

Abstract
This review focuses on pharmacophore approaches in researching protein interfaces that bind protein ligands. Pharmacophore descriptions of binding interfaces that employ molecular dynamics simulation can account for effects of solvation and conformational flexibility. In addition, these calculations provide an approximation to entropic considerations and as such, a better approximation of the free energy of binding. Residue-based pharmacophore approaches can facilitate a variety of drug discovery tasks such as the identification of receptor-ligand partners, identifying their binding poses, designing protein interfaces for selectivity, or defining a reduced mutational combinatorial exploration for subsequent experimental engineering techniques by orders of magnitudes.
Keywords: residue-based pharmacophore modeling, Protein-protein interactions, protein engineering
Introduction
Protein-protein interactions
Protein-protein interactions constitute the largest and most diverse type of macromolecular interactions that play role in all cellular functions, such as development, reproduction, metabolism, signal transduction or apoptosis. The number of possible protein-protein interactions in eukaryotic cells has been estimated to be between 130,000[1] and 650,000[2], or in general, every protein is assumed to interact with 3–10 other proteins [3]. Identifying and manipulating these interactions is a central goal of many studies. Novel insights into protein-protein interactions can facilitate answering various questions, such as (1) identifying the binding site on the receptor and ligand interfaces(2) identifying the cognate pose between receptor and ligand proteins (3) recognizing a cognate binding partner from a target proteome (4) engineering protein interfaces for binding and selectivity. All these goals can provide a deeper understanding of regulation of biological processes and facilitate the design of practical applications for generating new reagents and developing drugs.
Most target proteins have 3D structures available
During the last few decades there was an unprecedented progress and advance in structural biology (X-ray crystallography[4], NMR spectroscopy[5] and cryo-electron microscopy[6]), boosted by 15 years of structural genomics efforts [7,8]. In the meantime, computational structure modeling techniques showed a similar impressive advance, in particular the template based protein structure modeling approaches [9], which maturated to the point where they are able to deliver computational models with comparable accuracy to that of experimental structures, as long as an accurate sequence alignment can be obtained with a template structure [10]. As a result of these advances, most proteins today have three dimensional descriptions, either in the form of an experimentally solved structure [11] or a high quality computational model [7].
Role of pharmacophores in computational drug discovery
Computer aided drug design (CADD) approaches have been assisting drug discovery efforts since the 1980s, with such notable early successes as the rational design of inhibitors for HIV protease (e.g. Saquinavir) [12], carbonic anhydrase II (Dorzolamide) [13], neuraminidase (Tamiflu) [14], or for thymidylate synthase (Ralitrexed)[15], among many others, for the treatment of various infectious diseases and cancer. CADD approaches include a variety of computational and bioinformatics methods, and can be broadly grouped into Structure Based Drug Discovery (SBDD) and Ligand Based Drug Discovery (LBDD) branches [16]. One of the most common practices in SBBD is to screen a large number of small molecule compounds by docking them onto a target receptor structure (a protein or RNA) and evaluating their binding affinities[17,18]. A main limitation of these approaches is the difficulty in accurately predicting binding affinities[19,20], due to the limitations of current scoring functions and the inability to properly account for the flexibility of structures.
On the ligand side, some of the most prominent LBDD approaches include pharmacophore modeling [21]. A pharmacophore is an abstract description of key atoms, groups, charged regions, and their spatial interrelations that are essential for the biological activities of a drug molecule [23]. The pharmacophore describes a theoretically optimal ligand interface against which small molecule drug candidates can be screened. Once a set of low affinity known compounds (ligands) is identified, a spatial description of preferred properties (electrostatic, stereochemical, etc.), i.e., a “pharmacophore model,” can be obtained. Subsequently, one can search and identify, or engineer compounds that match the requirements of the pharmacophore model to achieve high receptor binding affinity. These approaches are very widely used in drug development with many notable successes [22]. Major challenges in this application are the inclusion of protein and compound flexibility in the pharmacophore calculation, the selection of an appropriate training set of compounds and the proper matching of the pharmacophore model with candidate compounds when applied for high throughput screening.
Pharmacophore modeling, or a related concept, the molecular interaction field (MIF) [23], can be used within SBDD applications as well, where the receptor structure is used as a starting point and a complementary pharmacophore interface is calculated. A molecular interaction field is a grid-based map reflecting the interaction energetics between a receptor and various generic moieties (e.g. water, methyl group, amine nitrogen, carboxyl oxygen, and hydroxyl). There are several approaches that use known receptor-ligand complexes as starting points to identify lead interacting compounds, such as Ligandscout[24] and Pocket [25].
Protein-based drugs
The field of recombinant therapeutic agents started with insulin in 1981 [26,27], and a few years later, the first monoclonal antibody was approved by the FDA in 1986 (Muromonab-CD3) for preventing kidney transplant rejection. Recombinant therapeutic agent discovery has seen a surge of drug development successes. As of today 178 such drugs have been approved [28]. Understanding protein-protein interactions can directly lead to drug development. For instance, the cell surface receptor-ligand interactions in the immune synapse (where the interacting cells are antigen-presenting cells and lymphocytes) provide co-stimulatory signals (inhibitory or stimulatory) that regulate the type, length and intensity of the immune response. Providing the wild type protein ligand, typically the soluble ectodomain of it, as a drug can shift the equilibrium of binding of the endogenous receptors and their ligands. One such example is abatacept [29], where the Fc region of the immunoglobulin IgG1 is fused to the extracellular domain of CTLA-4, a T-cell receptor. Abatacept will bind its cognate B7–1 and B7–2 ligands expressed on antigen-presenting cell and prevent these from binding to the CD28 T-cell receptor. CD28:B7–1/B7–2 interactions normally send stimulatory signals to the T-cell, but in the presence of therapeutic CTLA-4 (abatacept) this signal will be reduced. Hence, abatacept is successfully used in various autoimmune diseases.
While abatacept is essentially the wild type protein and in developing this drug the challenge was to understand the signaling network of CTLA-4 protein interactions, wild type protein interfaces can be used as a starting point in computational or experimental protein engineering experiments to induce selectivity or to increase affinity of binding. Belatacept is an engineered version of abatacept [30], with similar conceptual benefits of down regulating immune response, but primarily used to prevent rejection of kidney transplants. Belatacept differs by two mutations only in its protein interface compared to CTLA-4, which made it into a higher affinity binder to B7–1/B7–2 ligands.
Finally, these immune synapse interactions can also be manipulated by directly blocking protein interfaces with either small molecule drugs or monoclonal antibodies. One such drug is ipilimumab, a mAb that targets CTLA4 to stimulate the immune response against cancer cells [31].
Protein based therapeutics design usually follow the general concepts of designing proteins or peptides. These have two main branches; one is a challenging approach that requires the de novo prediction of a suitable backbone/scaffold protein [32,33]; alternatively, if a suitable backbone of a known template ligand is available, to predict a suitable amino acid sequence [34–36] with the desired binding specificity. The techniques involving these approaches have been reviewed elsewhere [37].
In the rapidly growing field of SBDD of protein-based drugs, pharmacophore approaches can play a critical role. These can include identifying cognate proteins from the target proteome, such as in the case of abatacept, or modifying existing protein interfaces for selectivity or affinity, as is case of belatacept or in mAb designing tasks [38]. For these protein-based SBDD applications, one has to modify the pharmacophore approaches developed for small molecules so that they can be applied to small peptides or full proteins.
Pharmacophore approaches utilizing small molecule sampling to study protein interfaces
The tendency of small organic molecules, both substrates and non-substrates, irrespective of their relevance to the target, to bind to similar, energetically favored sticky sites on a protein was reported in the 1980s. This has been shown by experimental studies when target proteins were soaked in organic solvents and their experimental crystal[39] or NMR[40] structure were subsequently examined. Small molecules were sticking to invariable, energetically favorable sites. Later on, computational methods emerged either illustrating or utilizing this phenomena, most notably, GRID[41] and the Multicopy Simultaneous Search (MCSS)[42], and some of the most competitive functional site search methods available today are also based on this concept [43,44].
GBPM stands for GRID-based pharmacophore model and is designed to identify protein interacting regions in three dimensional protein complexes [45], and serve as a general tool to study any kind of biomolecule. The GBPM approach is based on the re-parameterized GRID method [46], and proceeds by exploring three types of probes, a hydrophophic probe, and a hydrogen bond donor and acceptor probe on the interface of the interacting macromolecules. GBPM was used with interleukin-8 to successfully recognize itself as a potential ligand forming a homodimeric structure.
A newer development is GRAIL (GRids of phArmacophore InteractionfieLds) [47]. In contrast to GBPM it implements a pharmacophoric representation of the interface with a particular probe type, such as hydrogen-bond donor, hydrogen-bond acceptor, aromatic, positive or negative ionizable group, or hydrophobic group. GRAIL scores do not represent physical interaction energies but reflect how well geometric constraints (i.e., allowed distance and angle ranges) are met when the probe feature is placed at a grid point and how well the probe feature interacts with complementary features within the protein environment. It has the advantage that it is faster than grid-based calculations on atomistic levels. GRAIL implements dynamic information obtained by MD simulation of the limited probe repertoire, and its utility was demonstrated through the example of correctly ranking of various small molecule inhibitors of the oncology target Hsp90.
Extending the pharmacophore concept to protein like drugs
Here we discuss new developments where the receptor structure and its known or predicted interface are used to obtain a pharmacophoric description of protein binders using small molecule sampling approaches. Compared to grid based approaches, this development allows to account for structural flexibility both in the protein and in the ligand probe. Even in case of pharmacophoric approaches that target small molecules and not proteins, it has been shown recently that flexibility of protein structure can be taken into account through snapshots from molecular dynamic simulation. Methods that incorporate such approaches generate more accurate pharmacophores than those that explore a single crystal structure [48–50].
The SILCS approach (Site Identification by Ligand Competitive Saturation) is implementing the idea to explore small molecule binders[16]. SILCS aims at identifying small compounds with moderate affinity that can be further evolved or combined with crosslinks to produce larger, high affinity molecules. SILCS computationally solvates a target receptor protein in an explicit aqueous solution with a variety of possible small molecule compounds. Snapshots of a subsequent MD simulation are used to obtain a probability map (FragMAps) of fragment binding preferences. The advantage of the approach, in comparison to docking based screening methods, is that conformational changes, flexibility and solvent effects all can be taken into account. A challenge is to avoid aggregation of compounds; to that end, a minimalist repertoire of compounds is used to represent only four different interaction types, propane (hydrophobic aliphatic), benzene (aromatic) and water (hydrogen bond donor and acceptor). SILCS successfully recapitulated the binding preferences in the BCL-6:SMRT and BCL-6:BCOR protein-peptide complexes, despite some conformational changes induced in BCL6 after binding to either of the two ligands.
A conceptually similar approach was introduced in 2016, PROtein Lgand Interface Design (PROTLID)[51] (Fig. 1.). In contrast to small-molecule drug discovery methods and to SILCS, which use generic moieties and require a choice of possible probes, ProtLID uses the 20 natural amino acids as probes as it focuses on protein ligands only. This approach both limits the potential number of matches and reduces the combinatorial search space while providing an exhaustive exploration of protein like features. In PROTLID, 26 Functional Atom (FA) types are monitored for the 20 naturally occurring amino acid types, including, hydrogen-bonding capable side chain oxygen/nitrogen, and hydrophobic / aromatic centers. The optimal FA positions on the receptor interface are determined through exhaustive sampling of small, single-residue ligand probes using molecular dynamics (MD) in individual simulations. The resulting FA preferences constitute a unique spatial fingerprint, called the residue-specific (rs) pharmacophore. This designed rs-pharmacophore is then used in various subsequent applications.
Figure 1.
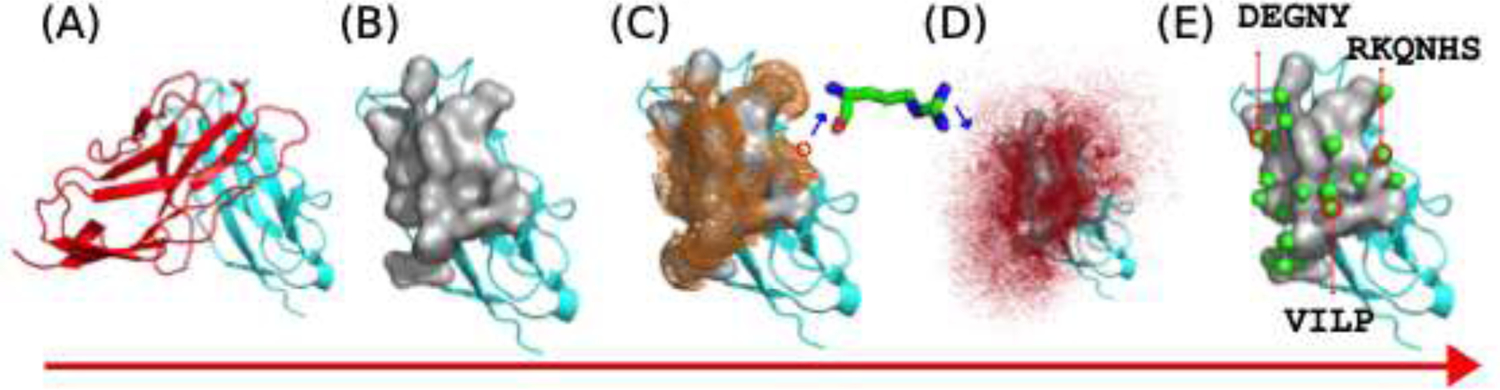
Reside specific pharmacophore generation using ProtLID. (A) identifying the protein binding interface. (B) Surface representation of the interface. (C) Mapping hypothetical starting points or reside probe sampling. (D) Residue binding preferences obtained from MD simulations (E) Predicted pharmacophore (complementary reside binding pattern, with locations (green spheres) and residue types (listed).
ProtLID has several conceptual advantages compared to other methods. First, the sampling-derived FA preferences are correlated to free energy, as opposed to empirical scores that are based on a single static snapshot, as in the case of most MIF- and pharmacophore-based methods. This is because FA preferences are quantified by the frequencies with which the FAs sample each region during all-atom molecular dynamics simulation, which are reflective of the Boltzmann distributions of thermally accessible receptor surface positions. Second, through the use of MD simulation, in addition to solvent effects, the flexibility of both the probes and the receptor are considered. The advantages of these features over both small-molecule and protein-protein docking methods were shown in practical applications when alternative structures of the same receptor were able to consistently locate the cognate ligands [51].
Applications of residue-based pharmacophores
Identifying cognate ligand partners
Once a rs-pharmacophore is derived for a protein interface, it can be used for a variety of subsequent applications. One possible application is to identify possible cognate ligand partners of a receptor from a collection of candidate ligands (a target sub-proteome) (Fig. 2.). For instance, in the immune synapse, about 500 different types of cell surface immunoglobulin receptors have been identified in the human genome [52], but only a few dozen known receptor-ligand complexes are established from the approximately 100,000 possible combinations. Meanwhile, many of these receptor-ligand interactions are implicated in managing the immune response or playing a role in neural development. An exhaustive exploration and prioritization of likely receptor ligand cognate pairs can greatly facilitate the process of identifying interacting protein pairs in this important sub-proteome[51].
Figure 2.
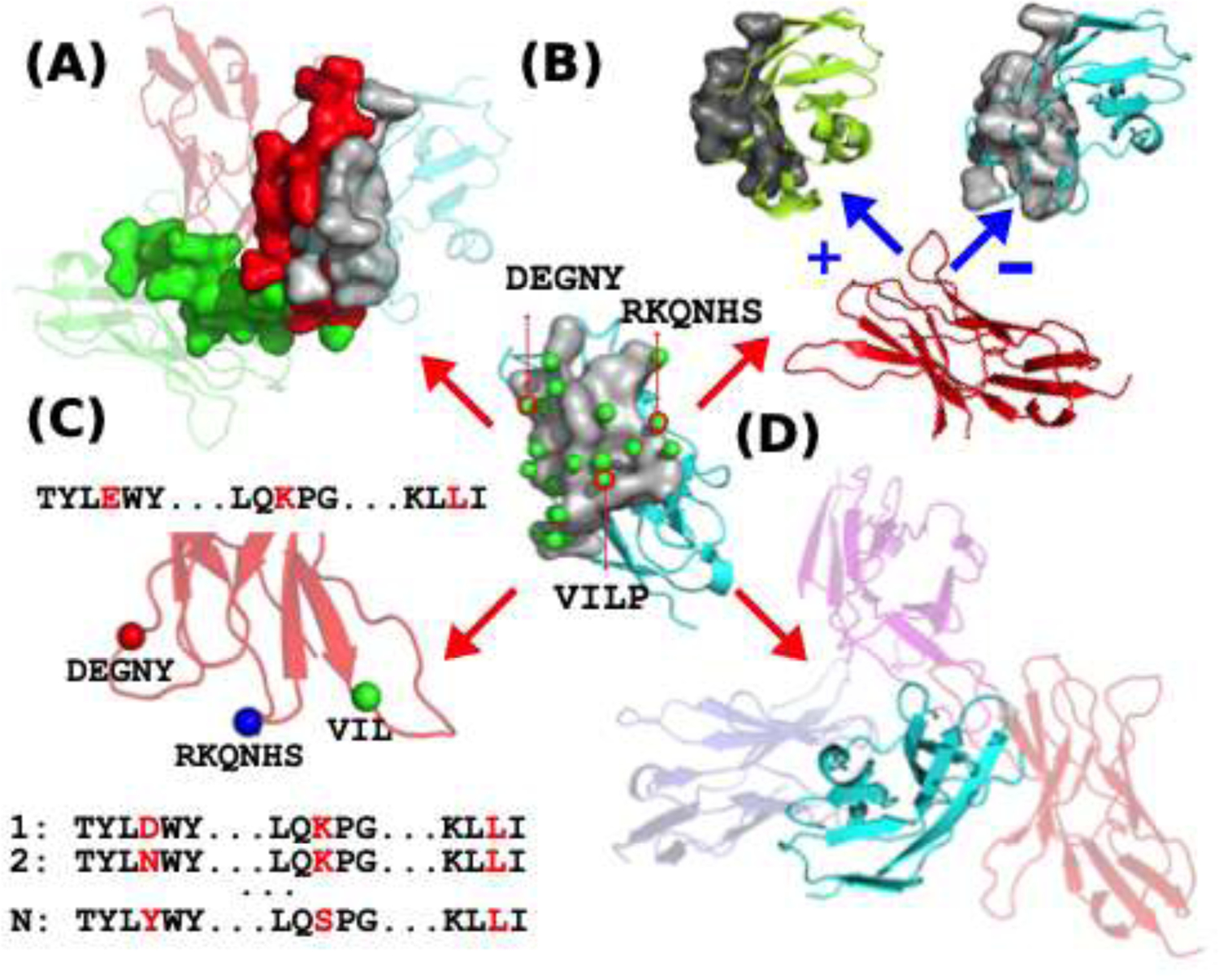
Possible uses of ProtLID in protein interface modeling. (A) Identifying cognate ligands for a receptor. Receptor (in cyan) interface is in grey. ProtLID predicts a residue based pharmacophore that fits (red interface) the best the cognate ligand among many candidates (such as green). (B) Redesigning protein interface for selectivity. Plus and minus signs refer to increased and decreased binding, as a function of better or poorer fit to the calculated pharmacophore. (C) Library design for phage display studies. Rs-pharmacophore directs the library construction (position and allowed mutations) for subsequent phage display studies (D) Docking two proteins. Randomly exploring possible binding interfaces for a cognate ligand.
Designing ligand specific interfaces
Another important application of rs-pharmacophores is to introduce or design selectivity for known protein interfaces. In signaling pathways, the same receptor frequently has multiple ligands that induce orthogonal signals, or can induce tissue specific responses depending on the expression levels of ligands in different cell types. These protein-protein interactions are usually transient in nature, with lower binding affinity, and as such, particularly suitable for engineering purposes [53,54]. Once a rs-pharmacophore is designed, it can be perceived as the optimal, high affinity complementary interface for a given target (Fig. 2). As such, it can be used as a template to guide the introduction of mutations on the ligand side that will either increase or decrease their binding affinity (i.e. the ligand interface will become more or less similar to the rs-pharmacophore). The promise of this approach has been already leveraged in two successful interface designs recently employing ProtLID method. In one application the biomedically critical PD1:PD-L1/PD-L2 receptor-ligand interactions were explored[55]. Chronic or persistent stimulation of the Programmed Cell Death-1 (PD-1) pathway prevents T cells from mounting anti-tumor and anti-viral immune responses. Blockade of this inhibitory checkpoint pathway has shown therapeutic importance by rescuing T cells from their exhausted state. Cognate ligands of the PD-1 receptor include the tissue-specific PD-L1 and PD-L2 proteins. ProtLID was utilized to custom-design a human PD-1 interface specific to human PD-L1 without any significant affinity to PD-L2. In subsequent cell assay experiments utilizing flow cytometry measurements, half of the single point mutant designs proved to introduce a statistically significant selectivity, with 9 of these maintaining a close-to wild type affinity to PD-L1 while virtually eliminating any detectable binding to PD-L2 [55].
In a subsequent application, the specificity of Herpes virus entry mediator (HVEM), a TNF receptor, was redesigned using rs-pharmacophore calculations with ProtLID[56]. HVEM regulates co-stimulation and co-inhibition signals for T-cell activation through co-signaling pathways. HVEM creates a unique cascade of molecular interactions in which it interacts with three ligands from two different superfamilies (TNF and IgSF) using two different binding interfaces, and generates functionally distinct bidirectional signaling pathways for controlling both inflammatory and inhibitory responses. The engagement of HVEM with ligands CD160 and BTLA, members of immunoglobulin superfamily, is associated with inhibitory signals, whereas inflammatory responses are regulated through its interaction with LIGHT from the TNF superfamily. The computationally redesigned HVEM recognition interface achieved a switchable binding specificity. In subsequent cell-based binding assays, the six types of new interfaces, designed with only single or double mutations, exhibited selective binding to only one or two out of the three cognate ligands[56].
Conclusion and future directions
A number of additional possible applications of rs-pharmacophore approaches can be envisioned. One interesting direction is to combine computational and experimental protein engineering techniques. For instance, in mAb design, the breadth and potency of a specific mAb can be enhanced using designed mutations[57]. Phage display discovery is often used for this purpose to randomly explore up to 1010 mutations simultaneously. However, a typical antibody-antigen interface with 15–20 residues presents 2015–2020 combinations of possible mutational variants, which translates into ~1019–1026 combinations, of which only a fraction can be sampled experimentally. As a result, phage display and other antibody combinatorial methods are reliant on restriction of either amino acid diversity or interfacial positions, which limits the effectiveness of the approach. Computational protein based pharmacophore methods can circumvent this limitation. A computationally designed residue-specific pharmacophore description of the antibody-antigen interface, or any scaffold or bioactive peptide, can be used to direct the library construction in subsequent phage display experiments, providing a restricted number of residue preferences in each position that undergoes combinatorial mutational studies (Fig. 2).
In other possible applications, a protein wide sampling of pharmacophore descriptions and a search for suitable binding ligands could identify both binding sites and cognate ligands, as an alternative to docking approaches.
To take advantage of protein based pharmacophore approaches it will be necessary to invest in the exploration of probe representation. For instance, instead of single residues one can employ di- or tripeptide sampling. In addition the definition of functionally important chemical groups can also affect the quality of the pharmacophore. Additional limitations of this approach are connected to the time consuming sampling step. This can be solved either by implementing advanced MD sampling techniques, such as replica exchange method [58], and/or taking advantage of the highly parallelizable nature of these jobs and employ clusters of computers.
Highlights.
-Structures are available for most known proteins
-Protein-based drugs are discovered at high rates
-Structure based pharmacophore approaches can be implemented for protein ligands
-MD snapshots based pharmacophores can consider structural flexibility and solvent effect
Acknowledgments
This review was partially based on our previous publications [51,55,56]. This work was supported by National Institutes of Health (NIH) grants GM118709, GM136357 and AI141816.
Funding
No funding was received for this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
No conflict of interest exists.
Intellectual Property
We confirm that we have given due consideration to the protection of intellectual property associated with this work and that there are no impediments to publication, including the timing of publication, with respect to intellectual property. In so doing we confirm that we have followed the regulations of our institutions concerning intellectual property.
Research Ethics
We further confirm that any aspect of the work covered in this manuscript that has involved human patients has been conducted with the ethical approval of all relevant bodies and that such approvals are acknowledged within the manuscript.
IRB approval was obtained (required for studies and series of 3 or more cases)
Written consent to publish potentially identifying information, such as details or the case and photographs, was obtained from the patient(s) or their legal guardian(s).
Contact with the Editorial Office
This author submitted this manuscript using his/her account in Editorial Manager (EM).
We understand that this Corresponding Author is the sole contact for the Editorial process (including EM and direct communications with the office). He/she is responsible for communicating with the other authors about progress, submissions of revisions and final approval of proofs.
We confirm that the email address shown below is accessible by the Corresponding Author, is the address to which Corresponding Author’s EM account is linked, and has been configured to accept email from the editorial office of Current Opinion in Structural Biology:
Conflict of interest statement
Nothing declared.
References
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Stumpf MP, Thorne T, de Silva E, Stewart R, An HJ, Lappe M, Wiuf C: Estimating the size of the human interactome. Proc Natl Acad Sci U S A 2008, 105:6959–6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Villoutreix BO, Bastard K, Sperandio O, Fahraeus R, Poyet JL, Calvo F, Deprez B, Miteva MA: In silico-in vitro screening of protein-protein interactions: towards the next generation of therapeutics. Curr Pharm Biotechnol 2008, 9:103–122. [DOI] [PubMed] [Google Scholar]
- 3.Bork P, Jensen LJ, von Mering C, Ramani AK, Lee I, Marcotte EM: Protein interaction networks from yeast to human. Curr Opin Struct Biol 2004, 14:292–299. [DOI] [PubMed] [Google Scholar]
- 4.Zheng H, Handing KB, Zimmerman MD, Shabalin IG, Almo SC, Minor W: X-ray crystallography over the past decade for novel drug discovery - where are we heading next? Expert Opin Drug Discov 2015, 10:975–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quinn CM, Wang M, Polenova T: NMR of Macromolecular Assemblies and Machines at 1 GHz and Beyond: New Transformative Opportunities for Molecular Structural Biology. Methods Mol Biol 2018, 1688:1–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuhlbrandt W: Biochemistry. The resolution revolution. Science 2014, 343:1443–1444. [DOI] [PubMed] [Google Scholar]
- 7.Khafizov K, Madrid-Aliste C, Almo SC, Fiser A: Trends in structural coverage of the protein universe and the impact of the Protein Structure Initiative (vol 111, pg 3733, 2014). Proceedings of the National Academy of Sciences of the United States of America 2014, 111:5060–5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grabowski M, Niedzialkowska E, Zimmerman MD, Minor W: The impact of structural genomics: the first quindecennial. J Struct Funct Genomics 2016, 17:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Croll TI, Sammito MD, Kryshtafovych A, Read RJ: Evaluation of template-based modeling in CASP13. Proteins 2019, 87:1113–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fiser A: Protein structure modeling in the proteomics era. Expert Rev Proteomics 2004, 1:97–110. [DOI] [PubMed] [Google Scholar]
- 11.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE: The Protein Data Bank. Nucleic Acids Res 2000, 28:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roberts NA, Martin JA, Kinchington D, Broadhurst AV, Craig JC, Duncan IB, Galpin SA, Handa BK, Kay J, Krohn A, et al. : Rational design of peptide-based HIV proteinase inhibitors. Science 1990, 248:358–361. [DOI] [PubMed] [Google Scholar]
- 13.Baldwin JJ, Ponticello GS, Anderson PS, Christy ME, Murcko MA, Randall WC, Schwam H, Sugrue MF, Springer JP, Gautheron P, et al. : Thienothiopyran-2-sulfonamides: novel topically active carbonic anhydrase inhibitors for the treatment of glaucoma. J Med Chem 1989, 32:2510–2513. [DOI] [PubMed] [Google Scholar]
- 14.von Itzstein M, Wu WY, Kok GB, Pegg MS, Dyason JC, Jin B, Van Phan T, Smythe ML, White HF, Oliver SW, et al. : Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 1993, 363:418–423. [DOI] [PubMed] [Google Scholar]
- 15.Rutenber EE, Stroud RM: Binding of the anticancer drug ZD1694 to E. coli thymidylate synthase: assessing specificity and affinity. Structure 1996, 4:1317–1324. [DOI] [PubMed] [Google Scholar]
- *16. Yu W, MacKerell AD Jr.: Computer-Aided Drug Design Methods. Methods Mol Biol 2017, 1520:85–106. A review of the CADD approaches, with special emphasis on molecular dynamic based approaches. A detailed description of the Author’s own SILC Smethod that uses MD for probe sampling and calculating phramacophoric description of the protein interface.
- 17.Fradera X, Mestres J: Guided docking approaches to structure-based design and screening. Current Topics in Medicinal Chemistry 2004, 4:687–700. [DOI] [PubMed] [Google Scholar]
- 18.Smith GR, Sternberg MJ: Prediction of protein-protein interactions by docking methods. Curr.Opin.Struct.Biol 2002, 12:28. [DOI] [PubMed] [Google Scholar]
- 19.de Vries SJ, van Dijk ADJ, Bonvin AMJJ: WHISCY: What information does surface conservation yield? Application to data-driven docking. Proteins-Structure Function and Bioinformatics 2006, 63:479–489. [DOI] [PubMed] [Google Scholar]
- 20.Fleishman SJ, Whitehead TA, Strauch EM, Corn JE, Qin S, Zhou HX, Mitchell JC, Demerdash ON, Takeda-Shitaka M, Terashi G, et al. : Community-wide assessment of protein-interface modeling suggests improvements to design methodology. J Mol Biol 2011, 414:289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang SY: Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov Today 2010, 15:444–450. [DOI] [PubMed] [Google Scholar]
- 22.Mustata G, Follis AV, Hammoudeh DI, Metallo SJ, Wang H, Prochownik EV, Lazo JS, Bahar I: Discovery of novel Myc-Max heterodimer disruptors with a three-dimensional pharmacophore model. J Med Chem 2009, 52:1247–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodford PJ: A Computational-Procedure for Determining Energetically Favorable Binding-Sites on Biologically Important Macromolecules. Journal of Medicinal Chemistry 1985, 28:849–857. [DOI] [PubMed] [Google Scholar]
- 24.Wolber G, Langer T: LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Inf Model 2005, 45:160–169. [DOI] [PubMed] [Google Scholar]
- 25.Chen J, Lai L: Pocket v.2: further developments on receptor-based pharmacophore modeling. J Chem Inf Model 2006, 46:2684–2691. [DOI] [PubMed] [Google Scholar]
- 26.Chance RE, Kroeff EP, Hoffmann JA, Frank BH: Chemical, physical, and biologic properties of biosynthetic human insulin. Diabetes Care 1981, 4:147–154. [DOI] [PubMed] [Google Scholar]
- 27.Goeddel DV, Kleid DG, Bolivar F, Heyneker HL, Yansura DG, Crea R, Hirose T, Kraszewski A, Itakura K, Riggs AD: Expression in Escherichia coli of chemically synthesized genes for human insulin. Proc Natl Acad Sci U S A 1979, 76:106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *28. Usmani SS, Bedi G, Samuel JS, Singh S, Kalra S, Kumar P, Ahuja AA, Sharma M, Gautam A, Raghava GPS: THPdb: Database of FDA-approved peptide and protein therapeutics. PLoS One 2017, 12:e0181748. A manually curated database of FDA approved protein and peptide based drugs. The database comes with a user friendly search interface and has been annotated for sequences, chemical properties, composition, disease area, mode of activity, physical appearance, category or pharmacological class, pharmacodynamics, route of administration, toxicity, target of activity, etc.
- 29.Moreland L, Bate G, Kirkpatrick P: Abatacept. Nat Rev Drug Discov 2006, 5:185–186. [DOI] [PubMed] [Google Scholar]
- 30.Larsen CP, Pearson TC, Adams AB, Tso P, Shirasugi N, Strobert E, Anderson D, Cowan S, Price K, Naemura J, et al. : Rational development of LEA29Y (belatacept), a high-affinity variant of CTLA4-Ig with potent immunosuppressive properties. Am J Transplant 2005, 5:443–453. [DOI] [PubMed] [Google Scholar]
- 31.Wolchok JD, Hodi FS, Weber JS, Allison JP, Urba WJ, Robert C, O’Day SJ, Hoos A, Humphrey R, Berman DM, et al. : Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann N Y Acad Sci 2013, 1291:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koga N, Tatsumi-Koga R, Liu GH, Xiao R, Acton TB, Montelione GT, Baker D: Principles for designing ideal protein structures. Nature 2012, 491:222–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D: Design of a novel globular protein fold with atomic-level accuracy. Science 2003, 302:1364. [DOI] [PubMed] [Google Scholar]
- 34.Rothlisberger D, Khersonsky O, Wollacott AM, Jiang L, DeChancie J, Betker J, Gallaher JL, Althoff EA, Zanghellini A, Dym O, et al. : Kemp elimination catalysts by computational enzyme design. Nature 2008, 453:190–195. [DOI] [PubMed] [Google Scholar]
- 35.Jiang L, Althoff EA, Clemente FR, Doyle L, Rothlisberger D, Zanghellini A, Gallaher JL, Betker JL, Tanaka F, Barbas CF 3rd, et al. : De novo computational design of retro-aldol enzymes. Science 2008, 319:1387–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stiel AC, Feldmeier K, Hocker B: Identification of protein scaffolds for enzyme design using scaffold selection. Methods Mol Biol 2014, 1216:183–196. [DOI] [PubMed] [Google Scholar]
- *37. Roy A, Nair S, Sen N, Soni N, Madhusudhan MS: In silico methods for design of biological therapeutics. Methods 2017, 131:33–65. A detailed review of structure based drug design approaches as applied to proteins, peptides and nucleic acids. The review mainly focuses on conceptual approaches that focus on protein design based ideas. The review also includes overview of methods that predict the binding affinity, cell penetration ability, half-life, solubility, immunogenicity and toxicity of the designed therapeutics.
- 38.Shirai H, Prades C, Vita R, Marcatili P, Popovic B, Xu J, Overington JP, Hirayama K, Soga S, Tsunoyama K, et al. : Antibody informatics for drug discovery. Biochim Biophys Acta 2014, 1844:2002–2015. [DOI] [PubMed] [Google Scholar]
- 39.Ringe D: What makes a binding site a binding site? Curr Opin Struct Biol 1995, 5:825–829. [DOI] [PubMed] [Google Scholar]
- 40.Hajduk PJ, Huth JR, Fesik SW: Druggability indices for protein targets derived from NMR-based screening data. J Med Chem 2005, 48:2518–2525. [DOI] [PubMed] [Google Scholar]
- 41.Goodford PJ: A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J Med Chem 1985, 28:849–857. [DOI] [PubMed] [Google Scholar]
- 42.Miranker A, Karplus M: Functionality maps of binding sites: a multiple copy simultaneous search method. Proteins 1991, 11:29–34. [DOI] [PubMed] [Google Scholar]
- 43.Hall DR, Kozakov D, Vajda S: Analysis of protein binding sites by computational solvent mapping. Methods Mol Biol 2012, 819:13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Viswanathan R, Fajardo E, Steinberg G, Haller M, Fiser A: Protein-protein binding supersites. PLoS Comput Biol 2019, 15:e1006704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ortuso F, Langer T, Alcaro S: GBPM: GRID-based pharmacophore model: concept and application studies to protein-protein recognition. Bioinformatics 2006, 22:1449–1455. [DOI] [PubMed] [Google Scholar]
- 46.Carosati E, Sciabola S, Cruciani G: Hydrogen bonding interactions of covalently bonded fluorine atoms: from crystallographic data to a new angular function in the GRID force field. J Med Chem 2004, 47:5114–5125. [DOI] [PubMed] [Google Scholar]
- **47. Schuetz DA, Seidel T, Garon A, Martini R, Korbel M, Ecker GF, Langer T: GRAIL: GRids of phArmacophore Interaction fieLds. J Chem Theory Comput 2018, 14:4958–4970. Grail method is introduced that combines traditional grid-based approaches with the pharmacophore concept. The approach accounts for protein flexibility through the analysis of large amounts of coordinate sets obtained by long-time MD simulations. The method was applied to identify inhibitors for the heat shock protein 90.
- 48.Choudhury C, Priyakumar UD, Sastry GN: Dynamics based pharmacophore models for screening potential inhibitors of mycobacterial cyclopropane synthase. J Chem Inf Model 2015, 55:848–860. [DOI] [PubMed] [Google Scholar]
- *49. Spyrakis F, Benedetti P, Decherchi S, Rocchia W, Cavalli A, Alcaro S, Ortuso F, Baroni M, Cruciani G: A Pipeline To Enhance Ligand Virtual Screening: Integrating Molecular Dynamics and Fingerprints for Ligand and Proteins. J Chem Inf Model 2015, 55:2256–2274. Flexibility of protein structure is taken into account when calculating a pharmacophore decription using MD simulation snapshots. In a retrospective analysis this approach has been shown to outperform methods that rely on rigid structure derived phamacophore.
- 50.Wieder M, Garon A, Perricone U, Boresch S, Seidel T, Almerico AM, Langer T: Common Hits Approach: Combining Pharmacophore Modeling and Molecular Dynamics Simulations. J Chem Inf Model 2017, 57:365–385. [DOI] [PubMed] [Google Scholar]
- **51. Yap EH, Fiser A: ProtLID, a Residue-Based Pharmacophore Approach to Identify Cognate Protein Ligands in the Immunoglobulin Superfamily. Structure 2016, 24:2217–2226. The detailed introduction of ProtLID method and its application to idenetify cogante ligand partners from a target subproteome.
- 52.Gil N, Fajardo EJ, Fiser A: Discovery of receptor-ligand interfaces in the immunoglobulin superfamily. Proteins 2020, 88:135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rubinstein R, Ramagopal UA, Nathenson SG, Almo SC, Fiser A: Functional Classification of Immune Regulatory Proteins. Structure 2013, 21:766–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chattopadhyay K, Lazar-Molnar E, Yan Q, Rubinstein R, Zhan C, Vigdorovich V, Ramagopal UA, Bonanno J, Nathenson SG, Almo SC: Sequence, structure, function, immunity: structural genomics of costimulation. Immunol Rev 2009, 229:356–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *55. Shrestha R, Garrett SC, Almo SC, Fiser A: Computational Redesign of PD-1 Interface for PD-L1 Ligand Selectivity. Structure 2019, 27:829–836. The first, experimentally verified protein interface redesign application of the ProtLID approach. PD1 receptor interface was redesigned to selectively binding PD-L1 ligand without measurable affinity to PD-L2.
- *56. Shrestha R, Garrett-Thomson SC, Liu W, Almo SC, Fiser A: Redesigning HVEM Interface for Selective Binding to LIGHT, BTLA, and CD160. Structure 2020. ProtLID approach was used to successfully design a 6-way swith of the TNF receptor HVEM. The constructs were verified with cell based flow cytometry experiments.
- 57.Tharakaraman K, Robinson LN, Hatas A, Chen YL, Siyue L, Raguram S, Sasisekharan V, Wogan GN, Sasisekharan R: Redesign of a cross-reactive antibody to dengue virus with broad-spectrum activity and increased in vivo potency. Proc Natl Acad Sci U S A 2013, 110:E1555–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yoda T, Sugita Y, Okamoto Y: Protein folding simulations by generalized-ensemble algorithms. Adv Exp Med Biol 2014, 805:1–27. [DOI] [PubMed] [Google Scholar]