

Graphical abstract
Keywords: King scallop, Genome sequencing, Gene annotation, Neurotoxin tolerance
Highlights
-
•
Generation of a new chromosome-level reference genome for the king scallop.
-
•
Disapproval of previous claim of ‘gene-rich’ feature by improved genome annotation.
-
•
Neurotoxin tolerance potentially contributed by the expansion of GluF genes.
Abstract
The king scallop, Pecten maximus is a well-known, commercially important scallop species and is featured with remarkable tolerance to potent phytotoxins such as domoic acid. A high-quality genome can shed light on its biology and innovative evolution of toxin resistance. A reference genome has recently been published for P. maximus, however, it is suspicious that over 67,700 genes are annotated in this genome, which is unexpectedly larger than its close relatives of pectinids. Herein, we provide an improved high-quality chromosome-level reference genome assembly and annotation for the king scallop P. maximus. A final set of 26,995 genes is annotated after carefully checking and curation of the predicted gene models, which significantly improves the accuracy of gene structure information. The large number of gene duplicates in the previous genome is mainly distorted by the fragmented annotation. Through integrated genomic, evolutionary and transcriptomic analyses, we reveal that the Phi subfamily of ionotropic glutamate receptors (iGluRs) are well preserved in molluscs, and P. maximus experienced the rapid expansion of the Phi class of iGluR (GluF) gene family. The GluF genes exhibit ubiquitously high expression and altered sequence characteristics for ligand selectivity, which may contribute to the remarkable tolerance to neurotoxins in P. maximus. Taken together, our study disapproves the previous claim of the 'gene-rich' genome of this species and provides a high-quality genome assembly for further understanding of its biology and evolution of toxin resistance.
1. Introduction
The king scallop Pecten maximus is widely distributed along the Northeast Atlantic coast and is one of the major commercial fisheries in Europe [1]. In recent years, the king scallop has been ranked consistently in the top five fishery species in UK, with over 48,800 tonnes landing in 2018 [2]. Besides its important commercial value, the king scallop fishery also serves as an excellent model for the study of neurotoxin resistance. When king scallops are exposed to harmful algal blooms (HABs) in their habitat, they can accumulate and retain a fairly high level of domoic acid (DA) for a long period [3], which makes them distinguished from other bivalves (e.g., mussels) that usually possess a toxin ‘saturation’ point [4], [5]. It is suggested that genomic features may be present and contribute to its unparalleled toxin resilience [6].
The rapid developments of high-throughput sequencing technologies facilitate the accumulation of vast amount of genomic data in molluscs [7]. Nevertheless, gene information encoded by the genomic assembly needs to be accurately retrieved to help us understand the organism biology and the evolution in a broad picture. Despite that numerous genomic annotation methods have been developed to integrate evidences from multi-omic datasets, accurate genome annotation remains a major challenge for large and complex genomes [8], [9]. Recently, a reference genome sequence of P. maximus has been assembled and annotated with 67,741 genes, which is about 3-fold more than its close relatives of the Pectinidae family and was interpreted as unique ‘gene-rich’ genomic feature [6]. Considering that the genome size and gene cassette is generally conservative across the Pectinidae species [7], the suspicious over-inflated number of genes could be potentially resulted from the artifacts or errors in genome annotation.
As the accuracy of reference genome sequences is of vital importance for genetic and genomic analysis, we generated an de novo genome assembly of P. maximus and annotated a set of 26,995 genes after manual curation of automated gene structural prediction. The high-quality reference genome sequences and transcriptomes enable a comprehensive analysis of the ionic glutamate receptors (iGluRs) in the king scallop, and lay the foundation for deep understanding of the innovative evolution of molluscan toxicant resistance.
2. Results and discussion
2.1. Genome sequencing and assembly
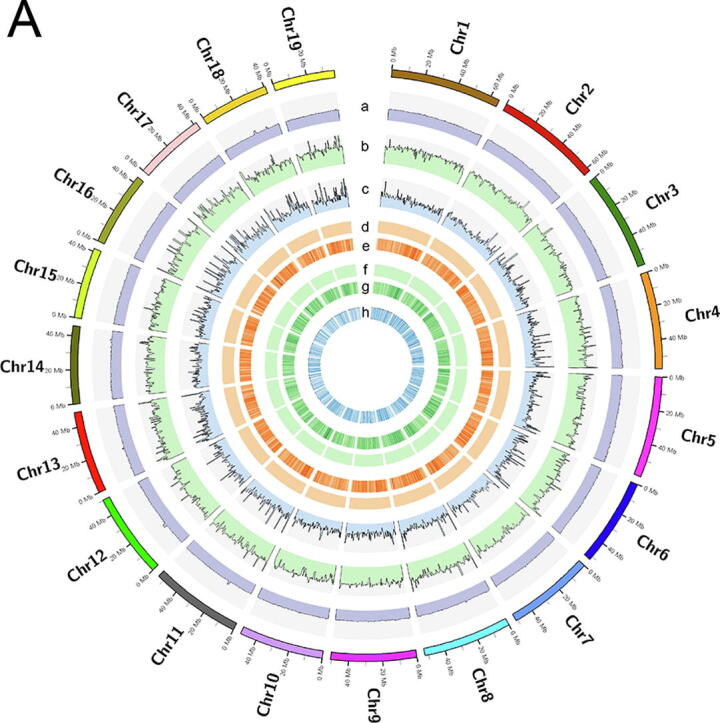
A total of 139.16 Gb bases were used for the assembly of the king scallop genome, including 46.62 Gb Illumina reads and 92.54 Gb Pacbio reads, representing ~ 128 × genome coverage (Supplementary Table S1). The genome of king scallop was estimated with a heterozygous ratio of 1.24% and repeat rate of 56.48%. We assembled a genome sequence with a total size of 1,059.85 Mb and a contig N50 of 997.79 Kb, which is ~100 Mb larger than the previous one and is more in line with the k-mer analysis (1,085.96 Mb) and previous assessments (1,025~1,150 Mb) [6] (Supplementary Fig. S1). The improved completeness was also revealed by the alignment rate of whole genomic sequencing reads. The mapping rate of Illumina and Pacbio reads reached 98.65% and 97.28% for the new assembly, which is higher than the previous one (~94%) [6]. We only detected 9,291 (0.0009%) conflicting sites in the genome assembly (Fig. 1A), indicating a high degree of consistency with the Illumina reads. The contigs were anchored into 19 chromosomes with a scaffold N50 of 50.6 Mb, which is consistent with the previous karyotype analysis of this scallop species [10]. The chromosomes of P. maximus exhibited a 1:1 perfect correspondence to that of Mizuhopecten yessoensis [11], despite that P. maximus has a closer phylogenetic relationship with the Argopecten genus where 16 haploid chromosomes were observed (Fig. 1B; Supplementary Fig. S2).
Fig. 1.

A: Global genome landscape of P. maximus. From outer to inner circles: GC content (a), depth of coverage of Illumina reads (b), depth of coverage of PacBio reads (c), distribution of homozygous (d) and heterozygous SNPs (e), distribution of homozygous (f) and heterozygous INDELs (g), distribution of genes (h). B: Macro-synteny of the P. maximus and A. purpuratus chromosomes to contemporary chromosomes of M. yessoensis. The conserved syntenic blocks are shown by the local fraction of genes from each M. yessoensis chromosomes. C: BUSCO evaluation on the predicted gene models. D: Distribution of number of exons per predicted gene model. E: Deviations of protein lengths for the single-copy orthologues in four scallops. F: Annotation of Hox1, Lox5, and Post2 gene structures in four scallop genomes. Exons from the same gene were annotated as distinct gene models in Kenny et al. (2020). Bins in grey indicate mis-annotated exons.
2.2. Evaluation of genome annotation
In Kenny et al. (2020), a BUSCO evaluation was performed in the genome mode. Despite that 95.5% of the BUSCO Metazoa dataset could be predicted in the genome assembly, whether or not they have been successfully annotated is unknown [6]. Therefore, we conducted the analysis in the protein mode with default parameters, and identified only 601 (61.4%) complete BUSCOs in the previous gene set (Fig. 1C). Notably, 314 (32.1%) of the BUSCOs were fragmented, suggesting that the previous adopted pipeline for gene structural annotation may encounter errors in defining gene boundaries [6]. In this study, we combined and manually curated the gene models predicted by ab initio prediction, homologous prediction and RNA-seq data alignment (Supplementary Table S5). A final set of 26,995 gene models were identified and 26,248 (97.23%) could be functionally annotated by at least one database (Supplementary Table S6). In addition, 1,289 miRNA, 2,373 tRNA, 102 rRNA, and 411 snRNA were identified in the P. maximus genome (Supplementary Table S7). The BUSCO analysis revealed a high-level of completeness by identifying 951 (97.2%) of the 978 single-copy orthologs in metazoan. The genome assembly and annotation files of P. maximus are stored at MolluscDB (http://mgbase.qnlm.ac/page/download/download) [12].
We checked the distribution of exon numbers per gene across seven molluscan species and noticed that the gene models of the former P. maximus genome assembly exhibited a distinguished pattern compared with the remaining sets (Fig. 1D). The number of genes with eight or more exons were obviously reduced in the previous annotation of P. maximus genomic assembly [6], while genes with four or fewer exons were sharply expanded, suggesting that a considerable number of large genes were mis-annotated as fragmented models. We further checked the protein length of 7,544 single copy orthologues in four scallops. It clearly shows that the previous version of gene models was incomplete (Fig. 1E). Even several well-conserved genes across metazoans, including Hox1, Lox5 and Post2, have been identified as two adjacent separate genes and mistaken as tandem duplication (Fig. 1F; Supplementary Fig. S4). We further checked the alignment of RNA-seq data from different tissues and developmental stages. The new version of reference genome sequences and annotation improved about 8% and 5% of the unique and overall mapping rate on average, respectively (Table S8).
2.3. Evolution of iGluR gene family
The king scallops can accumulate and tolerate potent neurotoxins such as DA, although the molecular mechanism of biotoxin resistance in scallops is not well understood. DA possess a typical structural feature resembling the principal excitatory neurotransmitter glutamate. It can attack the nervous system by acting as potent agonists of iGluRs on nerve cell membranes and cause excitotoxic effect [13]. The iGluR subunits have been previously classified into six classes based on studies in the vertebrates [14]. However, a recent study revealed a more complex evolution of iGluRs with diverse ligand selectivity across the animal kingdom [15].
We identified the full sets of iGluR family genes from the genomes of seven species covering major molluscan lineages, and performed a phylogenetic study with those identified from 28 additional representative species across the metazoans. The number of iGluR genes ranged from 19 (Hapalochlaena maculosa) to 36 (P. maximus) within the molluscs (Supplementary Table S9). Ramos-Vicente et al. (2018) revealed that the iGluR genes could be clustered into four major subfamilies that diverged prior to the split of metazoan lineages, including Lambda, N-methyl-D-aspartate (NMDA), Epsilon, and AKDF [15]. Our phylogenetic analysis recovered the monophyly of the groups defined by the four subfamilies and placed Lambda at the deepest lineage (Fig. 2A). Almost all the molluscan iGluR genes were grouped into the clade of NMDA and AKDF, suggesting the loss of Lambda and Epsilon in molluscs. Intriguingly, despite that the Phi class of iGluR genes (GluF) of the AKDF subfamily were previously considered to be restricted in Echinodermata, Hemichordate, and non-vertebrata Chordata, we identified the Phi class in all the seven molluscs. A novel class Zeta was identified exclusively in molluscs as a sister group of Phi, suggesting a molluscan specific expansion in the AKDF subfamily. Phylogenetics of GluF genes from 21 molluscs revealed four clades (subclass I to IV). Here we show that the subclass I and IV is present in most of the molluscs, whereas the subclass II and III is phylogenetically restricted in gastropods and bivalves, respectively (Fig. 2B). Notably, the king scallop possesses the largest number of GluF genes among the 21 analyzed molluscs. The flanking genes of two GluFIV genes are highly conservative, suggesting that the rapid expansion is driven by tandem duplication (Supplementary Fig. S5).
Fig. 2.

A: Maximum likelihood phylogenetic reconstruction reveals possible orthologous relationships among iGluR genes from representative mollusks and metazoan taxa. Numbers on the nodes represent support from 10,000 pseudoreplicates of the ultrafast bootstrap procedure. The branch of molluscan iGluR genes were denoted in light blue. The 36 iGluR genes of P. maximus were highlighted in red. B: Maximum likelihood phylogram of molluscan GluF genes. The branch of GluF genes of amphioxus, hemichordate, echinoderm, and molluscs were denoted in red, orange, yellow, and green, respectively. The six GluF genes of P. maximus were highlighted in red. Abbreviations of species, A. californica (Aca), T. granosa (Tgr), C. sinensis (Csi), E. chlorotica (Ech), P. f. martensii (Pfm), S. broughtonii (Sbr), S. constricta (Sco), B. platifrons (Bpl), C. gigas (Cgi), L. gigantea (Lgi), M. philippinarum (Mph), O. bimaculoides (Obi), O. minor (Omi), B. glabrata (Bgl), A. farreri (Afa), C. squamiferum (Csq), G. aegis (Gae), H. maculosa (Hma), P. canaliculata (Pca), P. maximus (Pma), M. yessoensis (Mye), Strongylocentrotus purpuratus (Spu), Saccoglossus kowalevskii (Sko), B. floridae (Bfl), A. thaliana (Ath). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Gene expression profiling facilitates the understanding of the function and evolution of iGluR families. Beside the king scallop, we collected another eight molluscs with the best availability of comprehensive transcriptome datasets to analyze the expression profiles in various adult tissues/organs (Fig. 3A). The iGluR genes generally exhibited diverse tissues/organs-preferential expression patterns across the nine molluscs. Interestingly, compared with the cephalopod and gastropods, the GluF genes in P. maximus and the other two scallops were ubiquitously expressed with the most prominent expression levels among all the iGluRs (Fig. 3A). It is suggested that GluF genes may be important receptors in response to neurotransmitters and control the signal transmission in the scallops.
Fig. 3.

A: Expression patterns of iGluR genes among different tissues in P. maximus and eight molluscs. B: Protein structure alignment of P. maximus GluFIII and R. norvegicus GluK2 (7KS3). C: Multiple protein alignment of Phi class of iGluR gene residues involved in ligand-binding. Numbers shown on top correspond to GluA2 of Rattus norvegicus (P19491). Residues involved in ligand binding are indicated by a black frame. Acid and basic amino acid residues were highlighted by red and light blue, respectively. The levels of conservation are denoted by blue background and a bar chart at the bottom. Agonists predicted for iGluRs with non-conservative residues at 653 and 655 were indicated with an asterisk. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Proteins of the GluF genes in molluscs present well-conserved transmembrane domains and residues for tetramerization (Supplementary Fig. S6). Three-dimensional models of the GluFIII of P. maximus indicate that its general fold is well preserved (Fig. 3B). Previous studies have revealed that the most important residues for fixing the amino acid backbone are Arg485 and an acidic residue at position 705 [16], [17], [18]. These two positions are well conserved in nine of the ten proteins of GluF genes in scallops, suggesting that their intrinsic ligand is an amino acid. Ligand selectivity of iGluRs is mainly determined by residues at 653 and 655 [19]. For typical glutamate-binding receptors, these two sites are occupied by glycine and threonine, whereas, for glycine-binding iGluRs, they are serine and a non-polar residue, respectively. In ctenophore, the residue 653 could be substitute to serine or threonine for glutamate-binding iGluRs, and to arginine for glycine-binding subunits [19]. Notably, despite that GluFI, GluFIV3, and GluFIV4 possess residues of typical glutamate-binding iGluRs, high variability was observed in residues 653 and 655 of the remaining GluF genes in P. maximus. The king scallop GluFIII, GluFIV1, and GluFIV2 genes, which possess a glycine or valine at residue 653, and a non-polar residue at residue 655, are thus candidates for glycine-binding receptors (Fig. 3C). As the span of domain opening is much smaller in the ligand-binding core of glycine-binding iGluRs, they usually are not affected by glutamate agonists, such as DA and kainic acid (KA) [20]. The presence of these glycine-binding iGluRs may explain scallop’s amazing ability to tolerate neurotoxin DA. Our bioinformatic analyses of GluF genes would provide important protein structural insights and guidance for further experiment-oriented investigations and deepen our understanding of their functional roles in the evolution of toxin resistant.
3. Conclusion
Our study provides a high-quality chromosome-level reference genome sequence for the king scallop P. maximus. A final set of 26,995 genes were annotated on the genome after carefully check and curation of the predicted gene models, which significantly improved the accuracy of gene structural information. We proved that the large numbers of gene duplicates in P. maximus were distorted by the fragmented annotation in the previous genomic study. The number of genes in P. maximus is actually comparable to those of other Pectinidae species with high-quality genomic assembly. Through integrated genomic, evolutionary and transcriptomic analyses, we revealed for the first time that the Phi subfamily of iGluRs were well preserved in molluscs, and the P. maximus experienced a rapid expansion in the GluF gene family. The GluF genes exhibited a ubiquitously high expression and altered sequence characteristics for ligand selectivity, which may contribute to the remarkable tolerance to neurotoxins in P. maximus. Taken together, our study disapproves the previous claim of the 'gene-rich' genome of this species (mostly due to inaccurate gene annotation) and provides a high-quality genome assembly for further understanding of its biology and evolution in toxin-resistance.
4. Materials and methods
4.1. Samples preparation and sequencing
The king scallops, P. maximus were purchased commercially from Seashell AS (Norddyrøy, Norway). The shell and DNA samples were deposited at the Key Laboratory of Marine Genetics and Breeding (Ministry of Education), Ocean University of China (Specimen code: OUC-MGB-2019-PMA-03). Soft tissues were dissected and frozen in liquid nitrogen immediately after sampling. Genomic DNA was extracted from the adductor muscle using phenol/chloroform alcohol method [21]. A paired-end Illumina library with an insert size of 350 bp was prepared and sequenced on an Illumina HiSeq X-Ten system. Genomic DNA from the same sample was also used to construct a library for PacBio sequencing on a PacBio Sequel Single-molecule Real-time (SMRT) platform. Mantle and kidney of three adult individuals were collected for transcriptome sequencing. Tissues were stored at −80 ℃ after being flash-frozen in liquid nitrogen. Total mRNA was extracted with phenol/chloroform alcohol and all the libraries were constructed by VAHTS Universal V6 RNA-seq Library Prep Kit for Illumina (Vazyme Biotech Co., Ltd) and sequenced on an Illumina HiSeq system. All of the sequencing data have been deposited at NCBI under the accession of PRJNA719586.
4.2. Genome size estimation and genome assembly
The Illumina reads were trimmed to remove adaptors and reads with >10% ambiguous first using Trimmomatic [22]. The paired reads were filtered when the number of low quality bases (Q < 5) in a single-ended sequencing read exceeds 20%. Then Jellyfish (version 2.2.5) [23] was used to count the k-mer frequency with a k-mer size of 17. Genome size was estimated according to the formula: genome size = k-mer number/k-mer depth [24]. Self-correction was first performed on the Pacbio data which were then assembled with Overlap-Layout-Consensus algorithm. Pacbio long reads were mapped to the contigs with Minimap2 (version 2.12) [25]. The assembly was then polished by Racon (version 1.4.13) [26] with default parameters. Illumina short reads were mapped to the polished assembly by Burrows-Wheeler Alignment tool (BWA, version 0.7.17) [27] and the results were used to conduct another round of polish by Pilon (version 1.23) [28]. Redundant haplotigs were purged with purge_Dups (v1.2.5) [29] using default parameters. To ensure high accuracy and integrity of the genome assembly, Illumina paired-end clean reads were aligned to it using BWA (version 0.7.17) [27]. The Core Eukaryotic Genes Mapping Approach (CEGMA) [30] was applied to evaluate the completeness of the gene set in the draft genome. The BUSCO (version 3) [31] was also performed with the parameters of “-i $inputfile -o $outputfile -l $metazoa_odb9 -m proteins -c 8”. Hi-C reads from the previous genome sequencing project of P. maximus were used for scaffolding with Juicer [6], [32]. Mis-joins, order and orient were corrected by 3D-DNA [33], and contigs were anchored into pseudo-chromosomes. Finally, the candidate assembly was manually corrected in Juicebox Assembly Tools [34], [35].
4.3. Genome annotation
Repetitive sequences in the genome assembly were identified through de novo identification and homologous alignment of sequences. De novo repetitive sequence database was built by LTR_FINDER [36], RepeatScout (version 1.0.5) and RepeatModeler (version 1.0.11). Repbase database [37] was used together for homology-based searches by Repeatmasker (version 4.0.9). The gene structure was predicted by de novo prediction, homology-based searches combined with transcriptome data alignment. Firstly, Augustus (version 3.3.2) [38], Glimmer HMM [39], Semi-HMM-based Nucleic Acid Parser (SNAP, v2013.11.29) [40], Geneid (version 1.4) [41] and Genescan (version 1.0) [42] were used to generate a de novo gene library based on the frequencies of codon usage and distribution of exons. Then protein coding sequences of M. yessoensis, Crassostrea gigas, Homo sapiens, Drosophila melanogaster, Octopus bimaculoides and Lottia gigantea, were aligned to the genome assembly using TBLATN and GeneWise [43]. The Illumina RNA-seq reads were also aligned to the assembly and all gene model evidences were integrated with EVidenceModeler [44]. The Program to Assemble Spliced Alignments (PASA) pipeline [45] was used to correct the integrated gene set. Finally, the predicted gene models were visualized via IGV and manually checked with TBtools [46]. The predicted proteins were aligned to public databases, including Swissprot, NCBI-NR and KEGG to perform gene functional annotation. InterProScan (version 4.8) [47] was used to search for domains or motifs in InterPro, Pfam and Gene Ontology (GO) database. The noncoding RNA genes, including rRNAs, tRNAs, snRNAs and miRNAs were annotated in the P. maximus genome. The miRNAs and snRNAs were screened using INFERNAL 1.1.2 against the Rfam database (version 14.1) [48] with default parameters. Transfer RNA were predicted by tRNAscan-SE (version 1.4) [49] with parameters for eukaryotes. The gene models of annotation used in this study and by Kenny et al. (2020) were compared with the other four scallops (M. yessoensis, Azumapecten farreri, A. irradians, and A. purpuratus) for quality evaluation. Briefly, orthologues from the six genome assemblies were identified with Orthofinder. All the single copy orthologues in M. yessoensis, A. farreri, A. irradians, and A. purpuratus were retrieved. Their corresponding orthologues were extracted from the two versions of P. maximus genome annotation, and the Z-scores were calculated for the protein lengths of each orthologue.
4.4. Gene family identification and phylogenetic analysis
The gene sets from 16 eumetazoan species were used to analyze the gene clustering, including Nematostella vectensis, Branchiostoma floridae, H. sapiens, O. bimaculoides, L. gigantea, Pomacea canaliculata, Aplysia californica, Biomphalaria glabrata, C. gigas, Saccostrea glomerata, Scapharca broughtonii, M. yessoensis, A. farreri, A. irradians, A. purpuratus and P. maximus. OrthoFinder (version 2.3.3) [50] was used to assign the gene family clusters with default parameters. The longest protein sequence was selected as representative when a gene possesses multiple splicing isoforms. Phylogeny of P. maximus was inferred by one-to-one orthologous gene families detected from the result of Orthofinder. Multiple alignments of the genes were performed using Mafft (version 7.221) [51] and the conserved blocks of alignments were selected by Gblocks [52] to concatenate a supergene for tree construction. Single-copy orthologues were used for maximum-likelihood (ML) phylogenetic relationships with IQ-TREE (version 1.6.12) [53] “$iqtree -s $inputfile -abayes -bb 1000 -nt AUTO”. An optimal substitution model was automatically selected with 10,000 bootstraps. Divergence time estimations were determined using MCMCTree (part of the PAML package) [54] with four reference divergence times obtained from TimeTree database [55]. Finally, gene family expansion and contraction were analyzed using CAFE (version 3) [56] with separated birth and death rates under a P value threshold of 0.01.
4.5. Phylogenetic analysis and classification of molluscan iGluRs
The iGluR genes were identified in the P. maximus genomes by HHsearch with an E-value threshold of 1e-5 against the iGluR hmm files (PF10613 and PF00060) from pfam database and were further confirmed by comparing to the Conserved Domains Database (http://www.ncbi.nlm.nih.gov/cdd) and SMART (https://smart.embl-heidelberg.de/). In addition, the iGluR genes were screened through BLAST searches and manual literature inspections. The identified iGluRs were classified based on molecular phylogeny and manual inspection of conserved residues. The same approach was applied to identify iGluRs genes in other six molluscan species, including Sinonovacula constricta, O. bimaculoides, B. glabrata, H. maculosa, P. canaliculata, M. yessoensis. Phylogenetic relationships of iGluR amino acid sequences were estimated using maximum-likelihood (ML) analyses with IQ-Tree (v1.6.12). Branch supports were evaluated with aBayes tests of 10,000 pseudo-replicates of the ultrafast bootstrap procedure [57]. ModelFinder from IQ-Tree was used to select the best-fitting model (LG + R9). Plant iGluRs from Arabidopsis thaliana were used as an outgroup. The sequence alignment file for phylogenetic analysis is stored on FigShare [58]. GluF of 21 molluscan species (A. californica, Tegillarca granosa, Cyclina sinensis, Elysia chlorotica, Pinctada fucata martensii, S. broughtonii, S. constricta, Bathymodiolus platifrons, C. gigas, L. gigantea, Modiolus philippinarum, O. bimaculoides, Octopus minor, B. glabrata, A. farreri, Chrysomallon squamiferum, Gigantopelta aegis, H. maculosa, P. canaliculata, M. yessoensis) were identified and then used to build phylogenetic tree with the same method mentioned above. The protein structures of iGluRs in the P. maximus were predicted by PHYRE2 [59]. Alignment of the amino acid sequences and protein structures were performed using Jalview (version 2.11.1.4) [60] and TM-align [61], respectively.
4.6. Transcriptome analysis
Transcriptome data from T. granosa, S. constricta, C. gigas, B. glabrata, A. farreri, C. squamiferum, G. aegis, H. maculosa, P. canaliculata and M. yessoensis were retrieved from the MolluscDB. Raw reads were first trimmed to remove those containing undetermined bases (“N”) and excessive numbers of low-quality positions (>10 positions with quality scores < 10). Hisat2 was used for the alignment of RNA-seq data [62]. The index for reference genomes were build with the annotation file using the default parameters. The high-quality reads were mapped to the reference index with the parameters of “$hisat2 -p 24 -x $index_filename -1 $forward_fq -2 $reverse_fq (-U $single_fq) --summary-file $summary_filename -S output_filename”. The counts of aligned read for each gene model were calculated with FeatureCounts [63], and were normalized by calculating the transcripts per kilobase of exon model per million mapped reads (TPM).
Acknowledgments
Acknowledgements
We acknowledge the grant support from National Natural Science Foundation of China (31871499 and 3197210450), Fundamental Research Funds for the Central Universities (202064008), Project of Sanya Yazhouwan Science and Technology City Management Foundation (SKJC-2020-02-009), and Taishan Scholar Project Fund of Shandong Province of China.
Authors' contributions
Q.Z., S.W. and L.B. designed and supervised the project. J.L. and C.W. collected scallop samples and conducted experiments. Q.Z., J.L., L.B. and S.W. performed the analysis. All authors participated in manuscript writing.
Conflict of interests
The authors declare that they have no competing interests.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2021.08.038.
Contributor Information
Lisui Bao, Email: baolisui@ouc.edu.cn.
Shi Wang, Email: swang@ouc.edu.cn.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Nicolle A., Moitié R., Ogor J., Dumas F., Foveau A., Foucher E. Modelling larval dispersal of Pecten maximus in the English Channel: A tool for the spatial management of the stocks. ICES J Sci. 2017;74:1812–1825. [Google Scholar]
- 2.Zedadra O, Guerrieri A, Jouandeau N, Seridi H, Fortino G, Spezzano G. Shellfish Stocks and Fisheries Review 2019: An assessment of selected stocks. Marine Institute & Bord Iascaigh Mhara; 2020. [Google Scholar]
- 3.Blanco J., Acosta C.P., Bermúdez de la Puente M., Salgado C. Depuration and anatomical distribution of the amnesic shellfish poisoning (ASP) toxin domoic acid in the king scallop Pecten maximus. Aquat Toxicol. 2002;60:111–121. doi: 10.1016/s0166-445x(01)00274-0. [DOI] [PubMed] [Google Scholar]
- 4.Bricelj V.M., Lee J.H., Cembella A.D., Anderson D.M. Uptake kinetics of paralytic shellfish toxins from the dinoflagellate Alexandrium fundyense in the mussel Mytilus edulis. Mar Ecol Prog Ser. 1990;63:177–188. [Google Scholar]
- 5.Borcier E., Morvezen R., Boudry P., Miner P., Charrier G., Laroche J. Effects of bioactive extracellular compounds and paralytic shellfish toxins produced by Alexandrium minutum on growth and behaviour of juvenile great scallops Pecten maximus. Aquat Toxicol. 2017;184:142–154. doi: 10.1016/j.aquatox.2017.01.009. [DOI] [PubMed] [Google Scholar]
- 6.Kenny N.J., McCarthy S.A., Dudchenko O., James K., Betteridge E., Corton C. The gene-rich genome of the scallop Pecten maximus. GigaScience. 2020;9:giaa037. doi: 10.1093/gigascience/giaa037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Z., Zhang L., Hu J., Wang J., Bao Z., Wang S. The evo-devo of molluscs: insights from a genomic perspective. Evol Dev. 2020;22:409–424. doi: 10.1111/ede.12336. [DOI] [PubMed] [Google Scholar]
- 8.Ejigu G.F., Jung J. Review on the computational genome annotation of sequences obtained by next-generation sequencing. Biology. 2020;9:295. doi: 10.3390/biology9090295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baptista R.P., Kissinger J.C., Sheppard D.C. Is reliance on an inaccurate genome sequence sabotaging your experiments? PLoS Pathog. 2019;15:e1007901. doi: 10.1371/journal.ppat.1007901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Insua A., López-Piñón M.J., Freire R., Méndez J. Karyotype and chromosomal location of 18S–28S and 5S ribosomal DNA in the scallops Pecten maximus and Mimachlamys varia (Bivalvia: Pectinidae) Genetica. 2006;126:291–301. doi: 10.1007/s10709-005-7408-7. [DOI] [PubMed] [Google Scholar]
- 11.Wang S., Zhang J., Jiao W., Li J., Xun X., Sun Y. Scallop genome provides insights into evolution of bilaterian karyotype and development. Nat Ecol Evol. 2017;1:1–12. doi: 10.1038/s41559-017-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu F., Li Y., Yu H., Zhang L., Hu J., Bao Z. MolluscDB: An integrated functional and evolutionary genomics database for the hyper-diverse animal phylum Mollusca. Nucleic Acids Res. 2021;49:D988–D997. doi: 10.1093/nar/gkaa918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cendes F., Andermann F., Frcp C., Carpenter S., Zatorre R.J., Cashman N.R. Temporal lobe epliepsy caused by domoic acid intoxication: evidence for glutamate receptor-mediated excitotoxicity in humans. Ann Neurol Off J Am Neurol Assoc Child Neurol Soc. 1995;37:123–126. doi: 10.1002/ana.410370125. [DOI] [PubMed] [Google Scholar]
- 14.Traynelis S.F., Wollmuth L.P., McBain C.J., Menniti F.S., Vance K.M., Ogden K.K. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramos-Vicente D., Ji J., Gratacòs-Batlle E., Gou G., Reig-Viader R., Luís J. Metazoan evolution of glutamate receptors reveals unreported phylogenetic groups and divergent lineage-specific events. Elife. 2018;7 doi: 10.7554/eLife.35774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mayer M.L. Crystal structures of the GluR5 and GluR6 ligand binding cores: Molecular mechanisms underlying kainate receptor selectivity. Neuron. 2005;45:539–552. doi: 10.1016/j.neuron.2005.01.031. [DOI] [PubMed] [Google Scholar]
- 17.Hald H., Naur P., Pickering D.S., Sprogøe D., Madsen U., Timmermann D.B. Partial agonism and antagonism of the ionotropic glutamate receptor iGLuR5: structures of the ligand-binding core in complex with domoic acid and 2-amino-3-[5-tert-butyl-3-(phosphonomethoxy)-4-isoxazolyl]propionic acid. J Biol Chem. 2007;282:25726–25736. doi: 10.1074/jbc.M700137200. [DOI] [PubMed] [Google Scholar]
- 18.Yao Y., Harrison C.B., Freddolino P.L., Schulten K., Mayer M.L. Molecular mechanism of ligand recognition by NR3 subtype glutamate receptors. EMBO J. 2008;27:2158–2170. doi: 10.1038/emboj.2008.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alberstein R., Grey R., Zimmet A., Simmons D.K., Mayer M.L. Glycine activated ion channel subunits encoded by ctenophore glutamate receptor genes. Proc Natl Acad Sci USA. 2015;112:E6048–E6057. doi: 10.1073/pnas.1513771112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swanson G.T., Sakai R. Ligands for ionotropic glutamate receptors. Marine Toxins as Research Tools. 2009;1:123–157. doi: 10.1007/978-3-540-87895-7_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sambrook J., Fritsch E.F., Maniatis T. Cold spring harbor laboratory press; 2001. Molecular cloning: a laboratory manual (No. Ed. 3) [Google Scholar]
- 22.Bolger A.M., Lohse M., Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marçais G., Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–770. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Varshney R.K., Chen W., Li Y., Bharti A.K., Saxena R.K., Schlueter J.A. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat Biotechnol. 2012;30:83–89. doi: 10.1038/nbt.2022. [DOI] [PubMed] [Google Scholar]
- 25.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vaser R., Sović I., Nagarajan N., Šikić M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017;27:737–746. doi: 10.1101/gr.214270.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walker B.J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE. 2014;9(11):e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guan D., McCarthy S.A., Wood J., Howe K., Wang Y. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics. 2020;36:2896–2898. doi: 10.1093/bioinformatics/btaa025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parra G., Bradnam K., Korf I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061–1067. doi: 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- 31.Waterhouse R.M., Seppey M., Simao F.A., Manni M., Ioannidis P., Klioutchnikov G. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol. 2018;35:543–548. doi: 10.1093/molbev/msx319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Durand N.C., Shamim M.S., Machol I., Rao S.S.P., Huntley M.H., Lander E.S. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 2016;3:95–98. doi: 10.1016/j.cels.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dudchenko O., Batra S.S., Omer A.D., Nyquist S.K., Hoeger M., Durand N.C. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 2017;356:92–95. doi: 10.1126/science.aal3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Durand N.C., Robinson J.T., Shamim M.S., Machol I., Mesirov J.P., Lander E.S. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 2016;3:99–101. doi: 10.1016/j.cels.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dudchenko O, Shamim M, Batra S, Durand N, Musial N, Mostofa R. The Juicebox Assembly Tools module facilitates de novo assembly of mammalian genomes with chromosome-length scaffolds for under $1000. BioRxiv. 2018:254797. [Google Scholar]
- 36.Xu Z., Wang H. LTR-FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007;35:265–268. doi: 10.1093/nar/gkm286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bao W., Kojima K.K., Kohany O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA. 2015;6:4–9. doi: 10.1186/s13100-015-0041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stanke M., Keller O., Gunduz I., Hayes A., Waack S., Morgenstern B. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006;34:435–439. doi: 10.1093/nar/gkl200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Majoros W.H., Pertea M., Salzberg S.L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20:2878–2879. doi: 10.1093/bioinformatics/bth315. [DOI] [PubMed] [Google Scholar]
- 40.Korf I. Gene finding in novel genomes. BMC Bioinf. 2004;5:59. doi: 10.1186/1471-2105-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blanco E., Parra G., Guigó R. Using geneid to identify genes. Curr Protoc Bioinformatics. 2007;18:4–13. doi: 10.1002/0471250953.bi0403s18. [DOI] [PubMed] [Google Scholar]
- 42.Burge C., Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- 43.Birney E., Clamp M., Durbin R. GeneWise and Genomewise. Genome Res. 2004;14:988–995. doi: 10.1101/gr.1865504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haas B.J., Salzberg S.L., Zhu W., Pertea M., Allen J.E., Orvis J. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008;9:1–22. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haas B.J., Zeng Q., Pearson M.D., Cuomo C.A., Wortman J.R. Approaches to fungal genome annotation. Mycology. 2011;2:118–141. doi: 10.1080/21501203.2011.606851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen C., Chen H., Zhang Y.i., Thomas H.R., Frank M.H., He Y. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13:1194–1202. doi: 10.1016/j.molp.2020.06.009. [DOI] [PubMed] [Google Scholar]
- 47.Jones P., Binns D., Chang H.-Y., Fraser M., Li W., McAnulla C. InterProScan 5: Genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kalvari I., Nawrocki E.P., Argasinska J., Quinones-Olvera N., Finn R.D., Bateman A. Non-coding RNA analysis using the Rfam database. Curr Protoc Bioinforma. 2018;62:1–44. doi: 10.1002/cpbi.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lowe T.M., Chan P.P. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016;44:W54–W57. doi: 10.1093/nar/gkw413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emms D.M., Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015;16:1–14. doi: 10.1186/s13059-015-0721-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Katoh K., Misawa K., Kuma K.I., Miyata T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2020;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 53.Nguyen L.T., Schmidt H.A., Von Haeseler A., Minh B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 55.Kumar S., Stecher G., Suleski M., Hedges S.B. TimeTree: A resource for timelines, timetrees, and divergence times. Mol Biol Evol. 2017;34:1812–1819. doi: 10.1093/molbev/msx116. [DOI] [PubMed] [Google Scholar]
- 56.De Bie T., Cristianini N., Demuth J.P., Hahn M.W. CAFE: A computational tool for the study of gene family evolution. Bioinformatics. 2006;22:1269–1271. doi: 10.1093/bioinformatics/btl097. [DOI] [PubMed] [Google Scholar]
- 57.Hoang D.T., Chernomor O., Von Haeseler A., Minh B.Q., Vinh L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol Biol Evol. 2018;35:518–522. doi: 10.1093/molbev/msx281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu J. Alignment used for phylogenetic analysis of iGluR families. figshare. 2021 doi: 10.6084/m9.figshare.15104712.v3. [DOI] [Google Scholar]
- 59.Kelley L.A., Mezulis S., Yates C.M., Wass M.N., Sternberg M.J.E. Europe PMC Funders Group The Phyre2 web portal for protein modelling, prediction and analysis. Nat Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Waterhouse A.M., Procter J.B., Martin D.M.A., Clamp M., Barton G.J. Jalview Version 2-A multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Y., Skolnick J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements Daehwan HHS Public Access. Nat Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liao Y., Smyth G.K., Shi W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.