

Abstract
The biological understanding of RNA has evolved since the discovery of catalytic RNAs in the early 1980s and the establishment of RNA interference (RNAi) in the 1990s. RNA is no longer seen as the simple mid-product between transcription and translation but as potential molecules to be developed as RNA therapeutic drugs. RNA-based therapeutic drugs have gained recognition because of their ability to regulate gene expression and perform cellular functions. Various nucleobase, backbone, and sugar-modified oligonucleotides have been synthesized, as natural oligonucleotides have some limitations such as poor low nuclease resistance, binding affinity, poor cellular uptake, and toxicity, which affect their use as RNA therapeutic drugs. In this review, we briefly discuss different RNA therapeutic drugs and their internal connections, including antisense oligonucleotides, small interfering RNAs (siRNAs) and microRNAs (miRNAs), aptamers, small activating RNAs (saRNAs), and RNA vaccines. We also discuss the important roles of RNA vaccines and their use in the fight against COVID-19. In addition, various chemical modifications and delivery systems used to improve the performance of RNA therapeutic drugs and overcome their limitations are discussed.
Keywords: ribonucleic acid therapy, AONs, siRNA, miRNA, RNA vaccine, therapeutic mechanism, chemical modification, delivery vehicle, COVID-19
Graphical abstract
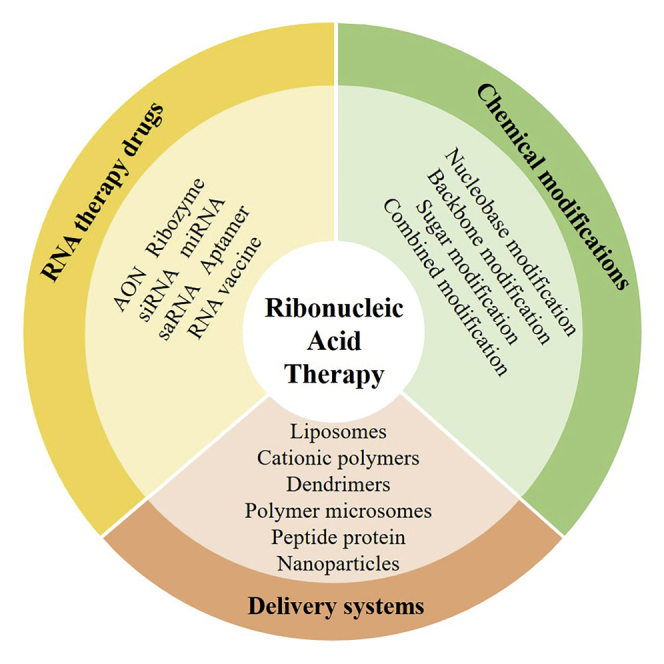
In this review, Zhao et al. introduce the mechanisms, applications, and development of six categories of RNA therapeutic drugs, as well as chemical modifications and delivery systems to challenge the hurdles before possible clinical application.
Introduction
As drugs, antibodies and small molecules mostly interact with proteins and display therapeutic activities toward diseases on either small or large scales. Representing two generations of drug therapy, antibodies and small molecules have exerted excellent effects and contributed considerably to the development of human health. However, antibodies can only recognize targets that are both druggable and secreted or extracellular, as intracellular delivery is deficient. In addition, antibody drugs and small-molecule drugs need to be administered every few weeks, which may stimulate immunological responses and cause side effects through unknown interactions. Furthermore, antibodies and small-molecule drugs have demonstrated limited effects on rare diseases, including hereditary transthyretin amyloidosis (hATTR) and Duchenne muscular dystrophy (DMD).1
RNA is considered an intermediate molecule between DNA and proteins that regulates the functions of genes and cells in all living organisms. The term RNA was updated when catalytic RNA was discovered in the 1980s by Guerrier-Takada et al.;2 later, RNA interference (RNAi) was discovered by Fire et al.3 in the 1990s, and the era of RNA therapy began. RNA can be chemically synthesized, and the manufacturing of RNA drugs is cost effective and more convenient than biologics, rendering batch-to-batch variability achievable and causing no adaptive immune responses as far as we know. Theoretically, antisense oligonucleotides (AONs) and small interfering RNAs (siRNAs) can be designed to suppress any gene, regardless of whether they are highly expressed or noncoding. Compared with antibodies that are involved in complicated processes either to work or to be interrupted, RNAs can be rapidly identified and antidotes are designed to specifically bind and inactivate the drug (Figure S1).
Considering pharmacology and drug development, it is important to develop highly efficient, low-toxicity, and specific drugs to treat diseases. The development of antisense therapeutics4 and RNAi techniques5 has led to a sudden increase in RNA therapeutic drugs that silence genes by targeting the corresponding mRNAs, which code for the proteins rather than the proteins themselves. However, RNA is inherently unstable, potentially immunogenic, likely to have off-target effects, and requires effective carrier substances for transport toward the targeted cells.
Many factors limit the efforts to improve the features of natural nucleic acids and develop them into drugs; these include poor ribonuclease (RNase) resistance, short biological half-life (t½), ineffective cellular uptake, and severe immune response. Various chemical modifications and effective delivery systems have been developed to overcome these limitations. RNA-based therapeutics can now be classified into six categories theoretically and chronologically, including AONs that inhibit mRNA translation, RNAi, aptamers that bind proteins and other molecular ligands, small activating RNAs (saRNAs) that activate genes, ribozymes that are catalytic RNAs, and RNA vaccines that have become research hotspots in recent decades.
As of April 2020, there were 431 RNA-targeting drug development programs worldwide, 63% of which were at the pre-investigational new drug (pre-IND) stage, 32% in early-stage clinical trials (phase I or II), 3% in phase III clinical trials, and five drugs awaiting regulatory decisions (Figure S2). Among all of the candidate drugs, oncology is the most popular target disease, encompassing 22% of ON candidates and 45% of mRNA candidates, followed by neurology, cardiology, and metabolic diseases, infectious diseases, hepatology, musculoskeletal and skin diseases, ophthalmology, and other diseases. The market capitalization of public ON companies has increased by 94.2% in the last 5 years, with three representative companies in the field attracting US $2.8 billion in private investment since 2015. Other pharmaceutical companies have also responded notably toward RNA therapy, as Novartis initiated a large phase III trial for TQJ230 in 2019 and acquired the developer of inclisiran for $9.7 billion, which was approved in December 2020. To date, the most commercially successful drug has been nusinersen, reaching sales of $4.7 billion by the end of 2019. In its first full year on the market, patisiran achieved sales of more than $150 million in 2019, which is expected to have doubled in 2020. Eteplirsen, which targets muscular diseases, is commercially successful as well, with $840 million in sales by the end of 2019, not adding expected investments.6
More than 30 years have passed since the transformation from traditional drugs to RNA-based therapy, with limitations being partly removed and new categories emerging. RNA therapy is a rapidly developing field with notable potential, and its evolution should be properly documented. In order to provide comprehensive summaries and unique insights into RNA therapy, we specifically analyze the development and mechanisms of RNA therapeutic drugs. In addition, we highlight the various chemical modifications and delivery systems that are undertaken to improve stability and efficiency. RNA drugs are listed in the order of discovery.
RNA therapeutic drugs
Antisense therapy
In 1977, the concept of antisense therapeutics was presented by Paterson et al.7 to introduce the function of nucleic acids in modulating gene expression. In 1978, Zamecnik and Stephenson8 demonstrated the inhibition of Rous sarcoma virus replication and cell transformation by a specific ON.
Antisense therapy refers to the technology in which DNA or RNA, called AONs, blocks mRNA transcription and translation through base pairing. DNA is synthesized in vitro and transported into cells. It later combines with target mRNA, and the DNA-RNA complex excites intracellular RNase H to cut the mRNA into small pieces (Figure 1A). In addition, DNA can be loaded to a virus or phage and enter cells indirectly, where its transcript can recognize and bind to target mRNAs, inhibiting mRNA from 5′ capping, 3′ polyadenylation, and splicing, ultimately terminating gene expression (Figure 1B). Broadly speaking, antisense therapy includes ribozymes, but these will be discussed later.9 According to a recent report, AONs also bind to and inhibit inefficient upstream open reading frames, subsequently activating translation and offering a treatment for diseases caused by inadequate gene expression. In addition, by inserting an RNA-cleaving ribozyme sequence near the targeted cleavage site, AONs can be designed to cleave specific sites in mRNAs.10
Figure 1.

Mechanism of antisense pathway and ribozyme
(A) DNA is synthesized in vitro and transported into cells, later combined with target mRNA, and the DNA-RNA complex excites intracellular RNase H to cut the mRNA into small pieces. (B) Virus- or phage-loaded DNA enters cells indirectly and is transcribed into RNA, which then recognizes and binds target mRNAs, inhibiting mRNA from 5′ capping, 3′ polyadenylation, and splicing. (C) Ribozymes conduct general acid-base reactions, with the 3,5-phosphodiester bond at the cutting site becoming cleaved after the reactions, and a covalent bond emerges between 2′-O− and 3′-phosphoric acid.
AONs possess more potential action mechanisms and are easier to deliver intracellularly. However, most AON drugs exert degradation effects on a one-to-one basis, which suggests that one AON drug can only interact with a single mRNA molecule. Currently, there are seven approved AON drugs (Table 1),6,11 and more than 50 candidates are in clinical trials.
Table 1.
Approved AON drugs
| Drugs | Inclinations | Year of approval | Companies |
|---|---|---|---|
| Vitravene (fomivirsen) | CMV | 1998 | Ionis, Novartis |
| Kynamro (mipomersen) | HoFH | 2013 | Ionis |
| Exondys 51 (eteplirsen) | DMD | 2016 | Sarepta Therapeutics |
| Spinraza (nusinersen) | SMA | 2016 | Ionis |
| Tegsedi (inotersen) | polyneuropathy in hATTR | 2018 | Ionis |
| Waylivra (volanesorsen) | FCS | 2019 | Ionis |
| Vyondys 53 (golodirsen) | DMD | 2019 | Sarepta |
HoFH, homozygous familial hypercholesterolemia.
Dysregulated editing of adenosine-to-inosine (A-to-I) RNA is related to various cancers. Tai Tay et al.12 have recently deciphered the secondary structure of antizyme inhibitor 1 (AZIN1) RNA, which is one of the best-studied A-to-I editing targets in cancer, by locating its editing site complementary sequence (ECS). The ECS was shown to be an ideal target for abolishing AZIN1 editing without affecting translation, while AONs targeting the editing region of AZIN1 caused substantial skipping of exon 11. The results demonstrated that AZIN1-targeting, AON-based therapeutics may be applicable to a wide range of tumor types.
Spliceosome-mediated RNA trans-splicing (SMaRT), an antisense therapy that intervenes at the posttranscriptional level, was first developed by Puttaraju et al.13 in 1999. SMaRT exploits the cell’s own splicing machinery to trans-splice therapeutic coding sequences into endogenous precursor (pre-)mRNA targets, joining exons from different pre-mRNA transcripts to generate a chimeric product.14 The therapeutic coding sequences are delivered by artificially engineered pre-mRNA trans-splicing molecules (PTMs). In order to induce the trans-splicing, rather than the cis-splicing, reaction to produce the desired product, a splicing domain and a binding domain must be contained in the PTM. The most well-described SmaRT approach is 3′ exon replacement (3′ER), which involves trans-splicing a therapeutic coding sequence downstream of a 5′ splice site to correct the distal exons of a target gene.15
SMaRT has been explored in a variety of therapeutic applications. It has been enthusiastically embraced in the development of genetic diseases treatments, especially in the correction of monogenic recessive mutations at the mRNA level, because even a single correction may lead to a heterozygous non-disease phenotype.15 DMD is the most common severe childhood muscular pathology, which can be treated by RNA trans-splicing 3′ER and internal exon replacement (IER) targeting on the dystrophin gene.16 Recessive dystrophic epidermolysis bullosa (RDEB), a severe autosomal recessive types of skin blistering disease, is functionally corrected by 3′ER, 5′ER, and IER SMaRT.17 Cystic fibrosis (CF) is a genetic disorder in which the lungs and the digestive system get clogged with mucus.18 Generating a mutant minigene target containing CF transmembrane conductance regulator (CFTR) exons 10–24 (ΔF508) and a mini-intron 10, also a pre-trans-splicing molecule containing CFTR exons 1–10 (+F508), researchers proved that 5′ER could generate functional CFTR for treatment.15 Spinal muscular atrophy (SMA) is a group of hereditary diseases that progressively destroys motor neurons caused by aberrant survival motor neuron-2 (SMN2) expression.19 Shababi et al.20 have discovered that the 3′ER SMaRT system can correct the mistake and increase the lifespan of mice with such disease.
SMaRT also finds its place in the treatment of infectious diseases. The diphtheria toxin A (DTA) chain has been trans-spliced onto the mRNA from the hepatitis C virus (HCV) through ribozyme-mediated 3′ER, which subsequently induces apoptosis.21 By activating mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK), herpes simplex virus type 1 thymidine kinase/ganciclovir (HSV-TK/GCV) can induce cytotoxicity of retinoblastoma cells through autophagy inhibition.22 GCV is a thymidine kinase while HSV-TK can phosphorylate GCV to GCV-triphosphate (TP), which functions as an inhibitor of DNA synthesis, competing with guanine-5′-triphosphate. This suicide gene therapy prevents DNA chain elongation, leading to cell death via apoptosis.23 Even in diseases with multifactorial etiologies, such as cancer, single gene repair has been able to target mutated genes, such as p16 and p53, for correction and restoration of the function.24,25
Although synthesized antisense nucleic acids preferentially select the promoter or translation initiation regions as their target sequences, their targets remain semi-random. The accessibility of different regions is not equivalent, which may be related to the high-level molecular folding of RNA molecules in the cell and the binding of RNA to protein factors. As it is not yet possible to accurately predict the high-level structure of mRNA in cells, researchers generally use the “gene-walk” method to design and synthesize a series of antisense nucleic acids that covers different regions of the mRNA molecule, and screen them through experiments.26
Ribozymes
Ribozymes, catalytic RNA molecules that induce specific degradation of target mRNAs, are also a category of RNA therapeutics. The discovery of catalytic RNAs was in the early 1980s, followed by a wave of research on nucleic acid-based inhibitors of gene expression. Simplified catalytic motifs were defined in the late 1980s and early 1990s, which rendered it possible to chemically synthesize ribozymes, enabling these molecules to function as target-specific inhibitors of gene expression.
Ribozymes act as enzymes that break or form covalent bonds, accelerating targeted reactions with or without proteins.27 There are more than 10 ribozymes that mostly conduct general acid-base reactions, although the detailed mechanisms vary.28 The general base induces deprotonation at 2′-OH to activate 2′-O−, whereas the general acid protonates the 5′-O of the leaving group. The 3,5-phosphodiester bond at the cutting site is cleaved after the reaction ends, with covalent bonds between 2′-O− and 3′-phosphoric acid emerging instead of the mRNA being cut into two pieces.29,30 Gene expression is inhibited at the level of translation, as mRNA is degraded (Figure 1C).
Ribozymes exist naturally; however, they can also be artificially engineered to express and target specific sequences in either the cis (on the same nucleic acid strand) or trans (a noncovalently linked nucleic acid) form.31 The term “hammerhead” ribozyme was given to a group of small self-cleaving ribozymes derived from single-stranded plant viroid RNAs. The hammerhead ribozyme is 30 nt, and its simple structure makes it an excellent choice for the design of trans-acting ribozymes. It can be engineered to cleave any target RNA by Watson-Crick base pairing.32,33 The “hairpin” or “paperclip” is another plant-derived and self-cleaving ribozyme identified in the negative strand of tobacco satellite RNA.34 The hairpin ribozyme generates 2′,3′-cyclic phosphate and 5′-hydroxyl termini, cleaving RNA substrates in a reversible reaction. It can also cleave and turn over numerous copies of different targets in the trans form (a noncovalently linked nucleic acid) by engineering. In addition, the hairpin ribozyme has been applied to catalyze ligation reactions.35
Both hammerhead and hairpin ribozymes downregulate specific cellular and viral targets in cells. In 1988, Haseloff and Gerlach36 synthesized a hairpin ribozyme that could be modified at the arms to cleave any target. It was later demonstrated that this hammerhead ribozyme had the potential to inhibit human immunodeficiency virus (HIV).27 The endoribonuclease RNase P is an enzyme that is widely found in humans and contains RNA and protein components. Under certain salt and ionic conditions, the RNA agents of several types of bacteria can induce cleavage at specific sites.37
Malaria is one of the world’s most daunting public health concerns, causing over 200 million infections and nearly half a million fatalities each year. Genetic modification of the Plasmodium parasite to attempt to break the transmission cycle of the pathogen and to cure those that are afflicted is limited by the availability of selectable markers and the time required to generate transgenic parasites.38,39 To solve this problem, Walker and Lindner40 developed a gene-editing system for Plasmodium based on Streptococcus pyogenes (Sp)Cas9 named CRISPR-RGR. This system utilizes a ribozyme-guide-ribozyme (RGR) single guide RNA (sgRNA) expression strategy with RNA polymerase II promoters. Catalytically dead SpCas9 (dSpCas9) is bound to the upstream region of a target gene, inducing a position-dependent but strand-independent reduction in gene expression. This robust gene-editing system has already generated both gene disruptions and coding sequence insertions in rodent-infectious Plasmodium, and it facilitates efficient genetic characterization of rodent-infectious Plasmodium species.
Ribozymes are molecules with catalytic activity. Similar to protein enzymes, they must be folded into a highly ordered tertiary structure to perform their functions. The nucleophilic attack group and the sensitive phosphorus atom of the nucleoside hydrolase ribozyme are located inside the same nucleotide, simplifying the formation of their active structure.41 For large ribozymes, however, the nucleophilic attack group and the sensitive phosphorus atom are either far away or in different molecules. As a result, the recognition of the cleavage site and appropriate positioning of the substrate in the active center are more complicated, commonly requiring the participation of metal ions or protein factors. For example, the magnesium ion is of great importance for the binding of the RNase P ribozyme to the substrate and stabilization of its active center.42 However, the specificity of ribozyme action is a major challenge that limits the application of ribozymes. Ribozymes usually recognize their cleavage sites by base pairing, but small mismatches can be tolerated by many ribozymes.
RNAi
Two decades after the appearance of AONs, the concept of RNAi was first described in Nature by Fire et al. in 1998.3 Three years later, RNAi technology was rated as one of the top 10 scientific advancements in 2001 and the breakthrough of 2002 by Science. RNAi is an evolutionarily conserved mechanism induced by the complex of small non-coding double-stranded RNA (dsRNA) and proteins, leading to the sequence-specific degradation of complementary mRNA or translation inhibition. Organisms naturally carry out RNAi silencing, which has allowed the identification of specific gene functions in plants, viruses, parasites, invertebrates, and protozoa. RNAi has quickly developed into a powerful biological tool for the silencing of specific genes, and in 2006, Andrew Fire and Craig Mello were awarded the Nobel Prize for Medicine and Physiology.
Specific silencing is triggered by RNAis, which are short dsRNA molecules, as well as siRNAs. In the cytoplasm of cells, RNAi is triggered by long pieces of dsRNA, and then dsRNA is cleaved into fragments known as siRNA (21–23 nt in length) by the enzyme Dicer.43 siRNA binds to the protein Argonaute (Ago) and forms a complex called the RNA-induced silencing complex (RISC),44 which unwinds siRNA, composed of two strands—the sense strand (or passenger strand) and the antisense strand (or guide strand)—and leads to the cleavage of the sense strand. The activated RISC containing the antisense strand then selectively degrades the complementary mRNA (Figure 2A).45 The activated RISC complex can also move on to destroy additional mRNA targets. Compared to AONs, siRNAs possess catalytic mechanisms that harness the RISC machinery, resulting in high potency and unusually sustained activity. By 2020, two siRNA-based drugs had been approved, with 20 candidates in clinical trials (Table 2).6
Figure 2.

RNAi pathway
(A) dsRNA is cleaved into siRNA (21–23 nt in length) by the enzyme Dicer. siRNA binds to the protein Argonaute (Ago) and forms siRISC, which unwinds the siRNA to the antisense strand and sense strand, which is cleaved. siRISC selectively degrades complementary mRNA. (B) Pre-miRNAs are processed by Dicer to obtain the mature miRNA and its complementary strand (miRNA∗). The complementary strand is degraded, and mature miRNA is incorporated into the miRISC complex, leading to mRNA cleavage.
Table 2.
siRNA drugs approved and in development
| Drugs | Indication | Status | Companies |
|---|---|---|---|
| Onpattro (patisiran) | hATTR | approved in 2018 | Alnylam |
| Givlaari (givosiran) | AHP | approved in 2019 | Alnylam |
| Lumasiran | hyperoxaluria | NDA | Alnylam |
| Inclisiran | dyslipidemia/ hypercholesterolemia | NDA | Alnylam/Novartis |
| QR-110 | Leber’s congenital amaurosis | phase III | ProQR |
| Vutrisiran | ATTR/hATTR | phase III | Alnylam |
| QP-1002 | renal disease/failure, delayed graft function | phase III | Quark |
| Tivanisiran (SYL1001) | dry eye | phase III | Sylentis |
| Fitusiran | hemophilia A and B | phase III | Alnylam/Sanofi Genzyme |
NDA, new drug application.
MicroRNA (miRNA), which is a class of small non-coding RNA, is another RNAi silencing trigger. Primary miRNAs (pri-miRNAs) are transcribed in the cell nucleus by RNA polymerase II and subsequently processed into mature miRNAs.46 This process is catalyzed by the core component of an RNase III-type endonuclease called Drosha, which releases precursor miRNA (pre-miRNA), a shorter hairpin of 70–100 nt.47 The exportin 5 complex conducts the transportation of the pre-miRNAs from the nucleus to the cytoplasm.48 Similar to siRNAs, pre-miRNAs are processed by Dicer to obtain mature miRNAs and their complementary strands. Subsequently, the complementary strand is degraded, and mature miRNAs are incorporated into a RISC-like complex. According to the extent of complementarity to the target mRNA, the RISC-like complex can lead to mRNA cleavage (near-perfect complementarity) or translational repression (imperfect complementarity) (Figure 2B).49 miRNAs are important post-transcriptional regulators of nearly every biological process in the cell and show remarkable tissue specificity, and therefore, they can be used as biomarkers for tracing the tissue of origin of cancers of unknown primary origins.50 Dysregulation of miRNAs has been associated with the pathogenesis of human diseases,51 resulting in the development of anti-miRNA ONs (AMOs). miRNA drugs are divided into miRNA mimics and antimiRs, with six candidates in phase II clinical trials, three candidates in phase I trials, and one candidate that has been terminated.
Compared to AONs and ribozymes, siRNAs and miRNAs show differences in structure and function. dsRNAs are cut into short-strand RNAs (20–25 nt), after which they bind to Ago, subsequently exerting an interference effect via the RISC complex (siRISC and miRISC). Meanwhile, AONs and ribozymes recognize and bind to target mRNAs directly using a single strand. Functionally, RNAi requires extremely strict recognition and degrades specific sequences that cannot be initiated with even one mistaken base. Thymine at 3′ overhangs makes RNAi of higher stability, reducing the possibility of degradation by ribozyme as well as the dependence on chemical modification. Additionally, siRNA exhibits obvious inhibition of target genes at a thousandth of the concentration of AONs or even less, and the effect increases with an increase in concentration.
A profound understanding of the cellular uptake and intracellular progress of therapeutic RNA delivery systems could greatly improve the development of siRNA-based therapeutics. Song et al.52 synthesized and discovered the delivery of mannose-modified trimethyl chitosan-cysteine/tripolyphosphate nanoparticles (MTC/TPP NPs).53 Nanoparticles carrying siRNA are taken in by macrophages via caveolae-mediated endocytosis (CvME), circumventing lysosomes. Researchers have identified the Golgi complex and endoplasmic reticulum as the key organelles for efficient delivery, and Syntaxin 6 (a target membrane-soluble N-ethyl-maleimide attachment protein receptor) as well as Niemann-Pick type C1 (NPC1) as indispensable regulators both in vivo and in vitro. Absence of Syntaxin 6 or NPC1 leads to substantial decreases in cellular uptake and gene silencing of the siRNA, resulting in poor therapeutic effectiveness against acute hepatic injury in mice. The CvME approach would provide ideas for designing optimal delivery vectors to facilitate the clinical translation of siRNA drugs.52
Off-target activity of siRNAs can lead to unexpected phenotypes and confuse the interpretation of the therapeutic effects. The off-target effects associated with siRNA delivery can be divided into three broad categories: siRNA-induced sequence-dependent regulation of unintended transcripts via partial sequence complementarity to their 3′ untranslated regions (UTRs), which are known as miRNA-like off-target effects; inflammatory responses caused by activation of Toll-like receptors, triggered by siRNAs and/or their delivery vehicles; and widespread effects on miRNA processing and function induced by exogenous siRNAs via saturation of the endogenous RNAi machinery.54
Aptamer
Soon after, the concept of aptamers was introduced by Ellington and Szostak55 in Nature in 1990 to describe single-stranded RNA molecules isolated from a group of random RNA molecules. In 2004, the first aptamer drug, Macugen (pegaptanib), was approved by the US Food and Drug Administration (FDA) to treat age-related macular degeneration (AMD). However, when the monoclonal antibody drug Avastin (bevacizumab) appeared on the market, it soon replaced Macugen, because it was more effective and safer.56 Currently, there are more than 10 aptamer candidates in clinical trials (Table 3; https://www.clinicaltrials.gov/).
Table 3.
Aptamers in development
| Drugs | Indication | Status | Companies | Locations (year) |
|---|---|---|---|---|
| Macugen (pegaptanib) | AMD | phase IV | NeXstar Pharmaceuticals | Marietta Eye Clinic, Murrieta, GA, USA (2019) |
| EYE001 | AMD | phase III | Eyetech Pharmaceuticals | Foundation for Fighting Blindness, Baltimore, MD, USA (2005) |
| E10030 | AMD | phase III | Ophthotec | Palmetto Retinal Center (2017) |
| ARC1905 | AMD | phase III | Archemix | Ophthotech, New York, NY, USA (2017) |
| REG1 | anticoagulation, ACS | phase II | Regado Biosciences | National Heart, Lung and Blood Institute (NHLBI), Bethesda, MD, USA (2008) |
| AS1411 | MRCC, AML | phase II | Antisoma | UCLA Los Angeles (2011) |
| ARC1779 | thrombosis, AMI | phase II | Archemix | Hackensack University Medical Center, Hackensack, NJ, USA (2010) |
| Nu172 | anticoagulation | phase II | ARCA Biopharma | Unknown (2011) |
| NOX-A12 | CLL, RMM | phase II | NOXXON Pharma | medical university, Innsbruck, Austria (2017) |
| NOX-E36 | T2DM, DN | phase II | NOXXON Pharma | Prague, Czech Republic (2014) |
| NOX-H94 | anemia | phase II | NOXXON Pharma | dialysis unit, Düsseldorf, Germany (2015) |
| ARC19499 | hemophilia | phase I | Baxalta | London, UK (2017) |
ACS, acute coronary syndrome; MRCC, metastatic renal cell carcinoma; AML, acute myeloid leukemia; AMI, acute myocardial infarction; CLL, chronic lymphocytic leukemia; T2DM, type 2 diabetes mellitus; ACR:albumin-creatinine ratio; RMM, relapsed multiple myeloma.
More specifically, aptamers are single-stranded DNA or RNA molecules that bind proteins with high specificity because of their stable three-dimensional shapes.57 DNA and RNA aptamers are typically generated through the process of systematic evolution of ligands by exponential enrichment (SELEX), which allows the selection of 20- to 100-nt aptamers from libraries, binding various protein families with high affinity to modulate their functions58 (Figure S3).
In addition, by combining with other types of therapeutic agents, aptamers can be utilized as delivery vehicles for siRNAs, enzymes, and anticancer drugs.58 Prostate-specific membrane antigen (PSMA) is an overexpressed receptor on the surface of prostate cancer cells and tumor vascular endothelium, which offers a binding site for aptamers. Meanwhile, siRNAs are responsible for the expression of survival genes. Aptamer-siRNA chimeric RNAs were identified and then combined with PSMA-expressing cells only, after which these RNAs were taken in and processed by Dicer, leading to the depletion of siRNA-targeted proteins and subsequent cell death. It has been proven that an aptamer-based siRNA delivery approach mediates tumor regression in a xenograft model.59
In 2011, Beisel et al.60 found that aptamers can be integrated into miRNA basal segments and that chimeric RNAs induce titratable control over gene silencing after binding to the ligand. Although no clinical applications have been developed, this technique is expected to play a role in the design of synthetic miRNA clusters, cis-acting miRNAs, and self-targeting miRNAs, which act both in cis and trans, leading to the feasibility of fine-tuning the regulatory strength and dynamics.
Aptamers have been widely applied in diagnostics and therapeutics, such as biological analysis, clinical medical tests, and directed/targeted therapeutics in the cardiovascular system. As an emerging chemical technology, it differs significantly from traditional drugs. First, the target is no longer restricted because aptamers fold themselves through complementary base pairing, electrostatic interactions, and hydrogen bonding, forming stable three-dimensional structures and tight binding to target molecules.61 Second, animal experiments are unnecessary considering the existence of DNA/RNA libraries, from which aptamers with low immunogenicity and high binding affinity are selected directly. Third, the small size not only promotes tissue penetration but also makes aptamers more accessible and easier to synthesize. In addition, high stability at extreme temperatures and in other harmful environments, a simple modification process, an inexpensive synthesis process, and low variation between batches render aptamer-based delivery systems achievable and successful.58
Although aptamers have shown good targeting performance in vitro, there are still many factors to be considered in order to apply aptamers in vivo. Aptamers bind to their targets by forming secondary or tertiary structures, and the formation of the correct structure can be further affected by the environment surrounding the target, such as temperature, pH, and ionic strength. Even the smallest changes in the internal environment are likely to affect the binding of aptamers to their targets, limiting the application of aptamers in vivo. In addition, the conformation of the target molecule in vivo may be different from that in vitro, possibly leading to a failure of correct recognition.62 Apart from their inherent properties, there are problems to be solved in the process of binding. Although aptamers can specifically bind to different targets, this targeting effect can only occur after aptamers complex with their targets. Aptamers must avoid removal by the reticuloendothelial system (RES) and the kidneys to allow their accumulation in tumor tissues. Aptamer-mediated entry of nano-drug-loaded particles into tumor cells can only take place after target receptor binding. Therefore, strategies combining aptamers and nanocarriers have to be designed very carefully, allowing the escape of the RES. Nano-drug delivery systems can be PEGylated to exert enhanced permeability and retention (EPR), which limits the drugs to the target area.63
saRNA
Prior to advanced activating RNA technologies, RNA therapy mostly centered on gene blockage and inhibition. In 1969, Britten and Davidson64 found that RNAs transcribed from redundant genomic regions could activate a group of protein-coding genes; thus, the theory of “activator” RNAs was proposed. However, their existence was not confirmed until Li et al.65 synthesized dsRNAs that successfully activated gene expression at the transcriptional level and named them saRNAs in 2006. A British company received authorization and initiated the first clinical trial on saRNA candidates worldwide in 2016. The candidate, MTL-CEBPA, can reverse the development of lung cancer and reduce symptoms; the clinical trial is currently in phase II.66 Broadly speaking, saRNAs include not only RNAs but also epigenetic activation conducted by piwi-interacting RNAs (piRNAs)67 and translational activation regulated by miRNAs.68 In a narrow sense, saRNAs are synthetic 21-nt dsRNAs targeting a specific sequence in the promoter region of target genes, delivered to cells by transfection. The effect of saRNA is defined as RNA activation (RNAa).69
Endogenous and external 21-bp dsRNAs recognize and bind to Ago (mainly Ago2), which unwinds the spiral structure and breaks hydrogen bonds. The sense strand is abandoned, whereas the remaining complex of the antisense strand and Ago penetrates the nucleus, subsequently identifying and combining with mRNA. Spatial conformation and modification status (acetylation and methylation) of specific sequences in mRNAs are influenced and transcription is initiated, activating the expression of target genes70, 71, 72 (Figure S4).
The specificity of saRNA is closely related to the length of the dsRNA, which is usually less than 30 nt. A study has shown that neither truncated nor extended dsRNA can activate translation.65 Additionally, the 5′ end of the saRNA is the key functional sequence. After replacing the antisense nucleic acid strand at the 5′ end of dsRNA with 5 bp, the activating effect was completely lost, whereas the same replacement of base pairs at the 3′ end and the middle region had no effect on activation.65 To induce translation, the interaction between saRNAs and genes is necessary, whereas simple identification has little effect. For example, Janowski et al.72 found that PR11 competes with PR8 and PR12 for the same site and induces different regulatory effects. By changing the composition and position of the binding protein in the promoter region, the interference effect was reversed to the activation effect by PR11. In addition, saRNA targets specific cells and sequences, which last longer than siRNAs, and the integrity of the double-stranded structure is of vital importance for saRNAs to function well.69
The advantages of saRNAs over traditional gene therapy methods in tumor treatment can be attributed to its specificity and potent effects. Researchers are currently focused on its activation of tumor suppressor genes in both cells and animals, with the aim of achieving anti-cancer effects by inducing apoptosis and cell cycle arrest, and inhibiting tumor cell invasion and migration.69
Similar to problems faced by other nucleic acid drugs, saRNAs have to challenge some hurdles before possible clinical application, including self-degradation and nuclease degradation in the circulatory system, off-target effects caused by failure to pair with the expected target, and natural immune system activation and immune cell phagocytosis.73
RNA vaccine
The appearance of mRNA drugs dates back to 1990, when Wolff et al.74 first found that the direct transfer of mRNA to mice through transfection could induce protein expression. Since then, researchers have been exploring the application of mRNA in vivo as a vaccine. The first success appeared in 1993, when Martinon et al.75 subcutaneously injected liposome (LP)-encapsulated mRNA encoding the nucleoprotein (NP) of influenza virus and later elicited NP-specific cytotoxic T lymphocytes (CTLs) after injections. Soon after that, researchers found that naked mRNA could trigger antigen-specific antibodies but could not protect animals against tumors. In 2000, Hoerr et al.76 introduced intradermal injection of mRNA, which did not require any transfection reagents, equipment, or a heterologous boost, yet could elicit an adaptive immune response, inducing antigen-specific antibodies and T cells with lytic activity against the model antigen β-galactosidase. It has been reported that mRNA-related companies such as CureVac, Arcturus, BioNTech, and Moderna received more than $4 billion in financing and cooperation orders in 2015 alone, representing a breakthrough year for mRNA therapeutics.
In 2017, Sahin et al.77 successfully created a personalized mRNA vaccine containing numerous new tumor antigens for patients with melanoma, and this achievement has redirected global attention to mRNA vaccines. Expressed regions (exons) and introns are parts of DNA that undergo splicing and transcription; the former is retained in the mRNA and is translated to peptides, ultimately expressing genes as proteins. By sequencing exons from patients, neoantigens that may lead to tumors can be predicted, and corresponding mRNAs that are transcribed to antigens that share the same antigenic determinants as neoantigens are designed. The mRNAs are delivered to cells and translated to proteins that function as antigens, which bind to major histocompatibility complex class I (MHC class I) and are presented to the cell surface. CD8+ T cells identify the complex and exert a CTL immune response, displaying anti-tumor activity.
The nucleic acid vaccine is a third-generation vaccine, as live attenuated vaccines and inactivated vaccines are of the first generation, and subunit vaccines are of the second generation. It contains several significant elements, such as the 5′ cap formed through a 5′-5′ linkage, which plays a role in the ribosomal recognition of mRNA during translation, 5′ UTR, open reading frame (ORF), 3′ UTR, and poly(A) tail.78 The cap is of great significance in maintaining mRNA stability, and it binds to the eukaryotic initiation factor eIF4F, influencing translation efficiency.79 The 5′ UTR is the main facilitator of translation modulation, whereas the 3′ UTR assembles a group of unstable elements. For example, AU-rich elements (AREs), GU-rich elements (GREs), and AUUUA repeats are the most common causes of mRNA instability in the 3′ UTR.80,81 After the cap at the 5′ end, the poly(A) tail is the most important structure for mRNA stability because most degradation starts here. In contrast to previous RNA drugs, which exert therapeutic effects directly via binding mRNAs and preventing gene expression, or attaching to biomacromolecules, mRNA vaccines undergo translation, and the products induce therapeutic function indirectly.
Compared to traditional vaccines, mRNA possesses outstanding features. Technically, it expresses more than one antigen at the same time, exceeding the number of antigens produced by the patient themselves (Figure 3A). In addition, the induced antigens are synthesized by host cells in patients and rarely cause non-specific tumor immune responses. In addition, there is no risk of external gene integration into the host DNA (Figure 3B). Financially, the costs are lower, and the production cycle is shorter. However, there are weak points to be improved considering its instability, low ribozyme resistance, and high immunogenicity.78
Figure 3.

Advantages of mRNA vaccine compared to traditional vaccine
(A) The numbers of antigens expressed by the RNA vaccine exceed those produced by the patient. (B) The induced antigens are synthesized by inner cells from patients and rarely cause non-specific tumor immune responses and external gene integration into the host DNA.
Currently, more than 60 RNA vaccine candidates are in clinical trials, mainly targeting the prevention and treatment of tumors and infectious diseases. Researchers either inject mRNA for direct administration or transfer it into dendritic cells to reinforce immunogenicity.82 Numerous RNA vaccines have been engineered and tested during the last two decades. According to a recent report in Nature, Sahin et al.83 tested a liposomal RNA (RNA-LPX) named melanoma FixVac (BNT111), which targets tumor-associated antigens. The vaccine mediates long-term responses in patients, who have experienced a checkpoint inhibitor (CPI) and still develop unresectable melanoma, whether applied individually or not. In January 2020, the first mRNA vaccine aimed at preventing cytomegalovirus (CMV) infection, mRNA-1647, entered the second phase of clinical trials.
Coronavirus disease 2019 (COVID-19) has caused a global pandemic since 2020. As of May 2021, more than 162.5 million people have been infected, and more than 3.3 million people have died because of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Since the outbreak of COVID-19 at the beginning of 2020, scientists worldwide have been searching for an effective vaccine to inhibit the virus and prevent more disaster.
Among vaccines types, RNA vaccine has raced to the forefront. Corbett et al.84 first synthesized mRNA-1273 and found that this vaccine candidate induced higher levels of antibody than did their recently tested human convalescent-phase serum. mRNA-1273 elicited a strong neutralizing reaction to SARS-CoV-2 in nonhuman primates, providing rapid protection in both upper and lower airways, causing no pathological changes in the lungs.
The key advantage of mRNA is that it is convenient to synthesize once researchers know the sequence of viral proteins; for example, most SARS-CoV-2 vaccines provoke an immune response that targets the coronavirus spike protein, which is found on the surface of the virus. According to the document “Draft landscape of COVID-19 candidate vaccines” published by the World Health Organization (WHO) in December 2020, seven potential RNA-based vaccines are in clinical trials, and two have already received emergency use authorization, BNT162b2 from Pfizer/BioNTech and mRNA-1273 from Moderna. Many more RNA vaccines, which are listed in Table 4,85 are in the pre-clinical investigation stage.
Table 4.
mRNA COVID-19 vaccines
| Drug | Sponsor/collaborator | Status | Latest update post |
|---|---|---|---|
| BNT162b2 | Pfizer/BioNTech | approved | May 13, 2021 |
| mRNA-1273 | Moderna | approved | April 29, 2021 |
| BNT162b2SA | BioNTech/Pfizer | phase III | May 4, 2021 |
| CVnCoV | CureVac | phase III | May 11, 2021 |
| BNT162b1 | BioNTech/Pfizer | phase II | May 4, 2021 |
| BNT162b3 | BioNTech | phase II | April 26, 2021 |
| ARCT-021 | Arcturus Therapeutics | phase II | March 29, 2021 |
| ChulaCov19 mRNA vaccine | Chulalongkorn University | phase I | October 6, 2020 |
| Unnamed | PLA/Walvax Biotech/Abogen Biosciences | phase I | July 7, 2020 |
mRNA should not be able to enter the nuclei of cells, thereby reducing the risk of unwanted integrations into the genome. It operates by simultaneously activating the MHC class I and MHC class II pathways to induce CD4+ and CD8+ T cell immune responses and also acts as an immunological adjuvant, which makes it a promising tool for preventing infectious disease pandemics.86 However, there are some challenges in the development of mRNA vaccines; for example, the safety and effectiveness of mRNA vaccine candidates differ significantly between animal models and humans. In addition, the candidates may cause side effects, such as local and systemic inflammation. Therefore, it is necessary to fully develop mRNA vaccines to improve their stability and antigen expression in the body and to develop better delivery tools. The mechanism of immune response initiation requires further elucidation, and new explorations are necessary.87
The RNA-based therapeutic drugs mentioned above have been introduced chronologically. Their potent regulatory effect on genes and proteins indicates that they have the potential to be developed into novel RNA therapeutic drugs for diseases such as virus infections and cancers. Natural nucleotides pose some limitations that affect their versatility as RNA therapeutic drugs, such as poor binding affinity and specificity with complementary ONs, which hinder delivery and treatment. Low nuclease resistance is likely to lead to degradation. Due to their anionic nature, natural RNAs show poor cell penetration and little interaction with albumin and other serum proteins, leading to rapid elimination. Furthermore, their high molecular weight and negative charges result in low membrane permeability.88 In order to overcome these limitations, ongoing studies have focused on chemical modifications of natural nucleotides and have made significant progress toward establishing ONs as optimal RNA therapeutic drugs.
Chemical modifications
Six decades ago, Michelson and Todd89 synthesized a thymidylate dinucleotide by the phosphate trimester method, which served as the starting point for the synthesis of ONs. Chemical modification is important for improving the affinity and nuclease resistance of these RNA therapeutic candidates. To gain high affinity and specificity for target nucleic acid sequences, a wide variety of chemical modifications on natural nucleotides have been studied, such as modifications in the nucleobase, backbone, and sugar moiety.
Nucleobase modifications
Modifications of the functional domain in the base sequence mainly include alterations in hydrogen bonding, electrostatic interaction, complexation, and the generalized acid-base interaction. Particular base modifications include hydroxymethylation, guanine oxidation, carboxyl substitution, cytosine methylation, deamination, and adenine methylation.90 Nucleobases are modified less often than the sugar-phosphate backbone, considering their significant role in maintaining structural stability. Changed hydrogen bonding may cause the loss of base pairing between strands, preventing the formation of certain base combinations. Despite this, base modifications potentially lend insight into the mechanism of gene silencing and result in novel methods for overcoming off-target effects that arise due to deleterious protein binding or mis-targeting of mRNA.91 Therefore, specific functions are induced when designed nucleobase analogs are introduced into ONs. Even a subtle change in size, electronic distribution, and so on can have a dramatic effect.
Ribavirin is an important antiviral nucleoside containing a triazole nucleoside. The latter is a type of nucleoside analog with 5′-amino transported to the imidazole ring in the biosynthesis of purine nucleoside, which gets widely aggregated and spliced in viral DNA duplication. However, possessing a structure similar to that of normal nucleosides, triazole nucleosides can be taken up by human cells, although they have toxic side effects. Increasing structural differences by chemical modification of the triazole ring offers a solution to toxicity reduction, and the improved compound is expected to play a role in anti-tumor and anti-cancer therapy.
Backbone modifications
Backbone modifications, including methylphosphonate, phosphorothioate (PS), and boranophosphate (BP) (Figure 4C), which can be broadly classified as neutral, anionic, or cationic internucleoside linkages, are designed to remove the physical and biological limitations of the natural phosphodiester linkage.92,93
Figure 4.

Overview of different chemical modifications for oligonucleotides
Diastereomeric PS linkages are created when sulfur takes the place of the non-bridging oxygen atom. PS-ONs are the major backbone modifications and have been used successfully in gene silencing. PS linkages confer sufficient nuclease degradation resistance, leading to higher bioavailability of ONs. In addition to nuclease resistance, PS-ONs also show improved absorption, distribution, and excretion profiles. It has been reported that PS modification can enhance binding affinity with plasma proteins, so that PS-ONs can be absorbed from the injection site into the bloodstream within a very short time,94,95 and they exhibit good uptake in several tissues, including the kidney, liver, spleen, lymph nodes, adipocytes, and bone marrow, but not in skeletal muscle or the brain. Once they arrive at the target organ, these PS-ONs can be quite stable due to chemical modifications and their half-life of 1–4 weeks. However, their binding affinity to the target ON sequences and their specificity are less satisfactory, as PS-modified molecules exhibit no miRNA inhibitory activity.96 Thus, selective substitution of phosphodiester bonds with PS bonds is optimal for increasing nuclease resistance while retaining the ability to bind target miRNAs. However, PS-ONs have increased the level of toxicity, which is a particular problem in siRNAs, such that more than half of the phosphodiester bonds have been replaced with PSs.97 It has also been reported in several studies that this type of modification damages RISC activity; thus, this approach is not recommended.98
Another efficient modification is the use of BPs, which have increased lipophilicity compared with PS analogs and twice the nuclease degradation resistance while causing low toxicity. As expected, BPs cannot be tolerated in the center of the guide strand.99
Other types of phosphodiester scaffolds have been introduced to reduce degradation by nucleases that target ONs. These peptide-substituted ON analogs, such as peptide nucleic acids (PNAs) and morpholino antisense oligonucleotides (morpholino ASOs), prevent gene expression by steric hindrance instead of RNase H-mediated mRNA cleavage. This method has been shown to exhibit specific targeting, improved delivery, and enhanced cellular uptake with little toxicity.100
The transactivator of transcription (TAT), which can conjugate to ONs and subsequently be loaded onto nanoparticles, behaves well in delivery. Chemically modified thiolated gelatin (tGel) and 50-terminal sulfhydryl-modified siRNA showed better affinity and binding to each other through disulfide bonds. The resulting polymeric siRNA can be encapsulated in self-assembled tGel nanoparticles.101,102
Additionally, Cell-penetrating peptides (CPPs) and ligands are normally conjugated to an ON-based drug delivery system. CPPs are a class of short peptides that efficiently penetrate biofilms and enter cells. In addition to being commonly used for the modification of nucleic acids and ONs, CPPs are loaded in nanocarriers for therapy. Cationic CPPs and anionic ONs form complexes through electrostatic interactions, promoting the entry of ON into cells and initiating RNAi, which leads to endogenous gene silencing.103 The combination of complexes and nanoparticles induces special cell binding and receptor-mediated endocytosis, performs well in gene delivery, and is expected to achieve long-term development in cancer gene therapy.104
Alnylam has recently introduced an enhanced stabilization chemistry plus (ESC+) version of siRNA, which significantly reduces off-target effects by introducing ethylene glycol nucleic acid (GNA) backbone structure at key sites. In the latest clinical trials, ALN-HBV02, using ESC+ technology, has reduced the level of hepatitis B virus antigen (HBsAg) by more than 200-fold.105
Sugar modifications
In recent years, there has been a sudden leap in the synthesis of nucleoside analogs through various modifications of the sugar moiety. The 2′-O modifications of ribose have several advantages, including ease of synthesis and increased RNA affinity, nuclease resistance, and cellular uptake.106 Thus, sugar modifications are mainly concentrated in the 2′ position, with 2′-O-methyl (2′-OMe), 2′-O-methoxyethyl (2′-OMOE), and 2′-fluoro (2′-F) as the major representatives.
The 2′-OMe or 2′-OMOE sugar modifications into selected nucleotides within AONs, siRNAs, and anti-miRNAs have been shown to confer excellent nuclease resistance and binding affinity.107, 108, 109 In the first reported success with 2′-MOE-modified ONs, Esau110 demonstrated decreased expression of miR-143 and increased expression of putative miR-143 target genes in cultured cells. Esau et al.111 later reported efficient inhibition of miR-122 in vivo by 2′-MOE ONs; miR-122 is a miRNA that is involved in the regulation of metabolic genes that regulate cholesterol synthesis, hepatic fatty acid synthesis, and oxidation in mouse hepatocytes. The perfect pairing between the 2′-OMOE PS modification of the 3′ overhangs of 21-nt siRNA and the pain-related cation-channel P2X3 resulted in successful gene targeting in vivo.112 However, after the modification of 2′-OMe or 2′-OMOE, ONs no longer support the degradation of mRNA through enzyme H, as the activity of siRNA is reduced.97
4′-Thio-modified ONs contain a sulfur atom attached to the 4′ carbon of the ribose ring. This modification enhances resistance to nuclease digestion when introduced within both the sense and antisense strands of siRNAs at various positions.100,113
Locked nucleic acid (LNA) and unlocked nucleic acid (UNA) (Figure 4D) are other sugar modifications that offer both enhanced binding affinity and good nuclease resistance. LNA is a bicyclic nucleic acid that tethers the 2′-oxygen to the 4′-carbon via a methylene bridge, locking the sugar structure into a 3′ endo conformation.114 It offers a great increase in binding affinity among all of the nucleic acid modifications. The use of LNA-modified anti-miRNAs to inhibit miRNA expression was first reported by Chan et al.115
UNA is an acyclic RNA mimic. Absence of a bond between the C2′ and C3′ atoms of the ribose ring is a characteristic modification, which renders UNA closely related to natural RNA monomers, simultaneously increasing flexibility.116 UNA monomers are compatible with RNase H activity, which is an important property for single-stranded antisense constructs. The application of UNA monomers in the design of superior siRNAs combines potent gene silencing with a dramatic reduction in off-target effects.117
2′-Deoxy-2′-fluoro-arabinonucleotide units (araF-N or FANA units) can be tolerated well throughout the sense strand of the siRNA duplex. In contrast, they can also bind to the 5′ or 3′ ends of the antisense strand. It has been reported that FANA unit-modified siRNAs are compatible with the intracellular RNAi system and induce specific target mRNA degradation. Modified siRNAs have substantially enhanced serum stability and activity. Compared to the unmodified siRNA, which has a short half-life in the serum (<15 min), a fully modified FANA strand has 4-fold increased potency and a longer half-life (∼6 h) when hybridized to an anti-RNA.118 Defects of PS-ONs such as poor binding affinity, lack of specificity, and low cellular uptake can be partially solved via sugar modifications.
Combining different modifications and others
Most research into these ON analogs focus on one type of modification in an attempt to observe the impact of that particular alteration. Combining different modifications that have different beneficial properties can be greatly advantageous, although their preparation is perhaps a little more costly and complicated.119
The 2′-F modification, which involves the introduction of a fluorine atom at the ribose 2′ position, results in exceptional affinity for target RNAs. However, 2′-F-modified ONs are not nuclease resistant, and the PS linkages must also be present to achieve good stability in the serum. In a comparison of the effect of 2′-sugar modifications on miR-21 inhibition, Davis et al.120 showed that 2′-F-modified ONs with a PS backbone outperformed both 2′-MOE-modified ONs with a PS backbone and 2′-OMe-modified ONs with a PS backbone.
Furthermore, fully LNA-modified anti-miRNAs are only moderately efficient at miRNA inhibition,121 possibly due to the tendency of LNA ONs to form dimers with exceptional thermal stability.122 Reducing the number and proximity of LNAs could potentially circumvent this problem; for example, a repeated pattern of two DNAs followed by one LNA could be used. Indeed, compared with other modified anti-miRNAs, ONs with LNA exhibit excellent miRNA inhibitory activity at doses as low as 5 nM, and the efficacy is further enhanced when the LNA substitutions are combined with other modifications, such as 2′-F.123
Recently, 2′-OMe-4′-thioRNA, a hybrid type of chemically modified ON, exhibited high binding affinity to complementary RNAs and high resistance to nuclease degradation. When evaluating 2′-OMe-4′-thioribonucleosides for chemical modification of AMOs, they could inhibit the expression of miR-21. Further investigation showed that PS modification contributed to long-term miR-122 inhibition by the 2′-OMe-4′-thioribonucleoside-modified AMO.124
Isonucleoside is a nucleoside analog, in which the nucleobase is moved from C-1 to other positions on ribose. Zhang et al.125 reported a novel isonucleoside containing a 5′-CH2-extended chain on the sugar moiety that forms a stable double helical structure with its complementary strand. Both its nuclease stability and RNase H activating ability are promoted compared with natural analogs. In siRNAs, passenger strands modified with isonucleoside at the 3′ or 5′ terminus have minimized passenger strand-specific off-target effects and retain their silencing activity.
siL2PT-1MA is a potential therapeutic siRNA that modifies PS and 2a,4a-BNA/LNA in the sense strand, along with 2a-methoxy (2a-OMe) nucleotides in the immunostimulatory motif. The duration of the RNAi effect is largely prolonged by the modification of 2a,4a-BNA/LNA and shortened by cholesterol conjugation. The application of this potential siRNA resulted in sustained reduction (24 days) of total cholesterol (TC) in the serum and apolipoprotein B (apoB) mRNA reduction in the liver without adverse effects. Serum interferon-A (IFN-A) levels induced by cholesterol-conjugated immunostimulatory siRNA (isRNA) are higher than those induced by nonmodified siRNA, indicating that cholesterol conjugation facilitates the immune reaction. A study by Wada et al.126 showed that a negative immune response is ameliorated with the modification of the adenosine residues complementary to the immunostimulatory motif and central 5a-UG-3a in the sense strand.
Furthermore, ON length as a parameter needs to be considered in the design of RNA therapeutic drugs,127 with the aim of generating potent and short ONs of sufficient target affinity, nuclease resistance, and cellular uptake, as well as low toxicity.
Compared with traditional drug molecules whose pharmacodynamic and pharmacokinetic properties are integrated, RNA drugs separate them, thereby determining the silencing effect on target genes and the absorption, distribution, metabolism, and elimination of drug molecules at the body level. Therefore, based on the general chemical modifications that improve pharmacokinetic properties, the enhanced pharmacodynamic properties of siRNA can be taken into consideration.
Currently, there are numerous chemical modifications focusing on the pharmacokinetic properties of siRNA, but few have focused on the improvement of the pharmacodynamic properties. With expanding clinical requirements and the continuous progress of the technology, more chemical modifications are required to provide more precise means of pharmacodynamic adjustment.
Delivery systems
In addition to chemical modifications, effective drug delivery is an approach to improve the ability of RNA therapeutic drugs to diagnose and treat diseases. A standard ON delivery system is biocompatible, biodegradable, and non-immunogenic. Carrier technology and drug delivery systems improve the biological activity and distribution of nucleic acid drugs in vivo, enhance the targeting effect and uptake by cells, increase the concentration and bioavailability of drugs in target tissues, and prevent RNA drugs from being absorbed by serum proteins or degraded by nucleases.128 Current nucleic acid drug delivery carriers mainly refer to liposomes (e.g., DOTAP and Lipofectamine), cationic polymer complexes (such as PEI and PAMAM), dendrimers (DENs), polymer microsomes, peptide protein delivery vehicles, and nanoparticles (Figure 5). Peptide protein-based vehicles are a hot area of research, and one of the basic carriers is cationic peptides, such as CPPs and protein transduction domains (PTDs). CPPs not only improve uptake efficiency significantly, but they also target specific cell receptors or body tissues accurately.
Figure 5.

Schematic depiction of nucleic acid carriers
(A) Polymeric nanoparticles containing RNA and cationic polymer. (B) Lipid nanoparticles.
For liposomes, ONs can be easily encapsulated into cationic liposomes by electrostatic absorption because of their negatively charged properties. Hence, cationic liposomes have been considered as ON delivery systems.129 Cationic lipids contain a hydrophobic chain and a cationic head, and the latter functions as the main agent to react with anionic ONs. Both have a significant influence on transfection efficiency and toxicity.130 Cationic lipids, the functional component of cationic liposomes, are categorized into monovalent lipids and multivalent lipids, and the former includes DODMA and DOTAP.131 Generally, cationic liposomes equipped with multivalent lipids exhibit higher transfection efficiency.128
Neutral liposomes consist of neutral lipids, including cholesterol, phosphatidylcholine (PC), phosphatidylethanolamine (PE), and DOPE. Compared to cationic liposomes and ionizable liposomes, neutral liposomes have excellent biocompatibility and pharmacokinetic characteristics. However, they do not interact with ONs to a great extent; therefore, neutral liposomes cannot efficiently absorb and encapsulate ONs.101
Ionizable liposomes are nanocarriers that carry more than a single type of charge and are protonated or deprotonated as the acidity of the environment changes. ON-loaded liposomes remained positively charged until administration but before entering cells. After the RNA drugs are released, the carriers again obtain positive charges.129 In addition, pH-sensitive ionizable liposomes were created and applied for successful delivery. Ionizable liposomes are composed of amino lipids with ionizable amine headgroups. The pKa value of the ionizable amino in liposomes is a significant factor in carrier design, as it determines the interaction of amino acids with cell membranes and serum proteins and subsequently affects the delivery effectiveness and toxicity.132
In contrast to other carriers, polymeric micelles have improved pharmacokinetics and biocompatibility, making them ideal carriers in drug delivery. Self-assembled amphiphilic polymers show improved stability in colloids and reduced non-specific interactions with other biomolecules. In addition, polymer micelles enhance the drug cycle time, change the drug release curve, and easily connect to targeted ligands.133 Gwak et al.134 produced a new cationic amphiphilic copolymer, poly(lactide-co-glycolide)-graft-polyethylenimine (PgP), which self-assembled from cationic polymeric micelles that deliver ONs to cure spinal cord injury.
The N-acetylgalactosamine (GalNAc) delivery approach is an alternative to nanoparticles, in which ligands are conjugated to RNA that permit them to enter target cells. Nair et al.135 found a tertiary conjugation between GalNAc and RNA, which is perhaps the most advanced application of this technique. In contrast to other intravenously infected nanoparticles, GalNAc is administered subcutaneously and targets the liver via systemic circulation.136 Other conjugates have been explored, such as vitamin E, cholesterol, and CPPs, but none has been successful.
The adeno-associated virus (AAV) genome was the first one cloned into a plasmid in 1982, and several studies later proved the success of viral vectors in the delivery of therapeutic genes in the early stages of this technology. However, only a few clinical studies were meaningful and significant immunotoxicities occurred. Viral vectors are effective DNA delivery vehicles because they evolved to target specific cell types and are the only system able to deliver payloads to the nucleus.137 Voretigene neparvovec-rzyl, also known as Luxturna, is an AAV serotype 2 (AAV2) vector loaded with cDNA encoding the human RPE65 gene, which is a target for inherited retinal dystrophy Leber’s congenital amaurosis (LCA) type 2 treatment. Visual tests have demonstrated that treatment with Luxturna improves light sensitivity, visual fields, and navigational ability in RPE65-mediated LCA patients, who were previously untreatable. These results led to Luxturna’s approval in 2017.138
Conclusions
This review summarizes different RNA therapeutics in terms of mechanisms, disease treatments, delivery systems, application methods, and shortage to improve. RNAi technology was popular and rated as one of the top 10 scientific advancements in 2001; however, public attention was continually distracted in the following decade because the technology provided few outcomes. A turning point occurred in 2016, when nusinersen (Spinraza) was approved by the FDA, and RNA therapy once again became a research focus. The outbreak of COVID-19 has greatly promoted research progress in related fields, and, fortunately, RNA-based COVID-19 vaccines emerged as a significant outcome for disease treatment and RNAi technology.
Natural RNA therapeutic drugs have some limitations, such as low binding affinity and poor nuclease stability. Through chemical modification, many ON analogs have been synthesized to overcome these limitations, and numerous clinical trials have been conducted to investigate this. RNA therapeutics are thought to be the most successful treatment for rare diseases, especially neurological and hepatic diseases. Currently, eight RNA therapeutic drugs have been approved by the FDA for clinical treatment in total, including Vitravene (fomivirsen), Kynamro (mipomersen), Macugen (pegaptanib), Exondys 51 (eteplirsen), Spinraza (nusinersen), Tegsedi (inotersen), Waylivra (volanesorsen), and Vyondys 53 (golodirsen). The most commercially successful drug is Nusinersen, reaching sales of $4.7 billion by the end of 2019.
Furthermore, with persistent clinical trials for these modified ONs, it is anticipated that more novel RNA therapeutic drugs will emerge for disease treatment and prevention in the near future.
Acknowledgments
This work was supported by the China Postdoctoral Science Foundation (2019M660240); the Postdoctoral Research and Development Funding of Sichuan University (2020SCU12016); the Research Funding for Talents Developing, West China Hospital of Stomatology Sichuan University (RCDWJS2020-14); and by the CAMS Innovation Fund for Medical Sciences (CIFMS, 2019-I2M-5-004).
Author contributions
Study design and supervision, R.S. and J.L.; manuscript writing, Y.Z.; figure design, Y.Z.; financial support, R.S. and J.L.; final approval of the manuscript, all authors.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.09.002.
Contributor Information
Rui Shu, Email: shurui@scu.edu.cn.
Jiang Liu, Email: liujiang@scu.edu.cn.
Supplemental information
References
- 1.Guan Y., Hao C., Cha D.R., Rao R., Lu W., Kohan D.E., Magnuson M.A., Redha R., Zhang Y., Breyer M.D. Thiazolidinediones expand body fluid volume through PPARγ stimulation of ENaC-mediated renal salt absorption. Nat. Med. 2005;11:861–866. doi: 10.1038/nm1278. [DOI] [PubMed] [Google Scholar]
- 2.Guerrier-Takada C., Gardiner K., Marsh T., Pace N., Altman S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell. 1983;35:849–857. doi: 10.1016/0092-8674(83)90117-4. [DOI] [PubMed] [Google Scholar]
- 3.Fire A., Xu S., Montgomery M.K., Kostas S.A., Driver S.E., Mello C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 4.Sharma V.K., Sharma R.K., Singh S.K. Antisense oligonucleotides: Modifications and clinical trials. MedChemComm. 2014;5:1454–1471. [Google Scholar]
- 5.Gaynor J.W., Campbell B.J., Cosstick R. RNA interference: A chemist’s perspective. Chem. Soc. Rev. 2010;39:4169–4184. doi: 10.1039/b920362c. [DOI] [PubMed] [Google Scholar]
- 6.Wang F., Zuroske T., Watts J.K. RNA therapeutics on the rise. Nat. Rev. Drug Discov. 2020;19:441–442. doi: 10.1038/d41573-020-00078-0. [DOI] [PubMed] [Google Scholar]
- 7.Paterson B.M., Roberts B.E., Kuff E.L. Structural gene identification and mapping by DNA-mRNA hybrid-arrested cell-free translation. Proc. Natl. Acad. Sci. USA. 1977;74:4370–4374. doi: 10.1073/pnas.74.10.4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zamecnik P.C., Stephenson M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA. 1978;75:280–284. doi: 10.1073/pnas.75.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weintraub H.M. Antisense RNA and DNA. Sci. Am. 1990;262:40–46. doi: 10.1038/scientificamerican0190-40. [DOI] [PubMed] [Google Scholar]
- 10.Jain M.L., Bruice P.Y., Szabó I.E., Bruice T.C. Incorporation of positively charged linkages into DNA and RNA backbones: A novel strategy for antigene and antisense agents. Chem. Rev. 2012;112:1284–1309. doi: 10.1021/cr1004265. [DOI] [PubMed] [Google Scholar]
- 11.Ma C.C., Wang Z.L., Xu T., He Z.Y., Wei Y.Q. The approved gene therapy drugs worldwide: from 1998 to 2019. Biotechnol. Adv. 2020;40:107502. doi: 10.1016/j.biotechadv.2019.107502. [DOI] [PubMed] [Google Scholar]
- 12.Tai Tay D.J., Song Y., Peng B., Toh T.B., Hooi L., Kaixin Toh D.F., Hong H., Tang S.J., Han J., Gan W.L. Targeting RNA editing of antizyme inhibitor 1: A potential oligonucleotide-based antisense therapy for cancer. Mol. Ther. 2021 doi: 10.1016/j.ymthe.2021.05.008. Published online May 28, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Puttaraju M., Jamison S.F., Mansfield S.G., Garcia-Blanco M.A., Mitchell L.G. Spliceosome-mediated RNA trans-splicing as a tool for gene therapy. Nat. Biotechnol. 1999;17:246–252. doi: 10.1038/6986. [DOI] [PubMed] [Google Scholar]
- 14.Hong E.M., Ingemarsdotter C.K., Lever A.M.L. Therapeutic applications of trans-splicing. Br. Med. Bull. 2020;136:4–20. doi: 10.1093/bmb/ldaa028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mansfield S.G., Clark R.H., Puttaraju M., Kole J., Cohn J.A., Mitchell L.G., Garcia-Blanco M.A. 5′ Exon replacement and repair by spliceosome-mediated RNA trans-splicing. RNA. 2003;9:1290–1297. doi: 10.1261/rna.5101903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lorain S., Peccate C., Le Hir M., Garcia L. Exon exchange approach to repair Duchenne dystrophin transcripts. PLoS ONE. 2010;5:e10894. doi: 10.1371/journal.pone.0010894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murauer E.M., Gache Y., Gratz I.K., Klausegger A., Muss W., Gruber C., Meneguzzi G., Hintner H., Bauer J.W. Functional correction of type VII collagen expression in dystrophic epidermolysis bullosa. J. Invest. Dermatol. 2011;131:74–83. doi: 10.1038/jid.2010.249. [DOI] [PubMed] [Google Scholar]
- 18.Liu X., Luo M., Zhang L.N., Yan Z., Zak R., Ding W., Mansfield S.G., Mitchell L.G., Engelhardt J.F. Spliceosome-mediated RNA trans-splicing with recombinant adeno-associated virus partially restores cystic fibrosis transmembrane conductance regulator function to polarized human cystic fibrosis airway epithelial cells. Hum. Gene Ther. 2005;16:1116–1123. doi: 10.1089/hum.2005.16.1116. [DOI] [PubMed] [Google Scholar]
- 19.Coady T.H., Baughan T.D., Shababi M., Passini M.A., Lorson C.L. Development of a single vector system that enhances trans-splicing of SMN2 transcripts. PLoS ONE. 2008;3:e3468. doi: 10.1371/journal.pone.0003468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shababi M., Glascock J., Lorson C.L. Combination of SMN trans-splicing and a neurotrophic factor increases the life span and body mass in a severe model of spinal muscular atrophy. Hum. Gene Ther. 2011;22:135–144. doi: 10.1089/hum.2010.114. [DOI] [PubMed] [Google Scholar]
- 21.Ryu K.-J., Kim J.H., Lee S.W. Ribozyme-mediated selective induction of new gene activity in hepatitis C virus internal ribosome entry site-expressing cells by targeted trans-splicing. Mol. Ther. 2003;7:386–395. doi: 10.1016/s1525-0016(02)00063-1. [DOI] [PubMed] [Google Scholar]
- 22.Yi Q.Y., Bai Z.S., Cai B., Chen N., Chen L.S., Yuan T., Mao J.H. HSV-TK/GCV can induce cytotoxicity of retinoblastoma cells through autophagy inhibition by activating MAPK/ERK. Oncol. Rep. 2018;40:682–692. doi: 10.3892/or.2018.6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tomicic M.T., Thust R., Kaina B. Ganciclovir-induced apoptosis in HSV-1 thymidine kinase expressing cells: Critical role of DNA breaks, Bcl-2 decline and caspase-9 activation. Oncogene. 2002;21:2141–2153. doi: 10.1038/sj.onc.1205280. [DOI] [PubMed] [Google Scholar]
- 24.Kastanos E., Hjiantoniou E., Phylactou L.A. Restoration of protein synthesis in pancreatic cancer cells by trans-splicing ribozymes. Biochem. Biophys. Res. Commun. 2004;322:930–934. doi: 10.1016/j.bbrc.2004.07.203. [DOI] [PubMed] [Google Scholar]
- 25.Kandoth C., McLellan M.D., Vandin F., Ye K., Niu B., Lu C., Xie M., Zhang Q., McMichael J.F., Wyczalkowski M.A. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Myers K.J., Dean N.M. Sensible use of antisense: How to use oligonucleotides as research tools. Trends Pharmacol. Sci. 2000;21:19–23. doi: 10.1016/s0165-6147(99)01420-0. [DOI] [PubMed] [Google Scholar]
- 27.Scherer L.J., Rossi J.J. Approaches for the sequence-specific knockdown of mRNA. Nat. Biotechnol. 2003;21:1457–1465. doi: 10.1038/nbt915. [DOI] [PubMed] [Google Scholar]
- 28.Nakano S., Chadalavada D.M., Bevilacqua P.C. General acid-base catalysis in the mechanism of a hepatitis delta virus ribozyme. Science. 2000;287:1493–1497. doi: 10.1126/science.287.5457.1493. [DOI] [PubMed] [Google Scholar]
- 29.Lilley D.M. Structure, folding and mechanisms of ribozymes. Curr. Opin. Struct. Biol. 2005;15:313–323. doi: 10.1016/j.sbi.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 30.Lilley D.M. The origins of RNA catalysis in ribozymes. Trends Biochem. Sci. 2003;28:495–501. doi: 10.1016/S0968-0004(03)00191-9. [DOI] [PubMed] [Google Scholar]
- 31.Wilson D.S., Szostak J.W. In vitro selection of functional nucleic acids. Annu. Rev. Biochem. 1999;68:611–647. doi: 10.1146/annurev.biochem.68.1.611. [DOI] [PubMed] [Google Scholar]
- 32.Haseloff J., Gerlach W.L. Simple RNA enzymes with new and highly specific endoribonuclease activities. 1988. Biotechnology. 1992;24:264–269. [PubMed] [Google Scholar]
- 33.Forster A.C., Jeffries A.C., Sheldon C.C., Symons R.H. Structural and ionic requirements for self-cleavage of virusoid RNAs and trans self-cleavage of viroid RNA. Cold Spring Harb. Symp. Quant. Biol. 1987;52:249–259. doi: 10.1101/sqb.1987.052.01.030. [DOI] [PubMed] [Google Scholar]
- 34.Buzayan J.M., Hampel A., Bruening G. Nucleotide sequence and newly formed phosphodiester bond of spontaneously ligated satellite tobacco ringspot virus RNA. Nucleic Acids Res. 1986;14:9729–9743. doi: 10.1093/nar/14.24.9729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hampel A., Tritz R., Hicks M., Cruz P. “Hairpin” catalytic RNA model: Evidence for helices and sequence requirement for substrate RNA. Nucleic Acids Res. 1990;18:299–304. doi: 10.1093/nar/18.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haseloff J., Gerlach W.L. Simple RNA enzymes with new and highly specific endoribonuclease activities. Nature. 1988;334:585–591. doi: 10.1038/334585a0. [DOI] [PubMed] [Google Scholar]
- 37.Phylactou L.A., Darrah C., Wood M.J. Ribozyme-mediated trans-splicing of a trinucleotide repeat. Nat. Genet. 1998;18:378–381. doi: 10.1038/ng0498-378. [DOI] [PubMed] [Google Scholar]
- 38.Jongco A.M., Ting L.M., Thathy V., Mota M.M., Kim K. Improved transfection and new selectable markers for the rodent malaria parasite Plasmodium yoelii. Mol. Biochem. Parasitol. 2006;146:242–250. doi: 10.1016/j.molbiopara.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 39.de Koning-Ward T.F., Janse C.J., Waters A.P. The development of genetic tools for dissecting the biology of malaria parasites. Annu. Rev. Microbiol. 2000;54:157–185. doi: 10.1146/annurev.micro.54.1.157. [DOI] [PubMed] [Google Scholar]
- 40.Walker M.P., Lindner S.E. Ribozyme-mediated, multiplex CRISPR gene editing and CRISPR interference (CRISPRi) in rodent-infectious Plasmodium yoelii. J. Biol. Chem. 2019;294:9555–9566. doi: 10.1074/jbc.RA118.007121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Selmer M., Dunham C.M., Murphy F.V., 4th, Weixlbaumer A., Petry S., Kelley A.C., Weir J.R., Ramakrishnan V. Structure of the 70S ribosome complexed with mRNA and tRNA. Science. 2006;313:1935–1942. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- 42.Harris M.E., Christian E.L. Recent insights into the structure and function of the ribonucleoprotein enzyme ribonuclease P. Curr. Opin. Struct. Biol. 2003;13:325–333. doi: 10.1016/s0959-440x(03)00069-1. [DOI] [PubMed] [Google Scholar]
- 43.Bernstein E., Caudy A.A., Hammond S.M., Hannon G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–366. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- 44.Rand T.A., Ginalski K., Grishin N.V., Wang X. Biochemical identification of Argonaute 2 as the sole protein required for RNA-induced silencing complex activity. Proc. Natl. Acad. Sci. USA. 2004;101:14385–14389. doi: 10.1073/pnas.0405913101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matranga C., Tomari Y., Shin C., Bartel D.P., Zamore P.D. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell. 2005;123:607–620. doi: 10.1016/j.cell.2005.08.044. [DOI] [PubMed] [Google Scholar]
- 46.Lee Y., Kim M., Han J., Yeom K.-H., Lee S., Baek S.H., Kim V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23:4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee Y., Ahn C., Han J., Choi H., Kim J., Yim J., Lee J., Provost P., Rådmark O., Kim S., Kim V.N. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- 48.Lund E., Güttinger S., Calado A., Dahlberg J.E., Kutay U. Nuclear export of microRNA precursors. Science. 2004;303:95–98. doi: 10.1126/science.1090599. [DOI] [PubMed] [Google Scholar]
- 49.He L., Hannon G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 50.Rosenfeld N., Aharonov R., Meiri E., Rosenwald S., Spector Y., Zepeniuk M., Benjamin H., Shabes N., Tabak S., Levy A. MicroRNAs accurately identify cancer tissue origin. Nat. Biotechnol. 2008;26:462–469. doi: 10.1038/nbt1392. [DOI] [PubMed] [Google Scholar]
- 51.Mendell J.T., Olson E.N. MicroRNAs in stress signaling and human disease. Cell. 2012;148:1172–1187. doi: 10.1016/j.cell.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song Y., Wu Y., Xu L., Jiang T., Tang C., Yin C. Caveolae-mediated endocytosis drives robust siRNA delivery of polymeric nanoparticles to macrophages. ACS Nano. 2021;15:8267–8282. doi: 10.1021/acsnano.0c08596. [DOI] [PubMed] [Google Scholar]
- 53.Chu S., Tang C., Yin C. Effects of mannose density on in vitro and in vivo cellular uptake and RNAi efficiency of polymeric nanoparticles. Biomaterials. 2015;52:229–239. doi: 10.1016/j.biomaterials.2015.02.044. [DOI] [PubMed] [Google Scholar]
- 54.Jackson A.L., Linsley P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010;9:57–67. doi: 10.1038/nrd3010. [DOI] [PubMed] [Google Scholar]
- 55.Ellington A.D., Szostak J.W. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 56.Ng E.W., Shima D.T., Calias P., Cunningham E.T., Jr., Guyer D.R., Adamis A.P. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat. Rev. Drug Discov. 2006;5:123–132. doi: 10.1038/nrd1955. [DOI] [PubMed] [Google Scholar]
- 57.Burnett J.C., Rossi J.J. RNA-based therapeutics: current progress and future prospects. Chem. Biol. 2012;19:60–71. doi: 10.1016/j.chembiol.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thevendran R., Sarah S., Tang T.H., Citartan M. Strategies to bioengineer aptamer-driven nanovehicles as exceptional molecular tools for targeted therapeutics: A review. J. Control. Release. 2020;323:530–548. doi: 10.1016/j.jconrel.2020.04.051. [DOI] [PubMed] [Google Scholar]
- 59.McNamara J.O., 2nd, Andrechek E.R., Wang Y., Viles K.D., Rempel R.E., Gilboa E., Sullenger B.A., Giangrande P.H. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat. Biotechnol. 2006;24:1005–1015. doi: 10.1038/nbt1223. [DOI] [PubMed] [Google Scholar]
- 60.Beisel C.L., Chen Y.Y., Culler S.J., Hoff K.G., Smolke C.D. Design of small molecule-responsive microRNAs based on structural requirements for Drosha processing. Nucleic Acids Res. 2011;39:2981–2994. doi: 10.1093/nar/gkq954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jayasena S.D. Aptamers: An emerging class of molecules that rival antibodies in diagnostics. Clin. Chem. 1999;45:1628–1650. [PubMed] [Google Scholar]
- 62.Yue Z., Shi-Ge X., Zhen W., Qing-He K., Yun L., Mei-Yi Y., Yan-Ping H., Yong J., Xiao-Gang C. Recent research of aptamer in target drug delivery. Prog. Biochem. Biophys. 2015;42:236–243. [Google Scholar]
- 63.Nicolas J., Mura S., Brambilla D., Mackiewicz N., Couvreur P. Design, functionalization strategies and biomedical applications of targeted biodegradable/biocompatible polymer-based nanocarriers for drug delivery. Chem. Soc. Rev. 2013;42:1147–1235. doi: 10.1039/c2cs35265f. [DOI] [PubMed] [Google Scholar]
- 64.Britten R.J., Davidson E.H. Gene regulation for higher cells: A theory. Science. 1969;165:349–357. doi: 10.1126/science.165.3891.349. [DOI] [PubMed] [Google Scholar]
- 65.Li L.C., Okino S.T., Zhao H., Pookot D., Place R.F., Urakami S., Enokida H., Dahiya R. Small dsRNAs induce transcriptional activation in human cells. Proc. Natl. Acad. Sci. USA. 2006;103:17337–17342. doi: 10.1073/pnas.0607015103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sarker D., Plummer R., Basu B., Meyer T., Ma Y.T., Evans J., Palmer D.H., Huang K.W., Chee C.E., Spalding D. First-in-human, first-in-class phase I study of MTL-CEBPA, a RNA oligonucleotide targeting the myeloid cell master regulator C/EBP-α, in patients with advanced hepatocellular cancer (HCC) Ann. Oncol. 2019;30(Suppl 5):v168–v169. [Google Scholar]
- 67.Yin H., Lin H. An epigenetic activation role of Piwi and a Piwi-associated piRNA in Drosophila melanogaster. Nature. 2007;450:304–308. doi: 10.1038/nature06263. [DOI] [PubMed] [Google Scholar]
- 68.Vasudevan S., Tong Y., Steitz J.A. Switching from repression to activation: MicroRNAs can up-regulate translation. Science. 2007;318:1931–1934. doi: 10.1126/science.1149460. [DOI] [PubMed] [Google Scholar]
- 69.Yupei S., Huojun H., Ge L., Yiling H. Research advances of saRNA in human malignant tumors cancer. Res. Prev. Treat. 2018;45:52–55. [Google Scholar]
- 70.Wang X., Arai S., Song X., Reichart D., Du K., Pascual G., Tempst P., Rosenfeld M.G., Glass C.K., Kurokawa R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature. 2008;454:126–130. doi: 10.1038/nature06992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li L.C. The multifaceted small RNAs. RNA Biol. 2008;5:61–64. doi: 10.4161/rna.5.2.5989. [DOI] [PubMed] [Google Scholar]
- 72.Janowski B.A., Younger S.T., Hardy D.B., Ram R., Huffman K.E., Corey D.R. Activating gene expression in mammalian cells with promoter-targeted duplex RNAs. Nat. Chem. Biol. 2007;3:166–173. doi: 10.1038/nchembio860. [DOI] [PubMed] [Google Scholar]
- 73.Jing Y., Yi-Ling H., Li-Ming H. Research progress of saRNA activated tumor suppressor gene p21 and applications of saRNA. Chin. Bull. Life Sci. 2020;32:574–580. [Google Scholar]
- 74.Wolff J.A., Malone R.W., Williams P., Chong W., Acsadi G., Jani A., Felgner P.L. Direct gene transfer into mouse muscle in vivo. Science. 1990;247:1465–1468. doi: 10.1126/science.1690918. [DOI] [PubMed] [Google Scholar]
- 75.Martinon F., Krishnan S., Lenzen G., Magné R., Gomard E., Guillet J.G., Lévy J.P., Meulien P. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur. J. Immunol. 1993;23:1719–1722. doi: 10.1002/eji.1830230749. [DOI] [PubMed] [Google Scholar]
- 76.Hoerr I., Obst R., Rammensee H.G., Jung G. In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur. J. Immunol. 2000;30:1–7. doi: 10.1002/1521-4141(200001)30:1<1::AID-IMMU1>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 77.Sahin U., Derhovanessian E., Miller M., Kloke B.P., Simon P., Löwer M., Bukur V., Tadmor A.D., Luxemburger U., Schrörs B. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547:222–226. doi: 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- 78.Kormann M.S., Hasenpusch G., Aneja M.K., Nica G., Flemmer A.W., Herber-Jonat S., Huppmann M., Mays L.E., Illenyi M., Schams A. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat. Biotechnol. 2011;29:154–157. doi: 10.1038/nbt.1733. [DOI] [PubMed] [Google Scholar]
- 79.Peng Z.H., Sharma V., Singleton S.F., Gershon P.D. Synthesis and application of a chain-terminating dinucleotide mRNA cap analog. Org. Lett. 2002;4:161–164. doi: 10.1021/ol0167715. [DOI] [PubMed] [Google Scholar]
- 80.Murray E.L., Schoenberg D.R. A+U-rich instability elements differentially activate 5′-3′ and 3′-5′ mRNA decay. Mol. Cell. Biol. 2007;27:2791–2799. doi: 10.1128/MCB.01445-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kozak M. At least six nucleotides preceding the AUG initiator codon enhance translation in mammalian cells. J. Mol. Biol. 1987;196:947–950. doi: 10.1016/0022-2836(87)90418-9. [DOI] [PubMed] [Google Scholar]
- 82.Kreiter S., Diken M., Selmi A., Türeci Ö., Sahin U. Tumor vaccination using messenger RNA: Prospects of a future therapy. Curr. Opin. Immunol. 2011;23:399–406. doi: 10.1016/j.coi.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 83.Sahin U., Oehm P., Derhovanessian E., Jabulowsky R.A., Vormehr M., Gold M., Maurus D., Schwarck-Kokarakis D., Kuhn A.N., Omokoko T. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature. 2020;585:107–112. doi: 10.1038/s41586-020-2537-9. [DOI] [PubMed] [Google Scholar]
- 84.Corbett K.S., Flynn B., Foulds K.E., Francica J.R., Boyoglu-Barnum S., Werner A.P., Flach B., O’Connell S., Bock K.W., Minai M. Evaluation of the mRNA-1273 vaccine against SARS-CoV-2 in nonhuman primates. N. Engl. J. Med. 2020;383:1544–1555. doi: 10.1056/NEJMoa2024671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Borah P., Deb P.K., Al-Shar’i N.A., Dahabiyeh L.A., Venugopala K.N., Singh V., Shinu P., Hussain S., Deka S., Chandrasekaran B., Jaradat D.M.M. Perspectives on RNA vaccine candidates for COVID-19. Front. Mol. Biosci. 2021;8:635245. doi: 10.3389/fmolb.2021.635245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pardi N., Hogan M.J., Porter F.W., Weissman D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018;17:261–279. doi: 10.1038/nrd.2017.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhen L., Fang Z., Fanjun C. Recent advances in mRNA vaccines for infectious diseases. Acta Med. Univ. Sci. Technol. Huazhong. 2021;50:234–239. [Google Scholar]
- 88.Watts J.K., Deleavey G.F., Damha M.J. Chemically modified siRNA: Tools and applications. Drug Discov. Today. 2008;13:842–855. doi: 10.1016/j.drudis.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 89.Michelson A.M., Todd. A.R. Nucleotides part XXXII: Synthesis of a dithymidine dinuleotide containing a 3′: 5′-internucleotidic linkage. J. Chem. Soc. 1955;1955:2632–2638. [Google Scholar]
- 90.Yi C., Pan T. Cellular dynamics of RNA modification. Acc. Chem. Res. 2011;44:1380–1388. doi: 10.1021/ar200057m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Peacock H., Kannan A., Beal P.A., Burrows C.J. Chemical modification of siRNA bases to probe and enhance RNA interference. J. Org. Chem. 2011;76:7295–7300. doi: 10.1021/jo2012225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Uhlmann E., Peyman A. Antisense oligonucleotides: A new therapeutic principle. Chem. Rev. 1990;90:543–584. [Google Scholar]
- 93.Micklefield J. Backbone modification of nucleic acids: Synthesis, structure and therapeutic applications. Curr. Med. Chem. 2001;8:1157–1179. doi: 10.2174/0929867013372391. [DOI] [PubMed] [Google Scholar]
- 94.Lennox K.A., Behlke M.A. Chemical modification and design of anti-miRNA oligonucleotides. Gene Ther. 2011;18:1111–1120. doi: 10.1038/gt.2011.100. [DOI] [PubMed] [Google Scholar]
- 95.Geary R.S. Antisense oligonucleotide pharmacokinetics and metabolism. Expert Opin. Drug Metab. Toxicol. 2009;5:381–391. doi: 10.1517/17425250902877680. [DOI] [PubMed] [Google Scholar]
- 96.Krützfeldt J., Rajewsky N., Braich R., Rajeev K.G., Tuschl T., Manoharan M., Stoffel M. Silencing of microRNAs in vivo with “antagomirs”. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 97.Amarzguioui M., Holen T., Babaie E., Prydz H. Tolerance for mutations and chemical modifications in a siRNA. Nucleic Acids Res. 2003;31:589–595. doi: 10.1093/nar/gkg147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Soutschek J., Akinc A., Bramlage B., Charisse K., Constien R., Donoghue M., Elbashir S., Geick A., Hadwiger P., Harborth J. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432:173–178. doi: 10.1038/nature03121. [DOI] [PubMed] [Google Scholar]
- 99.Hall A.H., Wan J., Shaughnessy E.E., Ramsay Shaw B., Alexander K.A. RNA interference using boranophosphate siRNAs: Structure-activity relationships. Nucleic Acids Res. 2004;32:5991–6000. doi: 10.1093/nar/gkh936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fattal E., Barratt G. Nanotechnologies and controlled release systems for the delivery of antisense oligonucleotides and small interfering RNA. Br. J. Pharmacol. 2009;157:179–194. doi: 10.1111/j.1476-5381.2009.00148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yuba E., Nakajima Y., Tsukamoto K., Iwashita S., Kojima C., Harada A., Kono K. Effect of unsaturated alkyl chains on transfection activity of poly(amidoamine) dendron-bearing lipids. J. Control. Release. 2012;160:552–560. doi: 10.1016/j.jconrel.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 102.Xie X., Yang Y., Lin W., Liu H., Liu H., Yang Y., Chen Y., Fu X., Deng J. Cell-penetrating peptide-siRNA conjugate loaded YSA-modified nanobubbles for ultrasound triggered siRNA delivery. Colloids Surf. B Biointerfaces. 2015;136:641–650. doi: 10.1016/j.colsurfb.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 103.Simeoni F., Morris M.C., Heitz F., Divita G. Insight into the mechanism of the peptide-based gene delivery system MPG: Implications for delivery of siRNA into mammalian cells. Nucleic Acids Res. 2003;31:2717–2724. doi: 10.1093/nar/gkg385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chaubey B., Tripathi S., Pandey V.N. Single acute-dose and repeat-doses toxicity of anti-HIV-1 PNA TAR-penetratin conjugate after intraperitoneal administration to mice. Oligonucleotides. 2008;18:9–20. doi: 10.1089/oli.2007.0088. [DOI] [PubMed] [Google Scholar]
- 105.Schlegel M.K., Foster D.J., Kel’in A.V., Zlatev I., Bisbe A., Jayaraman M., Lackey J.G., Rajeev K.G., Charissé K., Harp J. Chirality dependent potency enhancement and structural impact of glycol nucleic acid modification on siRNA. J. Am. Chem. Soc. 2017;139:8537–8546. doi: 10.1021/jacs.7b02694. [DOI] [PubMed] [Google Scholar]
- 106.Pattanayek R., Sethaphong L., Pan C., Prhavc M., Prakash T.P., Manoharan M., Egli M. Structural rationalization of a large difference in RNA affinity despite a small difference in chemistry between two 2′-O-modified nucleic acid analogues. J. Am. Chem. Soc. 2004;126:15006–15007. doi: 10.1021/ja044637k. [DOI] [PubMed] [Google Scholar]
- 107.Manoharan M. 2′-Carbohydrate modifications in antisense oligonucleotide therapy: importance of conformation, configuration and conjugation. Biochim. Biophys. Acta. 1999;1489:117–130. doi: 10.1016/s0167-4781(99)00138-4. [DOI] [PubMed] [Google Scholar]
- 108.Judge A.D., Bola G., Lee A.C., MacLachlan I. Design of noninflammatory synthetic siRNA mediating potent gene silencing in vivo. Mol. Ther. 2006;13:494–505. doi: 10.1016/j.ymthe.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 109.Bennett C.F., Swayze E.E. RNA targeting therapeutics: Molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010;50:259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- 110.Esau C.C. Inhibition of microRNA with antisense oligonucleotides. Methods. 2008;44:55–60. doi: 10.1016/j.ymeth.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 111.Esau C., Davis S., Murray S.F., Yu X.X., Pandey S.K., Pear M., Watts L., Booten S.L., Graham M., McKay R. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3:87–98. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 112.Dorn G., Patel S., Wotherspoon G., Hemmings-Mieszczak M., Barclay J., Natt F.J., Martin P., Bevan S., Fox A., Ganju P. siRNA relieves chronic neuropathic pain. Nucleic Acids Res. 2004;32:e49. doi: 10.1093/nar/gnh044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen X., Dudgeon N., Shen L., Wang J.H. Chemical modification of gene silencing oligonucleotides for drug discovery and development. Drug Discov. Today. 2005;10:587–593. doi: 10.1016/S1359-6446(05)03426-4. [DOI] [PubMed] [Google Scholar]
- 114.Petersen M., Bondensgaard K., Wengel J., Jacobsen J.P. Locked nucleic acid (LNA) recognition of RNA: NMR solution structures of LNA:RNA hybrids. J. Am. Chem. Soc. 2002;124:5974–5982. doi: 10.1021/ja012288d. [DOI] [PubMed] [Google Scholar]
- 115.Chan J.A., Krichevsky A.M., Kosik K.S. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65:6029–6033. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- 116.Pasternak A., Wengel J. Unlocked nucleic acid—An RNA modification with broad potential. Org. Biomol. Chem. 2011;9:3591–3597. doi: 10.1039/c0ob01085e. [DOI] [PubMed] [Google Scholar]
- 117.Snead N.M., Escamilla-Powers J.R., Rossi J.J., McCaffrey A.P. 5′ Unlocked nucleic acid modification improves siRNA targeting. Mol. Ther. Nucleic Acids. 2013;2:e103. doi: 10.1038/mtna.2013.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dowler T., Bergeron D., Tedeschi A.L., Paquet L., Ferrari N., Damha M.J. Improvements in siRNA properties mediated by 2′-deoxy-2′-fluoro-β-d-arabinonucleic acid (FANA) Nucleic Acids Res. 2006;34:1669–1675. doi: 10.1093/nar/gkl033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Prakash T.P., Allerson C.R., Dande P., Vickers T.A., Sioufi N., Jarres R., Baker B.F., Swayze E.E., Griffey R.H., Bhat B. Positional effect of chemical modifications on short interference RNA activity in mammalian cells. J. Med. Chem. 2005;48:4247–4253. doi: 10.1021/jm050044o. [DOI] [PubMed] [Google Scholar]
- 120.Davis S., Lollo B., Freier S., Esau C. Improved targeting of miRNA with antisense oligonucleotides. Nucleic Acids Res. 2006;34:2294–2304. doi: 10.1093/nar/gkl183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ørom U.A., Kauppinen S., Lund A.H. LNA-modified oligonucleotides mediate specific inhibition of microRNA function. Gene. 2006;372:137–141. doi: 10.1016/j.gene.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 122.Koshkin A.A., Nielsen P., Meldgaard M., Rajwanshi V.K., Singh S.K., Wengel J. LNA (locked nucleic acid): An RNA mimic forming exceedingly stable LNA:LNA duplexes. J. Am. Chem. Soc. 1998;120:13252–13253. [Google Scholar]
- 123.Lennox K.A., Behlke M.A. A direct comparison of anti-microRNA oligonucleotide potency. Pharm. Res. 2010;27:1788–1799. doi: 10.1007/s11095-010-0156-0. [DOI] [PubMed] [Google Scholar]
- 124.Takahashi M., Yamada N., Hatakeyama H., Murata M., Sato Y., Minakawa N., Harashima H., Matsuda A. In vitro optimization of 2′-OMe-4′-thioribonucleoside-modified anti-microRNA oligonucleotides and its targeting delivery to mouse liver using a liposomal nanoparticle. Nucleic Acids Res. 2013;41:10659–10667. doi: 10.1093/nar/gkt823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhang J., Chen Y., Huang Y., Jin H.W., Qiao R.P., Xing L., Zhang L.R., Yang Z.J., Zhang L.H. Synthesis and properties of novel l-isonucleoside modified oligonucleotides and siRNAs. Org. Biomol. Chem. 2012;10:7566–7577. doi: 10.1039/c2ob26219c. [DOI] [PubMed] [Google Scholar]
- 126.Wada S., Obika S., Shibata M.A., Yamamoto T., Nakatani M., Yamaoka T., Torigoe H., Harada-Shiba M. Development of a 2′,4′-BNA/LNA-based siRNA for dyslipidemia and assessment of the effects of its chemical modifications in vivo. Mol. Ther. Nucleic Acids. 2012;1:e45. doi: 10.1038/mtna.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Straarup E.M., Fisker N., Hedtjärn M., Lindholm M.W., Rosenbohm C., Aarup V., Hansen H.F., Ørum H., Hansen J.B., Koch T. Short locked nucleic acid antisense oligonucleotides potently reduce apolipoprotein B mRNA and serum cholesterol in mice and non-human primates. Nucleic Acids Res. 2010;38:7100–7111. doi: 10.1093/nar/gkq457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sun Y., Zhao Y., Zhao X., Lee R.J., Teng L., Zhou C. Enhancing the therapeutic delivery of oligonucleotides by chemical modification and nanoparticle encapsulation. Molecules. 2017;22:1724. doi: 10.3390/molecules22101724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang Y., Miao L., Satterlee A., Huang L. Delivery of oligonucleotides with lipid nanoparticles. Adv. Drug Deliv. Rev. 2015;87:68–80. doi: 10.1016/j.addr.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lam J.K., Chow M.Y., Zhang Y., Leung S.W. siRNA versus miRNA as therapeutics for gene silencing. Mol. Ther. Nucleic Acids. 2015;4:e252. doi: 10.1038/mtna.2015.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rabbani P.S., Zhou A., Borab Z.M., Frezzo J.A., Srivastava N., More H.T., Rifkin W.J., David J.A., Berens S.J., Chen R. Novel lipoproteoplex delivers Keap1 siRNA based gene therapy to accelerate diabetic wound healing. Biomaterials. 2017;132:1–15. doi: 10.1016/j.biomaterials.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 132.Zhang J., Fan H., Levorse D.A., Crocker L.S. Ionization behavior of amino lipids for siRNA delivery: Determination of ionization constants, SAR, and the impact of lipid pKa on cationic lipid-biomembrane interactions. Langmuir. 2011;27:1907–1914. doi: 10.1021/la104590k. [DOI] [PubMed] [Google Scholar]
- 133.Amjad M.W., Kesharwani P., Mohd Amin M.C.I., Iyer A.K. Recent advances in the design, development, and targeting mechanisms of polymeric micelles for delivery of siRNA in cancer therapy. Prog. Polym. Sci. 2017;64:154–181. [Google Scholar]
- 134.Gwak S.J., Nice J., Zhang J., Green B., Macks C., Bae S., Webb K., Lee J.S. Cationic, amphiphilic copolymer micelles as nucleic acid carriers for enhanced transfection in rat spinal cord. Acta Biomater. 2016;35:98–108. doi: 10.1016/j.actbio.2016.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nair J.K., Willoughby J.L., Chan A., Charisse K., Alam M.R., Wang Q., Hoekstra M., Kandasamy P., Kel’in A.V., Milstein S. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014;136:16958–16961. doi: 10.1021/ja505986a. [DOI] [PubMed] [Google Scholar]
- 136.Yu R.Z., Graham M.J., Post N., Riney S., Zanardi T., Hall S., Burkey J., Shemesh C.S., Prakash T.P., Seth P.P. Disposition and pharmacology of a GalNAc3-conjugated ASO targeting human lipoprotein (a) in mice. Mol. Ther. Nucleic Acids. 2016;5:e317. doi: 10.1038/mtna.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kulkarni J.A., Witzigmann D., Thomson S.B., Chen S., Leavitt B.R., Cullis P.R., van der Meel R. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021;16:630–643. doi: 10.1038/s41565-021-00898-0. [DOI] [PubMed] [Google Scholar]
- 138.Russell S., Bennett J., Wellman J.A., Chung D.C., Yu Z.F., Tillman A., Wittes J., Pappas J., Elci O., McCague S. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390:849–860. doi: 10.1016/S0140-6736(17)31868-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.