

Abstract
The emergence of the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic poses a never before seen challenge to human health and the economy. Considering its clinical impact, with no streamlined therapeutic strategies in sight, it is crucial to understand the infection process of SARS-CoV-2. Our limited knowledge of the mechanisms underlying SARS-CoV-2 infection impedes the development of alternative therapeutics to address the pandemic. This aspect can be addressed by modeling SARS-CoV-2 infection in the human context to facilitate drug screening and discovery. Human induced pluripotent stem cell (iPSC)-derived lung epithelial cells and organoids recapitulating the features and functionality of the alveolar cell types can serve as an in vitro human model and screening platform for SARS-CoV-2. Recent studies suggest an immune system asynchrony leading to compromised function and a decreased proportion of specific immune cell types in coronavirus disease 2019 (COVID-19) patients. Replenishing these specific immune cells may serve as useful treatment modality against SARS-CoV-2 infection. Here the authors review protocols for deriving lung epithelial cells, alveolar organoids and specific immune cell types, such as T lymphocytes and natural killer cells, from iPSCs with the aim to aid investigators in making relevant in vitro models of SARS-CoV-2 along with the possibility derive immune cell types to treat COVID-19.
Key Words: immune therapy, induced pluripotent stem cells, lung epithelial cells, NK cells, organoids, SARS-CoV-2, T cells
Graphical abstract
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has remarkably and very rapidly adapted to humans, its most recent host. Most of the mechanistic knowledge about the host-specific response and virus–host interplay is extrapolated from the knowledge obtained from infection by other members of the coronavirus family [1,2]. Lack of extensive understanding of the underlying infectivity and response between the virus and novel host (human being) is hindering development of appropriate treatment strategies. With the increase in SARS-CoV-2 infection and associated mortality across the globe, it is imperative to devise an in vitro SARS-CoV-2 infection model for providing the possibility of unraveling the strain virulence and the disease process therby facilitating drug screening and development of novel treatment strategies for improving clinical outcomes. Currently, immortalized cell lines are being utilized to model the permissiveness features of SARS-CoV-2 and propagate them [3,4]. However, these models lack physiological relevance, and immortalized cell lines are prone to containing genomic aberrations, limiting their scope as biologically relevant models of SARS-CoV-2 [5]. Although these cell lines are effective in their ability to propagate SARS-CoV-2, they are inept at recapitulating the features and function of the human airway epithelium, therefore emphasizing the need for a relevant and contextual in vitro system to study SARS-CoV-2 kinetics, trophism and host response [6]. Studies on the trophism of SARS-CoV-2 have demonstrated its specific affinity for the respiratory epithelium, which is also its primary route of entry because of high expression and activity of the angiotensin-converting enzyme 2 (ACE2) receptor, transmembrane protease serine 2 (TMPRSS) and cathepsin B [7]. ACE2 and TMPRSS2 have been detected in both nasal and bronchial epithelium [8]. Gene expression of ACE2 and TMPRSS2 has been reported to occur largely in alveolar epithelial type II (AT2) cells [9], which are central to SARS-CoV-2 pathogenesis. Human induced pluripotent stem cells (iPSCs) having the unique property of unlimited self-renewal capacity and can potentially differentiate into various types of somatic cells [10]. In addition, iPSCs offer the possibility of generating alveolar epithelial cells and three-dimensional (3D) lung organoids as an in vitro predictive model of SARS-CoV-2 infection and as a screening platform for coronavirus disease 2019 (COVID-19) therapeutics [11,12]. Apart from understanding the disease mechanism, such an iPSC-derived model of respiratory/alveolar tissue would serve as a platform for large-scale screening of anti-viral drugs [13]. With the large number of high-risk/immune-compromised COVID-19 patients around the globe, the current situation necessitates pursuing multiple approaches simultaneously to achieve the best outcomes for patients and society at large, in terms of curative and preventive care. Clinical findings in COVID-19 patients point to a dysregulated/exuberant immune response as a leading contributor to SARS-CoV-2-mediated pathology. Interestingly, the acute phase of SARS in humans is associated with a severe reduction in the number of T cells in the blood [14]. Thus far, only a limited number of studies have explored the role of the T-cell-mediated adaptive immune response in COVID-19 pathogenesis. A recent study showed that T-cell counts are reduced significantly, and those that survive are functionally exhausted in COVID-19 patients [15]. The study also suggested that the presence of persistently low CD8+ and CD4+ T-cell counts in non-intensive care unit COVID-19 patients would need aggressive intervention even in the immediate absence of severe symptoms to reduce the risk of further deterioration in the patient's condition. Based on these clinical insights, development of “off-the-shelf,” third-party, allogeneic, virus-specific T-cell therapies would be a powerful tool for treating COVID-19-associated lymphopenia, which is currently not amenable to standard therapeutic strategies. Similar to T cells, the role of natural killer (NK) cells in SARS-CoV-2 infections is currently being examined [16], [17], [18], [19], [20]. The rationale for using NK cells as a modality for immune therapy for COVID-19 is also being investigated. Here the authors discuss protocols for deriving respiratory epithelial cells and alveolar organoids with the aim of modeling the infection biology of the novel SARS-CoV-2 virus and investigating the possible applications of human iPSC (hiPSC)-derived T and NK cells as therapeutic intervention for this newly emerged pathogen.
Modeling SARS-CoV-2 Using iPSC-Derived Alveolar Cells and Organoids
Infectious viruses are often restricted to specific hosts and cell types that are the primary routes of entry into the host system. Modeling this host–virus interaction is challenging since most of the circulating human viruses are genetically different from viruses cultured under standard laboratory conditions. For example, the surface glycoproteins of human parainfluenza virus 3, which are critical for entry into the host cell, differ considerably from the surface glycoproteins cultured on immortalized monolayer cells in the laboratory [21] The SARS-CoV-2 infection begins with entry into the respiratory epithelial cells, which is mediated by the surface ACE2 receptor [7,22]. The SARS-CoV-2 virus propagates within the infected epithelial cells, including the AT2 cells, where, in some cases, the host innate immune response is triggered. Coincidently, most human pluripotent stem cell (hPSC) differentiation protocols have been directed toward obtaining AT2+ cell phenotypes because of their crucial physiological role in vivo along with the possibility of evaluating their functionality by measuring the expression and secretion of surfactant molecules [23,24]. These findings underscore the need for recapitulating the infection process in an in vitro model in which the features of the virus are mimicked well enough to infect its choice of cell type.
Directed differentiation of iPSCs toward lung cells faced considerable initial challenges [25]. To address these challenges, a rational stepwise approach mimicking mammalian lung development in vivo has essentially been followed in successfully differentiating PSCs to functional alveolar cells [[26], [27], [28], [29], [30]]. The key aim here is to obtain a pure population of lung epithelia with the ability to expand in vitro to perform disease modeling and harness this as a platform for novel drug development (Figure 1 ). Defined protocols have now been established to derive specific lung lineage cell phenotypes (Table 1 ). Bonafide lung epithelial cells were obtained by Huang et al. [31] by initially driving PSCs to a definitive endoderm fate and then further directing them to a FOXA2 and NKX2.1 (also known as thyroid transcription factor) lung epithelial progenitor cell phenotype. The study also demonstrated that FOXA2+NKX2.1+ double-positive progenitor cells gave rise to basal, club, goblet, ciliated, alveolar type I (AT1) and AT2 cells both in vivo and in vitro. Although yet to be established with hiPSCs, Garreta et al. [32], using mouse embryonic stem and iPSC cells, showed low oxygen tension significantly enhanced differentiation toward a lung progenitor phenotype. Recently, Surendran et al. [33] elucidated a protocol for generating lung epithelial cells from hiPSCs. Their protocol initially differentiated the iPSCs to a definitive endoderm fate followed by progression into anteriorized endoderm, which can give rise to both proximal and distal epithelial cells.
Figure 1.

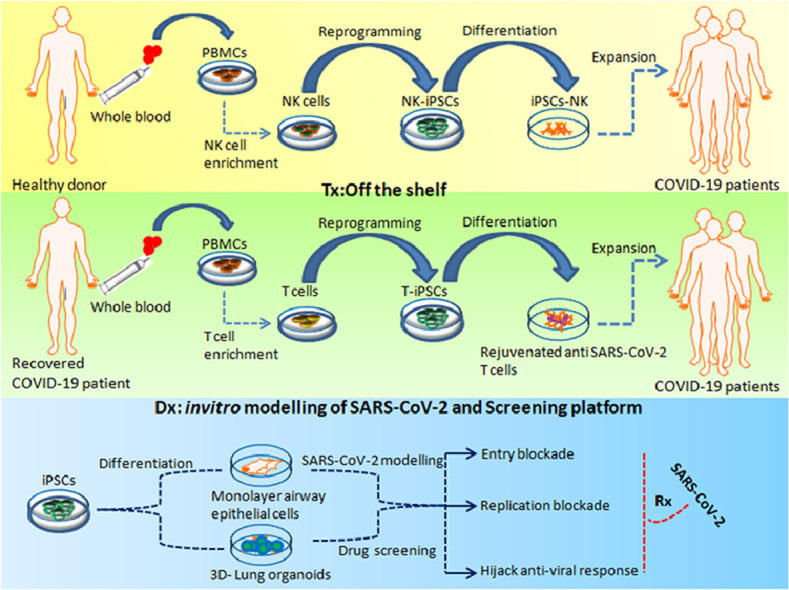
iPSC-based in vitro modeling of SARS-CoV-2 and iPSC-derived immune cellular therapy for COVID-19. Allogeneic NK cells can be derived from iPSCs, which can be administered to treat patients with COVID-19 with low NK counts. These iPSC-derived allogeneic NK cells may provide the necessary immune response, which can address the damage and control SARS-CoV-2 infection. T cells enriched from the primary PBMCs of recovered COVID-19 patients can be used to generate T-iPSCs, which can be differentiated to T cells to address patients with COVID -19 with lymphopenia. iPSC-derived airway epithelial cells and 3D lung organoids can be used to study the infection process of SARS-CoV-2 and as a screening platform for drug discovery and development. Dx, diagnosis; PBMCs, peripheral blood mononuclear cells; Rx, prescription; T-iPSCs, T lymphocyte iPSCs. (Color version of figure is available online.)
Table 1.
Derivation of lung cells and organoids from human pluripotent cells.
| Author | Stem cell type | Cell type derived | Time line in days | Culture conditions | Remarks |
|---|---|---|---|---|---|
| Ghaedi et al.[39] | iPSCs | Lung progenitor cells | 15–20+ | Differentiation was carried out on Matrigel. D0–D6: RPMI basal medium with activin A for first 48 h and then from D3, RPMI basal medium + activin A + B27 (50×) + sodium butyrate to direct differentiation to DE fate. D7–D9: Differentiate into AFE using IMDM basal medium + FBS + penicillin/streptomycin + L-glutamine + MEM non-essential amino acids for first 2 days and then introduce IMDM basal medium + Noggin + SB431542 for next 24 h. D10–D15: Differentiate into lung progenitor cells using IMDM basal medium + BMP4 + bFGF + Wnt3a + KGF + retinoic acid, etc. |
FOXA2 and NKX2.1 mRNA expression checked on day 15 using RT-PCR. Protocol is a three-step process to derive lung progenitor cells. Shortest protocol for deriving lung progenitor cells. |
| Firth et al.[28] | iPSCs | Lung epithelial cells | 45+ | Culture plates were coated with a combination of fibronectin, laminin and collagen IV gel for differentiation. D0–D3: iPSCs were differentiated into DE cells using RPMI basal medium + Wnt3A + activin A for 24 h. RPMI basal medium + 1% FBS and activin A added on day 2. D4–D9: DE cells were further differentiated into AFE. Ultra MEM-ITS + 2% FBS + SB431542 + Noggin added for 24 h. D5–D9, Noggin was removed and BMP4 added to inhibit Notch signal. D10–D17: Generation of lung progenitor cells using ultra MEM basal medium + 2% FBS + BMP4 + FGF2 + FGF10 + KGF, etc. D18–D42+: Differentiated cells into epithelial lung cells using DMEM/F12 + Ultroser G (serum substitute) + fetal clone II serum + insulin + bovine brain extract + transferrin + hydrocortisone + 3,3’,5-triiodo-L-thyronine sodium salt + epinephrine + retinoic acid + phosphorylethanolamine + ethanolamine. |
SOX17, FOXA2 and GATA6 transcript expression checked on day 4. Cells were stained for FOXA2, NKx2.1 and SOX2 protein expression. Cells were stained for Z0-1, E-cadherin and calmodulin protein expression. Protein expression of FOXJ1 and pericentrin evaluated on day 28. |
| Wong et al.[29] | ESCs/iPSCs | Lung epithelial cells | 40+ | Plates were pre-coated with human placental collagen. IV D0–D4: Cells were differentiated into DE using endoderm differentiation medium consisting of DMEM + GlutaMAX + NEAA penicillin/streptomycin + activin A + Wnt3A + mercaptoethanol. D5–D9: Differentiation of DE into AFE using endoderm differentiation medium + SHH + FGF2 D10–D20: Differentiation of AFE cells into immature lung epithelial cells. Cells were cultured with endoderm differentiation medium + FGF10 + KGF + BMP4 for 5 days and then further cultured with FGF10 + FGF7 + FGF18 for more 5 days. D20–D40+: Immature cells converted into mature cells using bronchial epithelial growth medium with FGF18 for 6–10 days followed by B-ALI medium for more than 15 days. |
FOXA2 and SOX17 gene expression was evaluated. Addition of FGF2 and SHH did not increase the number of NKx2.1+ cells. B-ALI medium was not added to the top of the well. Instead, it was added to the bottom of Transwell. |
| Huang et al.[31] | iPSCs/ESCs | Lung and airway epithelial cells | 40+ | iPSCs were treated with DMEM/F12 supplemented with N2 + B27 supplement + ascorbic acid + GlutaMAX + monothioglycerol + BSA + penicillin/streptomycin before forming into EBs. D0–D4: Primitive streak formation was achieved with BMP4 and Y-27632 (RI) added to the medium for 1 day. Cells were resuspended in DE induction medium. DMEM/F12 basal medium along with B27 + Y-27632 + BMP4 + bFGF + activin A added to culture for 3–5 days. D5–D6: Introduced AFE medium after dissociating EBs with trypsin and cultured on fibronectin-coated wells. Serum-free medium with dorsomorphin dihydrochloride + SB431542 introduced for 48 h along with Dkk1. After 48 h, switched to SB431542 and IWP2. D6–D15: Cells differentiated in lung progenitor medium for 8–10 days. CHIR99021 + FGF10 + FGF7 + BMP4 + EGF + ATRA added in SFD medium. D15–D40+: For further maturation of lung progenitors, cells were treated with CHIR99021 + FGF10 + FGF7 + BMP4 + ATRA with SFD medium. |
Cells were trypsinized prior to plating in low attachment plates to form EBs under serum-free conditions. CO2 5%/air environment. Differentiated cells were evaluated for expression of MUC1, ABCA3, MUC5B, FOXJ1, PDN and AQP5. |
| Garreta et al.[32] | iPSCs | Lung progenitor cells | 22+ | D0–D2: EBs were generated using hanging drop method under two different oxygen conditions (5% and 20%). EBs formed within 3 days. D2–D5: EBs plated on gelatin-coated plate in DE induction medium. The medium had DMEM + N2 + B27 + FBS + L-glutamine + penicillin/streptomycin + activin A and introduced for 72 h. D6: Medium changed to ADE medium composed of DMEM + N2 + B27 + FBS + L-glutamine + penicillin/streptomycin + Noggin + SB431542, etc. D7–D12: Medium was changed to lung progenitor induction medium composed of ADE medium without Noggin and SB431452. Wnt3A + hFGF10 + mFGF7 + BMP4 + hEGF + FGF2 and heparin sodium salt added for 5 days. D13–D23: Immature lung progenitor cells were grown in same medium for further 10 days to convert into mature cells. |
EBs were generated prior to differentiation into lung cells. NKx2.1+ PAX8 gene expression evaluated. Cells were incubated under 5% or 20% oxygen concentrations. Plate with 5% oxygen supply showed better maturation compared with 20% oxygen. |
| Surendran et al.[11] | iPSCs | Lung epithelial cells | 30+ | EBs generated before differentiating into lung epithelial cells. Cells were passaged in non-adherent plate. EBs introduced with basal medium along with activin A and cultured for next 2 days. D0–D5: EBs introduced with definitive endoderm medium with the help of activin A + B27 and BMP4 for 5 days D6–D10: Next step was to introduce anterior definitive medium. Basal medium + EGF + bFGF + SB431542 and Noggin (dual SMAD inhibitors) was used for next 5 days. D11–D20+: Generation of lung progenitor cells done with the help of differentiation basal medium. Cells differentiated into proximal lung epithelial cells using differentiation basal medium + BMP7 + FGF7 + PD032519 + retinoic acid + Noggin and CHIR99021 for next 7–10 days. For distal lung epithelial cell derivation, basal medium containing BMP2 + FGF10 + BMP4 + bFGF + Wnt3a was used for next 7–10 days. |
Expression of FOXA2 and SOX17 was evaluated. Nkx2.1 gene expression checked for proximal lung epithelial cells. NKX2.1 and SOX9 gene expression checked for distal lung epithelial cells. Shortest protocol for deriving lung epithelial cells. |
| Longmire et al.[30] | ESCs | Lung progenitor cells | 25+ | Before DE induction medium, EBs were generated. D0–D5: EBs in non-adherent plates. DE medium consisting of mixture of IMDM (75%) and Ham's medium (25%) with activin A was used for 5 days. D6: EBs transferred onto gelatin-coated plate. Anterior endoderm medium containing cSFDM medium supplemented with Noggin and SB431542 was used for next 24 h. D7–D22: Lung induction medium consisting of cSFDM supplemented with heparin + KFG + hFGF + BMP4 + Wnt3 + hEGE + FGF2 was used for next 8 days (D7–D15). For next 7 days, hFGF10 was introduced into medium. D22–D25+: Lung maturation medium consisting of Ham's F12 medium + KGF + transferrin + CaCl2 + insulin + sodium selenite + dexamethasone + 8-Br-cAMP + BSA, etc., for next 22–25 days. |
Cells sorted based on gene expression. Cells evaluated for SPC, CC10, CTFR and FOXJ1 expression. |
| Tamo et al.[26] | iPSCs | Lung alveolar epithelial cells |
30+ | Two-step protocol for derivation of iPSC-AECII-d alveolar epithelial cell line (LL)-iPSC-AECII. D0–D3: iPSC colonies trypsinized and cells cultured on Vitronectin XF and grown for 3 days in iPSC medium. D3–D8: Medium was replaced with STEMdiff DE kit for 5 days. D8–D29: Cells were grown in small airway epithelial cell growth medium (SAGM; Lonza) supplemented with 1% FBS for another 21 days. D30+: hTERT and hBmi protein introduced to lentiviral vector used for transduction, which was done in derived ACE II to generate long-lasting ACE II (LL-iPSC-ACE II). LL-iPSC-AECII was grown to confluency in a 24-well plate and DMEM medium with 10% FBS supplemented with 100 mM IWR-1 used for 7 days. |
Two-step protocol to derive ACEII cells expressing surfactant protein C, a specific AEC type II marker. AEC type I marker AQP5 did not show on ACE II type cells. AQP5 and hT1α used as ACE I markers. |
| Heo et al.[27] | hPSCs | Lung alveolar epithelial cell | 25+ | Cells seeded at low density in the plate. D0–D6: Generation of DE done with the help of activin A + CHIR99021 + sodium butyrate, etc. D6–D10: Cells were introduced to Noggin and SB431542 to form ADE and then directed toward alveolar epithelial cell fate. D10–D14: Alveolar commitment step in which BMP4 and CHIR99021 were used to form immature lung AECs. D14–D25+: These days considered AEC maturation days for cells, and maturation medium used along with FGF10 + dexamethasone + 8-Br-cAMP + IBMX + KGF. |
Cells were positive for NKX2.1, EPCAM and CPM expression. AECII differentiated to AECI, as determined by gene expression of T1 alpha. |
| Wang et al.[24] | hESCs | Lung alveolar epithelial type II (AE2) | 20+ | D0–D2: Generation of EBs done by splitting cells using collagenase IV and cultured on six-well non-coated plates in hES cell medium. D2–D6: EBs were cultured on fresh six-well non-coated plates with DM containing 80% DMEM + 20% FBS + 1% NEAA + 1 mM L-glutamine + 100 g/mL penicillin/streptomycin. D6–D20+: EBs cultured further on gelatin-coated plate in DM and allowed to expand. The selection of hES cell-derived ATII cells was started on day 6 by introducing 20 g/mL of G418. Alternate method used was without EB formation, where G418 added from D1 onward. |
The protocol allows derivation without formation of EBs. AT II cells were cultured on Matrigel along with G418 in the medium. Protocol is specific for derivation of AE2-type cells |
| Van Haute et al.[23] | hESCs | Lung epithelial cells | 28+ | D0–D4: Collagenase IV was used to split the cells with subsequent culture on 12-well plates with MEF feeders using hESC medium in liquid–liquid conditions. D4–D28: Differentiation started using differentiation medium (i.e., hESC medium without bFGF and β-mercaptoethanol) in liquid–liquid conditions for 4 days and then replaced with air–liquid interface culture for 20 days. Note: Two types of controls were used in the quantitative real-time RT-PCR experiments: (i) hESC on MEF feeders, which differentiated spontaneously in hESC medium, and (ii) hESC plated on porous membranes for 4 days in hESC medium followed by 24 days in differentiation medium in liquid–liquid conditions (named “NO ALI control”). |
Medium from upper compartment was removed. (referred to as ALI differentiation). SP-C and AQP5 had the highest expression after 20 days of culture. |
| Gotoh et al.[37] | hPSCs | Alveolar Epithelial Cells | 25+ | D0–D6: Single-cell enzymatic dissociation done to form DE cells from hESCs. D0–D2: Y-27632 used to avoid cell death. From D1 to D6, sodium butyrate + activin A + CHIR99021 used along with basal medium. D6–D10: AFE cells formed from DE using small molecules such as Noggin and SB431542. D10–D14: Ventralized AFE cells formed from AFE cells using BMP4 + ATRA + CHIR99021 D14–D25+: To D21, FGF10 used along with aforementioned small molecules and basal medium. After D21, dexamethasone + 8-Br-cAMP + IBMX + KGF, etc., added until D25 for better lung-related gene expression. |
CXCR4 + SOX17 and FOXA2 gene expression checked on D6. SOX2 and FOXA2 expression checked. NKX2.1 and GATA6 expression observed. HOPX, SOX9, NKX2.1 and GATA6 expression observed in cells. |
| Yamamoto et al.[38] | hiPSCs | Lung alveolar organoids | 35+ | D0–D6: From initial day onward, activin A + CHIR99021 + sodium butyrate used along with basal medium to form DE cells. D6–D10: Noggin and SB431542 used to form AFE cells from DE and processed for 4 days. D10–D14: Ventralized AFE generated using BMP4 + ATRA + CHIR99021, etc. D14–D21: NKX2.1+ progenitor cells identified on D21. CHIR99021 + FGF10 + KGF + DAPT used along with basal medium and isolation of CPM cells done on D21. D21–D35+: 3D co-cultured with human fetal lung fibroblasts and dexamethasone + 8-Br-cAMP + IBMX + KGF, etc., added until D35 to form alveolar organoids. |
2D differentiation (3D differentiation). Lamellar body-like organelles, a specific feature of AT2 cells, observed on D35. |
| Dye et al.[42] | hPSC | Lung organoids | 25+ | D0–D4: ESCs differentiated into DE cells using activin A along with RPMI 1640 medium. D4–D8: Cells were differentiated into AFE cells using DMEM/F12 + N2 + B27 supplement + 10 mM HEPES + L-glutamine + 1× penicillin/streptomycin + Noggin + SB431542 + FGF4 + CHIR99021 for 4 days To maintain culture for a long period of time, Noggin and SB431542 were removed from the medium. FGF2 + Sant-2 + SU5402 + SHH + SAG were present for 8 days. D8–D25+: Spheroids were formed after 4 days of growth factor treatment. Generated spheroids cultured on Matrigel-coated plate. Foregut medium with 1% FBS + growth factors + small molecules overlaid and replaced every 4 days. Formed organoids transferred into new Matrigel droplets every 10–15 days. |
SOX2 and FOXA2 expression checked. For short-term use, no need to use growth factor with small molecules. Organoids formed and medium replaced every 4 days. |
| Chen et al.[46] | hPSC | Lung cells (3D) | 25+ | D0–D3: MEFs introduced onto Matrigel for 24 h and supplied with hPSC medium and 5% CO2. After 24 h, EBs formed using embryoid formation medium and then switched to endoderm induction medium for 36–40 h. Activin A + BMP4 and bFGF used to form endoderm cells. For iPSC cell lines, endoderm cells purified using human CD184 MicroBead kit. D4–D6: AFE introduced on D4. EBs dissociated with trypsin/EDTA and cultured on fibronectin-coated plates. Cells introduced with anteriorization medium 1 for 1 day and then switched to anteriorization medium 2 for another day. On D4, Noggin and SB431542 used as small molecules. On D5, IWP + SB431542 used. D7–D25: Cells were treated with ventralization medium for 2 days and 3D clump formations observed. The suspended cells were LBOs. These organoids were introduced to branching medium and fed every other day until D25. CHIR99021 + BMP4 + FGF10 + KGF + retinoic acid used in branching medium for generation of lung-type cells. |
Endoderm expression determined using CXCR4 and C-KIT. FOXA1 and FOXA2 gene expression observed. SOX9 and NKX2.1 gene expression observed. |
ACEs, alveolar epithelial cells; ADE, anterior definitive endoderm; AFE, anterior foregut endoderm; AQP5, aquaporin 5; B-ALI, bronchial air–liquid interface; bFGF, basic fibroblast growth factor; BSA, bovine serum albumin; cSFDM, complete serum-free differentiation medium; D, day; DE, definitive endoderm; DMEM, Dulbecco's Modified Eagle's Medium; EDTA, ethylenediaminetetraacetic acid; EGF, epidermal growth factor; EPCAM, epithelial cell adhesion molecule; ESCs, embryonic stem cells; FBS, fetal bovine serum; hEGF, human epidermal growth factor; hESCs, human embryonic stem cells; hFGF, human fibroblast growth factor; IMDM, Iscove's Modified Dulbecco's Medium; KGF, keratinocyte growth factor; MEFs, mouse embryonic fibroblasts; MEM, Minimum Essential Medium; NEAA, non-essential amino acid; RT-PCR, reverse transcription polymerase chain reaction; 2D, two-dimensional.
Heo et al. [27] derived alveolar epithelial cells from hiPSCs to evaluate plumonary toxicity and inflammatory responses of toxins. In this protocol, the hiPSCs were differentiated for alveolar epithelial progenitor markers such as NKX2.1, epithelial cell adhesion molecule and carboxypeptidase M (CPM). Robust differentiation to alveolar epithelial cells was obtained, with more than 90% NKX2.1+ cells, whereas the percentage of cells co-expressing NKX2.1 and epithelial cell adhesion molecule was approximately 70%. The differentiated cells also expressed pulmonary surfactant-associated protein B and pulmonary surfactant-associated protein C, indicating typical functional signatures of AT2 cells. Since AT2 cells have the potential to trans-differentiate into an AT1 cell type during normal lung development [34] or after lung injury [35], the researchers determined the expression of T1 alpha/podoplanin, a cell membrane protein expressed specifically in AT1 cells [36], to distinguish the fate of iPSC-derived AT+ cells after 25 days of differentiation. The researchers also found comparable numbers of lung mesenchymal cells expressing PDGFR-β, CD90, NG2 and CD146 on day 25 of differentiation, suggesting that the differentiation protocol also generated the mesenchymal cell phenotypes found in alveolar niches.
Gotoh et al. [37] differentiated hiPSCs to form NKX2-1+ “ventralized” anterior foregut endoderm cells, from which cells expressing CPM were sorted for 3D co-culture with fetal human lung fibroblasts. The resulting CPM+ organoids, although devoid of club cells, contained mostly AT2 cells as well as some AT1 cells, ciliated cells and goblet cells. These iPSC-derived AT2 cells exhibited phenotypic properties similar to those of mature human AT2 cells, including expression of surfactant proteins and the possession of lamellar body-like structures. A study by Yamamoto et al. [38] demonstrated that pre-conditioning the NKX2-1+ endoderm progenitors significantly enhanced induction to an AT2 cell phenotype. However, a relatively homogeneous population of AT2 cells and AT1 cells was generated from hiPSCs reprogrammed from fetal or neonatal lung fibroblasts [39]. In addition, hiPSC-derived AT2 alveolospheres exhibited self-renewal capacity and immune responsiveness [40].
These studies clearly indicate that alveolar cell lineages produced from iPSC-derived lung organoids mimic the development of their in vivo counterparts (Figure 2 ) [41,42]. Incidentally, fibroblast growth factor signaling has been demonstrated to be crucial for promoting the induction of anterior foregut endoderm into human lung organoids encompassing the contextual cellular milieu [42]. Historically, a polarized human airway epithelium culture system has been used to represent the authentic airway for viral respiratory infection [43,44]. However, the human airway epithelium model represents few of the alveolar cell types, which limits its utility as a cellular model in the study of SARS-CoV-2. To address these issues, modeling viral respiratory infection using lung organoids has been explored [45], [46], [47], as this encompasses most of the pulmonary cell types and components present in the tissue counterpart.
Figure 2.

Schema of directed differentiation of iPSCs to lymphocytes, alveolar cells and lung organoids in vitro. hiPSCs in a monolayer are directed to an endoderm fate by inhibiting the TGFβ and BMP pathways and subsequently to early ventral anterior foregut cells by activating WNT and FGF signaling along with the TGFβ and BMP pathways. The anterior foregut cells are directed toward a lung progenitor cell fate by stimulating the WNT, FGF and RA signaling pathways. Downstream differentiation of lung progenitors can be achieved by employing protocols that activate FGF, WNT and RA signaling or by simultaneously activating the RA pathway and inhibiting the BMP pathway to derive lung organoids and alveolar cell types, respectively. By recapitulating key stages of lymphocyte development, T and NK cells can be derived from iPSCs. First, the iPSCs are induced to hematopoietic stem cell phenotypes, which undergo further specification, giving rise to hematopoietic progenitor cells using cytokines and factors activating the TGFβ and FGF signaling pathways. The hematopoietic progenitors can be directed toward a T-cell fate using a co-culture system in the presence of molecules that primarily activate the Notch pathway. NK cell derivation from hematopoietic progenitor cells requires the presence of various cytokines. The markers of each of the cell types are shown in blue. The signaling pathways are shown in red when inhibited and in green when stimulated. Small molecules and recombinant proteins used for directed differentiation processes are shown in purple. ATRA, all-trans retinoic acid; BMP, bone morphogenic protein; EGF, epidermal growth factor; FGF, fibroblast growth factor; OCT4, octamer-binding transcription factor 4; RA, retinoic acid; SAG, smoothened agonist; SCF, stem cell factor; SOX2, sex-determining region Y box; SSEA4, stage-specific embryonic antigen 4; TGFβ, transforming growth factor beta; VEGF, vascular endothelial growth factor. (Color version of figure is available online.)
Chen et al. [46] reported derivation of 3D lung organoids from hPSCs. These organoids recapitulated the developing lung tissue very nicely by containing cells with features of both the mesoderm and pulmonary endoderm. When grown on Matrigel, the lung bud organoids (LBOs) gave rise to branching colonies reminiscent of the structural features of the developing pulmonary tissue. The researchers further characterized the LBOs ex vivo by performing xenotransplantation in immune-deficient NOD-SCID gamma mice showing the typical features of lung development in vivo. The LBOs also recapitulated the hallmarks of respiratory syncytial virus infection in which detachment and shedding of infected cells into the lumens of the LBOs were observed to be reminiscent of respiratory syncytial virus in human lungs. With some modification of this protocol to generate lung organoids from PSCs, Han et al. [48] recently demonstrated the permissive nature of lung organoids derived from hPSCs to SARS-CoV-2 infection. The researchers further demonstrated that lung organoids, upon SARS-CoV-2 infection, produced chemokines very similar to what is observed in COVID-19 patients.
Porotto et al. [45] utilized hPSC-derived lung organoids to investigate the evolution and infection process of human parainfluenza virus type 3. The study elegantly demonstrated the viral evolution and pathogenesis in the distal lung, recapitulating the important features of human viral infection. A crucial advantage of PSC-derived lung organoids is the possibility of assessing any viral respiratory infection across the different stages of lung development. Clearly, this offers the opportunity to use lung organoids as a model system to assess the impact of viral respiratory infections and serves as an authentic model for respiratory viral pathogenesis for recapitulating viral respiratory infection in the host.
iPSC-Derived Lymphoid Cells to Treat COVID-19
As a perpetual source of PSCs, iPSCs can be directed to be differentiated toward the lymphoid lineage [49]. This aspect of the technology allows a continuous production of lymphocytes, thereby addressing the bottlenecks, such as cell numbers and dose limitations, seen with respect to primary lymphoid cells. Therefore, compared with a primary lymphoid source, iPSCs can be used to develop a robust and safe platform to develop “off-the-shelf” immune cell therapeutics to address COVID-19 (Figure 1). Thus far, iPSCs have been generated from almost all types of somatic cells [50]. However, selection of the initial iPSC source is critical for the latter derivation of lymphoid lineage cells. Reminiscent epigenetic memory of the cells is known to play a crucial role in deciding the differentiation fates of iPSCs [51,52]. How epigenetic memory is retained and paves the way for preferential differentiation to a specific cellular lineages has been reviewed before [49,51] and might explain why iPSCs generated from cells with hematopoietic lineage are more successfully differentiated toward lymphoid lineages.
T Cells and COVID-19
Incidentally, similar to what is observed in COVID-19 patients, SARS in humans is associated with a severe reduction in the number of T cells in the blood [53]. In addition, Zheng et al. [54] reported elevated exhaustion levels and reduced functional diversity of T cells in peripheral blood in COVID-19 patients, which was predictive of disease severity. The reason for T-cell exhaustion in COVID-19 patients can be attributed to their repeated activation, which also reduces the possibility of harvesting these cells from COVID-19 patients for adoptive cytotoxic T lymphocyte (CTL) therapy. CTLs play an important role in the immune system's defense against viral infections. CTLs interact with virus-infected cells, triggering a cascade that eventually kills the infected cells [55]. The key players in this cascade are the major histocompatibility complex (MHC) class I molecules, which present the antigenic peptides activating the CTLs, leading to the degranulation and subsequent death of the target cells [56]. This crucial mechanism, which protects us from virulent pathogens, takes effect by virtue of a very specific phenomenon known as MHC restriction in which the MHC allele is recognized by the patient's own T cells only. Therefore, MHC restriction is considered to be the primary constraint in adoptive T-cell therapy [57], especially with regard to employing this treatment in the context of COVID-19 in which the quality of the patient-derived primary CTLs may not be optimal. This aspect can undermine the expansion potential of T cells, limiting the ability to obtain enough CTLs to treat a large number of COVID-19 patients. However, hiPSCs, with their unique features of pluripotency and infinite propagation, can address this critical constraint by serving as an alternative source for the derivation of T cells for allogeneic “off-the-shelf” adoptive immunotherapy [58] to treat COVID-19 patients with lymphopenia (Figure 1). Some crucial insight in this direction has been obtained from the work of Nagano et al. [59], who successfully derived potent tumor antigen-specific CTLs from T-iPSCs in an allogeneic setting. More importantly, the researchers also underlined the issues that need to be addressed prior to the allogeneic application of CTLs in the clinic. In light of the aforementioned findings, it can be hypothesized that the ability to derive universal donor T cells from gene-modified PSC lines [60] may lead to the development of allogeneic virus-specific T-cell therapies to treat COVID-19-associated lymphopenia, which is currently not amenable to standard therapeutic strategies.
Generation of Anti-COVID-19 T Cells from iPSCs
Derivation of immunocompetent and therapeutic T cells from PSCs can replenish the T-cell reduction seen in severe COVID-19 cases [15]. To date, generation of fully functional and mature T cells from PSCs remains a challenge. Addressing these challenges, Montel-Hagen et al. [61] reported on a 3D PSC–artificial thymic organoid culture system that allows in vitro differentiation of hiPSCs to functional and mature T cells. The researchers harnessed the Notch ligand-expressing stromal cell line in serum-free conditions [62] to obtain CD4+CD8+ double-positive precursors toward directed differentiation of functional T cells. Interestingly, T cells reprogrammed to iPSCs and later driven to differentiate to T lymphocyte iPSCs were found to preserve the intricate and precise rearrangements of the α and β chains of the T-cell receptor (TCR) identical to the original T-cell clone [63]. Furthermore, these redifferentiated CD8 single-positive T cells from T lymphocyte iPSCs displayed increased telomere length suggestive of their rejuvenated status. In addition, the iPSC-derived T lymphocytes exhibited antigen-specific cytotoxicity and increased proliferative response, which can be attributed to their rejuvenation. The approach here was based on reprogramming CD8+ T cells into iPSCs (to retain the epigenetic memory of the parental cell [64]), which were called T lymphocyte iPSCs based on the cell type source, and the subsequent stepwise-directed differentiation to hematopoietic lineage T-cell progenitors, finally deriving functionally matured CD8+ T cells (Figure 2). These protocols [65], [66] provide useful in vitro tools for generating antigen-specific T cells for immunotherapy (Table 2 ). Nishimura et al. [67] demonstrated that the key to recognizing antigens by T cells is TCRs, which are encoded by uniquely rearranged genomic loci of the TCR α and ß chains. Generally, deriving T cells from iPSC lines has been shown to bear non-rearranged germline TCR loci, resulting in random rearrangement during differentiation. The researchers also confirmed that the specificity of the TCR rearrangement remains unchanged in T cells derived from T lymphocyte iPSCs.
Table 2.
Derivation of T cells from human pluripotent stem cells.
| Author | Stem cell Type | Cell type | Time line in days | Culture conditions | Remarks |
|---|---|---|---|---|---|
| Good et al.[65] | iPSCs | Antigen-specific T cells | 35+ | Note: Feeder OP9/DLL1 cells were readied a week prior on 0.1% gelatin. D0–D12: MEF feeder-dependent iPSCs were co-cultured on OP9/DLL1 with OP9 medium (MEM + FBS + penicillin/streptomycin). Increased the ratio of OP9 medium compared with iPSC medium. On day 9, multilayered center structure evolved with dome-like shape. D13–D35: Harvested hematopoietic progenitor-like cells were resuspended in OP9 medium with SCF + FLT3 + hIL-7. Cell passage was done every 5–7 days. D35+: Enriched CD4+ cells were cultivated in OP9 medium with IL-2 + IL-7 + CD3 antibody + CD28 antibody. Stimulated cells were collected after 7 days and mixed with irradiated HLA-A*02:01 + LCL loaded with MART-1 peptide in the presence of MEM + IL-7 + IL-21. Obtained CD8-αβ CTL cells. |
Generation of antigen-specific T cells are more effective when EBs are used. CD43+ cells were analyzed by flow cytometry. CD8-αβ, MART1 tetramer checked. Antigen-specific T cells were derived from iPSCs using this protocol. |
| Guo et al.[66] | hPSCs | T lymphocytes | 33+ | D0–11: EBs generated with basic differentiation medium (BDM) + BMP4 and cultured in 15-cm dish within 2.5 days. After 2.5 days, VEGF was added and cells were cultured for 6 days. On day 7, 2% of condition medium prepared from AFT024-mlL-3, AFT024-mlL-6, AFT024-hFIt3L and AFT024-mSCF was added. Doxycycline was added from day 6. Medium was replaced on alternate days. D12–D21: Hematopoietic maturation (iHPC) was carried out by seeding with OP9-DL1 cells in EM medium containing α-MEM + FBS + monothioglycerol + GlutaMAX + ascorbic acid + 2% conditioned medium (excluding AFT024-MIL-6). Cells sorted and EM medium used on alternate days. D21–D33+: Matured hematopoietic cells co-cultured with OP9-DL1 feeder cells in T-cell induction medium (TIM + α-MEM + FBS + GlutaMAX) supplemented with 2% conditioned medium derived from AFT024-hFIt3L and AFT024-Hil-7 cell culture for next 12 days. |
Hematopoietic differentiation: Conditioned medium added until day 11. CD31+CD45-CD41(LOW) analyzed with flow cytometer. Hematopoietic maturation step: Cells co-cultured with OP9-DL1. TIM medium changed every third day. CD3 and CD45.2 expression analyzed with flow cytometer. |
| Montel-Hagen et al.[61] | hPSCs | T cells | 67+ | D0–D3: Generation and isolation of hEMPs done using X-VIVO 15 medium along with rh activin A + rhBMP4 + rhVEGF + ROCK inhibitor Y-27632 dihydrochloride. Cells numbering 3 × 106 cells per 3 mL were cultured. Medium changed to X-VIVO 15 + rhBMP4 + rhVEGF + rhFGF. On D3.5, CD32–CD56+ cells were isolated using FACS D3–D10: Induction of hematopoietic lineage was done by T-cell differentiation. Embryonic mesodermal organoids were generated by aggregating hEMPs with MS5-HDLL4 cells. These cells were introduced into hematopoietic induction medium along with EGM2 + ROCK inhibitor + SB431543. On D4, 5 × 105 MS5-HDLL4 cells were combined with 0.5–1 × 104 purified hEMPs per PSC-ATO and centrifuged and cultured in hematopoietic induction medium in the presence of EGM2 + SB431542. Medium was changed twice in a week. D10–D17: Medium changed to EGM2 containing SB431542 along with cytokines rhTPO + rhFLT3L5 + rhSCF. D17–D67: PSC-ATOs were initiated by changing the medium to RPMI medium supplemented with B27 + ascorbic acid + rhSCF + rhFLT3L + rhIL-7. Medium was changed completely every 3–4 days for 50 more days. |
3D organoid formed before processing to next step. MS5-HDLL1 can also be used instead of MS5-HDLL4. PSC-ATO protocol employs PSC-ATOs to efficiently pattern hPSCs to T cells |
| Nishimura et al.[67] | T-iPSCs | T cells | 60+ | Regeneration of hiPSCs done using T cells or CTL clones. T cells were activated by a-CD3/CD28 antibody-coated beads or PHA-L. Activated cells reprogrammed and cultured with RPMI 1640 medium containing 10% AB serum + glutamine + penicillin + streptomycin. On D12, The RH10 medium was replaced with DMEM/F12 FAM + 20% KOSR + glutamine + NEAA + mercaptoethanol + bFGF. D0–D15: iPSCs were transferred to irradiated C3H10T1/2 feeder cells in EB medium containing IMDM + 15% FBS + human insulin + transferrin+ sodium selenite + glutamine + monothioglycerol + ascorbic acid + VEGF + SCF + FLT-3L. D16–D45: Derived hematopoietic progenitors were collected and grown on feeder OP9-DL11 cells. The medium consisted of MEM + 15% FBS + glutamine + penicillin + streptomycin + FLT-3L + IL-7. D46–D60: The T lineage cells was harvested and mixed with irradiated HLA-A24 PBMCs and co-cultured using RH10 medium in the presence of IL-7 and IL-15. |
iPSC clones transfected with small interfering RNA L527 using Lipofectamine RNAiMAX for removal of SeV vectors from cytoplasm. T cells differentiated on OP9-DL1 cells during co-culture in OP9 medium. CD45+ cells isolated using FACS. |
| Nagano et al.[59] | T-iPSCs | CTLs | D0–D12: Differentiation was done on stromal OP9 feeder cells in α-MEM supplemented with 20% FBS. D13–D17: Enriched CD34+ progenitors were plated on OP-DLL1 stromal feeder cells with OP9 medium containing hIL-7, hFlt and hSCF hematopoietic factors. D18–D40: Floating cells were collected and transferred to new OP9-DLL1 feeder cells with OP0 medium containing hematopoietic factors. Passage on to a new OP9-DLL1 feeder layer was done every week. |
T-cell-generating potential of T-iPSCs evaluated by frequency of CD4+CD8+ DP cells; once DP cells are generated, CD8-αβ SP cells are derived by stimulating isolated DP cells. |
bFGF, basic fibroblast growth factor; DMEM, Dulbecco's Modified Eagle's Medium; DP, double-positive; EB, embryoid body; FACS, fluorescence-activated cell sorting; FBS, fetal bovine serum; FGF, fibroblast growth factor; hSCF, human stem cell factor; hEMPs, human embryonic mesodermal progenitors; iHPCs, induced hematopoietic progenitor cells; IMDM, Iscove's Modified Dulbecco's Medium; KOSR, knockout serum replacement; MEF, mouse embryonic fibroblast; MEM, Minimum Essential Medium; NEAA, non-essential amino acid; PHA-L, Phaseolus vulgaris leukoagglutinin; PSC-ATO, PSC–artificial thymic organoid; SCF, stem cell factor; SeV, Sendai virus; SP, single-positive; T-iPSCs, T lymphocyte iPSCs; VEGF, vascular endothelial growth factor.
These studies provide crucial evidence of the possibility of deriving therapeutic anti-SARS-CoV-2-specfic T cells from T lymphocyte iPSCs generated from COVID-19 patients. Demonstrating the safety of iPSC-derived T cells, Ando et al. [68] reported enhancing the safety of iPSC-derived rejuvenated T cells by introducing inducible caspase 9 (iC9). The study demonstrated that activation of iC9 eliminated iPSC-derived rejuvenated CTLs without disturbing their antigen-specific killing activity, suggesting that the iC9/chemical inducer of dimerization safeguard system is a promising tool for future iPSC-mediated approaches to clinical therapy.
Derivation of NK Cells from iPSCs to Treat COVID-19
NK cells are lymphoid cells that originate from the same progenitor as T cells and B cells. They are an important part of the innate immune response, as they identify “non-self” cells independent of antigen presentation or recognition, thereby executing a rapid immune response. The broad cytotoxicity and rapid apoptosis induced by NK cells help contain virus-infected cells while the adaptive immune response is activated to produce cytotoxic T cells to clear the antigens. Thus far, primary NK cells have been the main source used for immunotherapy. Significant work is being done to obtain NK cells from other sources, such as PSCs, so that a perpetual supply of NK cells is available for therapy [69]. Primary NK cells are difficult to harvest and purify [70]. They are also difficult to standardize because of the heterogeneity of starting material from different donors [71]. In addition, the generation of large quantities of highly pure NK cells requires an extended manufacturing process, which can compromise the recovery of NK cells as well as their viability and potency [72].
NK cells derived from iPSCs have proven to be as effective as primary NK cells [73,74]. The possibility of generating NK cells from iPSCs may address some of the concerns related to adoptive NK cell therapy. Studies have demonstrated the possibility of deriving NK cells robustly from iPSCs, with their characteristic phenotypic features [75], [76], [77]. Most importantly, Knorr et al. [78] reported a clinical-grade, serum-free and feeder-free differentiation protocol for obtaining functional NK cells from iPSCs that involved a novel technique for generating the intermediate hematopoietic embryoid bodies using defined xenogeneic-free conditions and membrane-bound IL-21-expressing artificial antigen-presenting cells. According to the researchers, this method makes it possible to obtain a sufficient number of functional cytotoxic NK cells, derived from as few as 250 000 PSCs, to treat a single patient, thus facilitating the potential for its clinical application. NK cell-based COVID-19 treatment is currently being considered [79]. However, there is currently little insights into the signaling mechanims involved in attributing the functional properties of the iPSC-derived NK cells. Further studies are needed to understand the activation process of iPSC-derived NK cells in the generation of functionally efficient iPSC-derived NK cells. With our current understanding, these data suggest that iPSC-derived NK-based strategies (Table 3 ) combine the most attractive qualities of primary NK cells (e.g., high potential for cytotoxicity, including antibody-dependent cellular cytotoxicity, and potential for expansion and persistence in vivo after cryopreservation), making this a tempting immune cell candidate for COVID-19 therapy.
Table 3.
Derivation of natural killer cells from human pluripotent stem cells.
| Author | Stem cell type | Cell type derived | Time line in days | Culture conditions | Remarks |
|---|---|---|---|---|---|
| Hermanson et al.[73] | PSCs/iPSCs | NK cells | 32+ | Cells were cultured in 96-well plate format. EBs were derived prior to differentiation to NK cells. D0–D10: EBs inducted with SCF medium + VEGF + BMP4, etc., for hematopoietic differentiation to generate CD34+/CD43 NK cells. D11–D32+: EBs further differentiated into NK cells using IL-3 + IL-15 + IL-7 + SCF + FLT3 ligand. Medium change was done weekly for 28–32 days and harvested for APC expansion. |
KIR, CD16, NKp46, NKG2D and NKG2A analyzed with flow cytometry. |
| Woll et al.[69] | ESC | NK cells | 35+ | D0–D17: hESCs co-cultured with murine bone marrow stromal cell line M210-B4 along with medium containing RPMI 1640 + 15% FBS + glutamine + NEAA + penicillin/streptomycin + β-mercaptoethanol for next 17 days. D17–D20: CD34+CD45+ double-positive cells were isolated and single-cell suspension prepared. Used aforementioned medium (refer to article for more details). D20–D35+: Separated cells were co-cultured with murine fetal liver-derived stromal cell line AFT024 in medium containing DMEM/Ham's F12 + human serum AB (heat-inactivated) + L-glutamine + penicillin/streptomycin + sodium selenite + ethanolamine + β-mercaptoethanol + ascorbic acid + IL-3 (for 1 week) + IL-15 + SCF + IL-7 + tyrosine kinase 3 ligand for next 30–35 days. Medium was replenished every 5–7 days. |
M210-B4 cells found more efficiently compared with S17 cells. CD56/CD45 NK cells were found mostly when analyzed with flow cytometry |
| Zeng et al,[74] | iPSCs | NK cells | 40–47 | D0–D11: hPSCs were seeded on stromal feeder cells (OP9) in presence of α-MEM and 20% FBS for hematopoietic differentiation. D12–D47: Early hematopoietic progenitors were harvested and co-cultured with modified OP9 cell line expressing Notch ligand DLL1 first to increase hematopoietic progenitors and then driven toward lymphoid lineages. |
Clonality assays to detect rearranged TCRβ and TCRγ chain genes in PBC/iPSC lines were performed. Phenotypic characterization was carried out by flow cytometry and immunocytochemistry. |
| Zhu and Kaufman [75] | iPSCs | NK cells | D0–D7; D8–D14; D15–D43 |
Feeder-free adapted ESCs/iPSCs with ROCKi for EB formation. Hematopoietic cell (CD34+) derivation and NK cell differentiation under feeder-free conditions in the presence of IL-3, IL-6, IL-16, SCF and FLT3. NK cell expansion was carried out under feeder-free conditions in the presence of IL-2 and aAPCs. |
Phenotypic and functional characterization of hESC/iPSC-derived NK cells was done by flow cytometry. |
aAPCs, artificial antigen-presenting cells; APC, antigen-presenting cell; DMEM, Dulbecco's Modified Eagle's Medium; EB, embryoid body; ESCs, embryonic stem cells; FBS, fetal bovine serum; hESCs, human embryonic stem cells; MEM, Minimum Essential Medium; NEAA, non-essential amino acid; PBC, primary biliary cirrhosis; ROCKi, Rho kinase inhibitor; SCF, stem cell factor; VEGF, vascular endothelial growth factor.
Discussion
The challenges of using iPSC-derived cells for modeling and as an immune therapy strategy to address COVID-19 are intrinsic to the very biology of iPSCs (i.e., donor age, somatic cell type used for reprogramming and to what fate they are going to be differentiated) [80]. Retention of the epigenetic memory of the parental somatic source by iPSCs is a well-documented feature of the reprogramming process [81]. This is considered to be one of the primary reasons for their clonal heterogeneity deciding their potential differentiation fates [82]. Although longer iPSC passages have been shown to dilute epigenetic memory, they also increase the risk of incorporating random gene mutations, leading to genomic instability [83] of the lines, which is detrimental in the derivation of relevant cell types for modeling and therapy. In the past decade, rapid advances have been made in addressing the concerns relevant to the application of iPSCs [84]. Knowledge gained here has led to the formulation of criteria for defining good iPSC lines and their differentiated derivatives. However, there is a need and sufficient space to rethink existing strategies and bring forth improved and robust acceptable standards for clinical application of iPSCs and their derivatives [85]. The development of practical, reliable and cost-effective methods for obtaining iPSC-derived lung cells, T cells and NK cells is a vital prerequisite for their application in modeling and as a therapeutic prospect for addressing the COVID-19 pandemic. One of the major concerns regarding iPSC-based immune cell replacement therapy is risk of potential tumorigenicity from calcitrant iPSCs that have not undergone complete differentiation to the desired phenotype [86]. Here, any hint of stemness in even a single cell among the differentiated population poses an absolute risk of cellular transplantation. With continuous advancements in directed differentiation and purification protocols, such risks are being addressed [87,88]. However, translational work in animal models is necessary to assess if these assays and methods are sufficient to address the risk of teratomas upon transplantation of iPSC-derived immune cells in COVID-19 patients. Alveolar cells and lymphoid cells derived from iPSCs can be immunogenic, which remains a major issue for their therapeutic application [89,90]. However, long-term immunosuppression has been shown to be indispensable and prevents rejection of allogeneic PSC-derived products in animal studies [91]. This could be a possible route of intervention in humans receiving therapeutic allogeneic iPSC-derived cells [92]. By contrast, in the case of autologous iPSC-derived products, it is feasible to transplant the therapeutic cells without immune suppression [93]. The cost of developing iPSC-derived alveolar cells and lymphoid cells is significant, and this can be addressed to some extent by using allogeneic iPSC-derived immune cells to treat some cases of COVID-19. However, the use of immunosuppression furthers the risk of severe secondary infections in already disadvantaged patients [94]. In this scenario, the risk-versus-benefit ratio is highly debatable and akin to that seen when treating any disease with an allogeneic PSC-derived cell product. In the context of the current scenario and its urgency, a universal immune-tolerant iPSC-derived product will provide the necessary advantage for devising strategies to enable utilization of allogeneic iPSC-derived alveolar cells and lymphoid cells without the risk of immune rejection and other complications.
As of now, vaccines against SARS-CoV-2 have become the most viable global option, with several COVID-19 vaccines currently in trials and use [95]. Human cell lines have been routinely used to make vaccines against viruses [96,97]. Vaccines against viral diseases have been continuously evolving to provide a safer means of protection and to reduce possible side effects [98]. The choice of cell substrate is one of the most crucial components in viral vaccine manufacturing. Vaccines against chickenpox are being made using MRC5, a human fibroblast cell line [97]. Human embryonic kidney (HEK) 293, a cell line generated from aborted fetuses in the 1970s, is the cell substrate of choice for many COVID-19 vaccine manufacturers [99,100]. The immortalized HEK cells are harnessed to make the spike proteins of SARS-COV-2 [101] or to cultivate the recombinant packaging viruses needed for vaccine manufacturing [102]. Although the safety, pharmacology, toxicity and potency of HEK 293 cells as a platform for vaccine manufacture are well established [103], as an immortalized cell line of cancerous origin, HEK 293 cells run the inherent risk of chromosomal and genetic aberrations because of the countless number of passages they have undergone. Similar to immortalized cell lines, iPSCs under prolonged periods of culture have been shown to be capable of accumulating chromosomal and genetic aberrations [104]. However, this risk can be mitigated to a large degree by using differentiated lung- or alveolar-type cells derived from well-characterized iPSC lines. The iPSCs derived from chicken have been used to study viral infection and replication of avian viral diseases [105]. The use of galline iPSCs to make viruses replication-incompetent from their highly pathogenic form [106] adds to the immense application potential of such animal-specific iPSCs for vaccine production over chicken eggs and embryos, which are more prone to contamination risk, and can also aid in cutting down the cost of vaccine production.
In the current scenario, the iPSC-derived alveolar-type cells exposed to the SARS-CoV-2 virus would serve as an invaluable tool that is able to reproduce the genotypic and phenotypic aspects more closely associated with the infection [107]. Exosomes derived from iPSCs and their derivatives are being considered for packaging the messenger RNAs (mRNAs) encoding the SARS-CoV-2 antigen proteins [108]. The functionality of such exosome-packaged mRNAs will address some of the critical issues of vaccine scalability and stability faced by mRNA-based vaccines [109]. Apart from their utility as a “packaging factory” in vaccine manufacturing, iPSC-derived alveolar cells can significantly aid in novel target identification and repurposing of known and novel drug combinations against SARS-CoV-2 and lay the groundwork for enhanced preparedness for future viral pandemics.
Conclusions
This review is intended to aid researchers in streamlining and optimizing protocols for the generation of hiPSC-derived alveolar cells and lung organoids as a robust platform for studying respiratory viruses such as SARS-CoV-2 in the human context. Protocols for generating CTLs and NK cells from iPSCs have been discussed to enable the possibility of clinical utilization of these cells to treat cases of COVID-19 in which there is a need to replenish these immune cells. However, with our current limited knowledge of the role of immune cells such as CTLs and NKs in COVID-19 pathophysiology, it is imperative to rationalize their use as an immune therapy modality in COVID-19 patients. Here, it is crucial to have an understanding of the CTLs and NK cells status in individual COVID-19 patients and the timing of the therapeutic window, which would likely determine the balance between their beneficial antiviral and detrimental pathological action.
Declaration of Competing Interest
The authors have no commercial, proprietary or financial interest in the products or companies described in this article.
Acknowledgments
Funding
KC is funded by a BT/PR26190/GET/119/118/2017 grant from the Department of Biotechnology, Government of India, and the Narayana Nethralaya Foundation.
Author Contributions
Conception and design of the study: KC. Analysis and interpretation of data: KC, SA. Drafting or revising the manuscript: KC, RS, SA, DD and AG. All authors have approved the final article.
Acknowledgments
The authors thank the Narayana Nethralaya Foundation for its support.
References
- 1.Lim Y.X., Ng Y.L., Tam J.P., Liu D.X. Human Coronaviruses: A Review of Virus-Host Interactions. Diseases. 2016;4(3) doi: 10.3390/diseases4030026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fung T.S., Liu D.X. Human Coronavirus: Host–Pathogen Interaction. Annu Rev Microbiol. 2019;73:529–557. doi: 10.1146/annurev-micro-020518-115759. [DOI] [PubMed] [Google Scholar]
- 3.Matsuyama S., Nao N., Shirato K., Kawase M., Saito S., Takayama I., Nagata N., Sekizuka T., Katoh H., Kato F., Sakata M., Tahara M., Kutsuna S., Ohmagari N., Kuroda M., Suzuki T., Kageyama T., Takeda M. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc Natl Acad Sci U S A. 2020;117(13):7001–7003. doi: 10.1073/pnas.2002589117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bojkova D., Klann K., Koch B., Widera M., Krause D., Ciesek S., Cinatl J., Munch C. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature. 2020;583(7816):469–472. doi: 10.1038/s41586-020-2332-7. [DOI] [PubMed] [Google Scholar]
- 5.Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., Chen H.D., Chen J., Luo Y., Guo H., Jiang R.D., Liu M.Q., Chen Y., Shen X.R., Wang X., Zheng X.S., Zhao K., Chen Q.J., Deng F., Liu L.L., Yan B., Zhan F.X., Wang Y.Y., Xiao G.F., Shi Z.L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elbadawi M., Efferth T. Organoids of human airways to study infectivity and cytopathy of SARS-CoV-2. Lancet Respir Med. 2020;8(7):e55–e56. doi: 10.1016/S2213-2600(20)30238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoffmann M., Kleine-Weber H., Schroeder S., Kruger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A., Muller M.A., Drosten C., Pohlmann S. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181(2) doi: 10.1016/j.cell.2020.02.052. 271-280 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bertram S., Heurich A., Lavender H., Gierer S., Danisch S., Perin P., Lucas J.M., Nelson P.S., Pohlmann S., Soilleux E.J. Influenza and SARS-coronavirus activating proteases TMPRSS2 and HAT are expressed at multiple sites in human respiratory and gastrointestinal tracts. PLoS One. 2012;7(4):e35876. doi: 10.1371/journal.pone.0035876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zou X., Chen K., Zou J., Han P., Hao J., Han Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front Med. 2020;14(2):185–192. doi: 10.1007/s11684-020-0754-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma A., Sances S., Workman M.J., Svendsen C.N. Multi-lineage Human iPSC-Derived Platforms for Disease Modeling and Drug Discovery. Cell Stem Cell. 2020;26(3):309–329. doi: 10.1016/j.stem.2020.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Surendran H., Nandakumar S., Pal R. Human Induced Pluripotent Stem Cell-Derived Lung Epithelial System for SARS-CoV-2 Infection Modeling and Its Potential in Drug Repurposing. Stem Cells Dev. 2020;29(21):1365–1369. doi: 10.1089/scd.2020.0152. [DOI] [PubMed] [Google Scholar]
- 12.Katsura H., Sontake V., Tata A., Kobayashi Y., Edwards C.E., Heaton B.E., Konkimalla A., Asakura T., Mikami Y., Fritch E.J., Lee P.J., Heaton N.S., Boucher R.C., Randell S.H., Baric R.S., Tata P.R. Human Lung Stem Cell-Based Alveolospheres Provide Insights into SARS-CoV-2-Mediated Interferon Responses and Pneumocyte Dysfunction. Cell Stem Cell. 2020;27(6):890–904. doi: 10.1016/j.stem.2020.10.005. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han Y., Yang L., Duan X., Duan F., Nilsson-Payant B.E., Yaron T.M., Wang P., Tang X., Zhang T., Zhao Z., Bram Y., Redmond D., Houghton S., Nguyen D., Xu D., Wang X., Uhl S., Huang Y., Johnson J.L., Xiang J., Wang H., Pan F.C., Cantley L.C., tenOever B.R., Ho D.D., Evans T., Schwartz R.E., Chen H.J., Chen S. Identification of Candidate COVID-19 Therapeutics using hPSC-derived Lung Organoids. bioRxiv. 2020 [Google Scholar]
- 14.Zhou R., To K.K., Wong Y.C., Liu L., Zhou B., Li X., Huang H., Mo Y., Luk T.Y., Lau T.T., Yeung P., Chan W.M., Wu A.K., Lung K.C., Tsang O.T., Leung W.S., Hung I.F., Yuen K.Y., Chen Z. Acute SARS-CoV-2 Infection Impairs Dendritic Cell and T Cell Responses. Immunity. 2020;53(4) doi: 10.1016/j.immuni.2020.07.026. 864-877 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diao B., Wang C., Tan Y., Chen X., Liu Y., Ning L., Chen L., Li M., Liu Y., Wang G., Yuan Z., Feng Z., Zhang Y., Wu Y., Chen Y. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19) Front Immunol. 2020;11:827. doi: 10.3389/fimmu.2020.00827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soleimanian S., Yaghobi R. Harnessing Memory NK Cell to Protect Against COVID-19. Front Pharmacol. 2020;11:1309. doi: 10.3389/fphar.2020.01309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Eeden C., Khan L., Osman M.S., Cohen Tervaert J.W. Natural Killer Cell Dysfunction and Its Role in COVID-19. Int J Mol Sci. 2020;21(17) doi: 10.3390/ijms21176351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kothari A., Singh V., Nath U.K., Kumar S., Rai V., Kaushal K., Omar B.J., Pandey A., Jain N. Immune Dysfunction and Multiple Treatment Modalities for the SARS-CoV-2 Pandemic: Races of Uncontrolled Running Sweat? Biology (Basel) 2020;9(9) doi: 10.3390/biology9090243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maucourant C., Filipovic I., Ponzetta A., Aleman S., Cornillet M., Hertwig L., Strunz B., Lentini A., Reinius B., Brownlie D., Cuapio A., Ask E.H., Hull R.M., Haroun-Izquierdo A., Schaffer M., Klingstrom J., Folkesson E., Buggert M., Sandberg J.K., Eriksson L.I., Rooyackers O., Ljunggren H.G., Malmberg K.J., Michaelsson J., Marquardt N., Hammer Q., Stralin K., Bjorkstrom N.K., Karolinska C.-S.G. Natural killer cell immunotypes related to COVID-19 disease severity. Sci Immunol. 2020;5(50) doi: 10.1126/sciimmunol.abd6832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Masselli E., Vaccarezza M., Carubbi C., Pozzi G., Presta V., Mirandola P., Vitale M. NK cells: A double edge sword against SARS-CoV-2. Adv Biol Regul. 2020;77 doi: 10.1016/j.jbior.2020.100737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iketani S., Shean R.C., Ferren M., Makhsous N., Aquino D.B., des Georges A., Rima B., Mathieu C., Porotto M., Moscona A., Greninger A.L. Viral Entry Properties Required for Fitness in Humans Are Lost through Rapid Genomic Change during Viral Isolation. mBio. 2018;9(4) doi: 10.1128/mBio.00898-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wan Y., Shang J., Graham R., Baric R.S., Li F. Receptor Recognition by the Novel Coronavirus from Wuhan: an Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J Virol. 2020;94(7) doi: 10.1128/JVI.00127-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Haute L., De Block G., Liebaers I., Sermon K., De Rycke M. Generation of lung epithelial-like tissue from human embryonic stem cells. Respir Res. 2009;10:105. doi: 10.1186/1465-9921-10-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang D., Haviland D.L., Burns A.R., Zsigmond E., Wetsel R.A. A pure population of lung alveolar epithelial type II cells derived from human embryonic stem cells. Proc Natl Acad Sci U S A. 2007;104(11):4449–4454. doi: 10.1073/pnas.0700052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berical A., Lee R.E., Randell S.H., Hawkins F. Challenges Facing Airway Epithelial Cell-Based Therapy for Cystic Fibrosis. Front Pharmacol. 2019;10:74. doi: 10.3389/fphar.2019.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tamo L., Hibaoui Y., Kallol S., Alves M.P., Albrecht C., Hostettler K.E., Feki A., Rougier J.S., Abriel H., Knudsen L., Gazdhar A., Geiser T. Generation of an alveolar epithelial type II cell line from induced pluripotent stem cells. Am J Physiol Lung Cell Mol Physiol. 2018;315(6):L921–L932. doi: 10.1152/ajplung.00357.2017. [DOI] [PubMed] [Google Scholar]
- 27.Heo H.R., Kim J., Kim W.J., Yang S.R., Han S.S., Lee S.J., Hong Y., Hong S.H. Human pluripotent stem cell-derived alveolar epithelial cells are alternatives for in vitro pulmotoxicity assessment. Sci Rep. 2019;9(1):505. doi: 10.1038/s41598-018-37193-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Firth A.L, Dargitz C.T, Qualls S.J, Menon T, Wright R, Singer O, et al. Generation of multiciliated cells in functional airway epithelia from human induced pluripotent stem cells. Proc Natl Acad Sci U S A. 2014;111(17):1723–1730. doi: 10.1073/pnas.1403470111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong A.P, Bear C.E, Chin S, Pasceri P, Thompson T.0, Huan L.J, et al. Directed differentiation of human pluripotent stem cells into mature airway epithelia expressing functional CFTR protein. Nat Biotechnol. 2012;30(9):876–882. doi: 10.1038/nbt.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Longmire T.A, Ikonomou L, Hawkins F, Christodoulou C, Cao Y, Jean JC, et al. Efficient derivation of purified lung and thyroid progenitors from embryonic stem cells. Cell Stem Cell. 2012;10(4):398–411. doi: 10.1016/j.stem.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang S.X., Islam M.N., O'Neill J., Hu Z., Yang Y.G., Chen Y.W., Mumau M., Green M.D., Vunjak-Novakovic G., Bhattacharya J., Snoeck H.W. Efficient generation of lung and airway epithelial cells from human pluripotent stem cells. Nat Biotechnol. 2014;32(1):84–91. doi: 10.1038/nbt.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garreta E., Melo E., Navajas D., Farre R. Low oxygen tension enhances the generation of lung progenitor cells from mouse embryonic and induced pluripotent stem cells. Physiol Rep. 2014;2(7) doi: 10.14814/phy2.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Surendran H., Rajamoorthy M., Pal R. Differentiating Human Induced Pluripotent Stem Cells (iPSCs) Into Lung Epithelial Cells. Curr Protoc Stem Cell Biol. 2019;49(1):e86. doi: 10.1002/cpsc.86. [DOI] [PubMed] [Google Scholar]
- 34.Zhao L., Yee M., O'Reilly M.A. Transdifferentiation of alveolar epithelial type II to type I cells is controlled by opposing TGF-beta and BMP signaling. Am J Physiol Lung Cell Mol Physiol. 2013;305(6):L409–L418. doi: 10.1152/ajplung.00032.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olajuyin A.M., Zhang X., Ji H.L. Alveolar type 2 progenitor cells for lung injury repair. Cell Death Discov. 2019;5:63. doi: 10.1038/s41420-019-0147-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aspal M., Zemans R.L. Mechanisms of ATII-to-ATI Cell Differentiation during Lung Regeneration. Int J Mol Sci. 2020;21(9) doi: 10.3390/ijms21093188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gotoh S., Ito I., Nagasaki T., Yamamoto Y., Konishi S., Korogi Y., Matsumoto H., Muro S., Hirai T., Funato M., Mae S., Toyoda T., Sato-Otsubo A., Ogawa S., Osafune K., Mishima M. Generation of alveolar epithelial spheroids via isolated progenitor cells from human pluripotent stem cells. Stem Cell Reports. 2014;3(3):394–403. doi: 10.1016/j.stemcr.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamamoto Y., Gotoh S., Korogi Y., Seki M., Konishi S., Ikeo S., Sone N., Nagasaki T., Matsumoto H., Muro S., Ito I., Hirai T., Kohno T., Suzuki Y., Mishima M. Long-term expansion of alveolar stem cells derived from human iPS cells in organoids. Nat Methods. 2017;14(11):1097–1106. doi: 10.1038/nmeth.4448. [DOI] [PubMed] [Google Scholar]
- 39.Ghaedi M., Calle E.A., Mendez J.J., Gard A.L., Balestrini J., Booth A., Bove P.F., Gui L., White E.S., Niklason L.E. Human iPS cell-derived alveolar epithelium repopulates lung extracellular matrix. J Clin Invest. 2013;123(11):4950–4962. doi: 10.1172/JCI68793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jacob A., Morley M., Hawkins F., McCauley K.B., Jean J.C., Heins H., Na C.L., Weaver T.E., Vedaie M., Hurley K., Hinds A., Russo S.J., Kook S., Zacharias W., Ochs M., Traber K., Quinton L.J., Crane A., Davis B.R., White F.V., Wambach J., Whitsett J.A., Cole F.S., Morrisey E.E., Guttentag S.H., Beers M.F., Kotton D.N. Differentiation of Human Pluripotent Stem Cells into Functional Lung Alveolar Epithelial Cells. Cell Stem Cell. 2017;21(4) doi: 10.1016/j.stem.2017.08.014. 472-488 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nikolic M.Z., Sun D., Rawlins E.L. Human lung development: recent progress and new challenges. Development. 2018;145(16) doi: 10.1242/dev.163485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dye B.R., Hill D.R., Ferguson M.A., Tsai Y.H., Nagy M.S., Dyal R., Wells J.M., Mayhew C.N., Nattiv R., Klein O.D., White E.S., Deutsch G.H., Spence J.R. In vitro generation of human pluripotent stem cell derived lung organoids. Elife. 2015;4 doi: 10.7554/eLife.05098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang L., Bukreyev A., Thompson C.I., Watson B., Peeples M.E., Collins P.L., Pickles R.J. Infection of ciliated cells by human parainfluenza virus type 3 in an in vitro model of human airway epithelium. J Virol. 2005;79(2):1113–1124. doi: 10.1128/JVI.79.2.1113-1124.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh B.K., Li N., Mark A.C., Mateo M., Cattaneo R., Sinn P.L. Cell-to-Cell Contact and Nectin-4 Govern Spread of Measles Virus from Primary Human Myeloid Cells to Primary Human Airway Epithelial Cells. J Virol. 2016;90(15):6808–6817. doi: 10.1128/JVI.00266-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Porotto M., Ferren M., Chen Y.W., Siu Y., Makhsous N., Rima B., Briese T., Greninger A.L., Snoeck H.W., Moscona A. Authentic Modeling of Human Respiratory Virus Infection in Human Pluripotent Stem Cell-Derived Lung Organoids. mBio. 2019;10(3) doi: 10.1128/mBio.00723-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Y.W., Huang S.X., de Carvalho A., Ho S.H., Islam M.N., Volpi S., Notarangelo L.D., Ciancanelli M., Casanova J.L., Bhattacharya J., Liang A.F., Palermo L.M., Porotto M., Moscona A., Snoeck H.W. A three-dimensional model of human lung development and disease from pluripotent stem cells. Nat Cell Biol. 2017;19(5):542–549. doi: 10.1038/ncb3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim J., Koo B.K., Yoon K.J. Modeling Host-Virus Interactions in Viral Infectious Diseases Using Stem-Cell-Derived Systems and CRISPR/Cas9 Technology. Viruses. 2019;11(2) doi: 10.3390/v11020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Han Y., Duan X., Yang L., Nilsson-Payant B.E., Wang P., Duan F., Tang X., Yaron T.M., Zhang T., Uhl S., Bram Y., Richardson C., Zhu J., Zhao Z., Redmond D., Houghton S., Nguyen D.T., Xu D., Wang X., Jessurun J., Borczuk A., Huang Y., Johnson J.L., Liu Y., Xiang J., Wang H., Cantley L.C., tenOever B.R., Ho D.D., Pan F.C., Evans T., Chen H.J., Schwartz R.E., Chen S. Identification of SARS-CoV-2 Inhibitors using Lung and Colonic Organoids. Nature. 2021;589(7841):270–275. doi: 10.1038/s41586-020-2901-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nianias A., Themeli M. Induced Pluripotent Stem Cell (iPSC)-Derived Lymphocytes for Adoptive Cell Immunotherapy: Recent Advances and Challenges. Curr Hematol Malig Rep. 2019;14(4):261–268. doi: 10.1007/s11899-019-00528-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Raab S., Klingenstein M., Liebau S., Linta L. A Comparative View on Human Somatic Cell Sources for iPSC Generation. Stem Cells Int. 2014;2014 doi: 10.1155/2014/768391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Noguchi H., Miyagi-Shiohira C., Nakashima Y. Induced Tissue-Specific Stem Cells and Epigenetic Memory in Induced Pluripotent Stem Cells. Int J Mol Sci. 2018;19(4) doi: 10.3390/ijms19040930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brumbaugh J., Di Stefano B., Hochedlinger K. Reprogramming: identifying the mechanisms that safeguard cell identity. Development. 2019;146(23) doi: 10.1242/dev.182170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qin C., Zhou L., Hu Z., Zhang S., Yang S., Tao Y., Xie C., Ma K., Shang K., Wang W., Tian D.S. Dysregulation of Immune Response in Patients With Coronavirus 2019 (COVID-19) in Wuhan, China. Clin Infect Dis. 2020;71(15):762–768. doi: 10.1093/cid/ciaa248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zheng H.Y., Zhang M., Yang C.X., Zhang N., Wang X.C., Yang X.P., Dong X.Q., Zheng Y.T. Elevated exhaustion levels and reduced functional diversity of T cells in peripheral blood may predict severe progression in COVID-19 patients. Cell Mol Immunol. 2020;17(5):541–543. doi: 10.1038/s41423-020-0401-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Newton A.H., Cardani A., Braciale T.J. The host immune response in respiratory virus infection: balancing virus clearance and immunopathology. Semin Immunopathol. 2016;38(4):471–482. doi: 10.1007/s00281-016-0558-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wieczorek M., Abualrous E.T., Sticht J., Alvaro-Benito M., Stolzenberg S., Noe F., Freund C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front Immunol. 2017;8:292. doi: 10.3389/fimmu.2017.00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schendel D.J., Frankenberger B. Limitations for TCR gene therapy by MHC-restricted fratricide and TCR-mediated hematopoietic stem cell toxicity. Oncoimmunology. 2013;2(1):e22410. doi: 10.4161/onci.22410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iriguchi S., Kaneko S. Toward the development of true "off-the-shelf" synthetic T-cell immunotherapy. Cancer Sci. 2019;110(1):16–22. doi: 10.1111/cas.13892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nagano S., Maeda T., Ichise H., Kashima S., Ohtaka M., Nakanishi M., Kitawaki T., Kadowaki N., Takaori-Kondo A., Masuda K., Kawamoto H. High Frequency Production of T Cell-Derived iPSC Clones Capable of Generating Potent Cytotoxic T Cells. Mol Ther Methods Clin Dev. 2020;16:126–135. doi: 10.1016/j.omtm.2019.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Themeli M., Riviere I., Sadelain M. New cell sources for T cell engineering and adoptive immunotherapy. Cell Stem Cell. 2015;16(4):357–366. doi: 10.1016/j.stem.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Montel-Hagen A., Seet C.S., Li S., Chick B., Zhu Y., Chang P., Tsai S., Sun V., Lopez S., Chen H.C., He C., Chin C.J., Casero D., Crooks G.M. Organoid-Induced Differentiation of Conventional T Cells from Human Pluripotent Stem Cells. Cell Stem Cell. 2019;24(3) doi: 10.1016/j.stem.2018.12.011. 376-389 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seet C.S., He C., Bethune M.T., Li S., Chick B., Gschweng E.H., Zhu Y., Kim K., Kohn D.B., Baltimore D., Crooks G.M., Montel-Hagen A. Generation of mature T cells from human hematopoietic stem and progenitor cells in artificial thymic organoids. Nat Methods. 2017;14(5):521–530. doi: 10.1038/nmeth.4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kaneko S. In Vitro Generation of Antigen-Specific T Cells from Induced Pluripotent Stem Cells of Antigen-Specific T Cell Origin. Methods Mol Biol. 2016;1393:67–73. doi: 10.1007/978-1-4939-3338-9_6. [DOI] [PubMed] [Google Scholar]
- 64.Kim K., Doi A., Wen B., Ng K., Zhao R., Cahan P., Kim J., Aryee M.J., Ji H., Ehrlich L.I., Yabuuchi A., Takeuchi A., Cunniff K.C., Hongguang H., McKinney-Freeman S., Naveiras O., Yoon T.J., Irizarry R.A., Jung N., Seita J., Hanna J., Murakami P., Jaenisch R., Weissleder R., Orkin S.H., Weissman I.L., Feinberg A.P., Daley G.Q. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467(7313):285–290. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Good M.L, Vizcardo R, Maeda T, Tamaoki N, Malekzadeh P, Kawamoto H, et al. Using Human Induced Pluripotent Stem Cells for the Generation of Tumor Antigen-specific T Cells. J Vis Exp. 2019;24(152) doi: 10.3791/59997. [DOI] [PubMed] [Google Scholar]
- 66.Guo R, Hu F, Weng Q, Lv C, Wu H, Liu L, et al. Guiding T lymphopoiesis from pluripotent stem cells by defined transcription factors. Cell Res. 2020;30(1):21–33. doi: 10.1038/s41422-019-0251-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nishimura T., Kaneko S., Kawana-Tachikawa A., Tajima Y., Goto H., Zhu D., Nakayama-Hosoya K., Iriguchi S., Uemura Y., Shimizu T., Takayama N., Yamada D., Nishimura K., Ohtaka M., Watanabe N., Takahashi S., Iwamoto A., Koseki H., Nakanishi M., Eto K., Nakauchi H. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12(1):114–126. doi: 10.1016/j.stem.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 68.Ando M., Nishimura T., Yamazaki S., Yamaguchi T., Kawana-Tachikawa A., Hayama T., Nakauchi Y., Ando J., Ota Y., Takahashi S., Nishimura K., Ohtaka M., Nakanishi M., Miles J.J., Burrows S.R., Brenner M.K., Nakauchi H. A Safeguard System for Induced Pluripotent Stem Cell-Derived Rejuvenated T Cell Therapy. Stem Cell Reports. 2015;5(4):597–608. doi: 10.1016/j.stemcr.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Woll P.S, Grzywacz B, Tian X, Marcus R.K, Knorr D.A, Verneris M.R, et al. Human embryonic stem cells differentiate into a homogeneous population of natural killer cells with potent in vivo antitumor activity. Blood. 2009;113(24):6094–6101. doi: 10.1182/blood-2008-06-165225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yoon S.R., Lee Y.S., Yang S.H., Ahn K.H., Lee J.H., Lee J.H., Kim D.Y., Kang Y.A., Jeon M., Seol M., Ryu S.G., Chung J.W., Choi I., Lee K.H. Generation of donor natural killer cells from CD34(+) progenitor cells and subsequent infusion after HLA-mismatched allogeneic hematopoietic cell transplantation: a feasibility study. Bone Marrow Transplant. 2010;45(6):1038–1046. doi: 10.1038/bmt.2009.304. [DOI] [PubMed] [Google Scholar]
- 71.Koepsell S.A., Miller J.S., McKenna D.H., Jr. Natural killer cells: a review of manufacturing and clinical utility. Transfusion. 2013;53(2):404–410. doi: 10.1111/j.1537-2995.2012.03724.x. [DOI] [PubMed] [Google Scholar]
- 72.Lapteva N., Durett A.G., Sun J., Rollins L.A., Huye L.L., Fang J., Dandekar V., Mei Z., Jackson K., Vera J., Ando J., Ngo M.C., Coustan-Smith E., Campana D., Szmania S., Garg T., Moreno-Bost A., Vanrhee F., Gee A.P., Rooney C.M. Large-scale ex vivo expansion and characterization of natural killer cells for clinical applications. Cytotherapy. 2012;14(9):1131–1143. doi: 10.3109/14653249.2012.700767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hermanson D.L., Bendzick L., Pribyl L., McCullar V., Vogel R.I., Miller J.S., Geller M.A., Kaufman D.S. Induced Pluripotent Stem Cell-Derived Natural Killer Cells for Treatment of Ovarian Cancer. Stem Cells. 2016;34(1):93–101. doi: 10.1002/stem.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zeng J., Tang S.Y., Toh L.L., Wang S. Generation of "Off-the-Shelf" Natural Killer Cells from Peripheral Blood Cell-Derived Induced Pluripotent Stem Cells. Stem Cell Reports. 2017;9(6):1796–1812. doi: 10.1016/j.stemcr.2017.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhu H., Kaufman D.S. An Improved Method to Produce Clinical-Scale Natural Killer Cells from Human Pluripotent Stem Cells. Methods Mol Biol. 2019;2048:107–119. doi: 10.1007/978-1-4939-9728-2_12. [DOI] [PubMed] [Google Scholar]
- 76.Bock A.M., Knorr D., Kaufman D.S. Development, expansion, and in vivo monitoring of human NK cells from human embryonic stem cells (hESCs) and and induced pluripotent stem cells (iPSCs) J Vis Exp. 2013;(74):e50337. doi: 10.3791/50337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ueda T., Kumagai A., Iriguchi S., Yasui Y., Miyasaka T., Nakagoshi K., Nakane K., Saito K., Takahashi M., Sasaki A., Yoshida S., Takasu N., Seno H., Uemura Y., Tamada K., Nakatsura T., Kaneko S. Non-clinical efficacy, safety and stable clinical cell processing of induced pluripotent stem cell-derived anti-glypican-3 chimeric antigen receptor-expressing natural killer/innate lymphoid cells. Cancer Sci. 2020;111(5):1478–1490. doi: 10.1111/cas.14374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Knorr D.A., Ni Z., Hermanson D., Hexum M.K., Bendzick L., Cooper L.J., Lee D.A., Kaufman D.S. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl Med. 2013;2(4):274–283. doi: 10.5966/sctm.2012-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Market M., Angka L., Martel A.B., Bastin D., Olanubi O., Tennakoon G., Boucher D.M., Ng J., Ardolino M., Auer R.C. Flattening the COVID-19 Curve With Natural Killer Cell Based Immunotherapies. Front Immunol. 2020;11:1512. doi: 10.3389/fimmu.2020.01512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chhabra A. Derivation of Human Induced Pluripotent Stem Cell (iPSC) Lines and Mechanism of Pluripotency: Historical Perspective and Recent Advances. Stem Cell Rev Rep. 2017;13(6):757–773. doi: 10.1007/s12015-017-9766-9. [DOI] [PubMed] [Google Scholar]
- 81.Nashun B., Hill P.W., Hajkova P. Reprogramming of cell fate: epigenetic memory and the erasure of memories past. Embo J. 2015;34(10):1296–1308. doi: 10.15252/embj.201490649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Martinez-Fernandez A., Nelson T.J., Terzic A. Nuclear reprogramming strategy modulates differentiation potential of induced pluripotent stem cells. J Cardiovasc Transl Res. 2011;4(2):131–137. doi: 10.1007/s12265-010-9250-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rebuzzini P., Zuccotti M., Redi C.A., Garagna S. Achilles' heel of pluripotent stem cells: genetic, genomic and epigenetic variations during prolonged culture. Cell Mol Life Sci. 2016;73(13):2453–2466. doi: 10.1007/s00018-016-2171-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shi Y., Inoue H., Wu J.C., Yamanaka S. Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov. 2017;16(2):115–130. doi: 10.1038/nrd.2016.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Attwood S.W., Edel M.J. iPS-Cell Technology and the Problem of Genetic Instability-Can It Ever Be Safe for Clinical Use? J Clin Med. 2019;8(3) doi: 10.3390/jcm8030288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kawamata S., Kanemura H., Sakai N., Takahashi M., Go M.J. Design of a Tumorigenicity Test for Induced Pluripotent Stem Cell (iPSC)-Derived Cell Products. J Clin Med. 2015;4(1):159–171. doi: 10.3390/jcm4010159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Doi D., Magotani H., Kikuchi T., Ikeda M., Hiramatsu S., Yoshida K., Amano N., Nomura M., Umekage M., Morizane A., Takahashi J. Pre-clinical study of induced pluripotent stem cell-derived dopaminergic progenitor cells for Parkinson's disease. Nat Commun. 2020;11(1):3369. doi: 10.1038/s41467-020-17165-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sekine K., Tsuzuki S., Yasui R., Kobayashi T., Ikeda K., Hamada Y., Kanai E., Camp J.G., Treutlein B., Ueno Y., Okamoto S., Taniguchi H. Robust detection of undifferentiated iPSC among differentiated cells. Sci Rep. 2020;10(1):10293. doi: 10.1038/s41598-020-66845-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liu X., Li W., Fu X., Xu Y. The Immunogenicity and Immune Tolerance of Pluripotent Stem Cell Derivatives. Front Immunol. 2017;8:645. doi: 10.3389/fimmu.2017.00645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Garreta E., Sanchez S., Lajara J., Montserrat N., Belmonte J.C.I. Roadblocks in the Path of iPSC to the Clinic. Curr Transplant Rep. 2018;5(1):14–18. doi: 10.1007/s40472-018-0177-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Badin R.Aron, Bugi A., Williams S., Vadori M., Michael M., Jan C., Nassi A., Lecourtois S., Blancher A., Cozzi E., Hantraye P., Perrier A.L. MHC matching fails to prevent long-term rejection of iPSC-derived neurons in non-human primates. Nat Commun. 2019;10(1):4357. doi: 10.1038/s41467-019-12324-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Desgres M., Menasche P. Clinical Translation of Pluripotent Stem Cell Therapies: Challenges and Considerations. Cell Stem Cell. 2019;25(5):594–606. doi: 10.1016/j.stem.2019.10.001. [DOI] [PubMed] [Google Scholar]
- 93.Schweitzer J.S., Song B., Herrington T.M., Park T.Y., Lee N., Ko S., Jeon J., Cha Y., Kim K., Li Q., Henchcliffe C., Kaplitt M., Neff C., Rapalino O., Seo H., Lee I.H., Kim J., Kim T., Petsko G.A., Ritz J., Cohen B.M., Kong S.W., Leblanc P., Carter B.S., Kim K.S. Personalized iPSC-Derived Dopamine Progenitor Cells for Parkinson's Disease. N Engl J Med. 2020;382(20):1926–1932. doi: 10.1056/NEJMoa1915872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Segrelles-Calvo G., Araujo G.R.S., Llopis-Pastor E., Carrillo J., Hernandez-Hernandez M., Rey L., Rodriguez Melean N., Escribano I., Anton E., Zamarro C., Garcia-Salmones M., Frases S. Prevalence of opportunistic invasive aspergillosis in COVID-19 patients with severe pneumonia. Mycoses. 2021;64(2):144–151. doi: 10.1111/myc.13219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.WHO, COVID-19 vaccine tracker and landscape, 2021. https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines. (Accessed July 30th 2021).
- 96.Briggs D.J. The role of vaccination in rabies prevention. Curr Opin Virol. 2012;2(3):309–314. doi: 10.1016/j.coviro.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 97.Papaloukas O., Giannouli G., Papaevangelou V. Successes and challenges in varicella vaccine. Ther Adv Vaccines. 2014;2(2):39–55. doi: 10.1177/2051013613515621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rodrigues A.F., Soares H.R., Guerreiro M.R., Alves P.M., Coroadinha A.S. Viral vaccines and their manufacturing cell substrates: New trends and designs in modern vaccinology. Biotechnol J. 2015;10(9):1329–1344. doi: 10.1002/biot.201400387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bos R., Rutten L., van der Lubbe J.E.M., Bakkers M.J.G., Hardenberg G., Wegmann F., Zuijdgeest D., de Wilde A.H., Koornneef A., Verwilligen A., van Manen D., Kwaks T., Vogels R., Dalebout T.J., Myeni S.K., Kikkert M., Snijder E.J., Li Z., Barouch D.H., Vellinga J., Langedijk J.P.M., Zahn R.C., Custers J., Schuitemaker H. Ad26 vector-based COVID-19 vaccine encoding a prefusion-stabilized SARS-CoV-2 Spike immunogen induces potent humoral and cellular immune responses. NPJ Vaccines. 2020;5:91. doi: 10.1038/s41541-020-00243-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.ScienceMag.Org, Abortion opponents protest COVID-19 vaccines’ use of fetal cells, 2021. https://www.sciencemag.org/news/2020/06/abortion-opponents-protest-covid-19-vaccines-use-fetal-cells. (Accessed 12/08/2021 2021).
- 101.Wang D., Baudys J., Bundy J.L., Solano M., Keppel T., Barr J.R. Comprehensive Analysis of the Glycan Complement of SARS-CoV-2 Spike Proteins Using Signature Ions-Triggered Electron-Transfer/Higher-Energy Collisional Dissociation (EThcD) Mass Spectrometry. Anal Chem. 2020;92(21):14730–14739. doi: 10.1021/acs.analchem.0c03301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nguyen T.N.T., Sha S., Hong M.S., Maloney A.J., Barone P.W., Neufeld C., Wolfrum J., Springs S.L., Sinskey A.J., Braatz R.D. Mechanistic model for production of recombinant adeno-associated virus via triple transfection of HEK293 cells. Mol Ther Methods Clin Dev. 2021;21:642–655. doi: 10.1016/j.omtm.2021.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hu J., Han J., Li H., Zhang X., Liu L.L., Chen F., Zeng B. Human Embryonic Kidney 293 Cells: A Vehicle for Biopharmaceutical Manufacturing. Structural Biology, and Electrophysiology, Cells Tissues Organs. 2018;205(1):1–8. doi: 10.1159/000485501. [DOI] [PubMed] [Google Scholar]
- 104.Yoshihara M., Hayashizaki Y., Murakawa Y. Genomic Instability of iPSCs: Challenges Towards Their Clinical Applications. Stem Cell Rev Rep. 2017;13(1):7–16. doi: 10.1007/s12015-016-9680-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shittu I., Zhu Z., Lu Y., Hutcheson J.M., Stice S.L., West F.D., Donadeu M., Dungu B., Fadly A.M., Zavala G., Ferguson-Noel N., Afonso C.L. Development, characterization and optimization of a new suspension chicken-induced pluripotent cell line for the production of Newcastle disease vaccine. Biologicals. 2016;44(1):24–32. doi: 10.1016/j.biologicals.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 106.Sutton T.C. The Pandemic Threat of Emerging H5 and H7 Avian Influenza Viruses. Viruses. 2018;10(9) doi: 10.3390/v10090461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Huang J., Hume A.J., Abo K.M., Werder R.B., Villacorta-Martin C., Alysandratos K.D., Beermann M.L., Simone-Roach C., Lindstrom-Vautrin J., Olejnik J., Suder E.L., Bullitt E., Hinds A., Sharma A., Bosmann M., Wang R., Hawkins F., Burks E.J., Saeed M., Wilson A.A., Muhlberger E., Kotton D.N. SARS-CoV-2 Infection of Pluripotent Stem Cell-Derived Human Lung Alveolar Type 2 Cells Elicits a Rapid Epithelial-Intrinsic Inflammatory Response. Cell Stem Cell. 2020;27(6) doi: 10.1016/j.stem.2020.09.013. 962-973 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sabanovic B., Piva F., Cecati M., Giulietti M. Promising Extracellular Vesicle-Based Vaccines against Viruses, Including SARS-CoV-2. Biology (Basel) 2021;10(2) doi: 10.3390/biology10020094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liang Y., Duan L., Lu J., Xia J. Engineering exosomes for targeted drug delivery. Theranostics. 2021;11(7):3183–3195. doi: 10.7150/thno.52570. [DOI] [PMC free article] [PubMed] [Google Scholar]