

SUMMARY
Macroautophagy (hereafter referred to as autophagy) is a conserved process that promotes cellular homeostasis through the degradation of cytosolic components, also known as cargo. During autophagy, cargo is sequestered into double-membrane vesicles called autophagosomes, which are predominantly transported in the retrograde direction to the perinuclear region to fuse with lysosomes, thus ensuring cargo degradation1. The mechanisms regulating directional autophagosomal transport remain unclear. The ATG8 family of proteins associate with autophagosome membranes2 and play key roles in autophagy, including the movement of autophagosomes. This is achieved via the association of ATG8 with adaptor proteins like FYCO1, involved in the anterograde transport of autophagosomes toward the cell periphery1,3–5. We previously reported that phosphorylation of LC3B/ATG8 on threonine 50 (LC3B-T50) by the Hippo kinase STK4/MST1 is required for autophagy through unknown mechanisms6. Here, we show that STK4-mediated phosphorylation of LC3B-T50 reduces binding of FYCO1 to LC3B. In turn, impairment of LC3B-T50 phosphorylation decreases starvation-induced perinuclear positioning of autophagosomes as well as their colocalization with lysosomes. Moreover, a significantly higher number of LC3B-T50A-positive autophagosomes undergo aberrant anterograde movement to axonal tips in mammalian neurons and toward the periphery of mammalian cells. Our data support a role of a nutrient- sensitive STK4–LC3B–FYCO1 axis in the regulation of the directional transport of autophagosomes, a key step of the autophagy process, via the post-translational modification of LC3B.
Keywords: Autophagy, LC3B, Hippo kinases, STK4, trafficking, vesicle transport, FYCO1, starvation
eTOC blurb
Nieto-Torres et al. describe that phosphorylation of the autophagy protein LC3B is critical for the retrograde transport of autophagosomes within cells. Block of LC3B phosphorylation leads to aberrant autophagosome transport and association with lysosomes. This pathway may be relevant for autophagy-related pathologies, such as neurodegeneration.
Graphical Abstract
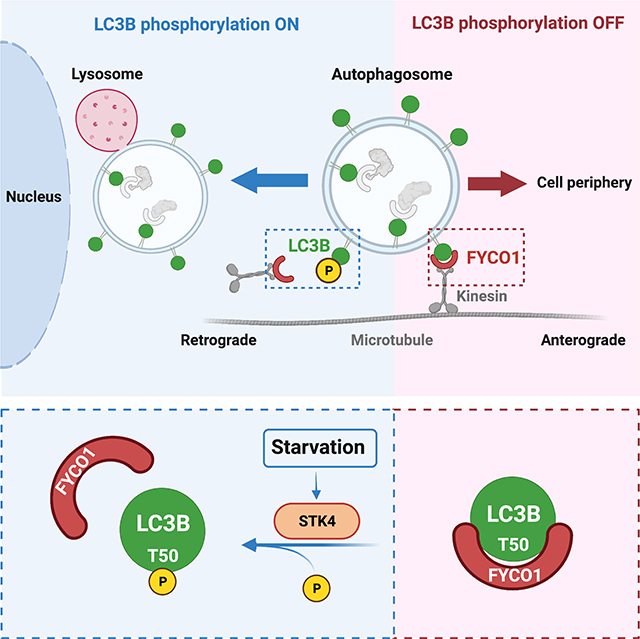
RESULTS
LC3B-T50 Phosphorylation Regulates the Interaction Between LC3B and FYCO1
We recently reported that phosphorylation of LC3B-T50 by STK4/MST1 is required for autophagy6. Specifically, depletion of Stk4 or expression of a non-phosphorylatable LC3B-T50A mutant causes accumulation of autophagosomes throughout the cell and clustering of lysosomes around the nucleus, leading to a block in autophagy in mouse embryonic fibroblasts (MEFs) and in C2C12 mouse myoblasts6. We hypothesized that phosphorylation of LC3B might modulate its interaction with autophagy adaptor proteins that bind LC3B and other ATG8 proteins on the outer surface of autophagosomes. To test this, we identified LC3B binding partners via affinity purification of HA-tagged LC3B proteins followed by mass spectrometry from human 293T cells transiently expressing HA-tagged wild-type (WT) LC3B or the phospho-mutant forms LC3B-T50A (T50A, phospho-deficient) and LC3B-T50E (T50E, phospho-mimetic) (Figure 1A and Data S1). Of the LC3B interactors identified, FYCO1, a known autophagy adaptor protein3,4, showed the most HA-LC3B-T50 phosphorylation-sensitive binding; specifically, FYCO1 binding was ~2-fold higher to the HA-T50A mutant and ~5-fold lower to the HA-T50E mutant compared with binding to HA-LC3B-WT (Figure 1B and Data S1).
Figure 1. LC3B phosphorylation modulates FYCO1–LC3B interactions.

(A) Schematic of pull-down assays performed in HEK 293T cells expressing HA-tagged WT (wild type), T50A (phospho-deficient mutant), and T50E (phospho-mimetic mutant) LC3B proteins to identify LC3B phosphorylation-dependent interactors by mass spectrometry. See also Data S1. (B) Quantification of FYCO1 binding to HA-LC3B proteins determined by mass spectrometry. Mean ± SD of three technical replicates. **p<0.0021, ***p<0.0002, ****p<0.0001 by one-way ANOVA. See also Data S1. (C) Representative western blot of FYCO1 co-immunoprecipitated with HA-tagged LC3B proteins expressed in HeLa LC3B-KO cells (n=3). See also Figures S2A, S2B and S3G. (D) Representative western blot of FYCO1 co-immunoprecipitated with HA-tagged LC3B proteins in Stk4−/−;Stk3−/+ and WT mouse embryonic fibroblasts (n=3). (E) Representative western blot of FYCO1 co-immunoprecipitated with non-phosphorylated or in vitro-phosphorylated GST-LC3B (n=3). GST-LC3B panel shows Coomassie staining. See also Figures S2C and S2D. (C-E) Values below blots are protein ratios in relative units. (F) Biolayer interferometry assay of His-tagged LC3B proteins binding to a peptide containing FYCO1-LIR and adjacent amino acids 4. Mean KD ± SEM of three technical replicates and R2 for binding curve fit are shown. *p<0.05 by one-way ANOVA. See Data S1 for quantification of all western blots.
These results were validated in LC3B knockout (LC3B-KO) HeLa cells generated using CRISPR-Cas9 technology and expressing HA-tagged-LC3B-WT, -T50A, or -T50E (Figure S1A). The HeLa LC3B-KO cell line background was used throughout the rest of the study, unless otherwise indicated. Consistent with earlier reports in MEFs6, HA-T50A phospho-mutant showed similar phenotypes, displayed increased numbers of autophagosomes that were insensitive to the autophagy inhibitor Bafilomycin A (Baf A), and exhibited LAMP1-positive vesicles that clustered in perinuclear regions (Figure S1B–1D). Immunoprecipitation of HA-LC3B (Figure 1C) or mCherry (mCh)-FYCO1 (Figure S2A) followed by western blotting for the reciprocal protein confirmed the differential interaction of FYCO1 with HA-LC3B phospho-mutants. Likewise, FYCO1–HA-LC3B binding was increased in Stk4−/−;Stk3−/+ MEFs6 (Figure 1D). Finally, in vitro phosphorylation of GST-LC3B on T50, verified with a phosphorylation-specific antibody6, reduced the GST-LC3B interaction with FYCO1 compared to non-phosphorylated GST-LC3B (Figure 1E). Notably, the LC3B phosphorylation-dependent binding profile of FYCO1 to LC3B (i.e., increased binding to HA-T50A, decreased binding to HA-T50E, as well as decreased binding to GST-pLC3B) was specific for FYCO1 since other known LC3B interactors KEAP1 and p62 bound to LC3B independently of T50 status (Figure S2B–2D, S3G and Data S1), whereas NDP52 showed increased binding to HA-T50A and increased but variable binding to HA-T50E (Figure S2B and S3G). Collectively, these data indicate that STK4-mediated phosphorylation of LC3B decreases its interaction with FYCO1, suggesting a role for FYCO1 in STK4–LC3B-mediated autophagy regulation.
LC3B is known to directly interact with FYCO1 through an extended LC3-interacting region (LIR), which contains a core LIR and several adjacent amino acids3,4. Within the LC3B–FYCO1 binding interface, amino acid T50 of LC3B is located in close proximity to two aspartic acid residues preceding the core FYCO1 LIR (Figure S2E). We therefore speculated that LC3B-T50 phosphorylation may directly affect FYCO1–LC3B binding via disruption of the binding interface. To test this, we performed biolayer interferometry to measure the binding affinities between His-LC3B-WT or phospho-mutants (bacterially expressed and purified) and a FYCO1 33 amino-acid peptide (amino acids 1265–1298) spanning the LIR domain and its adjacent aspartic acids4. The binding affinity of His-LC3B-WT and the FYCO1 peptide was in the micromolar range (KD ~4.1 × 10−6 M) (Figure 1F), similar to previous reports7. In contrast, the binding affinity between the FYCO1 peptide and phospho-mimetic His-T50E was significantly lower (KD ~2.8 × 10−5 M) than that of His-LC3B-WT (Figure 1F), suggesting that T50 phosphorylation may play a role in blocking the direct interaction between LC3B and the FYCO1-LIR region. Of note, His-T50A bound to the FYCO1 peptide with an affinity (KD ~6.0 × 10−6 M) in the same micromolar range as His-LC3B-WT (Figure 1F). The similar binding affinities displayed by His-LC3B-WT and the His-T50A phospho-null mutant could stem from the lack of STK4-dependent phosphorylation of the bacteria-derived LC3B proteins used for the binding experiments, as STK4 is not expressed in bacteria8. Thus, the affinity binding data and the co-immunoprecipitation results suggest that phosphorylation of LC3B-T50 directly decreases the LC3B binding affinity for FYCO1, revealing that phosphorylation of LC3B regulates the interaction between LC3B and FYCO1 and decreases LC3B-FYCO1 binding affinity.
Subcellular Localization of Autophagosomes is Regulated by LC3B-T50 Phosphorylation in a FYCO1-Dependent Manner
As FYCO1 associates with LC3B on autophagosomes to regulate their transport3,4, we next asked if LC3B-T50 phosphorylation status would affect FYCO1 localization to autophagosomes. LC3B is attached to autophagosomal membranes via lipidation of a C-terminal glycine9. Autophagosomes were defined as HA-LC3B-positive puncta, since lipidation-deficient versions (i.e., G120A) of WT and phospho-mutant LC3Bs cannot associate with autophagosomes membranes2, and as expected, were diffusely localized (Figure S2F). The G120A versions of WT and phospho-mutant HA-LC3Bs maintained their differential binding to FYCO1 (Figure S2G), suggesting that LC3B lipidation and by extension autophagosomal membrane association may not be essential for LC3B phosphorylation to regulate LC3B-FYCO1 binding. Notably, the HA-LC3B colocalization with FYCO1 was much higher in HA-T50A, and lower in HA-T50E, as compared to those in HA-LC3B-WT-expressing cells (Figure 2A and 2B), indicating that LC3B-T50 phosphorylation decreases whereas de-phosphorylation increases the association of FYCO1 with autophagosomes, consistent with the LC3B-FYCO1 interaction and binding-affinity data (Figure 1). Unlike FYCO1, p62, KEAP1 and NDP52 showed only slightly increased colocalization to HA-T50A compared to HA-LC3B, and none of these three control LC3B interactors displayed decreased colocalization to HA-T50E as observed for FYCO1 (Figure S2H), further supporting the notion that LC3B phosphorylation may affect FYCO1 binding in a specific manner.
Figure 2. Subcellular localization of LC3B phospho-mutants is differentially regulated by FYCO1.

(A) Representative immunofluorescence micrographs of HeLa-KO cells expressing HA-tagged LC3B-WT, T50A, or T50E co-stained for FYCO1. White arrowheads indicate LC3B:FYCO1 colocalization spots. (B) Quantification of HA-LC3B:FYCO1 colocalized voxels (volumetric pixels). Mean ± SEM, ****p<0.0001 by two-way ANOVA (n=17–27 cells from three experiments). See also Figure S2H. (C) Representative immunofluorescence micrographs of HA-tagged LC3B-WT, T50A, or T50E puncta in control (siNS) or siFYCOI-treated HeLa cells. Dashed line defines 10 μm from the outer nuclear edge. Scale bar = 10 μm. White arrowheads point to HA-LC3B-positive puncta in peripheral locations of the cell. (D) Average distance between HA-LC3B-positive puncta and the nucleus. Mean ± SEM of n=25–39 cells from three experiments. ****p<0.0001 by two-way ANOVA. See also Figure S3A.
As FYCO1 regulates anterograde transport of autophagosomes3, we next asked whether the LC3B phosphorylation-dependent association with FYCO1 affected the subcellular positioning of autophagosomes. Immunofluorescence microscopy and imaging analysis revealed that the average distance between LC3B-positive puncta and the nucleus was significantly shorter in HA-T50E-expressing cells (~4.0 μm), than in cells expressing HA-LC3B-WT (~8.8 μm), whereas HA-T50A showed a longer average distance (~11.6 μm) (Figure 2C, 2D and S3A). Notably, the dispersal of LC3B puncta to the cell periphery in HA-WT and HA-T50A-expressing cells was FYCO1-dependent, as siRNA-mediated FYCO1 knockdown (Figure S2I) resulted in perinuclear accumulation of HA-LC3B puncta (Figure 2C, 2D and S3A). These results show that the subcellular localization of LC3B puncta is regulated by LC3B phosphorylation in a FYCO1-dependent manner.
Starvation-Induced Perinuclear Clustering of Autophagosomes is Regulated Via an STK4–LC3B–FYCO1 Axis
Starvation is a robust inducer of autophagy and is known to stimulate perinuclear positioning of autophagosomes to promote fusion with lysosomes10. We investigated the effects of starvation on STK4–LC3B–FYCO1-mediated control of autophagosomal localization. Combined serum and amino-acid deprivation of HeLa cells resulted in a ~3-fold increase in the levels of phosphorylated (active) STK4 (STK4-pT183)11 (Figure 3A), consistent with previous findings that the Hippo pathway is activated following serum deprivation12. In turn, siRNA-mediated depletion of STK4 (Figure S3B) prevented starvation-induced perinuclear clustering of LC3B-positive puncta in HA-LC3B-WT–expressing cells (Figure 3B, 3C and S3C), suggesting that STK4-dependent phosphorylation of LC3B is required for starvation-induced perinuclear autophagosome positioning. Consistent with these results, starvation did not change the dispersed or perinuclear subcellular localization of LC3B-positive puncta in HA-T50A or HA-T50E cells, respectively (Figure 3D, 3E and S3D). Thus, starvation-induced STK4-mediated LC3B phosphorylation-dependent positioning of autophagosomes to the perinuclear region of mammalian cells. Because starvation-induced perinuclear positioning of autophagosomes promotes autophagosome-lysosome fusion13, we quantitated colocalization between HA-LC3B-WT or -LC3B phospho-mutants with either endolysosomal LAMP1 and lysosomal LAMP2 markers14, following starvation. Consistent with our previous observations in Stk4−/−;Stk3−/+ MEFs6, we observed that the starvation-induced increase in LAMP1/2 colocalization with HA-LC3B-WT was LC3B-phosphorylation dependent. Specifically, HA-T50A-LAMP1/2 colocalization was not induced by nutrient deprivation, whereas HA-T50E-LAMP1/2 colocalization was elevated compared to HA-WT in a starvation-independent manner (Figures 3F, 3G and S3E). These results indicate that LC3B phosphorylation can promote the association of autophagosomes with lysosomes.
Figure 3. Starvation induces perinuclear localization of LC3B puncta in an LC3B phosphorylation- and FYCOI-dependent manner.

(A) Representative western blot of total and phosphorylated STK4-T183 in HeLa LC3B-KO cells incubated in normal or starvation medium (n=3). Values indicate pSTK4:STK4 protein ratio normalized to α-tubulin. (B) Representative immunofluorescence micrographs of HA-LC3B puncta in HeLa LC3B-KO cells expressing control or STK4-targeting siRNA before or 1 h after incubation in starvation medium. Scale bar = 10 μm. White arrowheads point to HA-LC3B-positive puncta in peripheral locations of the cell. (C) Quantification of distance between LC3B puncta and the nucleus. Mean ± SEM of n=18–23 cells from three experiments. ***p<0.0002 by two-way ANOVA. See also Figure S3B. (D and E) Representative immunofluorescence micrographs (D) and quantification of distance from the nucleus (E) of LC3B puncta in HeLa cells expressing the indicated HA-tagged LC3B proteins. Scale bar = 10 μm. White arrowheads point to HA-LC3B-positive puncta in peripheral locations of the cell. Mean ± SEM of n=23–43 cells from three experiments. ****p<0.0001 by two-way ANOVA. See also Figure S3D. (F) Representative immunofluorescence micrographs of HeLa-KO cells expressing HA-tagged LC3B-WT, T50A, or T50E co-stained with antibodies against HA and LAMP2. Dotted insets are enlarged in bottom panels. Scale bars = 10 μm. White arrowheads point to HA-LC3B:LAMP1 colocalization events. (G) HA-LC3B-LAMP2 colocalized voxels. Mean ± SEM of n=30 cells from three experiments, *p<0.0332, ***p<0.0002, ****p<0.0001 by two-way ANOVA. See also Figure S3E. (H) Representative western blot of FYCO1 co-immunoprecipitated with HA-tagged LC3B proteins expressed in HeLa LC3B-KO cells before and 1 h after incubation in normal or starvation medium. Values under blots indicate percent decrease in HA-LC3B:FYCO1 protein ratio after starvation (n=5). See also Figures S3F and S3G. (I) HA-LC3B:FYCO1 colocalized voxels in HeLa LC3B-KO cells before or 1 h after incubation in starvation medium. Mean ± SEM of n=18–30 cells from three experiments. ****p<0.0001 by two-way ANOVA. See also Figure S2H. See Data S1 for quantification of all western blots.
Finally, we hypothesized that the starvation-induced perinuclear vesicle-positioning originated from a decrease in LC3B–FYCO1 binding. To test this directly, we performed co-immunoprecipitation experiments and immunofluorescence microscopy of HeLa LC3B-KO cells expressing HA-LC3B-WT or phospho-mutants grown under normal or starvation conditions to determine the degree of LC3B-FYCO1 association. In HA-LC3B-WT–expressing cells, starvation caused a ~50% reduction in the levels of FYCO1 protein that co-immunoprecipitated with HA-LC3B (Figure 3H), as well as >50% decrease in the percentage of HA-LC3B-positive puncta colocalizing with FYCO1 (Figure 3I). In contrast, starvation of cells expressing HA-T50A or HA-T50E only slightly reduced (~15%) the interaction between FYCO1 and either HA-LC3B phospho-mutant (Figure 3H) and had no significant effect on the colocalization of FYCO1 with HA-LC3B phospho-mutant–positive puncta (Figure 3I). Thus, starvation decreased the association between LC3B and FYCO1 in an LC3B-phosphorylation dependent manner. In contrast, starvation strongly decreased both the binding and colocalization of KEAP1 and NDP52 to either HA-LC3B-WT, HA-T50A or HA-T50E (Figure S2H, S3F and S3G), whereas the effect of starvation on the association of p62 to the LC3B phospho-mutants was relatively modest with p62 binding largely unaffected (Figures S3F and S3G) and a general small decrease in colocalization (Figure S2H, see figure legend for further discussion). Overall, our results indicate that nutrient deprivation regulates an STK4–LC3B–FYCO1 axis to control the subcellular positioning of autophagosomes in the perinuclear area of the cell, promoting the colocalization of autophagosomes with lysosomes.
LC3B Phosphorylation Regulates the Directional Transport of Autophagosomes in Mammalian Cells and Neurons
Previous work indicated that FYCO1 promotes the anterograde transport of autophagosomes and endosomes via its interaction with LC3B and the motor protein kinesin-13,15. Because LC3B-T50 phosphorylation regulates both LC3B binding to FYCO1 (Figure 1), as well as autophagosome positioning (Figure 2), we next investigated whether LC3B-T50 phosphorylation plays a role in autophagosomal transport. By using super-resolution live-cell imaging in HeLa LC3B-KO cells, we found in two separate replicates that the majority of GFP-LC3B WT vesicles moved primarily in the retrograde direction towards the perinuclear region (Figure S4A, S4B and Video S1), consistent with previous reports16,17. In contrast, GFP-T50A vesicles were mainly transported in an anterograde direction towards the cell periphery (Figure S4A, S4B and Video S1). While the directional distribution of GFP-T50E particles was not significantly different from that of GFP-LC3B-WT (Figure S4A, S4B and Video S1), T50E vesicles displayed significant switches in directionality compared to GFP-LC3B WT or GFP-T50A vesicles, suggesting impaired coordination between anterograde and retrograde motors associated to those vesicles (Figure S4C, and Video S1). These data suggest that phosphorylation of LC3B is important for directional retrograde transport of autophagosomes, and perinuclear positioning.
To further investigate the directionality of movement, we also evaluated the role of LC3B phosphorylation on autophagosome transport in axons of polarized cultured primary hippocampal neurons (Figure 4A). In axons, kinesin and dynein translocate cargoes in anterograde (toward the synapse) and retrograde (toward the soma) directions, respectively18. Previous studies showed that autophagosomes form in distal axonal regions, and are transported retrogradely toward the soma, where they fuse with lysosomes to drive degradation18–20. Consistent with previous reports18,19, our single particle live imaging and quantitative analysis21 showed that the majority (~80%) of GFP-LC3B WT vesicles moved primarily in the retrograde direction (Figure 4B–4C and Video S2), and these vesicles spent most (~60%) of time moving in this direction, i.e., toward the soma (Figure 4D). In contrast, quantitative analysis of the movement of GFP-T50A particles indicated an overall attenuation of retrograde transport parameters (i.e., mobile segments, time in motion, and density of particles, Figure 4B–4E and Video S2), with a corresponding increase in their anterograde movement dynamics: Only ~60% of GFP-T50A particles moved in the retrograde direction, at the expense of a significant increase in the number of anterograde-moving GFP-T50A particles (~40%) (Figure 4B–4C and Video S2), and their time spent moving toward the soma was significantly decreased to ~40%, while spending more time moving in the anterograde direction (Figure 4D and Video S2). Furthermore, GFP-T50A vesicles displayed significantly decreased retrograde flux (Figure 4F), a measure of retrograde traffic of GFP-T50A particles toward the soma, as calculated by the rate of movement as particles move through a randomly assigned gate at a single point in time (Star Methods). Additional quantitative analysis showed that retrograde run lengths of GFP-T50A vesicles were significantly reduced, while anterograde run lengths were enhanced (Figure 4G and Video S2), consistent with overall impairments to the retrograde movement dynamics of GFP-T50A vesicles. Interestingly, the anterograde segmental velocities of GFP-T50A vesicles were also significantly increased, although our analysis did not reveal significant retrograde velocity changes (Figure S4D). Notably, more GFP-T50A vesicles were stationary compared to GFP-LC3B WT vesicles (Figure 4E), suggesting that in addition to an overall change in retrograde movement and enhanced anterograde transport dynamics, LC3B phosphorylation is required for the proper activation of LC3B vesicle motility.
Figure 4. Directional transport of LC3B puncta is regulated by the LC3B phosphorylation.

Schematic illustration of experimental setup primary mouse hippocampal neurons. Black boxes highlight the area of the cells imaged. Directional anterograde or retrograde movement of LC3B-positive vesicles is depicted. (B) Representative images of primary mouse hippocampal neurons axons (first frame pictures) and kymographs of the indicated GFP-positive vesicles (pseudo-black lines). (C) Quantification of percent anterograde (Antero.) versus retrograde (Retro.) motile segments of GFP-LC3B-positive vesicles in primary hippocampal axons. A total of 6921 motile segments were analyzed. Mean ± SEM of n=33–38 cells from three experiments. *p<0.05, **p<0.01 by one-way ANOVA and Šidák’s multiple comparison correction. (D) Quantification of percent time in anterograde (Antero.) or retrograde (Retro.) motion or paused of GFP-LC3B-positive vesicles in primary hippocampal axons. A total of 824 GFP-LC3B positive vesicles from 33–38 cells were analyzed. Mean ± SEM of n=255–292 GFP-LC3B positive particles from three experiments. ****p<0.0001 by one-way ANOVA and Šidák’s multiple comparison correction. (E) Densities as numbers of tracks/100 μm of GFP-LC3B-positive vesicles in anterograde, retrograde, reversal (Rev.) motion or stationary (Stat.) in primary hippocampal axons. Mean ± SEM of n=33–38 cells from three experiments. *p<0.05, **p<0.01, by one-way ANOVA and Sidak’s multiple comparison correction. (F) Quantification of flux as numbers of anterograde versus retrograde tracks of GFP-LC3B-positive vesicles that pass a gate in 5 minutes in primary hippocampal axons. Mean ± SEM of n=33–38 cells from three experiments. *p<0.05, by one-way ANOVA and Sidak’s multiple comparison correction. (G) 1-Cumulative distribution frequency graphs of anterograde and retrograde segmental run lengths of GFP-LC3B-positive vesicles in primary hippocampal axons. A total of 6974 segments were analyzed from 33–38 cells from three experiments. *p<0.05, **p<0.01, ***p<0.001 by Kolmogorov–Smirnov test adjusted with Bonferroni. Similar statistically significant comparisons were obtained when analyzing the data by Rank Sum test (data not shown). (H) Proportion of time spent in anterograde transport of GFP-LC3B-positive vesicles in mouse primary neurons with or without Fyco1 knockdown. A total of 765 GFP-LC3B positive vesicles from 28–33 cells were analyzed. Mean ± SEM of n=142–229 GFP-LC3B positive vesicles from three experiments. *p<0.05 by non-parametric permutation Student’s t-test. (I-J) Co-transport analysis of GFP-LC3B-positive vesicles and Magic Red in primary hippocampal axons. Mean ± SEM of n=14–16 axons from three independent experiments. ***p<0.001 by Student’s t-test. For all the LC3B-positive vesicles transport-related experiments see also Videos S1 and S2 and Figure S4.
To determine if the aberrant enhanced anterograde transport of GFP-T50A vesicles was dependent on FYCO1, transport was quantitated in neurons where Fyco1 function was down-regulated using shRNAs. Live imaging revealed that compared to scrambled shRNA controls, the amount of time GFP-T50A vesicles spent moving in the anterograde direction was decreased to levels similar to that of GFP-LC3B WT particles (Figure 4H). Moreover, the density of anterograde-moving as well as reversing GFP-T50A vesicles was significantly reduced in shRNA Fyco1 axons, while that of GFP-LC3B WT particles remained unchanged (Figure S4F). These data indicate that LC3B phosphorylation is critical for proper retrograde movement of autophagosomes, and suggest that its absence attenuates retrograde movement and enhances anterograde transport in mammalian axons via interactions with FYCO1.
Finally, we analyzed the transport of GFP-T50E particles which also displayed transport dynamics phenotypes, including a lower percentage of retrograde-moving vesicles, and significantly reduced retrograde run lengths, compared to GFP-LC3B WT vesicles (Figure 4B, 4C, 4G and Video S2). In addition, GFP-T50E particles showed significant impairments in anterograde run lengths and velocities (Figure 4G and S4D), and a significantly higher frequency of directionality switches (Figure S4G and Video S2), consistent with the increased numbers of switches observed in HeLa LC3B-KO cells (Figure S4C and Video S1). Overall, these data indicate that the state of LC3B phosphorylation and FYCO1 regulate the directional transport of autophagosomes in axons, and that phosphorylation is required for the proper retrograde movement of autophagosomes in mammalian axons.
Because retrograde transport of autophagosomes is critical for association with degradative lysosomes en route to the soma, where lysosomes are enriched and where the bulk of degradation occurs22,23, we tested whether LC3B phosphorylation plays a role in the fusion of autophagosomes with lysosomes. To this end, we performed live-imaging in the soma and axon of GFP-LC3B WT or T50A vesicles with Magic Red, a fluorogenic substrate that activates upon cleavage by the active Cathepsin B acidic hydrolase24. In axons, we observed that the majority (>80%) of GFP-LC3B WT vesicles co-migrated with Magic Red positive particles compared to T50A vesicles (~40%) (Figure 4I and 4J). Furthermore, immunofluorescence analysis in the soma revealed reduced colocalization between GFP-T50A vesicles and Cathepsin B compared to GFP-LC3B WT vesicles (Figure S4H–4J). Altogether, these results are consistent with our observations in HeLa LC3B-KO cells (Figure 3F, 3G and S3E), and in Stk4−/−;Stk3−/+ MEFs6, indicating that LC3B phosphorylation regulates autophagosome transport in mammalian axons to promote the association between autophagosome and degradative lysosomes.
DISCUSSION
In this study, we demonstrated that STK4-mediated phosphorylation of the autophagy protein LC3B is critical for regulating its interaction with the adaptor protein FYCO1 and for the regulation of directional autophagosomal transport and autophagosome-lysosome association in mammalian cells. We also showed that nutrient deprivation activates the STK4–LC3B–FYCO1 regulatory axis. These findings expand our fundamental understanding of the molecular regulation of autophagy with implications for numerous autophagy-dependent processes25.
STK4-mediated phosphorylation of LC3B at T50 reduced binding of FYCO1 to LC3B, consistent with a previous report26, as well as FYCO1 localization to autophagosomes3,4. This post-translational regulatory mechanism appeared specific for FYCO1, as the binding/association of KEAP1, p62, or NDP52 to LC3B was not affected in the same differential manner via LC3B phosphorylation. Compatible with the role of FYCO1 in anterograde transport of intracellular vesicles including autophagosomes3,4,15, we showed that lack of LC3B-T50 phosphorylation resulted in enhanced anterograde transport and aberrant positioning to the cell periphery of LC3B-positive vesicles in primary neurons as well as in HeLa LC3B-KO cells. Consequently, our data showed a decreased association of autophagosomes with lysosomes, which ultimately may explain the defects in autophagosome-lysosome fusion and the autophagy block observed in LC3B phosphorylation-deficient cells6. Based on these results, we speculate that in the absence of phosphorylation at T50, a switch occurs from a retrograde-moving LC3B complex to a LC3B-FYCO1-kinesin anterograde-moving one. Previous work has shown that retrograde autophagosome movement depends on the direct interaction between LC3B and JIP1, a scaffolding protein that recruits the dynein activator dynactin to these vesicles to drive their movement toward the soma of mammalian neurons5,27. Thus, we posit that dephosphorylation on T50 may mediate a shift from an LC3B-JIP1-dynein to an LC3B-FYCO1-kinesin complex to promote the anterograde movement of autophagosomes, impairing the retrograde movement and their fusion with lysosomes. In turn, STK4-mediated phosphorylation of LC3B could induce FYCO1 dissociation from mature autophagosomes to allow their retrograde JIP1-dependent transport to ensure lysosomal fusion and cargo degradation. This, as well as whether LC3B phosphorylation could similarly affect the binding of JIP1 or other retrograde transport-promoting adaptors remains to be investigated.
While our studies show that phosphorylation of LC3B was critical for driving the proper retrograde transport in LC3B T50A particles, T50E-positive vesicles revealed an unusually high frequency of directional switches in primary neurons and in HeLa LC3B-KO cells. T50E phospho-mutation has been suggested to affect LC3B conjugation to lipids26, which could indirectly affect LC3B-mediated recruitment of transport proteins to autophagosomes and ultimately the transport of these vesicles. However, in our experimental system, the T50E phospho-mutant showed proficient lipidation (Figure S1E, see figure legend for further discussion), suggesting that the observed transport phenotypes may instead be caused by an altered regulation of the transport machinery. Additional experiments will be critical to explain the directional switches produced by LC3B-T50E, as well as to identify potential LC3B-T50 phosphatase(s) and other ATG8 family members involved in autophagosomal trafficking and recruitment of essential transport machinery.
We observed that starvation-induced perinuclear positioning of autophagosomes was dependent on STK4-mediated LC3B phosphorylation. We propose that the autophagosome repositioning process is initiated by starvation-induced activation of STK4, followed by LC3B phosphorylation, dissociation of FYCO1, and inhibition of anterograde transport and/or promotion of retrograde transport. In support of this model, anterograde transport of autophagosomes has been shown to decrease in HeLa cells during starvation28. We note that while the regulatory consequences of the STK4-LC3B-FYCO1 axis are evident under nutrient starvation, this axis may operate both under nutrient-rich and nutrient-deprived conditions to ultimately facilitate the retrograde transport of autophagosomes and fusion with lysosomes, as well as preventing a premature retrograde transport of autophagosomes, as explained above. Nutrient deprivation may further boost this pathway to deal with the increased autophagosome population and support the increased autophagy flux triggered under these conditions. Activation of STK4 in low nutrient conditions may concomitantly reduce cell proliferation29 and increase autophagy in an effort to re-establish homeostasis and restore nutrient availability. To this end, the nutrient-sensing complex mTORC2 has recently been identified as a direct inhibitor of STK4 activity30 and it will be appealing to investigate its potential role as an upstream regulator of the STK4–LC3B–FYCO1 axis. Indeed, it will be of interest to explore the role of this STK4–LC3B–FYCO1 regulatory axis in different biological processes, including in the decline in autophagy observed during aging and in many disease states10,31,32, where deregulation of autophagosome transport could be a crucial factor.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Malene Hansen (mhansen@sbpdiscovery.org).
Materials availability
Plasmids and cell lines generated in this study are available upon request to the Lead Contact.
Data and code availability
The published article includes all datasets generated in this study (See Data S1 for LC3B interactors proteomics data). The KymoAnalyzer software used to quantitively analyze the intracellular transport of vesicles in neurons is an open-source ImageJ-based package that can be downloaded from the Encalada lab website: http://www.encalada.scripps.edu/kymoanalyzer.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mammalian Cell lines
See Key Resources Table for details of all cells and reagents employed. HEK 293T cells, HeLa cells (both human female origin), and N2A neuroblastoma cells (mouse male origin) were purchased from ATCC. Immortalized wild-type (WT) and STK4-deficient mouse embryonic fibroblasts (MEFs) were produced as described6. Cell lines were cultured at 37°C in DMEM (Corning) supplemented with 10% fetal bovine serum (FBS, Gibco), referred to as “normal medium”, and were routinely checked for mycoplasma using the MycoScope PCR detection kit according to the manufacturer’s instructions (GenLantis).
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-FYCO1 | Atlas Antibodies | HPA035526 |
| Rabbit monoclonal anti-LC3B (D11) XP | Cell Signaling Technologies (CST) | 3868S |
| Rabbit polyclonal anti-MST1 | GeneTex | GTX109294 |
| Rabbit polyclonal anti-LAMP1 | Abcam | ab24170 |
| Rabbit polyclonal anti-p-MST1(Thr183)/MST2(Thr180) | CST | 3681S |
| Rabbit polyclonal anti pLC3B (Thr50) | 21st Century Biochemicals | 6 |
| Mouse monoclonal anti-mCherry (1C51) | Novus Biologicals | NBP1-96752 |
| Mouse monoclonal anti-β-actin (C4) | Millipore Sigma | MAB1501 |
| Mouse monoclonal anti-HA (6E2) | CST | 2367S |
| Mouse monoclonal anti-α-tubulin (DM1A) | CST | 3873P |
| Rabbit polyclonal anti-LAMP2 | Thermo Fisher | PA1-655 |
| Rabbit polyclonal anti-KEAP1 | Proteintech | 10503-2-AP |
| Rabbit polyclonal anti-NDP52 | GeneTex | GTX115378 |
| Mouse monoclonal anti-GFP | DHSB | DHSB-GFP-12A6 |
| Goat polyclonal anti-Mouse cathepsin B | R&D systems | AF965 |
| Goat anti-Rabbit HRP | CST | 7074s |
| Goat anti-Mouse HRP | CST | 7076s |
| Goat anti-Rabbit Alexa Fluor 488 | Thermo Fisher | A11034 |
| Goat anti-Rabbit Alexa Fluor 568 | Thermo Fisher | A11011 |
| Goat anti-Mouse Alexa Fluor 488 | Thermo Fisher | A11001 |
| Goat anti-Mouse Alexa Fluor 568 | Thermo Fisher | A11004 |
| Bacterial and Virus Strains | ||
| DH5α competent E. coli | Thermo Fisher | 18265017 |
| One Shot Stbl3 E. coli | Thermo Fisher | C7373-03 |
| Rosetta™ 2(DE3)pLysS E. coli | Millipore Sigma | 71403 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dulbecco’s Modified Eagle Medium (DMEM) | Corning | 10-013-CV |
| Fetal bovine serum (FBS) | Thermo Fisher | 26140049 |
| DMEM (for primary neurons) | Thermo Fisher | 11965 |
| FBS (for primary neurons) | Thermo Fisher | 10082147 |
| Poly-l-lysine | Millipore Sigma | P5899 |
| Neurobasal-A medium | Thermo Fisher | 10888022 |
| B-27 | Thermo Fisher | 17504044 |
| GlutaMAX | Thermo Fisher | 35050061 |
| Earle’s Balanced Salt Solution (EBSS): starvation medium | Thermo Fisher | 24010043 |
| Lipofectamine 2000 | Thermo Fisher | 11668019 |
| Opti-MEM | Thermo Fisher | 31985-070 |
| Bafilomycin A1 | Millipore Sigma | 88899-55-2 |
| DAPI | Millipore Sigma | D9542 |
| RNAiMAX | Thermo Fisher | 13778150 |
| Saponin, from Quillaja pract. | Acros Organics | 74499-23-3 |
| N3500 Nonidet-P40 substitute | US Biological | 9036-19-5 |
| Bovine serum albumin (BSA), protease free | Akron | AK1391-0100 |
| cOmplete Protease Inhibitor Cocktail tablets | Millipore Sigma | 11697498001 |
| PhosSTOP | Millipore Sigma | 4906845001 |
| Pierce ECL Western Blotting Substrate | Thermo Fisher | 32209 |
| SuperSignal™ West Pico PLUS Chemiluminescent Substrate | Thermo Fisher | 34577 |
| SuperSignal™ West Femto Maximum Sensitivity Substrate | Thermo Fisher | 34095 |
| Prolong® Gold Antifade Reagent | CST | 9071 |
| FYCO1 peptide (1265–1298 amino acids of the human protein) | Biomatik | N/A |
| GST-LC3B | Viva Bioscience | V32470-0500 |
| His-STK4 | Millipore Sigma | 14-624 |
| Magic Red Cathepsin B assay | Immunochemistry | 937 |
| Critical Commercial Assays | ||
| Q5® Hot Start High-Fidelity 2X Master Mix | New England Biolabs | M0494S |
| Gibson Assembly® Master Mix | New England Biolabs | E2611L |
| MycoScope PCR Mycoplasma Detection Kit | Genlantis | MY01100 |
| Experimental Models: Cell Lines | ||
| Human: HEK 293T cells | ATCC | CRL-3216 |
| Human: HeLa cells | ATCC | CCL-2 |
| Human: HeLa cells lacking endogenous LC3B | This paper | N/A |
| Mouse: Immortalized MEFs | 6 | N/A |
| Mouse: Immortalized Stk4−/−;Stk3+/− MEFs | 6 | N/A |
| Mouse: Primary hippocampal neurons | Encalada lab, see mice details below | N/A |
| Mouse: Neuro2a (N2A) neuroblastoma cells | ATCC | CCL-131 |
| Experimental Models: Mice | ||
| C57BL/6 | Charles River Laboratories | 027 |
| Oligonucleotides | ||
| ON-TARGETplus Control Pool: Non-Targeting pool | Dharmacon | D-001810-10-05 |
| ON-TARGETplus FYCO1 siRNA | Dharmacon | LQ-014350-01-0002 |
| ON-TARGETplus SMARTpool: Human STK4 | Dharmacon | L-004157-00-0005 |
| See Table S1 for additional oligonucleotides | ||
| Recombinant DNA | ||
| Plasmid: pHA-tag | This paper | N/A |
| Plasmid: pHA-LC3B | This paper | N/A |
| Plasmid: pHA-LC3B-T50A | This paper | N/A |
| Plasmid: pHA-LC3B-T50E | This paper | N/A |
| Plasmid: pHA-LC3B-G120A | This paper | N/A |
| Plasmid: pHA-LC3B-T50A-G120A | This paper | N/A |
| Plasmid: pHA-LC3B-T50E-G120A | This paper | N/A |
| Plasmid: pHA-STK4 | This paper | N/A |
| Plasmid: pEGFP-LC3B | This paper | N/A |
| Plasmid: pEGFP-LC3B-T50A | 6 | N/A |
| Plasmid: pEGFP-LC3B-T50E | 6 | N/A |
| Plasmid: pBABE-mCherry-FYCO1 | Univ. of Dundee | DU45204 |
| Plasmid: pSpCas9(bb)-2A-GFP | Addgene | 48138 |
| Plasmid: pSpCas9(bb)-2A-GFP-LC3B Human A | This paper | N/A |
| Plasmid: pSpCas9(bb)-2A-GFP-LC3B Human B1 | This paper | N/A |
| Plasmid: pSpCas9(bb)-2A-GFP-LC3B Human B2 | This paper | N/A |
| pLKO.1-puro non-targeting (scramble) shRNA Control | Millipore Sigma | SHC016-1EA |
| pLKO.1-puro FYCO1 shRNA 1 | Millipore Sigma | TRCN0000184601 |
| pLKO.1-puro FYCO1 shRNA 2 | Millipore Sigma | TRCN0000183567 |
| pLKO.1-puro FYCO1 shRNA 3 | Millipore Sigma | TRCN0000179990 |
| pLKO.1-puro FYCO1 shRNA 4 | Millipore Sigma | TRCN0000178746 |
| pLKO.1-puro FYCO1 shRNA 5 | Millipore Sigma | TRCN0000183006 |
| Software and Algorithms | ||
| SAINTq software | SourceForge | http://saint-apms.sourceforge.net/Main.html |
| In house R script (ver. 3.5.1, 64-bit) | SBP: Proteomics | https://www.r-project.org/ |
| Octet software 10.0.1 | FortéBio | https://www.sartorius.com/en/products/protein-analysis/octet-systems-software |
| PyMOL 2.3 | Schrödinger | https://pymol.org/2/ |
| Imaris 9.3.0 | Bitplane | https://imaris.oxinst.com/ |
| ImageJ | NIH | https://imagej.net/Fiji |
| KymoAnalyzer | Encalada Lab | http://www.encalada.scripps.edu/kymoanalyzer |
| Zen lite processing software | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen-lite.html |
| Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Microsoft Office: Excel, PowerPoint, Word | Microsoft | https://www.microsoft.com/en-us/microsoft-365 |
| RStudio | RStudio Inc | https://www.rstudio.com/I |
| Image Lab | Thermo Fisher | https://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
| Other Reagents | ||
| Pierce Anti-HA magnetic beads | Thermo Fisher | 88836 |
| Anti-RFP magnetic beads | MBL Life Science | M165-11 |
| 1–1/2 Micro glass coverslips – 12 mm diameter | Electron Microscopy Sciences | 72230-01 |
| Premium microscope slides (Superfrost) | Thermo Fisher | 12-544-7 |
| Spin-X centrifuge tube filter 0.45 μm cellulose acetate | Corning Inc. | 8162 |
| NuPAGE 4–12% Bis-Tris gels 1 mm × 10 wells | Thermo Fisher | NP0321 BOX |
| Premium autoradiography film (5 × 7 in.) | Denville Scientific | E3012 |
| Immobilon-P membrane, PVDF, 0.45 μm, 8.5 cm × 10 m roll | Millipore Sigma | IPVH85R |
Mouse Primary hippocampal neurons
C57BL/6 wild-type mice used to isolate primary hippocampal neurons were originally obtained from Charles River Laboratories. Mice colonies were maintained according to the guidelines recommended by the Department of Animal Resources (DAR) at Scripps Research, which included free access to food and water, with a 12h light-dark cycle. All mouse protocols were reviewed and approved by Institutional Animal Care and Use Committee (IACUC) at The Scripps Research Institute (Scripps Research). Mouse hippocampal neurons were isolated from newborn mice and cultured as described33. Briefly, hippocampi were dissected from 1- or 2-day-old mice of either sex, treated with papain (Worthington) for 15 min, and disrupted by aspirating through a micropipette tip 7 to 10 times. Dissociated neurons were resuspended in normal medium and plated in 24-well plates containing 12-mm glass coverslips pretreated with 50 μg/ml poly-L-lysine (Sigma) in borate buffer at 37°C. The medium was exchanged for Neurobasal-A medium (Gibco) containing 2% B-27 (Gibco) and 0.25% GlutaMAX (Gibco) 1 h after plating.
METHODS DETAILS
Plasmids and oligonucleotides
Plasmids encoding EGFP-tagged WT LC3B and phospho-mutants (T50A and T50E) were obtained as described6. Plasmids encoding HA-tagged LC3B proteins were generated by replacing the EGFP sequence in the EGFP plasmids with an HA-tag followed by a Tobacco etch virus protease sequence recognition site and a Flag-Tag (Genewiz) using AgeI and HindIII (New England Biolabs). Lipidation-deficient LC3B proteins were generated by mutagenic PCR using Q5 Hot Start High-Fidelity Master Mix (New England Biolabs) to introduce the GGG to GCT change that leads to a glycine to alanine (G120A) mutation. Bacterial expression plasmids encoding His-tagged WT, T50A, and T50E LC3B proteins were produced as described6. A plasmid encoding mCherry-tagged FYCO1 was generated by cloning the mCherry-FYCO1 gene from a pBABE-puro-mCherry-FYCO1 plasmid (Dr. Dario Alessi, University of Dundee) into the plasmid backbone of EGFP-LC3B (Addgene) by amplification of fragments using Q5 Hot Start High-Fidelity Master Mix and ligation using a Gibson Assembly Master Mix (New England Biolabs). pLKO.1-puro plasmids encoding scrambled shRNA and five FYCO1-targeted shRNAs were purchased from Sigma Aldrich. For details on oligonucleotides refer to Table S1.
Generation of HeLa LC3B Knockout Cells
LC3B gene knockout (KO) was performed using the CRISPR-CAS9 system according to a published protocol34. Briefly, HeLa cells were transfected with two plasmids encoding GFP-CAS9 (Addgene) and an sgRNA targeting the first or the fourth exon of the LC3B gene. After 24 h, GFP-positive cells were sorted by FACS and single cells were placed in 96-well plates. Clones were grown for 2–3 weeks and analyzed for gene deletion by PCR. Gene-edited clones were expanded in 24-well plates, and LC3B protein deficiency was confirmed by western blotting and immunofluorescence microscopy (See Figure S1) LC3B-KO cells were selected, expanded, and frozen before use in experiments.
Gene Silencing, Transient Transfection, and Cell Treatment
Cells were reverse transfected with 10 nM of non-silencing control or specific gene-targeting siRNAs (siNS, siFYCO1, or siSTK4; Dharmacon) using Opti-MEM and lipofectamine RNAiMAX according to the supplier’s recommendations (Life Technologies). After 48 h, cells were collected and used for experiments. Transient plasmid transfections were performed using Opti-MEM and Lipofectamine 2000 (Life Technologies), and cells were collected for experiments 24 h after transfection. For experiments with cells expressing siRNAs and plasmids, cells were transfected with siRNAs for 24 h and then transfected with the plasmid of interest for an additional 24 h before analysis.
For autophagy flux assays, cells were washed three times with normal medium, normal medium containing the late-autophagy blocking compound bafilomycin A1, (BafA, Sigma) at 50 nM, or starvation medium (Earle’s Balanced Salt Solution (Life Technologies) before incubation in the same media for the indicated times at 37°C.
Affinity purification
HeLa LC3B-KO cells, 293T cells, MEFs, or N2A cells were transiently transfected with plasmids (6.6 μg/106 cells) encoding HA-tag, HA-LC3B WT, HA-T50A or HA-T50E for 24 h, incubated with normal medium or starvation medium for 1 h, and then lysed with IP lysis buffer (0.5% NP-40, 150 mM NaCl, 50 mM Tris HCl, pH 7.5, protease inhibitors [cOmplete, Roche], and phosphatase inhibitors [PhosStop, Roche]). Cell lysates were clarified by serial centrifugation (600 ×g for 3 min and 9300 ×g for 10 min) and diluted 1.25-fold with lysis buffer. An aliquot of the lysate (5%) was reserved as input for western blotting and the remaining lysate was incubated with anti-HA magnetic beads (Pierce) overnight at 4°C. Beads were washed seven times with wash buffer (0.1% NP-40, 150 mM NaCl, 50 mM Tris HCl, pH 7.5) and three times with 50 mM Tris HCl, pH 7.5. Samples were then processed for mass spectrometry or western blot analysis as described below. For immunoprecipitation of mCherry-FYCO1, the same protocol was performed using anti-RFP magnetic beads (MBL Life Science).
Mass Spectrometry
Mass spectrometry was performed at the Proteomics Core of Sanford Burnham Prebys Medical Discovery Institute. HA-tag (negative control), HA-LC3B WT, HA-T50A and HA-T50E proteins were immunoprecipitated (three experimental replicates: R1, R2 and R3) as described above and subjected to on-bead digestion as previously described35. The total peptide concentration was determined using a NanoDrop spectrophotometer (Thermo Fisher) and the samples were then analyzed by LC-MS/MS using a Proxeon EASY nanoLC system (Thermo Fisher Scientific) coupled to a Q-Exactive Plus mass spectrometer (Thermo Fisher Scientific). Peptides were separated using an analytical C18 Acclaim PepMap column (75 μm × 250 mm, 2 μm vesicles; Thermo Scientific) and a 180-min gradient at a flow rate of 300 μl/min: 1% to 5% B in 1 min, 5% to 20% B in 139 min, 20% to 35% B in 30 min, and 35% to 45% B in 10 min (A = formic acid 0.1%; B = 80% acetonitrile + 0.1% formic acid). Mass spectrometry settings were as described previously 35. Mass spectra were analyzed with MaxQuant software version 1.5.5.1. Peptides were searched against the Homo sapiens Uniprot protein sequence database (downloaded July 2018) and GPM cRAP sequences (commonly known protein contaminants), as described35. Statistical analysis for the identified peptides was performed using an in-house R script (version 3.5.1) and SAINTq. Identified LC3B protein interactors and corresponding intensity, as well as average normalized intensity ratios interactor/LC3B are shown in Data S1 (Excel sheets show raw (A) and normalized data (B), respectively). Benjamini-Hochberg correction with 10% false discovery rate, >1.5 fold was applied to identify protein interactors binding either more or less to LC3B phospho-mutants. NDP52 was not identified in this mass spectrometry analysis.
Western blot
Reserved input lysate (20 μg of total protein) or immunoprecipitated materials from affinity purifications were analyzed by standard western blotting protocols. Briefly, proteins were separated in Novex 4–12% acrylamide Bis-Tris gels (NuPage) and transferred to PVDF membranes. Membranes were blocked in Tris-buffered saline containing 0.05% Tween-20 (TBST) and either 5% milk or 3% bovine serum albumin (BSA), according to the antibody manufacturer’s recommendations, and then incubated for 2 h at room temperature with primary antibodies diluted in 1% milk or 3% BSA in TBST with gentle rocking. Blots were washed three times (total 30 min) and incubated with horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit secondary antibodies (Cell Signaling Technologies) for 1 h at room temperature with gentle rocking. Blots were washed again and developed with Pierce ECL Western Blotting substrate (Base, SuperSignal West Pico/Femto, depending on protein levels) and visualized using a Bio-Rad ChemiDoc Imaging System or with HyBlot CL autoradiography films (Denville). Quantification of band intensity was carried out using ImageJ (National Institutes of Health) software. Details of western blot quantification are shown in Data S1 sheet C.
In Vitro Phosphorylation of LC3B and Co-immunoprecipitation of FYCO1
Aliquots of 3 μg of GST-LC3B (Viva Bioscience) were incubated alone or with 250 ng of His-STK4 (Millipore) in in vitro kinase reaction buffer (10 mM MgCl2, 1 mM EGTA, 1 mM dithiothreitol, 0.5 μm ATP, 20 mM Tris, pH 7.5) for 1 h at 30°C. The in vitro kinase reaction mixtures were incubated with 350 μl clarified lysate of HeLa LC3B-KO cells (8×105 cell equivalents, prepared as described above) and glutathione-sepharose beads (Bioworld) for 3 h at 4°C on a rotator. As a negative control, the same procedure was performed with a mock in vitro reaction buffer lacking GST-LC3B and His-STK4. Sepharose beads were washed seven times with wash buffer (0.1% NP-40, 150 mM NaCl, 50 mM Tris HCl, pH 7.5) and eluted by heating in Laemmli sample buffer at 95°C for 10 min. Samples were passed through a 0.45 μm cellulose acetate filter (Costar) and then subjected to western blot analysis.
Biolayer Interferometry
Cultures (500 ml) of Rosetta™ 2(DE3) pLysS E. coli (Millipore) expressing human LC3B-WT, T50A, or T50E6 were grown to an optical density of 0.4–0.6 at 600 nm, and recombinant protein expression was induced by addition of 0.5 mM isopropyl-β-D-thiogalactopyranoside (VWR) for 16–18 h at 25°C. Bacteria were harvested by centrifugation and lysed with a microfluidizer in 50 mM Tris-HCl (pH 8.0), 300 mM NaCl, 30 mM imidazole, and 2 mM β-mercaptoethanol (BME). The lysates were clarified by centrifugation, filtered, incubated with Ni-NTA beads for 1 h, and eluted in the same buffer containing 250 mM imidazole. The sample was then dialyzed overnight in Snakeskin dialysis tubing (3500 kDa pore size) in 50 mM Tris-HCl (pH 8.5), 100 mM NaCl, and 5 mM BME. The protein was concentrated using Amicon-3.5K filters and subjected to size exclusion chromatography on a Superdex 200 column (GE Healthcare). The concentration of eluted proteins was measured at 280 nm using a Nanodrop spectrophotometer and the proteins were verified by Coomassie Blue staining and western blotting. Purified His- LC3B-WT, T50A, or T50E proteins were diluted to 3 μg/ml in Dulbecco’s phosphate-buffered saline (PBS; Gibco) containing 0.1% BSA and 0.02% Tween-20 and immobilized on NTA capture sensors (FortéBio). Human FYCO1 peptide (amino acids 1265–1298, containing the LIR and adjacent residues)4 was synthesized by Biomatik and reconstituted at 2 mg/ml in DMSO. The binding reaction was performed using two-fold serial dilutions of FYCO1 peptide (0.5–20 μM). The association and dissociation phases were analyzed for 60 s and the curves were inverted to fit them to a 1:1 model. Affinity constants were calculated from the kinetic constant after normalization for the buffer control. All binding measurements were performed on an Octet Red instrument (FortéBio) and the results were processed using Octet software 10.0.1 (FortéBio).
Structural Representation of the LC3B–FYCO1-LIR Complex
The crystal structure of a Mus musculus complex between LC3B and FYCO1-LIR peptide (PDB 5WRD)36 was represented in cartoon form using Pymol software. Distance measurements between T50 in LC3B and D1235 and D1236 adjacent to FYCO1-LIR (homologous to amino acids D1276 and 1277 in Homo sapiens FYCO1) were performed with Coot (Crystallographic Object-Oriented Toolkit) software and were found to be consistent with hydrogen bonding distances. The Mus musculus FYCO1-LC3B complex structure was chosen for representation because coverage of the FYCO1 peptide sequence was greater than in the human structure.
Fluorescence and Confocal Microscopy
Cells (2×105) were seeded in 24-well plates containing 12-mm glass coverslips, transiently transfected with the appropriate plasmids, and treated with normal or starvation medium as described above. After treatment, cells were fixed with 4% paraformaldehyde in PBS for 20 min and processed for fluorescence microscopy using standard protocols (https://www.cellsignal.com). Briefly, cells were incubated with primary antibodies against FYCO1 or autophagosomal and lysosomal proteins (see Key Resources table) diluted in PBS containing 10% FBS and 0.2% saponin for 2 h at room temperature. Cells were then washed three times with PBS and incubated with Alexa Fluor 488- or 568-labeled secondary antibodies (Thermo Fisher) diluted at 1:1000 in PBS/10% FBS/0.2% saponin for 1 h at room temperature. Cells were washed three times in PBS, incubated with 4’,6-diamidino-2-phenylindole (DAPI) to label nuclei, and mounted with Prolong® Gold Antifade Reagent. Imaging was performed using a Zeiss LSM 710 NLO confocal microscope. For each condition, 15 to 20 images were acquired in randomly selected fields with a PlanApchromat 63X/1.4 DIC oil objective lens. An average of 10 to 15 stacks of 0.3 μm thickness were acquired.
Live-Cell Imaging in HeLa Cells
HeLa LC3B-KO cells (2×105) were seeded in 35-mm glass-bottom dishes (MatTek) for 24 h and transfected with GFP-tagged LC3B expression plasmids as described above. The medium was then replaced and the cells were imaged 2 h later with a Zeiss LSM 880 Rear Port Laser Scanning Confocal and Airyscan FAST microscope equipped with a stage pre-heated to 37°C. Approximately 9 to 10 cells per condition were randomly selected for imaging using a PlanApchromat 63X/1.4 DIC oil objective lens with single photon lasers emitting at 488 nm. Each cell was imaged for 3 min (360 frames, 2 frames/s) and, on average, 8–10 slices were acquired through a total cell thickness of ~2.0–2.5 μm to ensure the complete punctum was captured for particle tracking in four dimensions.
Live-cell imaging in Mouse Hippocampal Neurons
Hippocampal neurons were plated on 12-mm glass coverslips in 24-well plates, cultured for 8 to days, and then transfected using 2 μL of Lipofectamine 2000 (Life Technologies) and 1.6 μg of DNA per well with the GFP-LC3B expression plasmids with or without scrambled shRNA or a mixture of five FYCO1-targeting shRNAs (Sigma Aldrich). The efficiency of shRNAs targeting Fyco1 in mouse cells were verified in N2A neuroblastoma cells (data not shown), a proxy for primary neurons in which LC3B phospho-mutants displayed autophagy phenotypes similar to other cell types (Figure S4E). At 48 h post-transfection, the mid-axon region of neurons was imaged using a Nikon Ti-E Perfect Focus inverted microscope equipped with a total internal reflection fluorescence (TIRF) setup, an Andor iXon + DU897 EM camera, and a 100X/1.49 NA objective. A 488 nm laser was used for detection of GFP. Lasers were positioned at an angle for pseudo-TIRF acquisition. Each neuron was imaged for 5 min (300 frames, 1 frame/s). Axons were distinguished from dendrites by morphology33, and the polarity of axons was determined by identification of soma and axonal termini for each movie.
QUANTIFICATION AND STATISTICAL ANALYSIS
Vesicle number and positioning quantification in HeLa cells
LC3B-positive autophagosomes were quantified using Imaris 9.3.0 (Bitplane, Zurich). Confocal images of individual cells were processed using the software “spots” feature with an estimated diameter of 0.5 μm and then corrected using the “different spot sizes: region growing” feature. Thresholds were set using the “Quality” feature and determined manually. The local contrast threshold for region-growing spots was then manually set and maintained. Puncta were counted manually in 2–3 cells per condition and compared with the Imaris software counts to verify the accuracy and precision of measurements. The “surface” feature was used to reconstruct the nucleus of each cell using DAPI staining. Following generation of spots and surface objects, distance transformation was performed between the outer nuclear surface and the LC3B-positive puncta. Distances for individual puncta were exported to Excel (Microsoft) and processed. The distance from the nucleus of each punctum were averaged for each cell and the collective average of all puncta from all cells were represented as a single data point. Additionally, all distances for each condition were pooled and represented as relative frequency distributions with a best-fit line (non-linear regression, Gaussian curve) for interpretation. The same procedures were performed for LAMP-1 (endolysosomal marker; estimated punctum diameter = 0.33 μm).
Colocalization quantification
Colocalization data for LC3B protein interactors and LC3B were processed using Imaris software. LC3B interactor- or LC3B-labeled channels were provided appropriate colocalization thresholds in the software “coloc” feature using the “Polygon” tool. The same channel thresholds were maintained for all cells, and the most intense fluorescent overlap was quantified by Imaris as voxels (i.e., overlap between volumetric pixels in either channel) as described37. The voxel value for each cell was averaged for each condition and used as a single data point for statistical analysis. Colocalization for GFP-LC3B and Cathepsin B in primary neuronal soma was done in Coloc 2 Image J plugin.
Particle tracking quantification in HeLa Cells
Following raw image acquisition as described in the methods section, a maximum Z-projection of the movies taken from HeLa LC3B KO cells expressing GFP-LC3B proteins was generated using Zen Processing software. The movie files were further processed prior to particle tracking using Imaris image processing functions. The background subtraction function was set at 0.33 pm and a Gaussian-smoothing operation was used to remove the cytosolic GFP signal and to sharpen the visible puncta to improve particle tracking. GFP-positive puncta were marked using the spots feature of Imaris, with an estimated diameter of 0.5 μm for an individual punctum. An appropriate threshold was set to label all puncta visualized and was maintained for all movies analyzed. For particle tracking, the autoregressive motion algorithm was selected with 5–10 μm of maximum “predicted distance” and a maximum “gap distance” between 2 and 4 μm. Initial tracks were generated by Imaris and then manually curated and corrected to ensure each particle was assigned to the appropriate track for the duration of the movie. A reference frame was added over the nucleus and the general tracking statistics were exported to Excel for full processing. The final displacement relative to the reference frame labeled nucleus was subtracted from the initial displacement and each individual track was designated either “anterograde” or “retrograde.” The percentage of anterograde or retrograde particles was pooled for each cell and the mean was taken as a single data point for statistical analysis. To quantify directional switches in particle movement, the complete track and trajectories showed by every single particle were displayed and directional switches were arbitrarily determined and quantified by two independent people using visual inspection of individual tracks.
Particle tracking quantification in primary neurons
Axonal transport analysis was performed using KymoAnalyzer, a freely available ImageJ-based macro21. Briefly, kymographs were generated from time-lapse movies and particle trajectories were manually assigned from the kymograph images. Transport parameters, including direction, velocity, run length, time in motion, switches, and pauses, were automatically calculated by KymoAnalyzer. A detailed description of all the transport parameters used in this study can be found in21. Flux is measured by assigning a “flux analysis gate” in the middle of the kymographs to count the particles that cross the gate over the entire timespan of the kymograph and calculate the rate of movement of particles through the assigned gate as a function of time. Co-transport experiments were done by near-simultaneous two-color imaging. Red-green images were collected with exposure time of 75 ms with a 50 ms delay between them using Nikon’s proprietary LAMBDA 10–3 optical filter switch, for 30 sec long and at 5 frames/sec (5 Hz) and 5 min (300 sec) long at 1 frame/sec (1 Hz). Co-transport quantitation was done in ImageJ by creating individual and merged color kymographs to evaluate the degree of overlap between two channels.
Statistical analysis
Figure legends contain all information regarding statistical tests used, number of biological replicates, dispersion measures used, and specific p values. Statistical analysis and graph generation was conducted using Prism 8.0 software (GraphPad). One-way and two-way ANOVA were performed to analyze single-grouped datasets (Figures 1B, 1F, 2B, S1D, S4B and S4C) and double-grouped datasets (Figures 2D, 3C, 3E, 3G, 3I, S1C, and S3E), respectively. For all data sets, Tukey’s comparison test with a single pooled variance and Geisser–Greenhouse correction was performed. P values are summarized using the GraphPad reporting method: 0.1234 (ns), 0.0332 (*), 0.0021 (**), 0.0002 (***), and <0.0001 (****). Student’s t-test was used in figures S2D, and S4J 0.05 (*), and 0.01 (**). For the analysis of LC3B transport in primary hippocampal neurons, most axonal transport parameters did not follow a normal distribution. One-way ANOVA and Šidák’s multiple comparison correction was used for Figures 4C, 4D, 4E, 4F, and S4G. For figures 4H, and S4F the non-parametric permutation t test (rndttest function of MATLAB [Mathworks] was used. For Figures 4G and S4D, parameter differences were analyzed using both the Rank Sum test (MATLAB [Mathworks]) and the Kolmogorov–Smirnov test (RStudio) adjusting p-values for multiple comparisons using Bonferroni.
Supplementary Material
Data S1. Protein interactors of LC3B phospho-mutants and immunoblot quantifications. Related to STAR Methods and Figures 1 and 3. (A and B) show the identified LC3B protein interactors and corresponding intensity, as well as average normalized intensity ratios interactor/LC3B are shown (Excel sheets show raw and normalized data, respectively). Benjamini-Hochberg correction with 10% false discovery rate, >1.5 fold was applied to identify protein interactors binding either more (highlighted in green) or less (highlighted in red) compared to LC3B phospho-mutants. FYCO1, p62 and KEAP1 results are shown in bold letters. NDP52 was not identified in this mass spectrometry analysis. (C) shows the quantification of the replicates for the immunoblots (IB) presented in this study. IB quantifications were performed using ImageJ. IB replicates 1 (values within blue cells) are displayed in the manuscript.
Video S1. Directional transport of LC3B-WT, T50A, and T50E vesicles in HeLa LC3B-KO cells. Related to Figure 4.
Video S2. Directional transport of LC3B-WT, T50A, and T50E vesicles in mouse primary neurons. Related to Figure 4.
HIGHLIGHTS.
STK4-mediated LC3B phosphorylation lowers LC3B binding to the transport protein FYCO1
LC3B phosphorylation inhibition decreases retrograde transport of autophagosomes
Block of LC3B phosphorylation also compromises autophagosome-lysosome association
The STK4-LC3B-FYCO1 axis is a nutrient-sensitive, autophagy-regulatory pathway
ACKNOWLEDGEMENTS
We thank Lee R. Lee for assistance with the affinity-purification experiments, Dr. Caroline Kumsta for help with data analysis and figure preparation, members of the Hansen and Encalada labs for helpful discussions and/or critical reading of the manuscript, Drs. Tony Hunter and Jill Meisenhelder (Salk Institute) for helpful discussions, Dr. Alex Rosa Campos (Sanford Burnham Prebys Proteomics Core) for support with the mass-spectrometry analysis, Leslie Boyd (Sanford Burnham Prebys Cell Imaging Core) and Salk Institute Biophotonics Core personnel for help with image acquisition and analysis, Dr. Erica Ollmann-Saphire (La Jolla Institute for Immunology) for help with the binding-affinity assays, and Dr. Dario Alessi (University of Dundee) for providing pBABE-mCherry-FYCO1 plasmid. Graphical abstract and part of Figure 1A were created using Biorender.com. We thank Nora Lyang (Hansen Lab) for help with graphical abstract design. J.N.T. was supported by a Fundacion Ramon Areces Postdoctoral Fellowship and a K99/R00 pathway to independence NIH grant (K99AG062774), R.C. was supported by the George E. Hewitt Foundation for Medical Research, and T.C. was supported by a Royal Thai Government Scholarship from the Development and Promotion of Science and Technology Talents Project (DPST). This work was funded by the following grants to S.E.E.: R01AG049483; The Glenn Foundation for Medical Research Glenn Award for Research in Biological Mechanisms of Aging; a New Scholar in Aging Award from the Lawrence Ellison Foundation; and The Baxter Family Foundation; and to M.H.: R01 GM117466.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Søreng K, Neufeld TP, and Simonsen A (2018). Membrane Trafficking in Autophagy. In International Review of Cell and Molecular Biology, pp. 1–92. [DOI] [PubMed] [Google Scholar]
- 2.Rawet Slobodkin M, and Elazar Z (2013). The Atg8 family: Multifunctional ubiquitin-like key regulators of autophagy. Essays Biochem. 55, 51–64. [DOI] [PubMed] [Google Scholar]
- 3.Pankiv S, Alemu EA, Brech A, Bruun JA, Lamark T, Overvatn A, Bjorkoy G, and Johansen T (2010). FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol 188, 253–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olsvik HL, Lamark T, Takagi K, Larsen KB, Evjen G, Øvervatn A, Mizushima T, and Johansen XT (2015). FYCO1 contains a C-terminally extended, LC3A/B-preferring LC3-interacting region (LIR) motif required for efficient maturation of autophagosomes during basal autophagy. J. Biol. Chem. 290, 29361–29374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fu MM, and Holzbaur EL (2014). Integrated regulation of motor-driven organelle transport by scaffolding proteins. Trends Cell Biol 24, 564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilkinson DS, Jariwala JS, Anderson E, Mitra K, Meisenhelder J, Chang JT, Ideker T, Hunter T, Nizet V, Dillin A, et al. (2015). Phosphorylation of LC3 by the Hippo Kinases STK3/STK4 Is Essential for Autophagy. Mol Cell 57, 55–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng X, Wang Y, Gong Y, Li F, Guo Y, Hu S, Liu J, and Pan L (2016). Structural basis of FYCO1 and MAP1LC3A interaction reveals a novel binding mode for Atg8-family proteins. Autophagy 12, 1330–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halder G, and Johnson RL (2011). Hippo signaling: growth control and beyond. Development 138, 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanida I, Ueno T, and Kominami E (2004). Human light chain 3/MAP1LC3B Is cleaved at its carboxyl-terminal Met 121 to expose Gly120 for lipidation and targeting to autophagosomal membranes. J. Biol. Chem. 279, 47704–47710. [DOI] [PubMed] [Google Scholar]
- 10.Bejarano E, Murray JW, Wang X, Pampliega O, Yin D, Patel B, Yuste A, Wolkoff AW, and Cuervo AM (2018). Defective recruitment of motor proteins to autophagic compartments contributes to autophagic failure in aging. Aging Cell 17, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galan JA, and Avruch J (2016). MST1/MST2 Protein Kinases: Regulation and Physiologic Roles. Biochemistry 55, 5507–5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plouffe SW, Meng Z, Lin KC, Lin B, Hong AW, Chun JV, and Guan KL (2016). Characterization of Hippo Pathway Components by Gene Inactivation. Mol. Cell 64, 993–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Korolchuk VI, Saiki S, Lichtenberg M, Siddiqi FH, Roberts EA, Imarisio S, Jahreiss L, Sarkar S, Futter M, Menzies FM, et al. (2011). Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 13, 453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saftig P, and Klumperman J (2009). Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 10, 623–635. [DOI] [PubMed] [Google Scholar]
- 15.Raiborg C, Wenzel EM, Pedersen NM, Olsvik H, Schink KO, Schultz SW, Vietri M, Nisi V, Bucci C, Brech A, et al. (2015). Repeated ER-endosome contacts promote endosome translocation and neurite outgrowth. Nature 520, 234–238. [DOI] [PubMed] [Google Scholar]
- 16.Jahreiss L, Menzies FM, and Rubinsztein DC (2008). The itinerary of autophagosomes: From peripheral formation to kiss-and-run fusion with lysosomes. Traffic 9, 574–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kimura S, Noda T, and Yoshimori T (2008). Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct Funct 33, 109–122. [DOI] [PubMed] [Google Scholar]
- 18.Maday S, Wallace KE, and Holzbaur ELF (2012). Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol. 196, 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maday S, and Holzbaur ELF (2014). Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev. Cell 30, 71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dong A, Kulkarni VV, and Maday S (2019). Methods for imaging autophagosome dynamics in primary neurons. In Methods in Molecular Biology, pp. 243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neumann S, Chassefeyre R, Campbell GE, and Encalada SE (2017). KymoAnalyzer: a software tool for the quantitative analysis of intracellular transport in neurons. Traffic 18, 71–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cabukusta B, and Neefjes J (2018). Mechanisms of lysosomal positioning and movement. Traffic 19, 761–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kulkarni VV, and Maday S (2018). Neuronal endosomes to lysosomes: A journey to the soma. J. Cell Biol. 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boonacker E, and Van Noorden CJF (2001). Enzyme cytochemical techniques for metabolic mapping in living cells, with special reference to proteolysis. J. Histochem. Cytochem. 49, 1473–1486. [DOI] [PubMed] [Google Scholar]
- 25.Levine B, and Kroemer G (2019). Biological Functions of Autophagy Genes: A Disease Perspective. Cell 176, 11–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shrestha BK, Rasmussen MS, Abudu YP, Bruun JA, Larsen KB, Alemu EA, Sjøttem E, Lamark T, and Johansen T (2020). NIMA-related kinase 9 –mediated phosphorylation of the microtubule-associated LC3B protein at Thr-50 suppresses selective autophagy of p62/sequestosome 1. J. Biol. Chem. 295, 1240–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fu M. meng, Nirschl JJ, and Holzbaur ELF (2014). LC3 Binding to the scaffolding protein jip1 regulates processive dynein-driven transport of autophagosomes. Dev. Cell 29, 577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Geeraert C, Ratier A, Pfisterer SG, Perdiz D, Cantaloube I, Rouault A, Pattingre S, Proikas-Cezanne T, Codogno P, and Pous C (2010). Starvation-induced hyperacetylation of tubulin is required for the stimulation of autophagy by nutrient deprivation. J. Biol. Chem. 285, 24184–24194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu FX, and Guan KL (2013). The Hippo pathway: regulators and regulations. Genes Dev 27, 355–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sciarretta S, Zhai P, Maejima Y, DelRe DP, Nagarajan N, Yee D, Liu T, Magnuson MA, Volpe M, Frati G, et al. (2015). MTORC2 regulates cardiac response to stress by inhibiting MST1. Cell Rep. 11, 125–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hansen M, Rubinsztein DC, and Walker DW (2018). Autophagy as a promoter of longevity: insights from model organisms. Nat. Rev. Mol. Cell Biol. 19, 579–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tammineni P, Ye X, Feng T, Aikal D, and Cai Q (2017). Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer’s disease neurons. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Encalada SE, Szpankowski L, Xia CH, and Goldstein LSB (2011). Stable kinesin and dynein assemblies drive the axonal transport of mammalian prion protein vesicles. Cell 144, 551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bauer DE, Canver MC, and Orkin SH (2015). Generation of genomic deletions in mammalian cell lines via CRISPR/Cas9. J. Vis. Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hutt DM, Loguercio S, Campos AR, and Balch WE (2018). A Proteomic Variant Approach (ProVarA) for Personalized Medicine of Inherited and Somatic Disease. J. Mol. Biol. 430, 2951–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sakurai S, Tomita T, Shimizu T, and Ohto U (2017). The crystal structure of mouse LC3B in complex with the FYCO1 LIR reveals the importance of the flanking region of the LIR motif. Acta Crystallogr. Sect. Struct. Biol. Commun. 73, 130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coutu DL, Kokkaliaris KD, Kunz L, and Schroeder T (2017). Three-dimensional map of nonhematopoietic bone and bone-marrow cells and molecules. Nat. Biotechnol. 35, 1202–1210. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Protein interactors of LC3B phospho-mutants and immunoblot quantifications. Related to STAR Methods and Figures 1 and 3. (A and B) show the identified LC3B protein interactors and corresponding intensity, as well as average normalized intensity ratios interactor/LC3B are shown (Excel sheets show raw and normalized data, respectively). Benjamini-Hochberg correction with 10% false discovery rate, >1.5 fold was applied to identify protein interactors binding either more (highlighted in green) or less (highlighted in red) compared to LC3B phospho-mutants. FYCO1, p62 and KEAP1 results are shown in bold letters. NDP52 was not identified in this mass spectrometry analysis. (C) shows the quantification of the replicates for the immunoblots (IB) presented in this study. IB quantifications were performed using ImageJ. IB replicates 1 (values within blue cells) are displayed in the manuscript.
Video S1. Directional transport of LC3B-WT, T50A, and T50E vesicles in HeLa LC3B-KO cells. Related to Figure 4.
Video S2. Directional transport of LC3B-WT, T50A, and T50E vesicles in mouse primary neurons. Related to Figure 4.
Data Availability Statement
The published article includes all datasets generated in this study (See Data S1 for LC3B interactors proteomics data). The KymoAnalyzer software used to quantitively analyze the intracellular transport of vesicles in neurons is an open-source ImageJ-based package that can be downloaded from the Encalada lab website: http://www.encalada.scripps.edu/kymoanalyzer.