

Abstract
Cotton leaf curl Multan virus (CLCuMuV) and its associated satellites are a major part of the cotton leaf curl disease (CLCuD) caused by the begomovirus species complex. Despite the implementation of potential disease management strategies, the incessant resurgence of resistance-breaking variants of CLCuMuV imposes a continuous threat to cotton production. Here, we present a focused effort to map the geographical prevalence, genomic diversity, and molecular evolutionary endpoints that enhance disease complexity by facilitating the successful adaptation of CLCuMuV populations to the diversified ecosystems. Our results demonstrate that CLCuMuV populations are predominantly distributed in China, while the majority of alphasatellites and betasatellites exist in Pakistan. We demonstrate that together with frequent recombination, an uneven genetic variation mainly drives CLCuMuV and its satellite’s virulence and evolvability. However, the pattern and distribution of recombination breakpoints greatly vary among viral and satellite sequences. The CLCuMuV, Cotton leaf curl Multan alphasatellite, and Cotton leaf curl Multan betasatellite populations arising from distinct regions exhibit high mutation rates. Although evolutionarily linked, these populations are independently evolving under strong purifying selection. These findings will facilitate to comprehensively understand the standing genetic variability and evolutionary patterns existing among CLCuMuV populations across major cotton-producing regions of the world.
Keywords: Cotton leaf curl Multan virus, satellite molecules, evolution, genetic diversity, mutation, selection, recombination
1. Introduction
Genetic diversity among viral populations predominantly drives viral evolution, immune escape, pathogenicity, and resistance to chemicals (Sanjuán and Domingo-Calap 2019). Although viruses possess small genomes with error-prone replication processes, they display extraordinary adaptation capacities owing to the higher magnitudes of genetic variations (Simmonds, Aiewsakun, and Katzourakis 2019). Viruses undergo complex evolutionary processes (Koonin, Dolja, and Krupovic 2015; Solé 2016; Dolja and Koonin 2018; Shi et al. 2018; Wolf et al. 2018), which consequently enable them to incorporate numerous dynamic features such as diverse genome assortment, replication mechanisms, and gene expression strategies (Baltimore 1971; Gale, Tan, and Katze 2000; Elena 2016). Thus, genetic variation acts as a conjoint process for viruses to provide the raw materials for their successful adaptation to variable environments (Domingo 2020).
Several viral populations are known to exist in closely related complexes. The existence of these viral genomic variants is attributed to rapid replicative kinetics, large population size, and a high mutation rate (Domingo and Schuster 2016). DNA viruses might be genetically complex and possess several uniquely evolved viral genes (Villarreal 2008). DNA viruses are known to replicate through proofreading DNA-dependent DNA polymerases. Nonetheless, several studies have reported that single-stranded DNA (ssDNA) viruses can undergo as quick evolutionary events as RNA viruses (Drake 1991; Shackelton et al. 2005; Shackelton and Holmes 2006; Duffy and Holmes 2008). One possible thought supporting these findings is that the mutation and substitution rates among RNA and ssDNA viruses seem to be more similar than previously believed (Duffy, Shackelton, and Holmes 2008; Cuevas, Duffy, and Sanjuán 2009). A high level of within-host genetic variability has been proposed for ssDNA plant-infecting viruses of Geminiviridae and Nanoviridae families (Ge et al. 2007; Eric et al. 2008; Grigoras et al. 2010). Interestingly, the substitution rates for whitefly-vectored begomoviruses have been reported to be similar to those of RNA viruses (Duffy and Holmes 2008, 2009). Although mutation is a major factor that drives the diversity of viral populations (Roossinck 1997; García-Arenal, Fraile, and Malpica 2001; Balol et al. 2010), it does not account for all current genetic diversity like other evolutionary factors (such as recombination) (Martin et al. 2011). Among ssDNA viruses, it has been suggested that they achieve higher recombination rates via recombination-dependent replication processes (Jeske, Lütgemeier, and Preiß 2001).
Cotton (Gossypium spp. L., family Malvaceae) is a cultivated shrub and globally the largest source of natural fibers. The cotton is grown commercially in ∼150 countries and provides substantial economic returns for >100 million families (Tarazi, Jimenez, and Vaslin 2019). Cotton crop is vulnerable to a variety of insect pests and pathogens, among which cotton leaf curl disease (CLCuD, caused by begomoviruses complex) is the most damaging factor (Mansoor, Zafar, and Briddon 2006; Sattar et al. 2013; Sohrab et al. 2014). CLCuD was reported the first time in 1967 from Multan, Pakistan (Hussain and Ali 1975). In the following decades, it decreased cotton production, raising the concerns of agricultural specialists and farmers. Later in 1992–97, the yield loss in cotton reached up to 29 per cent (Briddon and Markham 2000), and meanwhile, in 1993, the CLCuD was first reported from India (Kumar, Kumar, and Khan 2010). The CLCuD comprises of a begomoviral species complex containing five viruses: Cotton leaf curl Multan virus (CLCuMuV), Cotton leaf curl Kokhran virus, Cotton leaf curl Gezira virus, Cotton leaf curl Alabad virus, and Cotton leaf curl Bangalore virus (Saleem et al. 2016).
The monopartite genomes of CLCuD-associated begomoviruses (CABs) are circular ssDNAs of ∼2.7 kb in size. The genomes encode a total of seven open reading frames (ORFs), of which five (C1, C2, C3, C4, and C5) are on the complementary strand, whereas two (V1 and V2) belong to the virion sense. The ORFs on the complementary strand are involved in replication, transcription activation, RNA silencing suppression, and DNA replication (Hanley-Bowdoin et al. 2013), whereas the ORFs on the virion sense contribute to encapsidation. Additionally, a non-coding common region or intergenic region of approximately 200 nucleotides (nts) contains a highly conserved nanonucleotide sequence (TAATATTAC) with an origin of replication (Ashraf et al. 2013). Furthermore, in bipartite begomoviruses, the DNA-B encodes for a movement protein (MP) and nuclear shuttle protein (NSP) (Fondong 2013). The NSP is known to facilitate the intracellular transportation of viral DNA between the nucleus and the cytoplasm and interacts with the MP to assist cell-to-cell movement of the viral DNA to healthy cells (Martins et al. 2020). The old world begomoviruses are known to have a frequent association with ssDNA helper molecules designated as alphasatellites and betasatellites (Zhou 2013) and more recently characterized non-coding deltasatellites with unknown function (Fiallo-Olivé, Tovar, and Navas-Castillo 2016; Lozano et al. 2016). Alphasatellites encode for a replication-associated protein (Rep), which is dependent on the helper virus for movement and whitefly-vectored transmission to host plants (Kumar et al. 2015). They are known to regulate the severity of begomoviral–betasatellites disease complex (Idris et al. 2010; Shweta and Khan 2018). On the other hand, betasatellites encode a protein βC1, which plays a crucial role in symptom determination and pathogenicity of begomoviruses (Saeed et al. 2005). It is well documented that the emergence, establishment, and evolution of new geminiviral species are mainly driven by recombination, which involves an exchange of genetic material and consequently regulates the evolution of viruses (Silva 2014).
As a major part of the CLCuD complex, CLCuMuV along with satellite molecules is a key limiting factor to cotton production in several regions of South Asia (Sattar et al. 2013; Zhou 2013). Also, in 2008, it has been reported to infect Chinese hibiscus in the Southern part of China (Mao et al. 2008). Notably, in 2017, a resurgence of resistant-breaking virulent strains of CLCuMuV has been reported from Punjab, India (Datta et al. 2017). Thus, in addition to the aspects of epidemiological shifting, the rebound of CLCuMuV indicates a possible pandemic threat in the future. The current information about geographical distribution, genetic diversity, and evolutionary dynamics of CLCuMuV and associated satellites remain insufficient or not up to date. Moreover, there are no data available regarding comparative analysis of genetic variability and evolutionary aspects to understand the virus and satellite populations arising from different geographical locations. Since these data are critical for designing sustainable disease management strategies, we have rigorously analyzed the current biodiversity, genomic variability, and evolutionary endpoints to get further insights into the complexity and molecular variability of CLCuMuV and accompanying satellites. Further, considering the role of satellites in CLCuMuV-mediated infections and disease complexes, we performed a parallel evolutionary analysis of DNA-A (CLCuMuV), alphasatellites (Cotton leaf curl Multan alphasatellite (CLCuMuA)), and betasatellites (Cotton leaf curl Multan betasatellite (CLCuMuB)) to get a better picture of begomoviral evolution expanded over two decades.
2. Results
2. 1. Current geographical distribution and phylogenetics of CLCuMuV and satellite molecules
The analyzed full-length CLCuMuV genome consisted of ∼2,700 nts, and its accompanying satellites were approximately half the size of the viral genome, i.e. alphasatellites comprised of ∼1,360 nts, whereas betasatellites consisted of ∼1,350 nts (Fig. 1A–C). Currently, the virus and satellites are known to be present in five countries, including China, Pakistan, India, the Philippines, and Thailand (Fig. 1D). Among a total of 121 globally known isolates of CLCuMuV, 50 virus isolates have been reported from China, followed by Pakistan (35), India (32), the Philippines (3), and Thailand (1). On the contrary, CLCuMuA has been recovered from cotton collected predominantly from Pakistan (153/162) followed by a few isolates from India (9/162), whereas there are no reports of CLCuMuA presence in other countries. Moreover, a total of 447 CLCuMuB global isolates, the majority of cotton-infecting CLCuMuB populations, have been reported from Pakistan (330), with a second contribution from India (65), followed by China (49) and the Philippines (3) (Fig. 1E). Notably, to date, there are no reports of CLCuMuV-associated satellites from Thailand.
Figure 1.
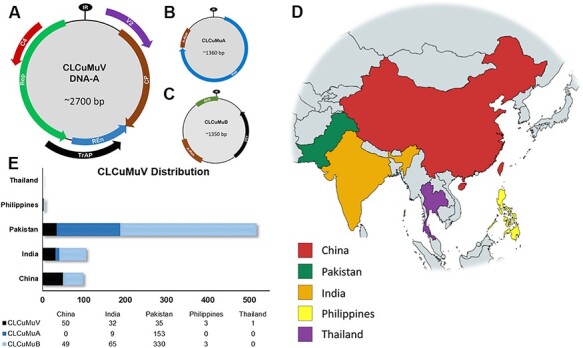
Genome organization and the geographical distribution of CLCuMuV and associated satellites. (A) CLCuMuV has a genome size of 2,700 nts with four ORFs on the complementary strand encoding Rep, TrAP, REn, and C4 proteins; the virion sense strand encodes CP and MP proteins. (B) Alphasatellites (CLCuMuA) possess genomes of ∼1,360 nts and encode for Rep proteins. Additionally, they have a non-coding A-rich region. (C) CLCuMuB encompasses a genome of ∼1,350 nts with only one ORF encoding for the βC1 protein; the betasatellite contains conserved, non-coding satellite conserved region (SCR) and A-rich regions. (D) Map representing the current geographical distribution of CLCuMuV and associated satellite molecules in six countries. (E) Horizontal bar graph denoting numbers of CLCuMuV, alphasatellites, and betasatellites reported from five countries.
2.2. Molecular phylogenetics of CLCuMuV, CLCuMuA, and CLCuMuB
2.2.1. CLCuMuV
Next, to assess the standing evolutionary relatedness among these populations, we performed molecular phylogenetic analysis of CLCuMuV, CLCuMuA, and CLCuMuB using full-genome sequences reported from different regions. The phylogenetic analysis of CLCuMuV sequences revealed the formation of nine unique groups (Fig. 2A). These groups divided the CLCuMuV populations from five countries based on the type of host plants they infect. For instance, a majority (thirty-nine isolates) of Chinese CLCuMuV populations were found to be associated with hibiscus, followed by cotton (four isolates), okra (three isolates), and Passiflora (one isolate) (Fig. 2A), whereas three isolates were observed to have missing host information (Fig. 2A). Chinese isolates of CLCuMuV clustered together and exhibited even distribution (Fig. 2A). However, three isolates were exceptional: NC004607 shared the same node with an isolate from Pakistan (AJ002447), having 100 per cent sequence homology; MK482365 grouped with Pakistani isolate (AJ496287), showing 100 per cent sequence similarity (Supplementary Table S2); and AJ002459 shared a node with an Indian isolate (JN558352), having 95.7 per cent sequence identity (Supplementary Table S2). Interestingly, for the Pakistani isolates of CLCuMuV, cotton was found to be the most common host (infected by twenty-eight isolates), followed by seven isolates with unknown host information. A total of fourteen Indian isolates of CLCuMuV were observed to infect cotton, followed by hollyhock (five isolates), hibiscus (four isolates), and papaya (one isolate), while the origin of eight isolates remained unknown due to missing information of the host plant. Additionally, three CLCuMuV isolates from the Philippines remained associated with hibiscus, while the only CLCuMuV isolate from Thailand was found to infect Emilia (Fig. 2A). The CLCuMuV isolates from the Philippines and Thailand were grouped with Chinese isolates, which indicates the high sequence homology of their genomes. On the contrary, CLCuMuV isolates from Pakistan and India exhibited a diverse pattern of distribution, showing that these populations are genetically more identical.
Figure 2.
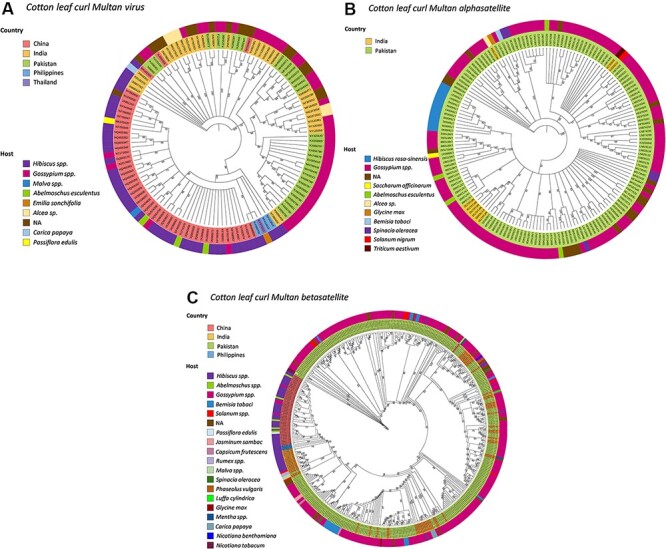
Mid-point rooted, Bayesian phylogenetic analyses based on full-length nucleotide sequences of CLCuMuV and associated satellites. The phylogenetic trees show the evolutionary relationship of (A) Cotton leaf curl Multan virus (CLCuMuV), (B) Cotton leaf curl Multan alphasatellite (CLCuMuA), and (C) Cotton leaf curl Multan betasatellite (CLCuMuB). The inner ring indicates the country of origin and the outer ring the host.
2.2.2. CLCuMuA
The phylogenetic tree based on full-length genomes of CLCuMuA divided a total of 153 Pakistani isolates into 9 unique groups. We found that 113 of these isolates were originating from cotton, followed by Malus (11 isolates), okra (4 isolates), Spinacia (4 isolates), Saccharum (1 isolate), soybean (1 isolate), and Bemisiatabaci (1), whereas 18 isolates lacked the information regarding host plant. Further, of nine Indian CLCuMuA isolates, six were associated with cotton. As for other plants like wheat, hollyhock, and Lycopersicon esculentum, each host was associated with one isolate (Fig. 2B). Further analysis of their phylogeny together with Pakistani isolates revealed that six of Indian CLCuMuA isolates (MF141732, MF141733, MF141734, MF141735, MF141740, and KY783480) grouped with an isolate from Pakistan (LN829161), whereas isolate MG373554 shared a clade with the Pakistani isolate (MN922310) (Fig. 2B), sharing 68.4 per cent sequence identity (Supplementary Table S3). Besides, KC305093 and KJ028212 Indian isolates clustered together with Pakistani isolate HE599398 (Fig. 2B), sharing sequence identities of 69 and 68.4 per cent, respectively (Supplementary Table S3).
2.2.3. CLCuMuB
Finally, analysis of the betasatellite populations (CLCuMuB) divided Pakistani isolates into twelve distinct groups. Among these groups, the largest one (with 266 isolates) consisted of cotton-infecting betasatellites, followed by B. tabaci (14), L. esculentum (7), Jasmine (2), spinacia (2), luffa (1), chili (1), bean (1) soybean (1), malvacerum (1), Nicotiana benthamiana, Nicotiana tabacum (1), whereas a total of 33 isolates of CLCuMuB were grouped together without available host information. Similarly, the Indian populations were subdivided into eight unique groups. The largest group (thirty-nine isolates) consisted of the betasatellites that were reported from cotton, followed by hibiscus (thirteen), okra (five), papaya (three), rumex (two), L. esculentum (two), mentha (one) and Passiflora (one), while only three isolates had an unknown origin (Fig. 2C). Additionally, a similar pattern of homology-based grouping among Indian and Pakistani isolates (Fig. 2C) was observed, representing that they exhibit a high sequence similarity of betasatellite genomes. Chinese CLCuMuB isolates formed a distinct group except for one isolate (JQ716368) that shared the same clade with the Pakistani isolate (HF565180) and shared a low sequence homology of 18.4 per cent (Supplementary Table S4). Further, the CLCuMuB isolates from the Philippines were clustered together and one isolate (KF413619) exhibited 79.2 per cent sequence similarity with the Indian isolate (AY704661), whereas another isolate (KF413617) remained closer to the Chinese isolate (JQ963630), sharing sequence homology of 96.9 per cent (Supplementary Table S4).
2. 3. Comparison of genetic variability among CLCuMuV, CLCuMuA, and CLCuMuB populations
We analyzed all datasets comprising CLCuMuV (121 sequences), CLCuMuA (162 sequences), and CLCuMuB (447 sequences) to compare the standing molecular diversity among viral and satellite populations arising from different geographical locations. Despite the existence of discrepancy among sample sizes, we were able to calculate average pairwise nucleotide diversities (π) for the aforementioned datasets. Interestingly, the average pairwise nucleotide differences were higher for Indian CLCuMuV isolates (π = 0.07984), followed by Pakistan (π = 05567), the Philippines (π = 0.009288), and China (π = 0.00711), respectively (Fig. 3A and Table 1). As for the alphasatellites (CLCuMuA) sequences, the nucleotide diversity remained higher among Indian populations (π = 0.09532) followed by Pakistani isolates with a lower value (π = 0.04415). Captivatingly, the genetic variation between CLCuMuB isolates from India was higher (π = 0.14640) as compared to those originating from Pakistan (π = 0.04415), China (π = 0.03451), and the Philippines (π = 0.01414) (Fig. 3A and Table 1).
Figure 3.
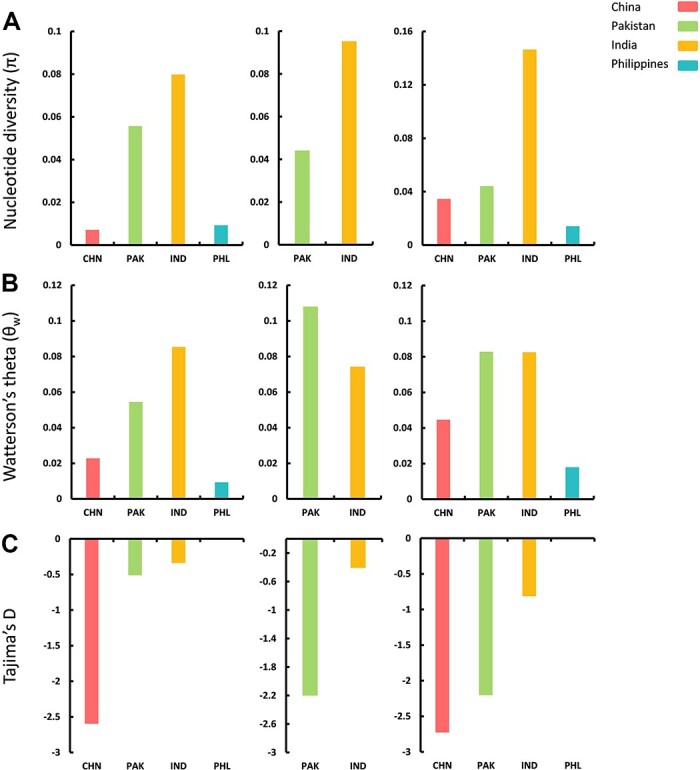
Estimation of genetic diversity was performed for CLCuMuV, CLCuMuA, and CLCuMuB populations separated by geographical origin. The calculated population genetic parameters include (A) nucleotide diversity (π), (B) Watterson’s theta (θw), and (C) Tajima’s D.
Table 1.
Molecular diversity among CLCuMuV, CLCuMuA, and CLCuMuB populations, distributed in different geographical regions.
| Genome type | Region | S | Eta | H | H d | π | θ w | Tajima’s D |
|---|---|---|---|---|---|---|---|---|
| CLCuMuV | China | 297 | 312 | 43 | 0.996 | 0.00711 | 0.022816 | −2.59713 |
| Pakistan | 596 | 694 | 29 | 0.988 | 0.05567 | 0.054456 | −0.51231 | |
| India | 922 | 1123 | 31 | 0.998 | 0.07984 | 0.085385 | −0.33961 | |
| Philippines | – | – | 03 | 1.000 | 0.00928 | 0.009288 | N/A | |
| CLCuMuA | Pakistan | 93 | 157 | 47 | 0.876 | 0.04415 | 0.108018 | −2.20125 |
| India | 347 | 391 | 09 | 1.000 | 0.09532 | 0.074281 | −0.40729 | |
| CLCuMuB | China | 283 | 330 | 32 | 0.965 | 0.03451 | 0.044666 | −2.72646 |
| Pakistan | 3 | 5 | 08 | 0.089 | 0.04415 | 0.082853 | −2.20125 | |
| India | 200 | 333 | 61 | 0.998 | 0.14640 | 0.082530 | −0.81456 | |
| Philippines | – | – | 03 | 1.000 | 0.01414 | 0.017919 | N/A |
S, number of polymorphic (segregating) sites; Eta, total number of mutations; H, number of haplotypes; Hd, haplotype diversity; π, nucleotide diversity; θw, Watterson’s theta.
Furthermore, the average number of segregating sites (θw) was remarkably higher among Indian isolates of CLCuMuV (θw = 0.085385), while these values were lower for isolates from Pakistan (θw = 0.05456). Conversely, the CLCuMuA populations from Pakistan exhibited a higher segregation rate (θw = 0.108018) as compared to Indian isolates (θw = 0.074281). Notably, the CLCuMuB isolates from Pakistan and India exhibited almost similar levels of segregating sites (θw = 0.082853 and θw = 0.082530, respectively) (Fig. 3B and Table 1). Likewise, we found Tajima’s D values highly negative in Chinese populations of CLCuMuV (−2.59713), indicating the presence of excessive polymorphic sites among these isolates. On the contrary, Tajima’s D was highly negative for CLCuMuA isolates from Pakistan (−2.20125) as compared to Indian populations (−0.40729). Finally, Tajima’s D values for CLCuMuB isolates remained highly negative for the Chinese population (−2.72646) followed by a closer value among Pakistani isolates (−2.20125) trailed by Indian isolates (−0.81456), whereas the sample size of the Philippines population was too low to calculate Tajima’s D value for CLCuMuV and CLCuMuB groups (Fig. 3C and Table 1).
2. 4. Recombination events have a clear correlation with genome variation of CLCuMuV, alphasatellites and betasatellites
To investigate the presence of recombination events among geographically isolated populations of CLCuMuV, CLCuMuA, and CLCuMuB, we used RDP4. For all analyzed datasets, only the unique recombination events detected at least by four methods supported by a P-value of <0.001 were considered reliable.
The results revealed that 55 per cent of isolates had detectable recombination events (Fig. 4); CLCuMuV populations originating from Pakistan were more prone to recombinational changes. Recombination event at positions 690–2191 (without gaps) was detected among 18/35 sequences (P-value = 1.645 × 10−24) pertaining to CLCuMuV (Table 2). Indian populations of CLCuMuV followed with a recombination detection percentage of 3.2 (P-value = 1.887 × 10−29) at genomic positions 998–1459 (no gaps). While only a smaller percentage of CLCuMuV sequences (3.2 per cent) was detected (P-value = 1.239 × 10−19) to have recombination (at positions 2190–2278) among Chinese populations (Fig. 4, Table 2). Due to the low number of sequences, the analysis was not performed for sequences from the Philippines and Thailand. The detailed recombination events detected in CLCuMuV populations can be found in Supplementary Table S5.
Figure 4.
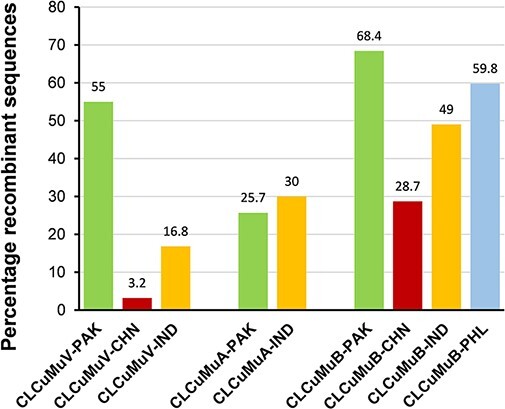
Estimation of percentage sequences evolving through recombinational variation. All analyzed datasets (CLCuMuV, CLCuMuA, and CLCuMuB) were further divided based on origin of the sequences from different geographical locations.
Table 2.
Frequently detected recombination events by RDP in CLCuMuV, CLCuMuA, and CLCuMuB datasets.
| Sequences | Recombination breakpoints | Parental sequences | ||||||
|---|---|---|---|---|---|---|---|---|
| Group | detected with recomb. event | Recombinanta | Begin | End | Major | Minor | Detection Methodsb | P-valuec |
| CLCuMuV-PAK | 18 | FJ218486 PAK | 690 (750) | 2191 (2284) | AJ496461 PAK | EU384573 PAK | RGBMCS3 | 1.645 × 10−24 |
| CLCuMuV-CHN | 1 | AJ002459 CHN | 2190 (2701) | 2278 (2912) | Unknown | NC004607 CHN | RGBMCSP | 1.239 × 10−19 |
| CLCuMuV-IND | 13 | JN678803 IND | 998 (1057) | 1459 (1521) | MN127818 IND | KY561820 IND | RGBM3 | 1.887 × 10−29 |
| CLCuMuA-PAK | 76 | LN874310 PAK | 174 (202) | 420 (994) | Unknown | LN874310 PAK | RGBMCS3 | 1.046 × 10−08 |
| CLCuMuA-IND | 1 | KJ028212 IND | 892 (1055) | 1286 (1807) | MF141735 IND | MF141732 IND | GMS3 | 2.203 × 10−06 |
| CLCuMuB-PAK | 65 | EU384594 PAK | 23 (66) | 953 (1826) | Unknown | LT549464 PAK | RGBMCS3 | 1.367 × 10−37 |
| CLCuMuB-CHN | 1 | JQ716368 CHN | 315 (1144) | 532 (2107) | HQ455359 CHN | Unknown | RGBMCS | 8.623 × 10−14 |
| CLCuMuB-IND | 20 | JF502396 IND | 202 (389) | 866 (1702) | JF502398 IND | JF502390 IND | RGBMCS3 | 2.787 × 10−19 |
| CLCuMuB-PHL | 1 | KF413617 PHL | 239 (464) | 1049 (2041) | KF413619 PHL | KX068710 PHL | RGBM3 | 3.222 × 10−05 |
Numbering starts at the first nucleotide after the cleavage site at the origin of replication and increases clockwise.
R, RDP; G, GeneConv; B, Bootscan; M, MaxChi; C, CHIMAERA; S, SisScan; 3, 3SEQ.
The described P-value corresponds to the program in bold, underlined type, and is the lowest P-value calculated for the event in question.
As for the CLCuMuA dataset from Pakistan, the most frequently detected recombination event was found in 76/153 sequences (P-value = 1.046 × 10−08) at positions 174–420 (without gaps) (Table 2). The total percentage of CLCuMuA recombinant sequences from Pakistan remained 25.7 per cent (Fig. 4), whereas Indian populations of CLCuMuA were detected to have recombination events (P-value = 2.203 × 10−06) only in one sequence out of nine, although the percentage of recombinant sequences remained higher (30 per cent) (Fig. 4). The recombination breakpoint was found at positions 892–1286 in alphasatellite genome. The detailed recombination events detected among alphasatellite populations can be found in Supplementary Table S6. Since alphasatellite has only been reported from Pakistan and India to date, no sequences were available for recombination analysis from other countries.
Finally, the highest number of CLCuMuB sequences (65/330) from Pakistan were found to have the most frequent recombination breakpoint (P-value = 1.367 × 10−37) at positions 23–953 (Table 2). On the other hand, twenty out of sixty-five CLCuMuB sequences from India were found to have the most common recombination event (P-value = 2.787 × 10−19) at positions 202–866 (Table 2). As for the Chinese populations of CLCuMuB, a smaller portion of the betasatellite sequence was found to have a recombination breakpoint at positions 315–532. Interestingly, CLCuMuB populations from the Philippines were detected to have recombination (P-value = 3.222 × 10−05) covering a larger genomic sequence (239–1049) (Table 2). The detailed information on recombination breakpoints detected for CLCuMuB can be found in Supplementary Table S7. Additionally, the percentage of recombinant sequences remained higher for Pakistani isolates (68.4 per cent), followed by the Philippines (59.8 per cent), India (49 per cent), and China (28.7 per cent) (Fig. 4).
2. 5. Recombination breakpoints display variable distribution among analyzed sequences
Further, in efforts to map the distribution of recombination breakpoints among analyzed sequences, we found that a great variation exists among viral and satellite populations. For example, in the case of frequently detected recombination events in the Pakistani recombinant isolate (FJ218486-PAK), we observed that recombination covered almost 55 per cent of the CLCuMuV genome (Fig. 5A). The TrAP and REn genes had complete recombinant sequence, while coat protein (CP), Rep, and AC4 had partial sequence undergone recombination. On the contrary, the Chinese recombinant isolate of CLCuMuV (AJ002459-CHN) showed recombination only on 3.2 per cent sequence with partial coverage of AC4 and Rep genes (Fig. 5A), whereas among Indian populations of CLCuMuV, frequent recombination was detected in 16.8 per cent of the genome sequence. The REn gene was completely covered by recombination, while only a part of the TrAP gene sequence was under recombinational variations. Interestingly, none of the sequences showed any evidence of recombination affecting the MP (Fig. 5A).
Figure 5.
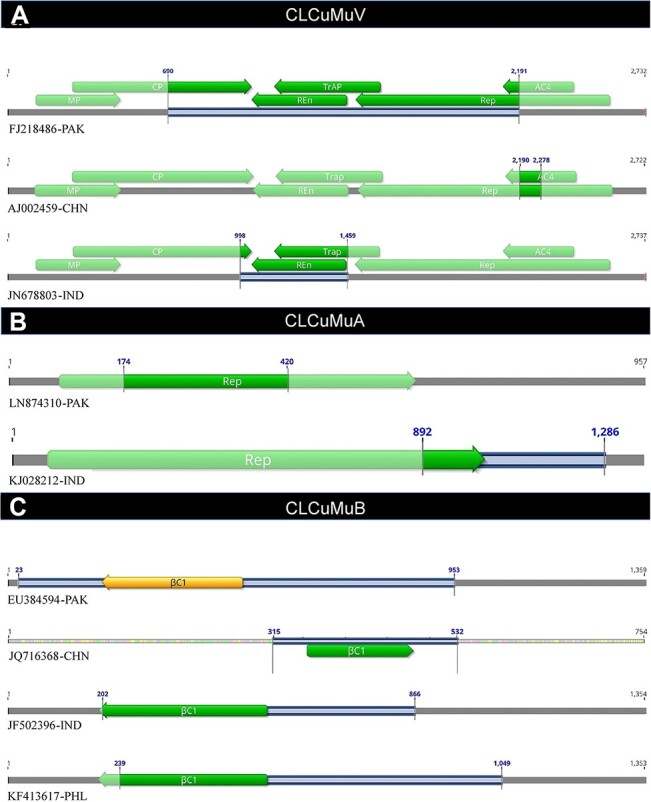
Patterns of most frequently detected recombination breakpoints distributed among populations of (A) CLCuMuV, (B) CLCuMuA, and (C) CLCuMuB. Each line represents a recombinant isolate and the genome area exhibiting recombination is highlighted by blue lines.
Additionally, analysis of CLCuMuA sequences revealed a contrasting pattern of recombination among recombinant sequences. For instance, recombinant CLCuMuA (LN874310-PAK) had 25.7 per cent of genomic sequence influenced by recombination, and interestingly, the recombination breakpoints were restricted to the coding sequence of Rep gene only (Fig. 5B). On the contrary, the Indian isolate of recombinant CLCuMuA (KJ028212-IND) had 30 per cent genomic sequence under recombination, and the breakpoints were distributed to Rep gene and the non-coding region of the alphasatellite.
Although the CLCuMuB recombinant sequences showed a somewhat similar pattern of recombination, the difference among the distribution of breakpoints was obvious. Remarkably, the Pakistani recombinant betasatellite (EU384594-PAK) had 68.4 per cent sequence under recombination with complete coverage of betasatellite-associated pathogenicity gene βC1 (Fig. 5C). On the other hand, Chinese (JQ716368-CHN) and Indian (JF502396-IND) isolates exhibited recombinant sequences covering 28.7 and 49.0 per cent, respectively. Although the Philippines recombinant isolate (KF413617-PHL) had comparatively a larger portion (59.8 per cent) of the genomic sequence under recombination, the βC1 gene was not completed covered (Fig. 5C).
2. 6. Negative selection pressure predominantly governs the standing genomic variation of CLCuMuV and associated satellite molecules
To further understand the possible role of selection pressure on the variable genomic variation observed between analyzed datasets, we compared the non-synonymous to synonymous substitutions (dN/dS) for each gene of the genomic component. The average dN/dS ratio for all genes (Rep, TrAP, REn, and AC4) of the CLCuMuV genome remained <1, indicating that the observed genomic variation is mainly being driven by the negative selection (Table 3). The average dN/dS ranged between 0.100 and 0.243, with the lowest value observed for the CP while the highest value was observed for the MP (Fig. 6; Table 3). A total of 249/280 negatively selected sites were found in the Rep gene sequence, and the values ranged between 0.195 and 0.939. Interestingly, few sites were also detected with positive selection although the proportion remained lower (3/280) than the negatively selected sites. The values of positively selected sites in the Rep gene ranged between 1.038 and 1.302 (Fig. 6, Table 3). Likewise, a pattern was displayed by MP, which had 101/114 negatively selected positions. The average value of negative selection for the MP remained 0.202, with the least being 0.011 and the highest at 0.899. There were two positively selected sites detected in MP. With an average positive selection value of 2.492, the minimum value remained 1.469 and the maximum value was 3.514 (Table 3). Furthermore, REn exhibited only one positively selected site with a value of 1.081, while this gene showed 104/115 negatively selected sites with 0.131 and 0.972 being minimum and maximum values, respectively. Remarkably, AC4 and CP did not show any positively selected site; however, the ratio of negatively sites were 81/88 and 223/243 for AC4 and CP genes, respectively.
Table 3.
Estimation of average dN/dS ratios and positive and negative selection for coding sequences of CLCuMuV, CLCuMuA, and CLCuMuB.
| Positive selection | Negative selection | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| ORF | Avg. dN/dS ratio | Total sites | Avg. | Min. | Max. | Total sites | Avg. | Min. | Max. |
| CLCuMuV | |||||||||
| Rep | 0.205 | 3 | 1.128 | 1.038 | 1.302 | 249 | 0.195 | 0.007 | 0.939 |
| TrAP | 0.130 | 1 | 1.040 | 1.040 | 1.040 | 117 | 0.122 | 0.007 | 0.912 |
| REn | 0.139 | 1 | 1.081 | 1.081 | 1.081 | 104 | 0.131 | 0.007 | 0.972 |
| AC4 | 0.130 | 0 | – | – | – | 81 | 0.130 | 0.007 | 0.927 |
| CP | 0.100 | 0 | – | – | – | 223 | 0.100 | 0.007 | 0.871 |
| MP | 0.243 | 2 | 2.492 | 1.469 | 3.514 | 101 | 0.202 | 0.011 | 0.899 |
| CLCuMuA | |||||||||
| Rep-A | 0.313 | 0 | – | – | – | 154 | 0.284 | 0.023 | 0.967 |
| CLCuMuB | |||||||||
| βC1 | 0.331 | 0 | – | – | – | 41 | 0.331 | 0.087 | 0.652 |
Figure 6.
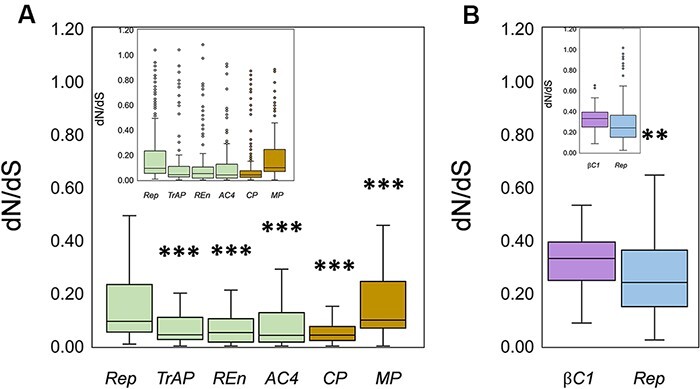
Estimation of selection pressure was performed by calculation of non-synonymous to synonymous substitution ratios (dN/dS). Box plots correspond to (A) dN/dS ratio calculated for the CLCuMuV-encoded genes on the complementary sense strand (Rep, TrAP, Ren, and AC4) and virion sense strand (CP and MP), (B) dN/dS ratio for CLCuMuB-encoded βC1 gene and CLCuMuA-encoded Rep gene. The horizontal lines inside the box represent the median values, while inset displays the data with outliers denoted by small circles. Asterisks represent significance: **P < 0.01; ***P < 0.001.
Notably, the Rep gene of CLCuMuA did not exhibit any positive selection, while the ratio of negative selection was 143/159. The average value of negatively selected sites was 0.284, with a minimum of 0.023 and a maximum of 0.967. Interestingly, on the other hand, the CLCuMuB-encoded βC1 gene showed that all the tested sites were under negative selection pressure (41/41), with an average value of 0.331 (Fig. 6, Table 3). In conclusion, all the analyzed genes were evolving under strong negative selection pressure. The percentage contribution of negative selection pressure for CLCuMuV-encoded genes remained ∼90, whereas for CLCuMuA-encoded Rep, it was 96 per cent and CLCuMuB-encoded βC1 exhibited evolution under 100 per cent negative selection pressure (Supplementary Fig. S1).
2. 7. Comparative analysis of negative selection pressure for individual genes isolated by country of origin
Next, we compared the extent of negative selection pressure between all genes encoded by CLCuMuV, CLCuMuA, and CLCuMuB. Interestingly, the genes encoded by Chinese isolates of CLCuMuV displayed a relatively higher percentage of sequences evolving under purifying selection pressure, whereas a slightly less percentage was seen for the Pakistani and Indian groups comprising Rep, TrAP, REn, AC4, MP, and CP genes (Fig. 7A). Further analysis of CLCuMuA-encoded Rep displayed that Pakistani populations exhibited a higher negative selection pressure (80.17 per cent) compared to a much lower negative selection pressure (13.73 per cent) in Indian populations. Although the difference remains clear, the low number of isolates from Indian populations cannot be neglected for their impact on the results. Additionally, a similar pattern was displayed by the βC1 gene for which the sequences from Pakistan exhibited a higher percentage of negative selection (90.9 per cent), followed by China (65.38 per cent) and India (58.75) (Fig. 7B). Notably, the alphasatellite from Pakistan and betasatellite from India had very few (4 and 1) sites under positive selection pressure (Fig. 7C).
Figure 7.
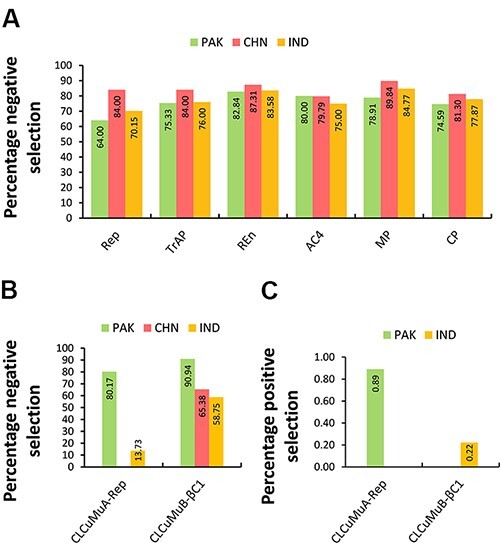
Percentage of sites evolving under negative/positive selection pressure. The datasets include (A) comparison between CLCuMuV-encoded 6 genes reported from Pakistan, China, and India, (B) percentage of negatively selected sites in CLCuMuA-Rep and CLCuMuB-βC1 genes, and (C) percentage of positively selected sites among CLCuMuA-Rep and CLCuMuB-βC1 genes from Pakistan and India, respectively.
To evaluate a region-based discrimination of dN/dS ratios, we subjected the data for each gene to statistical analysis using Mann–Whitney U-test. Interestingly, we found that for Rep and TrAP genes from Pakistani isolates of CLCuMuV, the negative selection pressure was significantly higher than that of Chinese and Indian populations. On the contrary, a significantly higher negative selection was observed in the case of REn gene populations from China and India. The difference remained non-significant for the AC4 gene, while it was statistically significant for MP and CP genes from Pakistan (Fig. 8A). We further compared the Rep and βC1 genes of alpha and betasatellites, respectively. To our surprise, the Rep populations from China appeared to have a stronger negative selection pressure as compared to those from Pakistan. Similarly, in the case of the βC1 gene, the Pakistani populations were evolving under low negative selection pressure as compared to Indian and Chinese βC1 genes (Fig. 8B). Furthermore, for the same datasets, a visual explanation of the variation between non-synonymous and synonymous substitution rates (dN − dS) is given in Supplementary Fig. S2.
Figure 8.
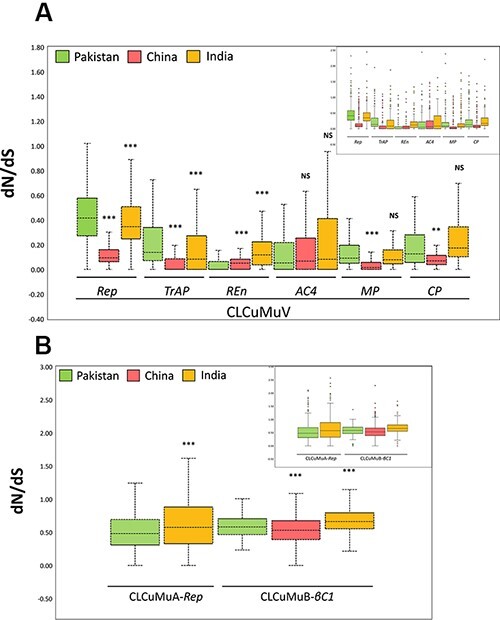
Estimation of comparative evolutionary pressure by calculation of dN/dS ratios. The coding sequences of (A) Rep, TrAP, REn, AC4, MP, and CP genes were divided into three groups, while that of (B) CLCuMuA-encoded Rep and CLCuMuB-encoded βC1 were divided into three and two groups, respectively. Significant values from Mann–Whitney U-test relative to the genome are indicated with asterisks: **P < 0.01; ***P < 0.001. The horizontal lines inside the box represent the median values, while inset displays the data with outliers denoted by small circles.
3. Discussion
Viruses are well known for their dynamic evolution and rapid adaptation (Roossinck 1997; Simmonds, Aiewsakun, and Katzourakis 2019). The adaptation and evolvability of DNA viruses mainly rely on the minor accumulations of genome-associated changes and more often on recombination events (Szpara and Van Doorslaer 2021). The whitefly-vectored begomoviruses possessing DNA genomes are destructive crop pathogens and exhibit extreme genomic plasticity, which not only enhances their virulence but also enables them to rapidly evolve under different cropping systems with an extended host range (Seal, vandenBosch, and Jeger 2006). Similar to other plant viruses, novel begomoviruses emerge and undergo rapid evolution through events of recombination and recurrent mutations (Padidam, Sawyer, and Fauquet 1999; Seal, vandenBosch, and Jeger 2006; Lefeuvre and Moriones 2015). Often accompanied by alpha or betasatellite molecules, CLCuMuV is a potentially damaging viral pathogen associated with CLCuD complex (Sattar et al. 2013; Zhou 2013). Although few studies are describing the recombination-driven evolution of CLCuMuV or its satellites, there is no comprehensive research on the most recent geographic distribution of these viral components. Also, previous studies focus only on CLCuMuV, neglecting its associated satellites and limited to a specific country/region (Sohrab et al. 2014; Saleem et al. 2016; Qadir et al. 2019). Here, we demonstrate that CLCuMuV isolates are mainly distributed in five countries, alphasatellites (CLCuMuA) are present only in Pakistan and India, while betasatellites (CLCuMuB) are distributed among four countries, except Thailand (Fig. 1D). Interestingly, the fact that three of them (India, China, and Pakistan) are among the top five cotton-producing countries of the world (STATISTA 2021) highlights that our study is well in time and much needed to explore the recent biodiversity and extent of molecular evolution. Currently, the CLCuMuV is predominantly distributed in China followed by Pakistan and India, while the majority of CLCuMuA and CLCuMuB is mainly present in Pakistan (Fig. 1E). Remarkably, while discussing the distribution of CLCuMuV and associated satellites, the role of sampling bias cannot be neglected. Although the basal lineage of CLCuMuV is thought to be from Pakistan (Datta et al. 2017), its continual rebound (Zubair et al. 2017) and introduction to new regions have become a matter of concern. For instance, in 2020, three isolates of CLCuMuV (MN127817.1, MN127818.1, and MN127819.1) were reported from India, while one (MN698836.1) isolated was reported from China (Supplementary Table S1). In the same year, the sequences of one CLCuMuA isolate (MN850796.1) and one CLCuMuB (MN910267.1) from Pakistan were deposited to the NCBI database (Supplementary Table S1). Despite the existence and implementation of quarantine and disease management strategies, the ongoing resurgence of CLCuMuV and its satellites to previously uninfected areas impose a raging threat to cotton production (Datta et al. 2017; Kajal et al. 2020). Notably, the isolates from Pakistan and India tend to infect cotton more frequently. This is contrary to the CLCuMuV isolates from China that have been reported to commonly infect hibiscus plants (Fig. 2A). Importantly, the observation of a commonly infected host or disease prevalence might be attributed to the possible effects of several factors like sampling biasness (Lacroix et al. 2016), virus–vector–host interactions, and the environmental heterogeneity among others (McLeish, Fraile, and García-Arenal 2021). The host range of alphasatellites (CLCuMuA) from Pakistan and India varied greatly, although the most commonly infected host was observed to be cotton (Fig. 2B). Furthermore, the Pakistani and Indian isolates of CLCuMuB were found to commonly infect cotton. On the other hand, the isolates from China and the Philippines remained abundant in hibiscus. The abundance of CLCuMuV in the Philippines and Southeastern China might be attributed to their high-level trade of agricultural products as compared to other regions (She et al. 2017). Most likely, the virus was introduced to China through the trade of hibiscus cuttings (Saleem et al. 2016). Together with movement/trade of infected plant materials, the presence of fecund B. tabaci biotypes in China (Guo et al. 2021), especially biotype Asia II 7 as an efficient vector of CLCuMuV (Chen et al. 2019), might have contributed to the increased viral spread in this region. We speculate that the standing diversity and abundance of CLCuMuV and its satellites might be a combinatorial outcome of several factors like agricultural intensification, abundance of susceptible hosts, viral genomic variations, selection pressure of vector/host, co-evolution, and specific virus–vector–plant interactions. We observed that although these isolates shared same clades based on geographical origin, there was a great variability in terms of the host plants. From an evolutionary perspective, the ability of plant viruses to infect a wide range of hosts highlights the practical effect (Roossinck 1997). Therefore, while expanding the host range of viruses is imperative to assess their evolutionary mechanisms, the diversity and genetic structure of viral populations in a single host are equally important to explain the evolutionary patterns (Jridi et al. 2006).
Analysis of homology and phylogeny further highlighted the evolutionary relatedness among CLCuMuV populations arising from different countries. For instance, Chinese CLCuMuV isolates clustered together, except AJ002459 that grouped with the Indian isolate (JN558352) showing 95.7 per cent sequence homology. Furthermore, the emergence of CLCuMuV in Thailand (KY373212.1 in 2017) and more recently from China (MK482365.1 in 2019 and MN698836.1 in 2020) is quite alarming for cotton growers and researchers. On the other hand, although the role of B. tabaci for the local spread of CLCuMuV has been well documented (Datta et al. 2017; Masood and Briddon 2018), there is no evidence of data suggesting the migration of B. tabaci from Pakistan to the Philippines, China, or other countries. Other studies suggest that particular biotypes of B. tabaci (Asia II7) might be more efficient in CLCuMuV transmission than others (Chen et al. 2019). The dominance of alpha and betasatellites in Pakistan as compared to other regions might be attributed to the presence of suitable hosts and efficient transmission vectors (Conflon et al. 2018). To date, there is no evidence of satellite emergence from Thailand. However, three isolates of CLCuMuB have already been reported from the Philippines (Supplementary Table S1); thus, the possible emergence of CLCuMuV-associated satellites in future cannot be neglected. Over the past few years, multiple infections of CLCuMuA were found in CLCuD complex, and additionally, alphasatellites were found in co-existence with betasatellites (Siddiqui et al. 2016). It might not be surprising to find multiple and co-existence of satellites due to the commonly observed mixed infection phenomenon among begomoviruses (Padidam, Sawyer, and Fauquet 1999). Perhaps, during whitefly-mediated transmission, satellite molecules might become associated with other viruses forming new complexes and introduced to disease-free regions.
Since variation in the genetic information ultimately affects the viral emergence (Padidam, Sawyer, and Fauquet 1999), evolution, and transmission by vectors (Pan et al. 2020), we further analyzed the standing genetic diversity of CLCuMuV, CLCuMuA, and CLCuMuB to understand the extent of existing genomic variation among these datasets. Remarkably, the Indian isolates of CLCuMuV exhibited the highest genetic diversity as compared to the isolates from Pakistan and China. Likewise, a similar pattern with a higher nucleotide diversity was observed for the Indian isolates of CLCuMuA and CLCuMuB (Fig. 3A). Additionally, the average number of segregating sites (θw) (Watterson 1975) was higher for the Indian isolates of CLCuMuV. On the contrary, it remained lower in the case of CLCuMuA, and interestingly, nearly similar values of θw were observed for CLCuMuB isolates from Pakistan and India (Fig. 3B). Finally, negative values of Tajima’s D variable among all analyzed datasets at varying degrees indicated that these populations are evolving under purifying selection. Consequently, the observed negative Tajima’s D explained the presence of excessive low-frequency alleles as a result of population expansion (Biswas and Akey 2006). It is important to mention that our datasets included full-length genomic sequences arising from different regions. Therefore, the existence of demographic events (population contraction/expansion) and population admixture cannot be ruled out, which might affect the standing genomic diversity by producing the artifactual negative selection. Nevertheless, these factors might (partly) help to explain the genetic variation of the analyzed datasets. Estimation of the population summary statistics provided compelling evidence that the evolution of CLCuMuV, CLCuMuA, and CLCuMuB is mainly driven by negative selection pressure.
The evolution and genomic diversification of the begomoviruses are well known to be driven by recombination events (Lefeuvre et al. 2007; Monci et al. 2002; Lima et al. 2017). Although some of the studies have analyzed the role of recombination in the genomic variation of CLCuMuV, most of these studies represent the populations that are geographically limited to a specific country/region (Saleem et al. 2016; Qadir et al. 2019; Chakrabarty et al. 2020). Additionally, no study combines the global populations of CLCuMuV and associated satellites (CLCuMuA and CLCuMuB) for analysis of their genomic diversity. In our results, although all sequences of CLCuMuV, CLCuMuA, and CLCuMuB were detected to have recombination breakpoints, the positions and pattern of recombination varied greatly (Fig. 4 and Table 2). Among all datasets, Pakistani isolates exhibited a greater number of detected recombination events (Table 2), perhaps due to the larger sample size as compared to other regions. Our findings are in agreement with a previous study that reports higher recombination frequency in different cotton-infecting ssDNA geminivirus (Saleem et al. 2016). Notably, detection of a higher recombination rate might explain a possible mechanism by which these populations can overcome the selective pressure and successfully adapt to new hosts and variable environments (Pérez-Losada et al. 2015). Interestingly, we found that sequences with randomly distributed recombinant breakpoints originating from different countries contained parental sequences from the same regions (Fig. 5 and Table 2). It shows that the recombination breakpoints among these populations are not conserved. Thus, the origin-based non-random distribution of recombination breakpoints could be attributed to the mechanistic aspects of recombination as implicated for ssDNA viruses (Lefeuvre et al. 2009). Also, it implicates that the exchange of genetic material between populations restricted to a particular geographical region might be responsible for the resurgence of new recombinant strains. Likewise, the pattern was observed in CLCuMuA and CLCuMuB isolates. Furthermore, complete absence or less detected recombination events in some sequences (e.g. CLCuMuV from China or the Philippines) implicate that for various reasons, we might have been unable to detect the recombinational changes. While recombination breakpoints remained variable among analyzed CLCuMuV sequences, the most frequently detected recombination events were found at variable positions depending upon the country of sequence origin (Fig. 5). Importantly, no evidence of recombination was found in the MP-coding region of CLCuMuV. Notably, the percentage of genome sequence under recombination was higher in Pakistani isolates of CLCuMuV and CLCuMuB. Whereas in the case of CLCuMuA, Indian populations had a larger area under recombination (Fig. 4). Previous studies show that recombination and pseudo-recombination are well-known phenomena that govern the rapid evolution of plant viruses, especially of begomoviruses (Harrison and Robinson 1999; Seal, vandenBosch, and Jeger 2006; Silva et al. 2014). Several factors including enhanced viral replication, mixed infections, increased host range, and vector are known to significantly regulate the recombination (Qadir et al. 2019). High frequencies of recombination detection in CP and Rep gene have been previously reported for cotton-infecting begomoviruses (Saleem et al. 2016), while for others, CP has been the hot spot of recombination (Yogindran et al. 2021). Recently, it has been demonstrated that several resistant-breaking variants of CLCuMuV and other cotton-infecting begomoviruses displayed significant recombination events not only in the viral genomes but also in the associated betasatellites (Chakrabarty et al. 2020). Owing to the known role of CP in virus transmission, changes in the CP sequence might subsequently affect the altered efficiency of vector-mediated viral transmission (Höhnle et al. 2001; Pan et al. 2020). We also detected a high frequency of recombination events in βC1 of betasatellite and Rep of alphasatellite. This finding is in agreement with previous reports that describe the βC1 (Datta 2017; Vinoth et al. 2017) and Rep (Vinoth et al. 2017) as recombination hotspots. Although our findings show that CLCuMuV and its associated satellites are independently evolving, to better understand the diversified speciation of variant isolates, co-existence of parental isolates and their evolution over time should be regularly monitored with reference to location.
Furthermore, our findings revealed that CLCuMuV, CLCuMuA, and CLCuMuB populations are evolving mainly under purifying selection pressure. This is in accordance with the previous study that concludes that the coding regions of an okra-infecting begomovirus were evolving under strong negative selection pressure (Kumar et al. 2017). To gain an in-depth understanding of this selection factor at the gene level, we opted to estimate dN/dS ratios for Rep, TrAP, REn, and AC4 genes of CLCuMuV and CLCuMuA-encoded Rep and CLCuMuB-encoded βC1 genes. While our results showed that with an average dN/dS ratio of <1, the majority of the codons remained under negative selection (Fig. 6, Table 3), the overall contribution of negatively selected sites remained >90 per cent. Remarkably, in the case of the βC1 gene, 100 per cent of codons were negatively selected (Supplementary Fig. S1). Likewise, an investigation on bipartite begomovirus revealed that MP and βC1 displayed substantial negative selection pressure. This effect also included the introduction of tyrosine phosphorylation site in the MP (Ho, Kuchie, and Duffy 2014). Previously, it has been shown that relatively a higher degree of purifying selection acts on the coding regions of a cotton geminivirus (Sanz et al. 1999). These findings provide critical information on how geminiviruses employ different mechanisms to evolve in different environments. Although there were few exceptions with positively selected sites, e.g. Rep (three codons), REn (one codon), TrAP (one codon), and MP (two codons), all the analyzed genes appeared to evolve under negative/purifying selection. However, the presence of overlapping coding sequences (existing in DNA-A only) might accumulate elevated levels of non-synonymous substitutions or limit the synonymous substitutions, ultimately increasing the dN/dS ratio (Simon-Loriere, Holmes, and Pagán 2013). Therefore, our observations of higher dN/dS ratios in codons of Rep, REn, TrAP, and MP might be artifactual findings owing to the fact that these genes are overlapping (Fig. 6). Nonetheless, with overlapping ORFs, a higher proportion of mutations leads to amino acid changes that consequently govern fitness trade-off and purifying selection (Xavier et al. 2020). Finally, the comparative analysis of all (geographically isolated) genes encoded either by CLCuMuV or by satellites demonstrated a significant difference in terms of evolutionary pressure. Overall, the frequency and pattern of gene variability might explain that different isolates might evolve through diverse mechanisms that not only facilitate their compatible interactions with hosts (plant and/or vector) but also enhance their virulence, make them more competent/resilient, assist them to successfully adopt a variety of ecosystems, and ultimately make them challenging pathogens in terms of management.
4. Materials and methods
4.1. Nucleotide sequences
A total of 730 sequences were used in this study, including 121 full-length DNA-A sequences of CLCuMuV, 162 alphasatellites, and 447 complete sequences of betasatellites associated with CLCuMuV. The sequences of all CLCuMuV isolates reported from five countries between 1998 and 2020 were retrieved on 23 February 2021 from the GenBank database (www.ncbi.nlm.nih.gov). Details of sequences used in this study are provided in Supplementary Table S1. All sequences were aligned so that they begin with the invariant nanonucleotide (5ʹ-TAATATT//AC-3ʹ) at the nicking position.
4.2. Multiple sequence alignment and phylogenetic analysis
To prepare multiple sequence alignments, full-length nucleotide sequences of CLCuMuV DNA-A encoding coat protein (CP), replication protein (Rep), trans-activating protein (TrAP), replication enhancer protein (REn), MP, AC4, alphasatellite, and betasatellite full-length genome sequences were aligned using MUSCLE option in MEGA-6 program (Tamura et al. 2013). All alignments were manually checked and adjusted when necessary, followed by subsequent analyses.
The phylogenetic tree construction was performed using Bayesian inference with MrBayes v.3.0b4 (Huelsenbeck and Ronquist 2001). To select the best-suited nucleotide substitution model for each dataset, MrModeltest v.2.2 was used in Akaike Information Criterion (Nylander 2004). The analyses were performed using 10 million generations, and the first 2.5 million generations were excluded as burn-in. Visualization and editing of phylogenetic trees were carried out using FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
4. 3. Nucleotide diversity and haplotype variability indices
The average pairwise number of nucleotide differences per site (nucleotide diversity, π) was estimated using DnaSP v.5 (Librado and Rozas 2009). The statistically significant differences among the mean nucleotide diversity from all datasets were estimated by calculating their 95 per cent bootstrap confidence intervals. Also, the nucleotide diversity was calculated on a 100-nucleotide sliding window, with a step size of 10 nucleotides across the full-length DNA-A, alphasatellite, and betasatellite nucleotide sequences. The number of haplotypes (H), the number of segregating sites (S), and haplotype diversity (Hd) were also calculated for all datasets using DnaSP v.5 (Rozas et al. 2003).
4.4. Recombination analysis
The full-length sequences of CLCuMuV DNA-A, alphasatellites, and betasatellites were separately analyzed for the occurrence of recombination. The recombination analysis was carried out using Geneconv, Chimaera, Rdp, Bootscan, SisterScan, 3Seq, and maximum Chi-square methods implemented in the recombination detection program v.4 (Martin et al. 2015). Alignments for all methods were performed using default settings. Statistical significance was inferred by P-values lower than a Bonferroni-corrected cutoff of 0.05. The recombination events detected by at least four different methods were considered reliable.
4.5. Detection of positive and negative selection
Potential negatively and positively selected sites in the coding regions of CP, Rep, TrAP, Ren, MP, and AC4 were identified using four distinct approaches: fixed-effects likelihood, single-likelihood ancestor counting (SLAC), random-effects likelihood, and partitioning for robust inference of selection (Scheffler, Martin, and Seoighe 2006). All methods were implemented in the Datamonkey web server (www.datamonkey.org) (Pond and Frost 2005). To avoid misleading results, Genetic Algorithm Recombination Detection (GARD) (Kosakovsky Pond et al. 2006) was implemented to search for the recombination breakpoints in all datasets. The dN/dS ratios for the aforementioned genes from all datasets were estimated using the SLAC method based on inferred GARD-corrected phylogenetic trees.
5. Conclusions
While most of the previously done CLCuMuV-related studies are confined to a particular region/host, we present a bioinformatics-based population evolutionary perspective to explain the current standing biogeographic and genetic diversification of the CLCuMuV and its associated satellites. Together with the expanded host range, the higher molecular variability of CLCuMuV might be attributed to a rampant evolutionary trajectory in its DNA genome, mainly driven by a high frequency of the recombinational changes. Moreover, the diversified distribution and pattern of recombinational changes display the existence of a greater molecular variability among the analyzed viral populations. The variable frequency and random distribution of the recombination breakpoints implicate that geographically separated viral/satellite isolates employ different mechanisms to overcome selection pressure and to successfully adapt to new hosts/environments. We show that CLCuMuV, CLCuMuA, and CLCuMuB are independently evolving under a strong negative selection pressure. Although a very small fraction of codons might undergo positive selection, there is compelling evidence to show that the evolution of CLCuMuV, CLCuMuA, and CLCuMuB is predominantly being driven by purifying selection. The continuous resurgence of new recombinant strains of CLCuMuV or its satellites might lead to resistance breaking, expansion of host range, and efficient vector transmission, thus being a threat to crop production and disease management. In the future, it would be imperative to study the host-dependent and vector/human-mediated dispersal and evolution of CLCuMuV and its associated satellites for an in-depth understanding of the expanding virosphere.
Supplementary Material
Contributor Information
Tahir Farooq, Plant Protection Research Institute and Guangdong Provincial Key Laboratory of High Technology for Plant Protection, Guangdong Academy of Agricultural Sciences, Guangzhou 510640, P.R. China.
Muhammad Umar, Tasmanian Institute of Agriculture, New Town Research Laboratories, University of Tasmania, 13 St. Johns Avenue, New Town, TAS 7008, Australia.
Xiaoman She, Plant Protection Research Institute and Guangdong Provincial Key Laboratory of High Technology for Plant Protection, Guangdong Academy of Agricultural Sciences, Guangzhou 510640, P.R. China.
Yafei Tang, Plant Protection Research Institute and Guangdong Provincial Key Laboratory of High Technology for Plant Protection, Guangdong Academy of Agricultural Sciences, Guangzhou 510640, P.R. China.
Zifu He, Plant Protection Research Institute and Guangdong Provincial Key Laboratory of High Technology for Plant Protection, Guangdong Academy of Agricultural Sciences, Guangzhou 510640, P.R. China.
Data availability
All data is available and present either in the publication or in the indicated databases.
Supplementary data
Supplementary data is available at Virus Evolution online.
Funding
This work was funded by the National Natural Science Foundation of China (31871937, 32072392), the Guangdong Basic and Applied Basic Research Foundation (2019A1515012150), the President Foundation of Guangdong Academy of Agricultural Sciences, China (grant no: BZ202005), and Discipline team building projects of Guangdong Academy of Agricultural Sciences in the 14th Five-Year Period (202105TD).
Conflict of interest
The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Author contributions
T.F. and M.U.: conceptualization, data mining, and analysis; T.F., Y.T. X.S., and Z.H.: original draft preparation; Z.H. and Y.T.: review and editing, supervision, and funding acquisition. All authors have read and agreed to the published version of the manuscript.
References
- Ashraf M. et al. (2013) ‘Molecular Characterization and Phylogenetic Analysis of a Variant of Highly Infectious Cotton Leaf Curl Burewala Virus Associated with CLCuD from Pakistan’, Australian Journal of Crop Science, 7: 1113–22. [Google Scholar]
- Balol G. et al. (2010) ‘Sources of Genetic Variation in Plant Virus Populations’, Journal of Pure and Applied Microbiology, 4: 803–8. [Google Scholar]
- Baltimore D. (1971) ‘Expression of Animal Virus Genomes’, Bacteriological Reviews, 35: 235–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas S., and Akey J. M. (2006) ‘Genomic Insights into Positive Selection’, Trendsin Genetics, 22: 437–46. [DOI] [PubMed] [Google Scholar]
- Briddon R. W., and Markham P. G. (2000) ‘Cotton Leaf Curl Virus Disease’, VirusResearch, 71: 151–9. [DOI] [PubMed] [Google Scholar]
- Chakrabarty P. K. et al. (2020) ‘Recombinant Variants of Cotton Leaf Curl Multan Virus Is Associated with the Breakdown of Leaf Curl Resistance in Cotton in Northwestern India’, Virusdisease, 31: 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T. et al. (2019) ‘Transmission Efficiency of Cotton Leaf Curl Multan Virus by Three Cryptic Species of Bemisia tabaci Complex in Cotton Cultivars’, PeerJ, 7: e7788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conflon D. et al. (2018) ‘Accumulation and Transmission of Alphasatellite, Betasatellite and Tomato Yellow Leaf Curl Virus in Susceptible and Ty-1-resistant Tomato Plants’, VirusResearch, 253: 124–34. [DOI] [PubMed] [Google Scholar]
- Cuevas J. M., Duffy S., and Sanjuán R. (2009) ‘Point Mutation Rate of Bacteriophage PhiX174’, Genetics, 183: 747–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S. et al. (2017) ‘Rebound of Cotton Leaf Curl Multan Virus and Its Exclusive Detection in Cotton Leaf Curl Disease Outbreak, Punjab (India), 2015’, Scientific Reports, 7: 17361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolja V. V., and Koonin E. V. (2018) ‘Metagenomics Reshapes the Concepts of RNA Virus Evolution by Revealing Extensive Horizontal Virus Transfer’, Virus Research, 244: 36–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo E. (2020) ‘Molecular Basis of Genetic Variation of Viruses: Error-prone Replication’, Virus as Populations, 35–71. [Google Scholar]
- Domingo E., and Schuster P. (2016) ‘What Is a Quasispecies? Historical Origins and Current Scope’. In: Domingo, E., and Schuster P. (eds) Quasispecies: From Theory to Experimental Systems, pp. 1–22. Cham: Springer International Publishing. [DOI] [PubMed] [Google Scholar]
- Drake J. W. (1991) ‘A Constant Rate of Spontaneous Mutation in DNA-based Microbes’, Proceedings of the National Academy of Sciences of the United States of America, 88: 7160–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy S., and Holmes E. (2009) ‘Validation of High Rates of Nucleotide Substitution in Geminiviruses: Phylogenetic Evidence from East African Cassava Mosaic Viruses’, The Journal of General Virology, 90: 1539–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy S., and Holmes E. C. (2008) ‘Phylogenetic Evidence for Rapid Rates of Molecular Evolution in the Single-Stranded DNA Begomovirus Tomato Yellow Leaf Curl Virus’, Journal of Virology, 82: 957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy S., Shackelton L. A., and Holmes E. C. (2008) ‘Rates of Evolutionary Change in Viruses: Patterns and Determinants’, Nature Reviews: Genetics, 9: 267–76. [DOI] [PubMed] [Google Scholar]
- Elena S. F. (2016) ‘Evolutionary Transitions during RNA Virus Experimental Evolution’, Philosophical Transactions of the Royal Society B: Biological Sciences, 371: 20150441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eric V. D. W. et al. (2008) ‘Experimental Observations of Rapid Maize Streak Virus Evolution Reveal a Strand-specific Nucleotide Substitution Bias’, Virology Journal, 5: 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiallo-Olivé E., Tovar R., and Navas-Castillo J. (2016) ‘Deciphering the Biology of Deltasatellites from the New World: Maintenance by New World Begomoviruses and Whitefly Transmission’, The New Phytologist, 212: 680–92. [DOI] [PubMed] [Google Scholar]
- Fondong V. N. (2013) ‘Geminivirus Protein Structure and Function’, Molecular Plant Pathology, 14: 635–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale M., Tan S.-L., and Katze M. G. (2000) ‘Translational Control of Viral Gene Expression in Eukaryotes’, Microbiology and Molecular Biology Reviews, 64: 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Arenal F., Fraile A., and Malpica J. M. (2001) ‘Variability and Genetic Structure of Plant Virus Populations’, Annual Review of Phytopathology, 39: 157–86. [DOI] [PubMed] [Google Scholar]
- Ge L. et al. (2007) ‘Genetic Structure and Population Variability of Tomato Yellow Leaf Curl China Virus’, Journal of Virology, 81: 5902–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoras I. et al. (2010) ‘High Variability and Rapid Evolution of a Nanovirus’, Journal of Virology, 84: 9105–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C.-L. et al. (2021) ‘Invasion Biology and Management of Sweetpotato Whitefly (Hemiptera: Aleyrodidae) in China’, Journal of Integrated Pest Management, 12: 1. [Google Scholar]
- Hanley-Bowdoin L. et al. (2013) ‘Geminiviruses: Masters at Redirecting and Reprogramming Plant Processes’, Nature Reviews: Microbiology, 11: 777–88. [DOI] [PubMed] [Google Scholar]
- Harrison B., and Robinson D. (1999) ‘Natural Genomic and Antigenic Variation in Whitefly-Transmitted Geminiviruses (Begomoviruses)’, Annual Review of Phytopathology, 37: 369–98. [DOI] [PubMed] [Google Scholar]
- Ho E. S., Kuchie J., and Duffy S. (2014) ‘Bioinformatic Analysis Reveals Genome Size Reduction and the Emergence of Tyrosine Phosphorylation Site in the Movement Protein of New World Bipartite Begomoviruses’, PLoS One, 9: e111957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höhnle M. et al. (2001) ‘Exchange of Three Amino Acids in the Coat Protein Results in Efficient Whitefly Transmission of a Nontransmissible Abutilon Mosaic Virus Isolate’, Virology, 290: 164–71. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck J. P., and Ronquist F. (2001) ‘MrBayes: Bayesian inference of phylogenetic trees’, Bioinformatics, 17(8): 754–5. [DOI] [PubMed] [Google Scholar]
- Hussain T., and Ali M. (1975) ‘A Review of Cotton Diseases of Pakistan’, The Pak Cottons, 19: 71–86. [Google Scholar]
- Idris A. et al. (2010) ‘An Unusual Alphasatellite Associated with Monopartite Begomoviruses Attenuates Symptoms and Reduces Betasatellite Accumulation’, The Journal of General Virology, 92: 706–17. [DOI] [PubMed] [Google Scholar]
- Jeske H., Lütgemeier M., and Preiß W. (2001) ‘DNA Forms Indicate Rolling Circle and Recombination-Dependent Replication of Abutilon Mosaic Virus’, The EMBO Journal, 20: 6158–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jridi C. et al. (2006) ‘Distinct Viral Populations Differentiate and Evolve Independently in a Single Perennial Host Plant’, Journal of Virology, 80: 2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajal K. B. et al. (2020) ‘Dominance of Recombinant Cotton Leaf Curl Multan-Rajasthan Virus Associated with Cotton Leaf Curl Disease Outbreak in Northwest India’, PLoS One, 15: e0231886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin E. V., Dolja V. V., and Krupovic M. (2015) ‘Origins and Evolution of Viruses of Eukaryotes: The Ultimate Modularity’, Virology, 479–480: 2–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond S. L. et al. (2006) ‘GARD: A Genetic Algorithm for Recombination Detection’, Bioinformatics, 22: 3096–8. [DOI] [PubMed] [Google Scholar]
- Kumar R. V. et al. (2017) ‘Molecular Genetic Analysis and Evolution of Begomoviruses and Betasatellites Causing Yellow Mosaic Disease of Bhendi’, Virus Genes, 53: 275–85. [DOI] [PubMed] [Google Scholar]
- Kumar A., Kumar J., and Khan J. A. (2010) ‘Sequence Characterization of Cotton Leaf Curl Virus from Rajasthan: Phylogenetic Relationship with Other Members of Geminiviruses and Detection of Recombination’, Virus Genes, 40: 282–9. [DOI] [PubMed] [Google Scholar]
- Kumar J. et al. (2015) ‘Cotton Leaf Curl Burewala Virus with Intact or Mutant Transcriptional Activator Proteins: Complexity of Cotton Leaf Curl Disease’, Archives of Virology, 160: 1219–28. [DOI] [PubMed] [Google Scholar]
- Lacroix C. et al. (2016) ‘Methodological Guidelines for Accurate Detection of Viruses in Wild Plant Species’, Applied and Environmental Microbiology, 82: 1966–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefeuvre P. et al. (2007) ‘Begomovirus ‘melting pot’ in the South-west Indian Ocean Islands: Molecular Diversity and Evolution through Recombination’, Journal of General Virology, 88: 3458–68. [DOI] [PubMed] [Google Scholar]
- —— (2009) ‘Widely Conserved Recombination Patterns among Single-Stranded DNA Viruses’, Journal of Virology, 83: 2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefeuvre P., and Moriones E. (2015) ‘Recombination as a Motor of Host Switches and Virus Emergence: Geminiviruses as Case Studies’, Current Opinion in Virology, 10: 14–9. [DOI] [PubMed] [Google Scholar]
- Librado P., and Rozas J. (2009) ‘DnaSP V5: A Software for Comprehensive Analysis of DNA Polymorphism Data’, Bioinformatics, 25: 1451–2. [DOI] [PubMed] [Google Scholar]
- Lima Alison T. M. et al. (2017) ‘The diversification of begomovirus populations is predominantly driven by mutational dynamics’, Virus Evolution, 3(1): vex005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano G. et al. (2016) ‘Characterization of Non-coding DNA Satellites Associated with Sweepoviruses (Genus Begomovirus, Geminiviridae) - Definition of a Distinct Class of Begomovirus-Associated Satellites’, Frontiers in Microbiology, 7: 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansoor S., Zafar Y., and Briddon R. W. (2006) ‘Geminivirus Disease Complexes: The Threat Is Spreading’, Trends in Plant Science, 11: 209–12. [DOI] [PubMed] [Google Scholar]
- Mao M. J. et al. (2008) ‘Molecular Characterization of Cotton Leaf Curl Multan Virus and Its Satellite DNA that Infects Hibiscus rosa-sinensis’, Bing Du Xue Bao, 24: 64–8. [PubMed] [Google Scholar]
- Martin D. P. et al. (2011) ‘Complex Recombination Patterns Arising during Geminivirus Coinfections Preserve and Demarcate Biologically Important Intra-Genome Interaction Networks’, PLoS Pathogens, 7: e1002203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- —— (2015) ‘RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes’, VirusEvolution, 1: vev003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins L. G. C. et al. (2020) ‘A Begomovirus Nuclear Shuttle Protein-Interacting Immune Hub: Hijacking Host Transport Activities and Suppressing Incompatible Functions’, Frontiers in Plant Science, 11: 398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masood M., and Briddon R. W. (2018) ‘Transmission of Cotton Leaf Curl Disease: Answer to a Long-Standing Question’, Virus Genes, 54: 743–5. [DOI] [PubMed] [Google Scholar]
- McLeish M. J., Fraile A., and García-Arenal F. (2021) ‘Population Genomics of Plant Viruses: The Ecology and Evolution of Virus Emergence’, Phytopathology, 111: 32–9. [DOI] [PubMed] [Google Scholar]
- Monci F. et al. (2002) ‘A Natural Recombinant between the Geminiviruses Tomato Yellow Leaf Curl Sardinia Virus and Tomato Yellow Leaf Curl Virus Exhibits A Novel Pathogenic Phenotype and Is Becoming Prevalent in Spanish Populations’, Virology, 303: 317–26. [DOI] [PubMed] [Google Scholar]
- Nylander J. (2004) ‘MrModeltest V2. Program Distributed by the Author’, Bioinformatics, 24: 581–3. [Google Scholar]
- Padidam M., Sawyer S., and Fauquet C. M. (1999) ‘Possible Emergence of New Geminiviruses by Frequent Recombination’, Virology, 265: 218–25. [DOI] [PubMed] [Google Scholar]
- Pan L.-L. et al. (2020) ‘Mutations in the Coat Protein of a Begomovirus Result in Altered Transmission by Different Species of Whitefly Vectors’, Virus Evolution, 6: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Losada M. et al. (2015) ‘Recombination in Viruses: Mechanisms, Methods of Study, and Evolutionary Consequences’, Infection, Genetics and Evolution: Journal of Molecular Epidemiology and Evolutionary Genetics in Infectious Diseases, 30: 296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond S. L., and Frost S. D. (2005) ‘Datamonkey: Rapid Detection of Selective Pressure on Individual Sites of Codon Alignments’, Bioinformatics, 21: 2531–3. [DOI] [PubMed] [Google Scholar]
- Qadir R. et al. (2019) ‘Diversity and Recombination Analysis of Cotton Leaf Curl Multan Virus: A Highly Emerging Begomovirus in Northern India’, BMC Genomics, 20: 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roossinck M. J. (1997) ‘Mechanisms of Plantvirus Evolution’, Annual Review of Phytopathology, 35: 191–209. [DOI] [PubMed] [Google Scholar]
- Rozas J. et al. (2003) ‘DnaSP, DNA Polymorphism Analyses by the Coalescent and Other Methods’, Bioinformatics, 19: 2496–7. [DOI] [PubMed] [Google Scholar]
- Saeed M. et al. (2005) ‘A Single Complementary-sense Transcript of A Geminiviral DNA Beta Satellite Is Determinant of Pathogenicity’, Molecular Plant-Microbe Interactions: MPMI, 18: 7–14. [DOI] [PubMed] [Google Scholar]
- Saleem H. et al. (2016) ‘Diversity, Mutation and Recombination Analysis of Cotton Leaf Curl Geminiviruses’, PLoS One, 11: e0151161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuán Rafael., and Domingo-Calap Pilar. (2021) ‘Genetic Diversity and Evolution of Viral Populations’, in Dennis H. Bamford and Mark Zuckerman (eds.), Encyclopedia of Virology (Fourth Edition) (Oxford: Academic Press; ), 53–61. [Google Scholar]
- Sanz A. I. et al. (1999) ‘Genetic Variability of Natural Populations of Cotton Leaf Curl Geminivirus, a Single-Stranded DNA Virus’, Journal of Molecular Evolution, 49: 672–81. [DOI] [PubMed] [Google Scholar]
- Sattar M. N. et al. (2013) ‘Cotton Leaf Curl Disease - An Emerging Threat to Cotton Production Worldwide’, Journal of General Virology, 94: 695–710. [DOI] [PubMed] [Google Scholar]
- Scheffler K., Martin D. P., and Seoighe C. (2006) ‘Robust Inference of Positive Selection from Recombining Coding Sequences’, Bioinformatics, 22: 2493–9. [DOI] [PubMed] [Google Scholar]
- Seal S., vandenBosch F., and Jeger M. J. (2006) ‘Factors Influencing Begomovirus Evolution and Their Increasing Global Significance: Implications for Sustainable Control’, Critical Reviews in Plant Sciences, 25: 23–46. [Google Scholar]
- Shackelton L. A. et al. (2005) ‘High Rate of Viral Evolution Associated with the Emergence of Carnivore Parvovirus’, Proceedings of the National Academy of Sciences of the United States of America, 102: 379–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelton L. A., and Holmes E. C. (2006) ‘Phylogenetic Evidence for the Rapid Evolution of Human B19 Erythrovirus’, Journal of Virology, 80: 3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She X. M. et al. (2017) ‘Molecular Characterization of Cotton Leaf Curl Multan Virus and Its Associated Betasatellite Infecting Hibiscus rosa-sinensis in the Philippines’, Journal of Plant Pathology, 99: 765–8. [Google Scholar]
- Shi M. et al. (2018) ‘The Evolutionary History of Vertebrate RNA Viruses’, Nature, 556: 197–202. [DOI] [PubMed] [Google Scholar]
- Shweta A., and Khan J. A. (2018) ‘Genome Wide Identification of Cotton (Gossypiumhirsutum)-Encoded microRNA Targets against Cotton Leaf Curl Burewala Virus’, Gene, 638: 60–5. [DOI] [PubMed] [Google Scholar]
- Siddiqui K. et al. (2016) ‘Diversity of Alphasatellites Associated with Cotton Leaf Curl Disease in Pakistan’, Virology Reports, 6: 41–52. [Google Scholar]
- Silva F. N. et al. (2014) ‘Recombination and Pseudorecombination Driving the Evolution of the Begomoviruses Tomato Severe Rugose Virus (Tosrv) and Tomato Rugose Mosaic Virus (Tormv): Two Recombinant DNA-A Components Sharing the Same DNA-B’, Virology Journal, 11: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds P., Aiewsakun P., and Katzourakis A. (2019) ‘Prisoners of War — Host Adaptation and Its Constraints on Virus Evolution’, Nature Reviews: Microbiology, 17: 321–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon-Loriere E., Holmes E. C., and Pagán I. (2013) ‘The Effect of Gene Overlapping on the Rate of RNA Virus Evolution’, Molecular Biology and Evolution, 30: 1916–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohrab S. S. et al. (2014) ‘Genetic Variability of Cotton Leaf Curl Betasatellite in Northern India’, SaudiJournal of Biological Sciences, 21: 626–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solé R. (2016) ‘The Major Synthetic Evolutionary Transitions’, Philosophical Transactions of the Royal Society B: Biological Sciences, 371: 20160175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STATISTA . (2021) Leading Cotton Producing Countries Worldwide in 2019/2020, <https://www.statista.com/statistics/263055/cotton-production-worldwide-by-top-countries/> accessed date: 10 February 2021.
- Szpara Moriah L., and Van Doorslaer K., Koenraad (2021) ‘Mechanisms of DNA Virus Evolution’, Encyclopedia of Virology, 71–78. [Google Scholar]
- Tamura K. et al. (2013) ‘MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0’, Molecular Biology and Evolution, 30: 2725–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarazi R., Jimenez J. L. S., and Vaslin M. F. S. (2019) ‘Biotechnological Solutions for Major Cotton (Gossypiumhirsutum) Pathogens and Pests’, Biotechnology Research and Innovation, 3: 19–26. [Google Scholar]
- Villarreal L. P. (2008) ‘Evolution of Viruses’, Encyclopedia of Virology, 174–84. [Google Scholar]
- Vinoth K. R. et al. (2017) ‘Molecular Diversity, Recombination and Population Structure of Alphasatellites Associated with Begomovirus Disease Complexes’, Infection, Genetics and Evolution, 49: 39–47. [DOI] [PubMed] [Google Scholar]
- Watterson G. A. (1975) ‘On the Number of Segregating Sites in Genetical Models without Recombination’, Theoretical Population Biology, 7: 256–76. [DOI] [PubMed] [Google Scholar]
- Wolf Y. I. et al. (2018) ‘Origins and Evolution of the Global RNA Virome’, mBio, 9(6), e02329–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier C. A. D. et al. (2020) ‘Evolutionary Dynamics of Bipartite Begomoviruses Revealed by Complete Genome Analysis’, bioRxiv, 2020.06.25.171728. [DOI] [PubMed] [Google Scholar]
- Yogindran S. et al. (2021) ‘Occurrence of Cotton Leaf Curl Multan Virus and Associated Betasatellites with Leaf Curl Disease of Bhut-Jolokia Chillies (Capsicum chinense Jacq.) In India’, Molecular Biology Reports, 48: 2143–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X. (2013) ‘Advances in Understanding Begomovirus Satellites’, Annual Review of Phytopathology, 51: 357–81. [DOI] [PubMed] [Google Scholar]
- Zubair M. et al. (2017) ‘Multiple Begomoviruses Found Associated with Cotton Leaf Curl Disease in Pakistan in Early 1990 are Back in Cultivated Cotton’, Scientific Reports, 7: 680. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is available and present either in the publication or in the indicated databases.