

ABSTRACT
Pneumococcal surface protein A (PspA) and pneumococcal surface protein C (PspC, also called CbpA) are major virulence factors of Streptococcus pneumoniae (Spn). These surface-exposed choline-binding proteins (CBPs) function independently to inhibit opsonization, neutralize antimicrobial factors, or serve as adhesins. PspA and PspC both carry a proline-rich domain (PRD) whose role, other than serving as a flexible connector between the N-terminal and C-terminal domains, was up to this point unknown. Herein, we demonstrate that PspA binds to lactate dehydrogenase (LDH) released from dying host cells during infection. Using recombinant versions of PspA and isogenic mutants lacking PspA or specific domains of PspA, this property was mapped to a conserved 22-amino-acid nonproline block (NPB) found within the PRD of most PspAs and PspCs. The NPB of PspA had specific affinity for LDH-A, which converts pyruvate to lactate. In a mouse model of pneumonia, preincubation of Spn carrying NPB-bearing PspA with LDH-A resulted in increased bacterial titers in the lungs. In contrast, incubation of Spn carrying a version of PspA lacking the NPB with LDH-A or incubation of wild-type Spn with enzymatically inactive LDH-A did not enhance virulence. Preincubation of NPB-bearing Spn with lactate alone enhanced virulence in a pneumonia model, indicating exogenous lactate production by Spn-bound LDH-A had an important role in pneumococcal pathogenesis. Our observations show that lung LDH, released during the infection, is an important binding target for Spn via PspA/PspC and that pneumococci utilize LDH-A derived lactate for their benefit in vivo.
KEYWORDS: pneumococcal surface protein A, pneumococcal surface protein C, Streptococcus pneumoniae, choline binding proteins, pathogenesis, pneumonia, virulence
INTRODUCTION
Streptococcus pneumoniae (Spn) is an opportunistic human pathogen capable of causing a wide spectrum of serious infectious diseases, including pneumonia, bacteremia, sepsis, and meningitis (1). The pneumococcus is the leading cause of community-acquired pneumonia and invasive disease worldwide (2). Spn is a Gram-positive bacterium with teichoic acid and lipoteichoic acid being major constituents of its cell wall and cell membrane, respectively (3). In Spn, and some but not all oral streptococci, these glycopolymers contain phosphorylcholine residues which serve as anchors for a family of surface proteins known as choline-binding proteins (CBPs). CBPs bind noncovalently to the phosphorylcholine moieties of teichoic and lipoteichoic acids via a choline-binding domain (CBD) composed of repeating, conserved choline-binding motifs at either their N or C terminus. CBPs play diverse and vital roles in the biology of the pneumococcus and are involved in cell wall remodeling, autolysis, biofilm formation, host cell adhesion and invasion, and resistance to antimicrobial factors such as complement and cationic antimicrobial peptides (4).
Pneumococcal surface protein A (PspA) and pneumococcal surface protein C (PspC; also known as CbpA) are among the most investigated CBPs (5, 6). PspA and PspC have loosely similar structural organizations with their N termini composed of a series of α-helical domains (Fig. 1A). PspA ranges from 65 to 95 kDa in size with an N-terminal signal peptide, followed by an α-helix domain (αHD) which contains what is known as the clade defining region (CDR). Next is the proline-rich domain (PRD), which is often interrupted by a highly conserved 22-amino-acid sequence called the nonproline block (NPB). At the C terminus is the CBD (6). PspC ranges from 59 to 105 kDa in size and also has an N-terminal signal peptide followed by a long multifunctional module that contains a factor H binding motif and two repeats of an R domain (R1 and R2), each of which is a three-α-helical raft-like structure (7). These are followed by a PRD and then the CBD (5, 6).
FIG 1.

Schematic diagrams of PspA, PspC, and representative proline-rich domains. (A) Schematic diagram of serotype 3 strain WU2 PspA and serotype 4 strain TIGR4 PspC. Shown for PspA is the N-terminal α-helix region, composed of the α-helix domain (αHD) and clade defining region (CDR), the proline-rich domain (PRD), and C-terminal choline-binding domain (CBD) composed of multiple choline-binding repeats (CBR). The α-helix region of PspC is composed of the factor H binding region (FH bind), two R domains (R1 and R2), and the PRD. This is followed by the CBD. (B) Schematic diagrams of PRDs from group 1, 2, and 3. Groups 1 and 2 are composed of different proline-repeating sequences. Group 3 shares aspects of both group 1 and group 2 but is characterized by the presence of the nonproline block (NPB). The red arrow indicates the NPB.
PspA has been demonstrated to bind to and prevent killing by the antimicrobial peptide lactoferricin. It has also been shown to block complement-activating host C-reactive protein from binding to exposed, i.e., unoccupied, phosphorylcholine residues on the bacterial cell wall (8, 9). PspC binds to polymeric immunoglobulin receptors and laminin receptors on host cells. These interactions have been shown to be responsible for Spn translocation across mucosal epithelial and vascular endothelial cell barriers, respectively (10, 11). Some, but not all, PspCs also bind serum factor H. The binding of factor H helps protect pneumococci against complement deposition on the bacterial surface (12, 13). Pertinent to this study, the role of PRD on either PspA or PspC was unknown, although the diversity of this domain had been explored for PspA since it has been shown to elicit protective antibody (14, 15). In 2018, we showed that PRD in PspA of different Spn isolates can be divided into 3 groups based on its amino acid sequence (14). Group 1 and 2 are divided based upon the arrangement of highly conserved proline-rich amino acid sequences. Group 3 shares aspects of both group 1 and group 2, but group 3 proline-rich sequences are interrupted by a highly conserved 22-amino-acid sequence called the highly conserved nonproline block (NPB) (sequence provided in Fig. 1B). Notably, ∼90% of Spn isolates have a version of PspA (48% to 56% of 123 strains) or PspC (77% of 43 strains) that is interrupted by the NPB (5, 6, 14–16), but a role for this sequence motif remained undiscovered.
Lactate dehydrogenase (LDH) is a key enzyme involved in cellular metabolism and is one of the most highly expressed proteins in mammalian cells. Mammalian LDH is a tetramer composed of LDH-A and/or LDH-B. The specific ratio of LDH-A to LDH-B is dependent on the cell type; in lung epithelial cells, they are in equal amounts (17–19). LDH-A converts pyruvate to lactate and generates NAD+ from NADH, whereas LDH-B converts lactate to pyruvate and reduces NAD+ to create NADH (20). Spn produces a variety of molecules that have been demonstrated to be cytotoxic to host cells. Primary among these are pneumolysin (Ply), a pore-forming toxin, and H2O2, which is produced by the enzyme streptococcal pyruvate oxidase (SpxB) as part of the bacterium’s own metabolism (21–23). During pneumococcal pneumonia, cytoplasmic LDH is released from dying host cells as result of exposure to Ply, SpxB-derived H2O2, and other cytotoxic molecules produced by the bacteria. Indeed, the amount of LDH present in tissue culture supernatants is frequently used as a measure of host cell death (22).
In this paper, we show that the NPB of both of PspA and PspC binds to mammalian LDH-A. Moreover, the generation of lactate by host LDH-A directly enhances the virulence of Spn as result of co-opted metabolic activity. Our results add to the increasing body of work that shows Spn appropriates host factors released by dying cells to exacerbate its virulence.
RESULTS
Spn binds to LDH released from dying cells.
Given that PspA is a highly abundant surface protein, we explored the possibility that it modulated Spn virulence in a yet-unknown fashion. To identify previously unknown host proteins that PspA interacted with, we performed pulldown experiments using His-tagged recombinant PspA from strain WU2 (PspAWU2), composed of poly-His-tagged PspA αHD and PRD, as bait for proteins in THP-1 human monocyte cell lysates. Among the top five host proteins identified following separation of eluted proteins by SDS-PAGE, band excision, and identification by mass spectroscopy was lactate dehydrogenase A (LDH-A) (see Fig. S1 and Table S1 in the supplemental material). LDH-A was particularly intriguing given that PspA also bound to LDH when tested by enzyme-linked immunosorbent assay (ELISA) using cell lysates from A549 human type II pneumocytes (see Fig. S2A) and because LDH is known to be released by dying host cells as result of Spn infection (22). Notably, and in comparison, PspA bound poorly to pneumococcal LDH as determined by ELISA (Fig. S2B). To further validate the interaction of PspA with host LDH, we repeated pulldown experiments using purified rabbit muscle LDH, consisting primarily of LDH-A, and purified lactoferrin, since it is known to bind PspA (9). PspAWU2 bound to a nickel column pulled down both these proteins, whereas they were not pulled down when PspAWU2 was absent (Fig. 2A). We also reaffirmed that host LDH was released from human lung cells in vitro and in vivo into the bronchi of mice following challenge or the induction of Spn pneumonia, respectively (Fig. 2B and C). Thus, PspA binds the bacterium to host LDH, which becomes accessible during the course of infection.
FIG 2.
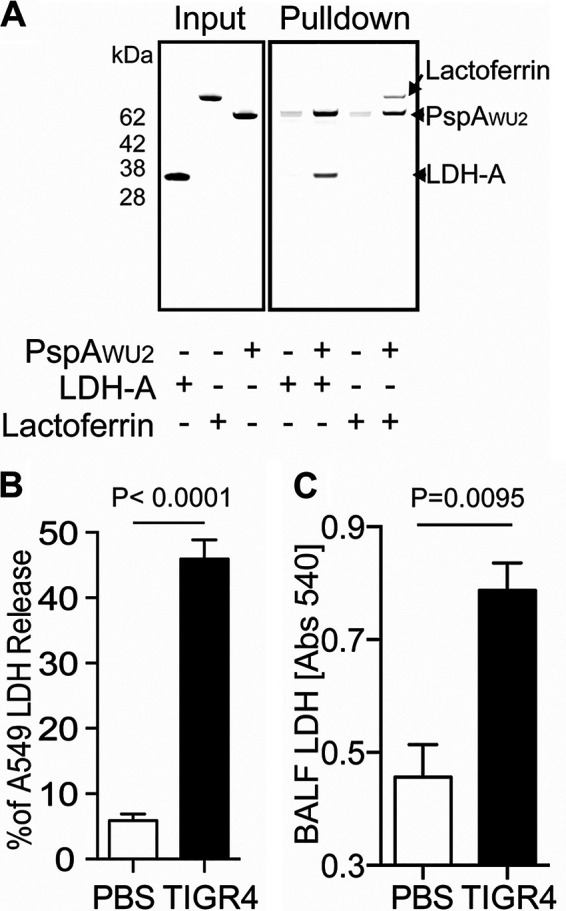
PspA binds to LDH from dying host cell via Spn infection. (A) Recombinant His-tagged PspAWU2 protein was pulled down with purified host LDH-A and human lactoferrin. The bound proteins were separated by SDS-PAGE and visualized by Coomassie blue dye. This experiment was performed two times. (B) Human A549 type II pulmonary epithelial cells were infected with Spn strain TIGR4 for 4 h at an MOI of 100. Cultured medium was collected to measure percent LDH release when normalized to that from uninfected cells and total lysis using lysis buffer for 30 min. (C) Six- to eight-week-old C57BL/6 mice were challenged intratracheally with Spn TIGR4 (5 × 105 CFU) or mock infected with PBS. One day after pneumococcal challenge, relative levels of LDH in bronchoalveolar lavage fluid (BALF) samples were determined by ELISA. Means and standard errors are shown. Statistical analyses were performed using a Mann-Whitney U test.
List of PspA interacted THP-1 proteins. Download Table S1, PDF file, 0.02 MB (16.7KB, pdf) .
Copyright © 2021 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Purified PspA binds to host LDH and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Recombinant His-tagged pneumolysin (Ply, green arrowhead) or PspAWU2 (yellow arrowhead) were pulled down with human macrophage THP-1 cell lysate using Ni-NTA resin. The input and bound proteins were separated by SDS-PAGE and visualized by Coomassie blue dye. PspA bound host proteins (red arrowheads) are listed on Table S1. Download FIG S1, PDF file, 1.0 MB (992.5KB, pdf) .
Copyright © 2021 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
PspA interaction with host and Spn LDH. (A) PspA binds to A549 type II pneumocyte cell line LDH. Interaction of PspA with A549 LDH was analyzed by ELISA using recombinant PspAWU2. (B) PspA has low affinity for Spn LDH compared to host LDH. Means and standard errors shown. Statistical analyses were performed using a t test. Download FIG S2, PDF file, 0.06 MB (56.2KB, pdf) .
Copyright © 2021 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
PspA binds to LDH-A via the NPB.
LDH tetramers within lung epithelial cells are composed equally of LDH-A and LDH-B (24). To determine if PspA had affinity for LDH-A, LDH-B, or both, we performed ELISAs with recombinant purified human LDH-A and LDH-B. Strong dose-response binding of LDH-A and PspAWU2 was observed, whereas LDH-B showed no binding (Fig. 3A). To identify the LDH-binding domain of PspA, a second series of pulldown assays was performed with increasingly smaller fragments (F) of PspAWU2: F-1, F-2, F-3, F-4, and F-5. Only full-length PspAWU2 (composed of αHD and PRD) pulled down LDH-A, suggesting that PRD was the responsible domain (Fig. 3B; see also Fig. S3A). These experiments were next repeated with recombinant PspA PRDs from strain TIGR4 (PRD group 1), strain EF6796 (PRD group 2), and WU2 (PRD group 3). Only WU2 PRD, which contains the NPB, captured LDH-A (Fig. 3C; Fig. S3B). Critically, we observed that wild-type WU2, but not an isogenic mutant deficient in PspA (WU2 ΔpspA) or an isogenic WU2 mutant carrying PspA with the NPB genetically deleted from the PRD (WU2 ΔpspA-NPB), bound to LDH-A in flow cytometry experiments (Fig. 3D). Thus, our collective experimental results indicate that the NPB within the PRD of PspA was responsible for specific binding to LDH-A.
FIG 3.

PspA binds to LDH-A via NPB. (A) Interaction of PspA with LDH-A or LDH-B was analyzed by ELISA under different PspAWU2 concentrations. (B) Diagram of PspAWU2 fragments (F-1 to F-5) used in the pulldown experiments, the corresponding truncated mutants created for testing of PspA binding to LDH-A and PRDs (see Fig. S3A in the supplemental material). (C) Different versions of PRD from designated Spn strains were tested for their ability to bind LDH-A in pulldown experiments (Fig. S3B). (D) Spn WU2, its isogenic pspA deletion mutant (WU2ΔpspA), and a isogenic NPB deletion mutant of PspA (WU2ΔNPB) were incubated with FITC-conjugated rabbit LDH-A. The LDH-A binding to pneumococci was measured by flow cytometry. Data are geometric means (means and standard errors shown) of the log fluorescence intensity measured on a flow cytometer (n = 3).
LDH binds to group 3 PRD motif. PspAWU2, fragments (F-1, F-2, F-3, F-4, and F-5) (A) and PRDs (WU2 PRD, EF6978 PRD and TIGR4 PRD) as bait (B) were pulled down with LDH and as visualized by Coomassie blue stains. Bound LDH were detected by immunoblotting using monoclonal anti-LDH antibody. The red arrows indicate LDH. Download FIG S3, PDF file, 0.6 MB (661.1KB, pdf) .
Copyright © 2021 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
NPB on PspC also binds to LDH-A.
As indicated, recombinant PRD from TIGR4 PspA did not interact with LDH-A (Fig. 3C). Yet, the annotated genome of TIGR4 indicates this strain carries a version of PspC with a group 3 PRD containing the NPB, suggesting TIGR4 should have the capacity to bind LDH-A. As expected, the NPB motif was detected in TIGR4 cell lysates by immunoblot using monoclonal antibody reactive with NPB (Fig. 4A). Furthermore, live TIGR4 was observed to bind LDH-A by flow cytometry (Fig. 4B). Notably, this was also not the case for EF3030, a serotype 19F strain whose versions of PspA and PspC do not have the NPB domain (Fig. 4A and B). Subsequent examination of a panel of 10 Spn clinical isolates showed that our detection of the NPB in bacterial lysates using monoclonal antibody via immunoblot was consistently positively correlated with LDH-A binding capacity of live pneumococci (Fig. 4C and D). Of note, and in contrast to other Spn strains, WU2 is unusual and does not carry PspC (5). Thus, our prior results with WU2 were fortuitously not confounded by the presence of a second PRD. To reaffirm that it was the NPB within the PRD that was responsible, EF3030 was mutated so as to carry distinct versions of PspA. Only the version of EF3030 expressing PspA carrying NPB within the PRD showed LDH binding by flow cytometry (Fig. 4E; see also Fig. S4). Finally, we incubated fluorescein isothiocyanate (FITC)-conjugated LDH-A with WU2 and clinical isolates of Staphylococcus aureus and uropathogenic Escherichia coli and then analyzed LDH-A binding ability by flow cytometry. Only WU2 showed affinity for LDH-A (Fig. 4F). Thus, LDH-A binding is not shared by other major pathogens.
FIG 4.

NPB on PspA and PspC binds to LDH. (A) Immunoblots of total bacterial cell lysates using anti-NPB monoclonal antibody PR-1A4.7 with total bacterial cell lysate was used to detect the NPB domain in WU2, TIGR4, and EF3030. (B) Spn WU2, TIGR4, or EF3030 was incubated with FITC-conjugated LDH. LDH binding to pneumococci was measured using flow cytometry. This experiment was performed two times with identical results. (C) Immunoblots using NPB monoclonal antibody of total Spn bacterial cell lysates from designated clinical isolates. The presence of PspA or PspC containing the NPB as inferred from publicly available genomic sequence data is indicated on the bottom. (D) The ability of these isolates to bind rabbit LDH-A was also measured using flow cytometry (n = 3). (E) EF3030, EF3030 isogenic pspA mutant (EF3030-01), EF3030 expressing PspAWU2 (EF3030-04), and EF3030 expressing PspAWU2 without NPB (EF3030-05) were incubated with FITC-conjugated rabbit LDH-A. The LDH-A binding to pneumococci was measured by flow cytometry. (F) Spn WU2, S. aureus, and urinary tract-pathogenic E. coli (UP E. coli) were incubated with FITC-conjugated rabbit LDH-A. The LDH binding to pneumococci was measured by flow cytometry. This experiment was performed two times with similar results.
PspA patterns of WU2 and EF3030 wild type or different isogenic mutants. Total cell lysates of Spn WU2, EF3030, isogenic pspA mutants (WU2ΔpspA and EF3030-01), isogenic pspA mutants (WU2ΔNPB), EF3030 expressing PspAWU2 (EF3030-04), and EF3030 expressing PspAWU2 without NPB (EF3030-05) were electrophorized on SDS-PAGE gels. Subsequently, expressed PspA were detected by using monoclonal anti-PspA antibody. Download FIG S4, PDF file, 0.3 MB (280.8KB, pdf) .
Copyright © 2021 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
LDH binding to pneumococci enhances Spn virulence in vitro.
The prevalence of the PRD containing NPB in both PspA and PspC, both of which are vital virulence determinants, suggests the NPB of PspC also contributes to Spn virulence. To test this, we challenged mice with WU2 or TIGR4. WU2 has NPB in PspA but does not have PspC. TIGR4 has an NPB in its PspC but not in its PspA. In each case, the inoculum was given in phosphate-buffered saline (PBS) with or without rabbit LDH-A. Strikingly, mixture of these bacteria with LDH-A prior to challenge resulted in >10-fold more pneumococci in the lungs of mice within a 24-h period and a greater likelihood of developing bacteremia (Fig. 5A and B). We also examined the virulence of EF3030 carrying versions of PspA with and without the NPB. EF3030 carrying PspA with the NPB was more virulent following bacterial mixture with LDH-A, whereas no LDH-A enhancement of virulence was observed for EF3030 with PspA lacking NPB (Fig. 5C). We conclude host LDH-A binding to PspA or PspC, via the NPB, enhanced the virulence of Spn.
FIG 5.

LDH-A enhanced Spn pathogenesis. Mice were infected (A) intratracheally with 5.0 × 106 CFU of WU2 or (B) 1.0 × 105 CFU of TIGR4 following coincubation and alongside rabbit LDH-A. (C) Mice were infected intratracheally with Spn preincubated with rabbit LDH-A. Mice received 1.0 × 106 CFU of EF3030, EF3030-04, or EF3030-05 intratracheally. Shown are bacterial titers in the lungs and blood 24 h after challenge. Shapes in panels A, B, and C represent bacterial burden for each mouse. Statistical analyses were performed using a Mann-Whitney U test or Kruskal-Wallis test with two-way ANOVA.
LDH-A enzymatic activity is required for Spn virulence enhancement.
We tested and determined that LDH-A binding did not impact Spn adhesion or invasion of healthy lung epithelial cells grown in vitro (see Fig. S5). We also found no impact of PspA binding on LDH-A enzymatic activity in vitro (see Fig. S6). Yet, pneumococci incubated with enzymatically inactive LDH-A, as result of a point mutation (25), failed to enhance Spn virulence in mouse challenge experiments (Fig. 6A; see also Fig. S7). We interpreted the latter finding as suggesting LDH-A confers its impact on Spn virulence via the generation of lactate or NAD+. An important role for exogenous lactate was confirmed by our observation of increased Spn burden in the lungs of mice challenged with Spn suspended in saline with lactate versus Spn in saline alone (Fig. 6B). In contrast, no effect on lung bacterial burden was seen when Spn was mixed with saline containing exogenous NAD prior to challenge (NAD 100 μM, 2.03 ± 0.26 × 106 CFU/g; control, 2.01 ± 0.46 × 106 CFU/g; ± standard error of the mean [SEM]; P = 0.979, n = 7/cohort). Finally, recent findings by Minhas et al. demonstrated that endogenous generation of NAD+ is vital to macrophage immune function (26). We therefore tested whether treatment with exogenous NAD had an influence on the cytokine and chemokine response of J774A.1 macrophages challenged with Spn or on their ability to take up Spn. We observed no difference in the production of tumor necrosis factor (NAD, 400 ± 11.6; control, 455.9 ± 45.2; ± SEM; P = 0.4206, n = 5/cohort) by infected macrophages that had been treated with NAD or evidence that pretreatment with exogenous NAD enhanced efficiency of bacterial uptake by macrophages after 1 h of coincubation (NAD, 3.7% ± 1.7% of initial inoculum; control, 3.4% ± 1.2% [SEM]; P > 0.999, n = 5/cohort). From these data, we conclude the observed contribution of PspA to virulence was most likely the result of co-opting host LDH-A enzymatic activity and the generation of lactate.
FIG 6.

LDH-A activity promotes pneumococcal virulence. (A) Mice were infected intratracheally with 5.0 × 106 CFU of WU2 alongside recombinant LDH-A or nonfunctional point mutated LDH-A (R106Q). (B) Mice were infected intratracheally with 1.0 × 106 CFU EF3030 alongside different concentrations of lactate. Shapes in panels represent bacterial burden for each mouse. Statistical analyses were performed using a Kruskal-Wallis test with Dunn’s multiple-comparison posttest.
LDH does not affect Spn adhesion and invasion. Preincubated Spn WU2 or EF3030-04 with LDH was incubated with A549 cells for 1 h and then washed (adhesion) 3 times by PBS or changed to DMEM medium with ampicillin-streptomycin (invasion) and incubated for 2 h and then washed 3× with PBS. Subsequently, A549 cells were detached by trypsin-EDTA, and then total bacterial CFU were calculated on blood agar plates (n = 3). Means and standard errors are shown. Download FIG S5, PDF file, 0.07 MB (72.3KB, pdf) .
Copyright © 2021 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
PspA does not affect LDH enzyme activity. Enzyme activity of preincubated LDH with different rations of PspAWU2 was measured using a LDH assay kit (n = 5). Means and standard errors shown. Download FIG S6, PDF file, 0.03 MB (32.7KB, pdf) .
Copyright © 2021 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Nonfunctional point mutant LDH loses enzyme activity. Enzyme activity of LDH-A or nonfunctional point mutant LDH-A (R106Q) was measured using an LDH assay kit (n = 3). Means and standard errors shown. Download FIG S7, PDF file, 0.03 MB (30.7KB, pdf) .
Copyright © 2021 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DISCUSSION
PspA and PspC are multidomain CBPs with clear and vital roles in pneumococcal pathogenesis. Prior to this report, they had been demonstrated to confer resistance to opsonization by various means (27, 28). The vital role of these proteins in pneumococcal biology is highlighted by the fact that most clinical isolates of Spn carry both proteins (5, 29, 30), and mutants deficient in PspA and PspC are markedly reduced in virulence in a variety of experimental models (31–34). PspA and PspC are among the most common and highly in vivo-expressed CBPs (5, 14, 29, 35, 36), and immunization with these proteins as antigens has been demonstrated to confer protection against invasive pneumococcal disease, especially when used in combination (31, 37, 38). Nonetheless, and despite >30 years of study on PspA and PspC, prior to this report, the role of the NPB in pneumococcal biology was undetermined. This report assigns function to group 3 PRD, which houses the NPB, and shows that it enhances the bacterium’s virulence as result of binding to enzymatically active LDH-A from host cells. The role of group 1 and group 2 PRDs remain an open question, although immunity against the proline-rich portion of the PRD appears to be protective (14, 15).
LDH, NADH, and the substrate pyruvate have all been shown to be released by host cells when they die (22, 39). LDH-A converts pyruvate to lactate and, in doing so, generates NAD+ from NADH. NAD+ is an important redox carrier and serves as the sole substrate for NAD-dependent enzymes (40). Thus, its sustained generation by LDH-A has the potential to catalyze other enzymatic events at the infection site to the bacterium’s benefit that were not tested. For example, multiple NAD+-dependent antioxidants are produced by the host (41). Sustained NAD+ production has the potential to benefit Spn by enhancing the neutralization of the profuse amounts of H2O2 produced as result of Spn’s own metabolism (23). In addition, exogenous NAD has also been shown to alter T-cell function and, in certain instances, induce T-cell death (42, 43). Exposure to exogenous NAD has also been shown to enhance Fcγ receptor-mediated endocytosis by macrophages (44). Thus, co-opted LDH-A activity may not exclusively benefit Spn. Importantly, and as our mice were naive to Spn and therefore lacked antibodies or pathogen-specific T cells against Spn, our experimental model does not reflect the potential impact of NAD+ generated by LDH-A on these adaptive immune system parameters. This is an important area to explore in the future, particularly with consideration that antibodies generated against PspA may block its binding to LDH-A altogether during Spn reinfection.
Incubation of Spn with lactate was, on its own, sufficient to confer virulence but only if the Spn carried a PspA with an NPB. Consistent with this, there are numerous ways exogenous lactate production might benefit Spn. Pneumococcal lactate oxidase converts lactate to pyruvate, and the latter has been demonstrated to be a vital energy source for Spn during aerobic growth (45, 46). As both the nasopharynx and the lungs are aerobic sites, PspA binding to host LDH may be a way for the bacterium to directly augment its energy supply. Binding of the bacterium to host cells via PspC’s other binding domains would, in this scenario, also enhance Spn’s ability to harvest pyruvate from damaged cells before it is diluted in the milieu. Lactate also has immunomodulatory properties which may benefit the bacterium. For example, lactate has been shown to reduce organ injury during Toll-like receptor and inflammasome-mediated inflammation of the gut. This was as a result of its interaction with Gi protein-coupled receptor GPR81 on host cells (47). Notably, Toll-like receptor and inflammasome activation are the principal ways by which the host detects Spn in the airway, and GPR81 is also present on lung epithelial cells (48, 49). Critically, the above described are speculation, and detailed investigative studies on how lactate generation helps Spn are now warranted to fully understand PspA’s contribution to the disease process.
Despite this lack of clarity on how LDH-A confers a beneficial effect to Spn, that PspA can enhance Spn virulence in the airway by binding to LDH-A is unequivocal. Our strongest evidence for this being that EF3030, a strain that lacks the NPB in either its PspA or PspC, had increased virulence when mutated to carry a version of PspA with the NPB. Likewise, that deletion of only the NPB from WU2 PspA starkly reduced virulence. The fact that LDH is released from dying cells and enhances pneumococcal virulence may be important under several other conditions known to enhance susceptibility to Spn. These include but are not limited to secondary infections to influenza virus and the increase in Spn susceptibility associated with smoking, cancer, and chronic obstructive pulmonary disease, all of which have been associated with increased LDH in BALF (50, 51). Importantly, LDH production is host-cell-type-dependent and affected by levels of glucose in the cell’s environment (52, 53). Thus, its contribution to Spn pathogenesis may be anatomical site specific or even change over the course of pneumonia, as capillary leakage increases glucose levels in the airway (54). Finally, it should be noted that PRD and/or other parts of PspA or PspC may bind to other metabolism-associated host factors, and these interactions may also influence pneumococcal pathogenesis. This would not be unexpected as PspA and PspC are on the bacterial surface and host metabolic enzymes are known to be released in large quantity by host cells under a variety of conditions (22, 55).
In summary, we have identified a role for the NPB carried by the version of the PRD that is most often found in both PspA and PspC. That role is binding to LDH-A. PspA binding to LDH-A was shown to enhance the bacterium’s virulence during pneumonia. To confer this phenotype, LDH-A must be enzymatically active, with local generation of lactate having beneficial effects for the bacterium. Our findings suggest Spn utilizes host proteins released from dying cells to its advantage and highlight the complex interactions between Spn and its host during disease. Finally, the fact that some pneumococci have versions of PspA without an NPB may indicate they have compensating virulence functions or, perhaps more likely, that different pneumococci have different host interaction strategies that allow them to survive and pass to a subsequent new host.
MATERIALS AND METHODS
Bacteria, cloning, and plasmids.
All bacterial strains, plasmids, and primers used in this study are listed in Table S2 in the supplemental material. Spn, S. aureus, and uropathogenic Escherichia coli (UPEC) were cultured on blood agar plates at 37°C within a candle jar (Spn) or in a 5% CO2 incubator. All three Spn strains have PRD in their PspA. Colony swabs were used to inoculate Todd-Hewitt broth (THB) with 0.5% yeast extract and grown until mid-exponential phase (optical density at 621 nm [OD621] of 0.3). Isogenic Spn mutants deficient in pspA were generated by insertion of a kanamycin resistance cassette using standard homologous recombination methods (56). Briefly, DNA corresponding to the flanking regions of pspA were amplified by PCR (primers listed in Table S2) and cloned adjacent to erm and kan (56) in pCR2.1 (pCR2.1 erm and pCR2.1 kan). This mutagenic construct was PCR amplified and used to transform Spn. Hybrid versions of pspA were generated using a PCR-based fusion method using primers in Table S2. In this instance, the mutagenic DNA products were cloned into pCR2.1 ermB (57) and used to transform the aforementioned pspA kanamycin-resistant isogenic mutant. In both instances, mutagenic Spn isolates were selected on blood agar plates with antimicrobials and validated by sequencing of amplified DNA. Human recombinant LDH-A and LDH-B expression vectors were purchased from Applied Biological Material (Richmond, BC, Canada). We removed the encoded N-terminal glutathione transferase (GST) tag on both LDH-A and LDH-B using restriction enzymes and cloned the genes into the pTEV5 expression vector, which added a removable version of the His tag (kindly provided by Michael Gray, The University of Alabama at Birmingham).
List of bacterial strains, plasmids, and primers used for this study. Download Table S2, PDF file, 0.05 MB (56.2KB, pdf) .
Copyright © 2021 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Expression and purification of recombinant proteins.
All protein expression vectors were transformed individually into E. coli NEBExpress Iq (New England BioLabs, Ipswich, MA) competent (PspAWU2 and PspA fragments) or E. coli BL21 for protein expression. After cultures reached on OD600 of 0.4 to 0.6, they were induced using 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 4 h at 37°C on shaker. Bacteria were harvested by centrifugation at 4,000 × g for 15 min. Cell pellets were resuspended in buffer A (50 mM Tris-HCl [pH 7.5] and 150 mM NaCl) with 1 mM phenylmethylsulfonyl fluoride (PMSF), a serine protease inhibitor, and then sonicated at 35% amplitude (2 s on/2 s off) for 30 min on ice for lysis and centrifuged at 12,000 × g for 30 min on 4°C. Overexpressed protein in the supernatant was purified using a cobalt resin column according to the manufacturer’s protocols for His tag purification. For LDH-A and LDH-B, the His tags were removed using tobacco etch virus protease and passage of the protein through a cobalt resin column to remove uncut protein. Purified proteins were loaded on the Superdex S200 or S75 columns using PBS. Collected protein fractions were checked by SDS-PAGE and concentrated by a centrifugal concentrator tube. The concentration of proteins was measured by UV 280 nm, and they were stored in Dulbecco’s phosphate-buffered saline (PBS) with 20% glycerol at −70°C.
Cell lines and growth conditions.
Low-passage-number stocks of THP-1, a human monocyte cell line (58), and A549, a human type II pneumocyte cell line (59), were obtained from ATCC and cultured as recommended. Cell lysates from monolayers at ∼80% confluence were generated using RIPA buffer (150 mM NaCl, 50 mM Tris-HCl, 1% Triton X-100, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate).
Ni-NTA pulldown assay.
Two micrograms of recombinant human lactoferrin (OPSA11754; AVIVA Systems Biology, San Diego, CA) and rabbit muscle LDH (LDH-A) (Sigma, St. Louis, MO) were mixed with 2 μg of 6×His-PspAWU2, fragments (F-1, F2, F-3, F-4, or F-5), or recombinant 6×His-PRDs (WU2PRD, TIGR4 PRD, or EF6796 PRD) in buffer A (PBS, 0.1% Triton X-100, 0.5 mM dithiothreitol [DTT], and 10 mM imidazole) and then incubated for 1 h at 4°C on rotator (6 rpm). Nickel-nitrilotriacetic acid (Ni-NTA) resin (Qiagen), 20 μl, was equilibrated in 1 ml of buffer A. Subsequently, Ni-NTA resin and mixtures of proteins were mixed together and incubated for 1 h at 4°C on a rotator (6 rpm). Incubated Ni-NTA resins were washed four times with 1 ml of buffer A containing 20 mM imidazole. SDS loading buffer (2×) was added to the Ni-NTA resins and then incubated at 90°C for 5 min. Proteins were separated on 4 to 12% gradient SDS-PAGE gels (Invitrogen) and visualized by Coomassie blue staining or immunoblots with an anti-LDH antibody (ab130923; Abcam).
ELISA.
Microtiter 96-well plates (Nunc, Weisbaden, Germany) were coated overnight at 4°C with 100 μl of different concentrations (0 to 4 μg/well) of recombinant human LDH-A, recombinant human LDH-B, or recombinant Spn PspA in PBS. Plates were washed, blocked with 200 μl 5% bovine serum albumin (BSA) in PBS for 2 h, washed three times by T-PBS (PBS with 0.5% Tween 20), and then incubated with 2 μg/well of PspAWU2 or A549 lysate at 37°C for 1 h. The plate was washed three times with T-PBS. Finally, the plate was incubated at room temperature for 1 h with mouse monoclonal anti-PspA (1b2.21) (60) or rabbit monoclonal anti-LDH (ab130923) antibody (1:1,000) in PBS with 5% BSA, washed three times with T-PBS, incubated for 1 h with 1:10,000 alkaline phosphatase-conjugated donkey anti-rabbit immunoglobulin antiserum IgG(H+L) (Southern Biotechnology Associates, Birmingham, AL), washed, and developed with p-nitrophenyl phosphate (Sigma) or 3,3′,5,5′ tetramethylbenzidine (TMB; BD Biosciences); the absorbance was read at 405 nm or 450 nm.
Detection of bound FITC-LDH on bacteria.
Pneumococci, S. aureus, or urinary tract-pathogenic E. coli isolates were grown as described above and resuspended to 1.0 × 108 CFU/ml in fluorescence-activated cell sorting (FACS) buffer (1× PBS with 3% fetal bovine serum [FBS] and 0.1% sodium azide). For the LDH binding assay, 1.0 × 106 CFU bacteria were incubated in 100 μl with 0.5 μg of FITC-conjugated LDH at 4°C for 30 min with shaking. Samples were washed 2 times with FACS buffer and then analyzed by flow cytometry using BD Accuri C6 (BD, Franklin Lakes, NJ).
LDH measurement.
Cell death was evaluated by detection of lactate dehydrogenase (LDH) in culture supernatants as previously described (61). For measurement of LDH in vivo, 6- to 8-week-old C57BL/6 mice were infected with Spn TIGR4 (5 × 105 CFU) in 40 μl saline by forced aspiration. One day after challenge, mice were euthanized, and then bronchoalveolar lavage (BAL) was performed using 1 ml of ice-cold PBS. Fifty microliters of BAL fluid (BALF) per mouse was transferred to a flat-bottom 96-well plate and used for detection of LDH. LDH in supernatants and BALF was measured using a Pierce LDH cytotoxicity assay kit (Thermo Fisher Scientific, Suwanee, GA). Plates were read using an iMark absorbance microplate reader (Bio-Rad Laboratories, Hercules, CA).
Immunoblotting.
Cultured Spn isolates were resuspended by RIPA buffer (Sigma) with proteinase inhibitor cocktail (Sigma) and lysed by the freeze-thaw method. Supernatants were collected, and the protein concentration was measured by bicinchoninic acid assay (Bio-Rad, Hercules, CA). Cell lysates (10 μg) were run on SDS-PAGE gels, and gels were electroblotted to a 0.45-μm-pore-size nitrocellulose membrane (Bio-Rad). The membrane was blocked with 3% bovine serum albumin in T-PBS (PBS containing 0.05% Tween 20) for 1 h at room temperature. The membranes were exposed to anti-NPB antibody (PR-1A4.7) (15) or anti-PspA antibody (1b2.21) for 1 h at room temperature, washed, and incubated with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (diluted 1:10,000 in PBS-T) for 1 h at room temperature. After washing, the membranes were developed with enhanced chemiluminescence (ECL) solution (Bio-Rad) and observed by using a ChemiDoc (Bio-Rad).
Mouse challenge.
Female 8- to 12-week-old BALB/c mice (The Jackson Laboratory) were used for protection studies. Briefly, Spn strains WU2, TIGR4, and EF3030 were incubated in THB until an OD621 of 0.3 (∼1.0 × 108 CFU/ml) and resuspended in PBS with or without of 5 μg/mouse LDH. Unbound LDH on EF3030 was removed by centrifugation, and the bacteria were resuspended in PBS. The mice were infected by forced aspiration with 40 μl of Spn WU2 (5.0 × 106 CFU), TIGR4 (1.0 × 105 CFU), or EF3030 (1.0 × 106 CFU). After 24 h, mice were euthanized, and then total CFU in the lungs were measured by serial dilution of homogenized lungs and extrapolation of colony counts on blood agar plates the next day.
Macrophage response assays.
The J774.1 macrophage cell line was seeded in a 12-well plate with 5 × 105 cells and grown in Dulbecco’s modified Eagle medium (DMEM) for 2 days. Cells were treated 1 h prior to infection with either medium alone or medium plus NAD at 50 μM. Cells were then washed with PBS three times to remove medium. Cells were then inoculated with Spn strain TIGR4 at a multiplicity of infection (MOI) of 10 (1 × 107) and spun down at 300 × g for 5 min to synchronize infection. After 1 h, the supernatant was collected for tumor necrosis factor (TNF) (DY-410) ELISA from R&D. Macrophages were washed and lysed with water, and associated Spn cells were enumerated for extrapolation of phagocytosis efficiency.
Statistical analyses.
Statistical analyses were performed using GraphPad Prism 6 software using nonparametric Mann-Whitney U test, Kruskal-Wallis test with Dunn’s multiple-comparison posttest, or two-way ANOVA.
Supplemental methods. Download Text S1, PDF file, 0.10 MB (102.1KB, pdf) .
Copyright © 2021 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
ACKNOWLEDGMENTS
Support for this project is through NIH grant AI118805 to D.E.B. and NIH grants AI114800, AI148368, AI146149, and AI156898 to C.J.O.
Footnotes
Citation Park S-S, Gonzalez-Juarbe N, Martínez E, Hale JY, Lin Y-H, Huffines JT, Kruckow KL, Briles DE, Orihuela CJ. 2021. Streptococcus pneumoniae binds to host lactate dehydrogenase via PspA and PspC to enhance virulence. mBio 12:e00673-21. https://doi.org/10.1128/mBio.00673-21.
Contributor Information
David E. Briles, Email: dbriles@uab.edu.
Carlos J. Orihuela, Email: corihuel@uab.edu.
N. Luisa Hiller, Carnegie Mellon University
REFERENCES
- 1.Shah SS, Ratner AJ. 2006. Trends in invasive pneumococcal disease-associated hospitalizations. Clin Infect Dis 42:e1–e5. doi: 10.1086/498745. [DOI] [PubMed] [Google Scholar]
- 2.Feldman C, Anderson R. 2020. The role of Streptococcus pneumoniae in community-acquired pneumonia. Semin Respir Crit Care Med 41:455–469. doi: 10.1055/s-0040-1702193. [DOI] [PubMed] [Google Scholar]
- 3.Gisch N, Kohler T, Ulmer AJ, Muthing J, Pribyl T, Fischer K, Lindner B, Hammerschmidt S, Zahringer U. 2013. Structural reevaluation of Streptococcus pneumoniae lipoteichoic acid and new insights into its immunostimulatory potency. J Biol Chem 288:15654–15667. doi: 10.1074/jbc.M112.446963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galán-Bartual S, Pérez-Dorado I, García P, Hermoso JA. 2015. Chapter 11. Structure and function of choline-binding proteins, p 207–230. In Brown J, Hammerschmidt S, Orihuela C (ed), Streptococcus pneumoniae. Academic Press, Waltham, MA. doi: 10.1016/B978-0-12-410530-0.00011-9. [DOI] [Google Scholar]
- 5.Brooks-Walter A, Briles DE, Hollingshead SK. 1999. The pspC gene of Streptococcus pneumoniae encodes a polymorphic protein, PspC, which elicits cross-reactive antibodies to PspA and provides immunity to pneumococcal bacteremia. Infect Immun 67:6533–6542. doi: 10.1128/IAI.67.12.6533-6542.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hollingshead SK, Becker R, Briles DE. 2000. Diversity of PspA: mosaic genes and evidence for past recombination in Streptococcus pneumoniae. Infect Immun 68:5889–5900. doi: 10.1128/iai.68.10.5889-5900.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luo R, Mann B, Lewis WS, Rowe A, Heath R, Stewart ML, Hamburger AE, Sivakolundu S, Lacy ER, Bjorkman PJ, Tuomanen E, Kriwacki RW. 2005. Solution structure of choline binding protein A, the major adhesin of Streptococcus pneumoniae. EMBO J 24:34–43. doi: 10.1038/sj.emboj.7600490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mukerji R, Mirza S, Roche AM, Widener RW, Croney CM, Rhee DK, Weiser JN, Szalai AJ, Briles DE. 2012. Pneumococcal surface protein A inhibits complement deposition on the pneumococcal surface by competing with the binding of C-reactive protein to cell-surface phosphocholine. J Immunol 189:5327–5335. doi: 10.4049/jimmunol.1201967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shaper M, Hollingshead SK, Benjamin WH, Jr, Briles DE. 2004. PspA protects Streptococcus pneumoniae from killing by apolactoferrin, and antibody to PspA enhances killing of pneumococci by apolactoferrin [corrected]. Infect Immun 72:5031–5040. doi: 10.1128/IAI.72.9.5031-5040.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orihuela CJ, Mahdavi J, Thornton J, Mann B, Wooldridge KG, Abouseada N, Oldfield NJ, Self T, Ala'Aldeen DA, Tuomanen EI. 2009. Laminin receptor initiates bacterial contact with the blood brain barrier in experimental meningitis models. J Clin Invest 119:1638–1646. doi: 10.1172/JCI36759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brock SC, McGraw PA, Wright PF, Crowe JE, Jr.. 2002. The human polymeric immunoglobulin receptor facilitates invasion of epithelial cells by Streptococcus pneumoniae in a strain-specific and cell type-specific manner. Infect Immun 70:5091–5095. doi: 10.1128/iai.70.9.5091-5095.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Janulczyk R, Iannelli F, Sjoholm AG, Pozzi G, Bjorck L. 2000. Hic, a novel surface protein of Streptococcus pneumoniae that interferes with complement function. J Biol Chem 275:37257–37263. doi: 10.1074/jbc.M004572200. [DOI] [PubMed] [Google Scholar]
- 13.Dave S, Brooks-Walter A, Pangburn MK, McDaniel LS. 2001. PspC, a pneumococcal surface protein, binds human factor H. Infect Immun 69:3435–3437. doi: 10.1128/IAI.69.5.3435-3437.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mukerji R, Hendrickson C, Genschmer KR, Park SS, Bouchet V, Goldstein R, Lefkowitz EJ, Briles DE. 2018. The diversity of the proline-rich domain of pneumococcal surface protein A (PspA): potential relevance to a broad-spectrum vaccine. Vaccine 36:6834–6843. doi: 10.1016/j.vaccine.2018.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daniels CC, Coan P, King J, Hale J, Benton KA, Briles DE, Hollingshead SK. 2010. The proline-rich region of pneumococcal surface proteins A and C contains surface-accessible epitopes common to all pneumococci and elicits antibody-mediated protection against sepsis. Infect Immun 78:2163–2172. doi: 10.1128/IAI.01199-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iannelli F, Oggioni MR, Pozzi G. 2002. Allelic variation in the highly polymorphic locus pspC of Streptococcus pneumoniae. Gene 284:63–71. doi: 10.1016/s0378-1119(01)00896-4. [DOI] [PubMed] [Google Scholar]
- 17.Gay RJ, McComb RB, Bowers GN, Jr.. 1968. Optimum reaction conditions for human lactate dehydrogenase isoenzymes as they affect total lactate dehydrogenase activity. Clin Chem 14:740–753. doi: 10.1093/clinchem/14.8.740. [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez Buitrago JM, Garcia Diez LC, de Castro S. 1981. Human semen lactate dehydrogenase isoenzymes in fertility studies (author's transl). Reproduccion 5:147–155. (In Spanish.) [PubMed] [Google Scholar]
- 19.Li SS, O'Brien DA, Hou EW, Versola J, Rockett DL, Eddy EM. 1989. Differential activity and synthesis of lactate dehydrogenase isozymes A (muscle), B (heart), and C (testis) in mouse spermatogenic cells. Biol Reprod 40:173–180. doi: 10.1095/biolreprod40.1.173. [DOI] [PubMed] [Google Scholar]
- 20.Burgner JW, II, Ray WJ, Jr.. 1984. On the origin of the lactate dehydrogenase induced rate effect. Biochemistry 23:3636–3648. doi: 10.1021/bi00311a010. [DOI] [PubMed] [Google Scholar]
- 21.Bryant JC, Dabbs RC, Oswalt KL, Brown LR, Rosch JW, Seo KS, Donaldson JR, McDaniel LS, Thornton JA. 2016. Pyruvate oxidase of Streptococcus pneumoniae contributes to pneumolysin release. BMC Microbiol 16:271. doi: 10.1186/s12866-016-0881-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gonzalez-Juarbe N, Bradley KM, Riegler AN, Reyes LF, Brissac T, Park SS, Restrepo MI, Orihuela CJ. 2018. Bacterial pore-forming toxins promote the activation of caspases in parallel to necroptosis to enhance alarmin release and inflammation during pneumonia. Sci Rep 8:5846. doi: 10.1038/s41598-018-24210-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spellerberg B, Cundell DR, Sandros J, Pearce BJ, Idanpaan-Heikkila I, Rosenow C, Masure HR. 1996. Pyruvate oxidase, as a determinant of virulence in Streptococcus pneumoniae. Mol Microbiol 19:803–813. doi: 10.1046/j.1365-2958.1996.425954.x. [DOI] [PubMed] [Google Scholar]
- 24.Papadopoulos NM. 1977. Clinical applications of lactate dehydrogenase isoenzymes. Ann Clin Lab Sci 7:506–510. [PubMed] [Google Scholar]
- 25.Clarke AR, Wigley DB, Chia WN, Barstow D, Atkinson T, Holbrook JJ. 1986. Site-directed mutagenesis reveals role of mobile arginine residue in lactate dehydrogenase catalysis. Nature 324:699–702. doi: 10.1038/324699a0. [DOI] [PubMed] [Google Scholar]
- 26.Minhas PS, Liu L, Moon PK, Joshi AU, Dove C, Mhatre S, Contrepois K, Wang Q, Lee BA, Coronado M, Bernstein D, Snyder MP, Migaud M, Majeti R, Mochly-Rosen D, Rabinowitz JD, Andreasson KI. 2019. Macrophage de novo NAD+ synthesis specifies immune function in aging and inflammation. Nat Immunol 20:50–63. doi: 10.1038/s41590-018-0255-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haleem KS, Ali YM, Yesilkaya H, Kohler T, Hammerschmidt S, Andrew PW, Schwaeble WJ, Lynch NJ. 2018. The pneumococcal surface proteins PspA and PspC sequester host C4-binding protein to inactivate complement C4b on the bacterial surface. Infect Immun 87:e00742-18. doi: 10.1128/IAI.00742-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J, Glover DT, Szalai AJ, Hollingshead SK, Briles DE. 2007. PspA and PspC minimize immune adherence and transfer of pneumococci from erythrocytes to macrophages through their effects on complement activation. Infect Immun 75:5877–5885. doi: 10.1128/IAI.00839-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hollingshead SK, Baril L, Ferro S, King J, Coan P, Briles DE, The Pneumococcal Proteins Epi Study G . 2006. Pneumococcal surface protein A (PspA) family distribution among clinical isolates from adults over 50 years of age collected in seven countries. J Med Microbiol 55:215–221. doi: 10.1099/jmm.0.46268-0. [DOI] [PubMed] [Google Scholar]
- 30.Crain MJ, Waltman WD, II, Turner JS, Yother J, Talkington DF, McDaniel LS, Gray BM, Briles DE. 1990. Pneumococcal surface protein A (PspA) is serologically highly variable and is expressed by all clinically important capsular serotypes of Streptococcus pneumoniae. Infect Immun 58:3293–3299. doi: 10.1128/IAI.58.10.3293-3299.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schachern PA, Tsuprun V, Ferrieri P, Briles DE, Goetz S, Cureoglu S, Paparella MM, Juhn S. 2014. Pneumococcal PspA and PspC proteins: potential vaccine candidates for experimental otitis media. Int J Pediatr Otorhinolaryngol 78:1517–1521. doi: 10.1016/j.ijporl.2014.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McDaniel LS, Yother J, Vijayakumar M, McGarry L, Guild WR, Briles DE. 1987. Use of insertional inactivation to facilitate studies of biological properties of pneumococcal surface protein A (PspA). J Exp Med 165:381–394. doi: 10.1084/jem.165.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berry AM, Paton JC. 2000. Additive attenuation of virulence of Streptococcus pneumoniae by mutation of the genes encoding pneumolysin and other putative pneumococcal virulence proteins. Infect Immun 68:133–140. doi: 10.1128/iai.68.1.133-140.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kerr AR, Paterson GK, McCluskey J, Iannelli F, Oggioni MR, Pozzi G, Mitchell TJ. 2006. The contribution of PspC to pneumococcal virulence varies between strains and is accomplished by both complement evasion and complement-independent mechanisms. Infect Immun 74:5319–5324. doi: 10.1128/IAI.00543-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosenow C, Ryan P, Weiser JN, Johnson S, Fontan P, Ortqvist A, Masure HR. 1997. Contribution of novel choline-binding proteins to adherence, colonization and immunogenicity of Streptococcus pneumoniae. Mol Microbiol 25:819–829. doi: 10.1111/j.1365-2958.1997.mmi494.x. [DOI] [PubMed] [Google Scholar]
- 36.D'Mello A, Riegler AN, Martinez E, Beno SM, Ricketts T, Foxman EF, Orihuela CJ, Tettelin H. 2020. An in vivo atlas of host pathogen transcriptomes during Streptococcus pneumoniae colonization and disease. Proc Natl Acad Sci U S A 117:33507–33518. doi: 10.1073/pnas.2010428117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ogunniyi AD, Grabowicz M, Briles DE, Cook J, Paton JC. 2007. Development of a vaccine against invasive pneumococcal disease based on combinations of virulence proteins of Streptococcus pneumoniae. Infect Immun 75:350–357. doi: 10.1128/IAI.01103-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cao J, Chen D, Xu W, Chen T, Xu S, Luo J, Zhao Q, Liu B, Wang D, Zhang X, Shan Y, Yin Y. 2007. Enhanced protection against pneumococcal infection elicited by immunization with the combination of PspA, PspC, and ClpP. Vaccine 25:4996–5005. doi: 10.1016/j.vaccine.2007.04.069. [DOI] [PubMed] [Google Scholar]
- 39.Labbe K, Saleh M. 2008. Cell death in the host response to infection. Cell Death Differ 15:1339–1349. doi: 10.1038/cdd.2008.91. [DOI] [PubMed] [Google Scholar]
- 40.Xiao W, Wang RS, Handy DE, Loscalzo J. 2018. NAD(H) and NADP(H) redox couples and cellular energy metabolism. Antioxid Redox Signal 28:251–272. doi: 10.1089/ars.2017.7216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Massudi H, Grant R, Guillemin GJ, Braidy N. 2012. NAD+ metabolism and oxidative stress: the golden nucleotide on a crown of thorns. Redox Rep 17:28–46. doi: 10.1179/1351000212Y.0000000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lischke T, Schumacher V, Wesolowski J, Hurwitz R, Haag F, Koch-Nolte F, Mittrucker HW. 2013. CD8-beta ADP-ribosylation affects CD8+ T-cell function. Eur J Immunol 43:1828–1838. doi: 10.1002/eji.201243231. [DOI] [PubMed] [Google Scholar]
- 43.Seman M, Adriouch S, Scheuplein F, Krebs C, Freese D, Glowacki G, Deterre P, Haag F, Koch-Nolte F. 2003. NAD-induced T cell death: ADP-ribosylation of cell surface proteins by ART2 activates the cytolytic P2X7 purinoceptor. Immunity 19:571–582. doi: 10.1016/S1074-7613(03)00266-8. [DOI] [PubMed] [Google Scholar]
- 44.Song EK, Lee YR, Yu HN, Kim UH, Rah SY, Park KH, Shim IK, Lee SJ, Park YM, Chung WG, Kim JS, Han MK. 2008. Extracellular NAD is a regulator for FcgammaR-mediated phagocytosis in murine macrophages. Biochem Biophys Res Commun 367:156–161. doi: 10.1016/j.bbrc.2007.12.131. [DOI] [PubMed] [Google Scholar]
- 45.Udaka S, Koukol J, Vennesland B. 1959. Lactic oxidase of pneumococcus. J Bacteriol 78:714–725. doi: 10.1128/JB.78.5.714-725.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taniai H, Iida K, Seki M, Saito M, Shiota S, Nakayama H, Yoshida S. 2008. Concerted action of lactate oxidase and pyruvate oxidase in aerobic growth of Streptococcus pneumoniae: role of lactate as an energy source. J Bacteriol 190:3572–3579. doi: 10.1128/JB.01882-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoque R, Farooq A, Ghani A, Gorelick F, Mehal WZ. 2014. Lactate reduces liver and pancreatic injury in Toll-like receptor- and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology 146:1763–1774. doi: 10.1053/j.gastro.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tomlinson G, Chimalapati S, Pollard T, Lapp T, Cohen J, Camberlein E, Stafford S, Periselneris J, Aldridge C, Vollmer W, Picard C, Casanova JL, Noursadeghi M, Brown J. 2014. TLR-mediated inflammatory responses to Streptococcus pneumoniae are highly dependent on surface expression of bacterial lipoproteins. J Immunol 193:3736–3745. doi: 10.4049/jimmunol.1401413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.LaRock CN, Nizet V. 2015. Inflammasome/IL-1beta responses to streptococcal pathogens. Front Immunol 6:518. doi: 10.3389/fimmu.2015.00518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Henderson RF. 1984. Use of bronchoalveolar lavage to detect lung damage. Environ Health Perspect 56:115–129. doi: 10.1289/ehp.8456115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu B, Bao L, Wang L, Li F, Wen M, Li H, Deng W, Zhang X, Cao B. 12 November 2019. Anti-IFN-gamma therapy alleviates acute lung injury induced by severe influenza A (H1N1) pdm09 infection in mice. J Microbiol Immunol Infect doi: 10.1016/j.jmii.2019.07.009. [DOI] [PubMed] [Google Scholar]
- 52.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F. 2015. Proteomics. Tissue-based map of the human proteome. Science 347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 53.Thul PJ, Akesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Bjork L, Breckels LM, Backstrom A, Danielsson F, Fagerberg L, Fall J, Gatto L, Gnann C, Hober S, Hjelmare M, Johansson F, Lee S, Lindskog C, Mulder J, Mulvey CM, Nilsson P, Oksvold P, Rockberg J, Schutten R, Schwenk JM, Sivertsson A, Sjostedt E, Skogs M, Stadler C, Sullivan DP, Tegel H, Winsnes C, Zhang C, Zwahlen M, Mardinoglu A, Ponten F, von Feilitzen K, Lilley KS, Uhlen M, Lundberg E. 2017. A subcellular map of the human proteome. Science 356:eaal3321. doi: 10.1126/science.aal3321. [DOI] [PubMed] [Google Scholar]
- 54.Sender V, Hentrich K, Pathak A, Tan Qian Ler A, Embaie BT, Lundstrom SL, Gaetani M, Bergstrand J, Nakamoto R, Sham LT, Widengren J, Normark S, Henriques-Normark B. 2020. Capillary leakage provides nutrients and antioxidants for rapid pneumococcal proliferation in influenza-infected lower airways. Proc Natl Acad Sci U S A 117:31386–31397. doi: 10.1073/pnas.2012265117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yamaji R, Chatani E, Harada N, Sugimoto K, Inui H, Nakano Y. 2005. Glyceraldehyde-3-phosphate dehydrogenase in the extracellular space inhibits cell spreading. Biochim Biophys Acta 1726:261–271. doi: 10.1016/j.bbagen.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 56.Dunny GM, Lee LN, LeBlanc DJ. 1991. Improved electroporation and cloning vector system for Gram-positive bacteria. Appl Environ Microbiol 57:1194–1201. doi: 10.1128/AEM.57.4.1194-1201.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Embry A, Hinojosa E, Orihuela CJ. 2007. Regions of diversity 8, 9 and 13 contribute to Streptococcus pneumoniae virulence. BMC Microbiol 7:80. doi: 10.1186/1471-2180-7-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chanput W, Mes JJ, Wichers HJ. 2014. THP-1 cell line: an in vitro cell model for immune modulation approach. Int Immunopharmacol 23:37–45. doi: 10.1016/j.intimp.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 59.Rasey JS, Grierson JR, Wiens LW, Kolb PD, Schwartz JL. 2002. Validation of FLT uptake as a measure of thymidine kinase-1 activity in A549 carcinoma cells. J Nucl Med 43:1210–1217. [PubMed] [Google Scholar]
- 60.Genschmer KR, Accavitti-Loper MA, Briles DE. 2013. A modified surface killing assay (MSKA) as a functional in vitro assay for identifying protective antibodies against pneumococcal surface protein A (PspA). Vaccine 32:39–47. doi: 10.1016/j.vaccine.2013.10.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gonzalez-Juarbe N, Bradley KM, Shenoy AT, Gilley RP, Reyes LF, Hinojosa CA, Restrepo MI, Dube PH, Bergman MA, Orihuela CJ. 2017. Pore-forming toxin-mediated ion dysregulation leads to death receptor-independent necroptosis of lung epithelial cells during bacterial pneumonia. Cell Death Differ 24:917–928. doi: 10.1038/cdd.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of PspA interacted THP-1 proteins. Download Table S1, PDF file, 0.02 MB (16.7KB, pdf) .
Copyright © 2021 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Purified PspA binds to host LDH and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Recombinant His-tagged pneumolysin (Ply, green arrowhead) or PspAWU2 (yellow arrowhead) were pulled down with human macrophage THP-1 cell lysate using Ni-NTA resin. The input and bound proteins were separated by SDS-PAGE and visualized by Coomassie blue dye. PspA bound host proteins (red arrowheads) are listed on Table S1. Download FIG S1, PDF file, 1.0 MB (992.5KB, pdf) .
Copyright © 2021 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
PspA interaction with host and Spn LDH. (A) PspA binds to A549 type II pneumocyte cell line LDH. Interaction of PspA with A549 LDH was analyzed by ELISA using recombinant PspAWU2. (B) PspA has low affinity for Spn LDH compared to host LDH. Means and standard errors shown. Statistical analyses were performed using a t test. Download FIG S2, PDF file, 0.06 MB (56.2KB, pdf) .
Copyright © 2021 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
LDH binds to group 3 PRD motif. PspAWU2, fragments (F-1, F-2, F-3, F-4, and F-5) (A) and PRDs (WU2 PRD, EF6978 PRD and TIGR4 PRD) as bait (B) were pulled down with LDH and as visualized by Coomassie blue stains. Bound LDH were detected by immunoblotting using monoclonal anti-LDH antibody. The red arrows indicate LDH. Download FIG S3, PDF file, 0.6 MB (661.1KB, pdf) .
Copyright © 2021 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
PspA patterns of WU2 and EF3030 wild type or different isogenic mutants. Total cell lysates of Spn WU2, EF3030, isogenic pspA mutants (WU2ΔpspA and EF3030-01), isogenic pspA mutants (WU2ΔNPB), EF3030 expressing PspAWU2 (EF3030-04), and EF3030 expressing PspAWU2 without NPB (EF3030-05) were electrophorized on SDS-PAGE gels. Subsequently, expressed PspA were detected by using monoclonal anti-PspA antibody. Download FIG S4, PDF file, 0.3 MB (280.8KB, pdf) .
Copyright © 2021 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
LDH does not affect Spn adhesion and invasion. Preincubated Spn WU2 or EF3030-04 with LDH was incubated with A549 cells for 1 h and then washed (adhesion) 3 times by PBS or changed to DMEM medium with ampicillin-streptomycin (invasion) and incubated for 2 h and then washed 3× with PBS. Subsequently, A549 cells were detached by trypsin-EDTA, and then total bacterial CFU were calculated on blood agar plates (n = 3). Means and standard errors are shown. Download FIG S5, PDF file, 0.07 MB (72.3KB, pdf) .
Copyright © 2021 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
PspA does not affect LDH enzyme activity. Enzyme activity of preincubated LDH with different rations of PspAWU2 was measured using a LDH assay kit (n = 5). Means and standard errors shown. Download FIG S6, PDF file, 0.03 MB (32.7KB, pdf) .
Copyright © 2021 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Nonfunctional point mutant LDH loses enzyme activity. Enzyme activity of LDH-A or nonfunctional point mutant LDH-A (R106Q) was measured using an LDH assay kit (n = 3). Means and standard errors shown. Download FIG S7, PDF file, 0.03 MB (30.7KB, pdf) .
Copyright © 2021 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
List of bacterial strains, plasmids, and primers used for this study. Download Table S2, PDF file, 0.05 MB (56.2KB, pdf) .
Copyright © 2021 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Supplemental methods. Download Text S1, PDF file, 0.10 MB (102.1KB, pdf) .
Copyright © 2021 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.