

Abstract
Background
Ceftriaxone is a β-lactam antibiotic used to treat central nervous system infections. Whether the neuroprotective effects of ceftriaxone after TBI are mediated by attenuating neuroinflammation but not its antibacterial actions is not well established.
Methods
Anesthetized male Sprague–Dawley rats were divided into sham-operated, TBI + vehicle, and TBI + ceftriaxone groups. Ceftriaxone was intraperitoneally injected at 0, 24, and 48 h with 50 or 250 mg/kg/day after TBI. During the first 120 min after TBI, we continuously measured heart rate, arterial pressure, intracranial pressure (ICP), and cerebral perfusion pressure. The infarct volume was measured by TTC staining. Motor function was measured using the inclined plane. Glutamate transporter 1 (GLT-1), neuronal apoptosis and TNF-α expression in the perilesioned cortex were investigated using an immunofluorescence assay. Bacterial evaluation was performed by Brown and Brenn’s Gram staining. These parameters above were measured at 72 h after TBI.
Results
Compared with the TBI + vehicle group, the TBI + ceftriaxone 250 mg/kg group showed significantly lower ICP, improved motor dysfunction, reduced body weight loss, decreased infarct volume and neuronal apoptosis, decreased TBI-induced microglial activation and TNF-α expression in microglia, and increased GLT-1 expression in neurons and microglia. However, the grades of histopathological changes of antibacterial effects are zero.
Conclusions
The intraperitoneal injection of ceftriaxone with 250 mg/kg/day for three days may attenuate TBI by increasing GLT-1 expression and reducing neuroinflammation and neuronal apoptosis, thereby resulting in an improvement in functional outcomes, and this neuroprotective effect is not related to its antibacterial effects.
Keywords: Traumatic brain injury, Ceftriaxone, Glutamate transporter, Glutamate receptor Neuroinflammation, Apoptosis
Introduction
After traumatic brain injury (TBI), excitotoxicity [1] and neuroinflammation [2] are two key secondary injuries that may induce subsequent neuronal cell death, including necrosis, autophagy or apoptosis [3, 4]. These injuries are associated with changes in certain parameters including glutamic acid transporter-1 (GLT-1) and the receptors NMDAR2A and NMDAR2B, microglia and astrocyte activation, and tumor necrosis factor α (TNF-α) production.
Thus, it is important to identify effective therapeutic drugs to overcome these secondary injuries. However, the development of new pharmacological drugs is a long and challenging process. Therefore, currently, drug repurposing is an alternative choice for novel drug discovery [5].
Ceftriaxone, a β-lactam antibiotic, is a long used and safe treatment for central nervous system CNS infection at an antibacterial dosage of 50–100 mg/kg [6]. It is permeable to the blood–brain barrier, has clear pharmacokinetic activity in the CNS [7], and is generally well tolerated, with a low incidence of adverse effects [8, 9]. Therefore, ceftriaxone is a potential drug candidate for other CNS insults.
Currently, ceftriaxone has been demonstrated to have beneficial effects in experimental Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, stroke, brain ischemia, seizure, drug/alcohol dependency and withdrawal [10] and TBI models [11–14]. It was thought to exert its neuroprotective effect by increasing the expression and activity of GLT-1 to decrease the synaptic excitatory glutamate concentration. However, the experimental dosage of 200–250 mg/kg was much higher than the usual clinical dosage of 50–100 mg/kg. Furthermore, its effect on the anti-neuroinflammatory-associated response is still unclear and warrants further study.
In the current study, we hypothesized that ceftriaxone may have neuroprotective effects and not by its antibacterial effects at different dosages. The aim of the present study was to determine the neuroprotective mechanism of ceftriaxone after TBI, especially the effects on intracranial pressure (ICP), GLT-1, glutamate receptors NMDAR2A (post-synaptic) and NMDAR2B (extra-synaptic), neuronal apoptosis, and neuroinflammation-associated effects (microglia and astrocyte activation and TNF-α expression), and to improve functional outcomes using the clinical dosage of 50 mg/kg and an experimental dosage of 250 mg/kg.
Methods
Experimental design
As shown in Fig. 1, in the first part, we tested the effects of ceftriaxone on physiological parameters. We continuously measured the mean arterial pressure (MAP), ICP, cerebral perfusion pressure (CPP) and heart rate for 120 min after TBI was induced. In the second part, ceftriaxone (50 mg/kg or 250 mg/kg) was given through the intraperitoneal route for 3 consecutive days. We attempted to elucidate whether these different dosages were associated with microglia and astrocyte activation, tumor necrosis factor α (TNF-α) expression, neuronal apoptosis, and cerebral injury in the ipsilateral injury side of the cortex using immunofluorescence staining and triphenyltetrazolium chloride (TTC) staining, while motor function was tested by the inclined plane at 72 h after TBI. The observational period set at 72 h after TBI was based on our previous study in TBI [15].
Fig. 1.
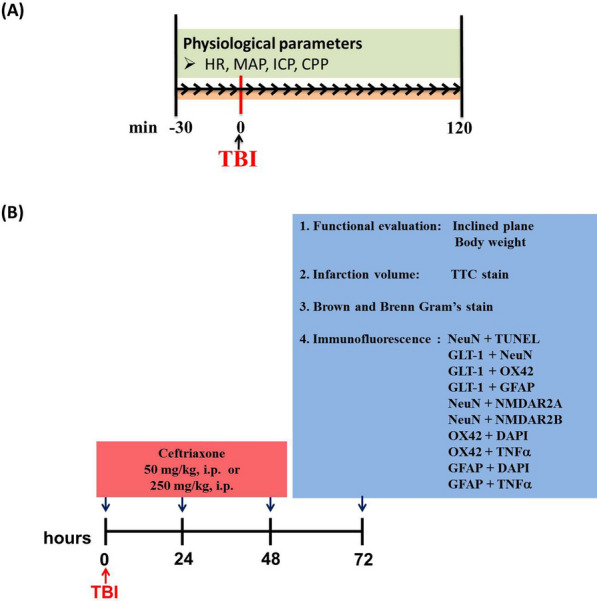
The overall experimental protocol in the current study
Animals
Adult male Sprague–Dawley (SD) rats were obtained commercially from BioLASCO Taiwan Co., Ltd. and used in this study. The animals were kept under a 12/12-h light/dark cycle and allowed free access to food and water. All of the experimental procedures were approved by the Chi Mei Medical Center’s Animal Care and Use Committee and conformed to the NIH guidelines (Approval No. 10808602), including minimizing discomfort to the animals during surgery and during the recovery period. At the end of the experiments, the rats were euthanized by an overdose of urethane (intraperitoneal injections of 2 ml of 500 mg/ml urethane solution) until deep unconscious condition determined by the absence of visible breathing. All the animal experiments were carried out in compliance with the ARRIVE guidelines. To avoid potential interrater variability, all rats were measured by the same laboratory experimenter.
Traumatic brain injury
Animals were anesthetized and intramuscularly injected with a mixture of ketamine (44 mg/kg, i.m.; Nankuang Pharmaceutical, Taiwan), atropine (0.063 mg/kg; Sintong Chemical Ind. Co., Taiwan), and Rompun (6.77 mg/kg, Bayer, Germany). Using a stereotaxic frame, a right craniectomy (with a radius of 2 mm) was performed 4 mm from the bregma and 3 mm from sagittal sutures. A fluid percussion device (VCU Biomedical Engineering, Richmond, VA, USA) was connected, and the brain was injured by rapidly injecting saline into the closed cranial cavity with a 2.2 atm plus 25 ms percussion. The detailed procedures are as described by Tsai et al. [15].
Surgery and physiological parameter monitoring
The right femoral arteries of rats were cannulated with polyethylene tubing (PE50) for blood pressure monitoring. MAP and heart rate (HR) were monitored continuously with a pressure transducer after induction of TBI. An ICP microsensor (Codman and Shurtlef, Inc., Rayman, MA, USA) was placed in the parenchyma of the left frontal lobe of each rat. ICP was monitored continuously, and ICP and CPP values were recorded at 5-min intervals in the first 120 min after TBI. The CPP value was defined as MAP-ICP [16]. Colon temperatures were measured with an electronic thermometer (model 43 TE; YSI, Inc, Yellow Springs, OH) and a temperature probe (series 400; YSI, Inc).
Treatment intervention
Anesthetized male SD rats were randomized and divided into sham-operated, TBI + vehicle, and TBI + ceftriaxone groups. Animals received three injections of ceftriaxone (50 mg/kg or 250 mg/kg) or vehicle intraperitoneally, including immediately, 24 h and 48 h after TBI. Vehicle for the TBI + vehicle group was normal saline (0.9%, Sintong, TAIWAN BIOTECH CO., LTD.), while the TBI + ceftriaxone-treated group received 50 mg/kg or 250 mg/kg ceftriaxone, dissolved in normal saline (Sintong TAIWAN BIOTECH CO., LTD.).
Motor function test
The animals were placed perpendicular to the 20 × 20-cm buffer ribbed surface of an inclined plane. The maximal angle at which an animal could remain on the inclined plane was recorded. Motor deficit measurements were performed at 72 h after TBI.
Cerebral injury volume assay
TTC staining was performed to measure the injury volume at 72 h following TBI [15]. The volume of injury, as revealed by negative TTC staining indicating dehydrogenase-deficient tissue, was measured in each slice and summed using computerized planimetry (PC-based Image Tools software, Image-Pro Plus Media Cybernetics, Inc., Rockville, MD, USA). The infarction volume was calculated as 2 mm (thickness of the slice) × [sum of the injury areas in all brain slices (mm2)] [17].
Immunofluorescence assay
Seventy-two hours after TBI, adjacent 6-μm sections corresponding to coronal coordinates 0.20 mm to 0.70 mm anterior to bregma were incubated with primary antibodies in PBS containing 0.5% normal bovine serum overnight at 4 °C. After being washed in PBS, the sections were incubated with secondary antibodies for 1 h at room temperature. The number of immunofluorescent-positive cells was calculated in two coronal sections from each rat using computerized planimetry corresponding to the perilesioned cortical region (400× magnification, Image-Pro Plus Media Cybernetics, Inc. Washington Street, Rockville, USA), and the number of positive cells was expressed as the mean number of positive cells in the region of interest. The details of the antibodies used are summarized in Table 1.
Table 1.
The detailed information of antibodies used in current study
| Antibody | Source | Catalog number | Working dilution |
|---|---|---|---|
| TUNEL kit | Clontech(Palo Alto, CA) | 630108 | |
| Mouse anti-NeuN | Abcam(Cambridge, UK) | ab104224 | 1:600 |
| Mouse anti-OX42 | Abcam(Cambridge, UK) | ab1211 | 1:200 |
| Mouse anti-GFAP | Cell signaling technology(Beverly, MA) | 3670 | 1:800 |
| Rabbit anti-GLT1 | Abcam(Cambridge, UK) | ab41621 | 1:100 |
| Rabbit anti-NMDAR2A | Chemicon international(Billerica, MA) | AB1555 | 1:600 |
| Rabbit anti-NMDAR2B | Abcam(Cambridge, UK) | ab65783 | 1:500 |
| Rabbit anti-TNFα | Abcam(Cambridge, UK) | ab6671 | 1:400 |
Histological and pathological staining at 72 h after TBI
Paraffin sections
The wet tissues were trimmed and dehydrated with serial alcohol solution. Next, the tissues were embedded in paraffin wax. The paraffin-embedded tissues were cut at a thickness of 3—5 μm.
The slide-mounted paraffin sections -were deparaffinized and hydrated with xylene and serial alcohol solutions. One ml 1% crystal violet was mixed with 5 drops 5% sodium bicarbonate and then added to slides held in a staining rack. The slides were gently agitated to ensure that each section was covered, and then the sections were stained for 1 min. The slides were rinsed in distilled water, flooded with Lugol’siodine for 1 min and rinsed again with water. The sections were blotted dry and breathed on, and then acetone was quickly added to the section until the color stopped running off. The slides were washed in tap water. Slides were placed on a staining rack, and basic fuchsin was applied to the tissue sections and stained for 3 min. The slides were then washed in tap water and blotted gently to remove excess fluid but not to completely dry. The slides were then dipped rapidly into acetone followed by picric acid-acetone and then acetone-xylene.
Gram-positive bacteria were stained blue; gram-negative bacteria were stained red. Cell nuclei were stained red to yellow. Backgrounds were stained yellow. All glass slides were digitized with a MoticEasyscan Digital Slide Scanner (Motic Hong Kong Limited, Hong Kong, China) at × 40 (0.26 μm/pixel) with high precision (high precision autofocus). MoticEasyscan whole-slide images were viewed with DSAssistant and EasyScanner software at Li-Tzung Biotechnology Inc. (Kaohsiung, Taiwan).
Statistical analysis
All of the data were analyzed using SigmaPlot, version 10.0 for Windows (Systat Software, San Jose, CA) or GraphPad Prism 6.0 (GraphPad Software, Inc., San Diego, CA, USA) in this study. The results are expressed as the mean with standard deviation of the mean for the experiments. Repeated measures analysis of variance (ANOVA) was conducted to test treatment-by-time interactions and the effect of treatment over time on each score. Two-way ANOVA followed by the post hoc Scheffe’s test was used to estimate the significant difference between these four groups. The box plots were used to present the distribution difference between four study groups. In addition, Wilcoxon ranked sum test was used to estimate the statistical difference. P-values < 0.05 were considered statistically significant.
Results
Basic data for the experimental rats
A total of 72 10-week-old male rats weighing 380 ± 22 g were used in the current study. The fluid percussion force was 2.20 ± 0.015 atm. The colonic temperature was controlled between 36 and 37 °C with lamp insulation during and up to 120 min after injury. No animal death or complications occurred over the course of the experiments.
Part I: Physiological parameter evaluation in the first 120 min after TBI
Ceftriaxone-treated groups had significantly lower ICP during the initial 120 min after TBI
Since a high ICP could affect the functional outcome [18], we first tested the effects of ceftriaxone on physiological parameters, including HR, MAP, ICP and CPP, during the initial 120 min after TBI. In the TBI + vehicle group, the ICPs were higher at 0–120 min after the start of TBI than they were for sham-operated controls. In contrast, the values for ICP in the TBI + ceftriaxone (250 mg/kg/day) group were significantly lower than those of the sham-operated control and vehicle + TBI groups, *p < 0.05, n = 6 in each group. Simultaneously, the TBI + ceftriaxone (250 mg/kg/d) group had a trend of higher CPP during the initial 120 min after TBI (Fig. 2).
Fig. 2.

Effects of ceftriaxone on TBI-induced a heart rate (HR), b mean arterial pressure (MAP), c intracranial pressure (ICP) and d cerebral perfusion pressure (CPP) in the initial 120 min after traumatic brain injury (TBI). ***p < 0.001, compared with the sham group; #p < 0.05, ##p < 0.01, compared with the TBI + vehicle group; $p < 0.05, compared with the TBI + ceftriaxone 50 mg/kg group, n = 6 in each group
Part II: Functional outcome and neuropathology investigation at 72 h after TBI
Ceftriaxone significantly improved body weight loss at 72 h after TBI
TBI in rats was characterized by body weight loss [19], and this correlated with injury severity [20]; therefore, we examined the ceftriaxone effect on body weight loss after TBI. The definition of body weight loss is the difference in body weight between 0 and 72 h after TBI. Figure 3 shows that TBI + vehicle rats had greater weight loss than sham-operated controls (***p < 0.001, n = 6 in each group). However, treatment with 250 mg/kg ceftriaxone after TBI significantly reversed the TBI-induced body weight loss (#p < 0.05, n = 6 in each group).
Fig. 3.
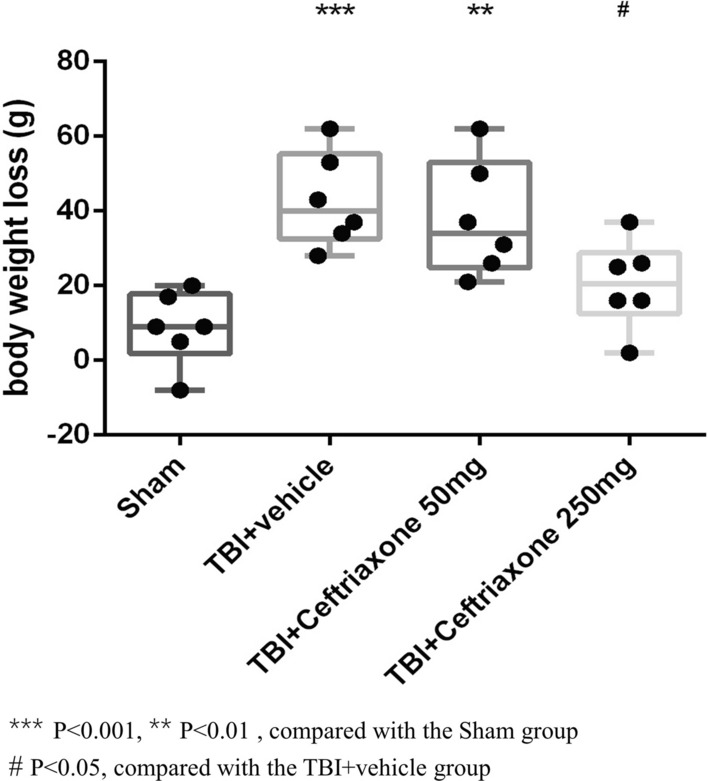
Effects of TBI-induced body weight losses at 72 h after TBI. ***p < 0.001, **p < 0.05 compared with the sham group; #p < 0.05, compared with the TBI + vehicle group; n = 6 in each group
Ceftriaxone significantly improved motor dysfunction at 72 h after TBI
Then, we tested whether ceftriaxone has beneficial effects on functional outcomes such as motor dysfunction after TBI. At 72 h after TBI, the maximal grip angle in inclined plane tests showed that vehicle-treated TBI rats had significantly lower motor performance than sham-operated controls (***p < 0. 001, Fig. 4). TBI-induced motor dysfunction was significantly attenuated by both 50 mg/kg and 250 mg/kg ceftriaxone treatment (n = 6 in each group, Fig. 4).
Fig. 4.
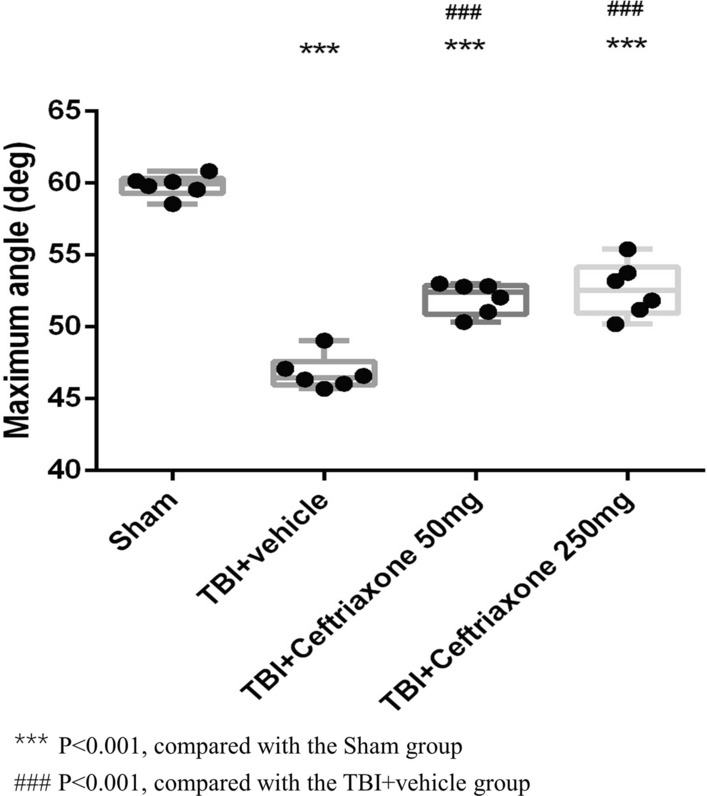
Effects of TBI-induced motor deficits at 72 h after TBI. ***p < 0.001, compared with the sham group; ###p < 0.001, compared with the TBI + vehicle group; n = 6 in each group
Ceftriaxone significantly decreased injury volume at 72 h after TBI
We examined whether the functional outcome improvement described above may relate to a decrease in injury volume after ceftriaxone treatment. At 72 h after TBI, the TTC-stained sections showed a significant increase in the infarct volume of the vehicle-treated TBI compared with those of sham TBI controls, (***p < 0.001, Fig. 5). The TBI-induced injury volume was significantly reversed by TBI treatment with 250 mg/kg ceftriaxone (#p < 0.05, n = 6 in each group, Fig. 5).
Fig. 5.
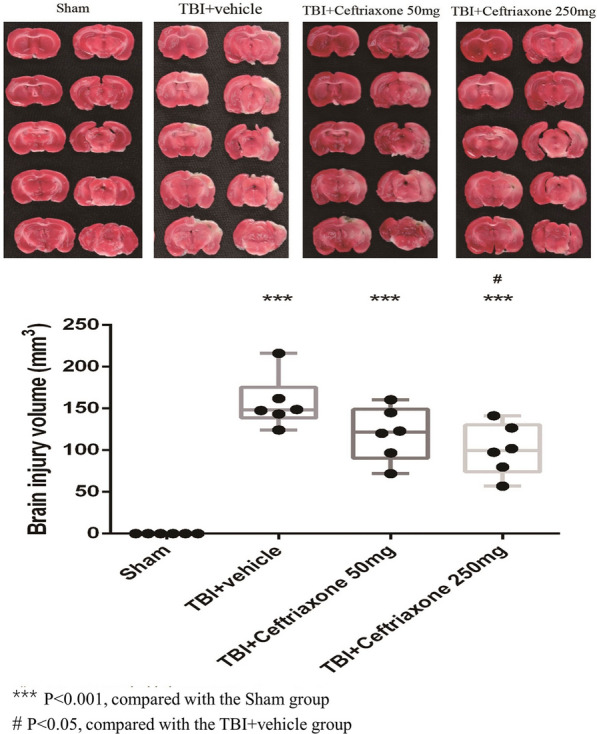
Effects of TBI-induced injury volumes in the perilesioned cortex at 72 h after TBI. ***p < 0.001, compared with the sham group; #p < 0.05, compared with the TBI + vehicle group; n = 6 in each group
Ceftriaxone significantly decreased neuronal apoptosis at 72 h after TBI
Based on the results of the infarct volume, we next evaluated the effect of ceftriaxone on TBI-induced neuronal apoptosis. At 72 h after TBI, vehicle-treated TBI rats had significantly higher numbers of NeuN plus TUNEL-positive cells in the perilesioned cortex than sham controls (***p < 0.001, Fig. 6). TBI-induced neuronal apoptosis was significantly decreased by TBI treatment with 250 mg/kg ceftriaxone ($$$p < 0.001, Fig. 6), n = 6 in each group.
Fig. 6.
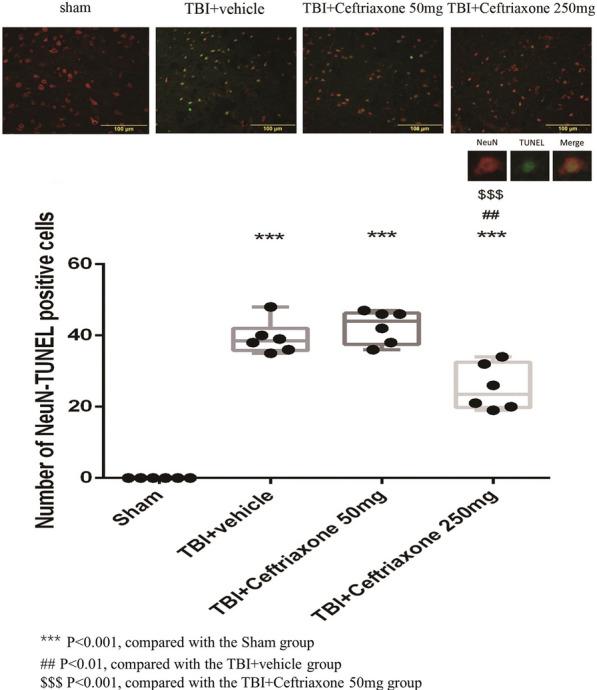
Effects of TBI-induced neuronal apoptosis in the perilesioned cortex at 72 h after TBI (markers NeuN plus TUNEL). The top panels depict representative NeuN plus TUNEL positive staining for one sham rat, one TBI + vehicle rat, one TBI + ceftriaxone (50 mg/kg)-treated rat, and one TBI + ceftriaxone (250 mg/kg)-treated rat. ***p < 0.001, compared with the sham group; ##p < 0.01, compared with the TBI + vehicle group; $$$p < 0.001, compared with the TBI + ceftriaxone 50 mg/kg group, n = 6 in each group
Antibacterial effects of ceftriaxone at 72 h after TBI
Next, we tested whether the neuroprotective effects of ceftriaxone were via antibacterial actions. Based on the morphology and staining, Fig. 7 shows that no obvious bacteria were found in sham-operated controls, TBI + vehicle and TBI + ceftriaxone (50 mg/kg or 250 mg/kg) rats.
Fig. 7.

Histopathological changes at 72 h after TBI with Brown and Brenn’s Gram’s staining (5.6×, 20× and 200× magnification), n = 6 in each group
Ceftriaxone further significantly increased GLT-1 expression in neurons and microglia but not astrocyte expression at 72 h after TBI
To clarify GLT-1 expression in specific cells in vivo after different treatments, we next tested the effects of ceftriaxone on GLT-1 expression in astrocytes, microglia and neurons using double immunofluorescence staining. As shown in Figs. 8, 9, and 10, the number of GLT-1-positive neurons, microglia and astrocytes in the perilesioned cortex was significantly increased compared with that in sham rats at 72 h after TBI. The number of GLT-1-positive neurons and microglia in the perilesioned cortex after TBI was significantly increased further by TBI treatment with 250 mg/kg ceftriaxone. However, GLT-1 expression in astrocytes was not significantly increased after ceftriaxone treatment.
Fig. 8.
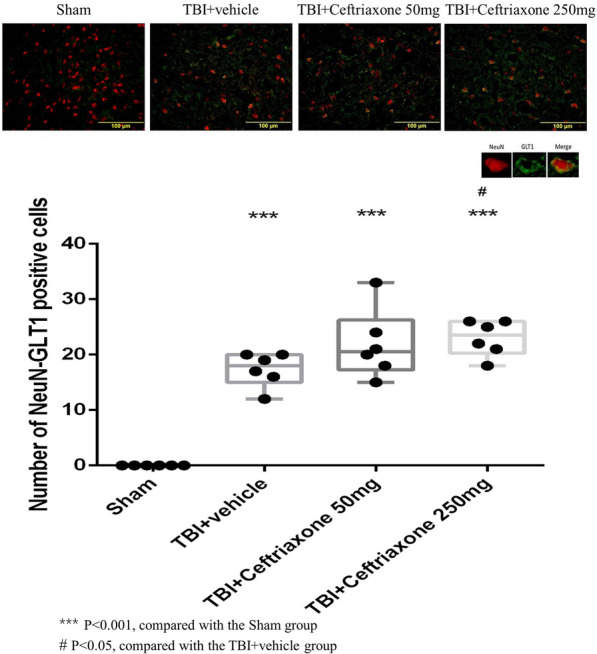
Effects of TBI-induced neuronal GLT-1 in the perilesioned cortex at 72 h after TBI (markers NeuN plus GLT-1). ***p < 0.001, compared with the sham group; #p < 0.05, compared with the TBI + vehicle group; n = 6 in each group
Fig. 9.
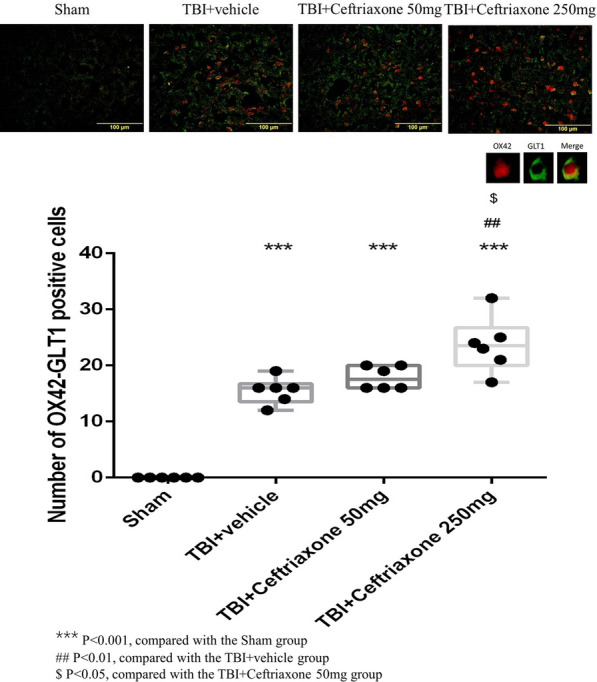
Effects of TBI-induced microglial GLT-1 in the perilesioned cortex at 72 h after TBI (markers OX42 plus GLT-1). ***p < 0.001, compared with the sham group; #p < 0.05, ##p < 0.01, compared with the TBI + vehicle group; ***p < 0.001, compared with the sham group; $p < 0.05, compared with the TBI + ceftriaxone 50 mg/kg group, n = 6 in each group
Fig. 10.

Effects of TBI-induced astroglial GLT-1 in the perilesioned cortex at 72 h after TBI (markers GFAP plus GLT-1). ***p < 0.001, compared with the sham group; #p < 0.05, compared with the TBI + vehicle group; ***p < 0.001, compared with the sham group, n = 6 in each group
Ceftriaxone significantly decreased NMDAR2B expression after TBI
Next, we examined the effects of ceftriaxone after TBI on the expression of the postsynaptic neuroprotective NMDAR2A subunit and the extra-synaptic neurodestructive NMDAR2B subunit. In the NMDAR2A plus NeuN staining assay, the number of positive neuronal NMDAR2A cells in the perilesioned cortex of vehicle-treated TBI rats (28 ± 2.44) was significantly increased compared with that in the sham controls (0 ± 0, ***p < 0.001, n = 6 in each group, Fig. 11). However, the TBI-induced increase in neuronal NMDAR2A-positive cells was not significantly changed by either 50 mg/kg or 250 mg/kg ceftriaxone therapy (n = 6 in each group, Table 2). When compared with the TBI + vehicle rats, the number of TBI-induced NMDAR2B plus NeuN stained cells was significantly decreased both at 50 mg/kg and 250 mg/kg in the TBI + ceftriaxone rats (n = 6 in each group, Fig. 12).
Fig. 11.
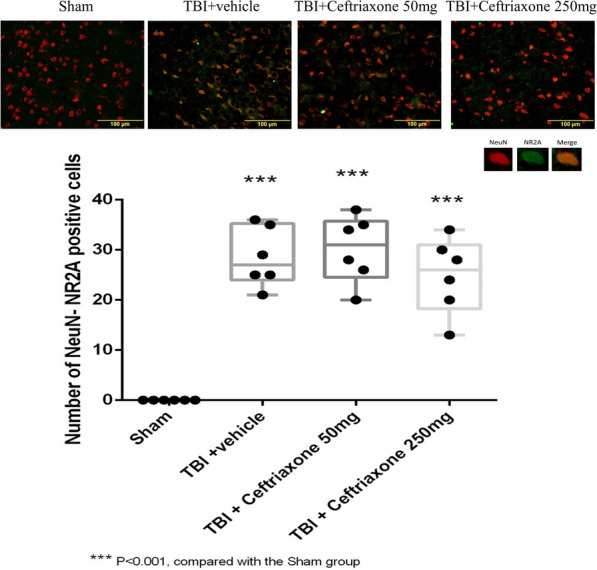
Effects of TBI-induced glutamate receptor subunits NMDAR2A at 72 h after TBI (marker Neu-N plus NMDAR2A). ***P < 0.001, compared with the Sham group, n = 6 in each group
Table 2.
Studies of Ceftriaxone effects on traumatic brain injury animal models
| Parameters/Authors | Species weight | Injury model severity | Dosage and course | Region observed | Parameters observed | Results |
|---|---|---|---|---|---|---|
| Wei et al. 2012 |
SD Rat 180–220 g |
Weight-drop 20 g/30 cm height |
200 mg/kg Single dose |
Cortex |
Brain edema TNF-α IL-6 IL-1β INF-r GLT-1 Learning and memory |
Increase expression of GLT-1 Improving cognitive function Alleviating brain edema Reducing excitotoxicity and neuroinflammation |
| Cui et al. 2013 |
SD rat 300–330 g |
Weight-drop 450 g/1.5 m height |
200 mg/kg/d × 5 days |
Hippocampus |
Brain edema Autophage GLT-1 Learning and memory |
Upper regulation of GLT-1 Suppression neuronal autophage Reducing brain edema |
| Goodrich et al. 2013 |
Long-Evans 367 ± 36 g |
Fluid percussion 2.3 ± 0.1 atm |
200 mg/kg/d × 7 days |
Cortex |
GLT-1 GFAP Seizure |
Increase GLT-1 protein expression Decrease GFAP expression Reduce seizure duration |
| Hameed et al. 2019 |
SD rat 367 ± 36 g |
Fluid percussion 2.3 ± 0.1 atm |
250 mg/kg/d × 7 days |
Cortex |
Interneuron GLT-1 GABA |
Prevent cortical inhibitory interneuron dysfunction Preserving GLT-1 expression |
| Present study 2020 |
SD rat 380 ± 22 g |
Fluid percussion 2.20 ± 0.01 atm |
50 mg/ kg/d or 250 mg/kg/d × 3 days |
Cortex |
TTC Motor TTC TUNEL GLT-1 OX42,TNF-α ICP |
Increase GLT-1 expression in microglia & neuron Improved motor dysfunction Decrease infarct volume, apoptosis Attenuated neuroinflammation Lower ICP in initial 120 min after TBI |
SD, Sprague–Dawley; FPI, fluid percussion injury
Fig. 12.

Effects of TBI-induced glutamate receptor subunits NMDAR2B at 72 h after TBI (marker Neu-N plus NMDAR2B). ***P < 0.001, compared with the Sham group, ##P < 0.01, compared with the TBI + vehicle group, n = 6 in each group
Ceftriaxone significantly decreased TBI-induced microglial activation and TNF-α expression in microglia in the perilesioned cortex at 72 h after TBI
Microglia is the major source of TNF-α production after TBI [21]; therefore, we examined whether the neuroprotective effect of ceftriaxone was anti-inflammatory. The OX42 plus 4',6-diamidino-2-phenylindole (DAPI) double staining showed that the number of positive cells in the perilesioned cortex of the vehicle-treated TBI rats (37 ± 1.15) was significantly higher than that in the sham-operated rats (0 ± 0, ***P < 0.001). Furthermore, TBI-induced positive OX42 in the DAPI-positive cells was significantly lower in the 250 mg/kg ceftriaxone treatment group compared with the TBI vehicle-treated rats (29 ± 18.6, ##P < 0.01, Fig. 13).
Fig. 13.
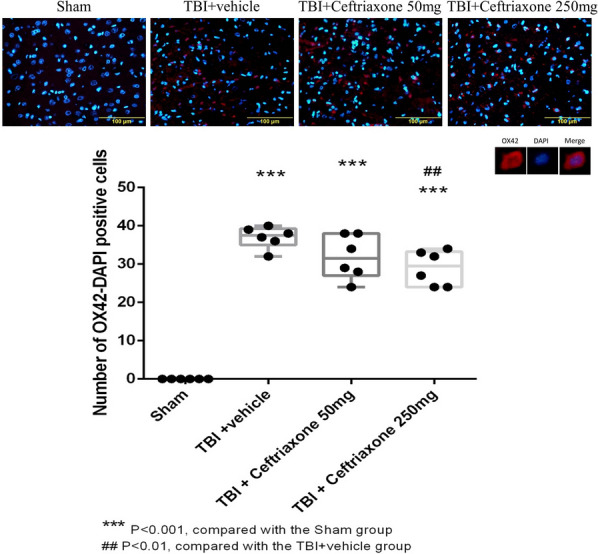
Effects of TBI-induced microglial expression in the perilesioned cortex at 72 h after TBI (markers OX42 plus DAPI). ***p < 0.001, compared with the sham group; ##p < 0.01, compared with the TBI + vehicle group; n = 6 in each group
Simultaneously, OX42 plus TNF-α double staining in the perilesioned cortex of the vehicle-treated TBI rats (35 ± 1.37) was significantly higher than that in the sham-operated rats (0 ± 0, ***P < 0.001). The TBI-induced increase was significantly reversed by TBI treatment with 250 mg/kg ceftriaxone (26 ± 1.82, ##p < 0.01, n = 6 in each group, Fig. 14). These results support that antineuroinflammation is one mechanism for the neuroprotective effects of ceftriaxone.
Fig. 14.
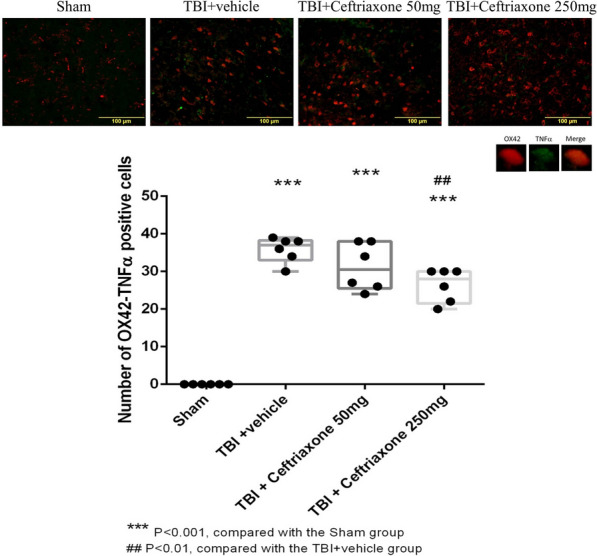
Effects of TBI-induced TNF-α expression in the microglia in the perilesioned cortex at 72 h after TBI (markers OX42 plus TNF-α). ***p < 0.001, compared with the sham group; ##p < 0.01, compared with the TBI + vehicle group; n = 6 in each group
Ceftriaxone significantly decreased TBI-induced astrocytic activation but not the TNF-α expression in astrocyte in the perilesioned cortex at 72 h after TBI
Similar to the microglial reaction,, TBI-induced positive GFAP in the DAPI-positive cells was significantly lower both in the 250 mg/kg and 50 mg/Kg ceftriaxone treatment group compared with the TBI vehicle-treated rats (n = 6 in each group, Fig. 15). However, the TBI-induced positive TNF-α in the GFAP-positive cells was not reversed by TBI treatment with 50 or 250 mg/kg ceftriaxone (Fig. 16).
Fig. 15.
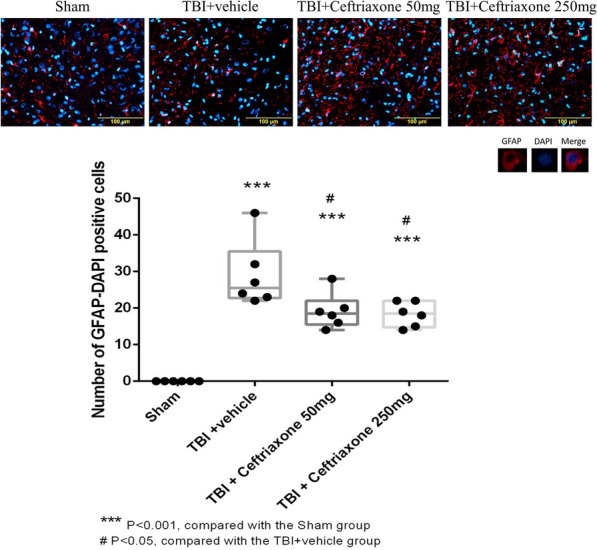
Effects of TBI-induced astrocytic expression in the perilesioned cortex at 72 h after TBI (markers GFAP plus DAPI). ***p < 0.001, compared with the sham group; #p < 0.05, compared with the TBI + vehicle group; n = 6 in each group
Fig. 16.
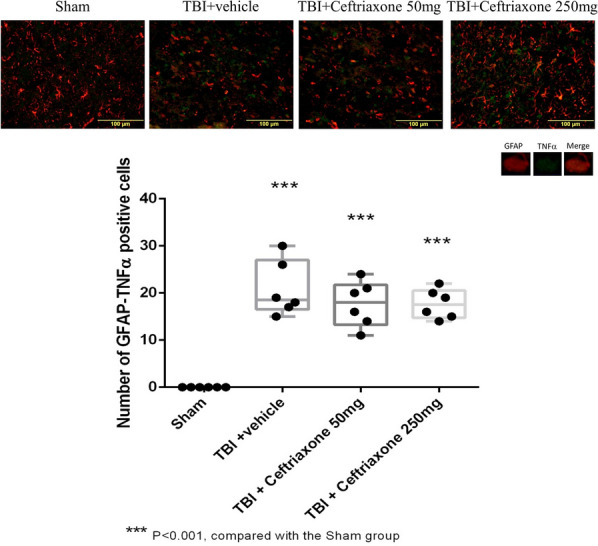
Effects of TBI-induced TNF-α expression in the astrocyte in the perilesioned cortex at 72 h after TBI (markers NeuN plus GLT-1). ***p < 0.001, compared with the sham group, n = 6 in each group
Discussion
Novelty of the current study
According to our review of the literature, this study is the first to demonstrate the effects of different dosages of ceftriaxone on TBI-induced glutamate receptor expression and neuroinflammation. We provide some new information on ceftriaxone in the field of neurocritical care, including that (1) the mechanism of neuroprotection of ceftriaxone is not related to its antibacterial effect; (2) treatment with 250 mg/kg/day ceftriaxone for 3 days may increase GLT-1 expression, decrease neuroinflammation injury volume and neuronal apoptosis, and improve motor dysfunction and body weight loss; and (3) treatment with 50 mg/kg/day or 250 mg/kg/day ceftriaxone for 3 days may attenuate extrasynaptic neurodestructive NMDAR2B subunit expression.
Determination of the dosage and time course of ceftriaxone administration
In the current study, the dosage of ceftriaxone administration (250 mg/kg/day) was in accordance with the Hameed et al. TBI animal model [14]. The administration time course immediately, 24 h and 48 h after TBI was based on the fact that TBI-induced cell apoptosis was maximal at 2–3 days after TBI [22] and significantly increased at 72 h after TBI compared with that of the sham group [15]. Based on our previous study, the observation end-point 72 h post-injury TBI were selected because lateral fluid percussion caused body weight loss, neuroinflammation and apoptosis compared with sham group in this time point [15].
Compared with the usual daily dose of ceftriaxone, which is 1–2 g administered once daily in adults and 50–100 mg/kg once daily in pediatric patients [6], the dose used in this study (250 mg/kg per day) is relatively high. However, in a preclinical study, the no-observed-adverse-effect dose level was 500 mg/kg/day in both sexes associated with the long-term administration of ceftriaxone for 6 months [23]. Therefore, the dosage and time course of ceftriaxone administration in our study may have potential for clinical applications in the future.
Early effects of ceftriaxone on physiological parameters during the initial 120 min after TBI
Consistent with previous studies [24, 25], in the first 10 min, our results showed an abrupt increase in ICP after TBI followed by a gradual decrease, including in the TBI-vehicle, low-dose and high-dose ceftriaxone treatment groups. The abrupt increase in ICP may be due to the sudden injection of a small amount of fluid into the cranial cavity due to the impact force, which then displaces or distorts the cerebral vessels, followed by an immediate decrease in cerebral blood flow and ultimately inadequate cerebral perfusion [24, 26]. We suggest that the gradual decrease in ICP within minutes after the peak may be related to cerebral hyperperfusion and effective autoregulation after TBI [26]. An undetected CSF leakage occurring at the ICP catheter insertion may be another cause of reduced ICP [24]. These phenomena were not associated with ceftriaxone treatment.
Compared to the TBI groups in our study, the ceftriaxone-treated group (250 mg/kg, i.p.) had a significantly lower ICP, mostly occurring approximately 10 min during the initial 120 min after TBI. We consider that these early beneficial effects of ceftriaxone on ICP may mitigate the development of secondary injury, such as neuroinflammation and excitotoxicity, and ultimately improve outcome [18] on the 3rd day after TBI according to our study. However, we did not measure whether ICP was altered as a result when ceftriaxone was treated alone in the absence of TBI. This issue needs clarified further.
Yuk et al. demonstrated that following a single intravenous injection of 2 gm ceftriaxone, the peak concentration of ceftriaxone was detected at 30 min post treatment [27]. However, we did not investigate the relationship between the brain ceftriaxone concentration and physiological parameter changes after TBI. This relationship warrants further investigation.
Based on the fact that increased ICP leads to increases in sympathetic activity, which is responsible for changes in gastrointestinal motility, water and electrolyte absorption [28], the relationship between ceftriaxone and gastrointestinal function warrants further investigation.
Ceftriaxone effects on body weight loss at 72 h after TBI
TBI rats are characterized by body weight loss [19], which is correlated with injury severity [20, 29]. The causes of body weight loss after TBI may relate to anorexia, enhancement of proteolysis, and distal intestinal atrophy [30] and affect gut microbial diversity [31, 32].
In our previous study, we found that TBI increased local and systemic TNF expression [33]. In the current study, ceftriaxone significantly decreased microglial activation and TNF-α expression in microglia after TBI. Since disruption of the brain-gut axis may lead to dysfunction of the gastrointestinal system [34], whether ceftriaxone may have beneficial effects on the gastrointestinal system, especially TNF-α expression in gut, and finally decrease the body weight loss after TBI is worthy of future investigation.
Ceftriaxone effects on motor deficiency after TBI
Following TBI, the mechanism of cell death includes necrosis, apoptosis and autophagy, which may affect functional outcomes [4, 35]. Cui et al. showed that treatment with ceftriaxone at 200 mg/kg/day for 5 days attenuated neuronal autophagy in the hippocampus after TBI12. In the current study, we added the new information that treatment with 250 mg/kg/day ceftriaxone for 3 days attenuated neuronal apoptosis in the perilesioned cortex and synergistically improved motor deficiency. However, treatment with 50 mg/kg/day ceftriaxone for 3 days did not attenuated neuronal apoptosis but improved motor dysfunction. Therefore, we speculate that neuronal autophagy may be involved in our model in higher dose of ceftriaxone treatment, but the mechanisms need to be clarified. Furthermore, the effect of ceftriaxone on the cell necrosis after TBI also needs to be investigated in the future.
Ceftriaxone effects on glutamate transporters at 72 h after TBI
GLT-1 is the major glutamate transporter responsible for glutamate uptake to decrease the accumulation of glutamate in the synaptic cleft and to reduce postsynaptic glutamate receptor overactivation [36]. GLT-1 is predominantly expressed in astroglia [36, 37] but is also present in neurons and in axon terminals [38]. Under physiological conditions, glutamate in the synaptic cleft is primarily transported by the glutamate transporter GLT-1 to astrocytes, where it is subsequently metabolized to glutamine and then safely transported back to neurons as glutamine.
However, under pathological conditions, the astrocyte glutamate transporter is downregulated and the microglia transporter is induced for glutamate clearance [39].
Our immunofluorescence assay results at 72 h after TBI show that neuronal GLT-1 levels were acutely elevated in the perilesioned cortex compared with sham rats and it is reasonable to believe that this is a protective effect to reduce the accumulation of glutamate in the synaptic cleft. Our results were consistent with Goodrich et al. in that they demonstrated that ceftriaxone treatment after TBI restored the expression of GLT-1 according to western blotting [13]. We further identified that treatment with 250 mg/kg ceftriaxone for 3 days after TBI further increased neuronal and microglial GLT-1 transporter but not astrocytic GLT1 transporter expression in the perilesioned cortex.
The possible reasons for the high expression of GLT-1 in microglia in the injured tissue may be related to the proliferation of microglia there [40] and up-regulated GLT-1 in microglia to transport glutamate from the extracellular space for glumate clearance [39]. The low expression in astrocytes may be due to the accumulation of glutamate in astrocyte vesicles that retards transportation and the inability of GLT-1 to reform the sealed cell membrane structure [41]. However, these speculative hypotheses require further experiments beyond the scope of this report.
Ceftriaxone effects on glutamate receptor subunits NMDAR2A and NMDAR2B at 72 h after TBI
Our results showed that treatment with 50 mg/kg/day or 250 mg/kg/day ceftriaxone for 3 days attenuated neurodestructive extrasynaptic NMDAR2B subunit expression. We speculate the decrease in NMDAR2B expression may be related to ceftriaxone increasing GLT-1 activity leading to the decreased presence of glutamic acid in the synaptic cleft and extrasynaptic region. However, the analysis of glutamate concentration in the extracellular space using microdialysis should be clarified in the future.
Ceftriaxone effects on neuroinflammation and apoptosis after TBI
Our study is consistent with Wei et al. who demonstrated that ceftriaxone administration, using a single dose of ceftriaxone (200 mg/kg i.p.) after weight-drop brain injury, can upregulate GLT-1 and reduce TNF-α expression [11]. We further contribute the new information that ceftriaxone up-regulated microglia GLT-1 expression and attenuated microglial activation and TNF-α expression in activated microglia and synergistically reduced neuronal apoptosis according to immunofluorescence staining in the perilesioned cortex. Therefore, we suggest that the clearance of extracellular glutamate by up-regulated microglia GLT-1 might synergistically reduce neuroinflammation. Since excitotoxicity and neuroinflammation play important roles in triggering and sustaining the neurodegenerative process after TBI and TNF-α is a link between excitotoxicity and neuroinflammation [42], we suggest that ceftriaxone administration in the acute stage may be a promising therapeutic strategy for critical care after TBI.
Ceftriaxone effects on neurons, microglia and astrocytes after TBI and possible molecular mechanisms
Our results showed that after TBI ceftriaxone increased neuronal GLT1 expression and attenuated NMDAR2B expression and neuronal apoptosis. Ceftriaxone also increased microglial GLT1 expression, microglial activation and TNF-α production. However, ceftriaxone attenuated astrocyte activation, but did not significantly alter TNF-α production and GLT1 expression in astrocytes. These results suggested that dysregulation of GLT-1 in different neuroglial cells is critically involved in inducing neuronal death after TBI. In the molecular transcriptional level, ceftriaxone treatment induced upregulation of GLT1 and GLT1 isoforms in association with activation of the Akt-NFκB signaling pathway and enhancement of the GLT-1 promoter activity and protein levels [43]. Whether the expression of Akt-NFκB signaling pathway is different in neurons, microglia and astrocytes, resulting in different GLT1 expression after TBI or ceftriaxone treatment, deserves further investigation in the future.
Comparison of our results with other experimental TBI models treated with ceftriaxone
Table 2 summarizes the previous and current studies in animal models of TBI treated with ceftriaxone. There have been several different administration dosages and course methods for ceftriaxone in the experimental TBI field, including 200 mg/kg/d1 [11–13] and 250 mg/kg/day [14], which were much higher than the usual clinical dosage of 50–100 mg/kg/day [6]. The time courses were variable from a single dose [11], or for 5 days 12 to 7 days [13, 14]. The differences in animal weight, injury models and severity, administration dosage and time course, and regional observations inevitably led to variable outcomes. Our results showed that TBI treatment with ceftriaxone at both 50 and 250 mg/kg/day for 3 days may improve motor deficits. Therefore, we propose that without its antibacterial effect, ceftriaxone may be a potential candidate for neuroprotection after TBI.
Limitations of the current study
Several limitations in our study should be mentioned. First, we only investigated the effects of GLT-1 and NMDA receptor subunits without detecting glutamate receptors such as α-amino–3-hydroxy-5 methyl-4-isoxazole propionic acid (AMPA), kainate, and metabotropic receptors, which may influence neuroinflammation and excitotoxicity [44]. Second, we only evaluated the perilesioned cortex. Whether ceftriaxone has a beneficial effect on the hippocampus, which is related to learning and memory performance and depression development after TBI, requires clarification using hippocampal histology staining and behavioral tests such as the Morris water maze and forced swimming test [45]. Third, we only investigated the short-term postinjury pharmacological intervention effects of ceftriaxone, while a 3-day time period assessment is useful to assess neuroinflammation-associated parameters of TBI-induced secondary insults [15]. Therefore, the longer-term neurological outcome and adverse reactions of ceftriaxone could be assessed in follow-up studies despite Ratti E showing no adverse effects of ceftriaxone when administered at 500 mg/kg/day for 6 months [23]. Fourth, we initiate ceftriaxone treatment right after impact has very little clinical relevance. Delayed treatment such as 30 min or 1 h or others after TBI to evaluate the effects of ceftriaxone on TBI should be performed in the future. Fifth, we only used a single functional method, an inclined plane test to measure motor functions of the upper and lower limbs after neural damage in rats. However, this test is not sufficient to exclude any possibility of motor weakness; therefore, multiple tests for motor function should be used in the future. Finally, to reinforce the essential impact of the results it would be evaluated the expression of the key caspases (i.e. casp-3/8/9, Bcl-proteins), the level of key pro- & anti-inflammatory cytokines/chemokines in the future.
Conclusions
In acute stage after TBI, the neuroprotective effect of ceftriaxone may relate to increase GLT-1 expression and reduce neuroinflammation and neuronal apoptosis resulting in an improvement in functional outcomes. This neuroprotective effect is not related to its antibacterial effects.
Acknowledgements
The authors thank all of the researchers who participated in this study.
Abbreviations
- TBI
Traumatic brain injury
- SD
Sprague–Dawley
- TTC
Triphenyltetrazolium chloride
- TNUEN
Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling
- GFAP
Glial fibrillary acidic protein
- DAPA
4', 6-Diamidino-2-phenylindole
- HR
Heart rate
- MAP
Mean arterial pressure
- ICP
Intracranial pressure
- CPP
Cerebral perfusion pressure
- GLT-1
Glutamate transporter 1
- TNF-α
Tumor necrosis factor α
- NR 2A
N-Methyl-d-aspartate receptor 2A
- NR2B
N-Methyl-d-aspartate receptor 2B
Authors' contributions
S-WL, H-CS, and C-CW conceived and designed the experiments. T-TEN and J-RK performed the experiments. S-WL and J-RK analyzed the data, H-CS and C-CC contributed reagents/materials/analysis tools, S-WL and C-CW wrote the paper. All authors read and approved the final manuscript.
Funding
Nil.
Availability of data and materials
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.
Declarations
Ethics approval and consent to participate
This study was approved by the Chi Mei Medical Center’s Animal Care and Use Committee and conformed to carry out in compliance with the ARRIVE guidelines with Approval No. 10808602.
Consent for publication
Not applicable.
Competing interests
The authors report no conflicts of interest concerning the materials or methods used in this study or the findings specified in this paper.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Sher-Wei Lim and Hui-Chen Su contributed equally to this work
Contributor Information
Sher-Wei Lim, Email: slsw0219@gmail.com.
Hui-Chen Su, Email: Michael.maggil@gmail.com.
Tee-Tau Eric Nyam, Email: ronaldowen@gmail.com.
Chung-Ching Chio, Email: chiocc@ms28.hinet.net.
Jinn-Rung Kuo, Email: kuojinnrung@gmail.com.
Che-Chuan Wang, Email: wangchechuan@gmail.com.
References
- 1.Zipfel GJ, Babcock DJ, Lee JM, Choi DW. Neuronal apoptosis after CNS injury: the roles of glutamate and calcium. J Neurotrauma. 2000;17:857–869. doi: 10.1089/neu.2000.17.857. [DOI] [PubMed] [Google Scholar]
- 2.Kumar A, Loane DJ. Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun. 2012;26:1191–1201. doi: 10.1016/j.bbi.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 3.Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain Pathol. 2004;14:215–222. doi: 10.1111/j.1750-3639.2004.tb00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Z, Handong W. Autophagy in traumatic brain injury: a new target for therapeutic intervention. Front MolNeurosci. 2018;11:190. doi: 10.3389/fnmol.2018.00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Corsello SM, Bittker JA, Liu Z, Gould J, McCarren P, Hirschman JE, et al. The Drug Repurposing Hub: a next-generation drug library and information resource. Nat Med. 2017;23:405–8. doi: 10.1038/nm.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Société de pathologieinfectieuse de langue française (SPILF) Practice guidelines for acute bacterial meningitidis (except newborn and nosocomial meningitis) Med Mal Infect. 2009;39:356–67. doi: 10.1016/j.medmal.2009.02.037. [DOI] [PubMed] [Google Scholar]
- 7.Patel IH, Chen S, Parsonnet M, Hackman MR, Brooks MA, Konikoff J, et al. Pharmacokinetics of ceftriaxone in humans. Antimicrob Agents Chemother. 1981;20:634–41. doi: 10.1128/AAC.20.5.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moskovitz BL. Clinical adverse effects during ceftriaxone therapy. Am J Med. 1984;77:84–88. doi: 10.1016/S0002-9343(84)80024-8. [DOI] [PubMed] [Google Scholar]
- 9.Sullins AK, Abdel-Rahman SM. Pharmacokinetics of antibacterial agents in the CSF of children and adolescents. Pediatr Drugs. 2013;15:93–117. doi: 10.1007/s40272-013-0017-5. [DOI] [PubMed] [Google Scholar]
- 10.Yimer EM, Hishe HZ, Tuem KB. Repurposing of the β-Lactam antibiotic, ceftriaxone for neurological disorders: a review. Front Neurosci. 2019;13:236. doi: 10.3389/fnins.2019.00236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wei J, Pan X, Pei Z, Wang W, Qiu W, Shi Z, et al. The beta-lactam antibiotic, ceftriaxone, provides neuroprotective potential via anti-excitotoxicity and anti-inflammation response in a rat model of traumatic brain injury. J Trauma Acute Care Surg. 2012;73:654–660. doi: 10.1097/TA.0b013e31825133c0. [DOI] [PubMed] [Google Scholar]
- 12.Cui C, Cui Y, Gao J, Sun L, Wang Y, Wang K, et al. Neuroprotective effect of ceftriaxone in a rat model of traumatic brain injury. Neurol Sci. 2014;35:695–700. doi: 10.1007/s10072-013-1585-4. [DOI] [PubMed] [Google Scholar]
- 13.Goodrich GS, Kabakov AY, Hameed MQ, Dhamne SC, Rosenberg PA, Rotenberg A. Ceftriaxone treatment after traumatic brain injury restores expression of the glutamate transporter, GLT-1, reduces regional gliosis, and reduces post-traumatic seizures in the rat. J Neurotrauma. 2013;30:1434–1441. doi: 10.1089/neu.2012.2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hameed MQ, Hsieh TH, Morales-Quezada L, Lee HHC, Damar U, MacMullin PC, et al. Ceftriaxone treatment preserves cortical inhibitory interneuron function via transient salvage of GLT-1 in a rat traumatic brain injury model. Cereb Cortex. 2019;29:4506–4518. doi: 10.1093/cercor/bhy328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsai YT, Kuo JR, Wang CC, Lin KC, Chio CC, Hu CY. Extracellular signal-regulated kinase 1/2 is involved in a tamoxifenneuroprotective effect in a lateral fluid percussion injury rat model. J Surg Res. 2014;189:106–116. doi: 10.1016/j.jss.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 16.The Brain Trauma Foundation. The American Association of Neurological Surgeons. The Joint Section on Neurotrauma and Critical Care Cerebral perfusion pressure thresholds. J Neurotrauma 2007; 24: S59–64.
- 17.Wang Y, Lin SZ, Chiou AL, Williams LR, Hoffer BJ. Glial cell line-derived neurotrophic factor protects against ischemia-induced injury in the cerebral cortex. J Neurosci. 1997;17:4341–4348. doi: 10.1523/JNEUROSCI.17-11-04341.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaur P, Sharma S. Recent advances in pathophysiology of traumatic brain injury. CurrNeuropharmacol. 2018;16:1224–1238. doi: 10.2174/1570159X15666170613083606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Charrueau C, Belabed L, Besson V, Chaumeil JC, Cynober L, Moinard C. Metabolic response and nutritional support in traumatic brain injury: evidence for resistance to renutrition. J Neurotrauma. 2009;26:1911–1920. doi: 10.1089/neu.2008.0737. [DOI] [PubMed] [Google Scholar]
- 20.Kuo JR, Lim SW, Zheng HX, Ho CH, Chang CH, Chio CC, et al. Triglyceride is a good biomarker of increased injury severity on a high fat diet rat after traumatic brain injury. Neurochem Res. 2020;45:1536–1550. doi: 10.1007/s11064-020-03018-x. [DOI] [PubMed] [Google Scholar]
- 21.Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;2(140):155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- 22.Marmarou A. Pathophysiology of traumatic brain edema: current concepts. Acta Neurochir Suppl. 2003;86:7–10. doi: 10.1007/978-3-7091-0651-8_2. [DOI] [PubMed] [Google Scholar]
- 23.Ratti E, Berry JD, Greenblatt DJ, Loci L, Ellrodt AS, Shefner JM, et al. Preclinical rodent toxicity studies for long term use of ceftriaxone. Toxicol Rep. 2015;2:1396–1403. doi: 10.1016/j.toxrep.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zwienenberg M, Gong QZ, Lee LL, Berman RF, Lyeth BG. ICP monitoring in the rat: comparison of monitoring in the ventricle, brain parenchyma, and cisterna magna. J Neurotrauma. 1999;16:1095–102. doi: 10.1089/neu.1999.16.1095. [DOI] [PubMed] [Google Scholar]
- 25.Fritz HG, Walter B, Holzmayr M, Brodhun M, Patt S, Bauer R. A pig model with secondary increase of intracranial pressure after severe traumatic brain injury and temporary blood loss. J Neurotrauma. 2005;22(7):807–2. doi: 10.1089/neu.2005.22.807. [DOI] [PubMed] [Google Scholar]
- 26.Wang CC, Kuo JR, Chen YC, Chio CC, Wang JJ, Lin BS. Brain tissue oxygen valuation by wireless near-infrared spectroscopy. Surg Res. 2016;200:669–675. doi: 10.1016/j.jss.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 27.Yuk JH, Nightingale CH, Quintiliani R. Clinical pharmacokinetics of ceftriaxone. Clin Pharmacokinet. 1989;17:223–35. doi: 10.2165/00003088-198917040-00002. [DOI] [PubMed] [Google Scholar]
- 28.Hartl R, Gerber LM, Ni Q, Ghajar J. Effect of early nutrition on deaths due to severe traumatic brain injury. J Neurosurg. 2008;109:50–56. doi: 10.3171/JNS/2008/109/7/0050. [DOI] [PubMed] [Google Scholar]
- 29.Faries PL, Simon RJ, Martella AT, Lee MJ, Machiedo GW. Intestinal permeability correlates with severity of injury in trauma patients. J Trauma. 1998;44:1031–5. doi: 10.1097/00005373-199806000-00016. [DOI] [PubMed] [Google Scholar]
- 30.Moinard C, Neveux N, Royo N, Genthon C, Marchand-Verrecchia C, Plotkine M, Cynober L. Characterizationof the alteration of nutritional state in brain injury inducedby fluid percussion in rats. Intensive Care Med. 2005;31:281–288. doi: 10.1007/s00134-004-2489-9. [DOI] [PubMed] [Google Scholar]
- 31.Matharu D, Dhotre D, Balasubramanian N, Pawar N, Sagarkar S, Sakharkar A. Repeated mild traumatic brain injury affects microbial diversity in rat jejunum. J Biosci. 2019;44:120. doi: 10.1007/s12038-019-9940-0. [DOI] [PubMed] [Google Scholar]
- 32.Rice MW, Pandya JD, Shear DA. Gut microbiota as a therapeutic target to ameliorate the biochemical, neuroanatomical, and behavioral effects of traumatic brain injuries. Front Neurol Front Neurol. 2019;10:875. doi: 10.3389/fneur.2019.00875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lim SW, Wang CC, Wang YH, Chio CC, Niu KC, Kuo JR. Microglial activation induced by traumatic brain injury is suppressed by postinjury treatment with hyperbaric oxygen therapy. J Surg Res. 2013;184:1076–1084. doi: 10.1016/j.jss.2013.04.070. [DOI] [PubMed] [Google Scholar]
- 34.Zhu CS, Grandhi R, Patterson TT, Nicholson SE. A review of traumatic brain injury and the gut microbiome: insights into novel mechanisms of secondary brain injury and promising targets for neuroprotection. Brain Sci. 2018;8:113. doi: 10.3390/brainsci8060113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Conti AC, Raghupathi R, Trojanowski JQ, McIntosh TK. Experimental brain injury induces regionally distinct apoptosis during the acute and delayed post-traumatic period. J Neurosci. 1998;18:5663–5672. doi: 10.1523/JNEUROSCI.18-15-05663.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maragakis NJ, Dietrich J, Wong V, Xue H, Mayer-Proschel M, Rao MS, et al. Glutamate transporter expression and function in human glial progenitors. Glia. 2004;45:133–143. doi: 10.1002/glia.10310. [DOI] [PubMed] [Google Scholar]
- 37.Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, et al. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 38.Chen W, Mahadomrongkul V, Berger UV, Bassan M, DeSilva T, Tanaka K, et al. The glutamate transporter GLT1a is expressed in excitatory axon terminals of mature hippocampal neurons. JNeurosci. 2004;24:1136–1148. doi: 10.1523/JNEUROSCI.1586-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Persson M, Rönnbäck L. Microglial self-defence mediated through GLT-1 and glutathione. Amino Acids. 2012;42:207–219. doi: 10.1007/s00726-011-0865-7. [DOI] [PubMed] [Google Scholar]
- 40.Chirumamilla S, Sun D, Bullock MR, Colello RJ. Traumatic brain injury induced cell proliferation in the adult mammalian central nervous system. J Neurotrauma. 2002;19:693–703. doi: 10.1089/08977150260139084. [DOI] [PubMed] [Google Scholar]
- 41.Rimmele TS, Rosenberg PA. GLT-1: the elusive presynaptic glutamate transporter. Neurochem Int. 2016;98:19–28. doi: 10.1016/j.neuint.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olmos G, Lladó J. Tumor necrosis factor alpha: a link between neuroinflammation and excitotoxicity. Mediat Inflamm. 2014;2014:861231. doi: 10.1155/2014/861231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rao PS, Saternos H, Goodwani S, Sari Y. Effects of ceftriaxone on GLT1 isoforms, xCT and associated signaling pathways in P rats exposed to ethanol. Psychopharmacology. 2015;232:2333–2342. doi: 10.1007/s00213-015-3868-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dingledine R, Borges K, Bowie D, Traynelis SF. Glutamate receptor ion channels. Pharmacol Rev. 1999;51:7. [PubMed] [Google Scholar]
- 45.Wu A, Molteni R, Ying Z, Gomez-Pinilla F. A saturated-diet aggravates the outcome of traumatic brain injury on hippocampal plasticity and cognitive function by reducing brain-derived neurotrophic factor. Neuroscience. 2003;119:365–375. doi: 10.1016/S0306-4522(03)00154-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.