

Synopsis:
Pancreatic cancer is the third leading cause of cancer death in the United States, with a five-year survival rate of 9%. Individuals with inherited pancreatic cancer syndromes are at increased risk for developing pancreatic cancer and may benefit from pancreatic cancer surveillance with the goal to detect and intervene upon early-stage cancer or high-risk precursor lesions. Given screening implications for family members and therapeutic implications for probands, all patients diagnosed with pancreatic cancer are recommended to undergo germline genetic testing.
Keywords: Hereditary, cancer risk, pancreatic cyst, surveillance
Introduction
In 2020, there will be an estimated 57,600 new cases and 47,050 deaths from pancreatic ductal adenocarcinoma (PDAC) in the United States.1 With a five-year survival rate of 9%, PDAC remains a highly lethal disease and often is diagnosed at an advanced, incurable stage.2 Despite this, routine surveillance of the general population is not recommended due to low incidence and lack of evidence for clinical benefit in asymptomatic individuals.3
While the lifetime risk for developing PDAC in the general population is only 1.6%,2 a subset of individuals is at increased risk based on inheritance of a germline pathogenic mutation and/or presence of familial PDAC, defined as families with at least two first-degree relatives (FDR) with PDAC without known genetic cause. An underlying hereditary susceptibility is identified in 10-20% of pancreatic adenocarcinoma patients,4-9 and professional society guidelines now recommend universal germline testing for all patients diagnosed with PDAC regardless of family history or age at cancer diagnosis.10 While testing may have implications for the proband’s cancer treatment, such as with poly (ADP-ribose) polymerase (PARP) inhibitors for BRCA1 or BRCA2 mutation carriers11 or anti-PD-1 antibodies for those with Lynch syndrome,12 results may also risk stratify relatives via cascade testing, which entails testing for the known pathogenic mutation in at-risk family members.
Individuals with inherited PDAC syndromes may benefit from surveillance with cross-sectional imaging studies or endoscopic ultrasound (EUS). The goal of routine screening of high-risk individuals is to improve cancer-associated survival by detecting and intervening early upon non-invasive precursor lesions or early-stage cancers. This review will discuss known inherited PDAC syndromes, goals and methods of screening, as well as surveillance outcomes among high-risk individuals.
Inherited PDAC syndromes
Hereditary Pancreatitis
Hereditary pancreatitis (HP) is a rare syndrome of acute recurrent pancreatitis that frequently leads to the development of chronic pancreatitis by young adulthood. HP is most commonly inherited in an autosomal dominant fashion due to pathogenic alterations in PRSS1 (cationic trypsinogen) (Table 1). Other genes that have been identified in families with hereditary or familial pancreatitis include SPINK1, CTRC, CFTR, CPA1 and CPB1. PRSS1 mutation carriers have a high penetrance of both pancreatitis and PDAC,13 although a recent study found only 7.2% of PRSS1 mutation carriers (83% of whom had pancreatitis) developed PDAC by age 70.14 Age at diagnosis may depend on time from first clinical episode of pancreatitis13 and history of cigarette smoking, with ever smokers more likely to develop PDAC, and doing so at a median of 20 years earlier than never smokers.15
Table 1:
Inherited PDAC syndromes associated with increased PDAC risk
| Syndrome | Gene name | Lifetime risk for pancreatic cancer | Genetic mutation frequency in pancreatic cancer cohorts | Screening recommendations* |
|---|---|---|---|---|
| Hereditary pancreatitis | PRSS1 | 7–40% 13,78 | < 0.1 | Start at age 40 years or 20 years after developing pancreatitis |
| Peutz-Jeghers syndrome | STK11 | ll-36%19,20 | < 0.1 | Start at age 30-35 years or 10 years younger than the earliest diagnosis in the family |
| Familial atypical mole and multiple melanoma syndrome | CDK2NA | 17%22 | 0.3-0.7%5,8,9,24 | Start at age 40 years or 10 years younger than the earliest diagnosis in the family |
| Hereditary breast and ovarian cancer |
BRCA1
BRCA2 |
3%79 5-10%30 |
0.3-1.3%5,8,9,31 1.3-3.6%5,8,9,31 |
If ≥ 1 FDR or SDR with PDAC, start at age 50 or 10 years younger than the earliest diagnosis in the family |
| Lynch syndrome | MLH1,MSH2,MSH6 | 4%36 | 1.3-3.6%5,8,9,31 | If ≥ 1 FDR or SDR with PDAC, start at age 50 or 10 years younger than the earliest diagnosis in the family |
| Ataxia telangiectasia | ATM | Unknown | 1.2-3.3%5,6,8,9 | If ≥ 1 FDR or SDR with PDAC, start at age 50 or 10 years younger than the earliest diagnosis in the family |
| Other | PALB2 | 2-3%43 | 0.2-0.4 %5,6,8,9 | If ≥ 1 FDR or SDR with PDAC, start at age 50 or 10 years younger than the earliest diagnosis in the family |
| Li-Fraumeni syndrome | TP53 | Unknown | 0.12-0.5%.5-7 | If ≥ 1 FDR or SDR with PDAC, start at age 50 or 10 years younger than the earliest diagnosis in the family |
| Familial PDAC- | Unknown | Varies with # FDRs with PDAC50 |
Not applicable | Start at age 50 years or 10 years younger than the earliest diagnosis in the family |
Long-standing pancreatitis is considered required for the subsequent development of PDAC in mutation carriers, although it may not be always symptomatic. In fact, a recent study of patients with PDAC and deleterious CPA1 and CPB1 variants demonstrated that many patients did not have a prior history of symptomatic pancreatitis.16 Currently, it is recommended to test for the presence of pancreatitis risk variants only in probands with a clinical and family history suggestive of HP as the population prevalence of HP is only 0.3/100,000 (with 68% of cases due to PRSS1 alterations).17
Peutz-Jeghers Syndrome
Peutz-Jeghers Syndrome (PJS) is an autosomal dominant hamartomatous polyposis syndrome most commonly arising in the setting of pathogenic alterations in the STK11/LKB1 gene, a tumor suppressor gene involved in multiple processes related to metabolic regulation. The clinical diagnosis requires 2 out of 3 criteria: at least 2 hamartomas in the gastrointestinal tract; mucocutaneous hyperpigmentation; and/or family history of PJS.
Patients with PJS are at increased risk of multiple gastrointestinal cancers (including colorectal, stomach, small intestine, pancreas), as well as female breast, gynecologic, testicular and lung cancers. A systematic review reported an overall lifetime risk of any cancer of 37-93% with a mean age at diagnosis of 42 years.18 One large study found the cumulative risk of PDAC to be 3%, 5%, 7%, and 11% at ages 40, 50, 60, and 70 years respectively,19 although a meta-analysis of 6 studies calculated an absolute rate of 118.6 cases of PDAC per 100,000 person-years, corresponding to a cumulative risk of 36% by age 64.20
Familial atypical mole and multiple melanoma syndrome
Familial atypical mole and multiple melanoma (FAMMM) syndrome is an autosomal dominant syndrome diagnosed in individuals with a personal history of >50 atypical nevi and a family history of melanoma. FAMMM is most commonly caused by pathogenic alterations in the cell cycle gene CDKN2A, a gene which encodes for p16 and p14ARF.
The association of FAMMM with PDAC was first reported in 1991, although families with coexisting melanoma and PDAC were reported 20 years earlier.21 An analysis of the Dutch FAMMM registry identified a specific 19 base-pair deletion in exon 2 of CDKN2A (“p16-Leiden”) which conferred a risk of PDAC of 17% by age 75.22 The age specific risk was < 1% for carriers at age 40 years and 4% at age 50 years. Smoking further modifies these risks.23
In one large PDAC cohort, the prevalence of CDKN2A mutations was 0.6%, including 3.3% among those with a FDR affected with PDAC, and 5.3% with FDR with melanoma.24 Consistent with this study, CDKN2A mutations have been identified in other unselected PDAC cohorts at a frequency of 0.3-0.7%.5,8,9
BRCA-associated cancer
BRCA-associated cancers include breast, ovarian, pancreatic and prostate cancer. BRCA-associated cancer is often referred to as hereditary breast and ovarian cancer (HBOC), an autosomal dominant syndrome caused by pathogenic alterations in BRCA1 and BRCA2. BRCA1 and BRCA2 mutations are present in about 1 out of every 40 Ashkenazi Jews25 and in about 1 out of every 300-465 women in the general population.26 Both BRCA1 and BRCA2 are tumor suppressor genes involved in homologous recombination, a form of DNA repair.
The association of BRCA alterations with PDAC was noted as early as 199627 with larger cohort studies subsequently identifying an increased risk for PDAC among both BRCA1 (2.3 fold)28 and BRCA2 (3.5-6 fold) mutation carriers.29,30 BRCA2 alterations are one of the most common cancer predisposition genes identified on germline genetic testing with rates of 1.3-3.6%5,6,8,9,31 among unselected patients with PDAC, and approximately double this rate32,33 among cohorts with familial PDAC and/or Ashkenazi Jewish ancestry. The frequency of BRCA1 mutation carriers identified in unselected PDAC cohorts is 0.3-1.3%.5,8,9,31
Among BRCA-associated cancers that have deficient homologous recombination, treatment with PARP inhibitors can selectively induce tumor cell death.11 PARP inhibitors are currently FDA-approved in specific settings for BRCA-associated ovarian (first approved in 2014), breast (2018), pancreatic (2019) and prostate (2020) cancers.
Lynch syndrome
Lynch syndrome is an autosomal dominant cancer predisposition syndrome caused by pathogenic alterations in mismatch repair genes (MLH1, MSH2, MSH6, PMS2) and EPCAM with an estimated population prevalence of 1 in 279.34 Lynch carriers are at increased lifetime risk for a wide spectrum of cancers, most commonly colorectal and endometrial cancer but also ovarian, urinary tract, gastric, small bowel, brain, sebaceous neoplasms and PDACs.35
In a study including 147 Lynch syndrome (MLH1, MSH2, MSH6) mutation carriers, 21% had at least one family member with PDAC.36 The same study found a 1.3% cumulative risk for developing PDAC by age 50, and 3.7% risk by age 70.
Lynch syndrome-associated cancers characteristically possess microsatellite instability (changes in lengths of repetitive DNA sequences) as a result of defective mismatch repair. Microsatellite instability-high (MSI-H) and mismatch repair deficient (MMR-D) tumors can be identified by evaluating tumor tissue with immunohistochemistry for the presence or absence of the MLH1, MSH2, MSH6 and PMS2 proteins, MSI testing, or next generation DNA sequencing.37-39 Patients with PDACs that are MSI-H or MMR-D are candidates for immune checkpoint inhibitors.12
ATM
ATM encodes for a protein important in DNA strand break repair. Homozygous ATM alterations lead to the syndrome of ataxia-telangiectasia, an autosomal recessive condition characterized by ataxia, increased sensitivity to radiation, and an over 100-fold increased risk of hematologic malignancies and other cancers.40 Germline ATM mutations are significantly more common in patients with familial PDAC compared to controls.41 Heterozygote ATM carriers are at increased risk for developing cancer, including a moderate increased risk of breast and PDAC; with possible risks for colon, prostate and ovarian cancer.41,42 One large case-control study found 5.7 fold increased odds of having ATM mutations in sporadic pancreatic ductal adenocarcinoma (PDAC) cases compared to controls.8 In cohorts of unselected PDAC patients, ATM variants have been identified in 1.2-3.3%.5,6,8,9
PALB2
PALB2 (partner and localizer of BRCA2) gene encodes a protein that interacts with and stabilizes the BRCA2 protein, with overlap in cancer predisposition between BRCA2 and PALB2 pathogenic variants. In an international study of 524 families with PALB2 alterations, the risk to age 80 years for female breast cancer was 53%, 5% for ovarian (RR 2.91), 2-3% for pancreatic (RR 2.37) and 1% for male breast cancer (RR 7.34).43 PALB2 mutations have been identified in 3-4% of familial44,45 and 0.2-0.4% of unselected PDAC cohorts.5,6,8,9
Li-Fraumeni syndrome
Li-Fraumeni syndrome (LFS) is an autosomal dominant highly penetrant syndrome caused by germline mutations in the TP53 gene, a tumor suppressor gene that regulates many processes involved in cell cycle and DNA repair. The estimated prevalence is about 1 in every 3,500-5,500 individuals, 46 and an analysis of 286 LFS patients in the National Cancer Institute (NCI) LFS study reported a cancer incidence of almost 100% for both sexes by age 70, but only 5 PDACs.47 In cohorts of unselected PDAC patients, TP53 mutations have been identified in 0.12-0.5%.5-7,48
Familial PDAC
Familial PDAC (FPC) kindreds are defined as those families with at least two FDRs with PDAC, without a known predisposing genetic mutation. In fact, in one study including 185 FPC patients, less than 15% were found to have informative germline testing.49 Even in the absence of an identifiable pathogenic mutation, FPC kindred patients are at increased risk for PDAC. An analysis of the prospective National Familial Pancreas Tumor Registry compared the number of PDACs observed to the expected number using National Cancer Institute (NCI) Surveillance Epidemiology and End Results (SEER) incidence rates and found that members of FPC kindreds with one FDR had 4.6 fold (95% CI 0.5-16.4) increased risk, 2 FDRs had a 6.4 fold (95% CI 1.8-16.4) increased risk, and those with 3 or more FDRs had a 32-fold (95% CI, 10.2-74.7) increased risk compared to SEER incidence rates.50
Goals of pancreatic surveillance
The International Cancer of the Pancreas Screening (CAPS) Consortium proposed the primary objectives for PDAC surveillance as the detection and treatment of stage I PDAC and PDAC precursor lesions with high-grade dysplasia, including pancreatic intraepithelial neoplasia (PanIN) and intraductal papillary mucinous neoplasms (IPMNs).51 Surveillance is associated with down-staging of diagnosed PDACs, especially among those who maintain an annual surveillance schedule.52,53 Recent analysis of NCI SEER data found survival of Stage I pancreatic ductal adenocarcinoma has been improving, with an 80% 5-year survival among patients who undergo pancreatic resection, highlighting the potential of early detection strategies.54
Whom to screen and when
The US Preventative Task Force recommends against screening for PDAC in asymptomatic adults in the general population, given no evidence for reduction in mortality and the potential for greater harm than benefit.3 Importantly, this recommendation does not apply to those individuals at high risk due to genetics and/or family history. Multiple professional society guidelines now recommend surveillance10,51,55,56 specifically for individuals with: (1) PJS, FAMMM or hereditary pancreatitis, (2) inherited pathogenic alterations in ATM, BRCA1, BRCA2, MLH1, MSH2, MSH6, PALB2, or TP53 and a family history of PDAC in at least one FDR51 or second-degree relative (SDR)10, and (3) familial PDAC with at least one FDR and one SDR with PDAC. The presence of a single FDR or SDR with PDAC (in the absence of a known genetic mutation or inherited PDAC risk syndrome) is not sufficient to recommend surveillance.
When PDAC screening is indicated, the optimal age to begin screening is not well established. Consensus guidelines from the CAPS Consortium51 and National Comprehensive Cancer Network10 recommend a gene/syndrome-specific approach. Carriers of high-risk genes or genetic syndromes (due to mutations in ATM, BRCA1, BRCA2, MLH1, MSH2, MSH6, PALB2, or TP53) with at least one FDR51 or SDR10 with PDAC may begin screening at age 50 years or 10 years younger than the earliest PDAC in the family (whichever is earliest). Screening should begin at a younger age for patients with PJS (age 30-35 years) and hereditary pancreatitis (age 40 years or 20 years after onset of pancreatitis) and CDKN2A mutation carriers (age 40 years or within 10 years of the earliest PDAC in the family). It is also not well established when or at what age screening should be discontinued, and this decision requires consideration of a patient’s comorbidities and life expectancy, perceived cancer risk, and personal preferences. In general, it is important that providers have an informed discussion with patients before starting a surveillance program.
Methods of screening
Visualization of the pancreas
Professional society guidelines recommend a combination of EUS and magnetic resonance imaging/magnetic resonance cholangiopancreatography (MRI/MRCP) to evaluate the pancreas in appropriate high-risk individuals.10,51,57 In a multicenter blinded study evaluating the diagnostic yield of EUS and MRI, the former was found to be more sensitive for the detection of small, solid lesions and chronic pancreatic type parenchymal changes, whereas the latter was more sensitive for detection of small, cystic lesions, suggesting an additive rather than duplicative effect of the two imaging techniques.58 In another study where patients underwent CT, MRI and EUS, CT imaging often missed subcentimeter pancreatic cysts detected by MRI and EUS.59 Given the complementary information provided by MRI and EUS, neither of which exposes patients to radiation, these modalities are preferentially used for routine high-risk pancreatic screening.
Among patients without concerning features on baseline imaging, alternating EUS and MRI screening may be performed annually (Figure 1).51 CAPS guidelines recommend that for concerning abnormalities (cysts with worrisome features, solid lesions, main pancreatic duct stricture and/or dilation of ≥6mm without a mass), EUS with fine needle aspiration (FNA) and/or CT imaging be obtained.51 Surveillance intervals should be shortened when worrisome features are present without clear evidence of cancer (3-6 months rather than every 12 months).51 Importantly, physician discretion and clinical concern based on patient characteristics and family history should be incorporated into the surveillance paradigm for all high-risk patients.
Figure 1: Approach to surveillance intervals for high-risk pancreatic cancer screening (per CAPS Consortium guidelines).
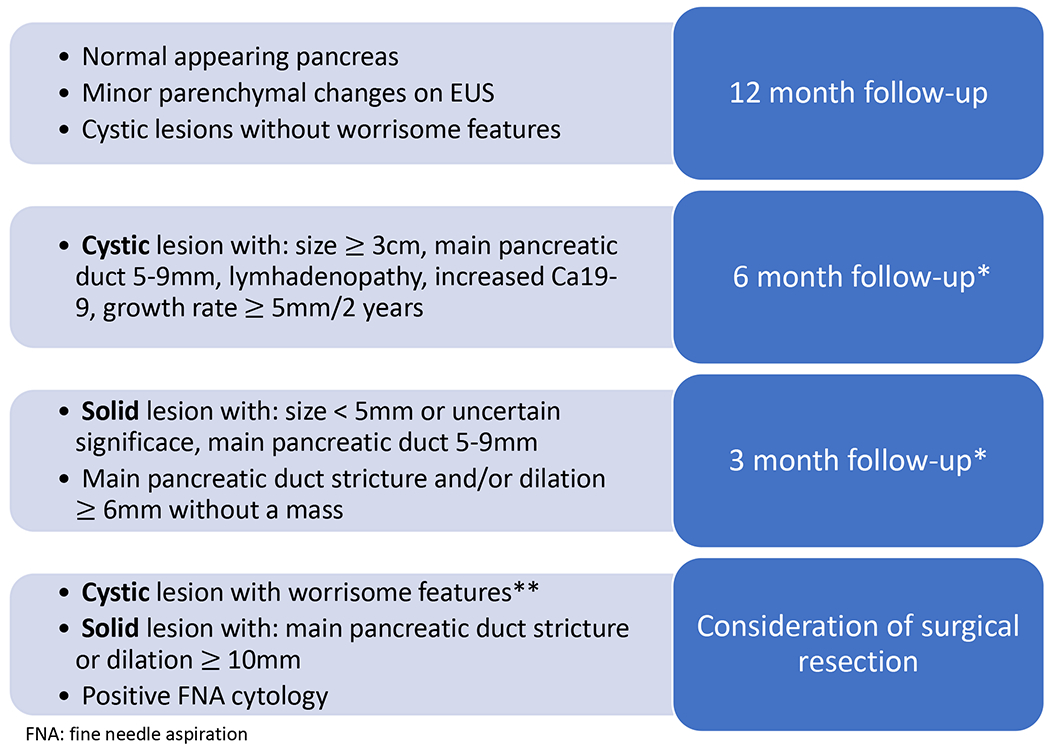
* For concerning features (including findings listed in the 6 month follow-up and 3 month follow-up boxes), fine needle aspiration and/or CT imaging should be considered to better characterize the lesion and assess risk for malignancy. In addition to MRI and EUS findings, patient specific risk factors (including clinical factors and family history) may also be incorporated into risk assessment and lead to earlier tissue sampling or more frequent follow-up per provider discretion.
** Worrisome features of cystic lesions include: mural nodule, enhanced solid component, thickened or enhanced cyst walls, abrupt main pancreatic duct caliber change or size ≥ 10mm, or patient symptoms (pancreatitis, jaundice, pain)
Biomarkers
In middle-aged and older adults, new-onset diabetes (NOD) can be a harbinger of PDAC. In a population based study of 2,122 individuals with diabetes diagnosed at ≥50 years of age, 0.85% were diagnosed with PDAC within 3 years of diabetes diagnosis.60 Similarly, in a prospective cohort study of 112,818 individuals from the Nurses’ Health Study and Health Professionals Follow-Up Study, NOD was associated with an increased risk of PDAC, and this risk was higher still in patients with NOD accompanied by unintentional weight loss and older age.61 While NOD and monitoring of fasting blood sugar or hemoglobin A1c have not been extensively evaluated in populations with inherited PDAC syndromes, CAPS consortium guidelines recommend serial measurement of fasting glucose and/or HgbA1c, with rising blood glucose suggestive of the need for closer surveillance.51
Carbohydrate antigen 19-9 (CA19-9) is a serum biomarker that has been associated with PDAC disease stage, overall survival time and response to treatment. The use of CA19-9 for general population screening is not recommended. However, in a multi-institutional analysis, elevation of CA19-9 had a sensitivity of 64% at 99% specificity for newly diagnosed resectable PDAC compared to healthy controls.62 Elevated CA19-9 levels were also detected in the year prior to PDAC diagnosis, with a sensitivity of 60% at 99% specificity within the 6 months prior to diagnosis.62 Similar results were found in a large study comparing subjects undergoing pancreatic surveillance with those with early-stage PDAC.63 The CAPS consortium recommends considering measurement of CA19-9 at the time of enhanced clinical concern for PDAC, such as with worrisome findings on imaging.51
Outcomes from surveillance
Results from imaging surveillance
Small cysts are common (> 40%) among inherited PDAC syndrome patients undergoing surveillance,59 although the vast majority are not worrisome or at significant risk of neoplastic transformation. The pattern of cysts or lesions may differ in size or progression risk depending on the underlying predisposition prompting screening,64 and germline mutation carriers are at higher risk for high grade dysplasia or cancer compared to FPC patients without known mutation.65
Imaging and clinical features associated with progression
Just as there are certain worrisome features seen on pancreas imaging among the general population that require monitoring or other intervention,66 certain imaging findings in inherited PDAC syndrome patients may necessitate shorter follow-up or intervention due to risk of neoplastic progression (Figure 1). In a CAPS analysis of 354 high-risk patients, 24 (7%) had progression (10 high-grade dysplasia, 14 pancreatic adenocarcinoma) over 16 years of evaluation.53 This corresponds to a 1.6% per year progression rate, with 93% of patients having a worrisome finding before being diagnosed with high-grade dysplasia or adenocarcinoma.53
Surgical outcomes and survival
The decision to pursue surgical intervention requires multidisciplinary review and evaluation of patient specific factors. As surgery remains the only treatment with the potential to cure PDAC, the goal of surveillance is to detect high-risk precursors or Stage I invasive cancer that can be removed surgically. In a series of 10 PDACs detected during surveillance of 354 high-risk patients, 9 had an R0 resection and 90% were alive at one year with 60% alive at 5 years.67 Another analysis of 71 patients who underwent pancreatic resection during surveillance found no difference in survival between those with low-risk and high-risk precursors, although survival was poorer in those diagnosed with pancreatic adenocarcinoma.68 A meta-analysis of high-risk cohort studies found 253 to 281 high-risk patients would need to be screened to prevent one death from PDAC,69 although further studies are needed to confirm these results.
Future investigations
New PDAC susceptibility genes
Germline genetic testing is now recommended for all patients diagnosed with PDAC,10 and the number of genes evaluated will continue to expand as researchers study PDAC prone families. For example, whole-genome sequencing of one such family led to the identification of a germline truncating mutation in the RABL3 gene, which alters RAS pathway regulation.70 Additionally, genome wide association studies (GWAS) have identified common genetic variants that predispose to PDAC with much lower penetrance, but may ultimately allow for clinically meaningful risk stratification using calculated genetic risk scores.71-73
New methods for earlier PDAC detection
The development of new circulating biomarkers for early detection of PDAC remains an active area of investigation. A number of tests are in development that measure circulating tumor DNA, proteins, metabolites and other types of markers in the blood.74,75 Other approaches are evaluating pancreatic juice, including with next generation DNA sequencing to detect mutations associated with high-grade dysplasia or invasive cancer.76 Molecular analysis of pancreatic cyst fluid is also being evaluated for its utility to distinguishing cysts that require resection, surveillance or neither.77
Summary.
For the subset of individuals at increased risk of PDAC due to an inherited PDAC syndrome, PDAC surveillance holds promise as a way to improve survival by detecting and intervening upon noninvasive dysplastic precursors or early cancers. Further studies are required to more clearly define the appropriate populations for screening and quantify the benefits and risks of screening programs.
Clinics Care Points.
PDAC has poor overall survival and is often diagnosed at an advanced stage.
Individuals with inherited PDAC syndromes may benefit from PDAC surveillance.
For most patients, surveillance involves annual pancreatic imaging utilizing EUS and MRI/MRCP which provide complementary views of the pancreas and are the current standard of care for PDAC screening.
The goal of PDAC surveillance is to detect and intervene early upon high-risk noninvasive precursors or early-stage cancers. While surveillance has not been definitively shown to improve survival, it is associated with down-staging of detected PDACs, and efforts are ongoing to refine surveillance approaches and understand their potential benefits.
Key points:
Individuals with inherited pancreatic cancer predisposition syndromes (both due to germline pathogenic alterations and family history of pancreatic cancer) may benefit from pancreatic cancer surveillance with endoscopic ultrasound and magnetic resonance imaging.
The goal of surveillance is to identify and intervene upon high-risk precursor lesions or early-stage cancers.
Annual pancreatic surveillance results in down-staging of pancreatic cancers that are detected.
Funding Support:
BMW is supported by the Hale Family Center for Pancreatic Cancer Research, Lustgarten Foundation Dedicated Laboratory program, NIH grant U01 CA210171, NIH grant P50 CA127003, Stand Up to Cancer, Pancreatic Cancer Action Network, Noble Effort Fund, Wexler Family Fund, and Promises for Purple. MG is supported by the National Cancer Institute (U01210170, CA62924 and R01CA176828 and by a Stand Up To Cancer-Lustgarten Foundation Pancreatic Cancer Interception Translational Cancer Research Grant (Grant Number: SU2C-AACR-DT25-17). Stand Up To Cancer is a program of the Entertainment Industry Foundation. SU2C research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C.
Disclosures:
BMW declares grant funding from Celgene and Eli Lilly, and consulting for BioLineRx, Celgene, and GRAIL.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Leah H. Biller, Department of Medical Oncology, Dana-Farber Cancer Institute and Harvard Medical School, 450 Brookline Ave, Boston, MA.
Brian M. Wolpin, Department of Medical Oncology, Dana-Farber Cancer Institute and Harvard Medical School, 450 Brookline Ave, Boston, MA.
Michael Goggins, Professor of Pathology, Medicine and Oncology, Johns Hopkins University, 1800 Orleans St Baltimore MD.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. [DOI] [PubMed] [Google Scholar]
- 2.National Cancer Institute. Cancer Stat Facts: Pancreatic Cancer. Available at: https://seer.cancer.gov/statfacts/html/pancreas.html. Accessed 12/5/2020.
- 3.U.S Preventive Services Task Force, Owens DK, Davidson KW, et al. Screening for Pancreatic Cancer: US Preventive Services Task Force Reaffirmation Recommendation Statement. JAMA. 2019;322(5):438–444. [DOI] [PubMed] [Google Scholar]
- 4.Roberts NJ, Norris AL, Petersen GM, et al. Whole Genome Sequencing Defines the Genetic Heterogeneity of Familial Pancreatic Cancer. Cancer Discov. 2016;6(2):166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yurgelun MB, Chittenden AB, Morales-Oyarvide V, et al. Germline cancer susceptibility gene variants, somatic second hits, and survival outcomes in patients with resected pancreatic cancer. Genet Med. 2019;21(1):213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shindo K, Yu J, Suenaga M, et al. Deleterious Germline Mutations in Patients With Apparently Sporadic Pancreatic Adenocarcinoma. J Clin Oncol. 2017;35(30):3382–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lowery MA, Wong W, Jordan EJ, et al. Prospective Evaluation of Germline Alterations in Patients With Exocrine Pancreatic Neoplasms. J Natl Cancer Inst. 2018;110(10):1067–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu C, Hart SN, Polley EC, et al. Association Between Inherited Germline Mutations in Cancer Predisposition Genes and Risk of Pancreatic Cancer. JAMA. 2018;319(23):2401–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brand R, Borazanci E, Speare V, et al. Prospective study of germline genetic testing in incident cases of pancreatic adenocarcinoma. Cancer. 2018;124(17):3520–3527. [DOI] [PubMed] [Google Scholar]
- 10.NCCN Guidelines Version 2.2021 Genetic/Family High-Risk Assessment: Breast, Ovarian, and Pancreatic. Available at: https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf. Accessed December 20, 2020. [Google Scholar]
- 11.Golan T, Hammel P, Reni M, et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N Engl J Med. 2019;381(4):317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marabelle A, Le DT, Ascierto PA, et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J Clin Oncol. 2020;38(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lowenfels AB, Maisonneuve P, DiMagno EP, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst. 1997;89(6):442–446. [DOI] [PubMed] [Google Scholar]
- 14.Shelton CA, Umapathy C, Stello K, Yadav D, Whitcomb DC. Hereditary Pancreatitis in the United States: Survival and Rates of Pancreatic Cancer. Am J Gastroenterol. 2018;113(9):1376. doi: 1310.1038/s41395-41018-40194-41395. Epub 42018 Jul 41318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lowenfels AB, Maisonneuve P, Whitcomb DC, Lerch MM, DiMagno EP. Cigarette smoking as a risk factor for pancreatic cancer in patients with hereditary pancreatitis. JAMA. 2001;286(2):169–170. [DOI] [PubMed] [Google Scholar]
- 16.Tamura K, Yu J, Hata T, et al. Mutations in the pancreatic secretory enzymes CPA1 and CPB1 are associated with pancreatic cancer. Proc Natl Acad Sci U S A. 2018;115(18):4767–4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rebours V, Boutron-Ruault MC, Schnee M, et al. The natural history of hereditary pancreatitis: a national series. Gut. 2009;58(1):97–103. [DOI] [PubMed] [Google Scholar]
- 18.van Lier MG, Wagner A, Mathus-Vliegen EM, Kuipers EJ, Steyerberg EW, van Leerdam ME. High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol. 2010;105(6):1258–1264; author reply 1265. [DOI] [PubMed] [Google Scholar]
- 19.Hearle N, Schumacher V, Menko FH, et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res. 2006;12(10):3209–3215. [DOI] [PubMed] [Google Scholar]
- 20.Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000;119(6):1447–1453. [DOI] [PubMed] [Google Scholar]
- 21.Lynch HT, Shaw TG. Familial atypical multiple mole melanoma (FAMMM) syndrome: history, genetics, and heterogeneity. Fam Cancer. 2016;15(3):487–491. [DOI] [PubMed] [Google Scholar]
- 22.Vasen HF, Gruis NA, Frants RR, van Der Velden PA, Hille ET, Bergman W. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden). Int J Cancer. 2000;87(6):809–811. [PubMed] [Google Scholar]
- 23.Potjer TP, Kranenburg HE, Bergman W, et al. Prospective risk of cancer and the influence of tobacco use in carriers of the p16-Leiden germline variant. Eur J Hum Genet. 2015;23(5):711–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McWilliams RR, Wieben ED, Rabe KG, et al. Prevalence of CDKN2A mutations in pancreatic cancer patients: implications for genetic counseling. Eur J Hum Genet. 2011;19(4):472–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roa BB, Boyd AA, Volcik K, Richards CS. Ashkenazi Jewish population frequencies for common mutations in BRCA1 and BRCA2. Nat Genet. 1996;14(2):185–187. [DOI] [PubMed] [Google Scholar]
- 26.McClain MR, Palomaki GE, Nathanson KL, Haddow JE. Adjusting the estimated proportion of breast cancer cases associated with BRCA1 and BRCA2 mutations: public health implications. Genet Med. 2005;7(1):28–33. [DOI] [PubMed] [Google Scholar]
- 27.Goggins M, Schutte M, Lu J, et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res. 1996;56(23):5360–5364. [PubMed] [Google Scholar]
- 28.Thompson D, Easton DF, Breast Cancer Linkage C. Cancer Incidence in BRCA1 mutation carriers. J Natl Cancer Inst. 2002;94(18):1358–1365. [DOI] [PubMed] [Google Scholar]
- 29.Breast Cancer Linkage C Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst. 1999;91(15):1310–1316. [DOI] [PubMed] [Google Scholar]
- 30.van Asperen CJ, Brohet RM, Meijers-Heijboer EJ, et al. Cancer risks in BRCA2 families: estimates for sites other than breast and ovary. J Med Genet. 2005;42(9):711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holter S, Borgida A, Dodd A, et al. Germline BRCA Mutations in a Large Clinic-Based Cohort of Patients With Pancreatic Adenocarcinoma. J Clin Oncol. 2015;33(28):3124–3129. [DOI] [PubMed] [Google Scholar]
- 32.Ferrone CR, Levine DA, Tang LH, et al. BRCA germline mutations in Jewish patients with pancreatic adenocarcinoma. J Clin Oncol. 2009;27(3):433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhen DB, Rabe KG, Gallinger S, et al. BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: a PACGENE study. Genet Med. 2015;17(7):569–577. doi: 510.1038/gim.2014.1153. Epub 2014 Nov 1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Win AK, Jenkins MA, Dowty JG, et al. Prevalence and Penetrance of Major Genes and Polygenes for Colorectal Cancer. Cancer Epidemiol Biomarkers Prev. 2017;26(3):404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dominguez-Valentin M, Sampson JR, Seppala TT, et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the Prospective Lynch Syndrome Database. Genet Med. 2020;22(1):15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kastrinos F, Mukherjee B, Tayob N, et al. Risk of pancreatic cancer in families with Lynch syndrome. JAMA. 2009;302(16):1790–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aguirre AJ, Nowak JA, Camarda ND, et al. Real-time Genomic Characterization of Advanced Pancreatic Cancer to Enable Precision Medicine. Cancer Discov. 2018;8(9):1096–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu ZI, Shia J, Stadler ZK, et al. Evaluating Mismatch Repair Deficiency in Pancreatic Adenocarcinoma: Challenges and Recommendations. Clin Cancer Res. 2018;24(6):1326–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grant RC, Denroche R, Jang GH, et al. Clinical and genomic characterisation of mismatch repair deficient pancreatic adenocarcinoma. Gut. 2020. [DOI] [PubMed] [Google Scholar]
- 40.Morrell D, Cromartie E, Swift M. Mortality and cancer incidence in 263 patients with ataxia-telangiectasia. J Natl Cancer Inst. 1986;77(1):89–92. [PubMed] [Google Scholar]
- 41.Roberts NJ, Jiao Y, Yu J, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012;2(1):41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Os NJ, Roeleveld N, Weemaes CM, et al. Health risks for ataxia-telangiectasia mutated heterozygotes: a systematic review, meta-analysis and evidence-based guideline. Clin Genet. 2016;90(2):105–117. [DOI] [PubMed] [Google Scholar]
- 43.Yang X, Leslie G, Doroszuk A, et al. Cancer Risks Associated With Germline PALB2 Pathogenic Variants: An International Study of 524 Families. J Clin Oncol. 2020;38(7):674–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jones S, Hruban RH, Kamiyama M, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324(5924):217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Slater EP, Langer P, Niemczyk E, et al. PALB2 mutations in European familial pancreatic cancer families. Clin Genet. 2010;78(5):490–494. [DOI] [PubMed] [Google Scholar]
- 46.de Andrade KC, Frone MN, Wegman-Ostrosky T, et al. Variable population prevalence estimates of germline TP53 variants: A gnomAD-based analysis. Hum Mutat. 2019;40(1):97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mai PL, Best AF, Peters JA, et al. Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li-Fraumeni syndrome cohort. Cancer. 2016;122(23):3673–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grant RC, Selander I, Connor AA, et al. Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology. 2015;148(3):556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chaffee KG, Oberg AL, McWilliams RR, et al. Prevalence of germ-line mutations in cancer genes among pancreatic cancer patients with a positive family history. Genet Med. 2018;20(1):119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klein AP, Brune KA, Petersen GM, et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res. 2004;64(7):2634–2638. [DOI] [PubMed] [Google Scholar]
- 51.Goggins M, Overbeek KA, Brand R, et al. Management of patients with increased risk for familial pancreatic cancer: updated recommendations from the International Cancer of the Pancreas Screening (CAPS) Consortium. Gut. 2020;69(1):7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vasen H, Ibrahim I, Ponce CG, et al. Benefit of Surveillance for Pancreatic Cancer in High-Risk Individuals: Outcome of Long-Term Prospective Follow-Up Studies From Three European Expert Centers. J Clin Oncol. 2016;34(17):2010–2019. [DOI] [PubMed] [Google Scholar]
- 53.Canto MI, Almario JA, Schulick RD, et al. Risk of Neoplastic Progression in Individuals at High Risk for Pancreatic Cancer Undergoing Long-term Surveillance. Gastroenterology. 2018;155(3):740–751.e742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blackford AL, Canto MI, Klein AP, Hruban RH, Goggins M. Recent trends in the incidence and survival of Stage 1A Pancreatic Cancer: A Surveillance, Epidemiology, and End Results analysis. J Natl Cancer Inst. 2020;112(5709818):1162–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stoffel EM, McKernin SE, Brand R, et al. Evaluating Susceptibility to Pancreatic Cancer: ASCO Provisional Clinical Opinion. J Clin Oncol. 2019;37(2):153–164. [DOI] [PubMed] [Google Scholar]
- 56.Stjepanovic N, Moreira L, Carneiro F, et al. Hereditary gastrointestinal cancers: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann Oncol. 2019;30(10):1558–1571. doi: 1510.1093/annonc/mdz1233. [DOI] [PubMed] [Google Scholar]
- 57.Aslanian HR, Lee JH, Canto MI. AGA Clinical Practice Update on Pancreas Cancer Screening in High-Risk Individuals: Expert Review. Gastroenterology. 2020;159(1):358–362. [DOI] [PubMed] [Google Scholar]
- 58.Harinck F, Konings IC, Kluijt I, et al. A multicentre comparative prospective blinded analysis of EUS and MRI for screening of pancreatic cancer in high-risk individuals. Gut. 2016;65(9): 1505–1513. [DOI] [PubMed] [Google Scholar]
- 59.Canto MI, Hruban RH, Fishman EK, et al. Frequent detection of pancreatic lesions in asymptomatic high-risk individuals. Gastroenterology. 2012;142(4):796–804; quiz e714-795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chari ST, Leibson CL, Rabe KG, Ransom J, de Andrade M, Petersen GM. Probability of pancreatic cancer following diabetes: a population-based study. Gastroenterology. 2005;129(2):504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yuan C, Babic A, Khalaf N, et al. Diabetes, Weight Change, and Pancreatic Cancer Risk. JAMA Oncol. 2020;6(10):e202948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fahrmann JF, Schmidt CM, Mao X, et al. Lead-time trajectory of CA19-9 as an anchor marker for pancreatic cancer early detection. Gastroenterology. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abe T, Kohi C, Kohi S, et al. Gene Variants That Affect Levels of Circulating Tumor Markers Increase Identification of Patients with Pancreatic Cancer. Clin Gastroenterol Hepatol. 2019: epub 10/29/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Konings IC, Harinck F, Poley JW, et al. Prevalence and Progression of Pancreatic Cystic Precursor Lesions Differ Between Groups at High Risk of Developing Pancreatic Cancer. Pancreas. 2017;46(1):28–34. [DOI] [PubMed] [Google Scholar]
- 65.Abe T, Blackford AL, Tamura K, et al. Deleterious Germline Mutations Are a Risk Factor for Neoplastic Progression Among High-Risk Individuals Undergoing Pancreatic Surveillance. J Clin Oncol. 2019;37(13):1070–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tanaka M, Fernandez-Del Castillo C, Kamisawa T, et al. Revisions of international consensus Fukuoka guidelines for the management of IPMN of the pancreas. Pancreatology. 2017;17(5):738–753. [DOI] [PubMed] [Google Scholar]
- 67.Canto MI, Kerdsirichairat T, Yeo CJ, et al. Surgical Outcomes After Pancreatic Resection of Screening-Detected Lesions in Individuals at High Risk for Developing Pancreatic Cancer. J Gastrointest Surg. 2020;24(5):1101–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Konings I, Canto MI, Almario JA, et al. Surveillance for pancreatic cancer in high-risk individuals. BJS Open. 2019;3(5):656–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Corral JE, Mareth KF, Riegert-Johnson DL, Das A, Wallace MB. Diagnostic Yield From Screening Asymptomatic Individuals at High Risk for Pancreatic Cancer: A Meta-analysis of Cohort Studies. Clin Gastroenterol Hepatol. 2019;17(1):41–53. [DOI] [PubMed] [Google Scholar]
- 70.Nissim S, Leshchiner I, Mancias JD, et al. Mutations in RABL3 alter KRAS prenylation and are associated with hereditary pancreatic cancer. Nat Genet. 2019;51(9):1308–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Klein AP, Wolpin BM, Risch HA, et al. Genome-wide meta-analysis identifies five new susceptibility loci for pancreatic cancer. Nat Commun. 2018;9(1):556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wolpin BM, Rizzato C, Kraft P, et al. Genome-wide association study identifies multiple susceptibility loci for pancreatic cancer. Nat Genet. 2014;46(9):994–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim J, Yuan C, Babic A, et al. Genetic and Circulating Biomarker Data Improve Risk Prediction for Pancreatic Cancer in the General Population. Cancer Epidemiol Biomarkers Prev. 2020;29(5):999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cohen JD, Li L, Wang Y, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359(6378):926–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu MC, Oxnard GR, Klein EA, et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Annals of Oncology. 2020;31(6):745–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Suenaga M, Yu J, Shindo K, et al. Pancreatic Juice Mutation Concentrations Can Help Predict the Grade of Dysplasia in Patients Undergoing Pancreatic Surveillance. Clin Cancer Res. 2018;24(12):2963–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Springer S, Masica DL, Dal Molin M, et al. A multimodality test to guide the management of patients with a pancreatic cyst. Sci Transl Med. 2019;11(501). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shelton CA, Umapathy C, Stello K, Yadav D, Whitcomb DC. Hereditary Pancreatitis in the United States: Survival and Rates of Pancreatic Cancer. Am J Gastroenterol. 2018;113(9):1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brose MS, Rebbeck TR, Calzone KA, Stopfer JE, Nathanson KL, Weber BL. Cancer risk estimates for BRCA1 mutation carriers identified in a risk evaluation program. J Natl Cancer Inst. 2002;94(18):1365–1372. [DOI] [PubMed] [Google Scholar]