

Summary
Processive exoribonucleases are executors of RNA decay. In humans, their physical but not functional interactions were thoughtfully investigated. Here we have screened cells deficient in DIS3, XRN2, EXOSC10, DIS3L, and DIS3L2 with a custom siRNA library and determined their genetic interactions (GIs) with diverse pathways of RNA metabolism. We uncovered a complex network of positive interactions that buffer alterations in RNA degradation and reveal reciprocal cooperation with genes involved in transcription, RNA export, and splicing. Further, we evaluated the functional distinctness of nuclear DIS3 and cytoplasmic DIS3L using a library of all known genes associated with RNA metabolism. Our analysis revealed that DIS3 mutation suppresses RNA splicing deficiency, while DIS3L GIs disclose the interplay of cytoplasmic RNA degradation with nuclear RNA processing. Finally, genome-wide DIS3 GI map uncovered relations with genes not directly involved in RNA metabolism, like microtubule organization or regulation of telomerase activity.
Subject areas: Molecular biology, Molecular microbiology, Transcriptomics
Graphical abstract
Highlights
-
•
Genetic interaction profiling of processive RNases: DIS3, DIS3L, EXOSC10, and XRN2
-
•
Network of positive interactions that buffer alterations in RNA degradation
-
•
Distinct functionalities of processive RNases
-
•
DIS3 mutation suppresses RNA splicing deficiency
Molecular biology; Molecular microbiology; Transcriptomics
Introduction
In eukaryotes, gene expression is regulated at numerous levels from chromatin structure throughout transcription, pre-RNA processing, and RNA localization up to translation. It is well established that RNA is an essential component of every living cell. It not only mediates the transfer of genetic information from DNA to proteins but also plays an important role in regulating gene expression. A plethora of different factors are involved in regulating the post-transcriptional life of RNA molecules. For example, pre-mRNA splicing requires five different snRNAs and more than 100 proteins (Wahl et al., 2009). More than 60 different helicases that control the biogenesis and decay of various RNA species are encoded in the human genome. Nucleotidyltransferases, such as poly(A) and poly(U) polymerases, add non-templated nucleotides to the 3′ end of RNA molecules and are involved in transfer RNA (tRNA), mRNA, and U6 snRNA maturation and decay. In the process of transcription, each cell synthesizes an excessive amount of RNA from different classes, and most of them are subjected to extensive processing or degradation. Additionally, stringent quality-control pathways exist at every step, thereby ensuring that aberrant transcripts are quickly degraded (Kilchert et al., 2016; Schmid and Jensen, 2010). Hence, it is evident that RNA decay is of high importance.
Ribonucleases (commonly called RNases) are responsible for RNA degradation and processing. They are divided into two main classes: endoribonucleases, cleaving – internal phosphodiester bonds in an RNA molecule, and exoribonucleases – cleaving mononucleotides from either end of RNA molecules. RNA molecules can be degraded or processed from the 5′ end by enzymes that belong to the XRN family of RNases (Nagarajan et al., 2013). There are two XRN proteins in humans. XRN1 is a cytoplasmic protein involved in bulk mRNA decay, a process that requires mRNA to be decapped by the DCP2/DCP1 complex. XRN2 is localized in the nucleus, but its function is less well described. The best-documented role of XRN2 is its involvement in the termination of RNAPII transcription termination (Eaton et al., 2018; Fong et al., 2015).
The 3′-to-5′ decay in humans involves the exosome complex or monomeric DIS3-like exonuclease 2 (DIS3L2) (Chlebowski et al., 2010, 2013; Lubas et al., 2013). The exosome is a large macromolecular assembly that is composed of the nine-subunit, ring-shaped, catalytically inactive core and differentially localized catalytic subunits that belong to the DIS3 and RRP6 protein families (DIS3, DIS3L, and EXOSC10/RRP6 in humans) (Chlebowski et al., 2013; Dziembowski et al., 2007; Lebreton et al., 2008; Tomecki et al., 2010). The RNA substrates of exosome complexes that contain DIS3 (nucleus) or DIS3L (cytoplasm) pass through the central channel of the exosome ring to reach the exoribonuclease active site (Drążkowska et al., 2013; Makino et al., 2013, 2015; Malet et al., 2010). Nuclear EXOSC10 is positioned at the top of the exosome ring (Wasmuth et al., 2014).
In simple and genetically tractable organisms, analyses of relationships between different pathways and the identification of novel factors and pathways are usually conducted through genetic screening. The development of high-throughput methods, such as synthetic genetic array (SGA) (Tong et al., 2001) or diploid-based synthetic lethality analysis on microarray (dSLAM) (Pan et al., 2007) for mapping and analyzing genetic interactions (GIs), was an important breakthrough for expanding our knowledge of GI networks on a genome-wide scale and broaden research possibilities in functional genomics. The yeast Saccharomyces cerevisiae is used for large-scale synthetic lethal screens due to its advantages in experimental ease. However, recent advances in gene silencing technology provided an experimental format for high-throughput GI mapping in higher organisms. This approach has already been used in a large number of studies of many basic cellular processes such as genome maintenance and the regulation of cell division (Kavanaugh et al., 2015; Mukherji et al., 2006; Paulsen et al., 2009; Sokolova et al., 2016; Stojic et al., 2020), but RNA metabolism has rarely been studied using this kind of approach, except nonsense-mediated decay (NMD), stress granule/P-body assembly, and ribosomal biogenesis (Badertscher et al., 2015; Ohn et al., 2008). To our knowledge, no studies have focused on such a fundamental process like RNA decay.
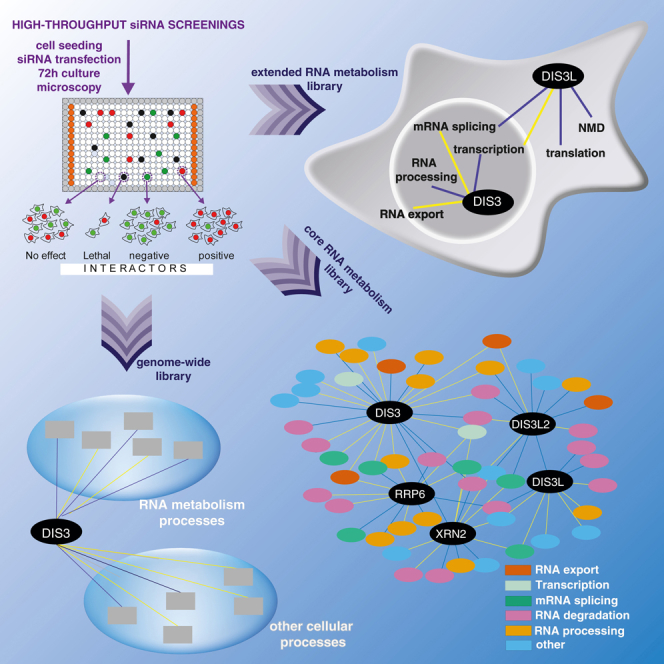
In the present study, we performed multiple high-throughput small interfering RNA (siRNA) screens using cells that were deficient in DIS3, DIS3L, DIS3L2, EXOSC10, and XRN2. All of the models were interrogated with a siRNA collection that targeted 280 genes that are involved in core RNA metabolism processes. We systematically analyzed genes that interact with query ribonucleases and comprise a network that gathers already known and newly identified functional associations, revealing the deep interplay between RNA decay and other RNA metabolism pathways. The observed unique GI profiles of the analyzed ribonucleases indicated that they play unique roles in RNA degradation. Shared GIs suggested some cooperation in the context of such processes as RNA splicing, nuclear export, and RNA transcription. Those findings prompted us to further elucidate the network of GIs for two of the mentioned ribonucleases, DIS3 and DIS3L, as they are both executors of exosome-mediated RNA degradation but within two different cellular compartments, nucleus and cytoplasm, respectively. This analysis was performed with a custom subset of a whole-genome siRNA library comprising genes related to RNA metabolism. These analyses revealed that DIS3 and DIS3L have divergent functions not only because of different cellular localization and substrate specificity but also at the level of functional associations. Nevertheless, we also observed some redundancy that corroborated RNA nuclear export. Notably, we found a deep interplay between cytoplasmic RNA degradation and nuclear RNA metabolism. Finally, we derived a network of all possible DIS3 interactions with cellular processes investigating the whole genome library excluding genes analyzed as extended RNA metabolism subset. Those results provide a unique overview of non-standard functional associations of DIS3 and offer a discovery tool that can be used to dissect the roles of DIS3 in biological processes beyond RNA decay.
Overall, the present study identified functional interactions of main mammalian ribonucleases with different cellular processes, thereby providing an expansive view of the interplay and coordinated action of different processes that buffer imbalances introduced by the dysfunction of RNA degradation.
Results and discussion
Design of high-throughput siRNA screening using competitive cell growth assay and stable cell lines expressing mutated versions of processive ribonucleases
We sought to identify functional interactions between proteins, pathways, and complexes involved in RNA metabolism based on high-throughput siRNA screens targeting the processive exoribonucleases DIS3, DIS3L, DIS3L2, EXOSC10, XRN1, and XRN2. All screens were based on a competitive growth assay in which the fitness of cells that expressed the mutated (MUT) version of the protein of interest was compared to that of cells that expressed wild type (WT) protein, an approach successfully used by us in previous projects (Szczesny et al., 2018; Tomecki et al., 2014). In brief, we exploited engineered human Flp-In 293 T-REx cell lines that, upon induction with tetracycline, simultaneously express a short hairpin RNA (shRNA) that silences an endogenous RNase gene and express an exogenous allele of that gene that is insensitive to the shRNA. Thus, an endogenous protein is replaced with a specific version that we choose. The construct also encodes a fluorescent protein that localizes to the nucleus, which serves as a shRNA expression marker (Figure 1A). For every RNase, we worked with two stable cell lines in which the endogenous protein was replaced with an exogenous overexpressed WT or MUT version, and that simultaneously expressed an enhanced green fluorescent protein (EGFP) or mCherry, respectively, so they could be cultured together and easily distinguished by fluorescence microscopy. The mutations we introduced completely inactivated DIS3L, DIS3L2, EXOSC10, and XRN2. In the case of DIS3, we introduced a mutation that was identified in multiple melanoma patients impairing but not destroying the exoribonucleolytic activity of the enzyme as that leads to cell death (Tomecki et al., 2014). Unfortunately, we were unsuccessful in generating cell lines with inducible dysfunction of XRN1; therefore, this ribonuclease was not further investigated in the present study.
Figure 1.

siRNA screens identified negative and positive genetic interactions of main mammalian processive ribonucleases
(A) Strategy to obtain a cellular model in which the endogenous copy of the analyzed gene (DIS3 – as an example, DIS3L, DIS3L2, XRN2, or EXOSC10) is replaced by a mutant version. Schematics of a HEK293 Flp-In T-Rex cell line-based model are shown, in which a vector was integrated that contained a bidirectional promoter that – upon tetracycline-mediated induction – drove expression of shRNA that silenced the endogenous version of the gene and WT or MUT sh-miRNA-insensitive exogenous gene variant.
(B) Schematic presentation of the screening procedure. HEK293 Flp-In T-Rex cells that expressed WT and MUT versions of the analyzed ribonuclease were co-cultured in 384-well plates and transfected in triplicate with a custom or genome-wide siRNA library. The number of green (WT) and red (MUT) cells was quantified by fluorescence microscopy. Subsequently, a custom computational workflow was used to calculate fitness scoring based on changes in red and green cell count balance. Two types of GIs were identified: (1) negative/exacerbating, and (2) positive/alleviating.
(C) Heatmap of genetic interactions of all of the analyzed ribonucleases, represented by summarized z∗ scores. The plots represent summarized z∗ scores for 81 of 280 tested genes that were identified as a significant hit for at least one query gene. Yellow represents positive hits. Blue represents negative hits. Black represents genes that had no interactions.
(D) Network diagram of identified genetic interactions between mammalian processive ribonucleases and known physical interactions. Black nodes indicate query ribonucleases. Pink nodes indicate genes that were identified as hits in the siRNA screen. White nodes indicate proteins that interacted with query proteins or screen hits according to the STRING database. Blue and yellow edges represent negative or positive genetic interactions, respectively. Gray edges represent known P-P interactions. Protein complexes that are known to interact with query ribonucleases are highlighted in colors on the net.
We performed the screen for each nuclease as illustrated in Figure 1B. Briefly, both cell lines were co-cultured in 384-well plates (for details, see STAR Methods) and transfected in triplicate with a custom siRNA library. As a quantified measure to explore GIs of query ribonucleases, we decided to use relative fitness. Therefore, we optimized the seeding conditions to have an equal number of cells expressing WT or MUT version of the ribonuclease after 72 hr of culture in control conditions. We measured the growth rate of cells that expressed the WT or MUT version of the ribonuclease using the live-cell imagining system, Incucyte. The analysis revealed that the seeding conditions are different, depending on the ribonuclease studied (Figure S1). Briefly, DIS3 MUT slows down the growth, which is in agreement with previous data (Tomecki et al., 2014). A decreased rate of proliferation was also observed for DIS3L. For the other ribonucleases, observable alterations in growth rate were not significant. Since our cellular model expressing mutated protein should be treated as a single mutation that limits cell growth substantially less than silencing of query gene with siRNA, it was suitable for measuring of GIs.
To quantify relative fitness scoring we calculated the relationship between the number of cells expressing WT and MUT version of the ribonuclease 72 hr after siRNA transfection. The number of green (WT) and red (MUT) cells was measured by fluorescence microscopy. To calculate fitness scoring based on changes in red and green fluorescence we used a custom computational workflow in which the relationship between the fitness of WT and MUT protein-expressing cells was expressed as robust z scores (z∗ scores) (for details see STAR Methods).
The GI definition can be generalized to any combination of genetic variants that deviates from the expected phenotype predicted by combining the effects that those genetic variants exert on phenotype alone. It is thus apparent that identification of GI depends on the prediction or calculation of an expected phenotype. Several mathematical models exist for estimating the expected phenotype and defining the GIs (Mani et al., 2008). However, the most popular method is the multiplicative model. We decided to use the Min mathematical function in which non-interacting genetic variants yield the expected phenotype in limiting cell growth on the level of a less fit single genetic variant. This model seemed suitable with our screening methodology as a cell line expressing a mutated version of a protein that was not exerting significant growth disturbances was screened with a library of siRNA that was expected to impact cell growth severely. Thus, based on alterations in relative fitness, we describe two types of GIs: (1) negative/aggravating, in which the siRNA-mediated silencing of a target gene resulted in the specific growth inhibition or death of cells that expressed MUT protein, and (2) positive/alleviating, in which the siRNA-mediated silencing of a target gene resulted in favoring the survival of cells that expressed MUT protein.
Depletion of nuclear exosome cofactors is synthetically lethal with exosome dysfunction, whereas cytoplasmic cofactors had the opposite effect
All of the cell lines were interrogated with a library of siRNAs that targeted 280 preselected genes (Figure 1B) involved in various aspects of RNA metabolism (e.g., polyadenylation, transcription, translation, nuclear import/export factors, and some confirmed or predicted nucleases; Table S1: Tab.1).
After screening all of the processive nucleases, we found 111 strong GIs that involved 81 of 280 target genes in the library (Figure 1C; Table S2). Among the identified GIs we found well-established physical interactors of the analyzed ribonucleases (Figure 1D). As expected for EXOSC10 and DIS3, a component of the exosome core (EXOSC3) was identified as a negative GI (Figure 1D). C1D, a protein that is well known to interact with the exosome (Costello et al., 2011; Garland et al., 2013; Schilders et al., 2007), was also classified as a hit for DIS3 and EXOSC10. Furthermore, a component of the protein complexes known to work as exosome activators the RNA helicase MTR4 encoded by the MTREX gene, was identified as a negative interactor with DIS3 and EXOSC10. MTR4 plays an essential role in all aspects of nuclear exosome functions by targeting various RNA substrates. This protein is part of the nucleolar-localized Trf4/Air2/Mtr4p Polyadenylation (TRAMP) and nuclear exosome targeting (NEXT) complexes (Lubas et al., 2011; Meola et al., 2016; Puno and Lima, 2018; Weick et al., 2018), As a part of TRAMP complex, MTR4 is involved in rRNA processing, in which EXOSC10 plays a crucial role as the exosome’s executive nuclease. As a part of the NEXT complex, it is responsible for recruiting a broader group of exosome substrates, such as PROMPTs, snRNAs, and pre-mRNAs, which DIS3 degrades in the nucleoplasm.
The fact that identified GIs included genes which products directly interact with the examined protein or work in the same complex was expected. These results indicate that there are true positive hits among identified GIs, which motivated us to investigate the rest of the observed GIs in depth.
Curiously, initial analysis of identified GIs indicated unusual compensation or cooperation possibilities in crosstalk between RNA decay pathways. For example, among the genes that aggravated the XRN2 mutation, we identified genes that work as exosome cofactors (RBM7 and C1D), implying that the exosome may work redundantly with XRN2 in the nuclear degradation of some RNA species. XRN2, similar to the exosome, is involved in the rRNA maturation process by processing 5′ ends and removing excised spacers (Preti et al., 2013; Sloan et al., 2013; Wang and Pestov, 2011). The processing of 5′ ends by XRN2 provides a quality check to decide whether the pre-rRNA should be further processed or degraded (Sloan et al., 2013; Wang and Pestov, 2011). A yeast homolog of C1D, Rrp74p, was shown to be required for exosome-mediated rRNA processing (Mitchell et al., 2003). This indicates the redundancy of 5′ and -3′ nuclear processing pathways for some stable RNAs is possible.
Interestingly, two superkiller (SKI) complex components (TTC37 and HBS1L), were found to be DIS3 mutation suppressors (Figure 1D). The SKI complex is a major cytoplasmic exosome cofactor that cooperates with DIS3L in cytoplasmic mRNA decay (Halbach et al., 2013; Schmidt et al., 2016). Similar to DIS3-SKI interactions, we tracked down another example of the positive interplay between cytoplasmic and nuclear RNA decay. We observed that the depletion of WDR61, another component of the SKI complex, has a suppressive effect on XRN2 mutations, another major ribonuclease in the nucleus. It is known that many genes involved in such suppressive interactions have close functional relationships (Leeuwen et al., 2016). Thus, this complex network of interactions implies that nuclear degradation may somehow counterbalance cytoplasmic RNA degradation pathways.
We sought to confirm observed suppressive effect phenotypes of mentioned SKI complex components depletion in cells expressing mutated nuclear exoribonucleases. As those interactions were never reported, their validation could corroborate the screen findings. Considering that our initial analysis in the screen consisted of scoring the fitness of both cells expressing WT or MUT version of examined ribonuclease after depletion of query gene, our validation method was to perform real-time cell proliferation assay. Briefly, the growth rates of cells that expressed the WT and MUT version of DIS3 or XRN2, transfected with siRNA targeting TTC37 and HBS1L or WDR61, respectively, were measured using the live-cell imagining system Incucyte (for details see STAR Methods). The main limitation of cell proliferation assays is that the cells should be in their exponential proliferation phase for meaningful results. However, in our set of experiments, we observed that treatment of cells with tested siRNAs influence the growth rate considerably such that 54 hr after transfection cells treated with siTTC37 and siHBS1L grew in a logarithmic fashion (Figures S2A and S2B), while cells treated with siWDR61 stayed in the linear phase of growth (Figure S2C). So, we concluded that we could not compare the growth rates in a meaningful way. To better reproduce conditions observed in the screen, we examined the number of WT and MUT cells after 72 hr of transfection. The most observable change was for siTTC37, confirming the suppressive effect observed in the primary screening. Alike in the screening, depletion of TTC37 in cells expressing WT DIS3 had a prominent impact on cell growth. After 72 hr after transfection, the number of cells is reduced to 60% of that of cells treated with control siRNA (Figures S2D and S2G). According to the positive GI definition in the minimal model, we observed that depletion of TTC37 is less severe in cells expressing MUT DIS3. The suppressive effect was also noticeable for silencing HBS1L (Figure S2E). Unfortunately, validation of the suppressive effect of WDR61 depletion on mutation in XRN2 was unsuccessful as both cell lines expressed MUT XRN2 and WT XRN2 growth at the same rate and had similar cell counts after 72 hr after transfection (Figure S2F). To exclude the possibility that the knockdown of query genes was insufficient, gene silencing was confirmed by RT-qPCR and Western blot analyses (Figure S3). The lack of validation of XRN2 and WDR61 GI had two possible explanations: (1) for the validation, we used different pools of siRNAs than we used in primary screening, so the effect observed in screening could be an off-target effect that is a known bias in RNAi screening; (2) as we applied a rather loose hit selection cutoff to favor false-positive hits over false-negative (see Statistical analysis and hit selection chapter in STAR Methods).
In conclusion, the custom single-plate screenings for DIS3, DIS3L, DIS3L2, EXOSC10, and XRN2 revealed biologically relevant functional interactions, although some of the hits may represent false positives.
Comparisons with yeast genetic screens revealed functional convergence of exosome executive ribonucleases
The structural and biochemical characterization of main executors of RNA processing, degradation, and turnover, including mechanistic details, were thoughtfully investigated, but their GIs have not been systematically explored previously. To date, the only knowledge of GIs has been available for yeast exosome-related ribonucleases (Dis3 and Rrp6) and can only be extracted from global genetic interaction networks for nonessential and essential budding yeast genes (Costanzo et al., 2016; Leeuwen et al., 2016; Usaj et al., 2017). Our first observation from the yeast screen was that Dis3 and Rrp6 GIs do not overlap in most cases, as observed in human cells (Figures 2A and 2B). We needed to create a list of human to yeast orthologs to dissect overlap between results from human and yeast screens for DIS3/Dis3 and EXOSC10/Rrp6. Notably, humans and yeast differ dramatically concerning cell organization and metabolism. Nevertheless, thousands of human genes have a trackable ortholog in yeast (Remm et al., 2001). For 280 genes tested in the core metabolism screen, we identified 190 pairs of orthologs (Table S3: Tab1). Curiously, the overlap between GIs for human and yeast exosome executor ribonucleases is minimal, as we were able to identify only individual overlapping hits (Figures 2C and 2D). Surprised by those observations, we performed GO enrichment analysis on human and yeast sets of hits. We concluded that although different as individual interactions, identified hits are mainly involved in RNA processing globally. The lack of overlap on the level of individual interactions but, at the same time, convergence on the functional level indicates the importance of performing screenings on each organism separately. Despite a high degree of conservation of human and yeast proteins and functional analogy, the compensations on GIs level may be achieved through different mechanisms, involving different players but with a similar final effect. Among many others, lineage-specific duplication of genes in both organisms and divergent roles of paralogs (like in humans for DIS3 and DIS3L) may explain different functional interaction networks.
Figure 2.

Limited overlap between yeast SGA screen for Dis3 and Rrp6 and human siRNA screenings on core RNA metabolism gene set
(A and B) Venn diagrams representation of overlap between genes that were identified as negative and positive genetic interactions for Dis3/DIS3 and Rrp6/EXOSC10 in yeast SGA (SC) (A) and human (HS) siRNA screen for core RNA metabolism gene set (B).
(C and D) Heatmap representation of genes that were identified as genetic interactions for Dis3/DIS3 (C) and Rrp6/EXOSC10 (D) in yeast SGA (SC) and human (HS) siRNA screen for core RNA metabolism gene set. The colors represent the strength of the hit score. Yellow represents positive hits. Blue represents negative hits. Black represents genes with no interactions, whereas gray means that the gene was not analyzed in this screen.
See also Table S3.
Limited overlap between genetic interactions of the main RNA degrading enzymes
Next, we compared GIs for DIS3, DIS3L, DIS3L2, EXOSC10, and XRN2 to gain further insights into the genetic interaction network in humans (Figure S4). Notably, they tended to have nonoverlapping GIs, suggesting distinct properties associated with their loss of function. We did not identify any gene that showed a genetic interaction with all five enzymes. However, a few genes interacted with three ribonucleases.
We identified five overlapping GIs (6% of 81 hits) for three ribonucleases (Figures S4A and S4B). The alpha subunit of RNAPII (POLR2A) displayed a strong positive GI with all nuclear ribonucleases (DIS3, EXOSC10, and XRN2), suggesting that the inhibition of transcription can counterbalance dysfunctional RNA decay. The effect was more pronounced for nuclear RNases because cytoplasmic DIS3L and DIS3L2 did not interact with POLR2A, although we did not study such interactions for XRN1.
Another very strong positive hit was DDX19B, a suppressor of DIS3, DIS3L, and DIS3L2 (Figure S4B). DDX19B is a highly conserved DEAD-box adenosine triphosphatase (ATPase) orthologous to yeast Dbp5p, an essential component of mRNA export machinery. Interestingly, Dbp5p works as an RNPase, rather than an RNA helicase promoting dissociation of Mex67p from mRNPs at the nuclear periphery (Lund and Guthrie, 2005; Tran et al., 2007). Consequently, disruption of the nuclear export process tends to have a suppressive effect on exosome-mediated alterations of RNA degradation. Intriguingly, the depletion of DDX19B can alleviate degradation defects mediated by all three DIS3 human paralogs, implying that paralog cooperativity might exist in some contexts.
Three additional genes, C1D, DDX56, and SNRPD3, were identified as interactors three times in our screen (Figures S4B, S4D, and S4E). However, these interactions were much more complex, without a tendency to be synthetically lethal or suppressing with a specific subset of ribonucleases. For example, SNRPD3 was synthetically lethal with mutated DIS3L but was a suppressor of mutations of EXOSC10 and XRN2.
Twenty genes had GIs with two nucleases, and the other 69% of the genes identified as hits in our screens were unique GIs (Figures S4A and S4C). Such a high number of uncommon GIs suggest unique functions of each of the examined ribonucleases, which is consistent with distinct substrate specificity (Davidson et al., 2012, 2019; Lubas et al., 2013; Staals et al., 2010; Szczepińska et al., 2015; Tomecki et al., 2010; Ustianenko et al., 2013), explained by their different modes of substrate selection, catalytic activity, and cellular compartmentalization in mammalian cells.
From the perspective of interrogated ribonucleases, the highest number of common interactors (eight) was observed between DIS3 and EXOSC10. This was unsurprising because these nucleases share the same core complex, are present in the closest cellular compartments, and have redundant functions in some contexts.
Reconstruction of a functional network revealed connections between RNA metabolic pathways
To further reinforce the implications of the identified GIs with functions of each ribonuclease, we performed Gene Ontology (GO) enrichment analysis.
DIS3 negative GIs showed significant enrichment of the rRNA processing term (Figure 3A; Table S4: Tab1), which is consistent with the description of the individual GIs mentioned above. In agreement with the cellular function of DIS3, two other biological processes related to rRNA processing were enriched: exonucleolytic trimming to generate the mature 3′-end of 5.8S rRNA from the tricistronic rRNA transcript and the maturation of 5.8S rRNA. Another significantly enriched term was nuclear-transcribed mRNA poly(A) tail shortening. Interestingly, genes that are ascribed to this biological process are a part of the CCR4-NOT complex (CNOT10 and CNOT4), and the third one (ZFP36) plays a role in recruiting CCR4 to this complex. CCR4-NOT is the major deadenylase that rapidly removes poly(A) tails from mRNAs that are to be degraded (Azzouz et al., 2009; Łabno et al., 2016). The exosome with DIS3 is the effector of this degradation pathway, so this GO term enrichment is consistent with established functional interactions of DIS3. Moreover, one of the GO terms enriched with negative interactors of DIS3 was gene silencing by RNA, which was enriched by three genes: TSNAX, CNOT10 and PRKRA. This was also consistent with known cellular functions of the exosome, which degrades the 5′ moiety of mRNAs that are cleaved by the RNA-induced silencing (RISC) complex during RNA interference in plants (Huntzinger and Izaurralde, 2011).
Figure 3.

Functional analysis of negative genetic interactions revealed connections of mammalian ribonucleases with RNA metabolic pathways
(A) Biological processes associated with genes that are synthetically lethal with mutations in DIS3, DIS3L2 and EXOSC10. Bar plots represent the number of identified negative hits associated with biological processes that were analyzed with the DAVID direct GO database.
(B–D) Functional interaction networks of DIS3 (B), DIS3L2 (C), and EXOSC10 (D) negative genetic interactors. Networks were constructed in R/Bioconductor based on the association of hit genes with biological processes according to the DAVID direct GO database. The colored rectangular labels show names of biological processes that are connected with gray edges to genes that are associated with them. No bar plots or network diagrams are shown for EXOSC10 and XRN2 because enrichment analyses of biological processes were non-significant.
See also Table S4.
Likewise, the GO enrichment analysis of negative interactors of EXOSC10 indicated that they are largely (four of nine) involved in rRNA processing (Figure 3A; Table S4: Tab7), consistent with the primary function of EXOSC10 in mammalian cells because it mainly resides in the nucleolus where the maturation of rRNA molecules occurs. Interestingly, the most enriched term was mRNA splicing via spliceosome, with six genes annotated to it: SNRNP200, EIF4A3, PNN, SNRPD1, SKIV2L2, and DDX16. EXOSC10 in budding yeast retains and degrades defective transcripts in the nucleus (Hilleren et al., 2001). However, the role of EXOSC10 in the surveillance of mRNA in mammalian cells has not been fully established. This suggests that EXOSC10 may play a role in cotranscriptional quality control in human cells, much like in yeast cells.
The GO enrichment analysis of negative interactors of DIS3L did not yield any significant terms because there were only four genes that were identified, and all of them are connected with RNA degradation. Likewise, GO enrichment analysis of XRN2 negative interactions did not yield any enriched biological processes.
Concerning negative GIs of DIS3L2, three biological processes were significantly enriched: histone mRNA metabolic process, termination of RNA polymerase II transcription, and gene silencing by RNA (Figure 3A; Table S4: Tab5). The enrichment of gene silencing by RNA and histone mRNA metabolic processes is consistent with known functions of DIS3L2 (Mullen and Marzluff, 2008). Enrichment of the termination of RNA polymerase II transcription term initially appeared surprising because DIS3L2 is a cytoplasmic protein, but genes that are ascribed to this process are also associated with the histone mRNA metabolic process term (Figure 3C). Notably, those genes are also connected with terms that are related to RNA splicing and mRNA export from the nucleus. These findings indicate that genes that are synthetic lethal with mutated DIS3L2 are involved in highly conected processes. Thus, speculating about which of these processes rely heavily on DIS3L2 to create strong GIs that were observed is difficult.
During mapping GIs, more emphasis is usually given to GIs that more severely decrease cell fitness than to suppression interactions. The reason for this may be the difficulty in explaining the underlying mechanisms between these types of interactions. However, Leeuwen et al. studied global-scale genetic suppression interactions in budding yeast and classified them into mechanistic categories (Leeuwen et al., 2016). They showed that a substantial part of positive GIs can be explained by already known functional interactions, such as when query genes and suppressors are from the same complex or the same or alternative pathway. However, ∼50% of cases of suppression interactions are with unknown, apparently functional relationships, thus providing a good source of compensatory pathway identification. The analysis of positive GIs identified by us in the context of biological processes revealed important interactions of RNA degradation in a broad biological context as positive hits are not directly involved in RNA degradation but rather other RNA metabolism processes, such as transcription, pre-mRNA splicing, and nuclear export.
Distinct functional interactions were specific to nuclear and cytoplasmic exosome
DIS3 and DIS3L are close paralogs with similar enzymatic activities sharing the same core complex essential for their function, suggesting at least partial redundancy. However, they have different localization between the nucleus and cytoplasm, respectively. Such compartmentalization limits substrate availability and may also determine different interactions with biological processes. DIS3 and DIS3L likely perform divergent functions in the cell, consistent with our preliminary screens that revealed almost no overlap in GIs between DIS3 and DIS3L. To elucidate this issue and determine the biological processes affected by the loss of function of these two genes, we again performed a high-throughput siRNA screen using the same system but expanded the scope of the screen. We used a custom set of 3904 siRNAs that targeted genes whose products were directly or indirectly related to RNA metabolism (see STAR Methods section for details; and Table S1: Tab2).
We identified 233 and 137 GIs specific to DIS3 and DIS3L, respectively, demonstrating that these nucleases play largely distinct roles (Figure S5A). Only 15 genes interacted positively with DIS3 and DIS3L, indicating that processes in which these enzymes work cooperatively are exceptions and not the rule. Moreover, 25 interactions were common for both nucleases but were suppressors of DIS3 mutation and synthetic lethal with MUT DIS3L. The complete list of GIs is provided in Table S5.
We analyzed 66 negative GIs specific to DIS3 concerning biological processes but with hardly any meaningful enrichment. First, there were very few genes with enriched GO terms with no statistical significance (Figure 4A; Table S6: Tab1). Second, these terms were not connected with one another, and the relationship between them and DIS3 is unclear (Figure 4A). A small set of genes implicated in rRNA processing (EXOSC6, PES1, NCL, RPL6, and DIMT1) is the exception. The limited enrichment of other than rRNA processing processes or pathways and a low number of negative GIs for DIS3 indicates that this enzyme is indispensable in its role. The importance of DIS3 as one of the main RNA degrading enzymes is confirmed by the fact that DIS3 is an essential gene in all model systems that have been studied to date and indicates that its activity cannot be replaced even by the other nuclear exosome catalytic subunit, EXOSC10.
Figure 4.

Distinct functional crosstalk between the nuclear or cytoplasmic exosome and RNA metabolism
(A–D) Functional classification of negative (A and B) and positive (C and D) genetic interactions between DIS3 (A and C) or DIS3L (B and D) and RNA metabolism genes. Bar plots represent enrichment (expressed as gene count) among the top 10 biological processes (ordered by Fischer’s exact p value). The enrichment analysis was performed based on biological processes in the DAVID direct GO database. Networks illustrate connections between biological ontologies. The colored nodes (blue and yellow) represent biological processes that are interconnected with genes associated with them in GO. The networks were constructed in R/Bioconductor based on the association of hit genes with biological processes according to the DAVID direct GO database.
See also Table S6.
In contrast, the negative GIs of DIS3L fall into very specific GO categories (Figure 4B; Table S6: Tab3). mRNA splicing and translation were among the highly enriched terms. The cytoplasmic exosome complex is involved in translation-dependent RNA surveillance pathways (Schaeffer et al., 2011; Tuck et al., 2020). Therefore, the enrichment of these GO terms corroborates its functions in RNA metabolism. Most of the enriched genes implicated in the translation process are ribosome complex constituents from small and large ribosome subunits. Genes enriched in the splicing category are constituents of the spliceosome from the second step of splicing and the U4/U6 x U5 snRNP complex. A high number of genes involved in splicing identified as synthetic lethal with MUT DIS3L suggests that the cytoplasmic degradation pathway by the exosome may somehow compensate for alterations of the mRNA maturation process. Notably, among negative GIs of DIS3L were genes involved in RNA transport from the nucleus (THOC2, ALYREF, NUP133, POP1, POP5, and EIF2S2). THOC2 and ALYREF are a part of the TREX complex (Chi et al., 2013), whereas NUP133 is a part of the nuclear pore (Boehmer et al., 2008), and all are involved in mRNP biogenesis and mRNA export (Fan et al., 2019; Vasu et al., 2001). These steps are subject to robust quality control. These results affirm the possibility that cytoplasmic degradation may compensate for the deregulation of nuclear mRNA metabolism.
Among the positive GIs were 15 that were common between DIS3 and DIS3L. Within this group, we found two interesting categories: (1) genes that are involved in RNA export from the nucleus that are part of the nuclear pore complex (RAN, TPR and NUP153) and (2) subunits of the RNA polymerase complex (POLR1C, POLR2G, POLR2H, and POLR2L).
Both paralogs interacted with RNA polymerases I and II, but DIS3 showed a more potent genetic interaction with RNA synthesis machinery, as almost all subunits of RNAPII (POLR2A, POLR2B, POLR2D, POLR2H, POLR2G, POLR2F, POLR2L, and POLR2I), one subunit of RNAPI (POLR1C), and one subunit of RNAPIII (POLR3K) were identified as positive GIs.
Remarkably, positive GIs that are specific to DIS3 fall into the most diverse GO categories. The highest number of genes were enriched in categories connected to mRNA splicing (Figure 4C; Table S6: Tab2). In contrast, for DIS3L, more GIs implicated in the splicing process were negative hits. However, among suppressors the mRNA splicing was also enriched (Figure 4D; Table S6: Tab4). Markedly, distinct sets of genes were enriched in the splicing process for DIS3 and DIS3L. One commonality was that most of them were constituents of the catalytic step 2 spliceosome.
Altogether, these results enclose that DIS3 and DIS3L possess unique functions in RNA degradation. Thus, it is an example of functional divergence, particularly subfunctionalization, in which both paralogs are separately specialized in several ancestral functions.
DIS3 dysfunction suppressed alterations of alternative splicing in cells with the depletion of U2 snRNP component
We were intrigued that genes implicated in splicing were the most abundant ones among MUT DIS3 suppressors. Moreover, the fact that constituents of U2 snRNP and U5 snRNP were the most enriched group among positive hits suggests that DIS3 dysfunction leads to alterations of splice site recognition. These results raised the possibility that the interplay is between alterations of splicing and the dysfunction of DIS3. Thus, we decided to dissect the effect of the depletion of one of the strongest positive hits. Among these were components of the splicing factor 3A subunit 1 (SF3a) complex and SF3b complex. The SF3a complex is an integral part of the pre-spliceosome and is required for complex A and B assembly (Brosi et al., 1993). SF3a interacts with U1 and U2 snoRNAs and pre-mRNA and plays a crucial role in initial steps of spliceosome assembly (Bertram et al., 2017; Fica and Nagai, 2017; Lardelli et al., 2010; Tanackovic and Krämer, 2005). We used one of its subunits (SF3A1) to study the interplay between the exosome and splicing. We examined how the dysfunction of DIS3 affects splicing after SF3A1 depletion.
To profile changes of alternative splicing (AS) patterns, we performed total RNA sequencing (RNA-Seq) under four conditions: Cells that expressed either WT DIS3 or the mutated version MUT DIS3 that were treated with either siRNA targeting the SF3A1 gene (siSF3A1) or control siRNA (siCTRL). To compare profiles of AS events, we employed JUM pipeline (Wang and Rio, 2018) because it allows the identification and estimation of global splicing patterns and differential analysis by combining many experimental conditions and considering biological replicates. JUM handles RNA-Seq reads that are mapped to splice junctions and first defines alternatively spliced junctions, assembles them into commonly known AS events (alternative 5′ splice site [A5SS], alternative 3′ splice site [A3SS], cassette exon [SE], mutually exclusive exon [MXE], and intron retention [IR]), and creates an additional separate category for complex AS isoforms (MIX). Expression levels of each identified AS are quantified by computing the change in percent spliced isoform (ΔPSI). Differential inclusion is usually restricted for |ΔPSI| > 0.1. Significantly different AS events can be identified by applying statistical tests (see STAR Methods). Based on this approach, we were able to identify condition-specific splice junction usage and check whether there are any significant differences in AS event frequencies between conditions.
The analysis revealed that SF3A1 depletion led to extensive alterations of multiple classes of AS events (Figure 5A). Importantly, significantly deferentially spliced events were not distributed equally between classes: SE was notable with 460 events compared with 180 events for IR, 151 events for A3SS, 147 events for A5SS, and 106 events for other classes.
Figure 5.

Suppression of alterations of alternative splicing that are caused by the depletion of a U2 snRNP component in the presence of DIS3 dysfunction
(A) Quantitative comparison of AS events with significant alterations of splicing patterns after SF3A1 depletion in cells that expressed WT DIS3 or MUT DIS3 (deferentially spliced events with |ΔPSI| > 0.1 and FDR <0.05). Analyzed AS events: alternative 5′ splice site (A5SS), alternative 3' splice site (A3SS), cassette exon (SE), mutually exclusive exon (MXE), intron retention (IR), and complex AS isoforms (MIX).
(B–D) Volcano plots representing the direction and significance of retained intron changes analyzed with JUM. Every dot is an identified IR event. Red dots are IR events that were significantly changed between SF3A1 depletion and control conditions (|ΔPSI| > 0.1 and FDR <0.05).
(E)ΔPSI violin plots of the distribution of calculated retained isoforms of introns that were significantly alternatively spliced under SF3A1 depletion conditions in a WT DIS3 background across other analyzed conditions (i.e., in cells that expressed MUT DIS3 and when comparing splicing between WT DIS3 and MUT DIS3 in siRNA control transfection [siCTRL] conditions).
(F) Gene Ontology enrichment analysis of biological process terms of JUM-identified transcripts with retained introns after SF3A1 depletion. Bar plots represent the number of identified genes that were associated with biological processes, analyzed with the DAVID direct GO database. The bar color code shows statistical significance, colored according to the value of negatively log10-transformed Fischer’s exact p value.
(G and I) Genome browser screen shots of genes that showed medium and high intron retention in cells transfected with SF3A1 siRNA SLC25A3 (G) and MRPL52 (I), respectively. Genes are shown in proximity to show the retained intron.
(H and J) RT-PCR validation of two genes that showed medium and high intron retention in cells transfected with SF3A1 siRNA, SLC25A3 (H), and MRPL52 (J), respectively. The intron retention level was measured in cells expressing WT or MUT version of DIS3 and transfected wit siRNA for SF3A1 or control siRNA for 72 hr. The rest of genes that were analyzed in the validation of intron retention are shown in Figure S6.
See also Figure S6.
Remarkably, DIS3 dysfunction strongly altered the effect of SF3A1 silencing on only two classes of AS events: IR and A5SS. Among 591 of the identified IR events, SF3A1 depletion in the presence of WT DIS3 significantly altered 180 introns, and 169 of these changes were more retained introns, meaning that retention frequency was higher than under control conditions (WT DIS3 cells treated with siCTRL) (Figure 5B). In the presence of MUT DIS3, only 78 IR events were significantly altered (Figure 5C), whereas mutation in DIS3 alone had no influence on intron retention (Figure 5D). Notably, for some of the introns SF3A1 depletion caused retention with a ΔPSI < −0.5 and median around −0.35 in the presence of WT DIS3 (Figure 5E). In the presence of MUT DIS3, the median ΔPSI was not below −0.25. More detailed analysis revealed that more than 30% of retained introns were the first intron of the transcript. Further evaluation, however, did not show any correlation between the presence of these introns in the 5′untranslated region (UTR) or coding sequence (CDS).
To verify the RNA-Seq results, we performed RT-PCR to detect accumulation of spliced and unspliced mRNA for four genes that sowed an increase in intron retention under SF3A1 depletion (SLC25A3, MRPL52, MRPS34, MRPS12) from high to a low level. We also included a test for genes that did not increase intron retention (GAPDH) and intronless gene (H2A17) for loading control. To detect both spliced and unspliced isoforms of the transcript, we designed primers for two adjacent exons that were separated by a retained intron (for details see STAR Methods). As the retained introns were relatively short, the spliced and unspliced isoform could be detected during one electrophoresis separation of RT-PCR products. As shown in Figures 5H and 5J we were able to detect unspliced isoforms of SLC25A3 and MRPL52 that showed moderate and high accumulation of retained intron in RNA-Seq experiment, respectively (Figures 5G and 5I). For genes MRPS12 and MRPS12 we were unable to detect isoforms with a retained intron (Figures S6A and S6B). It should be noted that the PSI was on a relatively low level for both of those genes. Moreover, the spliced isoforms were also accumulated on low levels, which suggests that the detection of those RNAs could be out of detection range for RT-PCR experiments. Unfortunately, because of the short and GC-rich sequence of those gene introns, we were not able to design primers for RT-qPCR experiments that are much more sensitive and could also be used in validation for intron retention.
To investigate the biological processes in which the transcripts that retained their introns may be implicated, we performed GO enrichment analysis. Interestingly, translation was the most enriched GO term (Figure 5F). The other significantly enriched terms were also related to translation. The precise examination of genes that were enriched in these GO terms revealed that they encode ribosomal proteins (RPs): RPL10, RPL10A, RPL24, RPL41, RPL27A, MRPL52, MRPS12, MRPS24, and MRPS34.
This suggests that DIS3 dysfunction alleviates SF3A1 depletion by rescuing appropriate isoforms of transcripts that encode ribosomal proteins, which in turn may influence translation and amend the balance between RNA processing, RNA degradation, and translation and help maintain cell homeostasis.
Several conclusions emerged from this experiment. First, SF3A1 depletion does not dysregulate splicing globally but rather leads to specific AS alterations, which is consistent with previous reports. The knockdown of individual splicing factors has noticeable but very distinct effects on AS (Papasaikas et al., 2015). Second, DIS3 dysfunction suppresses alterations of splice site selection upon SF3A1 depletion but only in IR and A5SS classes of alternative splicing, suggesting a particular mechanism of action rather than a global one. Third, in SF3A1-undepleted cells, the AS pattern was not significantly altered in MUT DIS3 compared with WT DIS3, and we did not observe the accumulation of specific pre-mRNAs.
In conclusion, our data indicate that DIS3 dysfunction alleviates the effects of SF3A1 depletion on a very specific subset of genes, partially explaining the suppressor interaction observed in the genetic screen. Further investigations are necessary to decipher the molecular mechanisms that determine why this particular subset of genes targets the DIS3-SF3A1 interaction. We can speculate that slowing down the nuclear RNA surveillance by DIS3 mutation allows for the execution of splicing events leading to the production of mRNA, which otherwise would be degraded.
Genome-wide siRNA screen of DIS3 identified important interactions of the nuclear exosome with processes that are involved in maintaining cell homeostasis
DIS3 is engaged in an interplay with different cellular pathways, and point mutations affecting its exoribonucleolytic activity are linked to multiple myeloma (Chapman et al., 2011; Tomecki et al., 2014). This prompted us to analyze GIs of DIS3 again, this time not only among genes connected to RNA metabolism, but the whole human genome. We used a siRNA library covering most human genes (Dharmacon ON-TARGETplus siRNA SMARTpool; 18,104 targets). Analysis of a subset of this library, including genes associated with RNA metabolism library (described in detail in section: distinct functional interactions were specific to nuclear and cytoplasmic exosome) revealed 274 GIs for DIS3 and extending the list of genes to whole-genome increased the number of observed interactions to 1112. Although substantial, the number is significantly lower than could have been expected given the size of the library. The complete list of GIs is provided in Table S7.
Functional analysis of the entire collection of genes that genetically interact with DIS3 revealed that the most enriched categories are consistent with the analysis of GIs identified with the custom RNA metabolism library (Figures 6A and 6B; Table S8: Tab1 and Tab2). Notably, the functional categories that were most extensive and represented at significantly higher frequencies than expected by chance were again connected with mRNA splicing, the regulation of RNAPII transcription, and mRNA export from the nucleus (Figure 6B). Nevertheless, we investigated whether genes that genetically interact with DIS3 are associated with biological processes other than RNA metabolism. We reevaluated the functional categorization of DIS3 GIs, excluding genes that were previously examined. The GO analysis of these positive and negative genetic interactors revealed that they fall into a very diverse group of functional categories that are important for cell maintenance (Figures 6A, 6B andS7). Importantly, the detailed examination of enriched categories revealed that they are enriched by a considerably low number of genes, meaning that genes are rather uniformly distributed among functional categories, with no category that is represented by a significant number of genes (Figures 6A and 6B). Analyses of the relationships between these genes did not yield a highly connected interaction network, which was in contrast to GIs that were identified among genes that are involved in extended RNA metabolism (Figure S7). Therefore, we subsequently performed an analysis of functional connections among enriched biological processes and cellular compartments. These analyses yielded some overrepresented functional categories that grouped together (Figures S8 and S9). These functional categories point to processes in which DIS3 may play an important role. We do not discuss all of these interactions in detail but highlight those that can provide essential information for further studies of the role of DIS3 in diverse biological processes.
Figure 6.

Genetic interactions between the nuclear exosome with genes that are involved in the maintenance of cell homeostasis
(A and B) Comparisons of biological processes associated with genetic interactions of DIS3 identified in the genome-wide screen, extended RNA metabolism screen, and hits from genome-wide subset, excluding hits identified in the extended RNA metabolism screen. Bar plots represent the number of identified hits that were associated with biological processes according to the enrichment analysis using the DAVID direct GO database. The fill color of the bar plots represents Fisher’s exact p value. Enrichment analyses were performed separately for (A) synthetic lethal interactions and (B) suppressors.
(C) Functional connections of synthetic lethal interactors and suppressors of DIS3 that are associated with microtubules. The hits of enriched cellular compartment categories that were connected to microtubules were extracted from the enrichment results using the DAVID direct GO database and visualized in Cytoscape as a network in which edges represent association of genes (blue nodes for synthetic lethal interactions and yellow nodes for suppressors) with functional categories (pink nodes).
See also Figures S7–S9, and Tables S7 and S8.
We used DIS3 with a mutation in the RNB domain that was reported to be present in MM patients. We sought to identify new GIs of DIS3 that could be relevant to MM. Of the genes that are reportedly involved in MM, only three had significant differential effects on cell viability in our screen: IRF4, CCND1, and MAFB. IRF4 encodes a transcription factor that is involved in the interferon response. It is often upregulated in MM because of t(6; 14) translocation and has been described as required for MM survival (Schaeffer et al., 2009). The silencing of this gene decreased the viability of cells that had the DIS3G766R mutation. CCND1 encodes a cyclin that is involved in G1/S progression, which is positively regulated by and interacts with RB1. It is frequently mutated in MM (Lawrence et al., 2013). The silencing of CCND1 confers an advantage for cells with MUT DIS3, which corroborates data in the literature. MAFB encodes a transcription factor that is involved in hematopoiesis. It is upregulated in MM cases with t (14; 20) translocation (Prideaux et al., 2014). The silencing of this gene negatively affected all of the cells, and cells that expressed WT DIS3 were affected more severely.
Additionally, we looked deeper into genes that to date have not been associated with MM. Among negative GIs of DIS3, we found genes that are involved in the regulation of telomerase activity (CCT2, NVL and TCP1) (Figure 6A; Table S8: Tab3). This was surprising because previous studies indicated that knockdown of the exosome rescued defects in telomerase activity and suggested that the exosome is a target for specific telomerase pathologies (Shukla et al., 2016). This inconsistency could be attributable to the fact that the exosome is involved in the degradation of the telomerase RNA component (hTR) (Gable et al., 2019; Nguyen et al., 2015; Tseng et al., 2015, 2018), and rescue by exosome inactivation restored defective telomerase activity through lower levels of hTR (Boyraz et al., 2016). In contrast, genes that were identified in our screen participate in the assembly of the telomerase holoenzyme. NVL can regulate telomerase assembly via its interaction with hTR (Her and Chung, 2012), whereas CCT2 and TCP1 are part of the TRiC complex required for TCAB1 protein folding (Freund et al., 2014). The depletion of TRiC subunits led to the mislocalization of hTR to nuclei and not to its global reduction (Chen et al., 2020). Moreover, hTR overexpression cannot compensate for TRiC or TABC1 loss. Although this explains why DIS3 dysfunction did not suppress the loss of proteins that regulate telomerase activity, it does not explain the observed synthetic lethality. Thus, the role of DIS3 in maintaining telomerase activity appears to be much more complex and should be further investigated.
Finally, analyses of GIs identified in the genome-wide screen revealed a substantial number of genes involved in microtubule organization (Figure 6C; Table S8: Tab3 and Tab4). DIS3 dysfunction reduces cell growth and division. Cells that lack DIS3 activity accumulate overcondensed chromosomes (Snee et al., 2016) and are blocked in mitosis (Murakami et al., 2007). Moreover, DIS3 mutants in yeast are synthetically lethal, with mutations that influence the kinetochore, anaphase-promoting complex, or spindle pole body (Milbury et al., 2019; Smith et al., 2011). The results of our screen indicated that the interplay between DIS3 deactivation and microtubule organization is much more diverse. We observed genes that belong to the microtubule or microtubule organizing center as both suppressors and synthetic lethal interactions. Comprehensive analyses of GIs that are engaged in microtubule organization showed some tendencies as synthetic lethal interactions include genes involved in the kinetochore and cell body, which is consistent with previous studies (Milbury et al., 2019; Smith et al., 2011). Positive interactors of DIS3 include genes involved in the motor function of microtubules and are also involved in the DNA damage response. This result provides valuable support to direct future studies of the involvement of DIS3 in microtubule maintenance.
Carrying out the genome-wide DIS3 high-throughput siRNA screen allowed us to functionally characterize this ribonuclease and provide insights into processes other than RNA decay in which it participates. The results of this screen may also be a rich resource for other investigators because we only described a small number of identified interactions.
Limitations of the study
In this article, we report functional associations between RNA decay and other RNA metabolism pathways deduced based on primary siRNA screenings. Since this work is of an exploratory nature, we performed limited validation of the results that can be expanded in the future case-specific studies. Additionally, we use three scales of siRNA screening for hit selection: (1) a one-plate library of 280 targets, (2) a custom library with 3004 targets, and (3) a genome-wide library. Due to technical reasons for one-plate screening and medium- or genome-wide screenings, we had to apply different normalization strategies (described in STAR Methods) that hampered comparison of respective results and led to smaller overlap of hits identified in DIS3 screens.
Notably, the main goal of this study was not focused on target search but on answering a more general biologic question to gain a comprehensive overview of possible processes interacting with RNA decay. This is also why we used loose hit scoring criteria, preferring to include false positives rather than miss identification of important interactions. Further investigation is necessary to gain more mechanistic knowledge of the identified hits, including validation of some newly discovered interactions. We intend this study to be a resource for other researches and we do encourage further studies to focus on the interplay of RNA decay with other processes as from our observations those screenings revealed some interesting cooperation between cellular processes.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-HBS1L | SigmaAldrich | catalog no. HPA029729; RRID:AB_10601228 |
| WDR61/REC14/SKI8 Polyclonal Antibody | BetylLabolatories | catalog no. A305-191A; RRID:AB_2631584 |
| Anti-α-Tubulin Mouse mAb | Sigma-Aldrich | catalog no. T5168; RRID:AB_477579 |
| Goat Anti-Mouse IgG, H&L Chain Specific | Millipore | catalog no. 401215; RRID:AB_10682749 |
| Goat Anti-Rabbit IgG, H & L Chain Specific Peroxidase Conjugate | Milipore | catalog no. 40139;3 RRID:AB_437797 |
| Chemicals, peptides, and recombinant proteins | ||
| Lipofectamine RNAiMAX | Invitrogen | catalog no. 13778-150 |
| TRI Reagent | Sigma-Aldrich | catalog no. 93289 |
| Phusion HF polymerase | Thermo Fisher Scientific | catalog no. F530 |
| SuperScript III Reverse Transcriptase | Invitrogen | catalog no. 18080085 |
| Viscolase | A&A Biotechnology | catalog no. 1010-100 |
| TURBO DNA-free Kit | Invitrogen | Catalog no. AM1907 |
| Random-primers | Thermo Fisher Scientific | catalog no. 48190011 |
| Critical commercial assays | ||
| Human Genome ON-TARGETplus siRNA Library-SMARTpool | Dharmacon | Drug Targets (G-104655-E2, Lot 11169), Druggable (G-104675-E2, Lot 11167), and Genome (G-105005-E2, Lot 11170) |
| KAPA Stranded RNA-Seq Library Preparation Kit | KAPA Biosystems | catalog no. KK8401 |
| Platinum SYBR Green qPCR SuperMix-UDG | Thermo Fisher Scientific | catalog no. 11733046 |
| Deposited data | ||
| Raw and analyzed data | This study | GEO: GSE155123 |
| Experimental models: Cell lines | ||
| 293 Flp-In T-REx cell lines stably transfected with construct expressing MUT or WT DIS3, DIS3L, DIS3L, XRN2 or EXOSC10 | (Szczepińska et al., 2015; Szczesny et al., 2018; Tomecki et al., 2014) | NA |
| Oligonucleotides | ||
| Silencer Select siRNA s20118 | Ambion | catalog no. 4392421 |
| Scrambled Silencer Select Negative Control siRNA | Ambion | catalog no. 4390844 |
| Stealth siRNA (set of 3) for TTC37 | Thermo Fisher Scientific | catalog no. TTC37HSS114433, TTC37HSS114434, TTC37HSS114435 |
| Stealth siRNA (set of 3) for WDR61 | Thermo Fisher Scientific | catalog no. WDR61HSS129640, WDR61HSS129641, WDR61HSS188452 |
| WDR61 FR primer for RT-qPCR GCAAGCCCATGATGATGCCATTTGG |
This study | NA |
| WDR61 RV primer for RT-qPCR AAGGCCAAAGTCCAGGCATCCAC |
This study | NA |
| TTC37 FR primer for RT-qPCR CTGTCTGCACAAGTGGATGG |
This study | NA |
| TTC37 RV primer for RT-qPCR CTGTTACCAGACGCACCAAG |
This study | NA |
| HBS1L FR primer for RT-qPCR GCAGCAGGCGATCATGTTAGTC |
This study | NA |
| HBS1L RV primer for RT-qPCR GATTCGGGCTCTGAAACGAGTG |
This study | NA |
| GAPDH FR primer for RT-qPCR | This study | NA |
| GAPDH RV primer for RT-PCR | This study | NA |
| SLC25A3 FR primer for RT-PCR CTTTCCAAGGGAGTGGTTGT |
This study | NA |
| SLC25A3 RV primer for RT-PCR AGATGTGGCGTGTTGAAGG |
This study | NA |
| MRPL52 FW primer for RT-PCR CTACCCCGGCTACTCCTG |
This study | NA |
| MRPL52 RV primer for RT-PCR CCAAGCGGCTACGCTG |
This study | NA |
| MRPS34 FR primer for RT-PCR CAAGTCCTGGCTGTGGC |
This study | NA |
| MRPS34 RV primer for RT-PCR TTGAAGGTCAGGATGCCC |
This study | NA |
| MRPS12 FR primer for RT-PCR GACCTCACCTTTAGGTCCTG |
This study | NA |
| MRPS12 RV primer for RT-PCR AAGTTAGGGACGTGTTGAGG |
This study | NA |
| GAPDH FR primer for RT-PCR | This study | NA |
| GAPDH RV primer for RT-PCR | This study | NA |
| H2AC17 FW primer for RT-PCR CACTTTCTGACTTAGGCC |
This study | NA |
| H2AC17 RV primer for RT-PCR AAGTTCAGCCCTTACTTGC |
This study | NA |
| Software and algorithms | ||
| Cutadapt 1.18 | (Martin, 2011) | https://cutadapt.readthedocs.io/en/stable/ |
| STAR 2.6.0c | (Dobin and Gingeras, 2015) | https://github.com/alexdobin/STAR |
| ScanR Acquisition software | Olympus | NA |
| ScanR Analysis software | Olympus | https://www.olympus-lifescience.com/en/microscopes/inverted/scanr/ |
| siscreenr | This study | https://github.com/olobiolo/siscreenr |
| R environment | (Gentleman et al., 2004) | https://www.r-project.org/ |
| magrittr | (Bache and Wickham, 2014) | https://www.rdocumentation.org/packages/magrittr/versions/1.5 |
| tidyr | (Wickham et al., 2020a) | https://www.rdocumentation.org/packages/tidyr/versions/0.8.3 |
| dplyr | (Wickham et al., 2020b) | https://www.rdocumentation.org/packages/dplyr/versions/0.7.8 |
| ggplot2 | (Wickham, 2016) | https://www.rdocumentation.org/packages/ggplot2/versions/3.3.2 |
| data.table | (Dowle et al., 2020) | https://www.rdocumentation.org/packages/data.table/versions/1.13.0 |
| reutils | (Schöfl, 2016) | https://www.rdocumentation.org/packages/reutils/versions/0.2.2 |
| JUM | (Wang and Rio, 2018) | https://github.com/qqwang-berkeley/JUM |
| clusterProfiler | (Yu et al., 2012) | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| Cytoscape | (Shannon et al., 2003) | https://cytoscape.org |
| Other | ||
| STRING protein-protein (P-P) | (Szklarczyk et al., 2019) | https://string-db.org/ |
| Database for Annotation, Visualization and Integrated Discovery | (Huang et al., 2009) | https://david.ncifcrf.gov/ |
| LightCycler 480 II | Roche | NA |
| scanR modular microscope-based imaging platform | Olympus | NA |
| NextSeq500 | Illumina | NA |
| Incucyte | Essen BioScience | NA |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andrzej Dziembowski (adziembowski@iimcb.gov.pl)
Materials availability
This study did not generate new materials.
Experimental model and subject details
Cellular model and cell line generation
Each screen was a competitive growth assay in which the fitness of cells that express the MUT protein of interest was compared with cells that expressed WT protein. We used an approach that was previously described by us to construct the cell lines (Szczepińska et al., 2015; Szczesny et al., 2018; Tomecki et al., 2014). Briefly, the parental 293 Flp-In T-REx cell line was stably transfected with a construct in which a bidirectional, tetracycline inducible promoter controls the expression of two transcription units. One transcription unit produces a bicistronic RNA that contains a miRNA precursor and mRNA for a fluorescent protein that localizes to the cell nucleus and serves as an expression marker. The other transcription unit contains the cDNA of the gene of interest with a C-terminal FLAG tag. The cDNA contains silent point mutations that render the resulting mRNA insensitive to the miRNA. Thus, the endogenous protein is replaced by a desired version of the protein that is encoded by the transgene and expressed at roughly the same levels. For every gene of interest, two stable cell lines were generated: one that expressed the WT version of the respective protein and EGFP as the expression marker and the other that expressed the protein of interest with mutations that impaired or abolished its catalytic activity with mCherry as the expression marker. This allowed the identification of cells that express the miRNA and distinction between WT and MUT cells. Procedures for construction of DNA plasmids encoding miRNA and RNAi-resistant gene of interest were described in details by Szczesny et al. (Szczesny et al., 2018). Sequences of miRNA cassettes and miRNA insensitive mRNAs were described in Łabno et al.(2016),Szczesny et al. (2018) and Tomecki et al. (2014) and summarized in Table S9.
Cell culture and transfection for siRNA screens
293 Flp-In T-REx cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS) without antibiotics at 37°C in a 5% CO2 atmosphere, with passages every 2-3 days. The cells that were used for transfection were obtained from 2-10 passages after thawing. Screening was performed in triplicate in 384-well plates (Greiner) with a poly-L-lysine-coated uClear bottom. Target siRNAs were stored at -80°C in 384-well plates. The day before transfection, they were thawed, and an aliquot was transferred to a master plate. Control siRNAs were added to the master plates by hand, and then the master plate was replicated to transfection plates. The plates were then sealed and stored at 4°C overnight. The next day, Lipofectamine siRNA MAX was added to the wells in OptiMEM. This was performed with a Multidrop Combi dispenser (ThermoFisher).
WT and MUT cells were grown separately. On the day of transfection, they were detached, counted, and mixed at a ratio where untreated cells reach roughly equal numbers (green:red) after 72 h of culture. This plating ratio was determined for each cell line and each batch of thawed cells. Once the cells were mixed, they were brought to an equal concentration, and 750 cells per well were added to the transfection plates with a Multidrop Combi dispenser. The plates were resealed and left at room temperature for 1 h to allow the cells to evenly attach to the plate surface. Afterward, they were moved to an incubator and cultured for an additional 71 h.
Seventy-two hours after transfection, 10% formaldehyde in phosphate-buffered saline (PBS) with 2 mg/ml Hoechst 33342 was added to each well, and the cells were fixed and stained for 20 min at room temperature. They were then washed three times with PBS and covered with 50 μl of PBS with 0.1% sodium azide. The plates were sealed and stored at 4°C until imaging but for not more than one week.
Cell culture and transfection for RNA isolation
Stably transfected derivatives of HEK293 FLP-In T-Rex cells that expressed WT DIS3 or the catatonically inactive version (G766R) of DIS3 were cultured for transfection in six-well format in DMEM (Gibco) supplemented with 10% FBS (Gibco) without antibiotics at 37°C in a 5% CO2 humidified atmosphere.
The expression of transgenes that coded for sh-miRNA-EGFP cassettes to silence endogenous DIS3 and sh-miRNA-insensitive FLAG-tagged exogenous DIS3 protein was induced with tetracycline at a final concentration of 25 ng/ml 24 h before siRNA transfection. For siRNA-mediated SF3A1 depletion, tetracycline-induced cells were subjected to Silencer Select siRNA s20118 transfection (Ambion, catalog no. 4392421). Scrambled Silencer Select Negative Control siRNA (Ambion, catalog no. 4390844) was used as a negative control. Transfections were performed using Lipofectamine RNAiMAX (Invitrogen, catalog no. 13778-150) and 20 nM siRNA according to the manufacturer’s recommendations and cultured for ∼72 h before harvesting.
Method details
RNAi screening
siRNA libraries
Human ON-TARGETplus SMARTpool siRNAs (Dharmacon) were used. A pool of four different siRNAs for each gene were used in a 384-well format. The core RNA metabolism library was designed by manually selecting 280 genes that are involved in various aspects of the RNA life cycle, including known and predicted RNA helicases that were described previously (Jankowsky et al., 2011). The extended RNA metabolism library was created by selecting 3904 siRNA pools from the Human Genome ON-TARGETplus siRNA Library-SMARTpool (Dharmacon). Genes that were targeted by the extended RNA metabolism library were chosen based on results of the experimental capture of RNA binding proteins (Baltz et al., 2012; Beckmann et al., 2015; Castello et al., 2012, 2016; Conrad et al., 2016; He et al., 2016; Kwon et al., 2013; Liao et al., 2016), the prediction of RNA binding proteins (Brannan et al., 2016; Ghosh and Sowdhamini, 2016; Malhotra and Sowdhamini, 2014), comprehensive overview articles (Cook et al., 2011; Gerstberger et al., 2014; Neelamraju et al., 2015; Sundararaman et al., 2016), and manual curation. For the genome-wide screening, we used the Human Genome ON-TARGETplus siRNA Library-SMARTpool (Dharmacon) that targeted 18104 genes. This library comprised three subsets: Drug Targets (G-104655-E2, Lot 11169), Druggable (G-104675-E2, Lot 11167), and Genome (G-105005-E2, Lot 11170).
Control siRNAs
Screen progress was monitored with seven control transfections: three positive controls to monitor transfection efficiency and four negative controls to ensure our competitive growth assay was unaffected by the transfection procedure itself and cells were in good condition. The positive controls were siRNAs that targeted the EGFP coding sequence and PLK1 and UCB1 genes. The siRNA that targeted EGFP should cause a large decrease in the green fraction (gf) because “green” cells would lose their fluorescent reporter and become “black.”PLK1 and UCB1are essential house-keeping genes. Their knockdown is lethal, and efficient transfection should greatly decrease cell number. We had no way of knowing a priori how it would affect gf. Thus, we focused on the strength of the effect and whether the effect was constant throughout the large screens.
In the genome-wide screen of DIS3, after processing the first batch of cells, we noticed that silencing of the PCF11 gene produces an extraordinarily low gf and decided to replace the UCB1 control with PCF11 in subsequent transfections.
For negative controls, we used the non-targeting siRNAs neg1, neg3, and neg4 and their pool (negp). Notably, the pool also contains neg2 siRNA. We decided to not use neg2 alone because it had a uniquely cytotoxic effect of considerable magnitude that was not replicated with negp.
Plate layout
In the siRNA libraries, the siRNAs that target particular genes usually occupy rows B to O and columns 3 to 22. These are called sample wells. We left rows A and P empty and plated cells in all columns in rows B to O. Control siRNAs were placed in columns 2 and 23, and cells in columns 1 and 24 were left non-transfected (nt).
RNA-Seq
Library preparation and sequencing
Total RNA was isolated with TRI Reagent (Sigma-Aldrich, catalog no. 93289) according to the manufacturer’s instructions. Following DNase treatment (Invitrogen, catalog no. 18080085), strand-specific RNA libraries were prepared using a KAPA Stranded RNA-Seq Library Preparation Kit (KAPA Biosystems, catalog no. KK8401) according to the manufacturer’s protocol. The libraries were sequenced using an Illumina NextSeq500 sequencing platform in the 150-nt paired-end mode. The RNA-Seq experiments were performed in duplicate.
Quality control and mapping of next-generation sequencing data
The Illumina sequencing reads were quality filtered using Cutadapt 1.18 (Martin, 2011) to remove Illumina adapter sequences, trim low-quality fragments (minimum Q score = 20), and remove reads that were shorter than 30 nt after trimming. For JUM alternative splicing analysis, reads were mapped to the human genome (hg38) using STAR 2.6.0c (Dobin and Gingeras, 2015) with the two-pass mode for better junction discovery. Before mapping, the genome was indexed with Gencode v28 basic annotation. For downstream analysis, only uniquely mapped reads were used.
Real-time cell proliferation assay
The proliferation rate of cells that expressed the WT or MUT version of the protein of interest was measured using the Live Cell Imaging System (Incucyte, Essen BioScience). Briefly, cells were seeded at a density of 8 × 104 cells/ml (1600 cells per well) into 384-well plate and imaged with 10x objective every 6 hours between 12-72h after cells seeding. We determined the cell counts per image during the recorded time using EGFP or mCherry signal as a marker of nuclei. Another variant of the cell proliferation assay included transfection of cells with siRNA before a microscopy observation. Reverse siRNA transfection was performed according to the procedure applied for the screening. Cell lines that expressed DIS3 WT or DIS3 MUT were reversely transfected in 384-well format using 20 nM siRNA targeting TTC37 (an equimolar pool of Invitrogen Stealth siRNA TTC37HSS114433, TTC37HSS114434, TTC37HSS114435 of the following sequences GGGUCUGCACAACAUUGCAAGAUAA, GAGGUGCCUUCUUACAUCAGCGAUU, GCUGAAAGUACAGAGGACCAGAAUA) or HBS1LV1 (r(CCAGUAGAUUCCCAGACAU)d(TT), described in Kalisiak et al.(2017). Similarly, cells expressing XRN2 WT or XRN2 MUT were transfected with an equimolar pool of siRNAs targeting WDR61 (Invitrogen Stealth siRNA WDR61HSS129640, WDR61HSS129641, WDR61HSS188452 of the following sequences ACUGCUUCAGAUGAUGGCUACAUCA, UGACCAACCAGUACGGUAUUCUCUU, GCCAUAGAUGGAAUCAUCAAUAUUU). Data for cell counts per image was calculated using the default software of the Incucyte. For all examined cell lines, technical repeats were performed.
Western blot analysis
For total protein isolation cell pellets were resuspended in 0.5xPBS containing 0.1 % NP-40, protease inhibitors cocktail and 0.2U/ml viscolase (A&A Biotechnology, catalog no. 1010-100) and lysed for 25 minutes at 37°C with shaking at 300 rpm. After incubation 10 μl of sample were saved for protein concentration measurements on NanoDrop 2000 station. The lysates were mixed with Laemmli buffer and denaturated for 5 min in 95°C. Further samples were separated on 10 or 12% SDS-PAGE gels, depending on the size of assayed proteins. In the next step proteins were transferred to nitrocellulose membranes (Amersham, catalog no. GE10600002). For loading control membranes were stained with 0.3% w/v Ponceau S in 3% v/v acetic acid and imagined. Next membranes were blocked by incubation in 5% w/v non-fat milk in TBST buffer for 1 hour. For the detection of assayed protein membranes were incubated with specific primary antibodies (see key resources table) overnight at 4°C. Then membranes were washed three times with TBST for 5 min at RT and further incubated with secondary antibody conjugated with HRP for 1 hour at RT. The detection was done with Clarity Western ECL Substrate (Bio-Rad, catalog no. 1705061)
Quantitative RT-PCR
Total RNA was isolated with TRI Reagent (Sigma-Aldrich, catalog no. 93289) according to the manufacturer’s instructions. Following DNase treatment (Invitrogen, catalog no. 18080085), the RNA was reverse transcribed with SuperScript III (Invitrogen; catalog no. 18080085), using random-primers (Thermo Fisher Scientific, catalog no. 48190011). The quantitative PCR was performed with Platinum SYBR Green qPCR SuperMix-UDG (Thermo Fisher Scientific; catalog no. 11733046) using LightCycler 480 II (Roche) device and appropriate primers listed in key resources table. Gene expression for each sample was normalized to GAPDH. Differences were determined using the 2−ΔΔC(t) calculation and % of KD was claculated.
RT-PCR
Total RNA was isolated with TRI Reagent (Sigma-Aldrich, catalog no. 93289) according to the manufacturer’s instructions. Following DNase treatment (Invitrogen, catalog no. 18080085), the RNA was reverse transcribed with SuperScript III (Invitrogen; catalog no. 18080085), using random-primers (Thermo Fisher Scientific, catalog no. 48190011). The PCR was performed using Phusion High-Fidelity DNA Polymerase (Phusion High-Fidelity DNA Polymerase, catalog no. F530) and appropriate primers listed in key resources table.
PCR products were resolved on agarose gels and visualized using GelGreen. The PCR primers were designed to amplify both spliced and unspliced isoform of mRNA as they were designed to map to both flanking regions of retained intron.
Quantification and statistical analysis
Quantification and statistics in RNAi screening
Image acquisition and analysis
Imaging of siRNA screening plates was performed using the ScanR system (Olympus) that was equipped with a MT20 illumination unit and 150 W mercury-xenon burner and a DAPI/FITC/Cy3/Cy5 quad Sedat filter set (Semrock) using a 20x/0.75 objective lens in the extended RNA metabolism screen and a 10x/0.35 PlnFLN lens in all of the other screens. Single images (not stacks) were taken from six fields of view in every well in the extended RNA metabolism screen and five fields of view in all of the other screens. In the genome-scale screen, the images were binned 2x2 to shorten acquisition times. ScanR Acquisition software was used for microscope control.
Primary image analysis was performed using ScanR Analysis software. Every channel was run through rolling-ball background subtraction. Segmentation was performed on the Hoechst channel using the Edge algorithm. The identified objects were gated based on a scatter plot of the circularity factor relative to the area. The objects that comprised the gate are hereinafter referred to as cells. The mean intensity of the EGFP and mCherry channels was calculated for all cells, and the two intensities were plotted on a scatter plot. Cells were classified into one of three groups: black (no reporter fluorescence), green (high EGFP fluorescence and no mCherry fluorescence), and red (high mCherry fluorescence and no EGFP fluorescence). The numbers of cells in each gate in each well were exported as tab-delimited text files.
Metrics
In our analysis, we noted two population metrics: general viability and relative fitness. General viability is the total number of cells in a particular well compared with nt cells in the respective plate. It is calculated plate-wise as relative viability (Equation 1) and reflects the cytotoxicity of particular siRNAs. Relative fitness reflects the direction and strength of the genetic interaction. Relative fitness is expressed as the fraction of cells that express the WT protein of interest among all cells that expressed a transgene (i.e., the fraction of green cells among all positive [green and red] cells). We called this metric the gf and calculated it well-wise (Equation 2).
| (Equation 1) |
where vi is relative viability in well i, Nti is the total number of positive cells in well i (green + red), and Ntntp is the mean total number of cells in all nt wells in the respective plate p. Negative cells (black; see below) were omitted for the purpose of this calculation because their number was always very low (not shown).
| (Equation 2) |
where gfi is the “green fraction” in well i, Ngi is the number of green cells in well i, and Nri is the number of red cells in well i.
Statistical analysis and hit selection
Downstream analysis was performed in the R environment (Gentleman et al., 2004) using home-written tools. The pipeline made use of the following packages: magrittr (Bache and Wickham, 2014), tidyr (Wickham et al., 2020a), dplyr (Wickham et al., 2020b), ggplot2 (Wickham, 2016), data.table (Dowle et al., 2020), and reutils (Schöfl, 2016).
Briefly, all of the result files were loaded and collated into a single data frame. gf was normalized by running Tukey’s median polish on each plate. The normalized values were then converted to robust z-scores (z∗ scores) according to Equation 3.
| (Equation 3) |
where z∗i is the z∗ score for well i, GFi is the normalized gf in well i, and MADGFref is the median absolute deviation of normalized gf in the reference set. z∗ scores were calculated globally and not plate-wise. The reference set in the genome-wide DIS3 screening was the sample wells because there were many sample siRNAs, and only a few were expected to produce a reaction. In the core RNA metabolism screens, the sample siRNAs targeted a small group of hand-picked genes with probable genetic interactions, which was likely to skew the baseline; therefore, we used untreated wells as a reference for gf. As a result, the median absolute deviation calculated for the whole-genome screen was higher. Notably, such normalization flattened the results, leading to a relatively low number of false positives but also lowered levels of statistically significant hits. As a result, a gene that was a hit in a single-plate screen may not be observed as a hit on the whole-genome screen. We lost many hits in the genome-wide screen that were identified in the core metabolism screen for DIS3 as their individual z∗ sores are lower (Figures S10A and S10C). But when we look at the raw data (green fraction), the reproducibility is very high (Figures S10B and S10D) (although the trend stays the same). As already mentioned, the same tendency for the normalized green fraction indicates the high technical reproducibility of the approach. Interestingly the overlap between identified positive hits was almost in 50%, whereas the trend in negative interactions was different (Figure S11). There were many more negative hits identified in the one-plate format screen than in the same subset of genes tested on a genome-wide scale, so it is hard to scale the overlap.
We set the z∗ score threshold for being considered a hit to a sufficiently low number to obtain a substantial amount of hits but sufficiently high to not obtain hits in nt wells and wells that were transfected with negative control siRNAs. Notably, the thresholds were set independently for each screen, ranging from 2 to 2.4.
Based on the z∗ scores, each well received a hit score: 1 if the z∗ score was higher than the threshold, -1 if the z∗ score was lower than the negative threshold, and 0 if the z∗ score fell between a negative and positive threshold. Hit scores were then summed across replicates to yield summarized hit scores. Wells with an absolute summarized hit score of 2 or more were considered hits in their respective screens. However, wells with relative viability less than 30% were disregarded when summarizing hit scores.
Since we decided to use z-score method we were aware that with this method we can control only false positive rate and we cannot easily calculate false negative rate (rate of interactions with substantial effect that are not identified as hit), as for this we would need to control not only sample size but also siRNA effect. As our aim was to perform functional analyses where higher number of tested hits also influence the significance of enrichment results, we wanted not to lose any real effect and fail to detect it. It is well known that there is inverse relationship between number of false negatives and false positives. With our attempt to increase possibility to identify important interaction we did increased the false positive rate. However, we know that, when using z-score of more than 2 for hit selection the false positive rate is 0.025 under the normal distribution assumption. Additionally, for core metabolism one plate screenings we applied the most stringent cut-off (2.4) that should give the false positive rate 0.005, that for 280 performed test would give less than 2 false positives. Surprisingly for some of tested ribonucleases we did not identified as many hits as expected, however still keeping false discovery rate (FDR) at medium level (Table below). Additionally, it should be noted, that we took advantage of repeating the screening experiments three times, and to increase the statistical power of our analysis we were considering hits scoring above threshold in at least two out of three experimental replicates.
| Type of screen | FPR/number of expected false positives | Number of significant hits | FDR |
|---|---|---|---|
| Core metabolism - DIS3 | 0.005/1.4 | 41 | 0.034 |
| Core metabolism - DIS3L | 0.005/1.4 | 18 | 0.078 |
| Core metabolism - DIS3L2 | 0.005/1.4 | 18 | 0.078 |
| Core metabolism – EXOS10 | 0.005/1.4 | 19 | 0.073 |
| Core metabolism - XRN2 | 0.005/1.4 | 15 | 0.093 |
| Extended RNA metabolism - DIS3 | 0.025/97.6 | 274 | 0.352 |
| Extended RNA metabolism - DIS3L | 0.025/97.6 | 152 | 0.642 |
| Genome-wide DIS3 | 0.025/452,6 | 1112 | 0.407 |
Differential alternative splicing analysis
Alternative splicing (AS) events were analyzed using JUM (Wang and Rio, 2018). For AS structure quantification, JUM uses RNA-Seq reads that are mapped to splice junctions. Only splice junctions that received more than five reads in each of the replicates of the siRNA-treated and control samples were considered valid junctions for downstream analysis. After profiling AS structures in each condition, JUM performs an analysis of differential AS structure usage between examined conditions. As a metric for quantifying differential splicing events between conditions, JUM calculates the ΔPSI. PSI represents the percentage of transcripts, including a particular exon or splice site, and is calculated from the read that is mapped to splice junctions. For the quantification of changes in AS, JUM compares the usage of each profiled AS by modeling the total number of reads that are mapped to AS sub-junctions with a negative binomial distribution and evaluating the overall change in the basal expression of AS between conditions. Only AS events that showed more than 10% of a difference (|ΔPSI|> 0.1) were considered differentially spliced. An Benjamini–Hochberg-adjusted p value of 0.05 was used as the statistical threshold for significance.
Gene Ontology enrichment analysis
The GO analyses were performed in the R/Bioconductor environment (Gentleman et al., 2004; Huber et al., 2015) using the clusterProfiler package (Yu et al., 2012) and data from the Database for Annotation, Visualization and Integrated Discovery (DAVID; https://david.ncifcrf.gov/home.jsp; Huang et al., 2009) with the RDAVIDWebService package (Fresno and Fernández, 2013). As a background for enrichment analysis, we used lists of genes that were tested in each of the analyzed libraries. The minimum number of genes in enriched processes was set to 2. The analyses were performed based on annotations of Biological Processes, Molecular Functions, and Cellular Compartments. The results are presented in the Tables S4, S6, and S8. The visualization of enrichment analysis was performed by plotting bar plots that represented the number of genes that were enriched in the top 10 categories, sorted by a Fischer’s exact test p value using the ggplot2 package.
Gene Ontology enrichment network analysis
The GO enrichment networks were created in the R/Bioconductor environment using the igrpah package, ggraph package, and home-written tools. To evaluate relationships between distinct biological processes from the GO analyses, we created networks that represented connections between these processes with shared genes that were assigned to them. The list of enriched functional categories and genes were extracted for the enrichment results and converted to graph format (igraph::graph.data.frame). In-network construction connections between genes and biological processes, represented as edges, were created based on the assignment of genes to extracted biological processes according to the DAVID GO library, with genes and biological processes as nodes. The networks were visualized with ggraph. The networks are presented in the Figures 3 and 4, and S6. Figures S7 and S8 were constructed so that edges represent the number of genes that are commonly assigned to connected functional categories that constitute nodes in those graphs.
Genetic interaction network generation
The resulting significant hits from the core RNA metabolism high-throughput siRNA screen were visualized in the network layout using Cytoscape (Shannon et al., 2003). For visualization, networks were defined as a list of nodes that represented query ribonucleases and genes that were defined as significant hits in the screen. The connection between nodes was based on the resulting genetic interaction that was identified in the screen and classified into two possible categories: synthetic lethal (blue edges) and suppressor (yellow edges). Additionally, all hits were analyzed in the STRING protein-protein (P-P) association database (Szklarczyk et al., 2019) to investigate overlap between known P-P interactions and identified genetic interactions. The extracted P-P interactions between query genes and hits are represented in the network with grey edges. To visualize interactions with known protein complexes, genes that interacted with screen hits and are known to from protein complexes were grouped together.
Acknowledgments
We acknowledge former and current members of AD lab for stimulating discussions and all support; Katarzyna Prokop and Dorota Adamska for technical support in NGS experiments; Michal Malecki for helpful discussions and suggestions on screening analyses. Financial support was provided by the TEAM program of the Foundation for Polish Science, financed by the European Union via the European Regional Development Fund (agreement no. TEAM/2016-1/3 to AD) and ERA Chair European Union's Horizon2020 program (agreement no.810425).
Author contributions
AD and RJS developed and directed the studies. AH-O set up the procedure of computational and bioinformatic analysis of normalized siRNA screen results, performed GSEA and network analyses and carried out RNA-Seq data analyses, performed validation experiments of siRNA screenings and alternative splicing, wrote the manuscript, and prepared all figures and tables with feedback from AD. AC performed high-throughput microscopy, image analysis, designed and programmed the siRNA screening analysis, and participated in writing the manuscript. JG performed cell culturing and transfections for validations experiments and real time cell proliferation assays. KA, KK, PO contributed to the technical component of screening implementation: established stable cell lines, performed cell culturing and transfections. RJS designed the siRNA screening procedure and subgenomic siRNA libraries, performed experiments with depletion of splicing component, and participated in writing the manuscript. AD provided funding and revised the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: September 24, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103036.
Contributor Information
Roman J. Szczesny, Email: rszczesny@ibb.waw.pl.
Andrzej Dziembowski, Email: adziembowski@iimcb.gov.pl.
Supplemental information
Data and code availability
-
•
RNA-Seq data have been deposited at GEO and are publicly aviable as of the date of publication. The assesion number is: GSE155123
-
•
The code for screen data analysis (development version) is available at GitHub (https://github.com/olobiolo/siscreenr)
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Azzouz N., Panasenko O.O., Colau G., Collart M.A. The CCR4-NOT complex physically and functionally interacts with TRAMP and the nuclear exosome. PLoS One. 2009;4:e6760. doi: 10.1371/journal.pone.0006760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bache S.M., Wickham H. 2014. magrittr: A Forward-Pipe Operator for R. [Google Scholar]
- Badertscher L., Wild T., Montellese C., Alexander L.T., Bammert L., Sarazova M., Stebler M., Csucs G., Mayer T.U., Zamboni N. Genome-wide RNAi screening identifies protein modules required for 40S subunit synthesis in human cells. Cell Rep. 2015;13:2879–2891. doi: 10.1016/j.celrep.2015.11.061. [DOI] [PubMed] [Google Scholar]
- Baltz A.G., Munschauer M., Schwanhäusser B., Vasile A., Murakawa Y., Schueler M., Youngs N., Penfold-Brown D., Drew K., Milek M. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell. 2012;46:674–690. doi: 10.1016/j.molcel.2012.05.021. [DOI] [PubMed] [Google Scholar]
- Beckmann B.M., Horos R., Fischer B., Castello A., Eichelbaum K., Alleaume A.-M., Schwarzl T., Curk T., Foehr S., Huber W. The RNA-binding proteomes from yeast to man harbour conserved enigmRBPs. Nat. Commun. 2015;6:10127. doi: 10.1038/ncomms10127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram K., Agafonov D.E., Dybkov O., Haselbach D., Leelaram M.N., Will C.L., Urlaub H., Kastner B., Lührmann R., Stark H. Cryo-EM structure of a pre-catalytic human spliceosome primed for activation. Cell. 2017;170:701–713.e11. doi: 10.1016/j.cell.2017.07.011. [DOI] [PubMed] [Google Scholar]
- Boehmer T., Jeudy S., Berke I.C., Schwartz T.U. Structural and functional studies of Nup107/Nup133 interaction and its implications for the architecture of the nuclear pore complex. Mol.Cell. 2008;30:721–731. doi: 10.1016/j.molcel.2008.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyraz B., Moon D.H., Segal M., Muosieyiri M.Z., Aykanat A., Tai A.K., Cahan P., Agarwal S. Posttranscriptional manipulation of TERC reverses molecular hallmarks of telomere disease. J. Clin. Invest. 2016;126:3377–3382. doi: 10.1172/JCI87547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannan K.W., Jin W., Huelga S.C., Banks C.A.S., Gilmore J.M., Florens L., Washburn M.P., Van Nostrand E.L., Pratt G.A., Schwinn M.K. SONAR discovers RNA-binding proteins from analysis of large-scale protein-protein interactomes. Mol. Cell. 2016;64:282–293. doi: 10.1016/j.molcel.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosi R., Hauri H.P., Krämer A. Separation of splicing factor SF3 into two components and purification of SF3a activity. J. Biol. Chem. 1993;268:17640–17646. [PubMed] [Google Scholar]
- Castello A., Fischer B., Eichelbaum K., Horos R., Beckmann B.M., Strein C., Davey N.E., Humphreys D.T., Preiss T., Steinmetz L.M. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- Castello A., Fischer B., Frese C.K., Horos R., Alleaume A.-M., Foehr S., Curk T., Krijgsveld J., Hentze M.W. Comprehensive identification of RNA-binding domains in human cells. Mol. Cell. 2016;63:696–710. doi: 10.1016/j.molcel.2016.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman M.A., Lawrence M.S., Keats J.J., Cibulskis K., Sougnez C., Schinzel A.C., Harview C.L., Brunet J.-P., Ahmann G.J., Adli M. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Roake C.M., Galati A., Bavasso F., Micheli E., Saggio I., Schoeftner S., Cacchione S., Gatti M., Artandi S.E. Loss of human TGS1 hypermethylase promotes increased telomerase RNA and telomere elongation. Cell Rep. 2020;30:1358–1372.e5. doi: 10.1016/j.celrep.2020.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi B., Wang Q., Wu G., Tan M., Wang L., Shi M., Chang X., Cheng H. Aly and THO are required for assembly of the human TREX complex and association of TREX components with the spliced mRNA. Nucleic Acids Res. 2013;41:1294–1306. doi: 10.1093/nar/gks1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chlebowski A., Tomecki R., López M.E.G., Séraphin B., Dziembowski A. Catalytic properties of the eukaryotic exosome. Adv. Exp. Med. Biol. 2010;702:63–78. [PubMed] [Google Scholar]
- Chlebowski A., Lubas M., Jensen T.H., Dziembowski A. RNA decay machines: the exosome. Biochim. Biophys. Acta (BBA) - Gene Regul. Mech. 2013;1829:552–560. doi: 10.1016/j.bbagrm.2013.01.006. [DOI] [PubMed] [Google Scholar]
- Conrad T., Albrecht A.-S., de Melo Costa V.R., Sauer S., Meierhofer D., Ørom U.A. Serial interactome capture of the human cell nucleus. Nat. Commun. 2016;7:11212. doi: 10.1038/ncomms11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook K.B., Kazan H., Zuberi K., Morris Q., Hughes T.R. RBPDB: a database of RNA-binding specificities. Nucleic Acids Res. 2011;39:D301–D308. doi: 10.1093/nar/gkq1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M., VanderSluis B., Koch E.N., Baryshnikova A., Pons C., Tan G., Wang W., Usaj M., Hanchard J., Lee S.D. A global genetic interaction network maps a wiring diagram of cellular function. Science. 2016;353:aaf1420. doi: 10.1126/science.aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello J.L., Stead J.A., Feigenbutz M., Jones R.M., Mitchell P. The C-terminal region of the exosome-associated protein Rrp47 is specifically required for box C/D small nucleolar RNA 3′-maturation. J. Biol. Chem. 2011;286:4535–4543. doi: 10.1074/jbc.M110.162826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson L., Kerr A., West S. Co-transcriptional degradation of aberrant pre-mRNA by Xrn2. EMBO J. 2012;31:2566–2578. doi: 10.1038/emboj.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson L., Francis L., Cordiner R.A., Eaton J.D., Estell C., Macias S., Cáceres J.F., West S. Rapid depletion of DIS3, EXOSC10, or XRN2 reveals the immediate impact of exoribonucleolysis on nuclear RNA metabolism and transcriptional control. Cell Rep. 2019;26:2779–2791.e5. doi: 10.1016/j.celrep.2019.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Gingeras T.R. Mapping RNA-seq reads with STAR. Curr. Protoc. Bioinform. 2015;51:11.14.1–11.14.19. doi: 10.1002/0471250953.bi1114s51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowle M., Srinivasan A., Gorecki J., Chirico M., Stetsenko P., Short T., Lianoglou S., Antonyan E., Bonsch M., Parsonage H. 2020. data.table: Extension of “data.frame”. [Google Scholar]
- Drążkowska K., Tomecki R., Stoduś K., Kowalska K., Czarnocki-Cieciura M., Dziembowski A. The RNA exosome complex central channel controls both exonuclease and endonuclease Dis3 activities in vivo and in vitro. Nucleic Acids Res. 2013;41:3845–3858. doi: 10.1093/nar/gkt060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziembowski A., Lorentzen E., Conti E., Séraphin B. A single subunit, Dis3, is essentially responsible for yeast exosome core activity. Nat. Struct. Mol. Biol. 2007;14:15–22. doi: 10.1038/nsmb1184. [DOI] [PubMed] [Google Scholar]
- Eaton J.D., Davidson L., Bauer D.L.V., Natsume T., Kanemaki M.T., West S. Xrn2 accelerates termination by RNA polymerase II, which is underpinned by CPSF73 activity. Genes Dev. 2018;32:127–139. doi: 10.1101/gad.308528.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J., Wang K., Du X., Wang J., Chen S., Wang Y., Shi M., Zhang L., Wu X., Zheng D. ALYREF links 3′-end processing to nuclear export of non-polyadenylated mRNAs. EMBO J. 2019;38:e99910. doi: 10.15252/embj.201899910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fica S.M., Nagai K. Cryo-EM snapshots of the spliceosome: structural insights into a dynamic ribonucleoprotein machine. Nat. Struct. Mol. Biol. 2017;24:791–799. doi: 10.1038/nsmb.3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong N., Brannan K., Erickson B., Kim H., Cortazar M.A., Sheridan R.M., Nguyen T., Karp S., Bentley D.L. Effects of transcription elongation rate and Xrn2 exonuclease activity on RNA polymerase II termination suggest widespread kinetic competition. Mol. Cell. 2015;60:256–267. doi: 10.1016/j.molcel.2015.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fresno C., Fernández E.A. RDAVIDWebService: a versatile R interface to DAVID. Bioinformatics. 2013;29:2810–2811. doi: 10.1093/bioinformatics/btt487. [DOI] [PubMed] [Google Scholar]
- Freund A., Zhong F.L., Venteicher A.S., Meng Z., Veenstra T.D., Frydman J., Artandi S.E. Proteostatic control of telomerase function through TRiC-mediated folding of TCAB1. Cell. 2014;159:1389–1403. doi: 10.1016/j.cell.2014.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gable D.L., Gaysinskaya V., Atik C.C., Talbot C.C., Kang B., Stanley S.E., Pugh E.W., Amat-Codina N., Schenk K.M., Arcasoy M.O. ZCCHC8, the nuclear exosome targeting component, is mutated in familial pulmonary fibrosis and is required for telomerase RNA maturation. Genes Dev. 2019;33:1381–1396. doi: 10.1101/gad.326785.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland W., Feigenbutz M., Turner M., Mitchell P. Rrp47 functions in RNA surveillance and stable RNA processing when divorced from the exoribonuclease and exosome-binding domains of Rrp6. RNA. 2013;19:1659–1668. doi: 10.1261/rna.039388.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstberger S., Hafner M., Tuschl T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014;15:829–845. doi: 10.1038/nrg3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P., Sowdhamini R. Genome-wide survey of putative RNA-binding proteins encoded in the human proteome. Mol. Biosyst. 2016;12:532–540. doi: 10.1039/c5mb00638d. [DOI] [PubMed] [Google Scholar]
- Halbach F., Reichelt P., Rode M., Conti E. The yeast Ski complex: crystal structure and RNA channeling to the exosome complex. Cell. 2013;154:814–826. doi: 10.1016/j.cell.2013.07.017. [DOI] [PubMed] [Google Scholar]
- He C., Sidoli S., Warneford-Thomson R., Tatomer D.C., Wilusz J.E., Garcia B.A., Bonasio R. High-resolution mapping of RNA-binding regions in the nuclear proteome of embryonic stem cells. Mol. Cell. 2016;64:416–430. doi: 10.1016/j.molcel.2016.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Her J., Chung I.K. The AAA-ATPase NVL2 is a telomerase component essential for holoenzyme assembly. Biochem. Biophys. Res. Commun. 2012;417:1086–1092. doi: 10.1016/j.bbrc.2011.12.101. [DOI] [PubMed] [Google Scholar]
- Hilleren P., McCarthy T., Rosbash M., Parker R., Jensen T.H. Quality control of mRNA 3′-end processing is linked to the nuclear exosome. Nature. 2001;413:538–542. doi: 10.1038/35097110. [DOI] [PubMed] [Google Scholar]
- Huang D.W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huber W., Carey V.J., Gentleman R., Anders S., Carlson M., Carvalho B.S., Bravo H.C., Davis S., Gatto L., Girke T. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods. 2015;12:115–121. doi: 10.1038/nmeth.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntzinger E., Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011;12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- Jankowsky A., Guenther U.-P., Jankowsky E. The RNA helicase database. Nucleic Acids Res. 2011;39:D338–D341. doi: 10.1093/nar/gkq1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalisiak K., Kuliński T.M., Tomecki R., Cysewski D., Pietras Z., Chlebowski A., Kowalska K., Dziembowski A. A short splicing isoform of HBS1L links the cytoplasmic exosome and SKI complexes in humans. Nucleic Acids Res. 2017;45:2068–2080. doi: 10.1093/nar/gkw862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanaugh G., Ye F., Mohni K.N., Luzwick J.W., Glick G., Cortez D. A whole genome RNAi screen identifies replication stress response genes. DNA Repair. 2015;35:55–62. doi: 10.1016/j.dnarep.2015.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilchert C., Wittmann S., Vasiljeva L. The regulation and functions of the nuclear RNA exosome complex. Nat. Rev. Mol. Cell Biol. 2016;17:227–239. doi: 10.1038/nrm.2015.15. [DOI] [PubMed] [Google Scholar]
- Kwon S.C., Yi H., Eichelbaum K., Föhr S., Fischer B., You K.T., Castello A., Krijgsveld J., Hentze M.W., Kim V.N. The RNA-binding protein repertoire of embryonic stem cells. Nat. Struct. Mol. Biol. 2013;20:1122–1130. doi: 10.1038/nsmb.2638. [DOI] [PubMed] [Google Scholar]
- Łabno A., Warkocki Z., Kuliński T., Krawczyk P.S., Bijata K., Tomecki R., Dziembowski A. Perlman syndrome nuclease DIS3L2 controls cytoplasmic non-coding RNAs and provides surveillance pathway for maturing snRNAs. Nucleic Acids Res. 2016;44:10437–10453. doi: 10.1093/nar/gkw649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lardelli R.M., Thompson J.X., Yates J.R., Stevens S.W. Release of SF3 from the intron branchpoint activates the first step of pre-mRNA splicing. RNA. 2010;16:516–528. doi: 10.1261/rna.2030510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M.S., Stojanov P., Polak P., Kryukov G.V., Cibulskis K., Sivachenko A., Carter S.L., Stewart C., Mermel C.H., Roberts S.A. Mutational heterogeneity in cancer and the search for new cancer genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebreton A., Tomecki R., Dziembowski A., Séraphin B. Endonucleolytic RNA cleavage by a eukaryotic exosome. Nature. 2008;456:993–996. doi: 10.1038/nature07480. [DOI] [PubMed] [Google Scholar]
- Leeuwen J.van, Pons C., Mellor J.C., Yamaguchi T.N., Friesen H., Koschwanez J., Ušaj M.M., Pechlaner M., Takar M., Ušaj M. Exploring genetic suppression interactions on a global scale. Science. 2016;354:aag0839. doi: 10.1126/science.aag0839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Castello A., Fischer B., Leicht S., Föehr S., Frese C.K., Ragan C., Kurscheid S., Pagler E., Yang H. The cardiomyocyte RNA-binding proteome: links to intermediary metabolism and heart disease. Cell Rep. 2016;16:1456–1469. doi: 10.1016/j.celrep.2016.06.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubas M., Christensen M.S., Kristiansen M.S., Domanski M., Falkenby L.G., Lykke-Andersen S., Andersen J.S., Dziembowski A., Jensen T.H. Interaction profiling identifies the human nuclear exosome targeting complex. Mol. Cell. 2011;43:624–637. doi: 10.1016/j.molcel.2011.06.028. [DOI] [PubMed] [Google Scholar]
- Lubas M., Damgaard C.K., Tomecki R., Cysewski D., Jensen T.H., Dziembowski A. Exonuclease hDIS3L2 specifies an exosome-independent 3′–5′ degradation pathway of human cytoplasmic mRNA. EMBO J. 2013;32:1855–1868. doi: 10.1038/emboj.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund M.K., Guthrie C. The DEAD-box protein Dbp5p is required to dissociate Mex67p from exported mRNPs at the nuclear rim. Mol. Cell. 2005;20:645–651. doi: 10.1016/j.molcel.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Makino D.L., Baumgärtner M., Conti E. Crystal structure of an RNA-bound 11-subunit eukaryotic exosome complex. Nature. 2013;495:70–75. doi: 10.1038/nature11870. [DOI] [PubMed] [Google Scholar]
- Makino D.L., Schuch B., Stegmann E., Baumgärtner M., Basquin C., Conti E. RNA degradation paths in a 12-subunit nuclear exosome complex. Nature. 2015;524:54–58. doi: 10.1038/nature14865. [DOI] [PubMed] [Google Scholar]
- Malet H., Topf M., Clare D.K., Ebert J., Bonneau F., Basquin J., Drazkowska K., Tomecki R., Dziembowski A., Conti E. RNA channelling by the eukaryotic exosome. EMBO Rep. 2010;11:936–942. doi: 10.1038/embor.2010.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra S., Sowdhamini R. Sequence search and analysis of gene products containing RNA recognition motifs in the human genome. BMC Genomics. 2014;15:1159. doi: 10.1186/1471-2164-15-1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani R., St.Onge R.P., Hartman J.L., Giaever G., Roth F.P. Defining genetic interaction. Proc. Natl. Acad. Sci. U. S. A. 2008;105:3461–3466. doi: 10.1073/pnas.0712255105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.J. 2011;17:10–12. [Google Scholar]
- Meola N., Domanski M., Karadoulama E., Chen Y., Gentil C., Pultz D., Vitting-Seerup K., Lykke-Andersen S., Andersen J.S., Sandelin A. Identification of a nuclear exosome decay pathway for processed transcripts. Mol. Cell. 2016;64:520–533. doi: 10.1016/j.molcel.2016.09.025. [DOI] [PubMed] [Google Scholar]
- Milbury K.L., Paul B., Lari A., Fowler C., Montpetit B., Stirling P.C. Exonuclease domain mutants of yeast DIS3 display genome instability. Nucleus. 2019;10:21–32. doi: 10.1080/19491034.2019.1578600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P., Petfalski E., Houalla R., Podtelejnikov A., Mann M., Tollervey D. Rrp47p is an exosome-associated protein required for the 3′ processing of stable RNAs. Mol.Cell Biol. 2003;23:6982–6992. doi: 10.1128/MCB.23.19.6982-6992.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherji M., Bell R., Supekova L., Wang Y., Orth A.P., Batalov S., Miraglia L., Huesken D., Lange J., Martin C. Genome-wide functional analysis of human cell-cycle regulators. PNAS. 2006;103:14819–14824. doi: 10.1073/pnas.0604320103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen T.E., Marzluff W.F. Degradation of histone mRNA requires oligouridylation followed by decapping and simultaneous degradation of the mRNA both 5′ to 3′ and 3′ to 5′. Genes Dev. 2008;22:50–65. doi: 10.1101/gad.1622708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami H., Goto D.B., Toda T., Chen E.S., Grewal S.I., Martienssen R.A., Yanagida M. Ribonuclease activity of Dis3 is required for mitotic progression and provides a possible link between heterochromatin and kinetochore function. PLoS One. 2007;2:e317. doi: 10.1371/journal.pone.0000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan V.K., Jones C.I., Newbury S.F., Green P.J. XRN 5′→3′exoribonucleases: structure, mechanisms and functions. Biochim. Biophys. Acta. 2013;1829:590–603. doi: 10.1016/j.bbagrm.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelamraju Y., Hashemikhabir S., Janga S.C. The human RBPome: from genes and proteins to human disease. J. Proteomics. 2015;127:61–70. doi: 10.1016/j.jprot.2015.04.031. [DOI] [PubMed] [Google Scholar]
- Nguyen D., Grenier St-Sauveur V., Bergeron D., Dupuis-Sandoval F., Scott M.S., Bachand F. A polyadenylation-dependent 3′ end maturation pathway is required for the synthesis of the human telomerase RNA. Cell Rep. 2015;13:2244–2257. doi: 10.1016/j.celrep.2015.11.003. [DOI] [PubMed] [Google Scholar]
- Ohn T., Kedersha N., Hickman T., Tisdale S., Anderson P. A functional RNAi screen links O -GlcNAc modification of ribosomal proteins to stress granule and processing body assembly. Nat. Cell Biol. 2008;10:1224–1231. doi: 10.1038/ncb1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X., Yuan D.S., Ooi S.-L., Wang X., Sookhai-Mahadeo S., Meluh P., Boeke J.D. dSLAM analysis of genome-wide genetic interactions in Saccharomyces cerevisiae. Methods. 2007;41:206–221. doi: 10.1016/j.ymeth.2006.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papasaikas P., Tejedor J.R., Vigevani L., Valcárcel J. Functional splicing network reveals extensive regulatory potential of the core spliceosomal machinery. Mol. Cell. 2015;57:7–22. doi: 10.1016/j.molcel.2014.10.030. [DOI] [PubMed] [Google Scholar]
- Paulsen R.D., Soni D.V., Wollman R., Hahn A.T., Yee M.-C., Guan A., Hesley J.A., Miller S.C., Cromwell E.F., Solow-Cordero D.E. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol. Cell. 2009;35:228–239. doi: 10.1016/j.molcel.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preti M., O’Donohue M.-F., Montel-Lehry N., Bortolin-Cavaillé M.-L., Choesmel V., Gleizes P.-E. Gradual processing of the ITS1 from the nucleolus to the cytoplasm during synthesis of the human 18S rRNA. Nucleic Acids Res. 2013;41:4709–4723. doi: 10.1093/nar/gkt160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prideaux S.M., Conway O’Brien E., Chevassut T.J. The genetic architecture of multiple myeloma. Adv. Hematol. 2014;2014:864058. doi: 10.1155/2014/864058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puno M.R., Lima C.D. Structural basis for MTR4–ZCCHC8 interactions that stimulate the MTR4 helicase in the nuclear exosome-targeting complex. PNAS. 2018;115:E5506–E5515. doi: 10.1073/pnas.1803530115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remm M., Storm C.E.V., Sonnhammer E.L.L. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons11Edited by F. Cohen. J. Mol. Biol. 2001;314:1041–1052. doi: 10.1006/jmbi.2000.5197. [DOI] [PubMed] [Google Scholar]
- Schaeffer D., Tsanova B., Barbas A., Reis F.P., Dastidar E.G., Sanchez-Rotunno M., Arraiano C.M., van Hoof A. The exosome contains domains with specific endoribonuclease, exoribonuclease and cytoplasmic mRNA decay activities. Nat. Struct. Mol. Biol. 2009;16:56–62. doi: 10.1038/nsmb.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer D., Clark A., Klauer A.A., Tsanova B., van Hoof A. Functions of the cytoplasmic exosome. Adv. Exp. Med. Biol. 2011;702:79–90. doi: 10.1007/978-1-4419-7841-7_7. [DOI] [PubMed] [Google Scholar]
- Schilders G., van Dijk E., Pruijn G.J.M. C1D and hMtr4p associate with the human exosome subunit PM/Scl-100 and are involved in pre-rRNA processing. Nucleic Acids Res. 2007;35:2564–2572. doi: 10.1093/nar/gkm082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid M., Jensen T.H. Nuclear quality control of RNA polymerase II transcripts. WIREs RNA. 2010;1:474–485. doi: 10.1002/wrna.24. [DOI] [PubMed] [Google Scholar]
- Schmidt C., Kowalinski E., Shanmuganathan V., Defenouillère Q., Braunger K., Heuer A., Pech M., Namane A., Berninghausen O., Fromont-Racine M. The cryo-EM structure of a ribosome–Ski2-Ski3-Ski8 helicase complex. Science. 2016;354:1431–1433. doi: 10.1126/science.aaf7520. [DOI] [PubMed] [Google Scholar]
- Schöfl G. 2016. Reutils: Talk to the NCBI EUtils. [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S., Schmidt J.C., Goldfarb K.C., Cech T.R., Parker R. Inhibition of telomerase RNA decay rescues telomerase deficiency caused by dyskerin or PARN defects. Nat. Struct. Mol. Biol. 2016;23:286–292. doi: 10.1038/nsmb.3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan K.E., Mattijssen S., Lebaron S., Tollervey D., Pruijn G.J.M., Watkins N.J. Both endonucleolytic and exonucleolytic cleavage mediate ITS1 removal during human ribosomal RNA processing. J. Cell Biol. 2013;200:577–588. doi: 10.1083/jcb.201207131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S.B., Kiss D.L., Turk E., Tartakoff A.M., Andrulis E.D. Pronounced and extensive microtubule defects in a Saccharomyces cerevisiae DIS3 mutant. Yeast. 2011;28:755–769. doi: 10.1002/yea.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snee M.J., Wilson W.C., Zhu Y., Chen S.-Y., Wilson B.A., Kseib C., O’Neal J., Mahajan N., Tomasson M.H., Arur S. Collaborative control of cell cycle progression by the RNA exonuclease Dis3 and Ras is conserved across species. Genetics. 2016;203:749–762. doi: 10.1534/genetics.116.187930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolova M., Turunen M., Mortusewicz O., Kivioja T., Herr P., Vähärautio A., Björklund M., Taipale M., Helleday T., Taipale J. Genome-wide screen of cell-cycle regulators in normal and tumor cells identifies a differential response to nucleosome depletion. Cell Cycle. 2016;16:189–199. doi: 10.1080/15384101.2016.1261765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staals R.H.J., Bronkhorst A.W., Schilders G., Slomovic S., Schuster G., Heck A.J.R., Raijmakers R., Pruijn G.J.M. Dis3-like 1: a novel exoribonuclease associated with the human exosome. EMBO J. 2010;29:2358–2367. doi: 10.1038/emboj.2010.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojic L., Lun A.T.L., Mascalchi P., Ernst C., Redmond A.M., Mangei J., Barr A.R., Bousgouni V., Bakal C., Marioni J.C. A high-content RNAi screen reveals multiple roles for long noncoding RNAs in cell division. Nat. Commun. 2020;11:1851. doi: 10.1038/s41467-020-14978-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundararaman B., Zhan L., Blue S.M., Stanton R., Elkins K., Olson S., Wei X., Van Nostrand E.L., Pratt G.A., Huelga S.C. Resources for the comprehensive discovery of functional RNA elements. Mol. Cell. 2016;61:903–913. doi: 10.1016/j.molcel.2016.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepińska T., Kalisiak K., Tomecki R., Labno A., Borowski L.S., Kulinski T.M., Adamska D., Kosinska J., Dziembowski A. DIS3 shapes the RNA polymerase II transcriptome in humans by degrading a variety of unwanted transcripts. Genome Res. 2015;25:1622–1633. doi: 10.1101/gr.189597.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczesny R.J., Kowalska K., Klosowska-Kosicka K., Chlebowski A., Owczarek E.P., Warkocki Z., Kulinski T.M., Adamska D., Affek K., Jedroszkowiak A. Versatile approach for functional analysis of human proteins and efficient stable cell line generation using FLP-mediated recombination system. PLoS One. 2018;13:e0194887. doi: 10.1371/journal.pone.0194887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D., Gable A.L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N.T., Morris J.H., Bork P. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanackovic G., Krämer A. Human splicing factor SF3a, but not SF1, is essential for pre-mRNA splicing in vivo. Mol. Biol. Cell. 2005;16:1366–1377. doi: 10.1091/mbc.E04-11-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomecki R., Kristiansen M.S., Lykke-Andersen S., Chlebowski A., Larsen K.M., Szczesny R.J., Drazkowska K., Pastula A., Andersen J.S., Stepien P.P. The human core exosome interacts with differentially localized processive RNases: hDIS3 and hDIS3L. EMBO J. 2010;29:2342–2357. doi: 10.1038/emboj.2010.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomecki R., Drazkowska K., Kucinski I., Stodus K., Szczesny R.J., Gruchota J., Owczarek E.P., Kalisiak K., Dziembowski A. Multiple myeloma-associated hDIS3 mutations cause perturbations in cellular RNA metabolism and suggest hDIS3 PIN domain as a potential drug target. Nucleic Acids Res. 2014;42:1270–1290. doi: 10.1093/nar/gkt930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong A.H.Y., Evangelista M., Parsons A.B., Xu H., Bader G.D., Pagé N., Robinson M., Raghibizadeh S., Hogue C.W.V., Bussey H. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- Tran E.J., Zhou Y., Corbett A.H., Wente S.R. The DEAD-box protein Dbp5 controls mRNA export by triggering specific RNA:protein remodeling events. Mol. Cell. 2007;28:850–859. doi: 10.1016/j.molcel.2007.09.019. [DOI] [PubMed] [Google Scholar]
- Tseng C.-K., Wang H.-F., Burns A.M., Schroeder M.R., Gaspari M., Baumann P. Human telomerase RNA processing and quality control. Cell Rep. 2015;13:2232–2243. doi: 10.1016/j.celrep.2015.10.075. [DOI] [PubMed] [Google Scholar]
- Tseng C.-K., Wang H.-F., Schroeder M.R., Baumann P. The H/ACA complex disrupts triplex in hTR precursor to permit processing by RRP6 and PARN. Nat. Commun. 2018;9:5430. doi: 10.1038/s41467-018-07822-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuck A.C., Rankova A., Arpat A.B., Liechti L.A., Hess D., Iesmantavicius V., Castelo-Szekely V., Gatfield D., Bühler M. Mammalian RNA decay pathways are highly specialized and widely linked to translation. Mol. Cell. 2020;77:1222–1236.e13. doi: 10.1016/j.molcel.2020.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usaj M., Tan Y., Wang W., VanderSluis B., Zou A., Myers C.L., Costanzo M., Andrews B., Boone C. TheCellMap.org: a web-accessible database for visualizing and mining the global yeast genetic interaction network. G3: Genes Genomes Genet. 2017;7:1539–1549. doi: 10.1534/g3.117.040220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ustianenko D., Hrossova D., Potesil D., Chalupnikova K., Hrazdilova K., Pachernik J., Cetkovska K., Uldrijan S., Zdrahal Z., Vanacova S. Mammalian DIS3L2 exoribonuclease targets the uridylated precursors of let-7 miRNAs. RNA. 2013;19:1632–1638. doi: 10.1261/rna.040055.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasu S., Shah S., Orjalo A., Park M., Fischer W.H., Forbes D.J. Novel vertebrate nucleoporins Nup133 and Nup160 play a role in mRNA export. J.Cell Biol. 2001;155:339–354. doi: 10.1083/jcb.200108007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl M.C., Will C.L., Lührmann R. The spliceosome: design Principles of a dynamic RNP machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- Wang M., Pestov D.G. 5′-end surveillance by Xrn2 acts as a shared mechanism for mammalian pre-rRNA maturation and decay. Nucleic Acids Res. 2011;39:1811–1822. doi: 10.1093/nar/gkq1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Rio D.C. JUM is a computational method for comprehensive annotation-free analysis of alternative pre-mRNA splicing patterns. PNAS. 2018;115:E8181–E8190. doi: 10.1073/pnas.1806018115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasmuth E.V., Januszyk K., Lima C.D. Structure of an Rrp6–RNA exosome complex bound to poly(A) RNA. Nature. 2014;511:435–439. doi: 10.1038/nature13406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weick E.-M., Puno M.R., Januszyk K., Zinder J.C., DiMattia M.A., Lima C.D. Helicase-dependent RNA decay illuminated by a cryo-EM structure of a human nuclear RNA exosome-MTR4 complex. Cell. 2018;173:1663–1677.e21. doi: 10.1016/j.cell.2018.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. Springer; 2016. ggplot2: Elegant Graphics for Data Analysis. [Google Scholar]
- Wickham H., Henry L., RStudio . 2020. tidyr: Tidy Messy Data. [Google Scholar]
- Wickham H., François R., Henry L., Müller K., RStudio . 2020. dplyr: A Grammar of Data Manipulation. [Google Scholar]
- Yu G., Wang L.-G., Han Y., He Q.-Y. clusterProfiler: an R Package for comparing biological themes among gene clusters. OMICS: J. Integr. Biol. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
RNA-Seq data have been deposited at GEO and are publicly aviable as of the date of publication. The assesion number is: GSE155123
-
•
The code for screen data analysis (development version) is available at GitHub (https://github.com/olobiolo/siscreenr)
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.