

ABSTRACT
Dengue is a mosquito-borne infectious disease that is highly endemic in tropical and subtropical countries. Symptomatic patients can rapidly progress to severe conditions of hemorrhage, plasma extravasation, and hypovolemic shock, which leads to death. The blood tests of patients with severe dengue typically reveal low levels of high-density lipoprotein (HDL), which is responsible for reverse cholesterol transport (RCT) and regulation of the lipid composition in peripheral tissues. It is well known that dengue virus (DENV) depends on membrane cholesterol rafts to infect and to replicate in mammalian cells. Here, we describe the interaction of DENV nonstructural protein 1 (NS1) with apolipoprotein A1 (ApoA1), which is the major protein component of HDL. NS1 is secreted by infected cells and can be found circulating in the serum of patients with the onset of symptoms. NS1 concentrations in plasma are related to dengue severity, which is attributed to immune evasion and an acute inflammatory response. Our data show that the DENV NS1 protein induces an increase of lipid rafts in noninfected cell membranes and enhances further DENV infection. We also show that ApoA1-mediated lipid raft depletion inhibits DENV attachment to the cell surface. In addition, ApoA1 is able to neutralize NS1-induced cell activation and to prevent NS1-mediated enhancement of DENV infection. Furthermore, we demonstrate that the ApoA1 mimetic peptide 4F is also capable of mediating lipid raft depletion to control DENV infection. Taken together, our results suggest the potential of RCT-based therapies for dengue treatment. These results should motivate studies to assess the importance of RCT in DENV infection in vivo.
IMPORTANCE DENV is one of the most relevant mosquito-transmitted viruses worldwide, infecting more than 390 million people every year and leading to more than 20 thousand deaths. Although a DENV vaccine has already been approved, its potential side effects have hampered its use in large-scale immunizations. Therefore, new treatment options are urgently needed to prevent disease worsening or to improve current clinical management of severe cases. In this study, we describe a new interaction of the NS1 protein, one of the major viral components, with a key component of HDL, ApoA1. This interaction seems to alter membrane susceptibility to virus infection and modulates the mechanisms triggered by DENV to evade the immune response. We also propose the use of a mimetic peptide named 4F, which was originally developed for atherosclerosis, as a potential therapy for relieving DENV symptoms.
KEYWORDS: apolipoprotein A1, dengue virus, immune evasion, nonstructural 1 protein, protein-protein interactions
INTRODUCTION
Dengue is the most important neglected tropical disease caused by an arbovirus. Nearly one-half of the world’s population lives in areas in which the disease is endemic. Each year, about 390 million infections are estimated to occur, leading to more than 20 thousand deaths and costs charged for billions of dollars worldwide (1). Dengue virus (DENV) belongs to the Flavivirus genus and has four different serotypes, which is a major challenge for vaccine production (2, 3). Symptomatic dengue is characterized by fever, aches, and rash, which can progress to severe dengue, showing signs of hemorrhage, plasma extravasation, and shock. There is no specific treatment for dengue, however, only oral or intravenous hydration (4, 5).
DENV nonstructural protein 1 (NS1) is the only nonstructural protein secreted from flavivirus-infected cells, and it can be found circulating in patients’ serum with the onset of symptoms (6, 7). Secreted NS1 has been related to immune evasion (8–12) and harmful inflammatory responses (13, 14), and elevated concentrations in patients’ serum have been implicated in dengue severity (15).
Severe cases of dengue are also associated with lower levels of high-density lipoprotein (HDL) (16, 17). HDL has anti-inflammatory properties and participates in innate immunity, particularly in Gram-negative lipopolysaccharide (LPS) binding and neutralization (18). Apolipoprotein A1 (ApoA1) is the major protein component of HDL, and it can also circulate in the serum in lipid-poor or lipid-free forms (19). Secreted ApoA1 interacts with membrane lipid transporters, such as those of the ATP-binding cassette (ABC) family and scavenger receptor class B type 1, to accumulate lipids and to form the mature HDL particle (20). HDL is responsible for reverse cholesterol transport (RCT) and the regulation of cholesterol levels in peripheral tissues (21). Intact cholesterol-rich domains on cell membranes are required for DENV infection, and it has been reported that chemical depletion of cholesterol inhibits virus entry and replication (22, 23).
Here, we describe a novel interaction between human ApoA1 and DENV2 NS1 protein and its role during DENV infection. We discovered that ApoA1 not only neutralizes the proinflammatory effects of NS1 but also promotes cholesterol depletion from the cell surface, thus inhibiting virus infection. Because ApoA1 seems to be downregulated in severe dengue patients, we propose the administration of the mimetic peptide 4F, which was originally developed for atherosclerosis treatment, as a potential therapy for relieving dengue symptoms.
RESULTS
NS1-treated RAW 264.7 cells accumulate lipid rafts on the cell membrane.
DENV NS1 has been described as one of the viral proteins responsible for immune activation, leading to a proinflammatory cytokine storm and endothelial damage (13, 24). During an acute inflammatory response, lipid rafts are likely to increase in size and number to allow proper docking of receptors and signaling molecules on the cell membrane (25, 26). To determine whether NS1-induced cell activation modulates lipid raft expression, RAW 264.7 cells were incubated with 50 μg/ml purified DENV2 NS1 protein. Accumulation of lipid rafts on the cell surface was assessed by the incorporation of fluorescently labeled cholera toxin B (CTB), and inflammatory activation was assessed by secretion of nitric oxide (NO) in the culture supernatant. The results demonstrated that enrichment of lipid rafts on the surface of NS1-treated cells (Fig. 1A) corresponded to increased concentrations of NO in the supernatant within 24 h after treatment (Fig. 1B). These results indicate that lipid raft accumulation is a consequence of macrophage inflammatory activation by NS1 protein. Lipid rafts can be regulated by lipid composition as well as protein-lipid interactions (27). To determine whether accumulation of lipid rafts on the surface of activated cells is a consequence of upregulation of cholesterol biosynthesis, RAW 264.7 cells were treated with purified NS1 protein and total cell cholesterol was quantified. Figure 1C shows that the level of total cholesterol was not altered by NS1-induced activation, indicating that lipid raft increase is not a consequence of de novo biosynthesis of cholesterol. These results suggest that cytoplasmic membrane lipid rafts are enriched with lipids redistributed from organelles, for example, during export of immune molecules such as the major histocompatibility complex class II (MHC-II). To address this, surface expression of MHC-II was quantified in RAW 264.7 cells activated with NS1 protein. The results indicated that MHC-II expression on the cytoplasmic membrane of NS1-activated RAW 264.7 cells correlated with lipid raft accumulation (Fig. 1D). Altogether, these results indicate that secreted NS1 protein activates macrophages and induces enrichment of lipid rafts on the cytoplasmic membrane.
FIG 1.
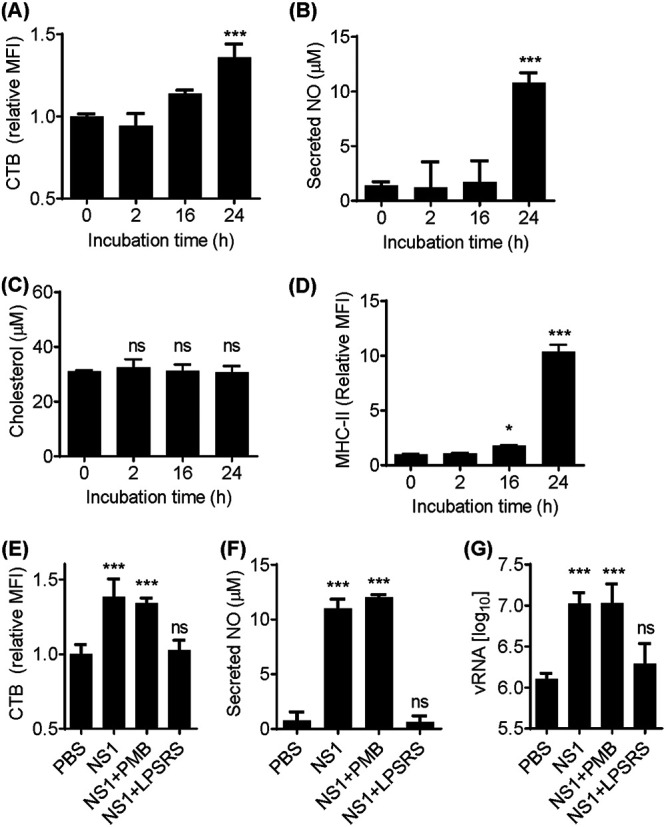
NS1-induced cell activation promotes lipid raft accumulation on the cell membrane and facilitates DENV attachment. (A) RAW 264.7 cells were incubated with 50 μg/ml NS1 for up to 24 h. Lipid rafts were quantified by flow cytometry using CTB-FITC, and the mean fluorescence intensity (MFI) was calculated relative to control cells incubated with PBS. (B) Immune activation was assessed by quantification of secreted NO in culture supernatants. (C) Total cholesterol was quantified in cell extracts from NS1-treated cells. (D) MHC-II expression on the cell surface was assessed by flow cytometry of NS1-treated cells. (E) Cells were treated with NS1 for 24 h in the presence of the LPS inhibitor PMB (100 μg/ml) or the LPS competitor LPS-RS (100 μg/ml). Lipid rafts were assessed by CTB -FITC incorporation. (F) Secreted NO was quantified in the supernatants by the Griess assay. (G) NS1-treated cells were challenged with DENV2 for 1 h at 4°C for viral attachment. Unbound virus was washed out with PBS, and total RNA was extracted. vRNA was quantified by qRT-PCR. Error bars represent the SDs of at least three biological replicates. Asterisks represent significant differences, compared to control. *, P < 0.1; ***, P < 0.001; ns, not significant.
NS1-induced lipid raft accumulation on cell membranes is mediated by TLR4.
DENV NS1 protein has been reported to induce macrophage activation via Toll-like receptor 4 (TLR4), a pattern recognition receptor known for recognizing bacterium-derived LPS (13, 24). To confirm that NS1-induced cell activation is mediated by TLR4, RAW 264.7 cells were incubated with purified NS1 protein for 24 h in the presence of the TLR4 antagonist LPS from Rhodobacter sphaeroides (LPS-RS). The results indicated that LPS-RS inhibited NS1-induced lipid raft accumulation (Fig. 1E), as well as inhibiting immune activation assessed by NO secretion (Fig. 1F). Additionally, to rule out the possibility of LPS contamination, purified NS1 protein was preincubated with the LPS inhibitor polymyxin B (PMB). PMB did not inhibit NS1-induced lipid raft accumulation on the cell surface (Fig. 1E) or inhibit NS1-induced TLR4-mediated immune activation (Fig. 1F). Thus, these results demonstrate that DENV2 NS1 induces macrophage activation via TLR4 and promotes accumulation of lipid rafts on the cell surface.
NS1-induced lipid raft accumulation facilitates DENV2 attachment to the cell membrane.
The main hypothesis explaining DENV-cell interaction is that the virus initially binds to nonspecific attachment factors on the cell surface to facilitate interactions with specific receptors (28). Several potential binding factors and receptors are associated with lipid rafts on the cell membrane (29), and lipid rafts are required for DENV entry in most cell lines (28). To determine whether NS1-induced lipid raft accumulation enhances DENV2 attachment to the cell membrane, RAW 264.7 cells were preincubated with NS1 for 24 h and then challenged with DENV2 for 1 h. Cell-bound virus particles were quantified by quantitative real-time PCR (qRT-PCR), and the results demonstrated that pretreatment with NS1 promoted a 1-log-unit increase in the number of DENV2 particles attached to the cell membrane, compared to control cells pretreated with phosphate-buffered saline (PBS) (Fig. 1G). Additionally, TLR4 inhibition by LPS-RS abolished the NS1 facilitation effect on DENV2 attachment to cell membrane. However, the presence of PMB did not inhibit this effect. Altogether, these experiments indicate that NS1-induced lipid raft accumulation facilitates DENV2 attachment to the cell membrane.
ApoA1 inhibits NS1-induced cell activation, lipid raft accumulation, and facilitation of DENV2 attachment to the cell membrane.
Unnecessary activation of the TLR4 inflammatory pathway can be prevented by several mechanisms in homeostasis. For example, serum ApoA1 binds and inhibits small amounts of LPS to avoid excessive inflammation (25). Thus, we hypothesized that ApoA1 could also inhibit NS1-induced macrophage activation and consequent lipid raft accumulation on the cell membrane, leading to neutralization of the NS1 facilitation effect on DENV2 attachment to the cell membrane. To determine whether ApoA1 inhibits NS1-induced activation, RAW 264.7 cells were incubated with NS1 for 24 h in the presence of different concentrations of purified human ApoA1. Immune activation was assessed by quantification of secreted NO in the cell culture supernatant. Results showed that ApoA1 inhibited NS1-induced cell activation in a concentration-dependent manner (Fig. 2A). To determine whether ApoA1 inhibits NS1-induced lipid raft accumulation, RAW 264.7 cells were incubated with NS1, with or without 100 μg/ml ApoA1, for 24 h. CTB quantification indicated that NS1-induced lipid raft accumulation on the cell membrane was inhibited in the presence of ApoA1 (Fig. 2B). Finally, to determine whether ApoA1 neutralizes the NS1 facilitation effect on DENV2 attachment to the cell membrane, RAW 264.7 cells were treated with NS1 in the presence of ApoA1 and then challenged with DENV2. Cell-bound virus quantification showed that the number of viral particles attached to NS1-treated cells was reduced in the presence of ApoA1 (Fig. 2C). Taken together, our results indicate that ApoA1 antagonizes NS1 capacity to activate macrophages and induce lipid raft accumulation, hence neutralizing the NS1 facilitation effect on DENV2 attachment to the cell membrane.
FIG 2.
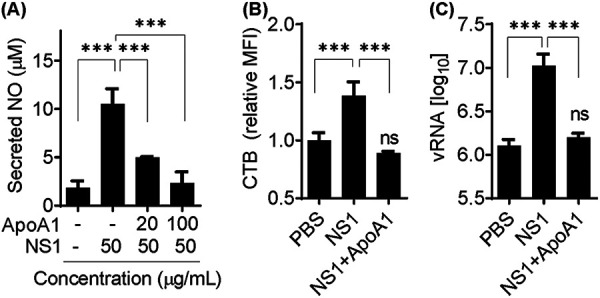
ApoA1 inhibits NS1-induced cell activation and the NS1 facilitation effect on DENV2 attachment to the cell membrane. (A) RAW 264.7 cells were incubated with NS1 for 24 h in the presence of different concentrations of ApoA1. Secreted NO was quantified in culture supernatants by the Griess assay. (B) Cells were fixed and labeled with CTB-FITC, and lipid rafts were analyzed by flow cytometry. The MFI of CTB was quantified relative to control cells incubated with PBS (pH 7.4). (C) Cells were preincubated for 24 h with 50 μg/ml NS1, in the presence or absence of 100 μg/ml ApoA1, and then challenged with DENV2 for 1 h at 4°C for virus attachment. Unbound virus was washed out with PBS, and total RNA was extracted. vRNA was quantified by qRT-PCR. Error bars represent the SDs of at least three biological replicates. Asterisks represent significant differences, compared to control. ***, P < 0.001; ns, not significant.
Human ApoA1 interacts with DENV NS1 protein.
To investigate whether secreted NS1 protein and human ApoA1 interact, recombinant hexameric NS1 protein was incubated with human serum from healthy donors and immunoprecipitated with anti-NS1 antibody covalently linked to NHS-HiTrap columns. Serum ApoA1 was coeluted with NS1 protein, suggesting an interaction between these proteins (Fig. 3A). Additionally, a direct binding assay was performed by enzyme-linked immunosorbent assay (ELISA) using the purified NS1 protein and the purified ApoA1 protein (Fig. 3B). In this experiment, two oligomeric forms of recombinant NS1 protein were used, i.e., glycosylated hexameric NS1 (expressed in Sf9 cells) and nonglycosylated dimeric NS1 (expressed in bacterial cells as described by Allonso et al. [30]). Our results showed that the two forms of NS1 bound ApoA1 with similar affinity, suggesting that the interaction does not depend on glycosylation. In order to assess the biophysical aspects of this interaction, another binding assay was performed by ELISA, in which increasing amounts of Triton X-100 or polyethylene glycol (PEG) were added to the solution. We observed that, in the presence of these reagents, the binding affinity decreased about 40% with 3% Triton X-100 and 50% with 25% PEG (Fig. 3C). This suggests that NS1 and ApoA1 interact through a nonpolar interface. It has been suggested that NS1 interacts with cell membranes by binding directly to lipids through the hydrophobic protrusion domain of the dimers (31). In order to evaluate whether ApoA1 inhibits NS1 interaction with the cell membrane, RAW 264.7 cells were incubated with NS1 in the presence of increasing concentrations of ApoA1, and cell-bound NS1 was quantified by flow cytometry. Data analysis showed that the amount of cell-bound NS1 decreased about 40% as the concentration of ApoA1 was increased to 100 μg/ml (Fig. 3D). Altogether, these results suggest that ApoA1 binds and inhibits NS1 interaction with cell membranes in a concentration-dependent manner.
FIG 3.

NS1 interacts with ApoA1 via nonpolar interfaces. (A) Purified NS1 was incubated with 10% human serum (HS) and immunoprecipitated with purified anti-NS1 antibody. The input (IN) and elution (E) fractions were analyzed by Western blotting with anti-NS1 and anti-ApoA1 monoclonal antibodies. (B) Microplates were coated with purified ApoA1 and incubated with increasing amounts of recombinant NS1 protein expressed in bacteria (NS1bac) or in Sf9 cells (NS1Sf9). Bound NS1 was detected using anti-NS1 polyclonal antibody and HRP-conjugated secondary antibody. OD490 values were normalized to negative-control (BSA) values. (C) ApoA1-coated microplates were incubated with 0.6 μM NS1bac in the presence of Triton X-100 or PEG. Bound NS1 was detected as described previously. (D) A monolayer of RAW 264.7 cells was fixed with 4% paraformaldehyde, blocked, and coincubated with purified ApoA1 and NS1Sf9. Bound NS1 was detected using anti-NS1 polyclonal antibody and HRP-conjugated secondary antibody. OD490 values were normalized to negative-control (BSA) values. Error bars indicate SDs of two independent experiments. *, P < 0.5; **, P < 0.05.
ApoA1-mediated lipid raft depletion inhibits DENV2 attachment to RAW 264.7 cells.
Lipid rafts are highly dynamic cholesterol-rich domains on the cell membrane that are required for docking specific transmembrane proteins such as immune molecules (27). During acute inflammation, cholesterol efflux from cell membranes is inhibited to avoid disruption of lipid rafts and to enhance inflammatory signaling (25). Several putative DENV receptors are localized to cholesterol-rich lipid rafts (32), and it is known that methyl-β-cyclodextrin (MβCD)-mediated cholesterol depletion inhibits DENV infection and replication (22, 23). Cholesterol efflux from the cell membrane to lipid-poor ApoA1 is mediated by ATP-binding cassette transporter A1 (ABCA1) (19). To investigate whether ApoA1 depletes lipid rafts from RAW 264.7 cell membranes, ABCA1 expression was induced by treatment with 0.3 mM 8-Br-cAMP (Fig. 4A), as described by Oram et al. (33). ABCA1-expressing cells were incubated with lipid-free ApoA1 for 2 h, and then lipid rafts were quantified by flow cytometry. Cells incubated with ApoA1 showed a 37% reduction in lipid rafts on the cell membrane, compared to cells incubated with PBS (Fig. 4B). As a control, RAW 264.7 cells were incubated with MβCD, and CTB quantification showed a 42% reduction in lipid rafts on the cell membrane, compared to cells incubated with PBS. Although our experiment did not assess the specific concentration of cholesterol on the cytoplasmic membranes, lipid raft depletion rates corroborated the gold standard quantification of [3H]cholesterol from MβCD-treated cells described by Mahammad and Parmryd (34). Furthermore, to determine whether ApoA1-mediated lipid raft depletion affects DENV attachment to the cell membrane, ABCA1-expressing RAW 264.7 cells were incubated with ApoA1 for 2 h and then challenged with DENV2 for 1 h. Cell-bound virus particles were quantified by qRT-PCR. Lipid raft-depleted cells demonstrated a 1-log-unit decrease in virus attachment, compared to control cells (Fig. 4C), confirming the importance of lipid rafts for DENV infection. Additionally, these results are in agreement with a previous report by Lee et al. (22), showing that cholesterol depletion inhibits flavivirus infection. Furthermore, we present evidence of the protective role of ApoA1-mediated lipid modulation during DENV infection.
FIG 4.
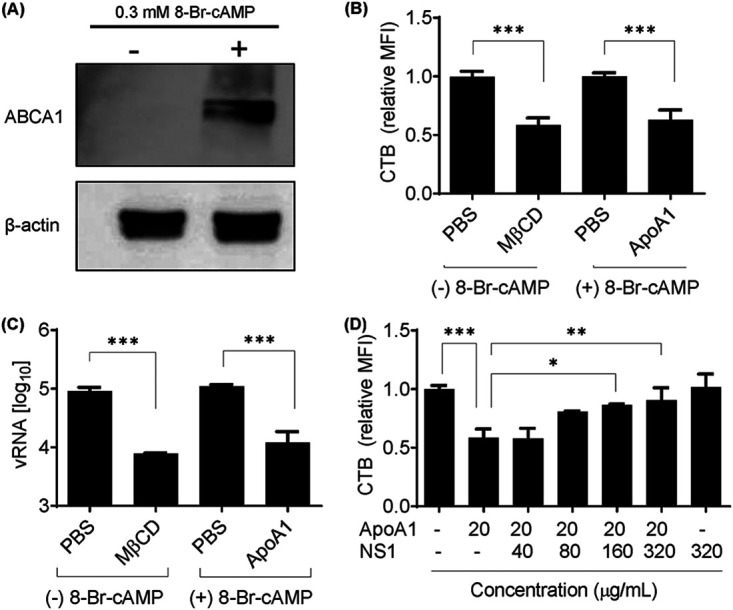
ApoA1-mediated lipid raft depletion from ABCA1-expressing cells inhibits DENV2 attachment to the cell membrane. (A) RAW 264.7 cells were treated with 0.3 mM 8-Br-cAMP for 16 h. Total cell lysate was analyzed by Western blotting with anti-ABCA1. (B) Lipid rafts were quantified by flow cytometry of cells labeled with CTB-FITC. Control cells were treated with PBS (pH 7.4) or depleted with 3 mM MβCD for 2 h. ABCA1-expressing cells were treated with 100 μg/ml ApoA1 for 2 h. MFI was calculated relative to control cells. (C) After induced lipid raft depletion, RAW 264.7 cells were challenged with DENV2 for 1 h at 4°C for viral attachment. Unbound virus was washed out with PBS, and total RNA was extracted. vRNA was quantified by qRT-PCR. (D) ABCA1-expressing cells were incubated with ApoA1 for 2 h in the presence of different concentrations of NS1. Lipid rafts were quantified by flow cytometry of CTB-FITC. Each bar represents the SD of at least three biological replicates. Asterisks represent significant differences, compared to control. *, P < 0.01; **, P < 0.01; ***, P < 0.001.
High concentrations of NS1 inhibit the ability of ApoA1 to deplete lipid rafts.
We hypothesized that the presence of NS1 could revert lipid raft modulation by ApoA1. To evaluate this hypothesis, ABCA1-expressing RAW 264.7 cells were incubated with 20 μg/ml ApoA1 for 2 h in the presence of increasing concentrations of NS1. CTB quantification showed that 320 μg/ml NS1 was able to completely inhibit ApoA1-mediated lipid raft depletion (Fig. 4D). It is important to note that the incubation of NS1 for only 2 h in the absence of ApoA1 was not able to induce lipid raft accumulation on the cell surface. Therefore, our results suggest that high concentrations of serum NS1 protein could inhibit ApoA1-mediated lipid raft depletion from the cell membrane.
ApoA1 concentrations are decreased in DENV-infected patients’ serum.
Modulation of lipid composition in the cell membrane is downregulated during acute inflammatory responses, such as virus infection. DENV-infected patients have lower levels of HDL and total serum cholesterol, compared to noninfected patients (16, 17). However, the actual concentration of ApoA1 in the serum of DENV-infected patients has not been described in the literature. To compare the serum concentrations of ApoA1 in dengue patients and non-dengue patients, we quantified ApoA1 in serum samples from patients from the University Hospital Clementino Chagas Filho and compared them with samples from healthy donors (Fig. 5A). Our results demonstrated that the average serum concentration of ApoA1 in DENV-infected patients was 0.10 ± 0.05 mg/ml, compared to 0.80 ± 0.20 mg/ml in non-dengue patients and 1.0 ± 0.20 mg/ml in healthy donors. Moreover, a significant difference between samples from dengue patients classified as having dengue fever (0.12 mg/ml ± 0.09 mg/m) versus severe dengue (0.08 mg/ml ± 0.01 mg/ml) was not observed (Fig. 5B). One of the major sources of ApoA1 is the liver, which is also one of the organs most injured during DENV infection (4). To assess whether hepatocytes infected with DENV2 downregulate expression of ApoA1 in vitro, mock or DENV2-infected HepG2 cells were analyzed by Western blotting. Forty-eight hours postinfection, the amount of intracellular ApoA1 in DENV2-infected cells was about 50% lower than that in mock-infected cells (Fig. 5C). Thus, our results suggest that ApoA1 expression in the liver is downregulated during DENV infection, and this could be a cause of low HDL and low cholesterol levels in the serum of dengue patients.
FIG 5.
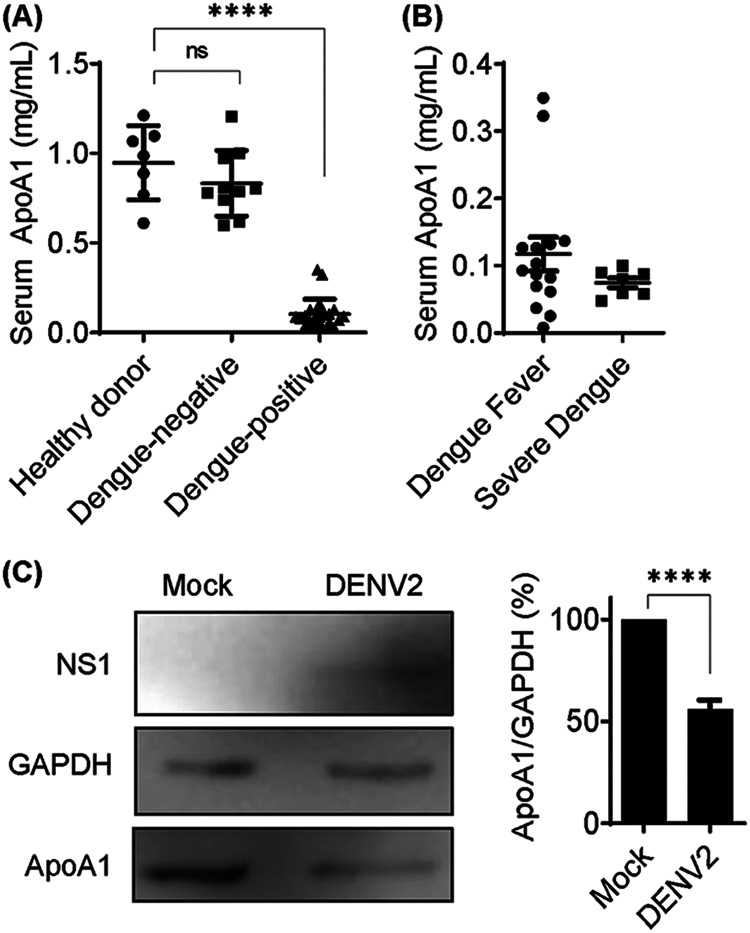
Serum ApoA1 concentrations are decreased in DENV-infected patients. (A) Total ApoA1 was quantified in human serum using the human ApoA1 ELISA kit (Thermo Fisher Scientific). Of samples from 33 patients, 23 were DENV positive and 10 were DENV negative. Samples from 7 healthy donors were also quantified as control samples. (B) Among DENV-positive patients, 16 were diagnosed with dengue fever and 7 were diagnosed with severe dengue. Bars represent mean and SDs. (C) HepG2 cells were mock infected or infected with DENV2 (multiplicity of infection of 1) for 48 h, and total cell lysates were analyzed by Western blotting. The ApoA1 band was quantified and expressed relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Bars represent means and SDs from three experiments. Asterisks represent significant differences, compared to mock-infected cells. ****, P < 0.005; ns, not significant.
ApoA1 mimetic peptide 4F inhibits proinflammatory and proviral effects of NS1.
Considering ApoA1 a protective factor in the case of DENV infection, it is plausible that the inflammatory response and DENV propagation could be controlled by increasing availability of ApoA1 in serum. Infusion of exogenous HDL, ApoA1, and mimetic peptides has been studied as a potential therapeutic approach for atherosclerosis (35). One of the most successful ApoA1 mimetics is peptide 4F (36). To evaluate the potential of the mimetic peptide in controlling the NS1 facilitation effect, RAW 264.7 cells were incubated with NS1 protein in the presence of peptide 4F for 24 h. The results demonstrated that peptide 4F inhibited NS1-induced immune activation in a concentration-dependent manner (Fig. 6A). Next, to determine whether peptide 4F depleted lipid rafts from the cell membrane, ABCA1-expressing RAW 264.7 cells were incubated with 50 μg/ml peptide 4F for 2 h. The results showed that peptide 4F induced 60% depletion of the lipid raft content in the cell membrane (Fig. 6B). Additionally, when these lipid raft-depleted cells were challenged with DENV2, we observed a 2.6-log-unit reduction in DENV2 attachment to the cell membrane (Fig. 6C). Altogether, these results indicate that peptide 4F is even more efficient than ApoA1 in controlling proinflammatory and proviral effects of NS1, as well as inhibiting DENV attachment to cells in vitro.
FIG 6.
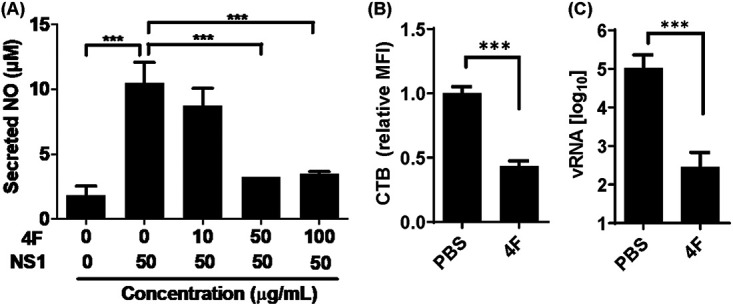
Peptide 4F-mediated lipid raft depletion from ABCA1-expressing cells inhibits DENV attachment to the cell membrane. (A) RAW 264.7 cells were incubated with NS1 for 24 h, in the presence of different concentrations of peptide 4F. Secreted NO was quantified from culture supernatants by the Griess assay. (B) ABCA1-expressing cells were incubated with 50 μg/ml peptide 4F for 2 h. Cells were fixed and labeled with CTB-FITC, and lipid rafts were analyzed by flow cytometry. The MFI of CTB was quantified relative to control cells incubated with PBS (pH 7.4). (C) ABCA1-expressing cells were incubated with peptide 4F for 2 h and then challenged with DENV2 for 1 h at 4°C for viral attachment. Unbound virus was washed out with PBS, and total RNA was extracted. vRNA was quantified by qRT-PCR. Error bars represent the SDs of at least three independent experiments. Asterisks represent significant differences, compared to control. ***, P < 0.001.
DISCUSSION
ApoA1 is the major protein component of HDL, and it is especially associated with RCT from peripheral tissues toward the liver (19). Deficient RCT may lead to cholesterol deposition on the endothelial wall and atherosclerotic development (37). Lipid rafts are highly dynamic cholesterol- and sphingolipid-rich domains on cell membranes that are stabilized by protein-protein or protein-lipid interactions (38). It is known that lipid rafts are important for DENV infection. Because clathrin-mediated endocytosis is dependent on these domains, it is likely that lipid raft depletion would inhibit DENV internalization (22). Moreover, several putative DENV receptors are residents on lipid raft domains (28, 32).
Cell membrane lipid rafts are regulated during acute inflammatory responses due to RCT inhibition in order to improve docking of inflammatory molecules on the cell membrane (25). The DENV NS1 protein is a proinflammatory antigen capable of inducing cell activation via TLR4, resulting in secretion of proinflammatory cytokines such as tumor necrosis factor alpha (TNF-α) and interleukin 6 (IL-6) (13, 15, 39). NS1 protein is the only flavivirus nonstructural protein secreted by infected cells. Therefore, it is likely that NS1 secretion from DENV-infected cells is particularly important to the pathogenesis of dengue. Our group previously described that secreted NS1 plays an important role in immune evasion by modulating the complement system (8, 9). Here, we demonstrate that soluble NS1 protein facilitates the attachment of DENV2 particles to noninfected macrophages by inducing accumulation of lipid rafts via TLR4 activation. Regulation of the lipid composition of the cell surface is crucial for innate immunity (40). Lipid raft stabilization and aggregation are associated with increased inflammatory activation, whereas depletion inhibits proper signaling transmission (41). Inflammatory activation via TLR4 inhibits cholesterol efflux and promotes redistribution of cellular lipids (42–44). Organelle-derived lipids are relocated to the cell membrane due to export of vesicles or immune molecules. MHC-II, for example, is a transmembrane complex localized to lipid rafts in antigen-processing compartments and is translocated to the cell membrane in response to inflammatory activation (45). Consequently, accumulation of lipid rafts on the cell membrane contributes to aggregation of signaling molecules that can potentially serve as receptors for DENV.
Additionally, we demonstrated that human ApoA1 could neutralize DENV NS1 proinflammatory effects. We showed that the addition of ApoA1 to RAW 264.7 cell cultures inhibited NS1-induced cell activation and lipid raft accumulation on the cytoplasmic membrane, as well as inhibiting the NS1-mediated facilitation effect on DENV2 attachment to the cell membrane. We also showed that DENV2 NS1 directly interacted and inhibited ApoA1 in a concentration-dependent manner. Serum concentrations of ApoA1 were significantly reduced in DENV-infected patients, compared to healthy individuals. This result suggests that inflammatory liver damage caused by DENV infection results in reduced ApoA1 availability. HDL and total cholesterol concentrations were significantly reduced in the serum of dengue patients, compared to that of non-dengue patients, which implies increased cholesterol accumulation in peripheral tissues (16, 17). It has been described that MβCD-induced cholesterol depletion inhibits both entry and early steps of flavivirus replication (22, 23). In our model, MβCD was able to induce lipid raft depletion, in agreement with cholesterol quantification previously reported by other groups (34). Accordingly, treatment of ABCA1-expressing cells with ApoA1 or peptide 4F also induced efficient lipid raft depletion. Although ABCA1 expression needed to be induced in immortalized cell lines, ABCA1 is constitutively expressed in primary cells, as well as in circulating macrophages and monocytes. Unpublished data from our group indicated that both ApoA1 and peptide 4F were able to deplete lipid rafts from human peripheral blood mononuclear cells without the need for 8-Br-cAMP preincubation (D. R. Coelho, P. H. Carneiro, and R. Mohana-Borges, unpublished data).
Lipid raft depletion mediated by MβCD inhibited DENV2 attachment to RAW 264.7 cell membranes, compared to controls. These results corroborate the data of Lee and colleagues that showed 1-log-unit inhibition of DENV infection in MβCD-treated N18 cells (22). Accordingly, ApoA1- and peptide 4F-mediated lipid raft depletion from ABCA1-expressing cells also inhibited DENV2 attachment to the cell membrane. We think that lipid raft depletion by ApoA1 or peptide 4F might impair proper docking of adhesion factors needed for DENV binding and entry and it might also disturb membrane remodeling during the early stages of DENV infection.
Although the pathogenesis of severe dengue is not well understood, systemic shock is likely a consequence of the acute inflammatory response, in which strong macrophage activation and a cytokine storm promote plasma leakage, hemorrhage, and low blood pressure. NS1 protein itself could be directly responsible for the acute inflammatory response and increased endothelial permeabilization, as described by Beatty et al. (14). In this scenario, RCT-oriented treatments, such as infusion of ApoA1 or peptide 4F, might be able to restore normal levels of HDL and to diminish acute inflammatory activation. The ApoA1 mimetic peptide 4F has the advantage of being stable and available for oral administration in substitution for intravenous injection of ApoA1 or reconstituted HDL (46). Moreover, peptide 4F has been shown very efficient in promoting cholesterol efflux in vitro (47), and it has demonstrated anti-inflammatory properties in vivo (48). Here, we showed that peptide 4F is also efficient in depleting lipid rafts from cultured macrophages and that it inhibits DENV attachment to cells.
Altogether, our studies demonstrate that DENV NS1-induced macrophage activation via TLR4 promotes accumulation of lipid rafts on the cell membrane and facilitates DENV attachment to noninfected cells. Moreover, we propose that ApoA1 has an important role in innate immunity against DENV, both in neutralizing NS1 proinflammatory effects and in maintaining low levels of lipid rafts in peripheral tissues. We propose that the reduced concentrations of ApoA1 in patients’ serum represent a mechanism of immune evasion that could be targeted by administering exogenous ApoA1 to patients with severe dengue. We also bring attention to the mimetic peptide 4F as a potential therapeutic agent to prevent severe dengue outcomes, due to its ability to deplete lipid rafts and to inhibit DENV attachment to the cell surface.
MATERIALS AND METHODS
Cell culture and virus.
RAW 264.7 cells (ATCC) were cultured in Dulbecco’s minimal essential medium (DMEM) from Invitrogen supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin from Gibco and were incubated at 37°C in a humidified chamber with 5% CO2. Sf9 cells were cultivated in Sf-900 III serum-free medium (SFM) (Invitrogen) supplemented with 10% FBS, penicillin, and streptomycin and were incubated in suspension at 28°C with gentle shaking. DENV2 strain 16681 was propagated in C6/36 cells cultured in minimal essential medium (MEM) (Invitrogen) supplemented with 10% heat-inactivated FBS, penicillin, and streptomycin and was incubated at 28°C.
NS1 protein purification.
Recombinant DENV2 NS1 protein was expressed in baculovirus-transduced Sf9 cells and purified as described by Akey et al. (31). Briefly, transduced cells were cultured in 1 liter of complete medium for 3 days. After centrifugation, the cell pellet was suspended in lysis buffer (50 mM Tris-HCl [pH 8.5], 50 mM ammonium sulfate, 10% glycerol, 0.5% Triton X-100) supplemented with 1× Halt protease inhibitor cocktail. The suspension was sonicated three times at 50% for 3 s in an ice bath, at 5-min intervals. After centrifugation at 15,000 × g, soluble NS1 protein was purified by ion-metal affinity chromatography (IMAC) using HisTrap HP columns (GE Healthcare), eluted with addition of 250 mM imidazole. The first IMAC step was followed by size exclusion chromatography with Superdex 200 (GE Healthcare) in lysis buffer. The fractions containing purified NS1 protein were processed in a second step of IMAC, in which the immobilized protein was washed with at least 500 ml Tris-buffered saline-glycerol (TBSG) buffer (50 mM Tris-HCl [pH 8.5], 150 mM NaCl, 10% glycerol) to remove traces of detergent. Finally, NS1 protein was eluted in 3 column volumes of TBSG plus 250 mM imidazole and dialyzed against TBSG buffer.
ApoA1 purification.
Recombinant ApoA1 was expressed in Escherichia coli strain BL21(DE3)pLys transformed with plasmid pET21(b)_apoa1, which was kindly provided by Michael Oda (Children’s Hospital Oakland Research Institute, Oakland, CA, USA), and was purified as described by Ryan et al. (49). Briefly, inclusion bodies were solubilized in 6 M guanidine hydrochloride, and ApoA1 was purified by IMAC in a HisTrap column. The purified ApoA1 was refolded by dialysis against Tris-buffered saline (TBS) (50 mM Tris-HCl [pH 8.0], 150 mM NaCl).
Flow cytometry.
RAW 264.7 cells were detached with a cell scraper, fixed in 4% paraformaldehyde for 45 min at 4°C, and stained with CTB conjugated to fluorescein isothiocyanate (FITC) (Sigma-Aldrich) or primary antibodies anti-MHC-II (BioLegends), anti-NS1 (30), or anti-ApoA1 (Abcam), with secondary antibodies conjugated to PECy7, Alexa Fluor 488, or Alexa Fluor 568 (Thermo Fischer Scientific), at 4°C for 1 h in the dark. At least 10,000 events/sample were analyzed by flow cytometry in a FACSCalibur system (BD).
Total cholesterol quantification.
One million RAW 264.7 cells/well were plated in 6-well plates and incubated with 50 μg/ml NS1 protein for up to 24 h. Total cholesterol was quantified with the Amplex Red cholesterol assay kit (Thermo Fisher Scientific), according to the manufacturer’s instructions.
Viral RNA quantification.
RAW 264.7 cells were challenged with DENV2 for 1 h at 4°C and then washed once with cold PBS (pH 7.4). Total RNA was purified from total cell lysate with an RNeasy kit (Qiagen), according to the instructions. Viral RNA (vRNA) was quantified from 1 μg total RNA by qRT-PCR with the SuperScript One-Step RT-PCR System kit and Platinum Taq DNA polymerase (Thermo Fischer Scientific), using the following primers: forward, TTG CGG TGT CAA TGG CTA ACA; reverse, CCA ATG CGT TCA ATC GGC T.
Coimmunoprecipitation.
Homemade mouse polyclonal antibody anti-NS1 (30) was conjugated to coimmunoprecipitation (CoIP) columns (Pierce) according to the manufacturer’s instructions. A control column was prepared using control IgG antibodies (Abcam). Recombinant NS1 protein was mixed with 10% human serum in PBS (pH 7.4) and incubated with the CoIP columns at 4°C overnight, with shaking. The CoIP columns were washed three times with PBS, and proteins were eluted with elution buffer (Pierce) and analyzed by Western blotting.
ELISA.
Recombinant ApoA1 was adsorbed in 96-well microplates (Nunc), blocked with 1% bovine serum albumin (BSA), and incubated with different concentrations of recombinant NS1 protein in PBS (pH 7.4) at 37°C for 2 h. After three washes with PBS, wells were incubated with anti-NS1 primary antibody and secondary antibody conjugated with horseradish peroxidase (HRP) (Thermo Fischer Scientific). Reactions were revealed with o-phenylenediamine dihydrochloride (OPD) and stopped with sulfuric acid. Microplates were analyzed with a SpectraMax M5 microplate reader.
NO quantification.
NO was quantified in cell supernatants with the Griess reagent system (Promega) according to the manufacturer’s protocol.
Human ApoA1 quantification in patients’ plasma.
Samples of human plasma from healthy donors, dengue-positive patients, and dengue-negative patients, from the Clementino Fraga Filho University Hospital, were quantified using the human ApoA1 ELISA kit (Thermo Fischer Scientific), according to the manufacturer’s instructions. Briefly, samples were diluted 1:1,000,000 in assay diluent and incubated in the ELISA microplate overnight at 4°C. Wells were washed with wash buffer and incubated with biotinylated primary antibody, followed by streptavidin conjugated to HRP. Reactions were revealed with 3,3′,5,5′-tetramethylbenzidine (TMB) substrate, stopped with stop solution, and analyzed by a SpectraMax M5 plate reader as the optical density (OD) at 450 nm and 550 nm.
Mimetic peptide 4F.
ApoA1 mimetic peptide 4F (acetyl-DWFKAFYDKVAEKFKEAF-NH2) was synthetized by GenOne (Rio de Janeiro, Brazil).
Statistical analysis.
Results shown in each figure were derived from two or three independent experiments with comparable findings; the data presented are means ± standard deviations (SDs), with the indicated P values of <0.001 being considered significant. Two-way comparisons were performed by two-tailed analysis of variance and an unpaired Student's t test. All analyses were performed using GraphPad Prism software version 4.0.
ACKNOWLEDGMENTS
Studies were supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (Brazil), the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) (Brazil), and the Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) (Brazil).
D.R.C. wrote the main manuscript text. All authors reviewed the manuscript.
We declare no financial competing interests and no nonfinancial competing interests.
Contributor Information
Ronaldo Mohana-Borges, Email: mohana@biof.ufrj.br.
Susana Lόpez, Instituto de Biotecnologia/UNAM.
REFERENCES
- 1.Guzman MG, Harris E. 2015. Dengue. Lancet 385:453–465. doi: 10.1016/S0140-6736(14)60572-9. [DOI] [PubMed] [Google Scholar]
- 2.Flipse J, Smit JM. 2015. The complexity of a dengue vaccine: a review of the human antibody response. PLoS Negl Trop Dis 9:e0003749. doi: 10.1371/journal.pntd.0003749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swaminathan S, Khanna N. 2019. Dengue vaccine development: global and Indian scenarios. Int J Infect Dis 84:S80–S86. doi: 10.1016/j.ijid.2019.01.029. [DOI] [PubMed] [Google Scholar]
- 4.World Health Organization. 2009. Dengue guidelines for diagnosis, treatment, prevention and control. World Health Organization, Geneva, Switzerland. [PubMed] [Google Scholar]
- 5.Chaloemwong J, Tantiworawit A, Rattanathammethee T, Hantrakool S, Chai-Adisaksopha C, Rattarittamrong E, Norasetthada L. 2018. Useful clinical features and hematological parameters for the diagnosis of dengue infection in patients with acute febrile illness: a retrospective study. BMC Hematol 18:20. doi: 10.1186/s12878-018-0116-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thiemmeca S, Tamdet C, Punyadee N, Prommool T, Songjaeng A, Noisakran S, Puttikhunt C, Atkinson JP, Diamond MS, Ponlawat A, Avirutnan P. 2016. Secreted NS1 protects dengue virus from mannose-binding lectin–mediated neutralization. J Immunol 197:4053–4065. doi: 10.4049/jimmunol.1600323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muller DA, Young PR. 2013. The flavivirus NS1 protein: molecular and structural biology, immunology, role in pathogenesis and application as a diagnostic biomarker. Antiviral Res 98:192–208. doi: 10.1016/j.antiviral.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 8.Silva EM, Conde JN, Allonso D, Nogueira ML, Mohana-Borges R. 2013. Mapping the interactions of dengue virus NS1 protein with human liver proteins using a yeast two-hybrid system: identification of C1q as an interacting partner. PLoS One 8:e57514. doi: 10.1371/journal.pone.0057514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conde JN, da Silva EM, Allonso D, Coelho DR, da Andrade IS, de Medeiros LN, Menezes JL, Barbosa AS, Mohana-Borges R. 2016. Inhibition of the membrane attack complex by dengue virus NS1 through interaction with vitronectin and terminal complement proteins. J Virol 90:9570–9581. doi: 10.1128/JVI.00912-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Avirutnan P, Zhang L, Punyadee N, Manuyakorn A, Puttikhunt C, Kasinrerk W, Malasit P, Atkinson JP, Diamond MS. 2007. Secreted NS1 of dengue virus attaches to the surface of cells via interactions with heparan sulfate and chondroitin sulfate E. PLoS Pathog 3:e183. doi: 10.1371/journal.ppat.0030183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Avirutnan P, Fuchs A, Hauhart RE, Somnuke P, Youn S, Diamond MS, Atkinson JP. 2010. Antagonism of the complement component C4 by flavivirus nonstructural protein NS1. J Exp Med 207:793–806. doi: 10.1084/jem.20092545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Avirutnan P, Punyadee N, Noisakran S, Komoltri C, Thiemmeca S, Auethavornanan K, Jairungsri A, Kanlaya R, Tangthawornchaikul N, Puttikhunt C, Pattanakitsakul S-N, Yenchitsomanus P-T, Mongkolsapaya J, Kasinrerk W, Sittisombut N, Husmann M, Blettner M, Vasanawathana S, Bhakdi S, Malasit P. 2006. Vascular leakage in severe dengue virus infections: a potential role for the nonstructural viral protein NS1 and complement. J Infect Dis 193:1078–1088. doi: 10.1086/500949. [DOI] [PubMed] [Google Scholar]
- 13.Modhiran N, Watterson D, Muller DA, Panetta AK, Sester DP, Liu L, Hume DA, Stacey KJ, Young PR. 2015. Dengue virus NS1 protein activates cells via Toll-like receptor 4 and disrupts endothelial cell monolayer integrity. Sci Transl Med 7:304ra142. doi: 10.1126/scitranslmed.aaa3863. [DOI] [PubMed] [Google Scholar]
- 14.Beatty PR, Puerta-Guardo H, Killingbeck SS, Glasner DR, Hopkins K, Harris E. 2015. Dengue virus NS1 triggers endothelial permeability and vascular leak that is prevented by NS1 vaccination. Sci Transl Med 7:304ra141. doi: 10.1126/scitranslmed.aaa3787. [DOI] [PubMed] [Google Scholar]
- 15.Rastogi M, Sharma N, Singh SK. 2016. Flavivirus NS1: a multifaceted enigmatic viral protein. Virol J 13:131. doi: 10.1186/s12985-016-0590-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Gorp ECM, Suharti C, Mairuhu ATA, Dolmans WMV, van Der Ven J, Demacker PNM, van der Meer JWM. 2002. Changes in the plasma lipid profile as a potential predictor of clinical outcome in dengue hemorrhagic fever. Clin Infect Dis 34:1150–1153. doi: 10.1086/339539. [DOI] [PubMed] [Google Scholar]
- 17.Biswas HH, Gordon A, Nuñez A, Perez MA, Balmaseda A, Harris E. 2015. Lower low-density lipoprotein cholesterol levels are associated with severe dengue outcome. PLoS Negl Trop Dis 9:e0003904. doi: 10.1371/journal.pntd.0003904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pirillo A, Catapano AL, Norata GD. 2015. HDL in infectious diseases and sepsis. Handb Exp Pharmacol 224:483–508. doi: 10.1007/978-3-319-09665-0_15. [DOI] [PubMed] [Google Scholar]
- 19.Zhou L, Li C, Gao L, Wang A. 2015. High-density lipoprotein synthesis and metabolism. Mol Med Rep 12:4015–4021. doi: 10.3892/mmr.2015.3930. [DOI] [PubMed] [Google Scholar]
- 20.Zannis VI, Chroni A, Krieger M. 2006. Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J Mol Med (Berl) 84:276–294. doi: 10.1007/s00109-005-0030-4. [DOI] [PubMed] [Google Scholar]
- 21.Feingold KR. 2000. Introduction to lipids and lipoproteins. In Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, Dungan K, Grossman A, Hershman JM, Hofland J, Kalra S, Kaltsas G, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrère B, McGee EA, McLachlan R, Morley JE, New M, Purnell J, Sahay R, Singer S, Stratakis CA, Trence DI, Wilson DP (ed), Endotext. MDText.com, Inc., South Dartmouth, MA. [Google Scholar]
- 22.Lee Y-R, Lei H-Y, Liu M-T, Wang J-R, Chen S-H, Jiang-Shieh Y-F, Lin Y-S, Yeh T-M, Liu C-C, Liu H-S. 2008. Autophagic machinery activated by dengue virus enhances virus replication. Virology 374:240–248. doi: 10.1016/j.virol.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soto-Acosta R, Mosso C, Cervantes-Salazar M, Puerta-Guardo H, Medina F, Favari L, Ludert JE, del Angel RM. 2013. The increase in cholesterol levels at early stages after dengue virus infection correlates with an augment in LDL particle uptake and HMG-CoA reductase activity. Virology 442:132–147. doi: 10.1016/j.virol.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 24.Diwaker D, Mishra KP, Ganju L, Singh SB. 2015. Protein disulfide isomerase mediates dengue virus entry in association with lipid rafts. Viral Immunol 28:153–160. doi: 10.1089/vim.2014.0095. [DOI] [PubMed] [Google Scholar]
- 25.Tall AR, Yvan-Charvet L. 2015. Cholesterol, inflammation and innate immunity. Nat Rev Immunol 15:104–116. doi: 10.1038/nri3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bukrinsky MI, Mukhamedova N, Sviridov D. 2020. Lipid rafts and pathogens: the art of deception and exploitation. J Lipid Res 61:601–610. doi: 10.1194/jlr.TR119000391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sezgin E, Levental I, Mayor S, Eggeling C. 2017. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat Rev Mol Cell Biol 18:361–374. doi: 10.1038/nrm.2017.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cruz-Oliveira C, Freire JM, Conceição TM, Higa LM, Castanho MARB, Da Poian AT. 2015. Receptors and routes of dengue virus entry into the host cells. FEMS Microbiol Rev 39:155–170. doi: 10.1093/femsre/fuu004. [DOI] [PubMed] [Google Scholar]
- 29.Fang L, Miller YI. 2019. Regulation of lipid rafts, angiogenesis and inflammation by AIBP. Curr Opin Lipidol 30:218–223. doi: 10.1097/MOL.0000000000000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Allonso D, da Silva Rosa M, Coelho DR, da Costa SM, Nogueira RMR, Bozza FA, Dos Santos FB, de Barcelos Alves AM, Mohana-Borges R. 2011. Polyclonal antibodies against properly folded dengue virus NS1 protein expressed in E. coli enable sensitive and early dengue diagnosis. J Virol Methods 175:109–116. doi: 10.1016/j.jviromet.2011.04.029. [DOI] [PubMed] [Google Scholar]
- 31.Akey DL, Brown WC, Dutta S, Konwerski J, Jose J, Jurkiw TJ, DelProposto J, Ogata CM, Skiniotis G, Kuhn RJ, Smith JL. 2014. Flavivirus NS1 structures reveal surfaces for associations with membranes and the immune system. Science 343:881–885. doi: 10.1126/science.1247749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smit J, Moesker B, Rodenhuis-Zybert I, Wilschut J. 2011. Flavivirus Cell Entry and Membrane Fusion. Viruses 3:160–171. doi: 10.3390/v3020160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oram JF, Lawn RM, Garvin MR, Wade DP. 2000. ABCA1 is the cAMP-inducible apolipoprotein receptor that mediates cholesterol secretion from macrophages. J Biol Chem 275:34508–34511. doi: 10.1074/jbc.M006738200. [DOI] [PubMed] [Google Scholar]
- 34.Mahammad S, Parmryd I. 2015. Cholesterol depletion using methyl-β-cyclodextrin. Methods Mol Biol 1232:91–102. doi: 10.1007/978-1-4939-1752-5_8. [DOI] [PubMed] [Google Scholar]
- 35.Leman LJ. 2015. The potential of apolipoprotein mimetic peptides in the treatment of atherosclerosis. Clin Lipidol 10:215–217. doi: 10.2217/clp.15.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stoekenbroek RM, Stroes ES, Hovingh GK. 2015. ApoA-I mimetics. Handb Exp Pharmacol 224:631–648. doi: 10.1007/978-3-319-09665-0_21. [DOI] [PubMed] [Google Scholar]
- 37.Savolainen J, Kautiainen H, Niskanen L, Mäntyselkä P. 2015. Decreasing cholesterol levels in the community: lifestyle change with statin? BMC Fam Pract 16:29. doi: 10.1186/s12875-015-0240-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pike LJ. 2006. Rafts defined: a report on the Keystone Symposium on Lipid Rafts and Cell Function. J Lipid Res 47:1597–1598. doi: 10.1194/jlr.E600002-JLR200. [DOI] [PubMed] [Google Scholar]
- 39.Modhiran N, Watterson D, Blumenthal A, Baxter AG, Young PR, Stacey KJ. 2017. Dengue virus NS1 protein activates immune cells via TLR4 but not TLR2 or TLR6. Immunol Cell Biol 95:491–495. doi: 10.1038/icb.2017.5. [DOI] [PubMed] [Google Scholar]
- 40.Varshney P, Yadav V, Saini N. 2016. Lipid rafts in immune signalling: current progress and future perspective. Immunology 149:13–24. doi: 10.1111/imm.12617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gay NJ, Symmons MF, Gangloff M, Bryant CE. 2014. Assembly and localization of Toll-like receptor signalling complexes. Nat Rev Immunol 14:546–558. doi: 10.1038/nri3713. [DOI] [PubMed] [Google Scholar]
- 42.Dennis EA, Deems RA, Harkewicz R, Quehenberger O, Brown HA, Milne SB, Myers DS, Glass CK, Hardiman G, Reichart D, Merrill AH, Sullards MC, Wang E, Murphy RC, Raetz CRH, Garrett TA, Guan Z, Ryan AC, Russell DW, McDonald JG, Thompson BM, Shaw WA, Sud M, Zhao Y, Gupta S, Maurya MR, Fahy E, Subramaniam S. 2010. A mouse macrophage lipidome. J Biol Chem 285:39976–39985. doi: 10.1074/jbc.M110.182915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carroll RG, Zasłona Z, Galván-Peña S, Koppe EL, Sévin DC, Angiari S, Triantafilou M, Triantafilou K, Modis LK, O'Neill LA. 2018. An unexpected link between fatty acid synthase and cholesterol synthesis in proinflammatory macrophage activation. J Biol Chem 293:5509–5521. doi: 10.1074/jbc.RA118.001921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li LC, Varghese Z, Moorhead JF, Lee CT, Chen JB, Ruan XZ. 2013. Cross-talk between TLR4-MyD88-NF-κB and SCAP-SREBP2 pathways mediates macrophage foam cell formation. Am J Physiol Heart Circ Physiol 304:H874–H884. doi: 10.1152/ajpheart.00096.2012. [DOI] [PubMed] [Google Scholar]
- 45.Anderson HA, Roche PA. 2015. MHC class II association with lipid rafts on the antigen presenting cell surface. Biochim Biophys Acta 1853:775–780. doi: 10.1016/j.bbamcr.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leman LJ, Maryanoff BE, Ghadiri MR. 2014. Molecules that mimic apolipoprotein A-I: potential agents for treating atherosclerosis. J Med Chem 57:2169–2196. doi: 10.1021/jm4005847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xie Q, Zhao S-P, Li F. 2010. D-4F, an apolipoprotein A-I mimetic peptide, promotes cholesterol efflux from macrophages via ATP-binding cassette transporter A1. Tohoku J Exp Med 220:223–228. doi: 10.1620/tjem.220.223. [DOI] [PubMed] [Google Scholar]
- 48.Dunbar RL, Movva R, Bloedon LT, Duffy D, Norris RB, Navab M, Fogelman AM, Rader DJ. 2017. Oral apolipoprotein A‐I mimetic D‐4F lowers HDL‐inflammatory index in high‐risk patients: a first‐in‐human multiple‐dose, randomized controlled trial. Clin Transl Sci 10:455–469. doi: 10.1111/cts.12487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ryan RO, Forte TM, Oda MN. 2003. Optimized bacterial expression of human apolipoprotein A-I. Prot Expr Purif 27:98–103. doi: 10.1016/s1046-5928(02)00568-5. [DOI] [PubMed] [Google Scholar]