

ABSTRACT
Global transcriptional regulators are prevalent in gram-positive pathogens. The transcriptional regulators of the Mga/AtxA family regulate target gene expression by directly binding to the promoter regions, that results in the coordinated expression of virulence factors. The spd_1587 gene of Streptococcus pneumoniae strain D39 encodes MgaSpn, which shares sequence similarity with global transcriptional regulators of the Mga/AtxA family. In this study, we demonstrated that MgaSpn regulates the biosynthesis of the capsule and phosphorylcholine, which play key roles in disease severity in S. pneumoniae infections. MgaSpn directly binds to the cps and lic1 promoters and affects the biosynthesis of the capsule and phosphorylcholine. MgaSpn binds to two specific sites on the promoter of cps, one of which contains the −35 box of the promoter, with high affinity. Consistently, low-molecular-weight capsule components were observed in the mgaSpn-null mutant strain. Moreover, we found that phosphorylcholine content was notably increased in the unencapsulated mgaSpn mutant strain. The mgaSpn null mutant caused more severe systemic disease than the parental strain D39. These findings indicate that the pneumococcal MgaSpn protein can inhibit capsule and phosphorylcholine production, thereby affecting the virulence of S. pneumoniae.
KEYWORDS: MgaSpn, gene regulation, capsule, phosphorylcholine, Streptococcus pneumoniae
Introduction
Streptococcus pneumoniae is a human commensal that asymptomatically colonizes the upper human respiratory tract. Infections caused by S. pneumoniae can lead to invasive diseases, including pneumonia, meningitis, and hypertoxicemia [1]. The transition from nasopharyngeal mucosal colonization to aggressive disease is a consequence of adaptation to the host and the coordinated expression of virulence factors [2]. Global transcriptional regulatory networks in pathogenic bacteria respond to external environmental alterations. They directly or indirectly regulate the expression of related virulence factors through these networks.
S. pneumoniae utilizes a large number of secreted and surface-exposed virulence factors during disease development. These include surface virulence factors such as capsular polysaccharides (CPS), teichoic acid (TA), and surface proteins [1,3]. CPS, the outermost of these virulence components, is covalently linked to the cell wall peptidoglycan. It forms a protective barrier that directly interacts with the host immune system [4–6]. Except for serotypes 3 and 37, CPS biosynthesis in S. pneumoniae generally follows the canonical Wzy-dependent pathway, first described by Avery and coworkers in serotype 2 strain D39 [7]. The cps gene cluster is composed of an upstream promoter and 17 downstream genes involved in capsule synthesis [8,9]. The activity of the cps promoter affects the expression of downstream genes, resulting in changes in capsule thickness [8–10]. In our previous study, we screened factors that regulate Pcps expression directly in strain D39, and a 58.6-kDa response regulator MgaSpn was identified [11].
MgaSpn or MgrA is an ortholog of the group A streptococcal Mga/AtxA family of global response regulators Mga [12] which transcriptionally activates numerous streptococcal genes that encode surface adhesion proteins, including emm (M protein), arp (M-like protein), scp1 and scpA (collagen-like proteins), and fba and sof (fibronectin binding proteins) as well as other virulence factors [13]. In strain R6, MgaSpn – encoded by spr1622 (equivalent to spd_1587) – activates the P1623B promoter in vivo [12]. The MgrA protein, encoded by sp1800, also a transcriptional repressor of rlrA pathogenicity islet genes in strain TIGR4, including rrgA, rrgB, and rrgC, is implicated in nasopharyngeal mucosal adhesion, whereas rlrA, rrgA, and srtD are required for mouse lung infection [2]. The rlrA pathogenicity islet genes are lacking in D39 and other pneumococcal strains. MgaSpn is a highly conserved protein in S. pneumoniae and is likely a regulator of other important virulence factors [12]. High-throughput transcriptome analyses revealed that mgaSpn is a conditionally expressed gene affected by the host environment [14]. However, the function of its encoded protein in strain D39 remains unknown.
Interestingly, MgaSpn can bind to the promoter of the lic1 operon involved in TA biosynthesis. TAs are involved in host epithelial cell interactions and possess a core structure of repeating units of Glc-AATGal-GalNAc (P-Cho)-GalNAc (P-Cho)-Rib-P, including the rare amino sugar 2-acetamido-4-amino-2,4,6-trideoxygalactose (AATGal) and two N-acetylgalactosamine (GalNAc) residues, each of which carries a phosphorylcholine (P-Cho) moiety that is unique to S. pneumoniae [15]. The P-Cho component provides an anchor for choline-binding proteins and recognizes the platelet-activating factor receptor, ultimately leading to increased cell adhesion and invasion [16]. The repeat unit is regulated by multiple operons: lic1/lic2, lic3, spr0091-spr0092, and spr1645-spr1655 [15]. The lic1 and lic2 operons are transcribed in opposite directions [17]. lic1 consists of five genes: tarI and tarJ, responsible for ribitol synthesis, and licA, licB, and licC required for choline uptake and activation. There are two promoters of lic1 genes (Plic1P1 and Plic1P2), and transcription from Plic1P1 is strongly dependent on the response regulator CiaR [18–20]. Although the structure of the lic1 operon has been studied extensively, the detailed regulatory mechanisms controlling P-Cho production in S. pneumoniae require further study.
Herein, we investigated the regulatory effect of MgaSpn on the virulence factor capsule and P-Cho of S. pneumoniae. The binding of the protective regions of MgaSpn to lic1 and cps promoters were analyzed by EMSA and DNase I footprint experiments. mgaSpn was knocked out to explore the changes in the expression levels of the downstream genes of the cps and lic1 operon and the changes in the contents of the capsule and P-Cho. Moreover, the effect of MgaSpn on the virulence of S. pneumoniae was confirmed by in vivo and in vitro experiments. The results provide novel insights on the role of MgaSpn in the virulence of S. pneumoniae.
Materials and methods
Bacterial strains and growth conditions
The bacterial strains and plasmids used in this study are listed in Table 1. S. pneumoniae D39 strain, and its derivatives were cultured in semi-synthetic casein hydrolyzate medium supplemented with 5% yeast extract (C + Y, pH 7.0) medium or blood agar plates at 37°C in a 5% CO2 atmosphere. Escherichia coli strains were grown in lysogeny broth (LB) with shaking or LB agar plates at 37°C [21]. Selective antibiotics were added when necessary.
Table 1.
Strains and plasmids used in this study
| Strain/plasmid | Relevant genotype/phenotype | Antibiotic (C, μg/ml) for strains selection | Reference /source |
|---|---|---|---|
| E. coli | |||
| DH5α | Sangon | ||
| BL21 | Sangon | ||
| S. pneumoniae | |||
| JH1900 |
S. pneumoniae D39 strain, Capsulated strain serotype 2, rpsl K56T |
Smr: 150 | lab stock [22] |
| JH0001 | D39ΔdexB-cps2A::JC1 | Kanr: 200 | present study |
| JH0002 | D39ΔdexB-cps2A | Smr: 150 | present study |
| JH1101 | D39ΔmgaSpn | ermr: 100 | present study |
| JH1102 | D39ΔdexB-cps2AΔmgaSpn | ermr: 100 | present study |
| JH1104 | D39ΔmgaSpn:: mgaSpn | specr: 50 | present study |
| JH1106 | D39ΔdexB-cps2AΔmgaSpn:: mgaSpn | specr: 50 | present study |
| TH7898 | D39s, ΔbgaA::Pef-tu-kan-rpsl | Kanr: 200 | lab stock [22] |
| Plasmids | |||
| pPEPZ-Plac | Cloning vector | specr: 50 | Addgene [56] |
| pPEPZ-mgaSpn | Contains mgaSpn | specr: 50 | present study |
| pET28-mgaSpn | Cloning vector | Kanr: 50 | present study |
Construction of unencapsulated mutant strains
The primers used in this study are listed in Table 2. The dexB-cps2A mutants (JH0002) of S. pneumoniae were generated in the streptomycin-resistant derivatives of strain D39 (JH1900) by allelic replacement with the counter-selectable Janus cassette as previously described [22,23]. The Janus cassette consists of a kanamycin resistance gene and a dominant wild-type rpsL+ allele encoding protein S12 of the small ribosomal subunit, a target for streptomycin [24,25]. The Janus cassette transforms JH1900 from streptomycin-resistant to streptomycin-sensitive. The upstream (UP-BA1) and downstream (DW-BA1) sequences of dexB-cps2A were amplified using the primer pairs Pr1901/Pr1902 and Pr1903/Pr1904 using JH1900 as the template. The Janus cassette (JC1) was amplified using primers Pr1905/Pr1906 with TH7898 as a template. A recombinant fragment (UP-BA1-JC1-DW-BA1) was obtained by overlap PCR using primers Pr1901/Pr1904. The resulting recombinant fragment was transformed into JH1900, and the transformed cells were inoculated onto kanamycin-containing (200 μg/mL) agar plates. Positive recombinant clones (JH1900ΔdexB-cps2A::JC1, JH0001) containing the UP-BA-JC1-DW-BA fragment were screened by PCR using the primers Pr1901/Pr1904. Transformation, colony selection, and identification were performed according to established protocols [26]. Similarly, the upstream (UP-BA2) and downstream (DW-BA2) sequences of dexB-cps2A were amplified using the primer pairs Pr1901/Pr1907 and Pr1908/Pr1904 using JH1900 as the template. The recombinant fragment (UP-BA2-DW-BA2) was obtained by overlap PCR using primers Pr1901/Pr1904. The resulting recombinant fragment was transformed into JH1900, and the transformed cells were inoculated on streptomycin-containing (150 μg/mL) agar plates. Positive recombinant clones (JH1900ΔdexB-cps2A, JH0002) containing the UP-BA2-DW-BA2 fragment were screened using the primers Pr1901/Pr1904.
Table 2.
Primers used in this study
| Name | Sequence (5′ – 3′) |
|---|---|
| Pr1901 | TTCCTGACGAGAAGGTAGTCAATAA |
| Pr1902 | AAAGCATAAGGAAAGGGGCCTTATAGTAATTCCACACAGA |
| Pr1903 | GGAGTTTTCAGCATTATCCTATAGGTGTTAATCATGAGTA |
| Pr1904 | GTCTAGATGGACATTCCCTACTGGG |
| Pr1905 | TCTGTGTGGAATTACTATAAGGCCCCTTTCCTTATGCTTT |
| Pr1906 | TACTCATGATTAACACCTATAGGATAATGCTGAAAACTCC |
| Pr1907 | ACTCATGATTAACACCTATACAAAAAGCACCTCAAAAAGGTATTACC |
| Pr1908 | CCTTTTTGAGGTGCTTTTTGTATAGGTGTTAATCATGAGT |
| Erm F | CCGGGCCCAAAATTTGTTTGAT |
| Erm R | AGTCGGCAGCGACTCATAGAAT |
| P1 | GTTCCAACACTAACTCCTGCT |
| P2 | ATCAAACAAATTTTGGGCCCGGGATGAGTAACCAAAAATAAC |
| P3 | ATTCTATGAGTCGCTGCCGACTCTCTCATGAATATCTTTC |
| P4 | GATTTTACCTGCCAAGAGACC |
| mgaSpn -BgI II | GGAAGATCTTTAAGGAGGCAAATATGAGAGATTTATTATCTAAAAAAAG |
| mgaSpn -XhoI | CCGCTCGAGTTACTCATCTAATCGAATAAAC |
| CPS2A F | TTGTCAGCTCTGTGTCGCTC |
| CPS2A R | TTATCAGTCCCAGTCGGTGC |
| CPS2B F | CTACCTCTCACCGTCGCAAG |
| CPS2B R | CAGCCCCGTAAGCAATGACT |
| CPS2C F | AAACAGCCAGAGGAAGCCAG |
| CPS2C R | GAAGGAGTCGTAGCTGGTCG |
| CPS2D F | TCCTGTCGGTGTCGTGATTG |
| CPS2D R | AAACGGCTTCCCTGTGTGTT |
| TarI F | GGTCAGCACCACCCTTTGTA |
| TarI R | CACACGCATGGGGATCAGTA |
| TarJ F | GTTCCGGTCGGGTCAGAAAT |
| TarJ R | CGTCCCAACTACATGGCTGT |
| licA F | CCGGTGTTTGGTCACTCTCA |
| licA R | CCAATGTGGGATTTGGCTGC |
| licB F | GAGTTGCAGCCACCACAAAG |
| licB R | ACGGAGTTCCTTTTGGCCTT |
| licC F | TTCGCCACTTGCATAAGCCT |
| licC R | CAAGGCAGGTCGCATCCTTA |
| gyrB F | GTTCGTATGCGTCCAGGGAT |
| gyrB R | ATACCACGCCCATCATCCAC |
| Pcps F | TACACATCTGCTTCTAAAATATTGT |
| Pcps R | TTAAAACGTCTACTCATGATTAACA |
| Pcps-bioR | TTAAAACGTCTACTCATGATTAACA (5′-biotinylated) |
| Plic1 F | GAATTCCTGCATAAATCATC |
| PlicP1 R | GAAGGGCAGTAAGTCAAGTA |
| Plic1P1-bioR | GAAGGGCAGTAAGTCAAGTA(5′-biotinylated) |
Construction of mgaSpn mutant strains
Long-arm homologous (LH)-PCR was used to construct an mgaSpn mutant using the genomic DNA of strain D39 (JH1900) as template [26]. In brief, primers P1/P2 and P3/P4 were used to amplify the upstream (438 bp) and downstream (432 bp) of mgaSpn, respectively. The erm-up/erm-down primer pair was used to amplify the erythromycin-resistance gene (780 bp). These amplicons were ligated to obtain a recombinant fragment that was transformed into JH1900 and JH0002. Colonies were selected on erythromycin-containing (100 μg/mL) agar plates and confirmed using PCR with primers P1 and P4 to obtain the erythromycin-resistant mgaSpn mutant strain JH1101 and unencapsulated mgaSpn mutant strain JH1102, respectively.
Mutant strains were complemented with plasmid pPEPZ containing a full-length copy of mgaSpn amplified by PCR and flanked by BgI II and XhoI restriction sites for subsequent cloning purposes. The resulting recombinant plasmid Ppepz-mgaSpn was used to transform JH1101 and then selected on spectinomycin-containing (50 μg/mL) agar plates to obtain the complemented strain JH1104.
Expression and purification of 6× His-MgaSpn
The mgaSpn sequence was amplified from strain D39 genomic DNA (JH1900). mgaSpn was cloned into the NdeI/XhoI restriction site of the pET28 vector and transformed into E. coli BL21 competent cells. The recombinant protein was expressed as a 6× His-tagged N-terminal fusion protein in log-phase growing cultures with 0.25 mM isopropyl β-D-thiogalactoside. Bacterial cells were resuspended in binding buffer (50 mM Tris-HCl, 50 mM NaCl) and subjected to sonication. The 6× His-MgaSpn was purified from the soluble fraction by affinity chromatography using a Ni2+-NTA sepharose column (GE Healthcare, Pittsburgh, PA, USA), and its purity was analyzed by 10% SDS-PAGE. Protein concentration was determined using a NanoDrop1000 Spectrophotometer (Thermo Fisher, Pittsburgh, PA, USA) and analyzed using ND-1000 V3.3.0 software.
Electrophoretic mobility shift assay (EMSA)
The 5′-biotin-labeled and unlabeled DNA fragments containing the promoter region of the Pcps and Plic1p1 probes were amplified from strain D39. First, DNA-binding assays were incubated at 25 °C for 10 min in a 10 μL reaction mixture containing 1 μL 10× binding buffer, 0.5 μg Poly (dI-dC), 0–4 μg 6× His-MgaSpn proteins, and 0.5 ng of the labeled probe. After incubation, the unlabeled probe in 200-fold excess was added as a specific competitor in the cold probe reaction system. Following incubation for 20 min at 25 °C and addition of 1 µL of gel loading buffer, the mixtures were electrophoresed on a 5% native-polyacrylamide gel using 1× TBE buffer. The subsequent step was carried out according to the protocol provided with the LightShift R Chemiluminescent EMSA Kit (Thermo Fisher).
DNase I footprinting
DNase I footprinting assays were performed as described previously [27]. Briefly, fluorescent FAM-labeled probes for cps (Pcps) and lic1p1 (Plic1p1) were amplified using PMD19-cps and PMD19-lic1p1 as templates with primers M13F-47 and M13R-48, respectively. Then, to a 40 µL reaction system, 350 ng probes were incubated with differing concentrations of MgaSpn protein at 25°C for 30 min. A 10-µL solution containing 0.015 units of DNase I (Promega, Madison, WI, USA) and 100 nM of freshly prepared CaCl2 was added to the system. After incubation at 37°C for 1 min, the reaction was stopped by adding 140 μL of terminator solution. The obtained samples were extracted with phenol and precipitated with ethanol. The pellets were dissolved in 30 µL of nuclease-free water. GeneScan-LIZ600 size standard (Applied Biosystems, Foster City, CA, USA) was used for electrophoresis.
RNA extraction and RT-PCR
Total RNA was extracted from 2 mL of log-phase bacterial culture grown in C + Y medium. Bacterial cells were treated with 200 μL of 15 mg/mL lysozyme and 10 μL proteinase K for 30 min at 25°C, and RNA was extracted using an RNAprep pure Cell/Bacteria Kit (Tiangen, Beijing, China). RNA (1 μg) was reverse-transcribed to cDNA using a PrimeScript first-strand cDNA synthesis kit (Takara, Japan). Real-time PCR was performed on CFX ConnectTM (BIO-RAD, Singapore). The gyrB gene was used as an internal reference for data processing. The primers used for PCR are listed in Table 2. The results of representative experiments are presented as the mean of three replicates ± standard deviation.
Enzyme-linked immunosorbent assay (ELISA)
ELISA was carried out using bacteria cultured in C + Y medium to an OD600 of 0.5. The bacterial solution (5 mL) was centrifuged at 13,800 × g for 5 min at 4°C. The supernatant was passed through a 0.22-μm filter and collected. The bacterial cells were washed twice with PBS, suspended in 1 mL PBS, and incubated in a 58°C water bath for 45 min to prepare cell wall samples. The resuspension solution was centrifuged as described above, and the pellets were resuspended in PBS. The samples were coated in a 96-well plate and incubated overnight at 4°C. Type-2 CPS polyclonal antibody (1:5000) (States Serum Institut, København, Denmark) and goat anti-rabbit IgG-HRP antibody were the primary and secondary antibodies used (1:8000) (KPL, Gaithersburg, MD, USA). After color development, the content of CPS on the bacterial surface was determined by measuring the absorbance at 450 nm. The results of representative experiments are presented as the mean of three replicates ± standard deviation.
Western blotting
S. pneumoniae culture suspension (5 mL, grown in C + Y medium to an OD600 of 0.5) was centrifuged and washed. Pellets were suspended in 200 µL SEDS lysis buffer (0.1% deoxycholate, 150 mM NaCl, 0.2% SDS, 15 mM EDTA at pH 8) and incubated at 37°C for 15 min for lysis. Samples were separated using 10% SDS-PAGE with Tris-glycine SDS buffer. The separated samples were transferred to PVDF membranes using the wet transfer method [11]. Membranes were incubated with primary antibodies overnight at 4°C and then washed thrice for 15 min each with TBST followed by incubation with the secondary antibody for 1 h at 37°C. The primary antibodies were directed against type 2 CPS (1:5000 Pneumococcus Type 2 serum), TAs (1:1000 rabbit anti-CWPS [cell wall polysaccharide] IgG), P-Cho (1:2000 mouse anti-TEPC-15 A mouse IgAκ monoclonal antibody purified from murine myeloma clone TEPC-15) [28–30], and MgaSpn (1:5000 mouse anti-MgaSpn). The secondary antibody used was goat anti-rabbit IgG (1:10,000) or goat anti-mouse IgA (1:8000).
Fluorescence activated cell sorting (FACS)
Briefly, 200 μL of S. pneumoniae cultured in C + Y medium to an OD600 of 0.5 was harvested by centrifugation at 9600 × g for 5 min. Pellets were washed with 500 μL of 0.05% PBST and centrifuged again. The washed pellets were suspended in 100 μL of 1% BSA. The samples were incubated with the respective primary antibodies for 1 h at 25°C. After incubation, the pellets were washed and resuspended as described above. The samples were stained with fluorescent-labeled secondary antibodies for 1 h at 25°C in the dark. Finally, the samples were suspended in 500 μL PBS and analyzed by technicians at the Children’s Hospital of Chongqing Medical University.
The primary antibodies along with their targets listed were as follows: TAs, 1:50 rabbit anti-CWPS; P-Cho, 1:25 mouse anti-TEPC IgA. The following secondary antibodies were used: TAs, 1:50 goat anti-rabbit IgG PE fluorescent antibody, and P-Cho, 1:50 goat anti-mouse IgA PE fluorescent antibody.
Adhesion assays
A549 cells were seeded in a 24-well plate at a density of 2 × 105/well and cultured at 37°C overnight. The bacterial suspensions were diluted with DMEM (Thermo Fisher) to 1 × 108 colony forming units (CFU)/mL, and 500 µL volumes were used at a multiplicity of infection (MOI) of 100 and incubated with A549 cells at 37°C for 30 min. The input bacteria were enumerated by plating serial dilutions [31]. Following incubation, the cells were gently rinsed five times with PBS. For adhesion assays, cells were lysed in sterile ddH2O. The lysates were serially diluted and plated on blood agar plates to determine intracellular and extracellular CFUs. For invasion assays, extracellular bacteria were treated with 200 mg/mL penicillin and 200 mg/mL gentamicin for 15 min. Intracellular CFUs were then determined as described above. Adhesion and invasion rates were calculated using the following formulas:
| (1) |
| (2) |
Animal experiments
Male C57BL/6 mice (6–8 weeks old, weighing ~20–21 g) were purchased from the Laboratory Animal Center of Chongqing Medical University. All animal experiments described in this study were approved by the Animal Care and Use Committee of Chongqing Medical University and were performed in strict accordance with the regulations of the Guide for the Care and Use of Laboratory Animals.
To investigate the effects of MgaSpn on the colonization of the mouse nasopharynx, we randomly divided the mice into three groups (n = 6 per group). Each mouse was inoculated with 2 × 107 CFU of bacteria through the nasal cavity. Nasal lavage fluid, heart blood, spleen, and lung tissues were collected and ground using a mechanical mortar and pestle. The samples were plated on blood agar plates after the appropriate dilutions to determine the CFU.
Furthermore, the role of MgaSpn in systemic infections was explored by dividing the mice into three groups (n = 12 per group). Each mouse was infected intranasally with 1 × 107 CFU of bacteria. The survival rate of the mice was monitored daily for 14 days. Moribund mice were not euthanized prior to the completion of the 14-day survival study. The lungs were first fixed with 4% paraformaldehyde for 48 h at 4°C, then embedded in paraffin and sectioned at 5-mm thickness, and finally stained with hematoxylin-eosin (H&E) using standard methods [32].
Statistical analysis
Statistical difference between groups were compared using Student’s t test, non-parametric Mann-Whitney, or Wilcoxon test using GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA). Survival data were analyzed using the log rank (Mantel-Cox) test. Statistical significance was defined as P < 0.05.
Results
MgaSpn binds the cps promoter region
To determine whether MgaSpn specifically binds to the cps promoter region in vitro, we purified a 6× His-tagged MgaSpn protein and used a 5′-biotin-labeled Pcps probe for EMSA. As the protein concentration increased, the bands for the probe and protein shifted, indicating that the binding of MgaSpn and Pcps was concentration-dependent. Two shifted bands were observed in each lane when > 2.0 μg of MgaSpn was added to the reaction system, indicating the presence of two binding sites for MgaSpn in the cps promoter. The mobility shift could be outcompeted with a 200-fold excess of an identical unlabeled Pcps probe (Figure 1(a)). These results indicate that MgaSpn specifically binds to the promoter region of the cps operon.
Figure 1.
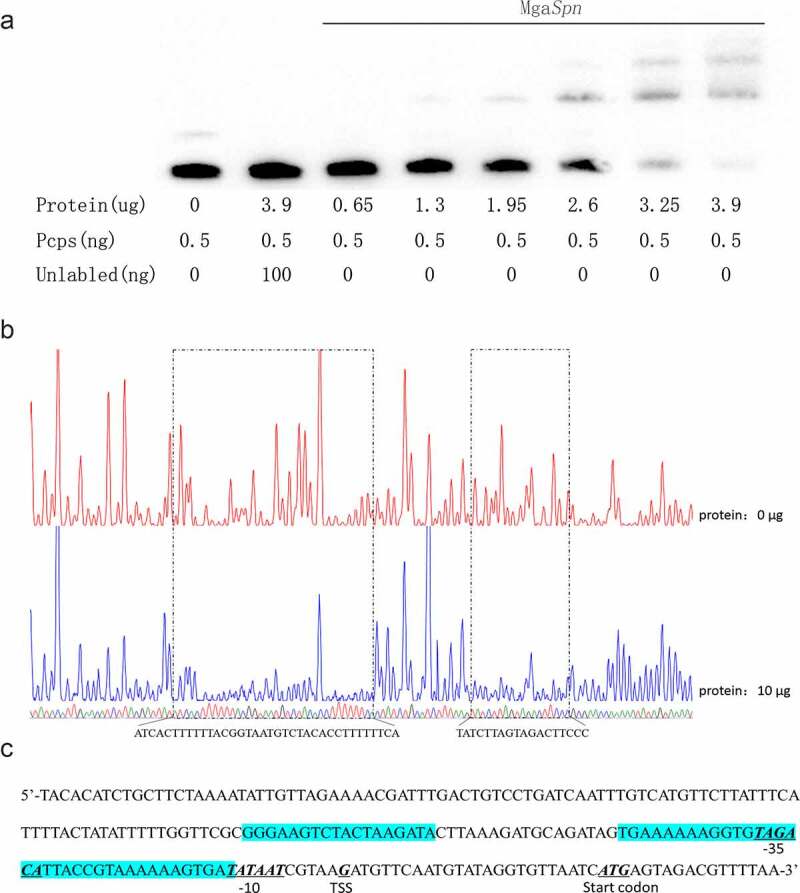
MgaSpn binds to cps probe
(a) EMSA of Pcps using the MgaSpn protein. (b) DNase I footprinting protection assay of MgaSpn. (c) Structural organization of the cps promoter-proximal region. The −10, −35, and predicted transcriptional start site (TSS) and start codon are underlined. Binding sites for MgaSpn are shown in blue.
To further determine the binding site more precisely, DNase I footprinting was performed using the same 218-bp long cps promoter fragment labeled with FAM. We identified two protection sites on the cps promoter. The first was a weak site of the 18-bp fragment and −64 to −81 bp relative to the transcription start site of the cps operon. The second was a stronger protected site of the 36-bp fragment (5′-TGAAAAAAGGTGTAGACATTACCGTAAAAAAGTGAT-3′) and contained the −35 box of the cps promoter (in bold italics) (Figure 1(b)).
MgaSpn is an inhibitor of cps operon expression and negatively regulates intracellular CPS production
qRT-PCR was performed to determine the relative mRNA levels of the first four genes (cps2A-D) of the cps operon to investigate whether MgaSpn could regulate the expression of cps operons. We replaced mgaSpn with an erm gene cassette to generate the knockout strain JH1101. Steady-state mRNA levels in JH1101 for cps2A/C/D were higher than those in the parental strain JH1900. Deletion of mgaSpn caused increased mRNA levels in the cps operon, indicating that MgaSpn may negatively regulate CPS biosynthesis (Figure 2(a)).
Figure 2.
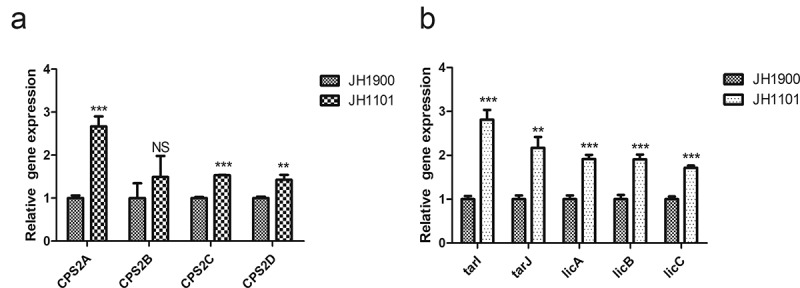
qPCR to analyze the relative mRNA levels of JH1900 and JH1101
(a) Relative mRNA levels of the first four genes cps2A-D downstream of the cps operon. (b) Relative mRNA levels of tarI, tarJ, licA, licB, and licC downstream genes in the lic1 operon. mRNA levels are expressed relative to that of gyrB. Data are presented as the mean ± SD from three independent experiments, each determined in duplicate. ***P < 0.001; **P < 0.01; NS, not significant as analyzed by unpaired two-tailed Student’s t-test.
The CPS of S. pneumoniae is important for its virulence; therefore, we determined the relative levels of CPS in the parental and mutant strains. We found that neither the cell wall nor the supernatant capsules were significantly altered between strains JH1900 and JH1101 (Figure 3(a,b)). These results suggest that the amount of CPS on the bacterial surface is not regulated by MgaSpn. However, the total CPS levels for JH1900, JH1101, and the complemented strain JH1104 were altered (Figure 3(c)). The levels were significantly increased in the mutant strain JH1101 and could be restored to the parental levels in the complemented strain JH1104. We also found additional small-molecular-weight bands in the mutant strain (Figure 3(c)). Collectively, these findings indicate that MgaSpn can modulate the biosynthesis of intracellular low-molecular-weight capsule components by repressing the expression of the cps operon in S. pneumoniae. We hypothesized that this regulation is possibly due to the inability of the newly synthesized low-molecular-weight precursor molecules to be released outside the cell.
Figure 3.
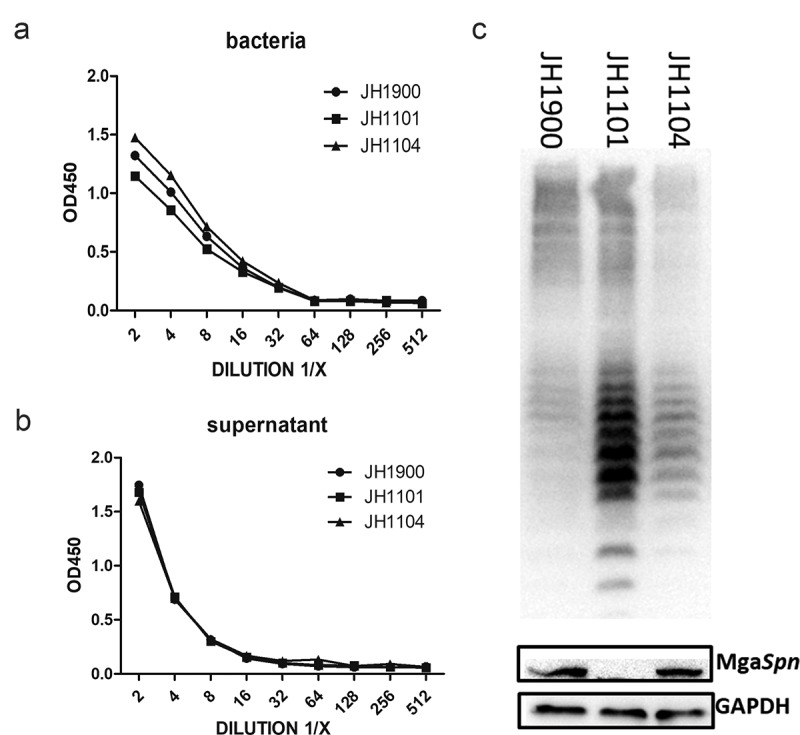
MgaSpn negatively regulates CPS production
(a) ELISA of CPS on bacterial surfaces. (b) CPS content in bacterial culture supernatants. (c) CPS banding patterns from whole-cell lysates for the indicated strains using western blotting. GADPH was used as a sample loading control. ELISA and western blots were probed with a rabbit anti-serotype 2 CPS polyclonal antibody. The results of representative experiments are presented as the mean of three replicates ± SD.
MgaSpn specifically binds to the lic1 promoter region
MgaSpn is a member of the Mga/AtxA family; thus, it may also regulate the biosynthesis of other virulence factors, similar to other members of this family. We utilized Plic1-F and Plic1P1-5′-bioR to amplify the 305-bp DNA fragment upstream of the lic1 operon as the lic1p1 probe. This amplicon contained two promoter sequences that regulate lic1 genes. The lic1p1 probe could readily be upshifted by adding MgaSpn to the EMSA reaction setup. The shifted band could compete with the 5′-end biotin-unlabeled lic1p1 DNA fragment, indicating that the binding was specific (Figure 4(a)).
Figure 4.
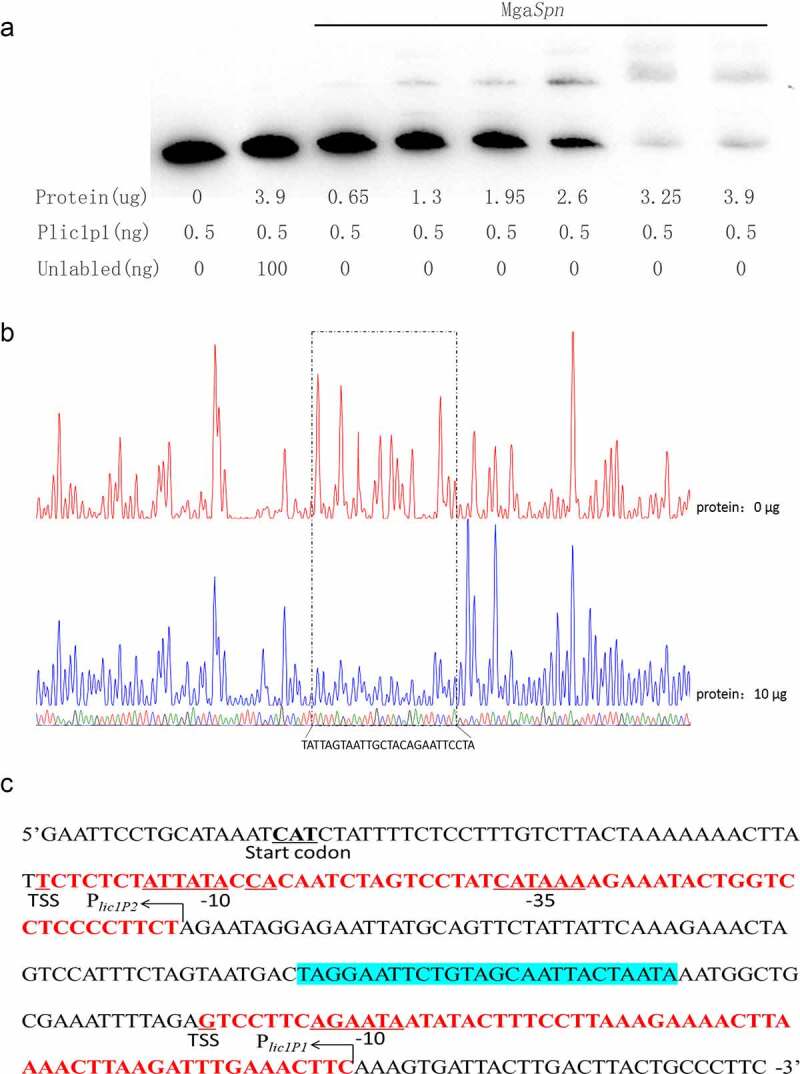
MgaSpn binds to lic1p1 probe
(a) EMSA of lic1p1 in the presence of MgaSpn protein. The biotin-labeled 305 bp Plic1p1 probe was incubated with the indicated amounts of 6× His-MgaSpn protein. For the competition analysis, the same but unlabeled DNA probe was included in a 200-fold concentration relative to the biotin-labeled probe. (b) DNase I footprinting protection assay of MgaSpn. (c) Structural organization of the lic1 promoter-proximal region. The −10 and −35 regions and TSS and start codon are underlined. Binding sites for MgaSpn are shown in blue. Plic1P1 and Plic1P2 are in red.
To further confirm the binding of MgaSpn to lic1 promoters, DNase I footprinting assays were performed to determine the specific recognition site of MgaSpn on the lic1p1 probe (Figure 4(b)). When 10 μg of MgaSpn protein was added to the system containing 450 ng of the lic1p1 probe, a protected region of 18 bp (5′-TAGGAATTCTGTAGCAATTACTAATA-3)′ appeared between Plic1P1 and Plic1P2 (Figure 4(c)). These data indicate that the MgaSpn recognition site lies between Plic1P1 and Plic1P2. Therefore, MgaSpn may regulate the co-transcription of lic1 genes.
MgaSpn represses P-Cho synthesis
Based on MgaSpn-specific binding to the lic1p1 probe, we hypothesized that MgaSpn could transcriptionally regulate TA biosynthesis. To test this, we quantified the mRNA levels of the lic1 operon for strains JH1900 and JH1101. The expression levels of tarI, tarJ, licA, licB, and licC were increased in the mgaSpn mutant strain (Figure 2(b)). This indicates that MgaSpn inhibited the downstream gene expression of the lic1 operon. In another experiment, the total P-Cho levels for JH1101 were higher than those for the parental JH1900 strain, whereas the TA content did not change (Figure 5(a)).
Figure 5.
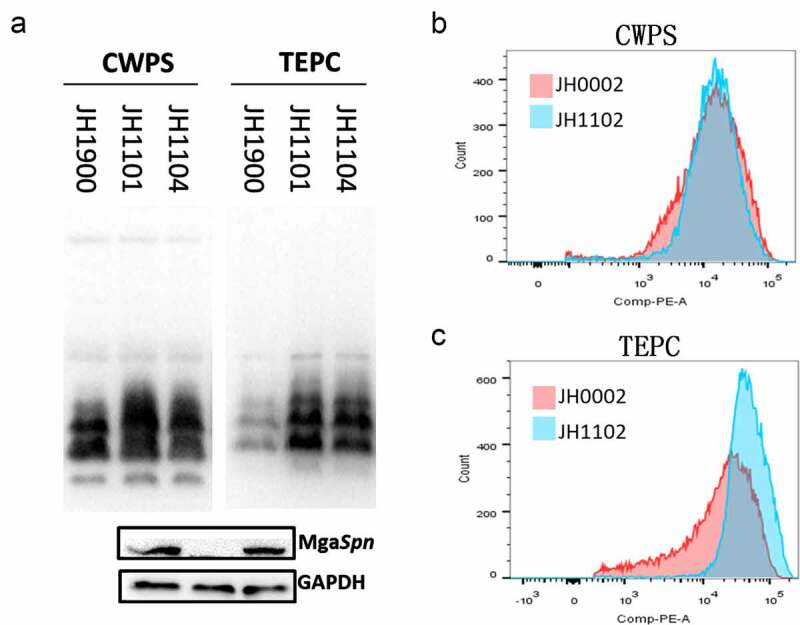
Detection of teichoic acids and phosphorylcholine
(a) TAs and P-Cho banding patterns from of whole-cell lysates of the indicated strains using western blotting. (b) Cell surface TAs determined with flow cytometry. (c) P-Cho content. CWPS was used as a TA probe and TEPC-15 was used as a P-Cho probe. GADPH was used as a sample loading control. The results of representative experiments are presented as the mean of three replicates ± SD.
Bacterial capsules shield TAs and P-Cho, and these components may be hindered by antibodies in encapsulated strains. Therefore, we knocked out the cps promoter region using JH1900 to construct the unencapsulated strain JH0002. Next, we constructed the unencapsulated defective strain JH1102 and the unencapsulated complemented strain JH1106. The surface TA content of JH1102 was similar to that of JH0002 (Figure 5(b)). However, the P-Cho fluorescence intensity of strain JH1102 was higher than that of JH0002, and the flow peak pattern of the mutant strain JH1102 was sharp and thin (Figure 5(c)). Such findings indicate that the knockout of mgaSpn might affect the mode of P-Cho modification to TA.
Deletion of mgaSpn influences adhesion and pathogenicity of S. pneumoniae
To understand how the increase in the intracellular capsule plays a role in the virulence of S. pneumoniae, the lung epithelial cell line A549 was used to determine the adhesion and invasion abilities of strains JH1900, JH1101, and JH1104. Strain JH1900 adhered to the epithelial cells at a higher level, but the mutant strain JH1101 possessed a greater invasive ability (Figure 6(a,b)). Following the removal of the capsule and exposure of more surface adhesion factors, we found that JH1102 was able to adhere and invade more efficiently than JH0002 (Figure 6(c,d)); most likely a consequence of the increased P-Cho levels. Cell infection experiments show that the loss of mgaSpn increased the invasiveness of S. pneumoniae D39.
Figure 6.
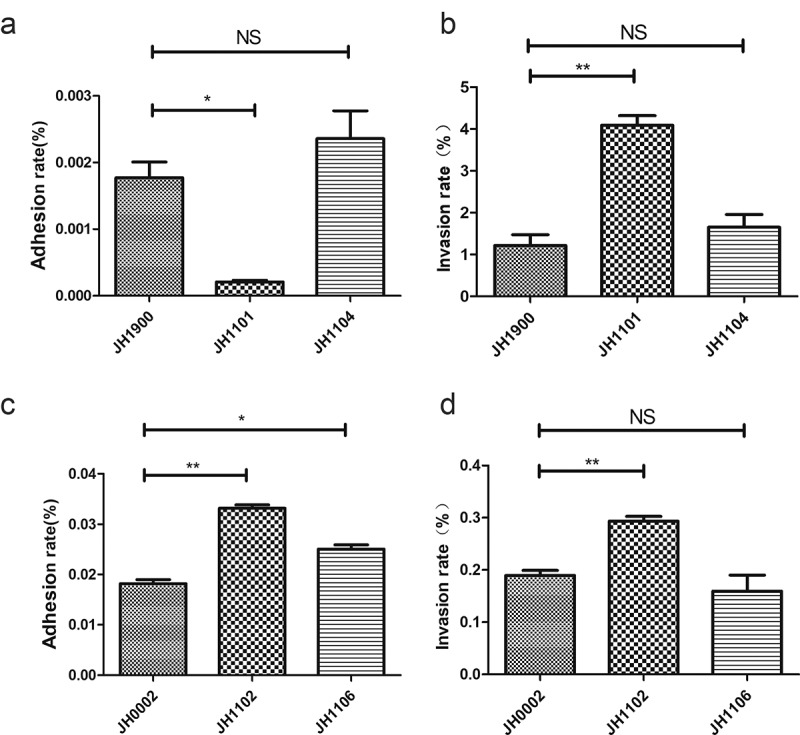
Infection of epithelial cells
Bacterial adherence and invasion of A549 cells. The cells were infected at an MOI of 100 with the indicated strains. (a, b) Adhesion and invasion rates of JH1900-, JH1101-, and JH1104-infected A549 cells. (c, d) Adhesion and invasion rates of unencapsulated JH1900-, JH1101-, and JH1104-infected A549 cells. ***P < 0.001; **P < 0.01; *P < 0.05; NS, not significant, analyzed by unpaired two-tailed Student’s t-test.
MgaSpn is involved in systemic virulence and nasopharyngeal colonization
Results of the in vitro experiments indicated that the encapsulated mutant strain JH1101 was similar to parental levels in its ability to phagocytose (data not shown), although its ability to invade cells increased. In addition, mgaSpn deletion resulted in increased P-Cho production, which increased invasion and adhesion to epithelial cells. These findings increased our interest in exploring the role of MgaSpn in systemic infections. Three groups of mice were challenged with strains JH1101, JH1900, and JH1104. The bacterial load after 48 h in the nasal lavage fluid of mice in the JH1101 infection group was higher than that in the JH1900 group (Figure 7(a)). We also observed that more bacteria invaded the blood and spleen in the JH1101 infected group, and the levels of bacterial load in the blood and spleen in the JH1900 and JH1104 groups were not statistically significant (Figure 7(c,d)). However, the lung colonization ability of JH1101 did not increase, as predicted (Figure 7(b)). However, the lung hematoxylin-eosin (H&E) staining demonstrated increased inflammatory cell infiltration and pathological injury in the JH1101 infected group (Figure 7(f)). These results indicated that JH1101 could colonize the nasal cavity and cause systemic infections better than the parental strain JH1900. Furthermore, mice challenged with JH1101 died within 4 days, whereas 20% of mice challenged with JH1900 and JH1104 survived (Figure 7(e)). Overall, these results were consistent with the in vitro virulence experiments, indicating that knockout of mgaSpn increases S. pneumoniae virulence.
Figure 7.
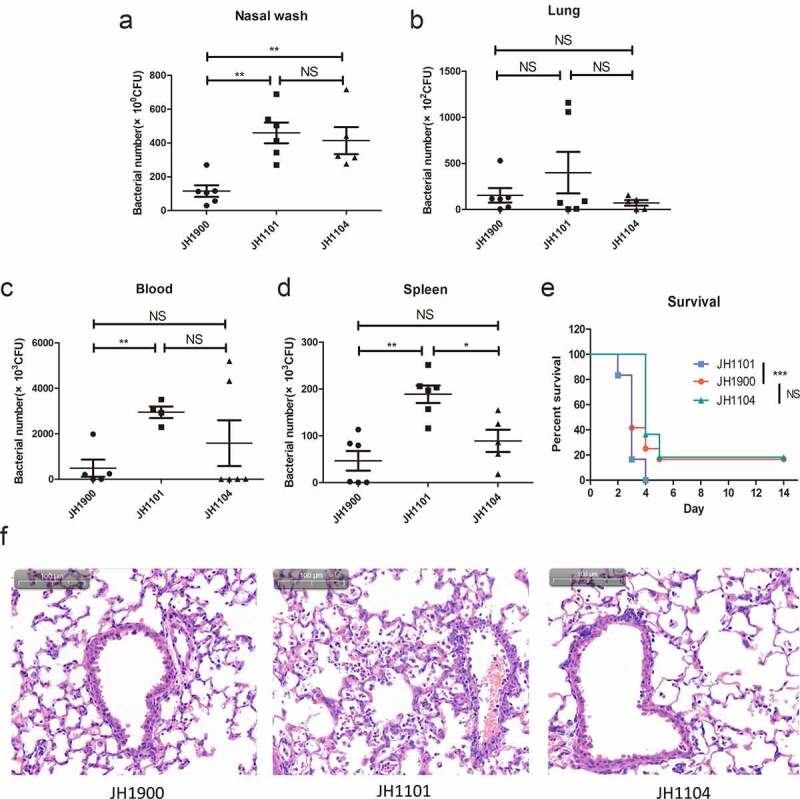
Bacterial colonization and survival of infected mice
(a) Nasopharyngeal colonization of mice (n = 6 per group) infected with the indicated strains at 2 × 107 CFU intranasally and collected from the nasopharyngeal lavage fluid 48 h post-infection. CFU of infected bacteria present in (b) lung homogenates, (c) blood, and (d) spleen homogenates. (e) Survival of mice (n = 12 for each group) after intranasal infection with 1 × 107 CFU. Survival was analyzed using log-rank comparisons. ***P < 0.001 (f) Photomicrographs of H&E-stained lung tissues of mice 48 h after challenge with the indicated strains with the same challenge dose as the colonization experiments.
Discussion
CPS of S. pneumoniae is a primary virulence factor, and its synthesis is regulated by the cps operon [8,33]. Even point mutations at key sites in the cps promoter are known to alter downstream gene expression and significantly alter the amount of CPS present on the cell surface [10,34]. In a previous study [11], a MgaSpn protein that could bind to the capsule promoter was screened by DNA pull-down assay and later found to play a role in the synthesis of the capsule and P-Cho. This study focused on the regulatory mechanism of MgaSpn on cell wall-associated surface capsules and P-Cho.
Both Mga and AtxA, members of the Mga/AtxA transcriptional regulation family, have been proven to be stand-alone response regulators capable of sensing signals from the peripheral environment [35,36]. When the pathogen is in a favorable growth environment, the transcription level of the Mga operon is activated, and the transcription level of the virulence genes regulated by Mga also changes correspondingly [36,37]. A subtle difference was noted in previous studies on whether MgaSpn affects the growth of S. pneumoniae: JH1900 and JH1101 reached the logarithmic stage simultaneously under normal culture conditions or at high glucose concentrations, but JH1101 often reached the logarithmic stage slowly compared with JH1900 at low glucose concentrations. Such findings suggest that MgaSpn can perceive changes in the concentration of external carbon sources. Unfortunately, we did not observe a statistical significance of this growth difference in a low-carbon environment, which may be because pathogenic bacteria quickly activate a compensation mechanism to eliminate this difference in an unfavorable environment [14,38,39]. In addition, the transcription level of mgaSpn is significantly increased during lung infection and nasopharyngeal colonization [14]. These findings indicate that MgaSpn plays an important role in the environmental adaptation of S. pneumoniae.
MgaSpn was originally defined as a transcriptional activator. This study and Hemsley et al.’s studies have demonstrated its repressive transcriptional function [2]. MgaSpn, a putative transcriptional regulator containing two conserved helix-turn-helix domains [33], regulates its target genes by direct DNA binding. EMSA and DNase I footprinting assay determined that MgaSpn specifically binds to the promoter regions of cps and lic1. Furthermore, MgaSpn negatively regulated the transcription of the cps and lic1 operons. In addition, ComE [11], RegM [40], ComX [41], CopY [42], CpsR [43], and GlnR [44] have been reported as possible transcription regulatory factors of the cps gene locus. At present, the research on the transcriptional regulation of the lic1 operon is scarce; CiaR is the only confirmed response regulator of lic1 operon, and thus this discovery sheds new light on transcriptional regulation of the lic1 operon [20,45].
However, the surface capsule of JH1101 did not show a significant difference when compared with JH1900, although the production of intracellular low-molecular-weight CPS was significantly increased in JH1101. The synthesis of CPS is divided into several stages: precursor synthesis, repeating unit polymerization, polymer inversion, and final positioning [46]. As the low-molecular-weight CPS accumulated in the mgaSpn mutant, we hypothesized that MgaSpn might be involved in the process of CPS precursor synthesis or repeating unit polymerization. Low-molecular-weight CPS has been previously observed in cps2C and cps2D null strains [47]. cps2B, cps2C, and cps2D constitute the tyrosine kinase phospho-regulatory system of S. pneumoniae [48]. Dephosphorylation of cps2D contributes to an increase in low-molecular-weight CPS [49,50]. The ExoP protein of the soil bacterium Sinorhizobium meliloti is a cps2D ortholog and a member of the tyrosine phosphatase system. Site-directed mutagenesis of the ExoP-encoding gene increased production of low-molecular-weight succinoglycans rather than high-molecular-weight succinoglycans [51], indicating polysaccharide polymer length is controlled by tyrosine kinase phospho-regulation. Thus, MgaSpn might affect the tyrosine phosphorylation process of cps2D, leading to an increase in low-molecular-weight CPS, but more experimental evidence is needed in this regard.
In the course of these studies, we found that the mutant strain JH1101 produced elevated levels of P-Cho relative to the parental strain. TA biosynthesis requires the synergy of multiple operons, in which the genes contained in the lic1 and lic2 operons play important roles in the formation of the core TA repeat unit [15]. We have demonstrated that MgaSpn is a negative regulator of the lic1 operon that is responsible for the production of activated ribitol and P-Cho [15]. Expression of Plic1 downstream genes increased the production of total P-Cho in strain JH1101. The genes contained in the lic2 operon were responsible for “decorating” TA precursors with P-Cho (licD1 and licD2) and transporting TA chains across the membrane (tacF) [15,52]. However, licD1/2 mRNA levels in strain JH1101 did not significantly differ from that of the parental strain, indicating that MgaSpn most likely regulates P-Cho biosynthesis primarily via the lic1 operon.
A thickened capsule enhances the virulence of S. pneumoniae and the resistance of phagocytes to phagocytosis. At the same time, an increase in the capsule thickness will also weaken the adhesion of S. pneumoniae to the upper respiratory tract mucosa [3]. In our study, strain JH1101 was not shown to be more resistant to phagocytosis, consistent with the similarity in the amount of CPS to the parental strain JH1900. We hypothesized that the change in small-molecular-weight capsules did not affect the phagocytic capacity of the bacteria. Furthermore, the adhesion ability of JH1101 to A549 cells was less efficient than that of the parental strain but invaded to a greater extent. The target cell preference of unencapsulated strains differs from that of encapsulated strains and is most likely an effect of cell wall-associated protein exposure [53]. Strain JH1102 possessed higher P-Cho levels but no capsule, which showed the highest adherence and invasive ability among all strains.
Capsular thickness and density are dynamic processes that are regulated according to the stage of colonization and infection [54]. To effectively colonize the nasal mucosa, S. pneumoniae needs to initially detach a part of the capsule at the adhesion site to expose more cell wall proteins [55]. In vivo experiments indicated that JH1101 was better at colonizing the nasal cavity than JH1900. This improved colonization may be related to increased P-Cho levels in strain JH1101; however, no statistically significant differences were observed in lung bacterial loads of JH1101 and JH1900. H&E staining of infected mouse lungs demonstrated higher levels of inflammatory cells in the lungs of JH1101 group mice, indicating severe lung damage. These results may indicate that JH1101 can quickly move to other organs through blood after causing lung inflammation. Consistently, the blood and spleen bacterial loads of the JH1101 infection group were significantly higher than those of the JH1900 infection group. In conclusion, both in vivo and in vitro experiments demonstrated that deletion of mgaSpn enhanced the invasion ability of S. pneumoniae.
Herein, we report for the first time that MgaSpn is a negative regulator of the cps and lic1 operons and can affect the biosynthesis of low-molecular-weight capsules and P-Cho. While our data indicated that mgaSpn deletion did not result in increased surface CPS levels, it did lead to the accumulation of low-molecular-weight capsule components. Furthermore, the mgaSpn mutant strain produced higher levels of P-Cho than the parental D39 strain. Finally, knockout of mgaSpn resulted in higher pathogenicity, which may be related to the increase in P-Cho production. An in-depth understanding of the microenvironment-induced transcriptional regulator MgaSpn warrants clarification on the occurrence and development of pneumococcal-mediated diseases and lead to new treatment strategies for infections caused by this pathogen.
Acknowledgments
This work was supported by the Projects of the National Natural Science Foundation of China (No. 81871698 and No. 81772153). The authors thank Prof. Shifang Dong for their technical assistance and the Experimental Animal Center of Chongqing Medical University for the feeding and care of the mice. We would like to thank Editage (www.editage.cn) for English language editing.
Funding Statement
This work was supported by the Projects of the National Natural Science Foundation of China under Grant [No. 81871698 and No.81772153].
Disclosure statement
No potential conflict of interest was reported by the authors.
Author contributions
YZ, XZ, YY, and JZ conceived and designed the experiments. SX, XG, and WS performed the experiments. SX and JZ analyzed the data. YZ, XZ, and JZ contributed reagents, materials, and analytical tools. SX and XZ wrote the paper. All authors have read and approved the final manuscript.
Data availability statement
The data that support the findings of this study are openly available in figshare at https://doi.org/ 10.6084/m9.figshare.14223077.v1.
References
- [1].Kadioglu A, Weiser JN, Paton JC, et al. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat Rev Microbiol. 2008;6(4):288–301. [DOI] [PubMed] [Google Scholar]
- [2].Hemsley C, Joyce E, Hava DL, et al. MgrA, an orthologue of Mga, acts as a transcriptional repressor of the genes within the rlra pathogenicity Islet in Streptococcus pneumoniae. J Bacteriol. 2003;185(22):6640–6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Engholm DH, Kilian M, Goodsell DS, et al. A visual review of the human pathogen Streptococcus pneumoniae. FEMS Microbiol Rev. 2017;41(6):854–879 [DOI] [PubMed] [Google Scholar]
- [4].Middleton DR, Paschall AV, Duke JA, et al. Enzymatic hydrolysis of pneumococcal capsular polysaccharide renders the bacterium vulnerable to host defense. Infect Immun. 2018;86(8). DOI: 10.1128/IAI.00316-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zafar MA, Hamaguchi S, Zangari T, et al. Capsule type and amount affect shedding and transmission of Streptococcus pneumoniae. mBio. 2017;8(4). DOI: 10.1128/mBio.00989-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Winkelstein JA.The role of complement in the host’s defense against Streptococcus pneumoniae. Rev Infect Dis. 1981;3(2):289–298. [DOI] [PubMed] [Google Scholar]
- [7].Lanie JA, Ng W-L, Kazmierczak KM, et al. Genome sequence of Avery’s virulent Serotype 2 Strain D39 of Streptococcus pneumoniae and comparison with that of unencapsulated laboratory strain R6. J Bacteriol. 2007;189(1):38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wen Z, Sertil O, Cheng Y, et al. Sequence elements upstream of the core promoter are necessary for full transcription of the capsule gene operon in Streptococcus pneumoniae strain D39. Infect Immun. 2015;83(5):1957–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yother J.Capsules of Streptococcus pneumoniae and other bacteria: paradigms for polysaccharide biosynthesis and regulation. Annu Rev Microbiol. 2011;65(1):563–581. [DOI] [PubMed] [Google Scholar]
- [10].Wang J,Xu X, Ma F, et al. Identification of a point mutation in the promoter region of cps operon responsible for capsular polysaccharide deficiency in Streptococcus pneumniae SPY1. 2015;55(6):780–787 [PubMed] [Google Scholar]
- [11].Zheng Y, Zhang X, Wang X, et al. ComE, an essential response regulator, negatively regulates the expression of the capsular polysaccharide locus and attenuates the bacterial virulence in Streptococcus pneumoniae. Front Microbiol. 2017;8:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Solano-Collado V, Espinosa M, Bravo A. Activator role of the pneumococcal Mga-like virulence transcriptional regulator. J Bacteriol. 2012;194(16):4197–4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hause LL, McIver KS. Nucleotides critical for the interaction of the Streptococcus pyogenes Mga virulence regulator with Mga-regulated promoter sequences. J Bacteriol. 2012;194(18):4904–4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Aprianto R, Holsappel S, Veening JW, et al. High-resolution analysis of the pneumococcal transcriptome under a wide range of infection-relevant conditions. Nucleic Acids Res. 2018;46(19):9990–10006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Denapaite D, Brückner R, Hakenbeck R, et al. Biosynthesis of teichoic acids in Streptococcus pneumoniae and closely related species: lessons from genomes. Microb Drug Resist. 2012;18(3):344–358. [DOI] [PubMed] [Google Scholar]
- [16].Cundell DR, Gerard NP, Gerard C, et al. Streptococcus pneumoniae anchor to activated human cells by the receptor for platelet-activating factor. Nature. 1995;377(6548):435–438. [DOI] [PubMed] [Google Scholar]
- [17].Hakenbeck R, Madhour A, Denapaite D, et al. Versatility of choline metabolism and choline-binding proteins in Streptococcus pneumoniae and commensal streptococci. FEMS Microbiol Rev. 2009;33(3):572–586. [DOI] [PubMed] [Google Scholar]
- [18].Marx P, Meiers M, Brückner R. Activity of the response regulator CiaR in mutants of Streptococcus pneumoniae R6 altered in acetyl phosphate production. Front Microbiol. 2014;5:772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Halfmann A, Schnorpfeil A, Müller M, et al. Activity of the Two-Component regulatory system CiaRH in Streptococcus pneumoniae R6. J Mol Microbiol Biotechnol. 2011;20(2):96–104. [DOI] [PubMed] [Google Scholar]
- [20].Halfmann A, Kovács M, Hakenbeck R, et al. Identification of the genes directly controlled by the response regulator CiaR in Streptococcus pneumoniae: five out of 15 promoters drive expression of small non-coding RNAs. Mol Microbiol. 2007;66(1):110–126. [DOI] [PubMed] [Google Scholar]
- [21].Bertani G. Lysogeny at mid-twentieth century: P1, P2, and other experimental systems. J Bacteriol. 2004;186(3):595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gutu AD, Wayne KJ, Sham L-T, et al. Kinetic Characterization of the WalRKSpn (VicRK) Two-Component System of Streptococcus pneumoniae : dependence of WalKSpn (VicK) Phosphatase Activity on Its PAS Domain. J Bacteriol. 2010;192(9):2346–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sung CK, Li H, Claverys JP, et al. An rpsL cassette, janus, for gene replacement through negative selection in Streptococcus pneumoniae. Appl Environ Microbiol. 2001;67(11):5190–5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ulijasz AT, Falk SP, Weisblum B. Phosphorylation of the RitR DNA-binding domain by a Ser-Thr phosphokinase: implications for global gene regulation in the streptococci. Mol Microbiol. 2009;71(2):382–390. [DOI] [PubMed] [Google Scholar]
- [25].Lu L, Ma Y, Zhang JR. Streptococcus pneumoniae recruits complement factor H through the amino terminus of CbpA. J Biol Chem. 2006;281(22):15464–15474. [DOI] [PubMed] [Google Scholar]
- [26].Wu K, Zhang X, Shi J, et al. Immunization with a combination of three pneumococcal proteins confers additive and broad protection against Streptococcus pneumoniae infections in mice. Infect Immun. 2010;78(3):1276–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wang Y, Cen X-F, Zhao G-P, et al. Characterization of a new GlnR binding box in the promoter of amtB in Streptomyces coelicolor Inferred a PhoP/GlnR competitive binding mechanism for transcriptional regulation of amtB. J Bacteriol. 2012;194(19):5237–5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Stuertz K, Merx I, Eiffert H, et al. Enzyme immunoassay detecting teichoic and lipoteichoic acids versus cerebrospinal fluid culture and latex agglutination for diagnosis of Streptococcus pneumoniae meningitis. J Clin Microbiol. 1998;36(8):2346–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kolberg J, Høiby EA, Jantzen E. Detection of the phosphorylcholine epitope in streptococci, Haemophilus and pathogenic Neisseriae by immunoblotting. Microb Pathog. 1997;22(6):321–329. [DOI] [PubMed] [Google Scholar]
- [30].Wu K, Huang J, Zhang Y, et al. A novel protein, RafX, is important for common cell wall polysaccharide biosynthesis in Streptococcus pneumoniae: implications for bacterial virulence. J Bacteriol. 2014;196(18):3324–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gupta R, Yang J, Dong Y, et al. Deletion of arcD in Streptococcus pneumoniae D39 impairs its capsule and attenuates virulence. Infect Immun. 2013;81(10):3903–3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gou X, Yuan J, Wang H, et al. IL-6 during influenza-streptococcus pneumoniae co-infected pneumonia-a protector. Front Immunol. 2019;10:3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].García E, Llull D, Muñoz R, et al. Current trends in capsular polysaccharide biosynthesis of Streptococcus pneumoniae. Res Microbiol. 2000;151(6):429–435. [DOI] [PubMed] [Google Scholar]
- [34].Shainheit MG, Mulé M, Camilli A. The core promoter of the capsule operon of Streptococcus pneumoniae is necessary for colonization and invasive disease. Infect Immun. 2014;82(2):694–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Dale JL, Raynor MJ, Ty MC, et al. A dual role for the bacillus anthracis master virulence regulator AtxA: control of sporulation and anthrax toxin production. Front Microbiol. 2018;9:482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Buckley SJ, Davies MR, McMillan DJ. In silico characterisation of stand-alone response regulators of Streptococcus pyogenes. PLoS One. 2020;15(10):e0240834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hondorp ER, McIver KS. The Mga virulence regulon: infection where the grass is greener. Mol Microbiol. 2007;66(5):1056–1065. [DOI] [PubMed] [Google Scholar]
- [38].Sztal TE, Stainier DYR. Transcriptional adaptation: a mechanism underlying genetic robustness. Development. 2020;147(15). DOI: 10.1242/dev.186452 [DOI] [PubMed] [Google Scholar]
- [39].Rossi A, Kontarakis Z, Gerri C, et al. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature. 2015;524(7564):230–233. [DOI] [PubMed] [Google Scholar]
- [40].Giammarinaro P, Paton JC. Role of RegM, a homologue of the catabolite repressor protein CcpA, in the virulence of Streptococcus pneumoniae. Infect Immun. 2002;70(10):5454–5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Luo P, Morrison DA. Transient association of an alternative sigma factor, ComX, with RNA polymerase during the period of competence for genetic transformation in Streptococcus pneumoniae. J Bacteriol. 2003;185(1):349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Reyes A, LEIVA A, CAMBIAZO V, et al. Cop-like operon: structure and organization in species of the Lactobacillale order. Biol Res. 2006;39(1):87–93. [DOI] [PubMed] [Google Scholar]
- [43].Wu K, Xu H, Zheng Y, et al. CpsR, a GntR family regulator, transcriptionally regulates capsular polysaccharide biosynthesis and governs bacterial virulence in Streptococcus pneumoniae. Sci Rep. 2016;6(1):29255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kloosterman TG, Hendriksen WT, Bijlsma JJE, et al. Regulation of glutamine and glutamate metabolism by GlnR and GlnA in Streptococcus pneumoniae. J Biol Chem. 2006;281(35):25097–25109. [DOI] [PubMed] [Google Scholar]
- [45].Johnston C, Hauser C, Hermans PWM, et al. Fine-tuning of choline metabolism is important for pneumococcal colonization. Mol Microbiol. 2016;100(6):972–988. [DOI] [PubMed] [Google Scholar]
- [46].Cartee RT, Forsee WT, Bender MH, et al. CpsE from Type 2 Streptococcus pneumoniae Catalyzes the Reversible Addition of Glucose-1-Phosphate to a Polyprenyl Phosphate Acceptor, Initiating Type 2 Capsule Repeat Unit Formation. J Bacteriol. 2005;187(21):7425–7433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Bender MH, Cartee RT, Yother J. Positive correlation between tyrosine phosphorylation of CpsD and capsular polysaccharide production in Streptococcus pneumoniae. J Bacteriol. 2003;185(20):6057–6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Morona JK, Miller D, Morona R, et al. The effect that mutations in the conserved capsular polysaccharide biosynthesis genes cpsA, cpsB, and cpsD have on virulence of Streptococcus pneumoniae. J Infect Dis. 2004;189(10):1905–1913. [DOI] [PubMed] [Google Scholar]
- [49].Nourikyan J, Kjos M, Mercy C, et al. Autophosphorylation of the Bacterial Tyrosine-Kinase CpsD Connects Capsule Synthesis with the Cell Cycle in Streptococcus pneumoniae. PLoS Genet. 2015;11(9):e1005518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ghosh P, Luong TT, Shah M, et al. Adenylate kinase potentiates the capsular polysaccharide by modulating Cps2D in Streptococcus pneumoniae D39. Exp Mol Med. 2018;50(9):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Niemeyer D, Becker A. The molecular weight distribution of succinoglycan produced by Sinorhizobium meliloti is influenced by specific tyrosine phosphorylation and ATPase activity of the cytoplasmic domain of the ExoP protein. J Bacteriol. 2001;183(17):5163–5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Zhang JR, Idanpaan-Heikkila I, Fischer W, et al. Pneumococcal licD2 gene is involved in phosphorylcholine metabolism. Mol Microbiol. 1999;31(5):1477–1488. [DOI] [PubMed] [Google Scholar]
- [53].Novick S, Shagan M, Blau K, et al. Adhesion and invasion of Streptococcus pneumoniae to primary and secondary respiratory epithelial cells. Mol Med Rep. 2017;15(1):65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Shak JR, Vidal JE, Klugman KP. Influence of bacterial interactions on pneumococcal colonization of the nasopharynx. Trends Microbiol. 2013;21(3):129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hammerschmidt S, Wolff S, Hocke A, et al. Illustration of pneumococcal polysaccharide capsule during adherence and invasion of epithelial cells. Infect Immun. 2005;73(8):4653–4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Keller LE, Rueff A-S, Kurushima J, et al. Three new integration vectors and fluorescent proteins for use in the opportunistic human pathogen Streptococcus pneumoniae. Genes (Basel). 2019;10(5):394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are openly available in figshare at https://doi.org/ 10.6084/m9.figshare.14223077.v1.