

Abstract
Introduction
Cortical thinning is a marker of neurodegeneration in Alzheimer's disease (AD). We investigated the age‐related trajectory of cortical thickness across the lifespan (9‐59 years) in a Colombian kindred with autosomal dominant AD (ADAD).
Methods
Two hundred eleven participants (105 presenilin‐1 [PSEN1] E280A mutation carriers, 16 with cognitive impairment; 106 non‐carriers) underwent magnetic resonance imaging. A piecewise linear regression identified change‐points in the age‐related trajectory of cortical thickness in carriers and non‐carriers.
Results
Unimpaired carriers exhibited elevated cortical thickness compared to non‐carriers, and thickness more negatively correlated with age and cognition in carriers relative to non‐carriers. We found increased cortical thickness in child carriers, after which thickness steadied compared to non‐carriers prior to a rapid reduction in the decade leading up to the expected age at cognitive impairment in carriers.
Discussion
Findings suggest that cortical thickness may fluctuate across the ADAD lifespan, from early‐life increased thickness to atrophy proximal to clinical onset.
Keywords: age‐related, cortical thickness, familial Alzheimer's disease, lifespan, presenilin1, trajectory
1. INTRODUCTION
Cortical thickness—the distance between the white matter and pial surfaces—has been proposed as a biomarker of Alzheimer's disease (AD)–related neurodegeneration.1, 2, 3 Studies of sporadic AD and autosomal dominant AD (ADAD)–etiologically distinct forms of AD with similar biological and clinical phenotypes4—show reduced cortical thickness across different brain regions ≈5 years before clinical symptom onset.5, 6, 7, 8, 9 Several studies have reported increased cortical thickness and gray matter volume in the preclinical stage of sporadic AD and ADAD.10, 11, 12, 13 A recent report from the Dominantly Inherited Alzheimer's Network (DIAN), for example, found elevated cortical thickness in a group of adult heterogenous ADAD carriers (ie, carriers of different mutations) relative to non‐carriers 20 to 15 years before the estimated age of clinical symptom onset, prior to the accelerated atrophy seen more proximal to clinical onset.14 What is not well understood, however, is the trajectory of cortical thickness in ADAD across a wider lifespan range, including childhood through clinical symptom onset. In typical neurodevelopment in childhood and adolescence, cortical thickness decreases, which has been argued to relate to normative pruning processes15, 16 or increases in myelination.17 Understanding how the cortical thickness trajectories of mutation carriers and non‐carrier family members differ across the lifespan is critical to elucidating the utility of this magnetic resonance imaging (MRI) measure as a marker for prognosis and disease monitoring in ADAD.
Using a previously defined AD cortical thickness signature comprising nine a priori brain regions,8 we characterized the cross‐sectional relationship of cortical thickness with age across the lifespan (9‐59 years) in presenilin1 (PSEN1) E280A ADAD mutation carriers and non‐carrier family members from a large Colombian kindred. Based on previous cortical thickness research in sporadic AD and ADAD,6, 13, 18 we hypothesized that PSEN1 E280A carriers would exhibit a stronger negative association between AD signature cortical thickness and age relative to non‐carriers, with evidence of slightly increased cortical thickness in early life, consistent with prior gray matter volume findings in PSEN1 E280A‐carrying children.13 Reduced cortical thickness was expected to be seen in carriers closer to the median expected age at onset of mild cognitive impairment (MCI) in the cohort. We also hypothesized that cortical thickness would track with cognitive performance in adult ADAD mutation carriers.
2. METHODS
2.1. Standard protocol approvals, registrations, and participant consent
Institutional review board committees at the University of Antioquia (Colombia) and the Partners Human Research Committee (Boston) approved this study. Participants provided written informed consent before participating, with child participants assenting and having the consent of their parents/guardians.
2.2. Participants and study design
A total of 211 members from Colombian families with the PSEN1 E280A mutation enrolled in two cross‐sectional studies were included: the Colombian Alzheimer's Prevention Initiative Biomarker Study of ADAD (API‐BIO, n = 124) and the Colombia‐Boston Biomarker Study of ADAD (COLBOS, n = 87). The demographic, cognitive, and imaging data for all participants included in this report (105 mutation carriers [16 with MCI or dementia due to ADAD], 106 non‐carriers) are detailed in Table 1 (a breakdown by study is provided in eTable1). Mutation carriers have a previously documented median expected age at onset of MCI at 44 years of age (95% confidence interval [CI] = 43–45 years) and dementia due to AD at 49 years of age (95% CI = 49–50 years).19 Participants and investigators were blinded to genetic status, and all participants in this study had at least one parent with the PSEN1 E280A mutation. Eligible individuals were screened for neurological and psychiatric disorders, and contraindications to MRI (e.g., metallic implants).
TABLE 1.
Demographic and neuroimaging characteristics of the sample
| Characteristic | Cognitively Unimpaired Carriers (n = 89) | Cognitively Impaireda Mutation Carriers (n = 16) | Non‐Carriers (n = 106) | P‐valueb | Cohen d |
|---|---|---|---|---|---|
| Age (y) | 28.87 (11.17) | 44.81 (4.64) | 29.55 (11.09) | .67 | 0.06 |
| Educational Attainment (y) | 9.87 (3.83) | 9.38 (4.67) | 9.87 (3.87) | .73 | 0.00 |
| Female, No. (%) | 54 (61) | 12 (75) | 60 (57) | .57c | |
| Tesla Strength, 3T (%) | 37 (42) | 6 (38) | 44 (42) | .99 c | |
| Bilateral AD Signature Thickness (Composite) | 2.84 (0.12) | 2.48 (0.24) | 2.80 (0.12) | .007** | 0.38 |
| MMSE Scored | 28.97 (1.12) | 22.37 (4.30) | 29.23 (0.84) | .09 | 0.26 |
| FAST Scored | 1.20 (0.40) | 3.69 (0.79) | 1.02 (0.15) | <.001*** | 0.60 |
| CERAD Word List Immediate Recalld | 18.99 (4.43) | 7.69 (4.53) | 19.86 (3.42) | .16 | 0.22 |
| CERAD Word List Delayed Recalld | 6.79 (1.99) | 1.25 (1.81) | 7.47 (1.32) | .012* | 0.40 |
| CERAD Constructional Praxis Copyd | 9.70 (1.61) | 3.81 (4.65) | 10.19 (1.25) | .036* | 0.34 |
| CERAD Constructional Praxis Delayed Recalld | 9.25 (2.49) | 5.44 (3.05) | 9.86 (1.50) | .06 | 0.30 |
Shown are the demographic data for the sample of cognitively unimpaired mutation carriers, impaired mutation carriers, and non‐carriers. Statistics are reported as mean (SD). Abbreviations: MMSE, Mini‐Mental State Examination; FAST, Functional Assessment Staging Scale; CERAD, Consortium to Establish a Registry for Alzheimer's Disease.
Cognitively impaired is defined by a Functional Assessment Staging Scale (FAST) Score of 3 or greater.
P‐value and Cohen d calculated for Independent T test for cognitively unimpaired PSEN1 E280A mutation carriers versus non‐carriers.
P‐value calculated for chi‐square test for presenilin 1 (PSEN1) E280A mutation carriers versus non‐carriers.
Cognitive data are not available for child participants (ages ≤17 years), as all cognitive tests used are designed and normed for administration to adults (ages ≥18). The results presented in this two‐sample t test are from cognitively unimpaired participants over the age of 18(n = 86 non‐carriers, n = 71 carriers).
The calculation of the difference in thickness in the bilateral AD signature was calculated using a general linear model adjusting for MRI scanner strength and age.
*P < .05.
** P < .01.
***P < .001.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using PubMed and Google Scholar. Although cortical thickness in Alzheimer's disease (AD) is widely studied, we found no AD‐related publications examining the trajectory of cortical thickness across the lifespan (ie, including childhood). Relevant citations about cortical thickness in sporadic AD and autosomal dominant AD (ADAD) literature are included.
Interpretation: Our findings align with emerging evidence of subtle increased cortical thickness in early preclinical ADAD mutation carriers relative to prototypically aging peers, followed by significant cortical atrophy more proximal to the onset of the AD clinical syndrome.
Future directions: Longitudinal staging of the rate of decline of cortical thickness in carriers of ADAD mutations relative to non‐carrier family members is needed, especially within the context of examining if genetic or lifestyle factors can slow or halt the trajectory of this neurodegenerative marker of AD.
2.3. Procedures
Participants lived in the region of Antioquia, Colombia. Clinical and cognitive testing was completed at the University of Antioquia. Spanish‐speaking neuropsychologists administered all cognitive tests, and clinical histories and neurological exams were performed by neurologists or physicians trained to assess neurodegenerative disorders. Ratings of clinical impairment in adult participants (≥18 years of age) were devised using the Functional Assessment Staging Scale (FAST20). FAST values of “1,” indicating absence of subjective or objective cognitive impairment, were imputed for child participants (9‐17 years of age; −35 to −27 estimated years until median age of expected onset [EYO] of MCI in this cohort19) to allow the inclusion of child participants in analyses with other cognitively unimpaired individuals. A Spanish version of the Mini‐Mental State Examination (MMSE21) and the Colombian‐normed version of the Consortium to Establish a Registry for Alzheimer's Disease (CERAD)22, 23 were administered to adult participants (≥18 years of age). The CERAD includes measures of verbal memory (word list learning immediate and delayed recall), and visual memory (constructional praxis copy and recall phase). Cognitive variables for child participants in this study were unavailable and would be uninterpretable in our analyses with tests designed for use with adults. Clinical data were stored in a secure database at the Neuroscience Group of Antioquia in Medellín, Colombia.
2.4. Imaging acquisition and processing
All participants underwent MRI on Siemens scanners at the Pablo Tobón Uribe Hospital in Medellín, Colombia. API‐BIO participants had a 1.5T scan, whereas COLBOS participants had a 3T scan. For participants who had a 1.5T MRI, the protocol included one three‐dimensional (3D) T1‐weighted MRI image (T1 fast‐field echo [FFE]; repitition time [TR] = 2530 ms, echo time [TE] = 3.39 ms; flip angle, 7°; field of view, 256 × 256; 1.0 × 1.0 × 1.0 mm; 176 slices). For participants who had a 3T MRI, the protocol entailed one 3D T1‐weighted MRI image (T1‐FFE; TR = 2400 ms, TE = 3.61 ms; flip angle = 8°; field of view, 240 × 240; 1.3 × 1.3 × 1.2 mm; 160 slices).
As described24 previously, MRI data were analyzed using the standard processing pipeline within FreeSurfer (FS) version 6.0. Surface segmentation was visually examined for quality and edited as necessary. An average of nine bilateral FS regions of interest (ROIs)—medial temporal lobe, inferior temporal gyrus, temporal pole, precuneus, superior parietal lobule, angular gyrus, supramarginal gyrus, superior frontal gyrus, and middle frontal gyrus—comprised a previously defined, widely studied, and sensitive measure of cortical thickness in AD.8 These ROIs were uncorrected for relative size and intracranial volume, as has been done previously8; non‐correction for intracranial volume has also been suggested as good practice for studies of cortical thickness.25, 26 In addition, for whole‐cortex analyses, individual FS maps of vertex‐wise cortical thickness were surface‐smoothed to an equivalent 20 mm (full‐width at half‐maximum) Gaussian filter and resampled to a common template surface (fsaverage) using FS.
2.5. Statistical analysis
Independent two‐sample t tests examined age and education differences between cognitively unimpaired carriers and non‐carriers. A general linear model (GLM) was conducted to examine differences in the AD cortical thickness signature and individual ROIs between cognitively unimpaired carriers and non‐carriers, specifically, after adjusting for MRI scanner strength and age. (Scanner strength was controlled for statistically because previous research suggested that 1.5T and 3T estimates of cortical thickness have decent reliability, but 3T MRI exhibits a slight bias toward larger thickness estimates.9) In addition, for all adult participants ≥18 years of age, independent two‐sample t tests were used to examine differences in neuropsychological test performance. Chi‐square tests were employed to assess if carriers and non‐carriers differed on the proportion of biological sex and groups scanned in a 3T versus 1.5T MRI scanner. Partial correlations adjusting for scanner strength as a categorical variable were employed to examine the association between the AD cortical thickness signature with age in all mutation carriers, cognitively unimpaired carriers, and non‐carriers, as well as the association between the AD cortical thickness signature with performance on all cognitive variables in all adult mutation carriers, adult cognitively unimpaired carriers, and adult non‐carriers. Fisher r‐to‐z calculations were conducted online (http://vassarstats.net/rdiff.html) to quantify differences in the magnitude of these aforementioned associations in carriers relative to non‐carriers. These analyses were conducted in SPSS version 26.
We also investigated the age trajectory of AD cortical thickness signature, examining the non‐linear relationships between bilateral AD signature and age in carriers as compared to non‐carriers by performing a piecewise linear regression. This approach allowed us to characterize the relation between thickness and age in early life, young adulthood, and close to the age at onset of the clinical symptoms in mutation carriers. The MATLAB (www.mathworks.com) non‐linear fitting function nlinfit and a modified version of the nlparci m‐function were used to perform the piecewise linear regression for PSEN1 E280A mutation carriers, estimating the location of two inflection points (one at the end of early neurodevelopment, and the other for the start of accelerated decline) as the unknown parameters. Other parameters included the slope and intercept of the first and third linear segments. To be unbiased, the same piece‐wise linear regression with two inflection points was also examined in non‐carriers. We statistically compared the slope (rate of change) differences before and after each inflection point within the carrier group and the differences of pre‐ or post‐inflection point slope, each in comparison with the respective slope in non‐carriers.
Based on the AD signature analyses, we also performed vertex‐wise analyses of thickness measurements across the entire cortex, as follows. Relationships between thickness and age were evaluated in three age subcohorts based on the two change‐points in mutation carriers identified by the aforementioned piecewise linear regression (see Results section for these change‐points). For each age subcohort, differences in thickness ∼ age slopes between carriers and non‐carriers were evaluated using models of thickness ∼ age*group (with scanner strength as covariate) and expressed as the β of the interaction term at each vertex. We report effect sizes (βs) and significance of these estimates on the surface at cluster‐wise P < .01 (uncorrected) and at P < .05 after applying false discovery rate (FDR) correction for multiple comparisons (FS implementation, minimum cluster extent = 100 mm2).
3. RESULTS
3.1. Demographics
Cognitively unimpaired mutation carriers and non‐carriers did not differ in age, education, biological sex proportion, or proportion scanned with 1.5T versus 3T MRI (Table 1). Relative to non‐carriers, cognitively unimpaired mutation carriers had a slightly, but significantly greater AD cortical thickness signature after controlling for scanner strength (d = 0.38); more scores of “2” on the FAST (ie, subjective cognitive concerns); slightly, but significantly worse performance on the CERAD Constructional Praxis Copy and CERAD Word List Delayed Recall; and a trend toward worse performance on the CERAD Constructional Praxis Recall (Table 1). Thickness was significantly greater in cognitively unimpaired carriers relative to non‐carriers in two of nine bilateral AD signature ROIs: the temporal pole and inferior temporal gyrus. Thickness in the other seven of nine AD signature ROIs did not differ significantly between cognitively unimpaired carriers and non‐carriers (Table 2).
TABLE 2.
Bilateral cortical thickness in AD signature regions of interest controlling for scanner strength and age
| Bilateral AD Signature Region of Interest | Cognitively Unimpaired Carriers (n = 89) | Cognitively Impaireda Mutation Carriers (n = 16) | Non‐Carriers (n = 106) | P‐valueb | Cohen d |
|---|---|---|---|---|---|
| Medial Temporal Cortex | 3.43 | 3.05 | 3.38 | .20 | 0.18 |
| Inferior Temporal Gyrus | 3.06 | 2.75 | 2.98 | .008* | 0.37 |
| Temporal Pole | 3.28 | 3.01 | 3.19 | .004* | 0.40 |
| Superior Parietal Lobule | 2.32 | 1.92 | 2.28 | .11 | 0.23 |
| Precuneus | 2.65 | 2.22 | 2.62 | .15 | 0.22 |
| Angular Gyrus | 2.63 | 2.21 | 2.61 | .21 | 0.25 |
| Supramarginal Gyrus | 2.73 | 2.36 | 2.73 | .95 | 0 |
| Superior Frontal Gyrus | 2.94 | 2.56 | 2.92 | .48 | 0.10 |
| Inferior Frontal Sulcus | 2.52 | 2.26 | 2.49 | .11 | 0.27 |
Shown are the thickness values (mm) in the nine previously defined bilateral AD signature ROIs8 in cognitively unimpaired PSEN1 E280A carriers, cognitively impaired carriers, and non‐carriers. The calculation of the difference in thickness in the bilateral ROIs was calculated using a general linear model adjusting for MRI scanner strength and age. Cohen d is calculated from the partial eta squared using a calculation described on the IBM forum here (https://www.ibm.com/support/pages/effect‐size‐relationship‐between‐partial‐eta‐squared‐cohens‐f‐and‐cohens‐d)
Cognitively impaired is defined by a Functional Assessment Staging Scale score of 3 or greater.
P‐value and Cohen d calculated for Independent t test for cognitively unimpaired PSEN1 E280A mutation carriers versus non‐carriers.
*P < .01.
3.2. Association between age and cortical thickness
Older age was associated with lower cortical thickness signature in all mutation carriers and non‐carriers, but the magnitude of the negative association between age and cortical thickness was significantly stronger in all mutation carriers (Table 3). The correlation between the cortical thickness signature and age in the cognitively unimpaired mutation carriers was also stronger than the relationship in non‐carriers (at trending significance).
TABLE 3.
Correlations between age and cortical thickness controlling for scanner strength
| Partial Correlation (Controlling for MRI Strength) | Cognitively Unimpaired Carriersa (n = 89) | All Mutation Carriers (n = 105) | Non‐Carriers (n = 106) | Fisher Z of Difference in Partial Correlation Strength | P‐value of the Fisher Z |
|---|---|---|---|---|---|
| Age x Bilateral AD Signature Thickness in all Participants | r = −.70, P < .001 | r = −.42, P < .001 | −3.00 | <.001* | |
| Age x Bilateral AD Signature Thickness in Cognitively Unimpaireda Participants Only | r = −.58, p < .001 | r = −.42, p < .001 | −1.46 | .07 |
Shown are the partial associations between age and cortical thickness, controlling for scanner strength (1.5T vs 3T) in cognitively unimpaired mutation carriers, all mutation carriers, and non‐carriers.
Cognitively impaired is defined by a Functional Assessment Staging Scale Score of 3 or greater.
*P‐value < .001.
3.3. Association between cortical thickness and cognition in adult participants (≥18 years of age)
Among cognitively unimpaired adult mutation carriers, the cortical thickness signature was significantly positively associated with CERAD constructional praxis delayed recall and trended with CERAD word list delayed recall (Table 4); these relationships were also trending stronger in cognitively unimpaired carriers relative to non‐carriers (Table 4). There were strong positive associations between the cortical thickness signature and cognitive variables in all mutation carriers, which were also all significantly stronger than those observed in non‐carriers (Table 4). In non‐carriers, the cortical thickness signature was only positively significantly associated with the CERAD constructional praxis copy score (Table 4).
TABLE 4.
Correlations between cortical thickness and cognition controlling for scanner strength and age in adult participants (18+ years old)
| Partial Correlation (Controlling for MRI Strength and Age) | Cognitively Unimpaired Adult Carriersa (n = 71) | All Adult Mutation Carriers (n = 87) | Adult Non‐Carriers (n = 86) | Fisher Z of Difference in Partial Correlation Strength | P‐value of the Fisher Z |
|---|---|---|---|---|---|
| MMSE x Bilateral AD Signature Thickness in all Participants | r = .70, P < .001** | r = ‐.11, P = .30 | −6.28 | <.001** | |
| MMSE x Bilateral AD Signature Thickness in Cognitively Unimpaireda Participants Only | r = .14, P = .26 | r = ‐.11, P = .30 | 0.19 | .42 | |
| CERAD Word List Learning x Bilateral AD Signature Thickness in all Participants | r = .48, P < .001** | r = .13, P = .26 | 2.56 | .005* | |
| CERAD Word List Learning x Bilateral AD Signature Thickness in Cognitively Unimpaireda Participants Only | r = .03, P = .83 | r = .13, P = .26 | −0.62 | .27 | |
| CERAD Word List Delayed Recall x Bilateral AD Signature Thickness in all Participants | r = .54, P < .001** | r = ‐.02, P = .84 | 4.04 | <.001** | |
| CERAD Word List Delayed Recall x Bilateral AD Signature Thickness in Cognitively Unimpaireda Participants Only | r = .23, P = .06 | r = ‐.02, P = .84 | 1.55 | .06 | |
| CERAD Constructional Praxis Copy x Bilateral AD Signature Thickness in all Participants | r = .58, P < .001** | r = .33, P = .002** | 2.09 | .019* | |
| CERAD Constructional Praxis Copy x Bilateral AD Signature Thickness in Cognitively Unimpaireda Participants Only | r = .03, P = .80 | r = .33, P = .002** | −1.91 | .028* | |
| CERAD Constructional Praxis Recall x Bilateral AD Signature Thickness in all Participants | r = .50, P < .001** | r = .02, P = .86 | 3.39 | <.001** | |
| CERAD Constructional Praxis Recall x Bilateral AD Signature Thickness in Cognitively Unimpaireda Participants Only | r = .27, P = .02* | r = .02, P = .86 | 1.57 | .05 |
Shown are the partial associations between cortical thickness and cognitive measures, controlling for scanner strength (1.5T vs 3T) and age in cognitively unimpaired mutation carriers, all mutation carriers, and non‐carriers.
Abbreviations: MMSE, Mini‐Mental State Examination; CERAD, Consortium to Establish a Registry for Alzheimer's Disease.
Cognitively impaired is defined by a Functional Assessment Staging Scale Score of 3 or greater.
*P‐value < .05.
**P‐value < .01.
3.4. Inflection point of the relationship between cortical thickness and age
The piecewise linear regression model, covarying for scanner strength, estimated the two change‐points of the negative association between age and the cortical thickness signature in carriers at 17.00 and 32.89 years (Figure 1, red points/line). In non‐carriers, there was a change‐point in this same relationship at 21.37 years and another (virtually indistinguishable) change‐point at 35.66 years (Figure 1, black points/line). The slope before their respective first change‐points in the relationship between age and AD signature thickness was significantly steeper in carriers than in non‐carriers (carrier slope = −0.0386, non‐carrier slope = −0.0181, P = .02). Carriers and non‐carriers, however, did not significantly differ in the rate of the relationship between AD signature thickness and age between the first and second change‐points (carrier slope = 0.0006, non‐carrier slope = −0.0012, P = .69). After their respective second change‐points, carriers again had a significantly stronger negative relationship between age and cortical thickness relative to non‐carriers (carrier slope = −0.0168, non‐carrier slope = −0.0011, P < .001).
FIGURE 1.
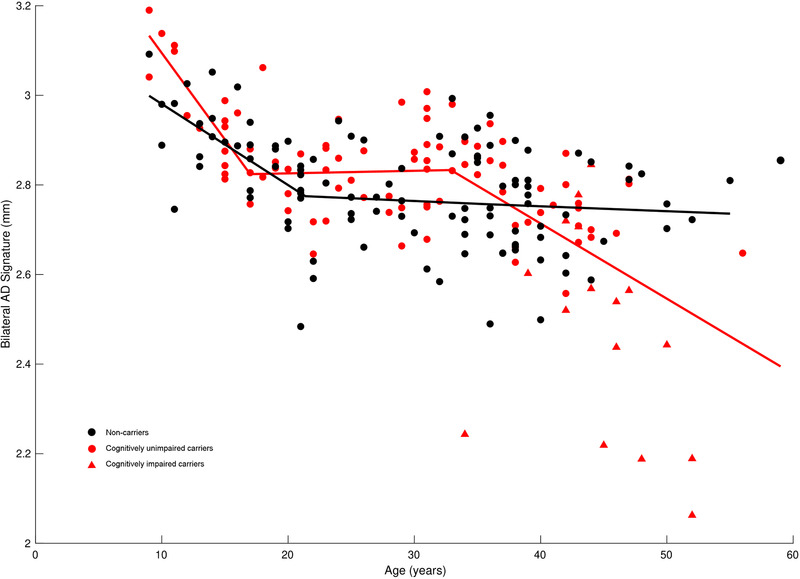
Piecewise linear curve fitting of the relationship between cortical thickness and age. Shown is the relationship of bilateral Alzheimer's disease (AD) signature thickness (mm) with age (in years) adjusted for magnetic resonance imaging (MRI) scanner strength in non‐carriers (black circles), as well as in cognitively unimpaired presenilin1 (PSEN1) E280A mutation carriers (red circles) and impaired mutation carriers (red triangles). The piecewise linear regression model estimated the two change‐points of the negative association between age and the cortical thickness signature in carriers as 17.00 years and 32.89 years; in non‐carriers, there was a change‐point at 21.37 years and another (which is virtually indistinguishable) at 35.66 years. The slopes before the groups’ respective first change‐points were significantly steeper in carriers than in non‐carriers (carrier slope = −0.0386, non‐carrier slope = −0.0181, P = .02). The groups, however, did not differ significantly in the rate of the relationship between AD signature thickness and age between the first and second change‐points (carrier slope = 0.0006, non‐carrier slope = −0.0012, P = .69). After their respective second change‐points, carriers exhibited a significantly stronger negative relationship between age and cortical thickness relative to non‐carriers (carrier slope = −0.0168, non‐carrier slope = −0.0011, P < .001)
3.5. Post hoc vertex‐wise modeling of the association between cortical thickness and age
Post hoc vertex‐wise models, using the change‐point ages from the piecewise regression in carriers as cutoffs, showed that child carriers (<18 years of age) exhibited a steeper negative relationship between age and cortical thickness when compared to non‐carriers, primarily in clusters localized to the left inferior temporal lobe, left medial frontal lobe, and bilateral lateral parietal lobes. These differences between carrier and non‐carrier children in the rate of the relationship between age and cortical thickness, however, did not survive after FDR correction (Figure 2, left panel). Carriers between the ages of 18 and 32 largely did not differ in the rate of the relationship between age and cortical thickness, with only a few small clusters reaching significance but not surviving FDR correction (Figure 2, center panel). In the oldest age bracket (>32 years), carriers exhibited a stronger negative relationship between age and AD signature thickness than non‐carriers at many bilateral clusters, although only left hemisphere clusters in the following regions survived FDR correction: precuneus, lateral parietal lobe, and medial and lateral frontal lobe (Figure 2, right panel).
FIGURE 2.
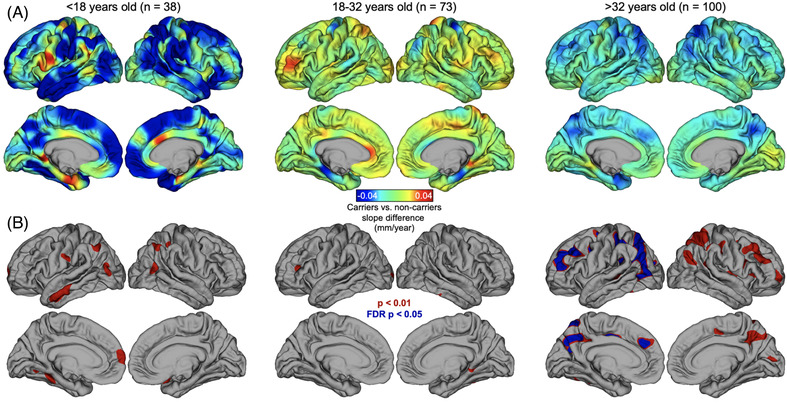
Vertex‐wise analysis of cortical thickness by age relationship. Shown is the vertex‐wise comparison of thickness ∼ age slopes in mutation carriers versus non‐carriers, covarying for scanner strength. (A) Effect size maps showing the difference in slopes between carriers and non‐carriers in age groups determined by the piecewise regression change‐points in carriers (left panel = prior to change‐point 1; center panel = between change‐points 1 and 2; right panel = after change‐point 2). Full model: vx‐thickness ∼ age*carrier_status + scanner. (B) Significant maps for vertex‐wise results: red areas are cluster‐wise P < .01; blue areas are P < .05 after false discovery rate (FDR) correction; minimum cluster extent = 100 mm2
4. DISCUSSION
This study describes the relationship between age and cortical thickness from childhood to dementia due to ADAD (9‐59 years) in PSEN1 E280A mutation carriers and non‐carriers hailing from Antioquia, Colombia. As hypothesized, in all mutation carriers there was a significantly stronger negative association between age and the AD cortical thickness signature relative to non‐carrier family members, after controlling for MRI scanner strength; the difference in the magnitude of this association was trending when non‐carriers were compared against cognitively unimpaired mutation carriers. Two cut‐points emerged in the age‐related trajectory of cortical thickness at ≈17 years (−27 EYO to median age of expected onset of MCI for the cohort19) and 32 years of age (−12 EYO to MCI). Non‐carriers also exhibited two change‐points in this relationship, with the first at 21 years of age and the second (which was virtually indistinguishable) occurring at 36 years of age. Before their respective first change‐points, child carriers had a significantly stronger negative relationship between AD signature cortical thickness and age than non‐carriers. Between their first and second change‐points, carriers and non‐carriers did not differ in the rate of the relationship between age and thickness. Finally, after their final change‐points, mutation carriers demonstrated a significantly stronger negative relationship between age and AD signature thickness relative to non‐carriers who, as a group, did not experience change in their rate of this relationship.
Analysis of the nine bilateral AD signature ROIs found that cognitively unimpaired carriers, relative to non‐carriers, had greater thickness in the temporal pole and inferior temporal gyrus. Vertex‐wise analysis based on the two mutation carrier change‐points revealed significant clusters in which carrier children had a stronger negative relationship between the AD signature and age than non‐carrier children, although none of these clusters survived correction for multiple comparisons. Carriers between the ages of 18 and 32 did not exhibit vertices with differing age‐related trajectories in thickness relative to age‐matched non‐carriers (none of the few significant vertices in this age group survived correction). In the oldest age bracket (>32 years), however, carriers exhibited a stronger negative relationship than non‐carriers between age and thickness, surviving correction in the left the hemisphere in regions including the precuneus, lateral parietal lobe, and medial and lateral frontal lobes. A prior study examined cortical thickness in 40 middle‐age individuals from this cohort (18 mutation carriers, mean age = 38.2 years [∼−6 EYO from expected MCI onset]), similarly identifying reduced cortical thickness in the precuneus and bordering medial parietal regions.6 Cortical thickening in these ROIs (and in the AD signature) in younger ADAD carriers may appear paradoxical but aligns with prior ADAD and sporadic AD research,10, 11, 12 including a report examining gray matter volume in child Colombian cohort members (18 mutation carriers, mean age = 13.0 years [≈31 EYO from expected MCI onset]), which found greater gray matter volume in the parahippocampal gyrus and temporal pole of child carriers relative to non‐carrier children,13 as well as in a study of cortical thickness in the DIAN where thickness was elevated in adult ADAD mutation carriers 20 to 15 years before expected clinical onset relative to non‐carriers.14 The medial temporal lobe is particularly vulnerable to early tau deposition in this ADAD cohort27 and sporadic AD.28 All of the aforementioned brain regions are susceptible to neurodegeneration early in ADAD due to PSEN1 E280A, and many are also implicated in memory encoding and retrieval processes, which are early cognitive symptoms of AD.29, 30 Relative to non‐carriers, we also found significantly stronger positive associations between the AD cortical thickness signature and memory performance in all adult carriers (and at trending significance in cognitively unimpaired carriers), suggesting a deleterious impact of lower cortical thickness on cognitive performance in preclinical and early symptomatic ADAD.
One potential driver of elevated cortical thickness in young mutation carriers could be a neuroinflammatory mechanism, including glial activation31 and nuclear hypertrophy.32 The direct neurotoxic effects of amyloid beta—known to be abnormally elevated in the plasma of child PSEN1 E280A carriers,13 perhaps as early as gestation33—may also correspond with a relative cortical thickness elevation in the earlier parts of the ADAD mutation carrier lifespan. The rate of neurodegeneration may then hasten in carriers in around the mid‐30s relative to a stabilization in cortical thickness in typically developed non‐carriers.
The AD cortical thickness signature may be a marker of the disease, especially in conjunction with other in vivo AD markers,34, 35 although preclinical differences in the thickness signature are relatively subtle from a neuroanatomical perspective (eg, 0.04 mm greater thickness, on average, in cognitively unimpaired ADAD carriers relative to non‐carriers). Clinically, it thus may be advisable to use cortical thickness as an outcome measure in larger AD studies, but not for single patient clinical diagnosis. Longitudinal staging of the sequence of amyloid beta, tau, and neurodegeneration in the PSEN1 E280A cohort is presently underway36 and will examine longitudinal changes in cortical thickness, particularly as carriers near the EYO for MCI. Research on how other genetic risk factors (eg, APOE ε437) or protective genetic factors (eg, APOE ε238, or the rare APOE ε3 Christchurch mutation39) contribute to cortical thickness across the lifespan is also needed.
4.1. Limitations
This study has several limitations, including the combination of individuals with 1.5T and 3T MRI studies in the same analysis. Scanner strength may contribute to the variance in cortical thickness values, given a slight bias toward larger thickness estimates in 3T relative to 1.5T MRI, athough thickness measurements across these scanning strengths have been found previously to be reliable.9 We take a conservative approach nonetheless and control for the effect of scanner strength in our analyses. The present study is also cross‐sectional, whereas only a longitudinal study could determine the true trajectory of cortical thickness within mutation carriers and non‐carriers across time. We posit, however, that the present findings would be similar to the findings of a longitudinal study in this cohort given the Colombian kindred's homogeneity and robust characterization of biological and cognitive profiles (see Fuller et al., 201940 for a review). In addition, although the homogeneity of a single‐mutation ADAD cohort provides a strong framework through which we can examine the lifespan trajectory of cortical thickness, we are limited in the generalization of our findings to other ADAD mutations, as well as sporadic AD—the most prevalent form of the disease.41 Finally, there are many factors that can influence cortical thickness measurements, such as brain cerebrovascular (and how it is influenced by positive and negative health behaviors) and myelination, which we do not have the data to examine in this report but are potential future avenues of cortical thickness research in ADAD.
In conclusion, across an age range spanning five decades we found that PSEN1 E280A mutation carriers exhibited a stronger negative relationship between age and cortical thickness relative to non‐carriers, which fluctuated across the lifespan. As a group, cognitively unimpaired ADAD carriers exhibited a small but significant elevation in the AD cortical thickness signature relative to non‐carriers. The rate of the negative relationship between age and AD signature thickness in PSEN1 E280A carriers was significantly stronger than non‐carriers in childhood before stabilizing in early adulthood. After ≈33 years of age, however, mutation carriers demonstrated a significantly stronger negative relationship between AD signature thickness and age (ie, neurodegeneration) relative to neurotypical non‐carriers, whose relationship between age and AD signature thickness did not change substantially from early adulthood into middle‐age. These findings align with the evidence of early amyloid beta overproduction in early life in ADAD mutation carriers, which may lead to a neuroinflammation‐induced elevation in cortical thickness decades before significant elevations of tau27 or clear neurodegeneration is evident. Future longitudinal research examining the trajectory of structural changes in the brains of ADAD carriers is needed, especially in the context of other genetic risk and protective factors.
CONFLICT OF INTEREST
Mr. Fox‐Fuller was supported by the National Institute on Aging (NIA F31AG062158) and was the member‐at‐large for the International Neuropsychological Society's Student Liaison Committee. Dr. Quiroz was supported by grants from the National Institutes of Health (NIH) Office of the Director (DP5OD019833), the NIA (R01 AG054671]), the Alzheimer's Association, and Massachusetts General Hospital Executive Committee on Research (ECOR). Mr. d'Oleire Uquillas was supported by the National Science Foundation, NSF GRFP (disbursed to institution) and the Ford Foundation National Academies of Sciences, Medicine and Engineering Predoctoral Fellowship (disbursed to institution). Dr. Kewei Chen was supported by the NIH (to the institute State of Arizona and to the institute) and was a paid consultant (unrelated to this manuscript) to the following: Green Valley Pharmaceutical, Shanghai, China; Beijing Normal University, Beijing, China; and The sixth people's hospital, Shanghai, China. In addition, Dr. Kewei Chen holds a patent regarding use of multi‐modal linkage (multi‐modal partial least square) to link and co‐analyze multi‐modal data (https://patents.google.com/patent/US20060074290). Dr. Su was supported on this work by R01AG055444 (funding was made to Banner Health) and was also supported by the following grants: NIH/NIA R01AG069453, NIH/NIA R21AG065942, NIH/NIA R42AG053149, NIH/NIA P30AG019610, NIH/NIA R01AG031581, NIH/NIA U19AG024904, NIH/NIBIB R21EB024366 BrightFocus ADR A2017272S, Alzheimer's Association AARG‐17‐532945 State of Arizona All grants were made to institution. Dr. Su is also a paid consultant (unrelated to this work) to Green Valley Pharmaceutical LLC. Dr. Jacobs received support (unrelated to this work) from the following: NIA‐NIH R01 AG 062559, R01 AG 068062‐01A1, and from Alzheimer Nederland (the Netherlands): AN‐19021. In addition, Dr. Jacobs is vice‐chair of the Neuromodulatory Subcortical Systems professional interest area of International Society to Advance Alzheimer's Research and Treatment (ISTAART; Alzheimer's Association). Dr. Guzmán‐Vélez was supported by funding from the National Institute on Aging (NIA) K23AG061276, the Massachusetts General Hospital ECOR Fund for Medical Discovery Clinical Fellowship Award, and the NIH Research Supplement for Diversity (DP5OD019833); she also received honoraria for a presentation at a Missouri University of Science and Technology departmental colloquium (unrelated to this work). Dr. Vila‐Castelar is supported by a grant from the Alzheimer's Association (2019A005859) and serves as co‐chair for the Sex Differences Special Interest Group, Diversity and Disparities Professional Interest Area‐PIA of ISTAART. Dr. Reiman has received support from the following grants: NIH grants R01 AG069453, R01 AG031581, P30 AG019610, R01 AG055444, R01 AG054671, R01 AG058468, U01 NS093334, OT2 OD026549, PO1 AG052350, U19 AG024904, U54 MD000507, U01 AG0169765 and AG016976, and R01 AG054671; Arizona Department of Health Services; and Banner Alzheimer's Foundation, Flinn Foundation, Anonymous Foundations, NOMIS Foundation, Alzheimer's Association, and GHR Foundation. Dr. Reiman's institution has received consulting fees for his work with: Alkahest, Alzheon, Aural Analytics, Denali, Green Valley, Retromer Therapeutics, and Vaxxinity. Dr. Reiman is an inventor, with a patent issued to Banner Health, related to biomarker strategies involved in the accelerated evaluation of AD prevention therapies. Dr. Reiman is on the Board of Directors, Flinn Foundation, and is a Member of the National Advisory Council on Aging (NACA). Dr. Reiman is also Cofounder and shareholder of ALZPath. Dr. Sperling was supported by the following grants: R01AG058825 from the NIH, payment made to Brigham and Women's Hospital (BWH); R01AG063689 from NIH, payment made to BWH R01AG053184; from NIH, payment made to BWH; P01AG036694 from NIH, payment made to Massachusetts General Hospital (MGH); R01AG063689 from NIH (subcontract with USC), payment made to MGH R01AG061848; from NIH, payment made to MGH R01AG058468; from NIH (subcontract with Banner Alzheimer's Institute), payment made to MGH; from a research grant from the Alzheimer's Association, payment made to MGH ; and research funding Eisai, Eli Lilly, Janssen, clinical research funding. Dr. Sperling received Honorarium payments (unrelated to this work) made directly to her from: Shionogi, Genentech, Oligomerix, Inc., Cytox, Prothena, Acumen, JOMDD, Renew, Alnylam Pharmaceuticals, Neuraly, Janssen, Neurocentria, AC Immune, Biogen, Eisai, Roche, and Takeda. Dr. Sperling also reports a financial relationship for my spouse Dr. Keith Johnson regarding the following: Cerveau, Janssen, AC Immune, Novartis (these are all honorarium consulting payments). Dr. Sperling received the following additional honoraria (unrelated to this work): 2018 Duke Lectureship; 2018 UCLA Talk; 2018 Alzheimer Research Roundtable; 2018 ADRC Talk; 2019 ACTC Scottdale; 2019 ADPD Symposium; 2019 University of Tokyo Roundtable; 2019 Biogen: ARIA State of the Art Meeting; 2019 University of Pittsburgh Distinguished Scientist Lecture; 2019 University of Cincinnati Lurie Lecture; 2019 CTAD San Diego; 2019 CT Research Academy Talk; 2020 UC Irvine Distinguished Lecture on the Brain; 2020 Virtual Rhode Island IDeA Symposium; 2020 IMPACT‐AD Course; 2020 University of Chicago DOM Weekly Grand Rounds Seminar Series; 2020 Adler Foundation Symposium; San Diego 2020 ACTC; and San Diego 2020 UC Irvine Lecture. Dr. Sperling has also received travel support for the following conferences (unrelated to this work): 2018 ACTC Atlanta; 2018 CTAD Spain; 2018 Harvard HUBWeek Panel; 2018 Japan JSNM; 2019 Palm Beach Reversing the Tide of Alzheimer's Fundraiser; 2019 HAI Meeting; 2019 ACTC Philadelphia; 2019 GHR Board of Directors in DC; 2019 Marr Family Race; 2019 ACTC St. Louis; 2019 FDA Type C Meeting; 2019 Marc Diamond UT Southwestern Symposium; and 2020 HAI Miami. Dr. Sperling reports the following leadership positions: 2014‐2018 National Institute on Aging, Standing Council Member; National Advisory Council on Aging 2018‐2019 Chair, Division of Neuroscience Review Committee; National Institute on Aging 2018‐Present Journal of Prevention of Alzheimer's Disease; Editorial Board 2020‐2021 Chair, Preclinical Alzheimer's Disease Biomarker Disclosure Workshop Committee, National Academies of Science, Engineering and Medicine. Dr. Lopera reports support from the COLBOS project to himself and his Institution and received funds from an NIH Subcontract with Massachusetts General Hospital. Dr. Lopera reports receiving funds from NIH to participate in various Alzheimer's‐related conferences. Dr. Keith Johnson reports research support from NIH (unrelated to this work) and received personal consulting fees from Novartis. Dr. Brad Dickerson reports research support from NIA, NIDCD, and NIMH, as well as royalties/licenses with Oxford University Press, Cambridge University Press, and Elsevier. Dr. Dickerson reports personal consulting fees received from: Acadia, Alector, Arkuda, Biogen, Novartis, Takeda, and Wave. Dr. Dickerson received honoraria from InTouch Medical and Biogen and was on the Data Safety/Advisory Boards for: Acadia, Alector, Arkuda, Biogen, Novartis, Takeda, and Wave. Dr. Dickerson is also Chair elect of the Association for FTD.
ROLE OF THE FUNDER/SPONSOR
The funding sources had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and the decision to submit the manuscript for publication.
Supporting information
Supporting material
ACKNOWLEDGMENTS
The authors thank the Colombian families with autosomal‐dominant Alzheimer's disease for contributing their valuable time and effort, without which this study would not have been possible. We also thank Francisco Piedrahita, Alex Navarro, and Claudia Ramos from Grupo de Neurociencias, Universidad de Antioquia in Medellín, Colombia for their help coordinating MRI visits for participants. We thank Dr. Feliza Restrepo for her help overseeing data collection at the Hospital Pablo Tobon Uribe in Medellin.
Fox‐Fuller JT, Torrico‐Teave H, Uquillas Fd'O, et al. Cortical thickness across the lifespan in a Colombian Cohort with autosomal‐dominant Alzheimer's disease: A cross‐sectional study. Alzheimer's Dement. 2021;13:e12233. 10.1002/dad2.12233
REFERENCES
- 1.Jack CR, Bennett DA, Blennow K, et al. NIA‐AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement J Alzheimers Assoc. 2018;14(4):535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dickerson BC, Stoub TR, Shah RC, et al. Alzheimer‐signature MRI biomarker predicts AD dementia in cognitively normal adults. Neurology. 2011;76(16):1395‐1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dickerson BC, Wolk DA. Biomarker‐based prediction of progression in MCI: comparison of AD signature and hippocampal volume with spinal fluid amyloid‐β and tau. Front Aging Neurosci. 2013;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lehtovirta M, Soininen H, Helisalmi S, et al. Clinical and neuropsychological characteristics in familial and sporadic Alzheimer's disease: relation to apolipoprotein E polymorphism. Neurology. 1996;46(2):413‐419. [DOI] [PubMed] [Google Scholar]
- 5.Weston PSJ, Nicholas JM, Lehmann M, et al. Presymptomatic cortical thinning in familial Alzheimer disease: a longitudinal MRI study. Neurology. 2016;87(19):2050‐2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quiroz YT, Stern CE, Reiman EM, et al. Cortical atrophy in presymptomatic Alzheimer's disease presenilin 1 mutation carriers. J Neurol Neurosurg Psychiatry. 2013;84(5):556‐561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacobs HIL, Van Boxtel MPJ, Jolles J, Verhey FRJ, Uylings HBM. Parietal cortex matters in Alzheimer's disease: an overview of structural, functional and metabolic findings. Neurosci Biobehav Rev. 2012;36(1):297‐309. [DOI] [PubMed] [Google Scholar]
- 8.Bakkour A, Morris JC, Dickerson BC. The cortical signature of prodromal AD: regional thinning predicts mild AD dementia. Neurology. 2009;72(12):1048‐1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dickerson BC, Fenstermacher E, Salat DH, et al. Detection of cortical thickness correlates of cognitive performance: reliability across MRI scan sessions, scanners, and field strengths. Neuroimage. 2008;39(1):10‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chételat G, Villemagne VL, Pike KE, et al. Larger temporal volume in elderly with high versus low beta‐amyloid deposition. Brain. 2010;133(11):3349‐3358. [DOI] [PubMed] [Google Scholar]
- 11.Fortea J, Sala‐Llonch R, Bartrés‐Faz D, et al. Increased cortical thickness and caudate volume precede atrophy in PSEN1 mutation carriers. J Alzheimers Dis JAD. 2010;22(3):909‐922. [DOI] [PubMed] [Google Scholar]
- 12.Fortea J, Vilaplana E, Alcolea D, et al. Cerebrospinal fluid β‐amyloid and phospho‐tau biomarker interactions affecting brain structure in preclinical Alzheimer disease. Ann Neurol. 2014;76(2):223‐230. [DOI] [PubMed] [Google Scholar]
- 13.Quiroz YT, Schultz AP, Chen K, et al. Brain imaging and blood biomarker abnormalities in children with autosomal dominant Alzheimer disease: a cross‐sectional study. JAMA Neurol. 2015;72(8):912‐919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Montal V, Vilaplana E, Pegueroles J, et al. Biphasic cortical macro‐ and microstructural changes in autosomal dominant Alzheimer's disease. Alzheimers Dement. n/a(n/a). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tamnes CK, Herting MM, Goddings A‐L, et al. Development of the Cerebral Cortex across Adolescence: a Multisample Study of Inter‐Related Longitudinal Changes in Cortical Volume, Surface Area, and Thickness. J Neurosci. 2017;37(12):3402‐3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frangou S, Modabbernia A, Williams SCR, et al. Cortical thickness across the lifespan: data from 17,075 healthy individuals aged 3‐90 years. Hum Brain Mapp. n/a(n/a). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Natu VS, Gomez J, Barnett M, et al. Apparent thinning of human visual cortex during childhood is associated with myelination. Proc Natl Acad Sci. 2019;116(41):20750‐20759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reiman EM, Quiroz YT, Fleisher AS, et al. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer's disease in the presenilin 1 E280A kindred: a case‐control study. Lancet Neurol. 2012;11(12):1048‐1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Acosta‐Baena N, Sepulveda‐Falla D, Lopera‐Gómez CM, et al. Pre‐dementia clinical stages in presenilin 1 E280A familial early‐onset Alzheimer's disease: a retrospective cohort study. Lancet Neurol. 2011;10(3):213‐220. [DOI] [PubMed] [Google Scholar]
- 20.Sclan SG, Reisberg B. Functional assessment staging (FAST) in Alzheimer's disease: reliability, validity, and ordinality. Int Psychogeriatr. 1992;4:55‐69. Suppl 1. [DOI] [PubMed] [Google Scholar]
- 21.Folstein MF, Folstein SE, McHugh PR. Mini‐mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189‐198. [DOI] [PubMed] [Google Scholar]
- 22.Morris JC, Heyman A, Mohs RC, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part I. Clinical and neuropsychological assessment of Alzheimer's disease. Neurology. 1989;39(9):1159‐1165. [DOI] [PubMed] [Google Scholar]
- 23.Aguirre‐Acevedo DC, Gómez RD, Moreno S, et al. [Validity and reliability of the CERAD‐Col neuropsychological battery]. Rev Neurol. 2007;45(11):655‐660. [PubMed] [Google Scholar]
- 24.Jacobs HIL, Hedden T, Schultz AP, et al. Structural tract alterations predict downstream tau accumulation in amyloid‐positive older individuals. Nat Neurosci. 2018;21(3):424‐431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwarz CG, Gunter JL, Wiste HJ, et al. A large‐scale comparison of cortical thickness and volume methods for measuring Alzheimer's disease severity. NeuroImage Clin. 2016;11:802‐812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barnes J, Ridgway GR, Bartlett J, et al. Head size, age and gender adjustment in MRI studies: a necessary nuisance?. NeuroImage. 2010;53(4):1244‐1255. [DOI] [PubMed] [Google Scholar]
- 27.Quiroz YT, Sperling RA, Norton DJ, et al. Association between amyloid and Tau accumulation in young adults with autosomal dominant Alzheimer disease. JAMA Neurol. 2018;75(5):548‐556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson KA, Schultz A, Betensky RA, et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol. 2016;79(1):110‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aguirre‐Acevedo DC, Jaimes‐Barragán F, Henao E, et al. Diagnostic accuracy of CERAD total score in a Colombian cohort with mild cognitive impairment and Alzheimer's disease affected by E280A mutation on presenilin‐1 gene. Int Psychogeriatr. 2016;28(3):503‐510. [DOI] [PubMed] [Google Scholar]
- 30.Bocanegra Y, Fox‐Fuller JT, Baena A, et al. Association between visual memory and in vivo amyloid and Tau pathology in preclinical autosomal dominant Alzheimer's disease. J Int Neuropsychol Soc JINS. 2020:1‐9. Published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Calsolaro V, Edison P. Neuroinflammation in Alzheimer's disease: current evidence and future directions. Alzheimers Dement J Alzheimers Assoc. 2016;12(6):719‐732. [DOI] [PubMed] [Google Scholar]
- 32.Iacono D, O'Brien R, Resnick SM, et al. Neuronal hypertrophy in asymptomatic Alzheimer disease. J Neuropathol Exp Neurol. 2008;67(6):578‐589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soto‐Mercado V, Mendivil‐Perez M, Velez‐Pardo C, Lopera F, Jimenez‐Del‐Rio M. Cholinergic‐like neurons carrying PSEN1 E280A mutation from familial Alzheimer's disease reveal intraneuronal sAPPβ fragments accumulation, hyperphosphorylation of TAU, oxidative stress, apoptosis and Ca2+ dysregulation: therapeutic implications. PLOS ONE. 2020;15(5):e0221669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.LaPoint MR, Chhatwal JP, Sepulcre J, Johnson KA, Sperling RA, Schultz AP. The association between tau PET and retrospective cortical thinning in clinically normal elderly. NeuroImage. 2017;157:612‐622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benzinger TLS, Blazey T, Jack CR, et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer's disease. Proc Natl Acad Sci. 2013;110(47):E4502‐E4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanchez JS, Hanseeuw B, Lopera F, et al. Longitudinal amyloid and Tau accumulation in autosomal dominant Alzheimer's disease: findings from the Colombia‐Boston (COLBOS) Biomarker Study. Alzheimers Res Ther. Published online In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.ε4 MichaelsonDMAPOE. The most prevalent yet understudied risk factor for Alzheimer's disease. Alzheimers Dement. 2014;10(6):861‐868. [DOI] [PubMed] [Google Scholar]
- 38.Wu L, Zhao L. ApoE2 and Alzheimer's disease: time to take a closer look. Neural Regen Res. 2016;11(3):412‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arboleda‐Velasquez JF, Lopera F, O'Hare M, et al. Resistance to autosomal dominant Alzheimer's disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019;25(11):1680‐1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fuller JT, Cronin‐Golomb A, Gatchel JR, et al. Biological and cognitive markers of Presenilin1 E280A autosomal dominant Alzheimer's disease: a Comprehensive Review of the Colombian Kindred. J Prev Alzheimers Dis. 2019;6(2):112‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.2019 Alzheimer's disease facts and figures ‐ ‐ 2019 ‐ Alzheimer's & Dementia ‐ Wiley Online Library . Accessed November 24, 2020. https://alz‐journals.onlinelibrary.wiley.com/doi/abs/10.1016/j.jalz.2019.01.010
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting material