss in a few of the tumors, we lacked information about the activity of the nonmutated alleles in most cases. In many cases, germline testing was used for eligibility, rather than analysis of tumor DNA. Furthermore, the clinical outcomes were likely limited because the majority of patients were heavily pretreated and were also resistant to prior therapy, including platinum-based chemotherapy and PARP inhibitors. Finally, higher ceralasertib doses with attenuated olaparib may ultimately be the optimal dosing schedule for this combination.
Despite these limitations and the limited ORR of 8.3%, we were able to confirm safety of ceralasertib combined with full-dose olaparib and generate signals of promising clinical benefit in both ATM-deficient and BRCA-mutated PARP inhibitor–resistant ovarian cancer. Further schedule optimization and testing of larger populations in appropriately powered single-arm and randomized trials are warranted.
ACKNOWLEDGMENT
H.M. is now at the University of Pittsburgh. D.D. is now at the Mount Sinai School of Medicine. D.S. is now at the University of Cincinnati. J.J. is now at Gilead Sciences Inc.
Haider Mahdi
Research Funding: Puma Biotechnology
Deborah Doroshow
Consulting or Advisory Role: Ipsen, Atheneum, Boston Healthcare Associates, Dedham Group, Guidepoint Global
Travel, Accommodations, Expenses: Ipsen
Davendra Sohal
Honoraria: Foundation Medicine
Consulting or Advisory Role: Perthera, Ability Pharma
Speakers' Bureau: Incyte
Research Funding: Celgene, Genentech, Bristol Myers Squibb, Incyte, Rafael Pharmaceuticals, Apexigen, Amgen
Vicki Keedy
Consulting or Advisory Role: Karyopharm Therapeutics, Daiichi Sankyo/Lilly
Research Funding: Medpacto, Plexxikon, Daiichi Sankyo, Lilly, Immune Design, GlaxoSmithKline, TRACON Pharma, Advenchen Laboratories, Adaptimmune, Deciphera, SpringWorks Therapeutics
Khanh T. Do
Employment: Moderna Therapeutics
Stock and Other Ownership Interests: Moderna Therapeutics
Consulting or Advisory Role: QED Therapeutics, The Jackson Laboratory
Patricia LoRusso
Honoraria: Five Prime Therapeutics
Consulting or Advisory Role: Genentech, CytomX Therapeutics, Roche/Genentech, Halozyme, Five Prime Therapeutics, Agenus, Agios, Cybrexa Therapeutics, Sotio, AbbVie, Genmab, Takeda, TYME, IQvia, Trial to Reduce IDDM in the Genetically at Risk (TRIGR), Pfizer, ImmunoMet, Black Diamond Therapeutics, GlaxoSmithKline, QED Therapeutics, AstraZeneca, EMD Serono, Shattuck Labs, Astellas Pharma, Salarius Pharmaceuticals, Silverback Therapeutics, Macrogenics, Kyowa Kirin International, Kineta, Zentalis, Molecular Templates, ABL Bio, SK Life Sciences, ST Cube, Bayer, I-Mab
Research Funding: Genentech
Travel, Accommodations, Expenses: Genentech
Juliane Jürgensmeier
Employment: Gilead Sciences
Stock and Other Ownership Interests: Gilead Sciences
Travel, Accommodations, Expenses: Gilead Sciences
Jeffrey Sklar
Stock and Other Ownership Interests: Precipio
Research Funding: Jilin Zixin
Colin Glover
Employment: AstraZeneca
Stock and Other Ownership Interests: AstraZeneca
Brunella Felicetti
Employment: AstraZeneca
Stock and Other Ownership Interests: AstraZeneca
Emma Dean
Employment: AstraZeneca/MedImmune
Stock and Other Ownership Interests: AstraZeneca/MedImmune, AstraZeneca
Travel, Accommodations, Expenses: AstraZeneca
Peter Mortimer
Employment: AstraZeneca
Stock and Other Ownership Interests: AstraZeneca
Travel, Accommodations, Expenses: AstraZeneca
Geoffrey I. Shapiro
Consulting or Advisory Role: G1 Therapeutics, Lilly, Pfizer, Roche, Merck Serono, Sierra Oncology, Cybrexa Therapeutics, Ipsen, Bayer, Fusion Pharmaceuticals, Bicycle Therapeutics, Almac Diagnostics, Astex Pharmaceuticals, Daiichi Sankyo, Angiex, Seattle Genetics, Artios, Boehringer Ingelheim, Concarlo, Atrin Pharmaceuticals, Syros Pharmaceuticals, Zentalis, CytomX Therapeutics
Research Funding: Pfizer, Genentech, Bayer, Immune Design, Vertex, Millennium, Puma Biotechnology, Tensha Therapeutics, Covidien, Novartis, Cellceutix, Sanofi, Cyclacel, Mirati Therapeutics, AstraZeneca, GlaxoSmithKline, Lilly, Aileron Therapeutics, PharmaMar, PTC Therapeutics, Roche, CanBas, Tesaro, Merck Serono, Sierra Oncology, Syros Pharmaceuticals, Curis, Merck, Array BioPharma, Seattle Genetics, Clovis Oncology, Exelixis, Boehringer Ingelheim, Esperas Pharma, Amgen, Bristol Myers Squibb
Patents, Royalties, Other Intellectual Property: Patent No.: 9872874, Title: Dosage regimen for sapacitabine and seliciclib, Issue Date: January 23, 2018; Provisional Patent No.:62/538,319, Title: Compositions and methods for predicting response and resistance to CDK4/6 inhibition, Filed: July 28, 2017
Travel, Accommodations, Expenses: Lilly, Pfizer, Bicycle Therapeutics, G1 Therapeutics, Sierra Oncology, Bayer
Joseph Eder
Honoraria: Roche Molecular Diagnostics
Consulting or Advisory Role: Roche/Genentech
No other potential conflicts of interest were reported.
PRIOR PRESENTATION
AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics, Boston, MA, October 26-30, 2019.
SUPPORT
Supported by AstraZeneca Inc. Also supported by the Dana-Farber/Harvard Cancer Center Specialized Program of Research Excellence (SPORE) in Ovarian Cancer, NIH grant P50 CA240243 to G.I.S.
AUTHOR CONTRIBUTIONS
Conception and design: Juliane Jürgensmeier, Brunella Felicetti, Peter Mortimer, Geoffrey I. Shapiro, Joseph Paul Eder
Administrative support: Patricia LoRusso
Provision of study materials or patients: Haider Mahdi, Navid Hafez, Davendra Sohal, Khanh T. Do, Patricia LoRusso, Geoffrey I. Shapiro
Collection and assembly of data: Haider Mahdi, Navid Hafez, Deborah Doroshow, Davendra Sohal, Vickie Keedy, Khanh T. Do, Patricia LoRusso, Manuel Avedissian, Jeffrey Sklar, Geoffrey I. Shapiro, Joseph Paul Eder
Data analysis and interpretation: Haider Mahdi, Navid Hafez, Deborah Doroshow, Davendra Sohal, Vickie Keedy, Patricia LoRusso, Juliane Jürgensmeier, Jeffrey Sklar, Colin Glover, Brunella Felicetti, Emma Dean, Geoffrey I. Shapiro, Joseph Paul Eder
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Haider Mahdi
Research Funding: Puma Biotechnology
Deborah Doroshow
Consulting or Advisory Role: Ipsen, Atheneum, Boston Healthcare Associates, Dedham Group, Guidepoint Global
Travel, Accommodations, Expenses: Ipsen
Davendra Sohal
Honoraria: Foundation Medicine
Consulting or Advisory Role: Perthera, Ability Pharma
Speakers' Bureau: Incyte
Research Funding: Celgene, Genentech, Bristol Myers Squibb, Incyte, Rafael Pharmaceuticals, Apexigen, Amgen
Vicki Keedy
Consulting or Advisory Role: Karyopharm Therapeutics, Daiichi Sankyo/Lilly
Research Funding: Medpacto, Plexxikon, Daiichi Sankyo, Lilly, Immune Design, GlaxoSmithKline, TRACON Pharma, Advenchen Laboratories, Adaptimmune, Deciphera, SpringWorks Therapeutics
Khanh T. Do
Employment: Moderna Therapeutics
Stock and Other Ownership Interests: Moderna Therapeutics
Consulting or Advisory Role: QED Therapeutics, The Jackson Laboratory
Patricia LoRusso
Honoraria: Five Prime Therapeutics
Consulting or Advisory Role: Genentech, CytomX Therapeutics, Roche/Genentech, Halozyme, Five Prime Therapeutics, Agenus, Agios, Cybrexa Therapeutics, Sotio, AbbVie, Genmab, Takeda, TYME, IQvia, Trial to Reduce IDDM in the Genetically at Risk (TRIGR), Pfizer, ImmunoMet, Black Diamond Therapeutics, GlaxoSmithKline, QED Therapeutics, AstraZeneca, EMD Serono, Shattuck Labs, Astellas Pharma, Salarius Pharmaceuticals, Silverback Therapeutics, Macrogenics, Kyowa Kirin International, Kineta, Zentalis, Molecular Templates, ABL Bio, SK Life Sciences, ST Cube, Bayer, I-Mab
Research Funding: Genentech
Travel, Accommodations, Expenses: Genentech
Juliane Jürgensmeier
Employment: Gilead Sciences
Stock and Other Ownership Interests: Gilead Sciences
Travel, Accommodations, Expenses: Gilead Sciences
Jeffrey Sklar
Stock and Other Ownership Interests: Precipio
Research Funding: Jilin Zixin
Colin Glover
Employment: AstraZeneca
Stock and Other Ownership Interests: AstraZeneca
Brunella Felicetti
Employment: AstraZeneca
Stock and Other Ownership Interests: AstraZeneca
Emma Dean
Employment: AstraZeneca/MedImmune
Stock and Other Ownership Interests: AstraZeneca/MedImmune, AstraZeneca
Travel, Accommodations, Expenses: AstraZeneca
Peter Mortimer
Employment: AstraZeneca
Stock and Other Ownership Interests: AstraZeneca
Travel, Accommodations, Expenses: AstraZeneca
Geoffrey I. Shapiro
Consulting or Advisory Role: G1 Therapeutics, Lilly, Pfizer, Roche, Merck Serono, Sierra Oncology, Cybrexa Therapeutics, Ipsen, Bayer, Fusion Pharmaceuticals, Bicycle Therapeutics, Almac Diagnostics, Astex Pharmaceuticals, Daiichi Sankyo, Angiex, Seattle Genetics, Artios, Boehringer Ingelheim, Concarlo, Atrin Pharmaceuticals, Syros Pharmaceuticals, Zentalis, CytomX Therapeutics
Research Funding: Pfizer, Genentech, Bayer, Immune Design, Vertex, Millennium, Puma Biotechnology, Tensha Therapeutics, Covidien, Novartis, Cellceutix, Sanofi, Cyclacel, Mirati Therapeutics, AstraZeneca, GlaxoSmithKline, Lilly, Aileron Therapeutics, PharmaMar, PTC Therapeutics, Roche, CanBas, Tesaro, Merck Serono, Sierra Oncology, Syros Pharmaceuticals, Curis, Merck, Array BioPharma, Seattle Genetics, Clovis Oncology, Exelixis, Boehringer Ingelheim, Esperas Pharma, Amgen, Bristol Myers Squibb
Patents, Royalties, Other Intellectual Property: Patent No.: 9872874, Title: Dosage regimen for sapacitabine and seliciclib, Issue Date: January 23, 2018; Provisional Patent No.:62/538,319, Title: Compositions and methods for predicting response and resistance to CDK4/6 inhibition, Filed: July 28, 2017
Travel, Accommodations, Expenses: Lilly, Pfizer, Bicycle Therapeutics, G1 Therapeutics, Sierra Oncology, Bayer
Joseph Eder
Honoraria: Roche Molecular Diagnostics
Consulting or Advisory Role: Roche/Genentech
No other potential conflicts of interest were reported.
REFERENCES
- 1.Tubbs A, Nussenzweig A: Endogenous DNA damage as a source of genomic instability in cancer. Cell 168:644-656, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knijnenburg TA, Wang L, Zimmermann MT, et al. : Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell Rep 23:239-254.e6, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heeke AL, Pishvaian MJ, Lynce F, et al. : Prevalence of homologous recombination-related gene mutations across multiple cancer types. JCO Precis Oncol 10.1200/PO.17.00286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lord CJ, Ashworth A: BRCAness revisited. Nat Rev Cancer 16:110-120, 2016 [DOI] [PubMed] [Google Scholar]
- 5.Robson M, Im SA, Senkus E, et al. : Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med 377:523-533, 2017 [DOI] [PubMed] [Google Scholar]
- 6.Pujade-Lauraine E, Ledermann JA, Selle F, et al. : Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol 18:1274-1284, 2017 [DOI] [PubMed] [Google Scholar]
- 7.Riaz N, Blecua P, Lim RS, et al. : Pan-cancer analysis of bi-allelic alterations in homologous recombination DNA repair genes. Nat Commun 8:857, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bell D, Berchuck A, Birrer M, et al. : Integrated genomic analyses of ovarian carcinoma. Nature 474:609-615, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coleman RL, Oza AM, Lorusso D, et al. : Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390:1949-1961, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ledermann J, Harter P, Gourley C, et al. : Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol 15:852-861, 2014 [DOI] [PubMed] [Google Scholar]
- 11.Rafii S, Gourley C, Kumar R, et al. : Baseline clinical predictors of antitumor response to the PARP inhibitor olaparib in germline BRCA1/2 mutated patients with advanced ovarian cancer. Oncotarget 8:47154-47160, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mirza MR, Pignata S, Ledermann JA: Latest clinical evidence and further development of PARP inhibitors in ovarian cancer. Ann Oncol 29:1366-1376, 2018 [DOI] [PubMed] [Google Scholar]
- 13.Oza AM, Tinker AV, Oaknin A, et al. : Antitumor activity and safety of the PARP inhibitor rucaparib in patients with high-grade ovarian carcinoma and a germline or somatic BRCA1 or BRCA2 mutation: Integrated analysis of data from Study 10 and ARIEL2. Gynecol Oncol 147:267-275, 2017 [DOI] [PubMed] [Google Scholar]
- 14.Penson RT, Valencia RV, Cibula D, et al. : Olaparib versus nonplatinum chemotherapy in patients with platinum-sensitive relapsed ovarian cancer and a germline BRCA1/2 mutation (SOLO3): A randomized phase III trial. J Clin Oncol 38:1164-1174, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalez-Martin A, Pothuri B, Vergote I, et al. : Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med 381:2391-2402, 2019 [DOI] [PubMed] [Google Scholar]
- 16.Moore K, Colombo N, Scambia G, et al. : Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med 379:2495-2505, 2018 [DOI] [PubMed] [Google Scholar]
- 17.Buisson R, Niraj J, Rodrigue A, et al. : Coupling of homologous recombination and the checkpoint by ATR. Mol Cell 65:336-346, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sorensen CS, Hansen LT, Dziegielewski J, et al. : The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol 7:195-201, 2005 [DOI] [PubMed] [Google Scholar]
- 19.Kim H, George E, Ragland R, et al. : Targeting the ATR/CHK1 axis with PARP inhibition results in tumor regression in BRCA-mutant ovarian cancer models. Clin Cancer Res 23:3097-3108, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yazinski SA, Comaills V, Buisson R, et al. : ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev 31:318-332, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim H, Xu H, George E, et al. : Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat Commun 11:3726, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Min A, Im SA, Jang H, et al. : AZD6738, A novel oral inhibitor of ATR, induces synthetic lethality with ATM deficiency in gastric cancer cells. Mol Cancer Ther 16:566-577, 2017 [DOI] [PubMed] [Google Scholar]
- 23.Rafiei S, Fitzpatrick K, Liu D, et al. : ATM loss confers greater sensitivity to ATR inhibition than PARP inhibition in prostate cancer. Cancer Res 80:2094-2100, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Bono JS, Tan DSP, Caldwell R, et al. : First-in-human trial of the oral ataxia telangiectasia and Rad3-related (ATR) inhibitor BAY 1895344 in patients (pts) with advanced solid tumors. J Clin Oncol 37, 2019. (15 suppl; abstr 3007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yap TA, Tan DSP, Terbuch A, et al. : First-in-human trial of the oral ataxia telangiectasia and RAD3-related (ATR) inhibitor BAY 1895344 in patients with advanced solid tumors. Cancer Discov 11:80-91, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmitt A, Knittel G, Welcker D, et al. : ATM deficiency is associated with sensitivity to PARP1- and ATR inhibitors in lung adenocarcinoma. Cancer Res 77:3040-3056, 2017 [DOI] [PubMed] [Google Scholar]
- 27.Perkhofer L, Schmitt A, Romero Carrasco MC, et al. : ATM deficiency generating genomic instability sensitizes pancreatic ductal adenocarcinoma cells to therapy-induced DNA damage. Cancer Res 77:5576-5590, 2017 [DOI] [PubMed] [Google Scholar]
- 28.de Bono J, Mateo J, Fizazi K, et al. : Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med 382:2091-2102, 2020 [DOI] [PubMed] [Google Scholar]
- 29.Mateo J, Carreira S, Sandhu S, et al. : DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med 373:1697-1708, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cecchini M, Walther Z, Sklar JL, et al. : Introduction to the Yale Precision Medicine Tumor Board. Lancet Oncol 19:19-20, 2018 [DOI] [PubMed] [Google Scholar]
- 31.Sholl LM, Do K, Shivdasani P, et al. : Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight 1:e87062, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sulkowski PL, Sundaram RK, Oeck S, et al. : Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nat Genet 50:1086-1092, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eisenhauer EA, Therasse P, Bogaerts J, et al. : New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer 45:228-247, 2009 [DOI] [PubMed] [Google Scholar]
- 34.Simon R: Optimal two-stage designs for phase II clinical trials. Control Clin Trials 10:1-10, 1989 [DOI] [PubMed] [Google Scholar]
- 35.Jung SH, Lee T, Kim K, et al. : Admissible two-stage designs for phase II cancer clinical trials. Stat Med 23:561-569, 2004 [DOI] [PubMed] [Google Scholar]
- 36.Yap TA, O'Carrigan B, Penney MS, et al. : Phase I trial of first-in-class ATR inhibitor M6620 (VX-970) as monotherapy or in combination with carboplatin in patients with advanced solid tumors. J Clin Oncol 38:3195-3204, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.D'Andrea AD: Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair (Amst) 71:172-176, 2018 [DOI] [PubMed] [Google Scholar]
- 38.Jonsson P, Bandlamudi C, Cheng ML, et al. : Tumour lineage shapes BRCA-mediated phenotypes. Nature 571:576-579, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cleary JM, Aguirre AJ, Shapiro GI, et al. : Biomarker-guided development of DNA repair inhibitors. Mol Cell 78:1070-1085, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]