

ABSTRACT
The cellular entry pathways and the mechanisms of Newcastle disease virus (NDV) entry into cells are poorly characterized. In this study, we demonstrated that chicken interferon-induced transmembrane protein 1 (chIFITM1), which is located in the early endosomes, could limit the replication of NDV in chicken macrophage cell line HD11, suggesting the endocytic entry of NDV into chicken macrophages. Then, we presented a systematic study about the entry mechanism of NDV into chicken macrophages. First, we demonstrated that a low-pH condition and dynamin were required during NDV entry. However, NDV entry into chicken macrophages was independent of clathrin-mediated endocytosis. We also found that NDV entry was dependent on membrane cholesterol. The NDV entry and replication were significantly reduced by nystatin and phorbol 12-myristate 13-acetate treatment, overexpression of dominant-negative (DN) caveolin-1, or knockdown of caveolin-1, suggesting that NDV entry depends on caveola-mediated endocytosis. However, macropinocytosis did not play a role in NDV entry into chicken macrophages. In addition, we found that Rab5, rather than Rab7, was involved in the entry and traffic of NDV. The colocalization of NDV with Rab5 and early endosome suggested that NDV virion was transported to early endosomes in a Rab5-dependent manner after internalization. Of particular note, the caveola-mediated endocytosis was also utilized by NDV to enter primary chicken macrophages. Moreover, NDV entered different cell types using different pathways. Collectively, our findings demonstrate for the first time that NDV virion enters chicken macrophages via a pH-dependent, dynamin and caveola-mediated endocytosis pathway and that Rab5 is involved in the traffic and location of NDV.
IMPORTANCE Although the pathogenesis of Newcastle disease virus (NDV) has been extensively studied, the detailed mechanism of NDV entry into host cells is largely unknown. Macrophages are the first-line defenders of host defense against infection of pathogens. Chicken macrophages are considered one of the main types of target cells during NDV infection. Here, we comprehensively investigated the entry mechanism of NDV in chicken macrophages. This is the first report to demonstrate that NDV enters chicken macrophages via a pH-dependent, dynamin and caveola-mediated endocytosis pathway that requires Rab5. The result is important for our understanding of the entry of NDV in chicken macrophages, which will further advance the knowledge of NDV pathogenesis and provide useful clues for the development of novel preventive or therapeutic strategies against NDV infection. In addition, this information will contribute to our further understanding of pathogenesis with regard to other members of the Avulavirus genus in the Paramyxoviridae family.
KEYWORDS: chicken macrophage, endocytic pathway, Newcastle disease virus, Rab5
INTRODUCTION
Newcastle disease virus (NDV) is a member of the Avulavirus genus belonging to the Paramyxoviridae family and is an economically important etiological agent of highly contagious poultry disease (1–3). NDV is an enveloped virus with a negative-strand, nonsegmented RNA genome, which contains six genes that encode nucleoprotein (NP), phosphoprotein (P), matrix protein (M), fusion protein (F), hemagglutinin-neuraminidase (HN), and large RNA-directed RNA polymerase (L), in which HN and F proteins are the two envelope glycoproteins required by infection of NDV (4–6). Generally, for the initiation of viral infection, the HN protein recognizes the cellular surface receptor, and then the F protein mediates the fusion of the viral envelope and the target cell membrane (4, 5, 7, 8).
To gain entry, enveloped viruses can penetrate cells through two main routes: (i) direct fusion between the viral envelope and the cellular plasma membrane and (ii) cellular endocytosis mechanism. For paramyxoviruses, the membrane fusion process is generally thought to take place in the plasma membrane under neutral pH conditions. In such cases, the attachment protein binding to cell receptors and subsequent interaction of an attachment protein with F protein triggers a series of irreversible conformational changes in the F protein, which leads to the fusion between viral and cellular membranes at neutral pH (9, 10). For the other enveloped viruses that enter cells through the endocytic pathways, after binding to cell surface adherence molecules or receptors, the viruses are internalized via a variety of endocytic processes (11, 12). After internalization via distinct routes, endocytosed enveloped viruses traverse the endosome network, the fusion is induced by the proper environmental conditions (e.g., low pH, proteases, ions, intracellular receptors, and lipid composition) in specific endosomal compartments (11). There is recent accumulating evidence indicating that in addition to the direct fusion with plasma membrane, paramyxoviruses can also enter into host cells through various endocytosis pathways (13–16). For instance, human metapneumovirus (HMPV) can be internalized into cells via clathrin-mediated endocytosis (16, 17). In addition, macropinocytosis is implicated in the entry of the Nipah virus (14). NDV has also been reported to utilize receptor-mediated endocytosis pathways for entry. Caveola-mediated endocytosis is involved in the entry of NDV into COS-7 and Hela cells; macropinocytosis and clathrin-mediated endocytosis are involved in the entry of NDV into DF-1 and dendritic cells. (18–21). These reports indicate the existence of multiple pathways of cell entry for NDV and that NDV may use alternative pathways to infect different target cells. However, the mechanisms of the endocytic entry of NDV into cells are not well characterized.
Macrophages are the first-line defenders of host defense against infection of pathogenic microorganisms. Chicken macrophages are considered one of the main types of target cells for NDV infection, and the productive infection of NDV in chicken macrophages has recently been confirmed (22–24). Macrophages are highly mobile and are present in multiple host tissues, and these cells are the key immunoregulatory agents against viral infection. The entry and replication of NDV in macrophages may favor the viral infection and pathogenicity via promoting the virus access to a wide variety of organs and altering the host immune responses (22, 23). The immune surveillance function of macrophages depends on specialized endocytic mechanisms, which allows them to engulf and digest invading pathogens. However, viruses also can hijack the intrinsic endocytic pathway of macrophages to achieve their infectious entry (25). Although NDV can infect and replicate in chicken macrophages (22, 23), whether NDV enters chicken macrophages via the endocytic mechanisms remains unclear.
In this study, we initially demonstrated that chicken interferon-induced transmembrane protein 1 (chIFITM1) can be located in the early endosomes and is able to limit the replication of NDV in chicken macrophage cell line HD11, suggesting the endocytic entry of NDV into chicken macrophages. Then, the mechanism of entry for NDV into chicken macrophages was elucidated. Chemical inhibitor treatment, RNA knockdown, and expression of dominant-negative (DN) proteins involved in the endocytosis pathways were conducted to explore which cellular factors are associated with the entry, especially the endocytic pathways of NDV into chicken macrophages. In addition, the entry of NDV into chicken primary macrophage and other chicken origin cell types were also assessed. Our results indicate that NDV can enter into chicken macrophages via a pH-dependent, dynamin and caveola-mediated endocytosis. In addition, Rab5 is required for the endocytic entry of NDV and subsequent traffic to early endosomes. The pathways and strategies may be different for the entry of NDV into different cell types.
RESULTS
NDV can be endocytosed in chicken macrophages.
To determine whether NDV could be endocytosed into chicken macrophages HD11 cells, purified NDV was labeled with a lipophilic fluorescent dye, DiOC, which can be quenched by the membrane-impermeable dye, trypan blue (13). HD11 cells were incubated with DiOC-labeled NDV F48E9 at 37°C for 1 h, and the noninternalized viruses were removed by washing with low-pH buffer (26, 27). Then the cells were treated or not treated with trypan blue. As shown in Fig. 1Aa, the green DiOC fluorescence could be observed in the cellular plasma membrane and cytoplasm of HD11 cells that were not treated with trypan blue. In contrast, the fluorescence in the cellular plasma membrane was quenched, and the green DiOC fluorescence was only observed in the cytoplasm of HD11 cells that were treated with trypan blue. The DiOC fluorescence in the cellular plasma membrane indicated that membrane fusion of DiOC-labeled NDV had occurred at the cell surface, whereas fluorescence in the cytoplasm, which was resistant to trypan blue, indicated that the intact virus particles including the viral lipid envelope were endocytosed.
FIG 1.

NDV undergoes endocytosis and chIFITM1 located in the early endosome restricts the NDV infection. (A) NDV can be endocytosed. HD11 cells were inoculated with DiOC-labeled NDV F48E9 at 37°C for 60 min. After incubation, cells were washed with low-pH buffer to remove noninternalized viruses, and then the cells were fixed and treated with trypan blue to quench the green fluorescence of DiOC in the plasma membrane surface. The fluorescence of DiOC was examined by confocal fluorescence microscopy (a). HD11 cells were inoculated with DiOC-labeled NDV F48E9 at 37°C for different time. The cells were treated with low-pH buffer to inactivate and remove noninternalized viruses and then collected and fixed in 4% paraformaldehyde. Each cell sample was divided into two. One sample was subjected directly for the flow cytometric analysis, and the other was treated with trypan blue. The fluorescence of DiOC was examined by flow cytometry. The fluorescence intensity that was resistant (b) and quenched (c) by trypan blue at 60, 90, and 120 min was calculated relative to that of 30 min (×100%). (B) Cellular localization of chIFITM1. HD11 cells were transfected with pCAGGS-HA-chIFITM1 or pCAGGS-HA. The cells were fixed at 24 h after transfection and then subjected to indirect immunofluorescence to detect HA-tagged chIFITM1 and early endosomes marker, EEA1, using anti-HA antibody (red) and anti-EEA1 antibody (green). The nuclei were stained with DAPI (blue). (C) chIFITM1 did not inhibit NDV adsorption. HD11 cells transfected with pCAGGS-HA-chIFITM1 or pCAGGS-HA were incubated with NDV F48E9 for 1 h at 4°C to allow for viral adsorption. Viral RNA was quantified by real-time RT-PCR. (D) chIFITM1 did not inhibit NDV internalization. HD11 cells transfected with pCAGGS-HA-chIFITM1 or pCAGGS-HA were incubated with DiOC-labeled NDV F48E9 for 1 h at 4°C to allow for viral adsorption. Cells were then washed and further incubated for 1 h at 37°C to allow for internalization. After incubation, the cells were washed with low-pH buffer to remove noninternalized viruses and then collected and fixed in 4% paraformaldehyde. Each cell sample was divided into two. One sample was subjected directly for the flow cytometric analysis, and the other was treated with trypan blue. The HA-chIFITM1 was detected by using an HA tag antibody. The green fluorescence of DiOC-labeled NDV that was resistant and quenched by trypan blue in HA-chIFITM1-positive and-negative cells was measured by flow cytometry. (E) chIFITM1 reduced the replication of NDV in HD11 cells. HD11 cells were transfected with pCAGGS-HA-chIFITM1 or pCAGGS-HA. At 48 h posttransfection, cells were infected with F48E9 at an MOI of 0.1. At 18 hpi, viral titers in the culture supernatants of infected cells were determined. The bars represent means ± the SD. Data were analyzed by using the Student t test (*, P < 0.05; **, P < 0.01).
Subsequently, the entry process of NDV in HD11 cells was examined by flow cytometry using DiOC-labeled NDV F48E9 and the fluorescence intensity of DiOC in HD11 cells which treated or not with trypan blue was determined at different time points. As shown in Fig. 1Ab, 65% of the DiOC fluorescence failed to be quenched by trypan blue after 30 min postinoculation compared to untreated HD11 cells. However, 90% of the DiOC fluorescence was resistant to the treatment of trypan blue at 60 min postinoculation for 120 min. Meanwhile, the fluorescence intensity of DiOC quenched by trypan blue was not significantly changed since 60 min postinoculation (Fig. 1Ac). These results indicated that both the membrane fusion at the cell surface and the endocytic entry of NDV were accomplished at 60 min postinoculation. Thus, all the internalization assay was performed at 60 min (1 h) postinoculation in this study.
chIFITM1 located in early endosomes restricts NDV infection.
Interferon-inducible transmembrane proteins (IFITMs) are broad antiviral factors that can restrict the entry of a wide range of viruses by blocking the fusion of the virus envelope with plasma membrane or the membrane of endosomes. The localization of IFITMs is closely associated with their antiviral activities (28, 29). A previous report indicated that chIFITM1 was diffusely expressed in the cytoplasm (30). Here, we further examined whether chIFITM1 colocalized with specific endocytic compartments. HD11 cells were transfected with pCAGGS-HA-chIFITM1 or pCAGGS-HA, then the cellular localization of chIFITM1 was examined via confocal fluorescence microscopy. The result showed that chIFITM1 was present in the cytoplasm and showed obvious colocalization with early endosomes marker EEA1 (Fig. 1B). The effects of chIFITM1 on the different stages of NDV infection were then evaluated. The cells transfected with pCAGGS-HA-chIFITM1 or pCAGGS-HA were incubated with NDV F48E9 for 1 h at 4°C to allow for viral adsorption but not internalization. Viral RNA was quantified by real-time reverse transcription-PCR (RT-PCR). No significant differences were observed for the NDV genomic RNA copies between pCAGGS-HA-chIFITM1- and pCAGGS-HA-transfected cells, indicating that the expression of chIFITM1 had no effects on NDV adsorption (Fig. 1C). Meanwhile, DiOC-labeled NDV F48E9 was adsorbed onto pCAGGS-HA-chIFITM1- and pCAGGS-HA-transfected cells, followed by incubation at 37°C to permit internalization, and noninternalized viruses were removed by washing with low-pH buffer. After treatment with trypan blue, the DiOC fluorescence was determined by flow cytometry. As shown in Fig. 1D, the mean fluorescence intensity of DiOC that was resistant to trypan blue in HA-chIFITM1-positive cells was comparable with that in HA-chIFITM1-negative cells. There was also no significant difference in the mean fluorescence intensities quenched by trypan blue in both kinds of cells. These results indicated that the expression of chIFITM1 had no effect on NDV internalization (both membrane fusion at the cell surface and endocytic entry). Notably, the titer of progeny virus in pCAGGS-HA-chIFITM1-transfected cells was significantly lower than in pCAGGS-HA-transfected cells at 18 h postinoculation (hpi), indicating that chIFITM1 restricted the replication of NDV (Fig. 1E). Since chIFITM1 could be located in the early endosomes and reduce the replication of NDV with no effects on the adsorption and internalization of NDV in HD11 cells, we inferred that the fusion of NDV in endosomal compartments was restricted by chIFITM1 as previously reported (28, 31), which reduced the release of viral content into the cytoplasm and the production of progeny virus. These results further indicated the endocytic entry of NDV into HD11 cells.
NDV can enter into HD11 cells in a pH-dependent manner.
Generally, the infectious entry of virus via the endocytic pathway requires an acidic condition in the endosomes (32, 33). Here, we tested whether the entry of NDV into chicken macrophages is pH dependent. The effects of two inhibitors of endosomal acidification, bafilomycin A1 and chloroquine, on the infection of NDV into HD11 cells were evaluated. The HD11 cells were first pretreated with various concentrations of inhibitors to evaluate the cytotoxic effect of inhibitors. The result of this cell viability assay showed that HD11 cells tolerated up to 5 μM bafilomycin A1 and 100 μM chloroquine, respectively (data not shown). Then, the cells were incubated with NDV F48E9 in the presence of inhibitors for 1 h at 4°C to allow viral adsorption but not internalization. Viral RNA was quantified by real-time RT-PCR. The result showed that there were no significant differences in the NDV genomic RNA copies between inhibitor-treated groups and the control group, indicating that both inhibitors had no effects on NDV adsorption (Fig. 2A). Meanwhile, DiOC-labeled NDV F48E9 was adsorbed onto HD11 cells, followed by incubation with indicated concentrations of inhibitors for 1 h at 37°C to permit internalization, and noninternalized viruses were removed by washing with low-pH buffer. After quenching the green fluorescence of DiOC in the plasma membrane surface with trypan blue as previously described (13), the green fluorescence of internalized virus was determined by confocal fluorescence microscopy and flow cytometry, respectively. As shown in Fig. 2Ba, the results of confocal fluorescence microscopy demonstrated that the mean fluorescence intensities of DiOC-labeled NDV F48E9 in inhibitor-treated cells were significantly reduced compared to the control cells. These data were verified by flow cytometry, since the mean fluorescence intensity that was resistant to trypan blue in inhibitor-treated cells was significantly lower than that in control cells (Fig. 2Bb). Meanwhile, there was no significant difference in the mean fluorescence intensities quenched by trypan blue in inhibitor-treated cells and control cells (Fig. 2Bb). These results indicated that the endocytic entry of NDV was inhibited by the two inhibitors of endosomal acidification, while the membrane fusion at the cell surface was not affected. Correspondingly, the progeny viral titers in both inhibitor-treated groups were also significantly lower than the control group (Fig. 2C). These results suggest that NDV could enter into HD11 cells in a pH-dependent manner.
FIG 2.

NDV entry into HD11cells requires acidic endosomal pH. (A) Bafilomycin A1 and chloroquine did not affect the adsorption of NDV. HD11 cells were treated with the indicated concentration of inhibitors for 1 h at 37°C. Cells were then incubated with NDV F48E9 in the presence of inhibitors for 1 h at 4°C to allow for viral adsorption. DMSO was included as a negative control. Viral RNA was quantified by real-time RT-PCR. (B) Bafilomycin A1 and chloroquine inhibited the internalization of NDV. DiOC-labeled NDV F48E9 was adsorbed onto HD11 cells and then incubated with indicated concentrations of inhibitors for 1 h at 37°C to permit internalization. After a low-pH buffer wash and quenching the green fluorescence of DiOC in the plasma membrane surface with trypan blue, the green fluorescence of internalized virus was determined by confocal fluorescence microscopy (a). Meanwhile, the cells with the same treatment were subjected to flow cytometry. Each cell sample was divided into two. One sample was subjected directly for the flow cytometric analysis, and the other was treated with trypan blue (b). The fluorescence intensity which resistant and quenched by trypan blue in inhibitor-treated cells was calculated relative to that of control cells (×100%). (C) Bafilomycin A1 and chloroquine reduced the replication of NDV. HD11 cells were infected with NDV F48E9 at an MOI of 0.1 in the presence of the inhibitors at 37°C for 1 h. DMSO was included as a negative control. Cells were then washed with PBS and incubated in medium containing 5% FBS at 37°C. At 18 hpi, viral titers in the culture supernatants of infected cells were determined. The bars represent means ± the SD. Data were analyzed by using the Student t test (*, P < 0.05; **, P < 0.01).
Entry of NDV into HD11 cells depends on dynamin.
The roles of dynamin in the entry of paramyxoviruses, including NDV, into some cell lines have been reported (16, 18). To examine whether dynamin is involved in the infection of NDV in chicken macrophages, dynasore, the inhibitor of GTPase activity of dynamin, was used to evaluate the effects of dynamin on the adsorption, internalization, and replication of NDV F48E9 in HD11 cells. First, HD11 cells were pretreated with increasing concentrations of dynasore, and the results showed that HD11 cells tolerated up to 50 μM dynasore (data not shown). Since dynamin is implicated in the clathrin- and caveolin-mediated endocytosis, the effect of dynasore on dynamin-dependent endocytosis was evaluated by uptake of fluorescently labeled transferrin (Tfn), which is a well-characterized cargo of the clathrin- and caveolin-mediated endocytic pathway and extensively used as a control in the endocytic entry of various viruses (27, 34–38). HD11 cells pretreated with dynasore were subsequently incubated with Tfn. Noninternalized (surface-bound) Tfn was removed after incubation by washing with low-pH buffer (26, 27), and the cells were then fixed and examined by fluorescence microscopy. Compared to the dimethyl sulfoxide (DMSO)-treated cells, the red fluorescence signals of Tfn were obviously reduced in 50 μM dynasore-treated HD11 cells, indicating the inhibition of Tfn uptake via dynamin-dependent endocytosis (Fig. 3A). Then, the effects of dynasore on the adsorption and internalization of NDV were evaluated as described above. As shown in Fig. 3Ba, the NDV genomic RNA copy numbers showed no significant difference between control and dynasore-treated groups, indicating that dynasore had no effect on NDV adsorption. In the internalization assay, the results of both confocal fluorescence microscopy and flow cytometry analyses revealed that the mean fluorescence intensity of DiOC that was resistant to trypan blue in the dynasore-treated cells was significantly lower than that in the control cells (Fig. 3Bb and c). Meanwhile, there was no significant difference in the mean fluorescence intensities quenched by trypan blue in dynasore-treated cells and control cells (Fig. 3Bc). These results indicated that the endocytic entry of NDV was inhibited by dynasore, whereas the membrane fusion at the cell surface was not affected. In line with this result, the titer of progeny virus in the dynasore-treated group was lower than the control group (Fig. 3C). The aforementioned results demonstrated that dynasore inhibited the endocytic entry of NDV in HD11 cells and suggested that dynamin was involved in NDV entry.
FIG 3.

NDV entry into HD11cells depends on dynamin. (A) Dynasore inhibited Tfn uptake. HD11 cells were treated with 50 μM dynasore or DMSO for 1 h at 37°C, followed by incubation with 10 μg/ml Tfn for 1 h at 4°C, and then shifted to 37°C for 1 h; after a wash with low-pH buffer, the cells were fixed and stained with DAPI. (B) Dynasore inhibited the internalization of NDV. HD11 cells were treated with the indicated concentration of inhibitors for 1 h at 37°C. The cells were then incubated with NDV F48E9 in the presence of inhibitors for 1 h at 4°C to allow for viral adsorption. DMSO was included as a negative control. Viral RNA was quantified by real-time RT-PCR (a). Alternatively, DiOC-labeled NDV F48E9 was adsorbed onto HD11 cells and then incubated with indicated concentrations of inhibitors for 1 h at 37°C to permit internalization. After a wash with low-pH buffer and quenching the green fluorescence of DiOC in the plasma membrane surface with trypan blue, the green fluorescence of internalized virus was determined by confocal fluorescence microscopy (b). Meanwhile, the treated cells were subjected to flow cytometry. Each cell sample was divided into two. One sample was subjected directly for the flow cytometric analysis, and another was treated with trypan blue (c). The fluorescence intensity which was resistant and quenched by trypan blue in inhibitor-treated cells was calculated relative to that of control cells (×100%). (C) Dynasore reduced the replication of NDV. HD11 cells were infected with NDV F48E9 at an MOI of 0.1 in the presence of the inhibitors at 37°C for 1 h. DMSO was included as a negative control. The cells were then washed with PBS and incubated in medium containing 5% FBS at 37°C. At 18 hpi, the viral titers in the culture supernatants of infected cells were determined. (D) Effects of dynamin II on Tfn uptake and NDV internalization. HD11 cells transfected with the EGFP-tagged WT or DN dynamin II were incubated with 10 μg/ml Tfn and DiI-labeled or unlabeled NDV F48E9 for 1 h at 4°C and then shifted to 37°C for 1 h. After a wash with low-pH buffer, the red fluorescence of Tfn in EGFP-positive cells was determined by confocal fluorescence microscopy and flow cytometry, respectively (a). The red fluorescence of DiI-labeled NDV in EGFP-positive cells was determined by flow cytometry (b). In addition, the level of HN protein on the cell surface in unlabeled NDV-infected EGFP-positive cells was determined using anti-HN antibody via flow cytometry (b). (E) Dynamin II knockdown inhibited the internalization of NDV. HD11 cells were transfected with siRNA targeting dynamin (siDynamin II) or a control siRNA (siControl). The effect of siRNA knockdown on dynamin expression was determined by Western blotting (a). Cell viability upon siDynamin II and siControl transfection was assessed as described in the text (b). At 48 h posttransfection, the internalization assay was performed with DiOC-labeled NDV F48E9 as described above. The green fluorescence of DiOC that was resistant and quenched by trypan blue was determined by confocal fluorescence microscopy (c) and flow cytometry (d), respectively. (F) Dynamin knockdown reduced the replication of NDV. HD11 cells were transfected with siDynamin II or siControl. At 48 h posttransfection, cells were infected with NDV F48E9 at an MOI of 0.1, and the viral titers in the culture supernatants of infected cells were determined at 18 hpi. The bars represent means ± the SD. Data were analyzed by using the Student t test (*, P < 0.05; **, P < 0.01).
Moreover, to further evaluate the role of dynamin in NDV entry, HD11 cells were transfected with plasmids expressing enhanced green fluorescent protein (EGFP)-tagged wild-type (WT) dynamin II and dominant-negative (DN) dynamin II (K44A). To determine the endocytosed virus in EGFP-positive cells, DiI (red fluorescence)-labeled and unlabeled NDV F48E9 was used for the internalization assay as described above, with Tfn uptake as a positive control. As shown in Fig. 3Da, the results of confocal fluorescence microscopy and flow cytometry demonstrated that the mean fluorescence intensity of Tfn in cells overexpressing DN dynamin II was significantly lower than that in the cells overexpressing WT dynamin II, indicating the inhibition of Tfn uptake by DN dynamin II. The effect of the WT and DN dynamin II on NDV internalization was also evaluated. The results of flow cytometry demonstrated that the mean fluorescence intensity of DiI in cells overexpressing DN dynamin II was significantly lower than that in cells overexpressing WT dynamin II (Fig. 3Db), indicating that the entry of NDV was inhibited in cells overexpressing DN dynamin II. In contrast, the mean fluorescence intensity of NDV HN protein on the cell surface was comparable in cells overexpressing WT and DN dynamin II (Fig. 3Db), suggesting the membrane fusion of NDV at the cell surface was not affected. These data indicated that the endocytic entry of NDV was inhibited by overexpression of DN dynamin II. To confirm these results, the small interfering RNA (siRNA)-mediated knockdown of dynamin II in HD11 cells was conducted. Western blotting demonstrated the efficiency of siRNA-mediated knockdown of dynamin (Fig. 3Ea), and the cell viability was not significantly affected by the siRNA transfection (Fig. 3Eb). The HD11 cells transfected with siRNA targeting dynamin II (siDynamin II) or a control siRNA (siControl) were incubated with DiOC-labeled NDV F48E9 at 37°C for 1 h to permit internalization, and noninternalized viruses were removed by washing with low-pH buffer. After treatment with trypan blue, the DiOC fluorescence was determined as described above. As shown in Fig. 3Ec and d, the results of confocal fluorescence microscopy and flow cytometry demonstrated that the mean fluorescence intensity of DiOC that was resistant to trypan blue in dynamin II knockdown cells was significant lower than that in siControl-transfected cells, whereas no significant change was observed for the mean fluorescence intensity quenched by trypan blue in dynamin II knockdown and siControl-transfected cells (Fig. 3Ed). Furthermore, the titer of progeny viruses in dynamin II knockdown HD11 cells was significantly lower than the control cells at 18 hpi, when the siRNA-transfected HD11 cells were infected with NDV (Fig. 3F). Taken together, these results indicated that dynamin had involved in the endocytic entry of NDV, without affecting the membrane fusion of NDV at the cell surface.
Entry of NDV into HD11 cells is clathrin independent.
We further examined whether NDV entered into chicken macrophages via clathrin-mediated endocytosis since reports showed that dynamin is implicated in both clathrin- and caveolin-mediated endocytosis, and some paramyxoviruses can use clathrin-mediated endocytosis to establish infection (9, 39, 40). Chlorpromazine is used because it is a commonly used inhibitor of clathrin-mediated endocytosis (12, 41). We found that cellular viability of HD11 did not significantly change against up to 5 μM chlorpromazine (data not shown). Next, the effect of chlorpromazine on endocytosis was evaluated by evaluating the uptake of Tfn. HD11 cells pretreated with chlorpromazine were subsequently incubated with Tfn. Noninternalized (surface-bound) Tfn was removed after incubation by washing with low-pH buffer, and the cells were fixed and examined by fluorescence microscopy. As shown in Fig. 4A, a significant reduction of the red fluorescence signals of Tfn was observed upon 5 μM chlorpromazine treatment, indicating the inhibition of Tfn uptake via endocytosis. Subsequently, the effects of chlorpromazine on the adsorption and internalization of NDV in HD11 cells were tested as described above. No significant difference in the NDV genomic RNA copy numbers was found between the control and inhibitor-treated groups when chlorpromazine was present during adsorption, indicating that chlorpromazine had no effect on NDV adsorption (Fig. 4Ba). Moreover, the results of confocal fluorescence microscopy and flow cytometry demonstrated that the mean fluorescence intensity of DiOC that was resistant to trypan blue in the chlorpromazine-treated group was comparable with that in the control group when chlorpromazine was present during internalization (Fig. 4Bb and c). There was no significant change in the mean fluorescence intensities quenched by trypan blue in inhibitor-treated and control groups (Fig. 4Bc). These findings suggested that chlorpromazine had no effects on adsorption, endocytic entry and membrane fusion of NDV on the surface of HD11 cells. Correspondingly, the viral titer in chlorpromazine-treated cells was comparable with that of control cells (Fig. 4C).
FIG 4.

Clathrin-mediated endocytosis is not required for NDV entry. (A) Chlorpromazine inhibited Tfn uptake. HD11 cells were treated with 5 μM chlorpromazine or DMSO for 1 h at 37°C, followed by incubation with 10 μg/ml Tfn for 1 h at 4°C, and then shifted to 37°C for 1 h; after a wash with low-pH buffer, the cells were fixed and stained with DAPI. (B) Chlorpromazine did not inhibit NDV adsorption and internalization. HD11 cells were treated with indicated concentration of inhibitors for 1 h at 37°C. Cells were then incubated with NDV F48E9 in the presence of inhibitors for 1 h at 4°C to allow for viral adsorption. DMSO was included as a negative control. Viral RNA was quantified by real-time RT-PCR (a). Alternatively, DiOC-labeled NDV F48E9 was adsorbed onto HD11 cells and then incubated with the indicated concentrations of inhibitors for 1 h at 37°C. The internalization assay was performed as described above. The green fluorescence of DiOC that was resistant and quenched by trypan blue was determined by confocal fluorescence microscopy (b) and flow cytometry (c), respectively. (C) Chlorpromazine did not affect the replication of NDV. HD11 cells were infected with NDV F48E9 at an MOI of 0.1 in the presence of the inhibitors at 37°C for 1 h. DMSO was included as a negative control. Cells were then washed with PBS and incubated in medium containing 5% FBS at 37°C. At 18 hpi, viral titers in the culture supernatants of infected cells were determined. (D) Effects of EPS15 on Tfn uptake and NDV internalization. HD11 cells transfected with the EGFP-tagged WT or DN EPS15 were incubated with 10 μg/ml Tfn and DiI-labeled or unlabeled NDV F48E9 for 1 h at 4°C and then shifted to 37°C for 1 h. After a wash with low-pH buffer, the red fluorescence of Tfn (a) and DiI-labeled NDV (b) and HN protein on the cell surface (panel b) was determined as described above. (E) Clathrin knockdown showed no effect on the internalization of NDV. HD11 cells were transfected with siRNA targeting clathrin heavy chain (siClathrinHC) or a control siRNA (siControl). The effect of siRNA knockdown on clathrin heavy chain expression was determined by Western blotting (a). Cell viability upon siClathrinHC and siControl transfection was assessed as described in the text (b). At 48 h posttransfection, the internalization assay was performed with DiOC-labeled NDV F48E9 as described above. The green fluorescence of DiOC that was resistant and quenched by trypan blue was determined by confocal fluorescence microscopy (c) and flow cytometry (d), respectively. (F) Clathrin knockdown showed no effect on the replication of NDV. HD11 cells were transfected with siClathrinHC or siControl. At 48 h posttransfection, the cells were infected with NDV F48E9 at an MOI of 0.1, and the viral titers in the culture supernatants of infected cells were determined at 18 hpi. The bars represent means ± the SD. Data were analyzed by using the Student t test (*, P < 0.05; **, P < 0.01).
In additional studies, we used EGFP-tagged WT and DN epidermal growth factor receptor pathway substrate 15 (EPS15), which has a large N-terminal deletion, to evaluate the potential roles of clathrin-mediated endocytosis in the entry of NDV because EPS15 is a key factor involved in clathrin-mediated endocytosis (27, 36, 42, 43). HD11 cells were transfected with plasmids expressing EGFP-tagged WT and DN EPS15 and subsequently underwent an internalization assay using DiI-labeled and unlabeled NDV F48E9; Tfn uptake was used as a positive control. As shown in Fig. 4Da, the results of confocal fluorescence microscopy and flow cytometry revealed that the mean fluorescence intensity of Tfn in cells overexpressing DN EPS15 was significantly lower than that in the cells overexpressing WT EPS15, indicating the inhibition of Tfn uptake by DN EPS15. The effect of the WT and DN EPS15 on NDV internalization was also evaluated here. As shown in Fig. 4Db, in comparison to cells overexpressing WT EPS15, no significant difference in the mean fluorescence intensity of DiI was detected in cells overexpressing DN EPS15, indicating that the entry of NDV was not affected by EPS15. In addition, the mean fluorescence intensity of NDV HN protein on the cell surface was comparable in cells overexpressing WT and DN EPS15 (Fig. 4Db), suggesting that the membrane fusion of NDV at the cell surface was not affected. Collectively, these data suggested that the EPS15 had no effect on the endocytic entry and membrane fusion of NDV on the surface in HD11 cells. To further evaluate the role of clathrin-mediated endocytosis during NDV entry, siRNA was used to knock down the expression of the clathrin heavy chain (CHC) in HD11 cells. Western blotting demonstrated the efficiency of siRNA-mediated knockdown of the CHC (Fig. 4Ea), and the cell viability was not significantly affected by siRNA transfection (Fig. 4Eb). An internalization assay was then performed as described above. The results of confocal fluorescence microscopy (Fig. 4Ec) and flow cytometry (Fig. 4Ed) demonstrated that the mean fluorescence intensity of DiOC that was resistant to trypan blue in CHC knockdown HD11 cells had not been significantly affected compared yo the siControl-transfected cells. There was also no significant change in the mean fluorescence intensity quenched by trypan blue in CHC knockdown and siControl-transfected cells (Fig. 4Ed). Besides, the titer of progeny viruses of NDV in siClathrinHC-transfected cells was comparable with the control siRNA-transfected cells (Fig. 4F). These data indicated that the entry of NDV into HD11 cells is not dependent on clathrin-mediated endocytosis.
Entry of NDV into HD11 cells depends on caveola-mediated endocytosis.
In addition to the clathrin-mediated endocytosis, dynamin also participated in caveola-mediated endocytosis, so the role of caveola-mediated endocytosis during NDV entry to chicken macrophage was assessed. Plasma membrane cholesterol is essential for the formation and maintenance of caveolae, and depletion or sequestration of cholesterol in the membrane can inhibit caveola-mediated endocytosis (12). In this study, methyl-β-cyclodextrin (MβCD), a reagent that can deplete the cholesterol from membrane, and nystatin, a steroid-binding antibiotic that can bind cholesterol and that can also used as an inhibitor of caveola-mediated endocytosis, were adopted to treat HD11 cells. The results of cellular viability assay showed that HD11 cells tolerated up to 2.5 mM MβCD and 1 μg/ml nystatin (data not shown). Previous studies have confirmed that the cholera toxin subunit B (CTB) is internalized through the caveola-mediated endocytosis and can serve as a marker for this endocytic pathway (27, 36, 44, 45). To test the effect of MβCD and nystatin on caveola-mediated endocytosis, HD11 cells were pretreated with 2.5 mM MβCD and 1 μg/ml nystatin, and a CTB uptake assay was performed. As shown in Fig. 5A, a significant reduction in the red fluorescence signals of CTB was observed in MβCD- and nystatin-treated cells compared to that in DMSO-treated cells, indicating the inhibition of CTB uptake. Since MβCD can disrupt the lipid raft microdomain, the reduced CTB uptake by MβCD treatment may also be attributed to the limited binding of CTB to the cells. Subsequently, the effects of MβCD on the adsorption and internalization of NDV in HD11 cells were tested, as described above. As shown in Fig. 5Ba, the NDV genomic RNA copies in HD11 cells were significantly reduced when MβCD was present during the adsorption of NDV, indicating that MβCD significantly inhibited NDV attachment to HD11 cells. In addition, DiOC-labeled NDV F48E9 was adsorbed onto HD11 cells, followed by incubation with MβCD for 1 h at 37°C to permit internalization. As demonstrated by confocal fluorescence microscopy and flow cytometry, the mean fluorescence intensity of DiOC that was resistant to trypan blue in the MβCD-treated cells was significantly lower than that in the control cells (Fig. 5Bb and c). Moreover, the mean fluorescence intensity quenched by trypan blue in MβCD-treated cells was significantly lower than that in the control cells (Fig. 5Bc), indicating the reduced level of membrane fusion at the cell surface which may be attributed to the limited attachment of virions. These data suggested the requirement of cellular membrane cholesterol for NDV entry. Next, the effects of nystatin on the adsorption and internalization of NDV in HD11cells were determined. Unlike MβCD, the presence of nystatin during adsorption did not result in significant differences in the NDV genomic RNA copy numbers in NDV-infected cells, indicating that nystatin did not affect the adsorption of NDV in HD11 cells (Fig. 5Ca). Furthermore, the results of an internalization assay obtained by confocal fluorescence microscopy and flow cytometry revealed that the mean fluorescence intensity of DiOC that was resistant to trypan blue in the nystatin-treated cells was significantly lower than that in the control cells (Fig. 5Cb and c), whereas the mean fluorescence intensity quenched by trypan blue in nystatin-treated cells was comparable to that in control cells (Fig. 5Cc). These results suggested that cholesterol was required for the internalization of NDV and indicated the potential of caveola-mediated endocytosis in the entry of NDV into HD11 cells. Since cholesterol had also involved in other lipid raft-associated endocytosis, phorbol 12-myristate 13-acetate (PMA), a specific inhibitor of caveola-mediated endocytosis which can decrease the level of caveolae at the cell surface, was used to further assess the role of caveola-mediated endocytosis in the entry of NDV. The results of the cellular viability assay showed that HD11 cells tolerated up to 0.1 μM PMA (data not shown). Evaluation of the effect of PMA on endocytosis via the CTB uptake assay demonstrated that treatment with PMA resulted in a significant reduction in the red fluorescence signals of CTB (Fig. 5A), indicating the inhibition of CTB uptake via caveola-mediated endocytosis. Consistent with the results obtained using nystatin, the adsorption of NDV in HD11cells was not significantly affected by the treatment of PMA (Fig. 5Da). The significant reduction of mean fluorescence intensity of DiOC that was resistant to trypan blue in PMA-treated cells indicated that the endocytic entry of NDV was inhibited by PMA (Fig. 5Db and c). In addition, the comparable level of the mean fluorescence intensity quenched by trypan blue in PMA-treated and control cells suggested that the membrane fusion of NDV at the cell surface was not affected (Fig. 5Dc). Moreover, as expected, the titers of progeny viruses in MβCD-, nystatin-, and PMA-treated cells were all lower than in control cells (Fig. 5E).
FIG 5.

Caveola-mediated endocytosis is required for NDV entry. (A) MβCD-, nystatin-, and PMA-inhibited CTB uptake. HD11 cells were treated with 2.5 mM MβCD, 1 μg/ml nystatin, 0.1 μM PMA, or DMSO for 1 h at 37°C, followed by incubation with 10 μg/ml CTB for 1 h at 4°C, and then shifted to 37°C for 1 h; a after wash with low-pH buffer, the cells were fixed and stained with DAPI. (B to D) Effects of inhibitors of caveola-mediated endocytosis on the adsorption and internalization of NDV. The effects of MβCD (B), nystatin (C), and PMA (D) on the adsorption and internalization of NDV were determined. HD11 cells were treated with the indicated concentrations of inhibitors for 1 h at 37°C. The cells were then incubated with NDV F48E9 in the presence of inhibitors for 1 h at 4°C to allow for viral adsorption. DMSO was included as a negative control. Viral RNA was quantified by real-time RT-PCR (subpanel a in panels B, C, and D). Alternatively, DiOC-labeled NDV F48E9 was adsorbed onto HD11 cells and then incubated with the indicated concentrations of inhibitors for 1 h at 37°C; the internalization assay was then performed as described above. The green fluorescence of DiOC that was resistant and quenched by trypan blue was determined by confocal fluorescence microscopy (subpanel b in panels B, C, and D) and flow cytometry (subpanel c in panels B, C, and D), respectively. (E) MβCD, nystatin, and PMA reduced the replication of NDV. HD11 cells were infected with NDV F48E9 at an MOI of 0.1 in the presence of the inhibitors at 37°C for 1 h. DMSO was included as a negative control. Next, the cells were washed with PBS and incubated in medium containing 5% FBS at 37°C. At 18 hpi, the viral titers in the culture supernatants of infected cells were determined. (F) Effects of caveolin-1 on CTB uptake and NDV internalization. HD11 cells transfected with plasmids expressing EGFP-tagged WT and DN caveolin-1 were incubated with 10 μg/ml CTB and DiI-labeled or unlabeled NDV F48E9 for 1 h at 4°C and then shifted to 37°C for 1 h. After a wash with low-pH buffer, the red fluorescence of Tfn (a) and DiI-labeled NDV (b) and HN protein (b) on the cell surface was determined as described above. (G) Caveolin-1 knockdown inhibited NDV internalization. HD11 cells were transfected with siRNA targeting caveolin-1 (siCaveolin-1) or siControl. The effect of siRNA knockdown on caveolin-1 expression was determined by Western blotting (a). Cell viability upon siCaveolin-1 and siControl transfection was assessed as described in the text (b). At 48 h posttransfection, an internalization assay was performed with DiOC-labeled NDV F48E9, as described above. The green fluorescence of DiOC that was resistant and quenched by trypan blue was determined by confocal fluorescence microscopy (c) and flow cytometry (d), respectively. (H) Caveolin-1 knockdown reduced the replication of NDV. HD11 cells were transfected with siCaveolin-1 or siControl. At 48 h posttransfection, the cells were infected with NDV F48E9 at an MOI of 0.1, and the viral titers in the culture supernatants of infected cells were determined at 18 hpi. (I) The colocalization of NDV with caveolin-1. HD11 cells were incubated with NDV F48E9 at an MOI of 10 at 4°C for 1 h and then shifted to 37°C for 1 h. The cells were then fixed and reacted with anti-caveolin-1 antibody and NDV HN antibody and visualized by confocal microscopy. The bars represent means ± the SD. Data were analyzed by using the Student t test (*, P < 0.05; **, P < 0.01).
To further investigate the effect of caveola-mediated endocytosis on the entry of NDV into HD11 cells, plasmids expressing EGFP-tagged WT and DN caveolin-1, which lack the first 81 amino acids, were transfected into HD11 cells (46, 47). Subsequently, the internalization assay using DiI-labeled and unlabeled NDV F48E9 was performed, and the CTB uptake was used as a positive control. As shown in Fig. 5Fa, the results of confocal fluorescence microscopy and flow cytometry demonstrated that the mean fluorescence intensities of CTB in cells overexpressing DN caveolin-1 was significantly lower than that in the cells overexpressing WT caveolin-1, indicating the inhibition of CTB uptake by DN caveolin-1. Next, the effects of the WT and DN caveolin-1 on NDV internalization were evaluated. As shown in Fig. 5Fb, the overexpression of DN caveolin-1 reduced the mean fluorescence intensities of DiI in comparison to cells overexpressing WT caveolin-1, indicating that the entry of NDV was reduced. Furthermore, no significant difference in the mean fluorescence intensities of NDV HN protein on the cell surface was observed in cells overexpressing WT and DN caveolin-1 (Fig. 5Fb), suggesting that the membrane fusion of NDV at the cell surface was not affected. These data indicated that the endocytic entry of NDV was inhibited by the overexpression of DN caveolin-1. These results were verified by knockdown of the expression of caveolin-1 using siRNA. Western blotting demonstrated the efficiency of siRNA-mediated knockdown of caveolin-1 (Fig. 5Ga), and cell viability was not significantly affected by siRNA transfection (Fig. 5Gb). HD11 cells transfected with siRNA targeting caveolin-1 (siCaveolin-1) or a control siRNA (siControl) were incubated with DiOC-labeled NDV F48E9 at 37°C for 1 h, and then an internalization assay was performed, as described above. As shown in Fig. 5Gc and d, significant reductions in the mean fluorescence intensities of DiOC that resistant to trypan blue were observed in caveolin-1 knockdown cells compared to that seen in siControl-transfected cells. Although no significant change in mean fluorescence intensities quenched by trypan blue was observed in caveolin-1 knockdown and siControl-transfected cells (Fig. 5Gd). These data further indicated that caveolin-1 is involved in the endocytic entry of NDV, with no effect on the membrane fusion of NDV at the cell surface. Correspondingly, in caveolin-1 knockdown of HD11 cells, the titer of progeny virus was significantly lower than that in the siControl-transfected cells (Fig. 5H). To directly evaluate the role of caveolae in the entry of NDV, HD11 cells were incubated with NDV at 4°C for 1 h to allow viral attachment and then shifted to 37°C for 1 h to permit internalization. Cells were then fixated and stained with antibodies against NDV HN and caveolin-1. The localization of NDV with caveolin-1 was analyzed by confocal microscopy. As shown in Fig. 5I, the colocalization of internalized NDVs and caveolin-1 was observed. These results indicated that the entry of NDV into chicken macrophages was dependent on caveola-mediated endocytosis.
Entry of NDV into HD11 cells is macropinocytosis independent.
Macropinocytosis has been reported as an alternative way for the entry of some paramyxoviruses (e.g., Nipah virus) in certain cells (13, 14). To examine the potential role of macropinocytosis in the entry of NDV in chicken macrophages, EIPA, a specific inhibitor of macropinocytosis that blocks Na+/H+ exchange, was used to treat HD11 cells. The results of a cellular viability assay showed that HD11 cells tolerated up to 80 μM EIPA (data not shown). Dextran is considered a specific fluid-phase marker of macropinocytosis (14, 48–50); to examine the effect of EIPA treatment on macropinocytosis in HD11 cells, HD11 cells were pretreated with EIPA, and a dextran uptake assay was performed. As shown in Fig. 6A, a significant reduction in the red fluorescence signals of dextran was observed in EIPA-treated cells compared to DMSO-treated cells, indicating the inhibition of dextran uptake via macropinocytosis. Subsequently, the effects of EIPA on the adsorption, internalization, and replication of NDV in HD11 cells were tested as described above. The results of adsorption assay suggested that EIPA had no effect on NDV adsorption (Fig. 6Ba). In addition, results of internalization assay demonstrated no significant difference in the mean fluorescence intensity of DiOC that was resistant to trypan blue between the EIPA-treated and control cells (Fig. 6Bb and c). Also, the mean fluorescence intensities quenched by trypan blue in EIPA-treated and control cells were comparable (Fig. 6Bc). As expected, there was no significant difference in viral titers among cells treated with EIPA and control cells (Fig. 6C). These findings suggested EIPA had no effects on adsorption, endocytic entry, and membrane fusion of NDV at the surface of HD11 cells. Moreover, phosphatidylinositide 3-kinase (PI3K) participates in several stages of macropinocytosis as an essential factor. Wortmannin, a potently inhibitor of PI3K, which can inhibit the uptake of cargo and internalization of virus via macropinocytosis (33, 51, 52), was used to treat HD11 cells. The result of the cellular viability assay showed that HD11 cells tolerated up to 5 μM wortmannin (data not shown), and the dextran uptake assay demonstrated that treatment with wortmannin inhibited the uptake of dextran via macropinocytosis (Fig. 6D). The effects of wortmannin on the adsorption, internalization, and replication of NDV in HD11 cells were examined as described above. Consistent with the results with EIPA, treatment with the PI3K inhibitor demonstrated no significant effects on the adsorption (Fig. 6Ea), endocytic entry (Fig. 6Eb and c), and membrane fusion of NDV at the cell surface (Fig. 6Ec) and on the viral titers of NDV in HD11 cells (Fig. 6F). These findings indicated that the endocytosis of NDV in HD11 cells was not dependent on macropinocytosis.
FIG 6.

Macropinocytosis is not required for NDV entry. (A and D) EIPA and wortmannin inhibited dextran uptake. HD11 cells were treated with 80 μM EIPA, 5 μM wortmannin, or DMSO for 1 h at 37°C, followed by incubation with 10 μg/ml dextran for 1 h at 4°C, and then shifted to 37°C for 1 h; after a wash with low-pH buffer, the cells were fixed and stained with DAPI. (B and E) EIPA and wortmannin did not inhibit NDV adsorption and internalization. HD11 cells were treated with indicated concentration of inhibitors for 1 h at 37°C. The cells were then incubated with NDV F48E9 in the presence of inhibitors for 1 h at 4°C to allow viral adsorption. DMSO was included as a negative control. Viral RNA was quantified by real-time RT-PCR (subpanel a in panels B and E). Alternatively, DiOC-labeled NDV F48E9 was adsorbed onto HD11 cells and then incubated with indicated concentrations of inhibitors for 1 h at 37°C. AN internalization assay was performed as described above. The green fluorescence of DiOC that was resistant and quenched by trypan blue was determined by confocal fluorescence microscopy (subpanel b in panels B and E) and flow cytometry (subpanel c in panels B and E), respectively. (C and F) EIPA and wortmannin did not affect the replication of NDV. HD11 cells were infected with NDV F48E9 at an MOI of 0.1 in the presence of the inhibitors at 37°C for 1 h. DMSO was included as a negative control. The cells were washed with PBS and incubated in medium containing 5% FBS at 37°C. At 18 hpi, the viral titers in the culture supernatants of infected cells were determined. The bars represent means ± the SD. Data were analyzed by using the Student t test (*, P < 0.05; **, P < 0.01).
Rab5, rather than Rab7, is required for NDV endocytosis and subsequent traffic to early endosomes.
Rab proteins play important roles in various aspects of membrane traffic and have been found to be involved in the entry of various viruses. The role of Rab proteins in the NDV entry into host cells was unknown, although it has been reported that NDV can enter cells via caveola-mediated endocytosis and locate to early endosomes (19). Rab5 and Rab7 are known as the key regulators of traffic to early and late endosomes; consequently, we examined the roles of both Rab5 and Rab7 in the entry of NDV into HD11 cells. HD11 cells were transfected with plasmids encoding EGFP-tagged WT and DN Rab5 and of WT and DN Rab7, respectively, and the Tfn uptake assay was performed as a control. As shown in Fig. 7Aa and b, the results of confocal fluorescence microscopy and flow cytometry demonstrated that the mean fluorescence intensities of Tfn in cells overexpressing DN Rab5 and Rab7 were significantly lower than those in cells overexpressing WT Rab5 and Rab7, respectively, which confirmed the functionality of WT and DN constructs of Rab5 and Rab7. Subsequently, an internalization assay using DiI-labeled and unlabeled NDV F48E9 was performed. As shown in Fig. 7B, the mean fluorescence intensities of NDV in cells overexpressing DN Rab5 were significantly lower than in cells overexpressing WT Rab5, whereas the mean fluorescence intensities of NDV were comparable in both kinds of cells, which overexpressed WT and DN Rab7, respectively. These data indicated that the entry of NDV was reduced in cells overexpressing DN Rab5. In addition, no significant difference in the mean fluorescence intensity of NDV HN protein on the cell surface was found between cells overexpressing WT and DN Rab5, as well as between cells overexpressing WT and DN Rab7 (Fig. 7B). These results further indicated that functional Rab5 rather than Rab7 was required for the endocytic entry of NDV, and both Rab5 and Rab7 did not affect the membrane fusion of NDV at the cell surface. To further confirm the roles of Rab5 and Rab7 in NDV entry, siRNA targeting Rab5 (siRab5) and Rab7 (siRab7) were used, respectively. Western blotting demonstrated the efficiency of siRNA-mediated knockdown of Rab5 and Rab7 (Fig. 7Ca), and cell viability was not significantly affected by siRNA transfection (Fig. 7Cb). Subsequently, HD11 cells transfected with siRab5, siRab7, or a control siRNA (siControl) were incubated with DiOC-labeled NDV F48E9 at 37°C for 1 h, and then an internalization assay was performed as described above. As shown in Fig. 7Cc and d, knockdown of the expression of Rab5, rather than Rab7, significantly reduced the mean fluorescence intensity of DiOC that was resistant to trypan blue compared to that in siControl-transfected cells. In addition, no significant change in the mean fluorescence intensities quenched by trypan blue was observed among Rab5, Rab7 knockdown, and siControl-transfected cells (Fig. 7Cd). Moreover, in Rab5-knockdown HD11 cells, the titers of progeny viruses were significantly lower than in siControl- and siRab7-transfected cells (Fig. 7D). In addition, the colocalization of NDV with Rab5 and Rab7 during NDV entry was examined by confocal microscopy. As shown in Fig. 7E, NDV was found to colocalize with Rab5, rather than Rab7, after entering the cells. These results indicated that NDV should be transported to early endosomes rather than late endosomes in a Rab5-dependent manner. Thus, the localization of NDV in endosomes during virus entry was further examined. As shown in Fig. 7F, colocalization of NDV with a marker of early endosomes, EEA1, was observed, whereas no obvious colocalization of NDV with a marker of late endosomes Lamp-1 was found. Collectively, our results demonstrated that Rab5 was required for NDV endocytosis and subsequent traffic to early endosomes and also indicated that NDV fusion should occur in early endosomes.
FIG 7.
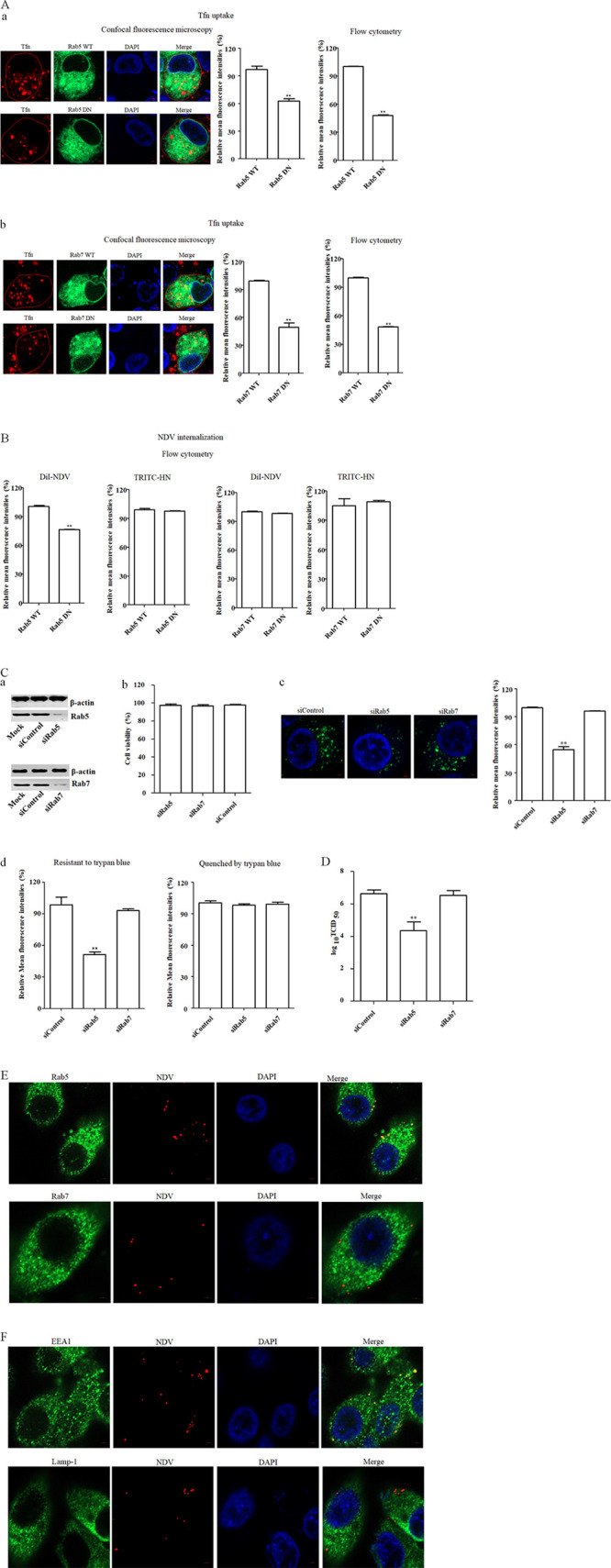
Effects of Rabs on NDV infection. (A and B) Effects of Rab5 and Rab7 on Tfn uptake (A) and NDV internalization (B). HD11 cells transfected with plasmids expressing EGFP-tagged WT or DN Rab5 and Rab7 were incubated with 10 μg/ml Tfn, and DiI-labeled or unlabeled NDV F48E9 for 1 h at 4°C and then shifted to 37°C for 1 h. After a wash with low-pH buffer, the red fluorescence of Tfn (subpanels a and b in panel A), DiI-labeled NDV (B), and HN protein (B) on the cell surface was determined as described above. (C) Rab5 knockdown reduced the internalization of NDV. HD11 cells were transfected with siRNA targeting Rab 5 (siRab5), Rab7 (siRab7), or siControl. The effect of siRNA knockdown of Rab5 and Rab7 expression was determined by Western blotting (a). Cell viability upon siRab5, siRab7, and siControl transfection was assessed, as described in the text (b). At 48 h posttransfection, an internalization assay was performed with DiOC-labeled NDV F48E9 as described above. The green fluorescence of DiOC that was resistant and quenched by trypan blue was determined by confocal fluorescence microscopy (c) and flow cytometry (d), respectively. (D) Rab 5 knockdown reduced the replication of NDV. HD11 cells were transfected with siRab5, siRab7, or siControl. At 48 h posttransfection, the cells were infected with NDV F48E9 at an MOI of 0.1, and the viral titers in the culture supernatants of infected cells were determined at 18 hpi. (E) Colocalization of NDV with Rab5. HD11 cells were incubated with NDV F48E9 at an MOI of 10 at 4°C for 1 h and then shifted to 37°C for 1 h. Cells were then fixed and reacted with anti-Rab5 antibody, anti-Rab7 antibody, and NDV HN antibody, respectively, and visualized by confocal microscopy. (F) Colocalization of NDV with the early endosome marker EEA1. HD11 cells were incubated with NDV F48E9 at an MOI of 10 at 4°C for 1 h and then shifted to 37°C for 1 h. The cells were fixed and reacted with anti-EEA1 antibody, anti-Lamp-1 antibody, and NDV HN antibody, respectively, and visualized by confocal microscopy. The bars represent means ± the SD. Data were analyzed by using the Student t test (*, P < 0.05; **, P < 0.01).
The caveola-mediated endocytosis is utilized by NDV to enter primary chicken macrophages.
To investigate whether caveola-mediated endocytosis was utilized by NDV to enter primary chicken macrophages, the highest concentration of inhibitors without causing obvious cytotoxic effects were used (data not shown), and the effects of these inhibitors on replication of NDV in primary chicken macrophages were determined. As shown in Fig. 8, the progeny viral titers in bafilomycin A1-, chloroquine-, dynasore-, MβCD-, nystatin-, and PMA-treated cells were significantly lower than in control cells, while the progeny viral titers in chlorpromazine-, EIPA-, and wortmannin-treated cells were comparable to titers measured in control cells. These results suggested that, identical to the results observed in HD11cells, NDV utilizes a pH-dependent, dynamin and caveola-mediated endocytosis for entry into primary chicken macrophages.
FIG 8.
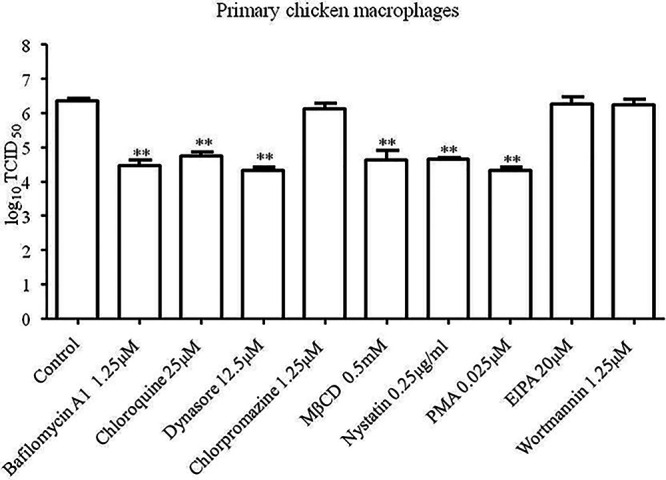
NDV utilizes caveola-mediated endocytosis to enter primary chicken macrophages. Primary chicken macrophages cultured in 24-well plates were treated with the highest concentrations of bafilomycin A1, chloroquine, dynasore, chlorpromazine, MβCD, nystatin, PMA, EIPA, and wortmannin without causing obvious cytotoxic effects for 1 h at 37°C. Cells treated with DMSO were included as a control. The cells were infected with F48E9 at an MOI of 0.1 in the presence of the inhibitors at 37°C for 1 h. After incubation, the cells were washed with PBS and incubated in medium containing 5% FBS at 37°C. At 24 hpi, the viral titers in the culture supernatants of infected cells were determined. The bars represent means ± the SD. Data were analyzed by using the Student t test (*, P < 0.05; **, P < 0.01).
Pathway utilized by NDV to enter other chicken origin cells.
A virus may exploit distinct pathways for entry into the different cell types. We further evaluated the pathways exploited by NDV to enter other chicken origin cells (DF-1 and LMH). The highest concentrations of inhibitors without causing obvious cytotoxic effects were used (data not shown), and the effects of these inhibitors on replication of NDV in DF-1 and LMH cells were determined. As shown in Fig. 9A, for DF-1 cells, the progeny viral titers in bafilomycin A1-, chloroquine-, dynasore-, and chlorpromazine-treated cells were significantly lower than titers in control cells, while the progeny viral titers in MβCD-, nystatin-, and PMA-treated cells were comparable to the titers in control cells. In addition, compared to the control cells, treatment of EIPA and wortmannin also reduced the progeny viral titers. These results indicated that different from the entry pathway in HD11 cells, NDV can enter DF-1 cells via a pH-dependent, dynamin and clathrin-mediated endocytosis pathway. In addition, macropinocytosis is also involved in the entry of NDV in DF-1 cells. These findings are in agreement with a previous report (21). In the case of LMH cells, the progeny viral titers in bafilomycin A1-, chloroquine-, dynasore-, MβCD-, nystatin-, and PMA-treated cells were significantly lower than the titers in control cells, while the progeny viral titers in chlorpromazine-, EIPA-, and wortmannin-treated cells were comparable to titers in control cells (Fig. 9B). These results indicated that similar to the entry pathway in HD11 cells, NDV entry into LMH cells was dependent on acidic pH, dynamin, and caveolae but not clathrin or macropinocytosis. Taken together, our results suggest that NDV may enter different cell types using different pathways and strategies.
FIG 9.
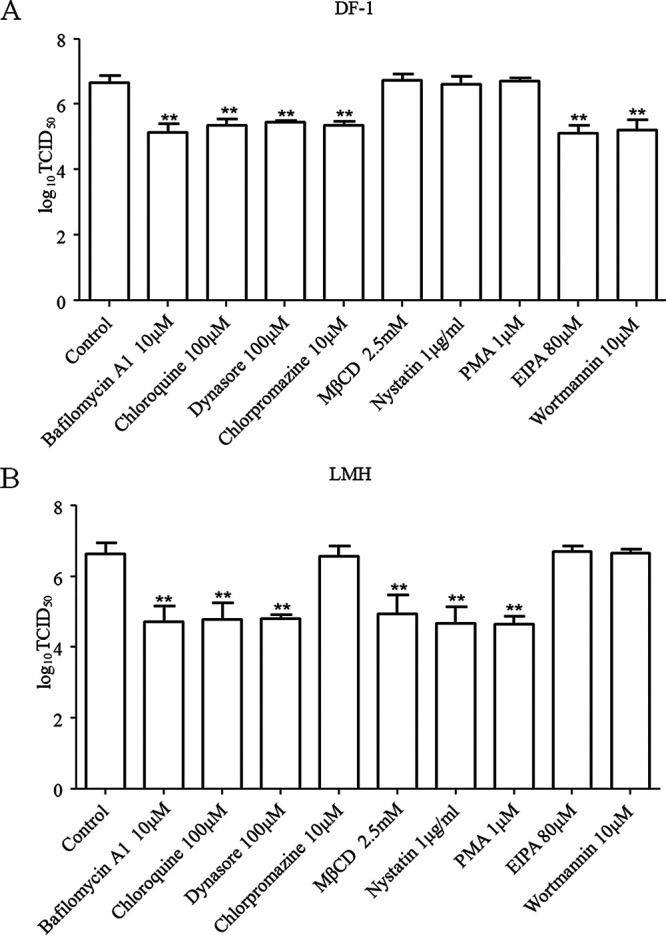
Effects of inhibitors on the replication of NDV in DF-1 (A) and LMH (B) cells. DF-1 and LMH cells cultured in 12-well plates were treated with the highest concentrations of bafilomycin A1, chloroquine, dynasore, chlorpromazine, MβCD, nystatin, PMA, EIPA, and wortmannin without causing obvious cytotoxic effects for 1 h at 37°C. Cells treated with DMSO were included as a control. The cells were infected with F48E9 at an MOI of 0.1 in the presence of the inhibitors at 37°C for 1 h. After incubation, the cells were washed with PBS and incubated again in medium containing 5% FBS at 37°C. At 18 hpi, the viral titers in the culture supernatants of infected DF-1 and LMH cells were determined. The bars represent means ± the SD. Data were analyzed by using the Student t test (*, P < 0.05; **, P < 0.01).
DISCUSSION
In order to establish infection, viruses can employ various strategies for entry into host cells. For enveloped paramyxovirus, complex mechanisms of cell entry have been identified (9, 10). In addition to entering target cells through direct penetration at the plasma membrane, some members of the Paramyxoviridae family can hijack the cellular intrinsic endocytic mechanisms to achieve their infectious entry into host cells (16, 19, 53). Previous studies have shown that NDV can infect multiple cell types and utilize both direct fusion with the plasma membrane and endocytic pathways for entry into host cells (18–21, 54). Although NDV is able to infect and replicate in chicken macrophages (22–24), one of the main target cells for NDV in vivo, little information is available about the invasion pathway of NDV for entering chicken macrophages.
In this study, we initially demonstrated that chIFITM1 which is located in the early endosomes can specifically limit the replication of NDV in chicken macrophages (HD11), indicating the endocytic entry of NDV in this cell type. Since we showed that chIFITM1 has no effects on the adsorption and internalization of NDV in HD11 cells but reduces the replication of NDV, these findings suggest that this host restriction factor should restrict NDV entry by blocking the fusion of viral envelop with endosomal membrane as previously reported (28). Indeed, we showed that the internalization of NDV in HD11 cells is sensitive to the treatment of inhibitors of endosomal acidification. The pH-dependent entry further indicates that NDV can be internalized in HD11 cells via endocytosis. Then, we found that dynamin is required in the entry of NDV into HD11 cells, indicating the potential roles of clathrin-mediated and caveola-mediated endocytosis in the entry of NDV. Further results showed that the entry of NDV in HD11 cells is dependent on caveola-mediated endocytosis rather than clathrin-mediated endocytosis. Recent evidences reveal that, except for entry at the plasma membrane, paramyxovirus is also able to utilize the endocytosis pathways for entry. The clathrin-mediated endocytosis and macropinocytosis have been implicated in the entry of HMPV and Nipah virus, respectively (9, 10, 14). Based on our results and a previous report (19), NDV is the only member of paramyxoviruses that known thus far that utilizes caveola-mediated endocytosis for entry into chicken macrophages. The caveola-mediated endocytosis is ligand triggered, and the formation of primary endocytic vesicles depends on cholesterol, lipid rafts, dynamin II, and a complex signaling pathway associated with the activation of several tyrosine and other kinases (55). Cholesterol is an important component of lipid rafts and is essential for the formation of caveolae. As such, the sensitivity to cholesterol depletion is a characteristic of caveola-mediated endocytosis (12, 55). Here, we showed that treatment with cholesterol depletion reagent MβCD and cholesterol binding reagent nystatin significantly reduced the level of NDV internalization, suggesting that membrane cholesterol is required for NDV internalization in HD11 cells. In addition, we found that except for the internalization, the adsorption of NDV in cholesterol-depleted HD11 cells treated with MβCD was also significantly reduced. The possible explanation is that the treatment of MβCD may disrupt the integrity of cholesterol-rich membrane microdomains which contain potential NDV receptors and consequently interfere with the attachment of NDV to receptor molecules. Previous studies have shown the cholesterol dependence of NDV entry into avian and mammalian cells (19, 56, 57); therefore, membrane cholesterol should be a requirement for NDV entry to host cells. Although cholesterol dependence is considered to be a marker of the caveola-mediated endocytosis (58), membrane cholesterol is also involved in other endocytic pathways, including clathrin-mediated endocytosis, macropinocytosis, lipid rafts, and phagocytosis (12). Thus, three criteria were used in this study to further confirm our results: (i) the internalization of NDV was significantly reduced in HD11 cells treated with PMA, which is a specific inhibitor of caveola-mediated endocytosis; (ii) the entry of NDV was reduced by the overexpression of DN caveolin-1 and knockdown of the expression of caveolin-1 in HD11 cells; and (iii) NDV was colocalized with caveolin-1 during entry into HD11 cells. Collectively, our results clearly indicate that NDV entry into HD11 cells is dependent on caveola-mediated endocytosis. Moreover, we demonstrated that this endocytic pathway is also utilized by NDV entering the primary chicken macrophages.
Macropinocytosis is another endocytosis pathway involved in the entry of several viruses (33, 59, 60). Nipah virus is reported to utilize the macropinocytosis pathway for entry into cells (14). In this study, the adsorption, the internalization, and the titers of progeny viruses of NDV in HD11 cells were not obviously affected by the treatment of inhibitors of macropinocytosis. Previous work indicated that the infection of NDV in BHK-21 cells is moderately reduced by pretreatment with MβCD, while the inhibitor of macropinocytosis has no inhibitory effect on the infectivity of NDV (61). Here, we demonstrated that NDV entry into HD11 cells is dependent on acidic pH, dynamin, cholesterol, and caveolin but not clathrin or macropinocytosis. Similar results were observed in LMH cells. However, the dependence differs between HD11 and DF-1 cells; NDV could enter DF-1 cells via a pH-dependent, dynamin- and clathrin-mediated endocytosis pathway, and macropinocytosis is involved in the entry of NDV in DF-1 cells. These results suggest that NDV may enter different cell types using different pathways and strategies. Most paramyxoviruses enter host cells in a receptor-dependent manner. Sialic acid-containing compounds, such as gangliosides and N-glycoproteins, have been suggested as the receptors of NDV (62, 63). In addition, previous studies showed that gangliosides were the primary receptors of NDV, which is responsible for the initial virus attachment, whereas N-linked glycoproteins acted as the authentic receptors which induce the conformational changes of envelope glycoprotein and subsequently the fusion of viral and cellular membranes (64, 65). Thus, during the entry process, NDV should bind to gangliosides to enhance virus adsorption onto cells, and this initial attachment may not induce the fusion between the viral and cellular membranes as previously reported (66). In contrast, the NDV-ganglioside interaction may mediate the activation of certain cellular signaling pathways that are involved in the endocytosis. A prior study indicated that mouse polyomavirus activates the PI3K pathway, which is required for the virus endocytosis through binding to the ganglioside GD1a receptor (67). Ganglioside GM2 is essential for the cell entry of Bombyx mori cypovirus via clathrin-mediated endocytosis pathway (68), and the specific interaction with a peptide and ganglioside GM3 is required for its entry via the macropinocytosis (69). Notably, ganglioside GM1, the receptor of simian virus 40, is concentrated in caveolae and mediates the entry of virus via caveola-mediated endocytosis (70–72). Since NDV can bind to ganglioside GM1 (65) and pretreatment of NDV with ganglioside GM1 prior to exposure to HD11 cells significantly inhibited NDV infection (our unpublished data), we speculated that during the entry process of NDV in HD11 and LMH cells, NDV may bind to ganglioside GM1 located in the caveolae and activate the tyrosine kinase signaling cascade and eventually induce the caveola-mediated endocytosis (44, 73). Moreover, the types of gangliosides in DF-1 cells may differ to those in HD11 and LMH cells, since the expression patterns of gangliosides are varied in different cells (e.g., the expression of ganglioside GM3 is more abundant than GM1 in many nonneuronal tissues) (74, 75). In addition, previous studies have shown that murine polyomavirus can utilize different gangliosides as receptors for entry into different cell types (72, 76); therefore, we infer that the entry of NDV in DF-1 cells via the clathrin-mediated endocytosis pathway and macropinocytosis may be mediated by the interaction of NDV with specific types of ganglioside, rather than GM1, although this required further elucidation.
Caveola-mediated endocytosis pathway is involved in the internalization of membrane components, extracellular ligands, bacterial toxins, and various other viruses (37, 38, 44). Traditionally, the internalized cargos are transported to caveosome with neutral pH and further sorted to the endoplasmic reticulum, bypassing the acidic endosomes (55, 77). Viruses that use this pathway for cell entry can exploit the components of the endoplasmic reticulum for uncoating and penetration into the cytosol (12, 78). In addition to this caveosome-endoplasmic reticulum route, an increasing number of viruses that enter cells via caveola-mediated endocytosis pathway have been found to be able to be transported to acidic endosomes (37, 79, 80). The fusion of viral envelope with endosomal membranes is generally triggered by the acidic pH within the endosomal pathway (11). Though paramyxoviruses are believed to promote fusion under neutral pH conditions, the fusion of HMPV and NDV is enhanced at acidic pH, suggesting the requirement of a low pH for virus entry (16, 54). Here, we demonstrated that NDV can enter chicken microphages via the caveola-mediated endocytosis in a pH-dependent manner and are located in early endosomes (with a relatively high [∼6] pH), rather than late endosomes (with a relatively lower [∼5] pH). The relatively high pH dependency of NDV and the location of internalized NDV in early endosomes indicate that the fusion of NDV might occur in early endosomes, and the acidic pH condition in early endosomes might be sufficient to trigger the fusion of NDV with endosomes, as is the case for Semliki Forest virus and vesicular stomatitis virus (81, 82). In addition, upon binding to the ganglioside receptor, the NDV HN protein undergoes only certain conformational change that does not affect the F protein (64). Thus, we speculated that after the endocytic entry, conformational changes of the HN protein are required for fusion promotion and may be further triggered by the acidic pH condition in the early endosomes. Alternatively, F protein may be activated by the acidic pH without the requirement of an interaction with HN. Further investigation, such as the construction of HN mutant recombinant viruses deficient in interaction with the F protein, will be needed to clarify this point.
The viruses that enter host cells via the endocytosis pathway can hijack the Rab proteins and the small GTPases to achieve their traffic and localizations to specific subcellular compartments (12, 83). The small GTPases Rab5 and Rab7 are key determinants of transport to early and late endosomes and are involved in the cellular entry of several viruses (36, 37, 84, 85). In the present study, the effects of Rab5 and Rab7 on NDV entry into HD11 cells were evaluated by overexpressing DN mutants and knocking down the expression of Rab5 and Rab7 using siRNA. Our results indicated that Rab5 is implicated in the entry of NDV in HD11 cells, whereas the expression of Rab7 is not required. Colocalization of virions with Rab5 and EEA1 (early endosome markers), rather than Rab7 and Lamp-1 (late endosome markers), indicated that NDV is located in early endosomes. Thus, we hypothesize that after endocytosis, NDV is transported to early endosomes in a Rab5-dependent manner and the virus uncoating occurs at the early endosomes as reported for dengue virus, West Nile virus, and respiratory syncytial virus (15, 85).
In conclusion, we comprehensively investigated for the first time, the entry process of NDV in chicken macrophage cell line HD11. Our results suggest that NDV enters HD11 cells via a pH-dependent, dynamin and caveola-mediated endocytosis pathway and Rab5 is involved in NDV entry into HD11 cells. Notably, we further demonstrated that this endocytosis pathway is also utilized by NDV for entry into primary chicken macrophages, indicating the endocytic entry of NDV in vivo. Entry and replication of NDV in macrophages is a benefit for the infection and dissemination of virus in vivo and can alter the host immune responses to support the viral infection. In addition, by being endocytosed into endosomes, NDV could avoid leaving the viral components exposed on the plasma membrane, and thus delay the detection of host immunosurveillance. Clarification of the entry pathways of NDV will promote our understanding the cellular entry mechanism of paramyxoviruses and provide insight into the development of novel antiviral inhibitors.
MATERIALS AND METHODS
Cells, eggs, and viruses.
Chicken macrophage cell line HD11 was cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS). Chicken embryo fibroblast cells (DF-1) and chicken hepatoma cells (LMH) were cultured in Dulbecco modified Eagle medium (Sigma, Shanghai, China) supplemented with 10% FBS. The primary chicken macrophages were obtained from peripheral blood of specific-pathogen-free (SPF) chickens as described previously, with some modifications (22). The peripheral blood prepared from SPF chickens was layered on Ficoll (Sigma) and centrifuged at 1,000 × g for 20 min. Cells were removed from the Ficoll gradient and washed three times using phosphate-buffered saline (PBS). The obtained cells were resuspended in RPMI 1640 medium (Sigma) supplemented with 10% FBS and cultured in 24-well plates. Next, the nonadherent lymphocytes were removed, and adherent macrophages were reserved. SPF embryonated chicken eggs were obtained from the National Poultry Laboratory Animal Resource Center, Harbin Veterinary Research Institute, China. The genotype IX velogenic NDV strain F48E9 was used in this study (86, 87).
Plasmids and siRNAs.
The coding region removing the termination codon of chIFITM1 was amplified from RNA extracted from HD11 cells. The resulting PCR product was subcloned downstream of the HA tag in the pCAGGS-HA plasmid. The recombinant plasmid encoding HA-tagged chIFITM1 protein was designated pCAGGS-HA-chIFITM1. The coding regions removing the termination codons of wild-type (WT) caveolin-1, dynamin II, EPS15, Rab5, and Rab7 were amplified from the HD11 cells RNA and cloned into the pEGFP-N1 vector (Clontech, Palo Alto, CA). Dominant-negative (DN) caveolin-1 was constructed by removing the sequence coding the first 81 amino acids of caveolin-1 and replacing it with a start codon, as described previously (46, 47). The DN EPS15, which has a large N-terminal deletion, was constructed, as previously described (27, 36, 42, 43). DN dynamin II (K44A), DN Rab5 (S34N), and DN Rab7 (T22N) were constructed via site-directed mutagenesis using specific primers, as described previously (36, 37, 87). All constructs were sequenced to confirm the presence of these introduced mutations. The plasmids were transfected into HD11 cells using the TurboFect transfection reagent (Thermo Fisher Scientific, Waltham, MA), as we described previously (87).
The siRNA that specifically recognizes the mRNA sequence of caveolin-1 (5′-CCGAGCCACAGUACGGAAA-3′), clathrin heavy chain (5′-GCAUCAACCCAGCAAACAU-3′), dynamin II (5′-CCAUCGGUGUGAUCACCAA-3′), Rab5 (5′-GCAAGCAGCUAUAGUUGUA-3′), and Rab7 (5′-CCAGUAUGUGAACAAGAAA-3′) were designed, and a control siRNA that does not have a specific target site in chicken cells was used (87). HD11 cells were transfected with 100 nM siRNA target caveolin-1, clathrin heavy chain, dynamin II, Rab5, Rab7, or a control siRNA using N-TER nanoparticle siRNA transfection system (Sigma), as we described previously (87).
Inhibitors.
The inhibitors used in this study included bafilomycin A1, a specific inhibitor of vacuolar (V-type) proton ATPases (APExBIO, Houston, TX); chloroquine, an inhibitor of endosomal acidification (Sigma);, dynasore, an inhibitor of dynamin, which involved in both clathrin- and caveola-mediated endocytosis (Sigma); chlorpromazine, an inhibitor of clathrin-mediated endocytosis (Sigma); methyl-β-cyclodextrin (MβCD), an agent disrupts lipid rafts by depleting the cholesterol component (Sigma); nystatin, a steroid-binding antibiotic that binds cholesterol, used as an inhibitor of caveola-mediated endocytosis (Sigma); phorbol 12-myristate 13-acetate (PMA), an inhibitor of caveola-mediated endocytosis (Sigma); 5-(N-methyl-N-isobutyl)amiloride (EIPA), an inhibitor of macropinocytosis (Sigma); and wortmannin, an inhibitor of PI3K activation (Merck, Brandenburg, NJ).
Cell viability assay.
An evaluation of the cytotoxic effects of the inhibitors and siRNA on HD11 cells and the cytotoxic effects of the inhibitors on DF-1, LMH cells and primary chicken macrophages was made using a CellTiter 96 AQueous One Solution cell proliferation assay kit (Promega, Madison, WI) according to the manufacturer’s instructions. Briefly, HD11, DF-1, and LMH cells and primary chicken macrophages grown in 96-well plates were incubated with various concentrations of inhibitors for 2 h. After incubation, the inhibitors were removed by three washes with PBS. Next, the cells were supplemented with medium containing 5% FBS, followed by incubation at 37°C for 24 h. Alternatively, HD11 cells were transfected with siRNAs and incubated at 37°C for 48 h. Subsequently, 20 μl of CellTiter 96 AQueous One Solution reagent was added to the cells, followed by incubation for 4 h at 37°C, and the absorbance was measured at a wavelength of 490 nm. The highest concentration of each inhibitor with no significant effect on cell viability (>90%) was used for further study (data not shown).
DiOC/DiI labeling of virus.
The NDV F48E9 was purified by sucrose density gradient centrifugation. The purified virus was incubated with 10 μM DiOC (green) or DiI (red) fluorescence (Thermo Fisher Scientific) with gentle shaking for 30 min at room temperature, respectively. Upon completion of labeling, unincorporated dye was removed with an Amicon Ultra-15 centrifugal filters (10 kDa; Merck Millipore, Cork, Ireland) by centrifugation for 45 min. The titer of the labeled virus was determined in HD11 cells.
Adsorption, internalization, and replication assays.
For the adsorption assays, HD11 cells cultured in 12-well plates were treated with the indicated concentration of inhibitors for 1 h at 37°C. After three washes with cold PBS, the cells were inoculated with NDV F48E9 at a multiplicity of infection (MOI) of 1 in the presence of the inhibitors at 4°C for 1 h. After the incubation period, the inoculum was removed, and the cells were washed three times with cold PBS. The cells were then harvested and subjected to viral RNA extraction and quantification of viral load via real-time RT-PCR, as previously described (87, 88). Briefly, viral RNA was extracted from NDV-infected HD11 cells using TRIzol reagent (Invitrogen, Carlsbad, CA). Specific primers and TaqMan probes against NDV were selected for the detection of NDV (88). Real-time RT-PCR was performed on the LightCycler 480 real-time PCR system (Roche Diagnostics Deutschland GmbH, Mannheim, Germany) with a One-Step PrimeScript RT-PCR kit (TaKaRa, Dalian, China) according to the manufacturers’ instructions. Triplicate reactions were performed for each sample. Standard templates with known copy numbers were included in the quantitative analyses, and H2O was used as the negative template control. Samples with a threshold cycle (CT) value above 35 were considered to be negative, and the detection limit of this assay is 102 genome copies of NDV was ∼101 50% egg infective doses (88).
For the internalization assays, HD11 cells were treated with the indicated concentration of inhibitors for 1 h at 37°C. After three washes with cold PBS, the cells were inoculated with DiOC-labeled NDV F48E9 at an MOI of 50 at 4°C for 1 h. The inoculum was then removed, and the cells were washed three times with cold PBS. Cells were further incubated with medium in the presence of the inhibitors for 1 h at 37°C. Alternatively, HD11 cells were transfected with siRNAs or hemagglutinin (HA)-tagged plasmids. At 48 h posttransfection, the cells were inoculated with DiOC-labeled NDV F48E9 at an MOI of 50 at 4°C for 1 h. Then, the inoculum was removed, and the cells were washed three times with cold PBS. Cells were further incubated with medium for 1 h at 37°C. In addition, HD11 cells were transfected with plasmids that encode EGFP-tagged proteins. At 48 h posttransfection, the cells were inoculated with DiI-labeled or unlabeled NDV F48E9 at an MOI of 50 at 4°C for 1 h. The inoculum was removed, and the cells were washed three times with cold PBS. The cells were further incubated with medium for 1 h at 37°C. For all of the experiments, the cells were washed three times with PBS and subsequently treated with citrate buffer (pH 3) after incubation at 37°C to inactivate and remove noninternalized viruses, as previously described (26, 27). After a wash with PBS, the cells were fixed in 4% paraformaldehyde and subjected either to visualization by confocal fluorescence microscopy or to flow cytometric analysis.
For the replication experiments, after treatment with the indicated concentration of inhibitors, the cells were inoculated with NDV F48E9 at an MOI of 0.1 in the presence of the inhibitors at 37°C for 1 h. Alternatively, HD11 cells were transfected with siRNAs or plasmids. At 48 h posttransfection, cells were infected with F48E9 at an MOI of 0.1 at 37°C for 1 h. After the incubation period, the cells were washed with PBS, followed by incubation in medium containing 5% FBS at 37°C. At 18 hpi, viral titers in the culture supernatants of infected cells were determined by the Reed-Muench method, as previously described (87, 89).
Uptake assay.
Alexa Fluor 568-labeled human transferrin (Tfn), Alexa Fluor 594-labeled cholera toxin subunit B (CTB), and Alexa Fluor 594-labeled dextran (Thermo Fisher Scientific) were used for the uptake assays, as previously described (36, 37, 90). HD11 cells cultured in 30-mm cell culture dishes were treated with the indicated inhibitors for 1 h at 37°C. The cells were incubated with 10 μg/ml of Tfn, CTB, or dextran at 4°C for 1 h and then washed and incubated at 37°C for 1 h. After incubation at 37°C, the cells were treated with citrate buffer (pH 3) to remove noninternalized proteins as previously described (26, 27). After a wash with PBS, the cells were fixed with 4% paraformaldehyde, stained with DAPI (4′,6′-diamidino-2-phenylindole) for 10 min, and then observed by confocal fluorescence microscopy. Alternatively, HD11 cells were transfected with WT or DN plasmids. At 48 h posttransfection, the cells were incubated with 10 μg/ml of Tfn or CTB at 4°C for 1 h and then washed and incubated at 37°C for 1 h. After incubation at 37°C, the cells were treated with citrate buffer (pH 3) to remove noninternalized proteins as previously described (26, 27). The cells were then washed with PBS, fixed in 4% paraformaldehyde, and subjected either to visualization by confocal fluorescence microscopy or to flow cytometry for analysis of Tfn or CTB uptake in EGFP-positive cells as previously reported (27).
Confocal fluorescence microscopy.
For the uptake assay involving treatment of inhibitors, the mean fluorescence intensity of labeling proteins within the cells from different fields was determined. For the uptake assay involving EGFP-tagged proteins, the mean fluorescence intensity of labeling proteins within the EGFP-positive cells from different fields was determined. For the virus internalization assay involving treatment of inhibitors and siRNA, the fixed cells were treated with trypan blue with a final concentration of 0.02% to quench the green fluorescence of DiOC in the plasma membrane surface, as previously described (13). The cells were washed with PBS, stained with DAPI, and examined by confocal fluorescence microscopy, and the mean fluorescence intensity of DiOC-labeled virions within the cells from different fields was determined. All images associated with uptake of labeling proteins and virus internalization were acquired by using LSM800-ZEISS confocal laser scanning microscopy (LSM 800; Zeiss, Oberkochen, Germany). The mean fluorescence intensity of a single plane from z-stack cross sections was used to determine the labeling proteins within the cells.
For the localization of HA-chIFITM1, HD11 cells cultured in 30-mm cell dishes were transfected with pCAGGS-HA-chIFITM1 or pCAGGS-HA. At 24 h after transfection, the cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100. The cells were incubated with mouse anti-HA and rabbit anti-EEA1 antibody (Abcam, Cambridge, UK) overnight at 4°C and then with anti-mouse IgG-TRITC antibody and anti-rabbit IgG FITC antibody (Sigma). After a wash with PBS, the cells were stained with DAPI and examined by confocal fluorescence microscopy.
For the localization of NDV, HD11 cells cultured in 30-mm cell dishes were inoculated with NDV F48E9 at an MOI of 10 at 4°C for 1 h and then washed and incubated at 37°C for 1 h. Infected cells were then fixed and permeabilized, as described above. For colocalization of NDV with caveolin-1, cells were stained with chicken anti-NDV HN antibody (87) and mouse anti-caveolin-1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA). For colocalization of NDV with Rab5, Rab7, EEA1, and Lamp-1, cells were stained with mouse anti-NDV HN antibody, rabbit anti-Rab 5 antibody (LifeSpan BioSciences, Seattle, WA), rabbit anti-Rab 7 antibody (Thermo Fisher Scientific), rabbit anti-EEA1 antibody (Abcam), or rabbit anti-Lamp-1 antibody (Abcam), respectively, and examined by confocal fluorescence microscopy.
Flow cytometry.
For the virus internalization assay, HD11 cells cultured in 6-well plates were inoculated with DiOC-labeled NDV F48E9 at an MOI of 50 at 4°C for 1 h. The inoculum was removed, and the cells were washed three times with cold PBS to remove unbound viruses. Cells were further incubated with medium at 37°C for 30, 60, 90, and 120 min, respectively. After the desired times, the cells were washed three times with PBS and treated with citrate buffer (pH 3) to inactivate and remove noninternalized viruses, as previously described (26, 27). After three washes with PBS, the cells were collected from the well with 0.25% trypsin and fixed in 4% paraformaldehyde. Each cell sample was divided into two. One sample was subjected directly for the flow cytometric analysis, and the other was treated with trypan blue with a final concentration of 0.02% to quench the green fluorescence of DiOC in the plasma membrane surface, as previously described (13). The green fluorescence of 10,000 cells was measured with a Cytomics TM FC 500 flow cytometer (Beckman Coulter, Fullerton, CA). After incubation, the virions that had not entered the cells via either membrane fusion at the cell surface or endocytosis were removed by washing with low-pH buffer, as previously described (26, 27); thus, the fluorescence intensity of DiOC that was resistant to trypan blue treatment could be used for the determination of endocytosed virus, and the fluorescence intensity of DiOC quenched by trypan blue could be an indicator of membrane fusion of NDV at the cell surface.
For the virus internalization assay involving treatment of inhibitors or siRNA, each fixed cell sample was divided into two. One sample was subjected directly for the flow cytometric analysis, and another was treated with trypan blue as described above; the green fluorescence of 10,000 cells was measured with a Cytomics TM FC 500. For virus internalization assay involving HA-chIFITM1, after fixation and treatment with trypan blue as described above, the cells were stained with HA monoclonal antibody and incubated at 4°C for 1 h. The cells were washed three times with PBS and stained with anti-mouse IgG-TRITC/antibody for 1 h at 4°C. The cells were washed three times with PBS and resuspended in PBS with 1% BSA. The green fluorescence of 10,000 TRITC-positive cells was measured with a Cytomics TM FC 500. For virus internalization assay involving EGFP-tagged proteins, the cells inoculated with DiI-labeled NDV F48E9 were subjected directly to flow cytometric analysis, whereas the cells inoculated with unlabeled NDV F48E9 were stained with NDV HN monoclonal antibody and incubated at 4°C for 1 h. The cells were washed three times with PBS and stained with anti-mouse IgG-TRITC/antibody for 1 h at 4°C. The cells were then washed another three times with PBS and resuspended in PBS with 1% BSA. The red fluorescence of 10,000 EGFP-positive cells was measured with an MA900 flow cell sorter (Sony Biotechnology, San Jose, CA). The fluorescence intensity of DiI-labeled NDV F48E9 was used to quantify the virions entrance into cells via both membrane fusion at the cell surface and the endocytic pathway, whereas the fluorescence intensity of HN on the cell surface was adopted to evaluate the level of membrane fusion at the cell surface.
For the uptake assay of labeling proteins involving EGFP-tagged proteins, the fixed cells were subjected directly for the flow cytometric analysis, and the red fluorescence of 10,000 EGFP-positive cells was measured with an MA900 flow cell sorter.
SDS-PAGE and Western blotting.
SDS-PAGE and Western blotting were performed according to previously described procedures (87). HD11 cells were washed with PBS and lysed with radioimmunoprecipitation assay buffer (Beyotime, Shanghai, China). Protein samples were then separated by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were incubated with the indicated primary antibodies and the corresponding secondary antibodies; β-actin was included as a control.
Effects of inhibitors on the replication of NDV in primary chicken macrophages.
The cytotoxic effects of the inhibitors on primary chicken macrophages were first evaluated as described above. Primary chicken macrophages were treated with the highest concentrations of bafilomycin A1, chloroquine, dynasore, chlorpromazine, MβCD, nystatin, PMA, EIPA, and wortmannin without causing obvious cytotoxic effects for 1 h at 37°C. Cells treated with DMSO were included as a control. Primary chicken macrophages were then infected with F48E9 at an MOI of 0.1 in the presence of the inhibitors at 37°C for 1 h. After incubation, the cells were washed with PBS and incubated in medium containing 5% FBS at 37°C. At 24 hpi, viral titers in the culture supernatants of infected cells were determined by the Reed-Muench method (89).
Effects of inhibitors on the replication of NDV in other chicken origin cells.
The cytotoxic effects of the inhibitors on DF-1 and LMH cells were first evaluated, and then the DF-1 and LMH cells cultured in 12-well plates were treated with the highest concentrations of bafilomycin A1, chloroquine, dynasore, chlorpromazine, MβCD, nystatin, PMA, EIPA, and wortmannin without causing obvious cytotoxic effects for 1 h at 37°C. Cells treated with DMSO were included as a control. The DF-1 and LMH cells were then infected with F48E9 at an MOI of 0.1 in the presence of the inhibitors at 37°C for 1 h. After incubation, the cells were washed with PBS and incubated in medium containing 5% FBS at 37°C. At 18 hpi, viral titers in the culture supernatants of infected cells were determined by the Reed-Muench method (89).
Statistical analysis.
Data are expressed as means ± the standard deviations (SD). A Student t test was performed, where appropriate, using Prism for Windows 5 (GraphPad Software, La Jolla, CA). The differences were considered significant if the P values were <0.05. Statistical significance is indicated by asterisks in the figures (*, P < 0.05; **, P < 0.01).
ACKNOWLEDGMENTS
This study was supported by a National Natural Science Foundation of China grant (31872504), the National Key Research and Development Program of China (2018YFD0501302), the China Agriculture Research System (CARS-40-K18), and the National “Twelfth Five Year” Plan for Science and Technology Support (2015BAD12803).
Contributor Information
Junfeng Sun, Email: sunjunfeng@caas.cn.
Shengwang Liu, Email: liushengwang@caas.cn.
Rebecca Ellis Dutch, University of Kentucky College of Medicine.
REFERENCES
- 1.Alexander DJ. 2009. Ecology and epidemiology of Newcastle disease, p 19–26. In Capua I, Alexander DJ (ed), Avian influenza and Newcastle disease. Springer, New York, NY. [Google Scholar]
- 2.Ganar K, Das M, Sinha S, Kumar S. 2014. Newcastle disease virus: current status and our understanding. Virus Res 184:71–81. 10.1016/j.virusres.2014.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alexander DJ, Aldous EW, Fuller CM. 2012. The long view: a selective review of 40 years of Newcastle disease research. Avian Pathol 41:329–335. 10.1080/03079457.2012.697991. [DOI] [PubMed] [Google Scholar]
- 4.Cox RM, Plemper RK. 2017. Structure and organization of paramyxovirus particles. Curr Opin Virol 24:105–114. 10.1016/j.coviro.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.El Najjar F, Schmitt AP, Dutch RE. 2014. Paramyxovirus glycoprotein incorporation, assembly and budding: a three-way dance for infectious particle production. Viruses 6:3019–3054. 10.3390/v6083019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Noton SL, Fearns R. 2015. Initiation and regulation of paramyxovirus transcription and replication. Virology 479–480:545–554. 10.1016/j.virol.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen L, Gorman JJ, McKimm-Breschkin J, Lawrence LJ, Tulloch PA, Smith BJ, Colman PM, Lawrence MC. 2001. The structure of the fusion glycoprotein of Newcastle disease virus suggests a novel paradigm for the molecular mechanism of membrane fusion. Structure 9:255–266. 10.1016/s0969-2126(01)00581-0. [DOI] [PubMed] [Google Scholar]
- 8.Iorio RM, Field GM, Sauvron JM, Mirza AM, Deng R, Mahon PJ, Langedijk JP. 2001. Structural and functional relationship between the receptor recognition and neuraminidase activities of the Newcastle disease virus hemagglutinin-neuraminidase protein: receptor recognition is dependent on neuraminidase activity. J Virol 75:1918–1927. 10.1128/JVI.75.4.1918-1927.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang A, Dutch RE. 2012. Paramyxovirus fusion and entry: multiple paths to a common end. Viruses 4:613–636. 10.3390/v4040613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith EC, Popa A, Chang A, Masante C, Dutch RE. 2009. Viral entry mechanisms: the increasing diversity of paramyxovirus entry. FEBS J 276:7217–7227. 10.1111/j.1742-4658.2009.07401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.White JM, Whittaker GR. 2016. Fusion of enveloped viruses in endosomes. Traffic 17:593–614. 10.1111/tra.12389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mercer J, Schelhaas M, Helenius A. 2010. Virus entry by endocytosis. Annu Rev Biochem 79:803–833. 10.1146/annurev-biochem-060208-104626. [DOI] [PubMed] [Google Scholar]
- 13.Krzyzaniak MA, Zumstein MT, Gerez JA, Picotti P, Helenius A. 2013. Host cell entry of respiratory syncytial virus involves macropinocytosis followed by proteolytic activation of the F protein. PLoS Pathog 9:e1003309. 10.1371/journal.ppat.1003309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pernet O, Pohl C, Ainouze M, Kweder H, Buckland R. 2009. Nipah virus entry can occur by macropinocytosis. Virology 395:298–311. 10.1016/j.virol.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 15.Kolokoltsov AA, Deniger D, Fleming EH, Roberts NJ, Jr, Karpilow JM, Davey RA. 2007. Small interfering RNA profiling reveals key role of clathrin-mediated endocytosis and early endosome formation for infection by respiratory syncytial virus. J Virol 81:7786–7800. 10.1128/JVI.02780-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schowalter RM, Chang A, Robach JG, Buchholz UJ, Dutch RE. 2009. Low-pH triggering of human metapneumovirus fusion: essential residues and importance in entry. J Virol 83:1511–1522. 10.1128/JVI.01381-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cox RG, Mainou BA, Johnson M, Hastings AK, Schuster JE, Dermody TS, Williams JV. 2015. Human metapneumovirus is capable of entering cells by fusion with endosomal membranes. PLoS Pathog 11:e1005303. 10.1371/journal.ppat.1005303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanchez-Felipe L, Villar E, Munoz-Barroso I. 2014. Entry of Newcastle disease virus into the host cell: role of acidic pH and endocytosis. Biochim Biophys Acta 1838:300–309. 10.1016/j.bbamem.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cantin C, Holguera J, Ferreira L, Villar E, Munoz-Barroso I. 2007. Newcastle disease virus may enter cells by caveola-mediated endocytosis. J Gen Virol 88:559–569. 10.1099/vir.0.82150-0. [DOI] [PubMed] [Google Scholar]
- 20.Tan L, Zhang Y, Qiao C, Yuan Y, Sun Y, Qiu X, Meng C, Song C, Liao Y, Munir M, Nair V, Ding Z, Liu X, Ding C. 2018. NDV entry into dendritic cells through macropinocytosis and suppression of T lymphocyte proliferation. Virology 518:126–135. 10.1016/j.virol.2018.02.011. [DOI] [PubMed] [Google Scholar]
- 21.Tan L, Zhang Y, Zhan Y, Yuan Y, Sun Y, Qiu X, Meng C, Song C, Liao Y, Ding C. 2016. Newcastle disease virus employs macropinocytosis and Rab5a-dependent intracellular trafficking to infect DF-1 cells. Oncotarget 7:86117–86133. 10.18632/oncotarget.13345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cornax I, Diel DG, Rue CA, Estevez C, Yu Q, Miller PJ, Afonso CL. 2013. Newcastle disease virus fusion and haemagglutinin-neuraminidase proteins contribute to its macrophage host range. J Gen Virol 94:1189–1194. 10.1099/vir.0.048579-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang P, Ding Z, Liu X, Chen Y, Li J, Tao Z, Fei Y, Xue C, Qian J, Wang X, Li Q, Stoeger T, Chen J, Bi Y, Yin R. 2018. Enhanced replication of virulent Newcastle disease virus in chicken macrophages is due to polarized activation of cells by inhibition of TLR7. Front Immunol 9:366. 10.3389/fimmu.2018.00366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown C, King DJ, Seal B. 1999. Detection of a macrophage-specific antigen and the production of interferon gamma in chickens infected with Newcastle disease virus. Avian Dis 43:696–703. 10.2307/1592739. [DOI] [PubMed] [Google Scholar]
- 25.Mercer J, Greber UF. 2013. Virus interactions with endocytic pathways in macrophages and dendritic cells. Trends Microbiol 21:380–388. 10.1016/j.tim.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 26.Bayati A, Kumar R, Francis V, McPherson PS. 2021. SARS-CoV-2 infects cells following viral entry via clathrin-mediated endocytosis. J Biol Chem 296:100306. 10.1016/j.jbc.2021.100306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kalia M, Khasa R, Sharma M, Nain M, Vrati S. 2013. Japanese encephalitis virus infects neuronal cells through a clathrin-independent endocytic mechanism. J Virol 87:148–162. 10.1128/JVI.01399-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Basler CF, Desai TM, Marin M, Chin CR, Savidis G, Brass AL, Melikyan GB. 2014. IFITM3 restricts influenza a virus entry by blocking the formation of fusion pores following virus-endosome hemifusion. PLoS Pathog 10:e1004048. 10.1371/journal.ppat.1004048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith SE, Busse DC, Binter S, Weston S, Diaz Soria C, Laksono BM, Clare S, Van Nieuwkoop S, Van den Hoogen BG, Clement M, Marsden M, Humphreys IR, Marsh M, de Swart RL, Wash RS, Tregoning JS, Kellam P. 2018. Interferon-induced transmembrane protein 1 restricts replication of viruses that enter cells via the plasma membrane. J Virol 93:e02003-18. 10.1128/JVI.02003-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith SE, Gibson MS, Wash RS, Ferrara F, Wright E, Temperton N, Kellam P, Fife M. 2013. Chicken interferon-inducible transmembrane protein 3 restricts influenza viruses and lyssaviruses in vitro. J Virol 87:12957–12966. 10.1128/JVI.01443-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li K, Markosyan RM, Zheng YM, Golfetto O, Bungart B, Li M, Ding S, He Y, Liang C, Lee JC, Gratton E, Cohen FS, Liu SL. 2013. IFITM proteins restrict viral membrane hemifusion. PLoS Pathog 9:e1003124. 10.1371/journal.ppat.1003124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Butcher M, Raviprakash K, Ghosh HP. 1990. Acid pH-induced fusion of cells by herpes simplex virus glycoproteins gB an gD. J Biol Chem 265:5862–5868. 10.1016/S0021-9258(19)39442-6. [DOI] [PubMed] [Google Scholar]
- 33.Mercer J, Helenius A. 2009. Virus entry by macropinocytosis. Nat Cell Biol 11:510–520. 10.1038/ncb0509-510. [DOI] [PubMed] [Google Scholar]
- 34.Blight GD, Morgan EH. 1987. Receptor-mediated endocytosis of transferrin and ferritin by guinea-pig reticulocytes: uptake by a common endocytic pathway. Eur J Cell Biol 43:260–265. [PubMed] [Google Scholar]
- 35.Roberts RL, Fine RE, Sandra A. 1993. Receptor-mediated endocytosis of transferrin at the blood-brain barrier. J Cell Sci 104(Pt 2):521–532. [DOI] [PubMed] [Google Scholar]
- 36.Shi BJ, Liu CC, Zhou J, Wang SQ, Gao ZC, Zhang XM, Zhou B, Chen PY. 2016. Entry of classical swine fever virus into PK-15 cells via a pH-, dynamin-, and cholesterol-dependent, clathrin-mediated endocytic pathway that requires Rab5 and Rab7. J Virol 90:9194–9208. 10.1128/JVI.00688-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang YN, Liu YY, Xiao FC, Liu CC, Liang XD, Chen J, Zhou J, Baloch AS, Kan L, Zhou B, Qiu HJ. 2018. Rab5, Rab7, and Rab11 are required for caveola-dependent endocytosis of classical swine fever virus in porcine alveolar macrophages. J Virol 92:e00797-18. 10.1128/JVI.00797-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu YZ, Xu QQ, Wu DG, Ren H, Zhao P, Lao WG, Wang Y, Tao QY, Qian XJ, Wei YH, Cao MM, Qi ZT. 2012. Japanese encephalitis virus enters rat neuroblastoma cells via a pH-dependent, dynamin and caveola-mediated endocytosis pathway. J Virol 86:13407–13422. 10.1128/JVI.00903-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gutierrez-Ortega A, Sanchez-Hernandez C, Gomez-Garcia B. 2008. Respiratory syncytial virus glycoproteins uptake occurs through clathrin-mediated endocytosis in a human epithelial cell line. Virol J 5:127. 10.1186/1743-422X-5-127.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leser GP, Ector KJ, Ng DT, Shaughnessy MA, Lamb RA. 1999. The signal for clathrin-mediated endocytosis of the paramyxovirus SV5 HN protein resides at the transmembrane domain-ectodomain boundary region. Virology 262:79–92. 10.1006/viro.1999.9890. [DOI] [PubMed] [Google Scholar]
- 41.Kaksonen M, Roux A. 2018. Mechanisms of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol 19:313–326. 10.1038/nrm.2017.132. [DOI] [PubMed] [Google Scholar]
- 42.Benmerah A, Bayrou M, Cerf-Bensussan N, Dautry-Varsat A. 1999. Inhibition of clathrin-coated pit assembly by an Eps15 mutant. J Cell Sci 112:1303–1311. [DOI] [PubMed] [Google Scholar]
- 43.Benmerah A, Poupon V, Cerf-Bensussan N, Dautry-Varsat A. 2000. Mapping of Eps15 domains involved in its targeting to clathrin-coated pits. J Biol Chem 275:3288–3295. 10.1074/jbc.275.5.3288. [DOI] [PubMed] [Google Scholar]
- 44.Pelkmans L, Helenius A. 2002. Endocytosis via caveolae. Traffic 3:311–320. 10.1034/j.1600-0854.2002.30501.x. [DOI] [PubMed] [Google Scholar]
- 45.Damm EM, Pelkmans L, Kartenbeck J, Mezzacasa A, Kurzchalia T, Helenius A. 2005. Clathrin- and caveolin-1-independent endocytosis: entry of simian virus 40 into cells devoid of caveolae. J Cell Biol 168:477–488. 10.1083/jcb.200407113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harmon B, Schudel BR, Maar D, Kozina C, Ikegami T, Tseng CT, Negrete OA. 2012. Rift Valley fever virus strain MP-12 enters mammalian host cells via caveola-mediated endocytosis. J Virol 86:12954–12970. 10.1128/JVI.02242-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Macovei A, Radulescu C, Lazar C, Petrescu S, Durantel D, Dwek RA, Zitzmann N, Nichita NB. 2010. Hepatitis B virus requires intact caveolin-1 function for productive infection in HepaRG cells. J Virol 84:243–253. 10.1128/JVI.01207-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sánchez EG, Quintas A, Pérez-Núñez D, Nogal M, Barroso S, Carrascosa ÁL, Revilla Y. 2012. African swine fever virus uses macropinocytosis to enter host cells. PLoS Pathog 8:e1002754. 10.1371/journal.ppat.1002754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gimenez MC, Rodríguez Aguirre JF, Colombo MI, Delgui LR. 2015. Infectious bursal disease virus uptake involves macropinocytosis and trafficking to early endosomes in a Rab5-dependent manner. Cell Microbiol 17:988–1007. 10.1111/cmi.12415. [DOI] [PubMed] [Google Scholar]
- 50.Rasmussen I, Vilhardt F. 2015. Macropinocytosis is the entry mechanism of amphotropic murine leukemia virus. J Virol 89:1851–1866. 10.1128/JVI.02343-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Caì Y, Postnikova EN, Bernbaum JG, Yú SQ, Mazur S, Deiuliis NM, Radoshitzky SR, Lackemeyer MG, McCluskey A, Robinson PJ, Haucke V, Wahl-Jensen V, Bailey AL, Lauck M, Friedrich TC, O’Connor DH, Goldberg TL, Jahrling PB, Kuhn JH. 2015. Simian hemorrhagic fever virus cell entry is dependent on CD163 and uses a clathrin-mediated endocytosis-like pathway. J Virol 89:844–856. 10.1128/JVI.02697-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aleksandrowicz P, Marzi A, Biedenkopf N, Beimforde N, Becker S, Hoenen T, Feldmann H, Schnittler HJ. 2011. Ebola virus enters host cells by macropinocytosis and clathrin-mediated endocytosis. J Infect Dis 204(Suppl 3):S957–S967. 10.1093/infdis/jir326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Diederich S, Thiel L, Maisner A. 2008. Role of endocytosis and cathepsin-mediated activation in Nipah virus entry. Virology 375:391–400. 10.1016/j.virol.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.San Roman K, Villar E, Munoz-Barroso I. 1999. Acidic pH enhancement of the fusion of Newcastle disease virus with cultured cells. Virology 260:329–341. 10.1006/viro.1999.9841. [DOI] [PubMed] [Google Scholar]
- 55.Nabi IR, Le PU. 2003. Caveolae/raft-dependent endocytosis. J Cell Biol 161:673–677. 10.1083/jcb.200302028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sheng XX, Sun YJ, Zhan Y, Qu YR, Wang HX, Luo M, Liao Y, Qiu XS, Ding C, Fan HJ, Mao X. 2016. The LXR ligand GW3965 inhibits Newcastle diseasevirus infection by affecting cholesterol homeostasis. Arch Virol 161:2491–2501. 10.1007/s00705-016-2950-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martin JJ, Holguera J, Sanchez-Felipe L, Villar E, Munoz-Barroso I. 2012. Cholesterol dependence of Newcastle disease virus entry. Biochim Biophys Acta 1818:753–761. 10.1016/j.bbamem.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fielding CJ, Fielding PE. 2003. Relationship between cholesterol trafficking and signaling in rafts and caveolae. Biochim Biophys Acta 1610:219–228. 10.1016/s0005-2736(03)00020-8. [DOI] [PubMed] [Google Scholar]
- 59.Lee JH, Pasquarella JR, Kalejta RF. 2019. Cell line models for human cytomegalovirus latency faithfully mimic viral entry by macropinocytosis and endocytosis. J Virol 93:e01021-19. 10.1128/JVI.01021-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nanbo A, Imai M, Watanabe S, Noda T, Takahashi K, Neumann G, Halfmann P, Kawaoka Y. 2010. Ebolavirus is internalized into host cells via macropinocytosis in a viral glycoprotein-dependent manner. PLoS Pathog 6:e1001121. 10.1371/journal.ppat.1001121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wen Z, Zhao B, Song K, Hu X, Chen W, Kong D, Ge J, Bu Z. 2013. Recombinant lentogenic Newcastle disease virus expressing Ebola virus GP infects cells independently of exogenous trypsin and uses macropinocytosis as the major pathway for cell entry. Virol J 10:331. 10.1186/1743-422X-10-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Villar E, Barroso IM. 2006. Role of sialic acid-containing molecules in paramyxovirus entry into the host cell: a minireview. Glycoconj J 23:5–17. 10.1007/s10719-006-5433-0. [DOI] [PubMed] [Google Scholar]
- 63.Navaratnarajah CK, Generous AR, Yousaf I, Cattaneo R. 2020. Receptor-mediated cell entry of paramyxoviruses: mechanisms, and consequences for tropism and pathogenesis. J Biol Chem 295:2771–2786. 10.1074/jbc.REV119.009961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ferreira L, Villar E, Munoz-Barroso I. 2004. Conformational changes of Newcastle disease virus envelope glycoproteins triggered by gangliosides. Eur J Biochem 271:581–588. 10.1111/j.1432-1033.2003.03960.x. [DOI] [PubMed] [Google Scholar]
- 65.Ferreira L, Villar E, Munoz-Barroso I. 2004. Gangliosides and N-glycoproteins function as Newcastle disease virus receptors. Int J Biochem Cell Biol 36:2344–2356. 10.1016/j.biocel.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 66.Cosset FL, Lavillette D. 2011. Cell entry of enveloped viruses. Adv Genet 73:121–183. 10.1016/B978-0-12-380860-8.00004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.O’Hara SD, Garcea RL. 2016. Murine polyomavirus cell surface receptors activate distinct signaling pathways required for infection. mBio 7:e01836-16. 10.1128/mBio.01836-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhu L, Hu X, Kumar D, Chen F, Feng Y, Zhu M, Liang Z, Huang L, Yu L, Xu J, Xue R, Cao G, Gong C. 2018. Both ganglioside GM2 and cholesterol in the cell membrane are essential for Bombyx mori cypovirus cell entry. Dev Comp Immunol 88:161–168. 10.1016/j.dci.2018.07.011. [DOI] [PubMed] [Google Scholar]
- 69.Lim KJ, Sung BH, Shin JR, Lee YW, Kim DJ, Yang KS, Kim SC. 2013. A cancer-specific cell-penetrating peptide, BR2, for the efficient delivery of an scFv into cancer cells. PLoS One 8:e66084. 10.1371/journal.pone.0066084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Parton RG. 1994. Ultrastructural localization of gangliosides: GM1 is concentrated in caveolae. J Histochem Cytochem 42:155–166. 10.1177/42.2.8288861. [DOI] [PubMed] [Google Scholar]
- 71.Campanero-Rhodes MA, Smith A, Chai W, Sonnino S, Mauri L, Childs RA, Zhang Y, Ewers H, Helenius A, Imberty A, Feizi T. 2007. N-glycolyl GM1 ganglioside as a receptor for simian virus 40. J Virol 81:12846–12858. 10.1128/JVI.01311-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tsai B, Gilbert JM, Stehle T, Lencer W, Benjamin TL, Rapoport TA. 2003. Gangliosides are receptors for murine polyoma virus and SV40. EMBO J 22:4346–4355. 10.1093/emboj/cdg439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sverdlov M, Shajahan AN, Minshall RD. 2007. Tyrosine phosphorylation-dependence of caveolae-mediated endocytosis. J Cell Mol Med 11:1239–1250. 10.1111/j.1582-4934.2007.00127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cutillo G, Saariaho AH, Meri S. 2020. Physiology of gangliosides and the role of antiganglioside antibodies in human diseases. Cell Mol Immunol 17:313–322. 10.1038/s41423-020-0388-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Müthing J, Cacić M. 1997. Glycosphingolipid expression in human skeletal and heart muscle assessed by immunostaining thin-layer chromatography. Glycoconj J 14:19–28. 10.1023/a:1018552729572. [DOI] [PubMed] [Google Scholar]
- 76.Smith AE, Lilie H, Helenius A. 2003. Ganglioside-dependent cell attachment and endocytosis of murine polyomavirus-like particles. FEBS Lett 555:199–203. 10.1016/s0014-5793(03)01220-1. [DOI] [PubMed] [Google Scholar]
- 77.Pelkmans L, Kartenbeck J, Helenius A. 2001. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat Cell Biol 3:473–483. 10.1038/35074539. [DOI] [PubMed] [Google Scholar]
- 78.Schelhaas M, Malmström J, Pelkmans L, Haugstetter J, Ellgaard L, Grünewald K, Helenius A. 2007. Simian Virus 40 depends on ER protein folding and quality control factors for entry into host cells. Cell 131:516–529. 10.1016/j.cell.2007.09.038. [DOI] [PubMed] [Google Scholar]
- 79.Engel S, Heger T, Mancini R, Herzog F, Kartenbeck J, Hayer A, Helenius A. 2011. Role of endosomes in simian virus 40 entry and infection. J Virol 85:4198–4211. 10.1128/JVI.02179-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Eash S, Querbes W, Atwood WJ. 2004. Infection of Vero cells by BK virus is dependent on caveolae. J Virol 78:11583–11590. 10.1128/JVI.78.21.11583-11590.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Johannsdottir HK, Mancini R, Kartenbeck J, Amato L, Helenius A. 2009. Host cell factors and functions involved in vesicular stomatitis virus entry. J Virol 83:440–453. 10.1128/JVI.01864-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kielian MC, Marsh M, Helenius A. 1986. Kinetics of endosome acidification detected by mutant and wild-type Semliki Forest virus. EMBO J 5:3103–3109. 10.1002/j.1460-2075.1986.tb04616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rink J, Ghigo E, Kalaidzidis Y, Zerial M. 2005. Rab conversion as a mechanism of progression from early to late endosomes. Cell 122:735–749. 10.1016/j.cell.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 84.Macovei A, Petrareanu C, Lazar C, Florian P, Branza-Nichita N. 2013. Regulation of hepatitis B virus infection by Rab5, Rab7, and the endolysosomal compartment. J Virol 87:6415–6427. 10.1128/JVI.00393-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Krishnan MN, Sukumaran B, Pal U, Agaisse H, Murray JL, Hodge TW, Fikrig E. 2007. Rab 5 is required for the cellular entry of dengue and West Nile viruses. J Virol 81:4881–4885. 10.1128/JVI.02210-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shao Y, Sun J, Han Z, Liu S. 2018. Recombinant infectious laryngotracheitis virus expressing Newcastle disease virus F protein protects chickens against infectious laryngotracheitis virus and Newcastle disease virus challenge. Vaccine 36:7975–7986. 10.1016/j.vaccine.2018.11.008. [DOI] [PubMed] [Google Scholar]
- 87.Sun J, Han Z, Qi T, Zhao R, Liu S. 2017. Chicken galectin-1B inhibits Newcastle disease virus adsorption and replication through binding tohemagglutinin-neuraminidase (HN) glycoprotein. J Biol Chem 292:20141–20161. 10.1074/jbc.M116.772897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wise MG, Suarez DL, Seal BS, Pedersen JC, Senne DA, King DJ, Kapczynski DR, Spackman E. 2004. Development of a real-time reverse-transcription PCR for detection of Newcastle disease virus RNA in clinical samples. J Clin Microbiol 42:329–338. 10.1128/jcm.42.1.329-338.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Reed LJ, Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am J Epidemiol 27:493–497. 10.1093/oxfordjournals.aje.a118408. [DOI] [Google Scholar]
- 90.Sun X, Yau VK, Briggs BJ, Whittaker GR. 2005. Role of clathrin-mediated endocytosis during vesicular stomatitis virus entry into host cells. Virology 338:53–60. 10.1016/j.virol.2005.05.006. [DOI] [PubMed] [Google Scholar]